
Abstract
This chapter is focused on radiobiological aspects at the molecular, cellular, and tissue level which are relevant for the clinical use of ionizing radiation (IR) in cancer therapy. For radiation oncology, it is critical to find a balance, i.e., the therapeutic window, between the probability of tumor control and the probability of side effects caused by radiation injury to the healthy tissues and organs. An overview is given about modern precision radiotherapy (RT) techniques, which allow optimal sparing of healthy tissues. Biological factors determining the width of the therapeutic window are explained. The role of the six typical radiobiological phenomena determining the response of both malignant and normal tissues in the clinic, the 6R’s, which are Reoxygenation, Redistribution, Repopulation, Repair, Radiosensitivity, and Reactivation of the immune system, is discussed. Information is provided on tumor characteristics, for example, tumor type, growth kinetics, hypoxia, aberrant molecular signaling pathways, cancer stem cells and their impact on the response to RT. The role of the tumor microenvironment and microbiota is described and the effects of radiation on the immune system including the abscopal effect phenomenon are outlined. A summary is given on tumor diagnosis, response prediction via biomarkers, genetics, and radiomics, and ways to selectively enhance the RT response in tumors. Furthermore, we describe acute and late normal tissue reactions following exposure to radiation: cellular aspects, tissue kinetics, latency periods, permanent or transient injury, and histopathology. Details are also given on the differential effect on tumor and late responding healthy tissues following fractionated and low dose rate irradiation as well as the effect of whole-body exposure.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Radioresistance
- Radiation-induced toxicity
- Hypoxia
- Tumor microenvironment
- Normal tissue complication probability
- Tumor control probability
-
To understand that tumor targeting includes a proportion of healthy tissues.
-
To recognize that the radiobiology of tumors and healthy tissues is different.
-
To acknowledge that healthy tissues limit the possibilities of tumor control and are responsible for radiotherapy-associated toxicity.
-
To be able to recognize and make estimates about tumor growth as well as tumor control, both important factors for determining the response to radiotherapy.
-
To obtain knowledge about biological factors which determine the outcome of radiotherapy in cancer treatment.
-
To familiarize with the principles of dose fractionation and how different tissues respond to changes in fraction sizes, number of fractions and frequency.
-
To learn about the radiobiological effects of the dose rate and its clinical application.
-
To understand tumor hypoxia, its methods of evaluation, oxygen effect, and oxygen enhancement ratio (OER) as well as to familiarize with different therapeutic approaches to tumor hypoxia.
-
To understand the role of different factors in tumor radiation resistance and progression including those coming from the tumor microenvironment and as a result of cells with a stem cell phenotype.
-
To be able to define the role of the human microbiota in healthy or pathologic gut and to use radiotherapy effects as an example for the microbiota implication in disease.
-
To acknowledge the goals of palliative radiotherapy in contrast to radical radiotherapy; become aware of the applications of palliative radiotherapy and potential biological targets.
-
To become aware of the abscopal effect; the type of biological damage caused by ionizing radiation leading to abscopal effects; the main biological damages caused by radiation that are recognized by the immune system; how irradiated tumors can show an immune response; how systemic anti-tumor immune responses occur.
-
To be able to define and describe the acute/early adverse effects as well as late adverse effects of radiotherapy.
-
To recognize that if tissues are part of a heavily irradiated volume, late toxicity will occur.
-
To explain Normal Tissue Complication Probability (NTCP) and the advantages and drawbacks of using radiobiology modeling for NTCP as well as to give an overview of different NTCP models.
5.1 Principles of Tumor Radiotherapy
Box 5.1 Radiation Therapy in Cancer Treatment
-
RT is one of the cornerstones in cancer treatment.
-
The objective of RT resides in finding an optimal balance between chances of cure and risk of associated toxicity.
-
Differential effect of RT between tumors and normal tissues depends on multiple factors related to both malignant and healthy tissue radiobiology, but also on beam characteristics and treatment schedule.
-
Technical advances and sophistication of RT devices improve ballistic accuracy and allows unprecedented changes in treatment schedules, probably changing both malignant and healthy tissue radiobiology.
Radiation therapy (RT) is a locoregional treatment modality for cancer. Using radiation for therapeutic purposes began only a few months after the discovery of X-rays by Wilhelm Röntgen in 1895. The first “true” RT succeeded in managing a case of lupus erythematosus in 1897 by Eduard Schiff (1899). More than 120 years later, RT is still one of the cornerstones of tumor treatment, with more than half of all cancer patients treated by radiation in the course of their therapeutic management [1] (Box 5.1).
The interesting (but also dangerous) properties of ionizing radiation (IR) reside in its ability to penetrate more or less deeply in biological tissues, depending on its energy, and to react with the environment. These reactions consist in direct energy deposition and the generation of free radicals near and within living cells. The consequence of energy deposition is damage to the DNA structure leading to cell death if unrepairable [2]. Of course, all cells are concerned, tumor cells as well as cells in the healthy tissues, and within the irradiated volume, no difference is made between tumor and healthy cells. Treating a tumor would be easily achievable by the administration of a very high dose if it was not surrounded by the patient. Therefore, several strategies have been developed to ensure both the best tumor control and the least consequences ensuing from healthy tissues exposure, taking advantage of differences between tumor and healthy cells, known as the benefit/risk balance (Fig. 5.1).

