
Abstract
Flax is an important oilseed crop in North America and is mostly grown as a fibre crop in Europe. As a self-pollinated diploid with a small estimated genome size of ~370 Mb, flax is well suited for fast progress in genomics. In the last few years, important genetic resources have been developed for this crop. Here, we describe the assessment and comparative analyses of 1,506 putative simple sequence repeats (SSRs) of which, 1,164 were derived from BAC-end sequences (BESs) and 342 from expressed sequence tags (ESTs). The SSRs were assessed on a panel of 16 flax accessions with 673 (58 %) and 145 (42 %) primer pairs being polymorphic in the BESs and ESTs, respectively. With 818 novel polymorphic SSR primer pairs reported in this study, the repertoire of available SSRs in flax has more than doubled from the combined total of 508 of all previous reports. Among nucleotide motifs, trinucleotides were the most abundant irrespective of the class, but dinucleotides were the most polymorphic. SSR length was also positively correlated with polymorphism. Two dinucleotide (AT/TA and AG/GA) and two trinucleotide (AAT/ATA/TAA and GAA/AGA/AAG) motifs and their iterations, different from those reported in many other crops, accounted for more than half of all the SSRs and were also more polymorphic (63.4 %) than the rest of the markers (42.7 %). This improved resource promises to be useful in genetic, quantitative trait loci (QTL) and association mapping as well as for anchoring the physical/genetic map with the whole genome shotgun reference sequence of flax.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Flax (Linum usitatissimum L.) has been cultivated for several thousand years mainly for its seed oil and its high-quality stem fibres. In North America, flax is grown primarily as an oilseed crop used for food and feed as well as in bio-product applications such as linoleum flooring, paint and varnishes. Most oilseed flax varieties are rich in omega-3 (alpha linolenic acid, 55–57 %) fatty acid which has been functionally associated with numerous health claims. ‘Solin’ varieties with loss of function mutations in the fatty acid desaturase 3 (fad3) genes are low in omega-3 (2–3 %) and high in omega-6 (~70 %) fatty acids, characteristics required for margarine processing. Flax oil extraction generates a meal which is rich in protein and is sought after as animal feed.
Until recently, the stems of North American oilseed flax were considered undesirable because the persistence of straw in the field was problematic. However, in the last decade, the fibre industry has placed substantial effort into the development of high-value products from oilseed flax stems with applications in the pulp, technical fibre and bio-fuel industries. A shift from oilseed flax towards dual purpose or total utilization flax is currently occurring. To assist breeding efforts towards simultaneous improvements of seed and stem traits, breeders need a good grasp of the complexity of the genetic mechanisms underlying traits such as oil content, fatty acid composition, stem fibre content and fibre composition. Knowledge of the existing genetic diversity for these traits in primary and secondary gene pools is also essential to accelerate their introgression in breeding programs. Quantitative trait loci (QTL) and association mapping (AM) studies have the ability to provide some insights into genetic mechanisms of complex traits and provide molecular markers to implement marker-assisted breeding.
To date, only a limited number of useful markers have been developed in flax and, as a consequence, genetic maps and QTL studies remain limited (Cloutier et al. 2011; Oh et al. 2000; Spielmeyer et al. 1998). While some isozyme, RAPD and AFLP markers have been developed in flax (Everaert et al. 2001; Fu 2006; Krulickova et al. 2002; Spielmeyer et al. 1998), such marker systems are either labour-intensive or suffer from low reproducibility. Microsatellites or simple sequence repeats (SSRs) consist of tandemly repeated short motifs of 2–6 nucleotides. SSR markers are based on the amplification size polymorphism generated when lines have variable numbers of these short tandem repeats in a particular locus. The abundance, distribution, reproducibility and generally codominant nature of SSR markers make them highly suitable for linkage mapping and genetic diversity studies (Cloutier et al. 2009; Soto-Cerda et al. 2011a; Wiesner et al. 2001). SSR markers have been developed through SSR-enriched library screening and, more recently, through the more economical mining of EST or genomic sequence data. A total of 508 SSR markers have been reported as follows: 10 (Wiesner et al. 2001), 23 (Roose-Amsaleg et al. 2006), 35 (Deng et al. 2010), 60 (Soto-Cerda et al. 2011a), 38 (Deng et al. 2011), 9 (Kale et al. 2011), 20 (Rachinskaya et al. 2011), 42 (Bickel et al. 2011), 248 (Cloutier et al. 2009) and 23 (Soto-Cerda et al. 2011b). In addition, commercially available inter simple sequence repeat (ISSR) primers from the University of British Columbia (UBC) collection have been used, mostly in genetic diversity studies of L. usitatissimum L. or its wild progenitor L. bienne Mill. (Chen et al. 1998; Rajwade et al. 2010; Uysal et al. 2010; Wiesnerova and Wiesner 2004).