The benefit/risk balance. The objective of RT is to control the tumor while sparing normal tissues, to ensure the patient’s cure without unacceptable side effects
Some biological processes favor the benefit/risk ratio in RT and the differential effect between tumor and healthy cells. Except for some very radiosensitive or radioresistant tumors, healthy and tumor cells demonstrate quite similar radiation sensitivities. However, DNA damage is less efficiently repaired by tumors than by healthy cells. This is the basis for the dose fractionation principle, demonstrated by Claudius Regaud and applied in the clinic by Henri Coutard in 1934, and which still is used today in modern RT. The total dose necessary to control the tumor is generally delivered in a series of small daily doses. The time lapse between each fraction allows DNA damage to be repaired by healthy cells whereas tumor cells do not repair or do so to a lesser extent. The biological effectiveness (the chances of tumor control but also the risk of damage to normal tissues) is reduced when using fractionated doses due to DNA repair and cell repopulation in both tumors and healthy tissues [3]. Numerous parameters associated with fractionation regimens, such as the total dose, the dose per fraction, and the time between fractions and the total treatment time, will influence both tumor response and normal tissue damage and will be described in more detail below [4]. Another biological factor participating in the differential effect is radiation-induced cell death. In a majority of cases, the initial radiation exposure is not what kills cells but rather unrepaired DNA damage, which condemns them to death as soon as they re-enter in the cell cycle. Rapidly proliferating tumor tissues will suffer significant cell death under these conditions compared to slowly proliferating healthy tissues. However, some healthy tissues such as oral and intestinal mucosa or hematopoietic cells proliferate rapidly and may be susceptible to early mitotic cell deaths if present in the irradiated volume. Finally, tumor control will also depend on other factors such as tumor heterogeneity (the tumor cannot be simply considered as a cluster of tumor cells), oxygenation status before RT and variations during treatment, tumor vascularization, resident and recruited immune cells, and so forth. Considering all these biological factors, the objective of treatment planning in RT is to find the best compromise between chances of cure and risk of associated toxicity [5].
In an ideal world, RT may target only the tumor volume; however, in real life, this is never the case. For healthy tissues, besides dose and fractionation, the volume exposed is of paramount importance in determining the risk of developing toxicity. Technical advances in dose delivery, planning systems and associated imaging devices have helped to achieve ever increasing ballistic accuracy. Advanced technologies, such as volumetric modulated arc therapy (VMAT), image-guided radiotherapy (IGRT), stereotactic body radiotherapy (SBRT), heavy ions [6], or proton therapy have all contributed to progress [7]. Consequently, the use of highly focused beams reduces the volume of normal tissues present within the irradiated volume and can spare very sensitive organs, thus minimizing the risk of toxicity. Reducing the volume also permits changes in fractionation schedules. For example, SBRT uses hypofractionation, delivering ablative doses per fraction between 8 and 20 Gy instead of the conventional 2 Gy/fraction. The gain in biological effectiveness strongly increases tumor control as illustrated in early-stage primary lung cancer. These changes in fractionation schemes may also induce a “new” radiobiology of tumors and healthy tissues in response to very high doses of IR, an area that remains to be explored [8]. Finally, ultra-high dose rate FLASH RT demonstrates a very sharp differential effect between tumor and healthy tissues and is the subject of intense research for future clinical applications [9].
Technical advances have strongly contributed to the chances of cure for numerous cancers and increased patients’ survival. This increased life expectancy following cancer treatment, however, favors the emergence of side effects, especially long-term sequelae. Normal tissues can be divided into “early” and “late” responding tissues. Early-responding tissues (intestinal mucosa, hematopoietic system, skin, gonads) are characterized by the presence of cell proliferation compartments and are mostly implicated in acute radiation-induced toxicity. Late-responding tissues demonstrate no distinct cell proliferation compartment and are mostly implicated in late toxicity. For each normal tissue, dose constraints, which vary depending on the RT technique used, may be applied. These constraints help to minimize the risk of developing severe treatment-associated toxicity [10].
RT still plays a significant role in cancer cures. Its efficiency depends on numerous parameters related to both tumor and normal tissue radiobiology. The objective of cancer therapy, using modern RT often concurrently with other therapeutic strategies (surgery, chemotherapy, immunotherapy, etc.) is for the patients to survive without debilitating sequelae. This goal may be achieved using technological advances in RT, combined with strategic knowledge of both tumor and healthy tissue radiobiology.
5.2 Therapeutic Window and Therapeutic Ratio (Box 5.2)
Box 5.2 The Therapeutic Window and Therapeutic Ratio
-
Therapeutic window: The difference between tumor control probability (TCP) and normal tissue complication probability (NTCP) at identical irradiation dose.
-
Therapeutic ratio: The relation between TCP and NTCP or efficacy to toxicity ratio.
5.2.1 The Therapeutic Window
RT is one of the most effective treatment modalities in cancer therapy. However, despite modern precision RT, it is generally unavoidable to deposit IR to the tumor volume without risk of radiation injury to the surrounding healthy normal tissues or organs. Hence, the therapeutic effectiveness of radiation is dependent on the balance between tumor control and normal tissue adverse effects. In fact, the tolerance dose of the normal tissues or organs at risk determines the dose which can be safely applied to the tumor volume. For almost all normal tissues and organs, dose-volume constraints are well documented in the literature, for example, the QUANTEC (QUantitative Analysis of Normal Tissue Effects in the Clinic) data, as guidance in the clinical practice (see Sect. 5.13.6) [11]. The so-called therapeutic window is a conceptual window of opportunity between the tumor control probability (TCP) and normal tissue complication probability (NTCP) (Fig. 5.2).

Illustration of the therapeutic window. For an identical delivered dose, the curves show the difference between tumor control probability and normal tissue complication probability and methods to widen the window. (Reprinted from Drug radiotherapy combinations: review of previous failures and reasons for future optimism; Figure from Higgins et al. [12], with permission)
The ultimate aim of RT in the clinic is accomplished when the therapeutic window is large, with an optimized balance between benefits and risks, hence a treatment that is highly likely to be effective and safe. The shape and position of the dose–response curves for tumor control and toxicity to the normal tissues (Fig. 5.2) determines the probability that enough radiation is delivered to destroy the tumor cells without serious complications. The position of the curves determines the feasibility of the application of RT to the patient. The therapeutic window is large in radiosensitive tumor types like lymphoma, but small for other tumor types such as brain and pancreatic cancer. If the dose–response curve for normal-tissue toxicity is positioned at the left side of the tumor control curve or in case the curves are close together, the aimed tumor response could only be achieved at the cost of a high complication risk. The standard RT treatment is that one with tumor control probability (TCP) ≥0.5 and normal tissue complication probability (NTCP) ≤0.05 [13].
It is worth noting that Fig. 5.2 illustrates an ideal situation. The TCP curve might in particular however deviate for two main reasons. First, tumors are more heterogeneous compared with normal tissue; subsequently, the expression of the TCP curve becomes shallower than that of the NTCP curve. Secondly, not only the region of interest does contain the malignant cells, but there might be metastatic extensions outside the irradiation treatment volume. Hence, it is unlikely that the TCP curve for local control of specific tumors scores 100% [14, 15].
Several treatment parameters influence the therapeutic window. For example, when the overall treatment time is prolonged, the therapeutic window is narrowed (Fig. 5.3) [15, 16]. It is however difficult to practice this strategy because each complication translates the effect of a treatment parameter on the therapeutic window differently.

Prolongation of the overall treatment time narrows the therapeutic window. Conventional irradiation course in 6 weeks versus a split-course course in 10 weeks. (Adopted from [16])
Several methods can be used to widen the therapeutic window, to increase the probability of complication-free tumor control:
-
Fractionated RT. See Fig. 5.4. Decrease of the organ or tissue at risk volume using precision RT techniques allowing optimal dose distribution (e.g., stereotactic irradiation/particle irradiation).
-
Combination therapy with molecular targeting or immune-modulating drugs. Optimally, drugs should be carefully chosen to selectively sensitize tumor and not normal tissue cells, taking the 6R’s or Hallmarks of Radiobiology into account (see Sect. 5.4).