The ability to detect QTL using genetic maps of a segregating population or by linkage disequilibrium (LD) in AM studies depends on the marker saturation, the distribution and the accuracy of the phenotypic characterization of the traits. Single nucleotide polymorphism (SNP) markers promise to provide the high level of saturation (several thousands) that is paramount to QTL identification by AM in low LD regions of the genome. Developing and applying thousands of SSR markers would be comparatively costly due to the labour involved in their assessment. However, SSRs remain an excellent marker system for the construction of skeletal genetic maps onto which SNPs can be added (Allen et al. 2011). The aim of this project was to increase the number of publicly available SSR markers in flax to over 1,000, on par with other major crops. To realize that goal, we mined additional ESTs, sequenced more than 80,000 flax bacterial artificial chromosome (BAC) ends and mined them for the presence of SSRs. Polymorphism was assessed on a set of 16 flax genotypes and comparative analyses of EST-SSRs and gDNA-SSRs was performed.
Materials and methods
Plant materials
A set of 16 flax accessions were grown in a growth cabinet. The leaf and stem tissue of plantlets at the first branching stage were collected and DNA was extracted using a modified CTAB method (Cloutier et al. 2001). The DNA was quantified using a fluorometer and diluted to a 10 ng/μL working solution. The 16 genotypes represent oilseed types with different fatty acid profiles as well as fibre types (Table 1).
SSR design
A BAC library of cultivar CDC Bethune was constructed (Table 2). A total of 43,776 clones were sequenced from both ends by the BC Cancer Agency Genome Sciences Centre (Vancouver, Canada) using universal primers, Sanger’s dideoxy chain termination method with Big Dye V3.1 chemistry and resolved on an ABI 3730xl (Applied Biosystems, Foster City, USA). Trimmed high-quality BAC-end sequences (BESs) totalling ~56 Mb were mined with the Perl script MISA (Thiel et al. 2003) for the presence of putative SSRs using criteria of a minimum of nine repeats for dinucleotide, six repeats for trinucleotide and five repeats for tetra-, penta- and hexa-nucleotide motifs. Primers were designed from BES containing putative SSR motifs as previously described (Cloutier et al. 2009).
A total of 243,272 flax Expressed Sequence Tags (ESTs) from flax generated by the NAPGEN consortium, the TUFGEN project (Venglat et al. 2011) or publicly available in GenBank, were assembled into 34,156 unigenes comprising 14,374 contigs and 19,782 singletons using criteria previously described (Cloutier et al. 2009). A total of 33,163 unigenes of high quality and sufficient length were mined with the same criteria as described above for the BES. Putative SSRs previously detected and assessed from an original set of 146,611 assembled ESTs (Cloutier et al. 2009) were ignored and only the putative SSRs from novel contigs and/or singletons were retained for primer design performed using Primer3 (Rozen and Skaletsky 2000).
Polymorphism assessment
Each primer pair was assessed using DNA from the 16-genotype panel as previously described (Cloutier et al. 2009). Most amplicons were resolved using the GeneScan 500 ROX size standard (ABI) but amplicons larger than 450 bp were resolved using MapMarker 1000 (BioVentures Inc, Murfreesboro, USA) which gave improved sizing accuracy for larger fragments. Allele sizes were recorded for each genotype of the panel. Primer pairs were considered polymorphic if at least one of the 16 genotypes had a different allele size; monomorphic when all lines amplified the same size fragment; and failed when no consistent PCR product was observed after two additional PCR attempts at 58 and 49 °C, respectively. Primer pairs that amplified more than one polymorphic locus were scored independently. The polymorphic information content (PIC) value was estimated for each marker to determine their potential usefulness in determining the genetic variability of other Linum accessions (Botstein et al. 1980). To illustrate the genetic relationship of the 16 flax accessions, we constructed a dendrogram using the neighbour-joining method (Nei 1973) as implemented in PowerMarker (Liu and Muse 2005).
Results
A total of 1,660 BESs were identified to have at least one putative SSR, from which 1,164 primer pairs were designed. A total of 673 (57.8 %) primer pairs were polymorphic and detected 720 loci with 43 primer pairs detecting two polymorphic loci and two primer pairs detecting three polymorphic loci (Supplementary Table S1). The monomorphic BES-SSR markers totalled 478 (41.1 %) and only 13 (1.1 %) failed (did not work, DNW).
A total of 382 putative novel EST-SSRs were identified from the EST assembly and 342 primer pairs were designed. Roughly the same proportion of EST-SSR primer pairs was polymorphic (145; 42.4 %; Supplementary Table S2) and monomorphic (153; 44.7 %) while 44 failed (12.9 %). The 145 polymorphic EST-SSRs detected 149 loci.