Fractionation as an effective method to widen the therapeutic window. Curves schematically represent the probability of normal tissue side effects (NTCP, red curve), the probability of tumor control (TCP, blue curve) as well as the complication free tumor control curve (green) following single-dose radiation (a) and dose fractionation (b). (Figure from Shrieve and Loeffler [17], with permission from Wolters Kluwer Health, Inc.)
5.2.2 The Therapeutic Ratio
The therapeutic ratio or therapeutic index is an imperative measure used in the treatment planning to ensure that the RT course achieves its goals [18]. The ratio represents the difference between the TCP and NTCP curves for the same delivered dose at a fixed endpoint of NTCP [14]. Therefore, it represents the quantity used in the tumor treatment planning for the purpose of disease cure without complications. The ratio is defined as the relationship between TCP and NTCP, i.e., efficacy/toxicity ratio. Chang et al. stated that a common method used to calculate the therapeutic ratio which is the probability of cure without complications [19] and given by:
As the difference between TCP and NTCP becomes large it means that TR approaches 1 and treatment is fairly effective for tumor control than for causing normal tissue morbidity, but the pattern is reversed when the difference between TCP and NTCP becomes small. That is, TR approaches 0 and the treatment may fail and be relatively more toxic [14]. As explained above, there are many treatment parameters and methods that affect the therapeutic ratio, for example, combination therapy with a radiosensitizing agent or drug. This effect is revealed in practice as increasing tumor cure rate with improved quality of life as a result of a therapeutic gain [13, 16]. In this circumstance, the therapeutic ratio is the ratio of dose-modifying factors (DMFs) of tumor over that for normal tissues.
Finally, the therapeutic ratio differentiates between early and late responding normal tissues in terms of their response to concomitant RT and chemotherapeutics or targeted agents. While the therapeutic ratio of early responding tissue is usually <1, the therapeutic ratio of late responding tissues is >1 which reflects the advantageous consequence of concomitant RT and chemotherapy. This may lead to a high level of early injury, but a neutral level of late damage to late responding tissues. Fortunately, early side effects can be relieved by using either extensive supportive care or adaptation of the standard treatment. The combination of RT and chemotherapy may prove effective if selective radiosensitization of malignancy is obtained and the probability of late-responding normal tissue damage is not increased. However, early toxicities might also be a concern.
5.3 Tumor Growth and Tumor Control (Box 5.3)
Box 5.3 Tumor Response Following Radiotherapy
-
Tumor control probability (TCP) is guided by dose, tumor characteristics, and normal tissue radiation sensitivity.
-
Killing of clonogenic cells within a tumor partly explain TCP during RT, but the effect is also influenced by host factors, for example, immune cell attack.
5.3.1 Tumor Control
The main objective of curative RT is to successfully achieve local tumor control [16]. The relationship between TCP and radiation dose is shown in Fig. 5.5 which illustrates that there is poor tumor control with low dose, but high tumor control with high dose [20]. The steepness of the curve depends on differences in tumor size, tumor cell radiation sensitivity and repopulation as well as other factors. These factors give rise to variation in TCP of different tumors but also inter-patient variation in clinical practice. Subsequently, this improvement in tumor control is reflected in an increase in the life expectancy of cancer patients. To this end, it is preferable to evaluate RT success based on tumor control.

Tumor Control Probability (TCP) and radiation dose relationship. The scheme demonstrates the sigmoid relationship of probability of tumor control and normal tissue damage to radiation dose
Complete tumor control requires that every clonogenic cell is destroyed. Unfortunately, cell killing is randomly distributed within a population of tumor cells, and there are about 109 cells in each gram of tumor. A small fraction of these cells (about 1%), in reality, contains cells with clonogenic-forming ability; so, a human tumor could have billions of clonogenic cells; therefore, eliminating every such cell is a great challenge. The likelihood of obtaining tumor control is related to radiation dose, features of control probability of the tumor and the number of surviving clonogenic tumor cells (Fig. 5.6.).