The number of alleles detected at a single polymorphic locus ranged from 2 to 9 with an average of 2.76 in the BES-SSRs and ranged from 2 to 6 with an average of 2.36 in the EST-SSRs (Supplementary Tables S1 and S2). The mean polymorphism information content (PIC) value was 0.39 (0.12–0.85) and 0.34 (0.12–0.70) for the BES-SSRs and EST-SSRs, respectively. Frequency distribution of PIC values of SSR loci showed that nearly 25 % of the markers had PIC values greater than 0.5 (Supplementary Figure S1).
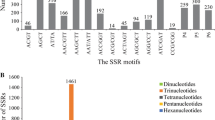
The total number of SSR markers and their associated polymorphism for each motif length is illustrated in Fig. 1. Regardless of the source of SSRs trinucleotide SSRs were the most abundant representing 54.6 and 68.7 % of the BES-SSRs and EST-SSRs, respectively. These trinucleotide motifs also displayed a higher proportion of monomorphic amplicons regardless of the source. Dinucleotide motifs were only 30.6 and 16.8 % of the two SSR categories but they represented 40.6 and 24.8 % of the total polymorphic SSRs. Compound SSRs represented sequences that had two SSR motifs within 100 bp. These motifs were generally different from one another and compound SSRs only represented 3.1 and 9.6 % of all BES-SSRs and EST-SSRs, respectively. In total, 77 and 63 % of the dinucleotide, 50 and 39 % of the trinucleotide and 39 and 43 % of the tetranucleotide motif SSRs were polymorphic in the BES and EST datasets, respectively.

Number of SSRs classified based on motif type, source (BES or EST) and polymorphism. DNW (did not work) represents primer pairs that failed to amplify reproducible amplicons
BES-SSRs tended to have a higher number of repeats per locus with 41.1 % having 9 or more repeats as compared to 26.9 % for EST-SSRs (Fig. 2a). SSRs with higher numbers of repeats tended to be more polymorphic. 41.1 % of the BES-SSRs with 9–25 repeats accounted for 56.1 % of the polymorphic BES-SSRs while 26.9 % of the EST-SSRs with nine or more repeats accounted for 38.3 % of the polymorphic EST-SSRs. SSR length is a measurement of the motif length and the number of repeats. Long SSRs (25–54 bp) represented a higher proportion of the total SSRs detected in BES (24.1 %) as compared to ESTs (15.9 %) and were more polymorphic than shorter SSR loci (Fig. 2b).

Number of SSRs and associated polymorphism based on a the number of repeats and b the SSR length (bp). SSRs are also classified as per their source (BES or EST). DNW (did not work) represents primer pairs that failed to amplify reproducible amplicons
Of the eight possible dinucleotide motifs, four, namely AT/AT, AG/CT, GA/TC and TA/TA, accounted for 99.4 and 98 % of the dinucleotide motifs from BES-SSRs and EST-SSRs, respectively (Fig. 3). The motifs AC/GT and GA/TC were represented by only four SSRs in total in both datasets and CG/CG and GC/GC were completely absent. Approximately twice as many GA/TC motifs were observed in EST-SSRs (28.8 %) as compared to BES-SSRs (15.6 %) while the opposite trend was true for the TA/TA motif. Of the 30 possible trinucleotide motifs, AAG/CTT, AGA/TCT and GAA/TTC were most frequent representing 36 and 38.7 % of the BES- and EST-SSRs, respectively (Fig. 3). Some motifs were represented by one or two SSRs only and motif ACG/CGT was not detected at all in either type of sequence. The proportion of SSRs originating from each motif was similar between the two datasets with a few exceptions such as AAT/ATT, TAA/TTA and TCA/TGA that were proportionately higher in BES-SSRs as compared to EST-SSRs. Frequency distributions for tetra-, penta- and hexa-nucleotide motif SSRs are more difficult to discern because they represent only a small proportion of the total SSRs detected. Nevertheless, there seem to be some biases, i.e., some motifs were present more frequently than expected by random distribution such as AAAG/CTTT and ATTA/TAAT which appeared ten and eight times in the BES-SSRs, respectively.

Percentage of dinucleotide and trinucleotide SSRs classified based on their source (BES or EST) and their motif
Polymorphism rates varied greatly among motifs. However, this type of data is only valid for high-frequency motifs because percent polymorphism associated with a rare motif does not provide an accurate estimate. Considering only motifs represented by at least 15 SSRs, a scatter plot of the total number of SSRs by motif against the percentage of polymorphic SSR per motif for 23 different motifs (including 4 dinucleotide and 19 trinucleotide motifs) showed that 2 dinucleotide (AT/TA and AG/GA) and 2 trinucleotide (AAT/ATA/TAA and GAA/AGA/AAG) motifs and their iterations contributed the majority (536) of the polymorphic SSRs (Fig. 4).

Diagram illustrating that two dinucleotide and two trinucleotide motifs and their respective iterations represent the majority of the SSRs in flax and are also the most polymorphic
Using all 869 SSR markers, a dendrogram of the 16 accessions was constructed (Supplementary Figure S2). Fibre and oilseed flax belonged to separate clades. Also, within the oilseed group, the low linolenic accessions clustered together.