The response of clonogenic tumor cells at 2 Gy/fraction as a function of the total dose. Assuming that each 2 Gy fraction reduces the clonogenic cell population with 50%, 30 fractions of 2 Gy will reduce 1010 clonogenic tumor cells to ten surviving cells. In order to eliminate each clonogenic tumor cell, additional fractions of 2 Gy are required to reach tumor control
5.3.2 Tumor Growth
The tumor growth rate can be used to determine how a cancer will respond to RT treatment by predicting or understanding the key features of the tumor tissue response to radiation. The tumor growth rate was developed for examining the capacity of clonogenic-forming cells of a tumor and assumes that the regrowth component is a function of repopulation by the surviving of cells with colony-forming ability [20, 21].
There are considerable differences in growth rate between different tumors due to differences in size and biology. Therefore, the tumor growth curve has exponential and non-exponential parts when plotted on a logarithmic scale. That is, the tumor volume doubling time (VDT), the duration of time required for the tumor to double in size, increases for small tumors because there is a sufficiency in nutrient and oxygen supply resulting in a reduction of cell cycle, a higher proportion of cycling cells or and a lower cell death rate. As a result, the slope of the growth curve, which reflects the doubling time of the cells, has an exponential pattern for small tumors. Conversely, VDT decreases for large tumors because of the limitation of nutrient and oxygen supply. This leads to a prolongation of cell-cycle progression but also a high rate of cell death. As a result, the slope of the growth curve has no exponential patterns for large tumors. The Gompertz equation describes such progressively slowing tumor growth:
where V0 is the volume at arbitrary zero time while A and B are parameters that determine the speed of growth [16]. VDTs are remarkably variable in human tumors, both between primary and metastatic lesions and among tumors with different histology (Table 5.1). Please also note that even within one tumor entity (localization, histology, and primary or metastasis similar) there is a range in VDT illustrating the problem of tumor heterogeneity.
5.3.2.1 Cell-Cycle Kinetics and Growth Fraction in Tumors
The growth fraction (GF) refers to the proportion of cycling cells that has highly colony-forming ability and is in the active process of cell cycling (omitting cells in G0 phase), with capacity of DNA duplication and cell division [22]. Similarly, as in normal tissue, some tumor cells are not involved in active proliferation for different reasons, for instance as a result of hypoxia, differentiation, and catabolic insufficiency. Moreover, it is estimated that about 50% of cells in a tumor are not neoplastic cells but are cells making up the tumor stroma. Therefore, it is clear that the cell population in tumors contains quiescent (Q) cells, and since GF is defined as the proportion of cycling cells, it can be calculated as stated by [13, 22]:
where p is proliferating cells.
For estimation of the cell-cycle kinetics (TC), three principal methods are used: (1) bromodeoxyuridine (BrdUrd) or thymidine analogues iododeoxyuridine (IdUrd), (2) 3H-thymidine for the synthesis of DNA and (3) positron emission tomography (PET) imaging of the tumors in vivo by radiolabeled 18F-fluoro-3′-deoxy-3′-l-fluorothymidine (FLT) [13, 16].
The first method includes labeling of the cells with BrdUrd or IdUrd. When cells pass through the S-phase, these labels are incorporated into the newly created DNA strand. An antibody against BrdUrd or IdUrd as well as a DNA-specific dye are used to stain a single-cell suspension prepared from a cell culture in vitro or a tumor biopsy, and the duration of the S phase (TS) and fraction of cells in S phase are assessed using flow cytometry.
In the second method, cell-cycle kinetics (TC) is estimated from labeled cells by measuring the duration of the cell cycle by either pulse or continuous labeling with 3H-thymidine. The labeling agent is incorporated into the DNA as cells progress through S-phase and the cell-cycle kinetics (TC) is estimated from the labeled cells [16].
The third principal method applies PET tracers to detect and evaluate tumor proliferation in vivo. In this method, radiolabeled 18F-fluoro-3′-deoxy-3′-l-fluorothymidine (FLT) is used. FLT is phosphorylated by thymidine kinases (TK) and since regulation of TK activity occurs in the S-phase, it means that metabolites of FLT (mono-, di-, and tri-phosphates) are reflecting the number of cells in S-phase and hence replication status. The FLT tracer activity in a tumor is subsequently evaluated by a PET scanner from which the cell-cycle kinetics can be estimated [16].
For estimation of the GF, according to literature the GF is obtained by assessment of two distinct cell subpopulations, one that does not grow and another which grows with a uniform cell-cycle distribution [13, 21]. This method includes the exposure of a growing culture of cells with 3H-thymidine for the synthesis of DNA, and then after the period of at least one complete cell cycle to ensure all cells producing DNA pass through the S-phase and are labeled, an autoradiography of tumor section is taken, and GF is calculated by:
There is also a possibility to take proliferation into account by immunohistochemistry assessment of tumor tissue sections by staining of the nuclear antigen Ki-67, which in tumors has different levels depending on the tumor proliferation. The method includes staining of tumor cell cultures or a tumor biopsy with a Ki-67 specific antibody followed by counting the number of positive tumor cells. The GF growth fraction is estimated from labeled cells by measuring the proportion of proliferating cells using continuous labeling. Although the method is frequently used to assess S-phase cells, recent results have indicated that Ki-67 also has different functions in other cell-cycle phases which may in fact influence proliferation estimations [23].
5.3.2.2 The Potential Doubling Time (Tpot)
“The potential doubling time (Tpot) of a tumor is defined as the cell doubling time without any cell loss.” There are two methods used to estimate Tpot. In the first method, DNA is labeled with thymidine analogues and then the cells fraction in S phase (LI) and the duration of the S phase (TS) are estimated by using flow cytometry to calculate Tpot by:
where λ is a correction parameter for the non-rectangular age distribution of growing cell populations, in the order of 0.7–1.
Different tumor tissues have different values of LI, but they have similar TS, in the range of 12 h. As a result, Tpot has a spectrum of values ranging from 4 to 34 days, as shown in Table 5.2. Of note, it has been demonstrated that in a clinical RT context, the pre-treatment Tpot does not predict outcome as one also needs to consider the repopulation rate of colony-forming cells [13].
5.3.2.3 Cell Loss in Tumors
Slow tumor growth is not only explained by the fact that not all cells within a tumor are proliferating but also due to considerable cell loss where multiple parameters regulate these two factors. If there was no cell loss and if every tumor cell was actively proliferating, the tumor doubling time would imitate the cell-cycle kinetics (TC). Therefore, when there is cell loss the TD is long and when there is reduced GF the Tpot of the tumor is longer than the time of cell cycle [14].
The net growth rate, or the VDT, of tumors results from the balance of cell production and cell loss. In clinical settings, the GF and knowledge of the cycle time of the individual cells does not reflect the speed of tumor growth; namely, the cycle time of the individual cells is much faster than the speed of tumor growth. Such discrepancy is attributed to cell loss which can be considered by calculating cell loss factor (CLF). The cell-loss factor refers the ratio of the cell loss rate to the production of new cells, and it can be calculated by:
where Tpot is the potential tumor doubling, and VDT is the tumor volume doubling time that is calculated by the essential time for the tumor to double its volume (V) by using:
When the cell loss factor is high, it means that there is loss of newly produced cells from the GF; thus, tumor growth is slow. Cell loss has been attributed to different parameters [16, 24]: (1) Cells are in the inactive phase of cell cycling (in G0 phase) which is a non-proliferative compartment, (2) There is inadequate nutrition and oxygen levels due to the tumor outgrowth that gives rise to pushing cells into areas at a distance from blood supply, (3) Metastasis, (4) Immune cell killing of the tumor, and (5) Exfoliation (the complete removal of a single epithelial cell or group of cells from a layer of epithelium by spontaneous or induced means). In animal models of tumors, this would not apply but could be a mechanism by which cells lose their integrity in carcinomas of the gastrointestinal tract, for example, where the epithelium is renewed rapidly (Box 5.4).
Box 5.4 Tumor Growth Kinetics
-
Tumor volume doubling time (VDT) is influenced by tumor localization site, primary or metastatic status as well as histology but also by intra-tumor heterogeneity factors.
-
There are differences in growth rates between different tumor types and metastatic lesions tend to grow faster than primary lesions.
-
A logarithmic scale can be used to judge treatment effectiveness. An estimate of tumor growth rate is determined by cell-cycle kinetics, the growth fraction, the cell loss rate (CL), and the potential doubling time (Tpot). Even among tumors of the same histological type, these parameters differ greatly. Cell kinetics cycle of cells and growth fraction in tumors can be monitored ex vivo using various DNA-labeling strategies and in vivo using PET tracers.
-
Ki67 immunohistochemistry staining of tumor biopsies is a method for assessing S-phase cell proportion.
-
The potential doubling time (Tpot) refers to the time it would take for volume to double without loss of cells. Consequently, the potential doubling time and the observed volume doubling time are different because tumors show a high amount of cell loss.
-
Tumor cell loss also contributes to VDT and may be attributed to limited nutrition and oxygen supply, metastatic propensity, immune cell killing, and tumor cell exfolia.
5.4 6R’s Concept
Box 5.5 The Hallmarks of Radiobiology
-
The Hallmarks of Radiobiology or 6R’s are six typical molecular, cellular, or tissue processes which determine the effects of radiation on both malignant and healthy tissues.
-
The 6R’s are: Radiosensitivity, Repair, Redistribution, Repopulation, Reoxygenation, Reactivation of the immune response.
The so-called 6R’s are six biological features which determine the outcome of RT in the clinic: the balance between the complication rate (side effects due to normal tissue injury) and the tumor control rate (palliation or curation due to tumor cell sterilization) (Fig. 5.7). These basic principles or Hallmarks of Radiobiology have been evolved from Withers’ 4R’s—“Recovery/repair, Redistribution, Repopulation and Reoxygenation”—[25] via Steels’ 5R’s—the addition of “intrinsic cellular Radiosensitivity” [26] to 6R’s by including “Reactivation of the immune response” [27] (Box 5.5).

The Hallmarks of Radiobiology, the 6R’s
The six Hallmarks of Radiobiology (Fig. 5.7) in brief:
-
Radiosensitivity: Intrinsic and acquired radioresistance of normal tissue cells and tumor cells to radiation, in particular cancer (stem) cells among the heterogenic tumor cell population.
-
Repair capacity, efficiency, and mechanisms of sublethal DNA damage repair, and related sensitivity to fractionated irradiation—which is high for most healthy tissues and low for most tumors.
-
Redistribution of cells in the cell-cycle affects their radioresistance. Cells in mitosis are most sensitive to radiation, while cells in the S-phase are radioresistant. Redistribution following irradiation will push radioresistant S-phase cells towards a radiosensitive cell-cycle phase.
-
Repopulation: Cell repopulation of—not by radiation eradicated cancer cells—involved in the (accelerated) repopulation of the tumor—which is detrimental—and beneficial repopulation of normal tissue cells recovering from acute injury.
-
Reoxygenation: Cells in hypoxic niches within the tumor are radioresistant. Reoxygenation between multiple radiation fractions given in a radiation course is an important phenomenon by which originally hypoxic tumor cells will be reoxygenated and hence radiosensitized.
-
Reactivation of the immune response: Local irradiation induces a systemic immune activation to attack distant tumor cell niches which can be located outside the irradiated volume (abscopal effect).
5.4.1 The 6R’s in Detail
5.4.1.1 Radiosensitivity
Many authors refer to the radiosensitivity as the degree of tumor and normal tissue regression following irradiation. There are many factors that determine the radiosensitivity which are the proportion of cells with clonogenic capacity, growth rate and reproduction rate, mitosis activity, metabolic rate, tissue type, radiation dose, inherent radiosensitivity, and hypoxia. For example, cells with fast growth or high metabolic rate are highly radiosensitive. Essentially, since the reproductive capacity of cancer cells is higher than the reproductive capacity of late responding normal tissue cells, cancer cells are more sensitive to radiation, but this depends on the cancer tissue type.
5.4.1.2 Repair
Tumor and normal cells differ in terms of repair after radiation-induced damage. Unlike normal cells, the repair process and mechanism of tumor cells are defective. While normal tissue cells do repair their radiation-induced DNA damage efficiently, malignant cells often cannot.
There are three types of radiation damage to mammalian cells:
-
1.
Potentially lethal damage (PLD): Cell death depends on the environmental conditions. In a normal situation, damaged cells will not repair and die, but in case of reformed environmental conditions, cells can repair their DNA damage.
-
2.
Sublethal damage (SLD): The death of a cell depends on the sublethal damage condition. DNA damage can be repaired if no extra injury is taking place. The recovery kinetics, the repair time, lies in the range of a few hours following DNA double strand break (DNA DSB) induction.
-
3.
Lethal damage (LD): Irreparable and irreversible damage leading to cell death.
The main reason for cell death is the production of the asystematic generation of chromosomal aberrations, including rings and di-centric aberrations that result from an interaction between more than one DNA DSB [28]. Details regarding the DNA Damage Response and repair pathways are given in Chap. 3.
5.4.1.3 Redistribution (Re-assortment)
Figure 5.8, panel a shows the distribution of eukaryotic cells over the four cell-cycle phases, which include the G1 phase, S-phase (synthesis phase), G2 phase (interphase phase), and M phase (mitosis and cytokinesis). Cells in the different phases of the cell cycle vary in radiation sensitivity. Cells in the S-phase are resistant to radiation (Fig. 5.8, panel a) while cells in the M and G2 phase are sensitive to radiation [14].

The cell-cycle phase and radiation sensitivity. Cell survival curves of V79 Chinese hamster cells irradiated at different phases of the cell cycle on the left side
When cells experience radiation-induced insult, three effects occur:
-
1.
Recruitment: Stem cells of some tumors are in the G0 phase, which is a radioresistant phase; therefore, they may repair their damage and survive. In order to kill these cells efficiently, these cells are recruited into the cell cycle so as to arrive in a radiosensitive phase.
-
2.
Cells are blocked in the radiosensitive phase (G2). Cells in G2 are highly likely to be sterilized by the first radiation dose.
-
3.
Cells are allowed to re-assort and progress to the radiosensitive phase. Cells in radio-resistant phases survive, yet since they continue to cycle, there is a likelihood that they will arrive at a sensitive phase and be sterilized by a second or later fraction.
5.4.1.4 Repopulation
The renewal capability of tissue clonogenic cells that follows the reduction of tissue cells, with clonogenic-forming capacity, is referred to as repopulation (regeneration). Following radiation-induced tissue injury, the tissues will react by repopulation of surviving clonogenic cells, i.e., compensation for the lost cells occurs relatively quickly, with decreasing clonogenic doubling times from 9.8 to 3.4 days. This will result in a larger number of tumor cells which is detrimental. For normal tissue injury, repopulation from the stem cell compartment will regenerate the damaged tissue, there with reducing early radiation toxicity [16].
Biologically, there are three reasons for accelerated repopulation. Firstly, when tissue is exposed to radiation, cell kinetics, which may be reminiscent of a normal epithelium is stimulated; thus, this response causes regenerative reaction of clonogenic cells to initiate repopulation by activating growth factors, such as keratinocyte growth factor (KGF). Secondly, reoxygenation will occur during the course of fractionated RT, facilitating tissue regeneration. Finally, signaling such as that via the epithelial growth factor receptor (EGFR) is activated after irradiation; hence, this signal works as the regulated regenerative response [22].
The onset of repopulation in many cases is thought to be about 3 weeks after the start of fractionated RT. Its mechanism and kinetics depend on tissue types and might be dose dependent [29] (see also Chap. 6). From the clinical point of view, the total dose should be delivered over a controlled period of time. Any reduction in overall time is limited by the radiation tolerance of acutely responding normal tissues, but an extended overall treatment time might lead to diminished tumor response due to the increase of cells as a result of repopulation.
5.4.1.5 Reoxygenation
The empirical observation that oxygen levels in tumors may be enhanced in the period after irradiation is known as reoxygenation. Reoxygenation of originally hypoxic tumor cells, besides exploiting differences in DNA repair between normal tissue cells and tumor cells, is an important mechanism and reason for fractionated RT. During the fractionation course, lethally damaged cells are removed, and the blood supply increases. Thereby, initially radioresistant hypoxic cells are gradually reoxygenated and become sensitive to radiation (Fig. 5.9).

Simplified illustration of the reoxygenation process. Tumor cell compartments include anoxic, hypoxic, and aerated cells. Most tumors show a heterogeneous pattern of hypoxia with gradients of oxygen pressure decreasing with increasing distance from blood vessels. The oxygen status of the tumor cells is not constant; it is a dynamic, constantly changing phenomenon. Following exposure to irradiation, well-oxygenated cells will be sterilized, but many hypoxic cells will not. During the course of fractionated RT hypoxic cells can be reoxygenated, and therefore become sensitive to radiation and can be sterilized. (Figure was adapted from [13])
The oxygen enhancement ratio and the role of oxygen in the radiation response has been explained in Chap. 2. If reoxygenation is efficient between dose fractions, the presence of hypoxic cells does not have a significant effect on the outcome of a multi-fractionation scheme. In a hypofractionation regimen, the time period to obtain full reoxygenation of hypoxic tumor cells might however be too short (discussed in Chap. 6).
5.4.1.6 Reactivation of the Immune System
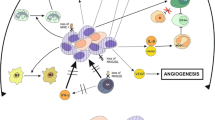
When irradiating a tumor, the tumor microenvironment (TME) will also be exposed. Such exposure of the TME might affect the immune system, both locally and systemically. Activation of an anti-tumor response depends on the treatment regimen, i.e., the fractionation schedule, dose, and timing because these factors disturb the balance between immunosuppressive and immune-stimulatory effects. As a result, a specific radiation treatment protocol can induce an anti-tumor immune reaction. When cells of tissues are exposed to radiation, the immune response to attack tumor cells is generated in a few steps (Fig. 5.10).