Discussion
Here, we described the development and analysis of 818 novel polymorphic SSR primer pairs in flax detecting 869 loci, of which 145 primer pairs were derived from ESTs and 673 from BESs, more than doubling the combined previously available flax SSR collections of 508 (Table 3). With a total of 1,326 SSR markers now publicly available, flax compares favourably to other major crops (Varshney et al. 2006b, 2010). Taken together, these resources should prove valuable in genetic, QTL and association mapping, for anchoring the physical map and integration of the whole genome shotgun sequence assembly.
Traditionally, SSRs were developed from SSR-enriched libraries which represented a major bottleneck (Kalia et al. 2011). However, with technological advances in generating large-scale sequence data (EST, exome, genomic surveys, BES, whole genome sequence, etc.) and their availability in public domain, in silico approaches to the identification of putative SSRs have become practical (Tang et al. 2008). Here, we capitalized on the availability of ESTs (Cloutier et al. 2009; Venglat et al. 2011) and BESs (Ragupathy et al. 2011) to develop the largest collection of flax SSRs to date.
The number of SSRs assessed herein is sufficiently large to provide general conclusions regarding source (EST vs. genomic DNA), motif type, length, sequence and evolution of SSR loci. Overall, no major difference existed between EST-SSRs and BES-SSRs with the exception of the failure rate that was higher in EST-SSRs, probably as a result of poor primer binding due to their design over a splice site, mismatches caused by poor sequence quality or the presence of a large intron hindering amplification (Tang et al. 2008).
Trinucleotide SSRs were the most abundant in Arabidopsis (Mun et al. 2006; Tian et al. 2004), Medicago (Mun et al. 2006), soybean (Hisano et al. 2007; Mun et al. 2006; Tian et al. 2004), rice (Mun et al. 2006), pea (Gong et al. 2010), sugarcane (Cordeiro et al. 2001; Parida et al. 2009a), chickpea (Choudhary et al. 2009), wheat (Peng and Lapitan 2005; Yu et al. 2004), barley (Thiel et al. 2003; Varshney et al. 2006a), pepper (Yi et al. 2006), Lotus japonicus (Mun et al. 2006) and citrus (Chen et al. 2006). These estimates could be construed as biased because in the majority of the above studies, SSRs were mined from ESTs which are known to display a prevalence of trinucleotides (Cavagnaro et al. 2010; Li et al. 2004; Morgante et al. 2002; Tian et al. 2004). Of the 12 published plant genomes analyzed by Ragupathy et al. (2011), only Brachypodium had a higher percentage of trinucleotide SSRs. In flax, trinucleotide SSRs were also the most abundant but dinucleotide SSRs were the most polymorphic, as reported for several other crops (Blair et al. 2009; Cavagnaro et al. 2010; Hisano et al. 2007; Mun et al. 2006). The above SSR motif results are not always consistent and readily comparable because the thresholds of parameters used for identification of the SSR loci, especially the number of repeats per motif, are not uniform across species and even across research reports of the same species. Mononucleotide repeats, often not accounted for, were reported to be the most abundant in Brachypodium, rice, sorghum, Arabidopsis, Medicago and Populus (Cardle et al. 2000; Gupta and Prasad 2009; Mun et al. 2006; Sonah et al. 2011).
In flax, trinucleotides were more numerous in ESTs (68.7 %) compared to BESs (54.6 %) likely because of the suppression of non-trimeric SSRs in coding regions which could result in changes in reading frames (Kalia et al. 2011). Polymorphism was also positively correlated with the number of repeats per locus and the overall locus length. Among sources, BES-SSRs (58 %, average PIC 0.39) displayed a significantly higher level of polymorphism than EST-SSRs (42 %, average PIC 0.34) as previously reported (Eujayl et al. 2002; Kalia et al. 2011). Even with only ~40 % of EST-SSRs being polymorphic, flax has a higher polymorphism than wheat, barley, soybean and cotton (Eujayl et al. 2002; Han et al. 2004; Hisano et al. 2007; Thiel et al. 2003; Varshney et al. 2006a). However, the polymorphism level can vary significantly across studies because it is also a reflection of the number of lines surveyed in the panel and its genetic diversity. Here, we used 16 flax oilseed and fibre accessions providing a good genetic diversity of the breeding material but not necessarily of Linum usitatissimum because all accessions investigated were varieties or advanced breeding lines.