Illustration of the steps of radiation-induced systemic immune activation contributing to attack on distant/metastatic tumor cells
Different radiation treatment schemes with respect to the total dose and fraction size have been shown to have diverse effects on the immune response, and therewith also on target expression with consequences for combination treatments like with immunotherapy. To obtain optimal modulation of the radiation response, specific immunomodulating or targeted drugs can be selected. The radiation-induced TME effects modulating the immune response requires further research to find the ideal immunotherapy and RT regimen [30].
The 6R’s offer options for modulation of the radiation response. Modulation strategies, such as via combination therapy with immunomodulating agents, should be aimed to widen the therapeutic window (Sect. 5.12) using approaches such as via radioprotection of the normal tissues thus decreasing the NTCP or by tumor radiosensitization by increasing TCP. Options for clinical application of such strategies are highly dependent on the tumor and normal tissue type included in the radiation treatment volume. Finally, to be noted is the close link between the Hallmarks of Radiobiology and the Hallmarks of Cancer [31] and therewith related therapeutic options, which have been discussed in detail elsewhere [32, 33].
5.5 Dose Fractionation (Box 5.6)
Box 5.6 Fractionation and the Dose Rate
-
Clinically used fractionation schemes are aimed at eradicating malignant tissue while sparing late responding healthy tissue.
-
The biological rationale of fractionated irradiation is based on the typical radiation response of the dynamic and heterogeneous exposed tissue and cell population.
-
Dose rates used in clinical RT vary from low dose rate with exposure times in hours-days to ultra-high dose rates with radiation dose delivery in the millisecond range.
-
The biological effect of radiation decreases with decreasing dose rate to a larger extent for normal tissue with low α/β ratio than for tumors with high α/β ratio.
-
Experimental data demonstrate that ultra-high dose rate irradiation might better spare late responding normal tissue.
-
The 6R’s of radiobiology are the biological processes involved in the dose rate effect.
5.5.1 Evolution of Fractionation
In the early years of RT, radiation oncologists soon realized that a radiation treatment course delivered in multiple fractions over several weeks resulted in better tumor control than a treatment course delivered in a single fraction and also reduced normal tissue toxicity [21]. The history of RT and fractionation is described in detail in Chap. 2. Generally spoken, a treatment course consisting of 30 daily 2 Gy fractions (total dose 60 Gy) is, at isoeffective normal tissue late response level, more effective in eradication of the tumor than a treatment course consisting of a few high dose fractions. Hence, if the total prescribed dose is divided into multiple small radiation fractions with a time interval between the fractions, tumor control could be enhanced at an acceptable level of associated morbidity, relative to a single large dose fraction [13]. However, modern RT techniques allow to give higher dose per fraction while sparing more efficiently the surrounding normal tissue. This may affect this fractionation concept further in future.
The irradiated cell population comprises the malignant tissue as well as acute and late responding healthy (“normal”) tissues. When an RT dose is delivered in several fractions, there are advantages and disadvantages in terms of tumor cell kill and normal tissue cell sparing, which are discussed in detail in Sect. 5.14.
5.5.2 Fractionation Parameters and Their Significance
Acute normal tissue effects of RT depend on both fraction size and the overall treatment time. The intensity of acute reactions depends on weekly applied total dose, i.e., the dose per fraction and number of fractions in a week. After an acute reaction has peaked, further stem cell killing cannot increase the intensity of acute reactions but can prolong the healing time. A persistent early response from severe depletion of regenerating cells is termed a consequential late injury [13]. In contrast, non-consequential late normal tissue effects depend predominantly on fraction size, while the overall treatment time has little influence. Therefore, during hypofractionation, late effects are severe while early effects are matched by appropriate dose adjustment, as discussed in detail in Chap. 6 (Sects. 6.2 and 6.3).
Another important parameter is the inter-fraction interval. Due to the slow repair kinetics of sublethal damage (SLD) in late responding tissues, a minimum of 8 h of inter-fraction interval is recommended for most tissues. The overall treatment time affects both acute effects and tumor control. Prolongation of the overall treatment time (within normal RT range) has a large sparing effect on early responding normal tissues but little sparing effect on late responding normal tissues. However, excessive prolongation of overall treatment time causes the surviving tumor cells to proliferate during treatment. For any prolongation in treatment time, extra dose is required to counteract tumor cell proliferation, due to the phenomenon of accelerated repopulation. For example, in head and neck cancer, after a lag period of 4 weeks during a course of RT, the tumor doubling rate could increase due to triggering of surviving clonogens to divide more rapidly as tumor shrinks after initiation of treatment. A dose of up to 0.6 Gy of each daily dose would be “wasted” due to increased tumor cell load [34]. When the overall treatment time is prolonged, for each extra day, local control would decrease by 1.4% (0.4–2.5%) due to accelerated repopulation.
5.5.3 Clinical Fractionation and the Dose Rate Effect
5.5.3.1 Clinical Fractionation
Differential responses of normal and cancerous tissues when fractionating RT doses can be explained by biological factors that are known as the 6R’s (see Sect. 5.4). During fractionation, tumor cells are redistributed and reoxygenated, causing further tumor damage. Moreover, the fractionation process will spare normal tissues by allowing repair of SLD between dose fractions and by allowing repopulation with new cells to occur over the overall treatment time. Therefore, a prolonged radiation treatment given over several weeks results in a greater therapeutic ratio than one or few short duration sessions because of tumor reoxygenation and early reacting normal tissue regeneration.
Radiation fractionation can lead to biologically optimal RT when the equi-effective total dose is related to the dose per fraction for tumors, early responding tissue, and late responding tissue. This relationship is determined by dose per fraction number, fraction number, tumor type, treatment site, and treatment plan. Using different normal tissues as models, it was found that with decreasing dose per fraction, the isoeffective total dose increases more rapidly than for acute effects or tumor response. This relationship can be described by the linear-quadratic (LQ) model. According to the LQ model, with appropriately chosen α/β values to represent isoeffect dose relationships at least at the 1–6 Gy dose range, a standard fractionation scheme with five small sized fractions per week over a few weeks would be beneficial regarding the tumor cure-normal tissue complication balance. Hence, deviation from standard fractionation affects the Biological Effective Dose (BED), which includes schedules with different fraction size and inter-fraction time as well as overall treatment duration. The BED is the total dose required to produce a particular effect in small dose fractions, used as the quantity to compare different fractionation regimens, see Table 5.3 for models that are used to deal with a deviation from standard fractionation.
5.5.3.2 The Dose Rate Effect
The dose rate is defined as the ratio of the radiation dose to the duration of the radiation exposure. The term should be used only in the context of short periods of time, for example, dose per second or dose per hour, the SI dose rate unit is Gy/h. Acute exposure refers to a high radiation dose delivered in seconds or minutes, and chronic exposure means that the radiation dose is delivered over a longer period of continuous exposure over hours to days to even months and years. The spectrum of dose rates used in radiation oncology is presented in Table 5.4.
Physical aspects of the dose rate are presented in Chap. 2. The application of low dose rate irradiation in brachytherapy in the clinic, is discussed in Chap. 6. FLASH is a novel RT treatment technique using ultra-high dose rates. Using FLASH, multiple studies indicate sparing of healthy tissue acute and late toxicities while maintaining tumor control, hence widening the therapeutic window (Sect. 5.2). FLASH is discussed in detail in Chap. 6. The radiation dose rate has a large biological impact on exposed cells and tissues. Both in vitro and in vivo experimental data revealed that, for a defined biological endpoint, for example, cell survival or a certain late normal tissue reaction like myelitis of the spinal cord, the biological effect decreases with decreasing dose rate. With decreasing dose rate, the total dose to obtain a certain isoeffective biological endpoint—for example, a probability of 50% loss of kidney function or reduction of the cell survival with a fraction of 0.4, is increased. Dose rate sparing is almost absent for acute responding normal tissues and tumors.
In terms of fractionation, the decrease in dose rate can be considered as lowering the fraction size of the total radiation dose to be delivered in external beam HDR radiotherapy (Fig. 5.11). Referring to the LQ model (Sect. 5.5) for comparison of biological effectiveness of different radiation treatment schemes, low dose rate irradiation could be considered as super-fractionation (Fig. 5.11 and Box 5.7).