A distinct bias towards certain motifs was evident: two dinucleotide and two trinucleotide motifs and their iterations accounted for 65.5 % (536/818) of all polymorphic motifs (Fig. 4). Of the 1,506 target sequences from which primers were designed, these motifs represented more than half (845) and their polymorphism level greatly exceeded that of all the other motifs taken together (63.4 vs. 42.7 %). At the opposite end of the spectrum, motif ACG/CGT and its iterations (CGA/TCG and GAC/GTC) represented only 14 of the 1,506 SSRs (<1 %) with only four being polymorphic while dinucleotide CG/GC was not detected in either flax ESTs or BESs (Supplementary Table 2). The biases observed were both in relative abundance and polymorphism level. Motif abundance seems to be species specific due to factors such as genome content and composition, variation in rate of mutation across genome including rate of slippage and codon usage (Buschiazzo and Gemmell 2006; Sonah et al. 2011). The trinucleotide motif AGC/GCA/CAG was the most abundant trinucleotide motif in 8 crops and AGG/GGA/GAG in 4 while AG/GA was the most abundant dinucleotide in 14 crops and AC/CA in 4 (Yu et al. 2009). In flax, GAA/AAG/AGA with 320 and AT/TA with 228 were the most abundant trinucleotide and dinucleotide motifs, respectively, indicative of its unique SSR genome composition as compared to other crops.
Repeat numbers of EST-SSRs tended to be lower than gDNA-SSRs (Morgante et al. 2002). This was particularly true in flax where 5–7 repeat SSRs represented 64.7 % of all EST-SSRs but only 51.6 % of the BES-SSRs. These short SSRs were less polymorphic than the longer ones regardless of the source as previously reported (Wierdl et al. 1997; Ellegren 2004; Cavagnaro et al. 2010; Blair et al. 2009) and somewhat in disagreement with Tang et al. (2008). However, in this latter case, they considered SSRs with as few as 4 or 5 repeats even for dinucleotides which was not the case in this study because the SSR identification was performed using ESTs and BES from a single genotype (CDC Bethune) while Tang et al. (2008) used multiple genotypes. Surprisingly, these short SSRs were more polymorphic than long SSRs, possibly because in this case, they were derived exclusively from ESTs where long SSRs can be deleterious (Sureshkumar et al. 2009; Tang et al. 2008).
Predominant distribution of long alleles of SSR loci in genomic regions containing both coding [~26.8 % in flax, Ragupathy et al. (2011)] and non-coding sequences compared to EST-SSRs in this study could be associated with factors contributing to SSR origin and evolution per se. For instance, a dinucleotide motif arrayed 3 or 4 times in a locus may originate from cryptically simple sequences by both substitutions and indel mutations (Buschiazzo and Gemmell 2006). These substrate sequences further expand to an array of repeating units in a given locus, mainly through two mechanisms: slippage during DNA replication in a repeat domain (stabilized with an inefficient DNA mismatch repair-MMR system of the host) and unequal crossing over (Ellegren 2004). Studies of SSR evolution in the human genome suggested a mutational bias leading to an increase in SSR length at an individual locus (expansion) rather than a decrease in repeat number (contraction) on the evolutionary timescale (Ellegren 2004). Also, neutrality of mutations in the SSR loci present in non-coding regions of the genome favour an increase in repeat length, however, in coding sequences selective constraints against frame shift mutations weed out expansion or contraction of motifs except for triplets (Li et al. 2004). Although repeats containing proteins are well characterized (Faux et al. 2007), expansion of amino acid homopolymer domains beyond a threshold length impacts protein functionality (Kashi and King 2006). Also, conserved regulatory roles associated with some genic SSR sites favoured by selective forces curtail expansion in EST-SSRs despite the potential adaptive advantage (Li et al. 2004; Parida et al. 2009b).
The SSR markers described herein promise to be useful to characterize the genetic variability of other Linum accessions. The number of alleles and their relative frequencies are both indicators of a marker’s usefulness (Shete et al. 2000) and are taken into account in the PIC value estimates. More than 200 of the markers described have PIC values greater than 0.5 (Supplementary Figure S1) which should make them particularly useful in characterizing Linum collections as illustrated by the relationship observed in the dendrogram of the 16 accessions (Supplementary Figure S2). Indeed, accessions of similar lineage clustered together as predicted by the type (fibre vs. oilseed) and end-use quality (conventional vs. low or high linolenic acid content).
Conclusion
Here, we described the development of the largest collection of SSRs in flax to date bringing the overall number to over 1,300, comparable to many other major crops. A comprehensive comparative analysis of the composition and polymorphism of SSRs developed from ESTs and BES was performed showing some important differences between flax and other crops. The SSR resource described herein will be useful in genetic, QTL and association mapping. Map-based cloning, physical anchoring of the WGS reference genome and other downstream applications in breeding such as marker-assisted selection are likely to benefit from this important resource, paving the way for genetic improvement of flax.
References
Åkerman Å, Nilsson F, Sylvén N, Fröier K (1951) Svensk växtförädling. Del 1 Åkerbruksväxterna. (Plant Breeding in Sweden. I. Field Crops). Stockholm, Natur och kultur, p 745
Allen AM, Barker GLA, Berry ST, Coghill JA, Gwilliam R, Kirby S, Robinson P, Brenchley RC, D’Amore R, McKenzie N, Waite D, Hall A, Bevan M, Hall N, Edwards KJ (2011) Transcript-specific, single-nucleotide polymorphism discovery and linkage analysis in hexaploid bread wheat (Triticum aestivum L.). Plant Biotechnol J 9:1086–1099
Bickel CL, Gadani S, Lukacs M, Cullis CA (2011) SSR markers developed for genetic mapping in flax (Linum usitatissimum L.). Res Rep Biol 2011:23–29
Blair MW, Torres MM, Pedraza F, Giraldo MC, Buendia HF, Hurtado N (2009) Development of microsatellite markers for common bean (Phaseolus vulgaris L.) based on screening of non-enriched, small-insert genomic libraries. Genome 52:772–782
Botstein D, White RL, Skalnick MH, Davies RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphism. Am J Hum Genet 32:314–331
Buschiazzo E, Gemmell NJ (2006) The rise, fall and renaissance of microsatellites in eukaryotic genomes. BioEssays 28:1040–1050
Cardle L, Ramsay L, Milbourne D, Macaulay M, Marshall D, Waugh R (2000) Computational and experimental characterization of physically clustered simple sequence repeats in plants. Genetics 156:847–854
Cavagnaro PF, Senalik DA, Yang L, Simon PW, Harkins TT, Kodira CD, Huang S, Weng Y (2010) Genome-wide characterization of simple sequence repeats in cucumber (Cucumis sativus L.). BMC Genomics 11:569
Chen Y, Hausner G, Kenaschuk E, Procunier D, Dribnenki P, Penner G (1998) Identification of microspore-derived plants in anther culture of flax (Linum usitatissimum L.) using molecular markers. Plant Cell Rep 18:44–48
Chen CX, Zhou P, Choi YA, Huang S, Gmitter FG (2006) Mining and characterizing microsatellites from citrus ESTs. Theor Appl Genet 112:1248–1257
Choudhary S, Sethy NK, Shokeen B, Bhatia S (2009) Development of chickpea EST-SSR markers and analysis of allelic variation across related species. Theor Appl Genet 118:591–608
Cloutier S, Rampitsch C, Penner GA, Lukow OM (2001) Cloning and expression of a LMW-i glutenin gene. J Cereal Sci 33:143–154
Cloutier S, Niu Z, Datla R, Duguid S (2009) Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor Appl Genet 119:53–63
Cloutier S, Ragupathy R, Niu Z, Duguid S (2011) SSR-based linkage map of flax (Linum usitatissimum L.) and mapping of QTLs underlying fatty acid composition traits. Mol Breed 28:437–451
Cordeiro GM, Casu R, McIntyre CL, Manners JM, Henry RJ (2001) Microsatellite markers from sugarcane (Saccharum spp.) ESTs cross transferable to erianthus and sorghum. Plant Sci 160:1115–1123
Deng X, Long S, He D, Li X, Wang Y, Liu J, Chen X (2010) Development and characterization of polymorphic microsatellite markers in Linum usitatissimum. J Plant Res 123:119–123
Deng X, Long S, He D, Li X, Wang Y, Hao D, Qiu C, Chen X (2011) Isolation and characterization of polymorphic microsatellite markers from flax (Linum usitatissimum L.). Afr J Biotechnol 10:734–739
Dribnenki JCP, Green AG, Atlin GN (1996) Linola™ 989 low linolenic flax. Can J Plant Sci 76:329–331
Dribnenki JCP, McEachern SF, Chen Y, Green AG, Rashid KY (2003) Linola™ 2047 low linolenic acid flax. Can J Plant Sci 83:81–83
Duguid SD, Kenaschuk EO, Rashid KY (2003) Macbeth flax. Can J Plant Sci 83:803–805
Ellegren H (2004) Microsatellites: simple sequences with complex evolution. Nat Rev Genet 5:435–445
Eujayl I, Sorrells ME, Baum M, Wolters P, Powell W (2002) Isolation of EST-derived microsatellite markers for genotyping the A and B genomes of wheat. Theor Appl Genet 104:399–407
Everaert I, Riek JD, Loose MD, Waes JV, Bockstaele EV (2001) Most similar variety grouping for distinctness evaluation of flax and linseed (Linum usitatissimum L.) varieties by means of AFLP and morphological data. Plant Var Seeds 14:69–87
Faux NG, Huttley GA, Mahmood K, Webb GI, Banda MG, Whisstock JC (2007) RCPdb: an evolutionary classification and codon usage database for repeat containing proteins. Genome Res 17:1118–1127
Fu YB (2006) Redundancy and distinctiveness in flax germplasm as revealed by RAPD dissimilarity. Plant Genet Resour 4:117–124