The dose rate effect seen as an extreme form of fractionation. Cell survival following fractionated HDR irradiation with increasing number of fractions (solid curves). With an infinite number of tiny fractions, and complete sublethal damage repair, the dose-squared β parameter of the LQ tends to zero, and only the dose-linear β parameter plays a role. Then, the Biologically Effective Dose (BED) is reached for a certain endpoint effect E. Similar sparing phenomenon with decreasing dose rate in continuous LDR exposure (dotted curves)
Box 5.7 The Dose Rate and the Biological Effective Dose
-
At extremely low dose rate, i.e., irradiation with an infinitely large number of infinitesimally small dose fractions, the theoretical total dose required to produce an isoeffect is the Biological Effective Dose (BED) of the LQ model.
Thus, as exposure is elongated, the shoulder of the cell survival curve tends to become shallower, this is because the α parameter of the linear-quadratic model does not change significantly, while the β parameter tends towards 0.
This situation also implies, dependent on the fractionation sensitivities of irradiated tumor and normal tissues involved (i.e., their repair capacity characteristics expressed in their α/β values) as well as of their DNA repair kinetics (the half time for sublethal damage repair T1/2), an optimal therapeutic ratio situation. For the LQ model adaptation to correct for the dose rate effect and incomplete repair, additional parameters are introduced (e.g., Joiner and van der Kogel [16]). The dose rate effect of continuous low dose irradiation is discussed in view of the 6R’s of radiobiology in Sect. 5.4 below.
The repair process of radiation-induced DNA lesions has been explained in depth in Chap. 3. DNA DSB, if not repaired, are lethal to the cell. A DNA DSB can either be induced by single-track action or double-track action. A single-track X-ray lesion is independent of dose rate and linearly proportional to dose (the contribution of α in the LQ model). In double-track action, the two interactive single strand DNA lesions are produced by different tracks of X-ray photons, and the formation of double strand lesions is therefore dependent on the dose rate and is proportional to the radiation dose squared (the contribution of β in the LQ model). In fact, the protracted delivery of a given radiation dose reduces the effect of double-track action because time offered between lesions is long enough for repair to occur [28] (Fig. 5.12).

Illustration single-track action and double-track action. In single-track action, the two interactive lesions are produced by a single track of ionization induced by an X-ray photon that subsequently produces a dose which is independent of dose rate and linearly proportional to dose. In double-track action, the two interactive lesions are produced by a different track of ionization induced by X-rays which subsequently produces a dose which is dependent on the dose rate (decreasing the dose rate reduces double-track action) and non-linearly proportional to the radiation dose squared
5.5.3.3 Repair and the LQ Model Parameters
Figure 5.13 shows that lowering the dose rate has greater effect on cells or a tissue with a low α/β ratio, for example, 3 Gy than with a high α/β ratio of, for example, 10 Gy. At a low α/β ratio, the curves are spread out more, implying that late responding normal tissues are particularly spared relative to tumors when decreasing the dose rate.

The effect of lowering the dose rate on the survival of cells. (a) Cells characterized with an α/β ratio of 10 Gy (typical for a tumor or early responding normal tissue). (b) Cells with an α/β ratio of 3 Gy (typical for a late responding normal tissue). See text for details. (Figure from Shrieve and Loeffler [17], with copyright permission from Wolters Kluwer Health, Inc.)
Also, for a tissue having an equivalent α/β ratio, larger sparing is obtained with decreasing tissue-specific half time (T1/2) for sublethal damage repair. Similarly, as with fractionated radiation, this can be attributed to incomplete repair between the “fractions” or during continuous exposure. Hence, at longer repair half time, low dose rate irradiation is causing more damage, and less discriminative between tissues with different α/β ratio.
It is well recognized that cells in the G2 or M phase of the cell cycle are more sensitive to radiation than cells in the G1, G0, or S cell-cycle phases (Sect. 5.4, Fig. 5.8). During continuous low dose rate irradiation, the process of redistribution would push initially relative radioresistant cells into a radiosensitive cell-cycle phase. This process is dependent on numerous cellular and tissue factors, and therefore difficult to predict.
Another phenomenon that might occur is the inverse dose rate effect, which represents a reversal of the typical pattern of the conventional sparing with decreasing dose rate. For the same radiation dose, radiation delivered at a certain specific lower dose rate increases the radiosensitivity of cells in comparison to radiation delivered at a higher dose rate. This is illustrated in Fig. 5.14.