Gong YM, Xu SC, Mao WH, Hu QZ, Zhang GW, Ding J, Li YD (2010) Developing new SSR markers from ESTs of pea (Pisum sativum L.). J Zhejiang Univ Sci B 11:702–707
Gupta S, Prasad M (2009) Development and characterization of genic SSR markers in Medicago truncatula and their transferability in leguminous and non-leguminous species. Genome 52:761–771
Han ZG, Guo WZ, Song XL, Zhang TZ (2004) Genetic mapping of EST-derived microsatellites from the diploid Gossypium arboretum in allotetraploid cotton. Mol Genet Genomics 272:308–327
Hisano H, Sato S, Isobe S, Sasamoto S, Wada T, Matsuno A, Fujishiro T, Yamada M, Nakayama S, Nakamura Y, Watanabe S, Harada K, Tabata S (2007) Characterization of the soybean genome using EST-derived microsatellite markers. DNA Res 14:271–281
Kale SM, Pardeshi VC, Kadoo NY, Ghorpade PB, Jana MM, Gupta VS (2011) Development of genomic simple sequence repeat markers for linseed using next-generation sequencing technology. Mol Breed. doi:10.1007/s11032-011-9648-9
Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK (2011) Microsatellite markers: an overview of the recent progress in plants. Euphytica 177:309–334
Kashi Y, King DG (2006) Simple sequence repeats as advantageous mutators in evolution. Trends Genet 22:253–259
Kenaschuk EO, Rashid KY, Gubbels GH (1996) AC Emerson flax. Can J Plant Sci 76:483–485
Krulickova K, Posvec Z, Griga M (2002) Identification of flax and linseed cultivars by isozyme markers. Biol Plant 45:327–336
Li YC, Korol AB, Fahima T, Nevo E (2004) Microsatellites within genes: structure, function and evolution. Mol Biol Evol 21:991–1007
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Morgante M, Hanafey M, Powell W (2002) Microsatellites are preferentially associated with nonrepetitive DNA in plant genomes. Nat Genet 30:194–200
Mun JH, Kin DJ, Choi HK, Gish J, Debellé F, Mudge J, Denny R, Endré G, Saurat O, Dudez A-M, Kiss GB, Roe B, Young ND, Cook DR (2006) Distribution of microsatellites in the genome of Medicago truncatula: a resource of genetic markers that integrate genetic and physical maps. Genetics 172:2541–2555
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Nat Acad Sci USA 70:3321–3323
Oh TJ, Gorman M, Cullis CA (2000) RFLP and RAPD mapping in flax (Linum usitatissimum). Theor Appl Genet 101:590–593
Parida SK, Kalia SK, Kaul S, Dalal V, Hamaprabha G, Selvi S, Pandit A, Singh A, Gaikwad K, Sharma TR, Srivastava PS, Singh NK, Mohapatra T (2009a) Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor Appl Genet 118:327–338
Parida SK, Dalal V, Singh AK, Singh NK, Mohapatra T (2009b) Genic non-coding microsatellites in the rice genome: characterization, marker design and use in assessing genetic and evolutionary relationships among domesticated groups. BMC Genomics 10:140
Peng JH, Lapitan NLV (2005) Characterization of EST-derived microsatellites in the wheat genome and development of eSSR. Funct Integr Genomics 5:80–96
Rachinskaya OA, Lemesh VA, Muravenko OV, Yurkevich OY, Guzenko EV, Bol’sheva NL, Bogdanova MV, Samatadze TE, Popov KV, Malyshev SV, Shostak MG, Heller K, Hotyleva LV, Zelenin AV (2011) Genetic polymorphism of flax Linum usitatissimum based on the use of molecular cytogenetic markers. Russ J Genetics 47:56–65
Ragupathy R, Rathinavelu R, Cloutier S (2011) Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genomics 12:217
Rajwade AV, Arora RS, Kadoo NY, Harsulkar AM, Ghorpade PB, Gupta VS (2010) Relatedness of Indian flax genotypes (Linum usitatissimum L.): an inter-simple sequence repeat (ISSR) primer assay. Mol Biotechnol 45:161–170
Roose-Amsaleg C, Cariou Pham E, Vautrin D, Tavernier R, Solignac M (2006) Polymorphic microsatellite loci in Linum usitatissimum. Mol Ecol Notes 6:796–799
Rowland GG (1991) An EMS-induced low-linolenic-acid mutant in McGregor flax (Linum usitatissimum L.). Can J Plant Sci 71:393–396
Rowland GG, Hormis YA, Rashid KY (2002) CDC Bethune flax. Can J Plant Sci 82:101–102
Rozen S, Skaletsky HJ (2000) Primer3 on the WWW for general users and for biologist programmers. In: Krawetz S, Misener S (eds) Bioinformatics methods and protocols: methods in molecular biology. Humana Press, Totowa, pp 365–386
Shete S, Tiwari H, Elston R (2000) On estimating the heterozygosity and polymorphism information content value. Theor Popul Biol 57:265–271
Sonah H, Deshmukh RK, Sharma A, Singh VP, Gupta DK, Gacche RN, Rana JC, Singh NK, Shama TR (2011) Genome-wide distribution and organization of microsatellites in plants: an insight into marker development in Brachypodium. PLoS ONE 6:e21298
Soto-Cerda BJ, Carrasco RA, Aravena GA, Urbina HA, Navarro CS (2011a) Identifying novel polymorphic microsatellites from cultivated flax (Linum usitatissimum L.) following data mining. Plant Mol Biol Rep 29:753–759
Soto-Cerda BJ, Urbina Saavedra H, Navarro C, Mora Ortega P (2011b) Characterization of novel genic SSR markers in Linum usitatissimum (L.) and their transferability across eleven Linum species. Electron J Biotechnol 14:6
Spielmeyer W, Green AG, Bittisnich D, Mendham N, Lagudah ES (1998) Identification of quantitative trait loci contributing to Fusarium wilt resistance on an AFLP linkage map of flax (Linum usitatissimum). Theor Appl Genet 97:633–641
Sureshkumar S, Todesco M, Schneeberger K, Harilal R, Balasubramanian S, Weigel D (2009) A genetic defect caused by a triplet repeat expansion in Arabidopsis thaliana. Science 323:1060–1063
Tang J, Baldwin SJ, Jacobs JME, van der Linden CG, Voorrips RE, Leunissen JAM, van Eck H, Vosman B (2008) Large-scale identification of polymorphic microsatellites using an in silico approach. BMC Bioinform 9:374
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422
Tian AG, Wang J, Cui P, Han YJ, Xu H, Cong LJ, Huang XG, Wang XL, Jiao YZ, Wang BJ, Wang YJ, Zhang JS, Chen SY (2004) Characterization of soybean genomic features by analysis of its expressed sequence tags. Theor Appl Genet 108:903–913
Uysal H, Fu Y-B, Kurt O, Peterson GW, Diederichsen A, Kusters P (2010) Genetic diversity of cultivated flax (Linum usitatissimum L.) and its wild progenitor pale flax (Linum bienne Mill.) as revealed by ISSR markers. Genet Resour Crop Evol 57:1109–1119
Varshney RK, Grosse I, Hähnel U, Siefken R, Prasad M, Stein N, Langridge P, Altschmied L, Graner A (2006a) Genetic mapping and BAC assignment of EST-derived SSR markers shows non-uniform distribution of genes in the barley genome. Theor Appl Genet 113:239–250
Varshney RK, Hoisington DA, Tyagi AK (2006b) Advances in cereal genomics and applications in crop breeding. Trends Biotechnol 24:490–499
Varshney RK, Glaszmann JC, Leung H, Ribaut J-M (2010) More genomic resources for less-studied crops. Trends Biotechnol 28:452–460
Venglat P, Xiang D, Qiu S, Stone S, Tibiche C, Cram D, Alting-Mees M, Nowak J, Cloutier S, Deyholos M, Bekkoui F, Sharpe A, Wang E, Rowland G, Selvaraj G, Datla R (2011) Gene expression analysis of flax seed development. BMC Plant Biol 11:74
Wierdl M, Donimska M, Petes TD (1997) Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics 146:769–779
Wiesner I, Wiesnerova D, Tejklova E (2001) Effect of anchor and core sequence in microsatellite primers on flax fingerprinting patterns. J Agric Sci 137:37–44
Wiesnerova D, Wiesner I (2004) ISSR-based clustering of cultivated flax germplasm is statistically correlated to thousand seed mass. Mol Biotechnol 26:207–214
Yi G, Lee JM, Lee S, Choi D, Kim BD (2006) Exploitation of pepper EST-SSRs and SSR-based linkage map. Theor Appl Genet 114:113–130
Yu JK, Dake TM, Singh S, Benscher D, Li W, Gill B, Sorrells ME (2004) Development and mapping of EST-derived simple sequence repeat markers for hexaploid wheat. Genome 47:805–818
Yu JW, Dixit A, Ma KH, Chung JW, Park YJ (2009) A study on relative abundance, composition and length variation of microsatellites in 18 underutilized crop species. Genet Resour Crop Evol 56:237–246
Acknowledgments
The Total Utilization Flax GENomic (TUFGEN) project was funded by Genome Canada and several co-funders. The Cloutier laboratory would like to acknowledge Genome Prairie for management of the project, the province of Manitoba, Flax Council of Canada and Manitoba Flax Growers Association for co-funding specific to this part of the project. Sincere thanks to Joanne Schiavoni for manuscript editing and Michael Shillinglaw for figure preparation.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by R. Visser.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Cloutier, S., Miranda, E., Ward, K. et al. Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.). Theor Appl Genet 125, 685–694 (2012). https://doi.org/10.1007/s00122-012-1860-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-012-1860-4