The inverse dose rate effect. When the dose rate delivered to HeLa cells is decreased from 1.54 to 0.37 Gy/h, the efficiency of cell killing increases, with damage generated similar to that from an acute exposure [35]. When cells are exposed to higher dose rates, they are kept in the phase of the cycle in which they are at the beginning of irradiation. However, use of lower dose rates may allow cells to continue cycling during irradiation. When cells are exposed to 0.37 Gy/h, cells tend to progress from other phases of the cell cycle and arrest in G2, which is a radiosensitive phase of the cycle. As a result, an enriched population of G2 cells is responsible for increasing the radiosensitivity of cells
Tumor cell repopulation during continuous low dose rate (LDR) exposure might negatively influence treatment outcome since a larger number of cells have to be sterilized if the repopulation rate outflows the duration of exposure, which might occur with fast repopulating tumor cells (e.g., cell doubling time of 24 h).
The impact of irradiation on the immune response has been shown to be dependent on the radiation dose (see Chap. 6), and the dose rate of exposure is likely to play a role [36]. The effect of low dose rate irradiation regarding reactivation of the immune response is however not well described.
Chronic low dose rate exposure will not cause oxygen depletion in initially well-oxygenated tumor cells. Initially, hypoxic cells might benefit from reoxygenation during long-term radiation exposure. However, as pointed out in Sect. 5.4, the kinetics of reoxygenation is very much dependent on the tumor type.
5.6 Whole-Body Irradiation
5.6.1 Introduction
Whole-body irradiation (WBI) or total body irradiation (TBI) refers to the therapeutic protocol in which a patient’s total body is irradiated with γ/X-rays. WBI is used as part of the conditioning regimen for transplantation of bone marrow or hematopoietic stem cells for lymphoma, leukemia, or multiple myeloma and as a palliative regimen in selected cases of lymphoma and leukemia [37]. WBI implicates irradiation of the total body, with reduction of the dose to the lungs, to lessen the hazard of radiation-induced lung toxicity [38, 39]. Historically, in the fifth and sixth decade of the last century scientists trying to reverse early responding tissue effects of radiation, demonstrated experimentally that bone marrow engrafted with hematopoietic stem cells from a donor animal “could recapitulate the blood system” and thus showed that previously irradiated bone marrow could be rescued. This contributed to the development of therapeutic techniques involving bone marrow ablation followed by bone marrow engraftment with hematopoietic stem cells for the treatment of some marrow cancers, for example, leukemia or multiple myeloma [37]. This procedure is mainly used to eliminate residual cancer cells in the transplant recipient, and to further suppress or destroy the immune system; subsequently, it serves to prevent immunologic rejection of blood stem cells or transplanted donor bone marrow. Thus, the chances of engraftment are increased, and the bone marrow stromal cells of the patient are spared [38, 39].
5.6.2 Details of Radiobiological Mechanisms of Whole-Body Irradiation
5.6.2.1 Leukemia
Since a characteristic of WBI is that it can sterilize small numbers of widely spread cells that are sensitive to radiation, this makes it a treatment option for (residual) marrow disease. Biologically, leukemia is associated with a spectrum of intrinsic cellular radiosensitivity that ranges from notable radiosensitivity to significant radioresistance, which determine the extent of leukemic cell killing. The molecular biology responsible for the variety in radiosensitivity of leukemia is not entirely known, but increased apoptosis seems to require functional p53, c-myc, and Bcl2 genes. Therefore, it seems that radiosensitivity results from the apoptosis retention after activating p53, c-myc, and Bcl2 genes by radiation [40]. RT in conjunction with a wide range of treatment modalities such as (myeloablative) chemotherapy and the subsequent graft-versus-tumor effect are therefore required to obtain significant eradication of malignant clones [22, Chap. 16].
5.6.2.2 The Normal Hematopoietic System
Bone marrow stem cells typically have D0 values ranging from 0.5 to 1.4 Gy. These cells are therefore intrinsically radiosensitive. Even though hematopoietic rescue (i.e., stem cells) could allow the delivery of high doses that eliminate the recipient’s marrow cells which in turn prepares the stem cell microenvironment for repopulation to occur, this procedure is associated with long-term or life-threatening consequences. Critical organs of concern in WBI are those described as late responding tissues. Fortunately, as effect on these tissues is dependent on total dose, dose rate and fractionation, appropriate scheduling of the treatment allows some protection. While a modest number of cancer cells being radiosensitive will be killed, complete cancer cell killing may not always be possible with radiation alone. Therefore, TBI often needs to be given together with chemotherapy. Moreover, incomplete bone marrow ablation may result in mixed chimerism of bone marrow after transplant [22, 40].
As a result of immunological mismatch between recipient and donor, rejection of donor stem cells may occur. In order to avoid this, TBI is used to prevent the recipient from rejecting donor stem cells.
Bone marrow transplantation results are influenced by the treatment schedule. Lymphoid cells repair a large amount of radiation-induced DNA damage during the time interval between fractions. Hence, the effectiveness of fractionated TBI is reduced significantly in comparison with single-dose TBI and results in more graft rejections. However, the fractionation effect is reversed for bone marrow stromal cells (“colony-forming unit fibroblasts”). The success of engraftment is based on the likelihood of sparing bone marrow stromal cells, and when treatment is delivered as single-dose TBI, the likelihood of damaging both bone marrow stromal cells and their progenitors increases. Importantly, the effectiveness of single-dose TBI is increased significantly in comparison with fractionated TBI, but at the cost of increased long-term toxicity [22].
5.6.2.3 Palliation
Unlike curative RT, palliative RT is used to control the symptoms of advanced, incurable cancer (the primary tumor or metastatic deposits) by slowing down tumor growth, controlling symptoms and causing cancer to regress [41]. WBI may be effective for palliation, especially for advanced leukemia or lymphoma, using rather low doses in the order of 0.1 Gy/fraction. In experiments with solid tumors, tumor cells with colony-forming abilities in both experiments of formation of artificial metastases and naturally developing metastases, these tumors could be suppressed with specific low doses [42]. It is assumed that either chronic TBI or low dose total body irradiation may stimulate the immune system to eliminate metastatic cancer cells. However, nowadays, TBI is only very rarely used for this indication. For further information, see tumor microenvironment changes and abscopal effect discussed in Sect. 5.15.
5.6.3 Fractionation Dose Effect in Whole-Body Irradiation
In WBI, the doses delivered for transplantation of bone marrow or stem cells are in the range of 10–12 Gy [43]. To reduce long-term complications in the recipient, this dose is typically divided into 2 Gy fractions [14, 44]. In the so-called reduced conditioning regimens, single fractions of 2 Gy or two fractions of 2 Gy are given. When WBI is split into multiple small fractions and spread over a period of time, outcomes are generally improved, and toxicity is diminished. While the former is due to the fact that the dose is still adequate to eradicate both any cells of residual malignant tissue and the recipient’s bone marrow, the latter is explained in Sect. 5.2 [45].
5.6.4 Dose Rate Effect in Whole-Body Irradiation
The dose rate in RT influences the effectiveness of the radiation exposure. An explanation of how a survival curve may become shallower at low dose rates was discussed in Sect. 5.5. In WBI, the pattern is different from localized RT because the effect of radiation dose depends on the tissue type. When low dose rate is used, the incidence of normal tissue toxicity is decreased in comparison with high dose rates [46,47,48]. However, changing the radiation dose rate from the low dose rate to high does not affect the probability of engraftment success [49, 50].
5.7 Prediction of Radiation Response of Tumors (Box 5.8)
Box 5.8 Tumor Response Prediction
-
Functional parameters including reoxygenation, redistribution, repopulation, repair, and radiosensitivity are traditionally used to define radio-responsiveness.
-
The role of repopulation also proved to be a robust predictive marker as illustrated in head and neck cancer where increased tumor expression of EGFR indicates efficacy of accelerated radiation.
-
DNA, RNA, and proteins have recently been identified to define tumor RT responsiveness yet few of them have attained clinical validation and/or application.
5.7.1 Principles of Prediction of Radiation Response of Tumors
Radiation treatment has been improved greatly over the last two decades by integrating 3-D anatomy into planning systems, and developing of image-guided (IGRT), intensity-modulated (IMRT) and intensity modulated arc radiation therapy (IMAT) techniques, resulting in individualization of treatment. Radiation treatment portals and arcs are much more tailored to the anatomy of each patient’s tumor and normal tissue. Today medical professionals prescribe RT taking into account the type of primary tumor, its grade, stage, location, size, biological characteristics, and concomitant treatments. However, these clinical parameters do not give an accurate prediction of the effect of RT since wide variations in response occur between patients given the same treatment and having similar tumors [51,52,53]. Furthermore, diverse treatment options are now available. Moreover, several tumor RT sensitizing strategies using different drugs which can enhance the effects of RT, especially those that target the molecular pathways, are becoming available now. Treatments frequently employ combined approaches. One such avenue currently underway involves trials that combine chemotherapy/RT with immunotherapy.
RT can differentially affect tumor responses due to a variety of radiobiological factors, which are referred to as the 6R’s. Among them, hypoxia, proliferation, and radiosensitivity have proved to be fairly good predictive markers [51,52,53,54,55,56,34]. Beyond these well-known classical biomarkers (BMs), there are also a number of promising candidate molecular biomarkers currently being tested in preclinical and clinical studies such as genetic and epigenetic factors as discussed in Sect. 5.10. In this section, classical and modern BMs as well as their role in predictive assays will be discussed in addition to the available methods used to detect them.
5.7.2 Classical Factors
RT constitutes approximately 60% of cancer treatment. If a clinical assay could successfully predict the RT response, it would have wide-ranging clinical implications. A broad group of old-fashioned radiobiological variables that affect RT outcome including tumor oxygen status, the degree of repopulation or proliferation rate, intrinsic radiosensitivity, and both individual and tumor radiosensitivity is shown in Fig. 5.15.

Review of classical biomarkers used to obtain information on relevant features of radiobiology
Predictive factors in therapy may relate more directly to primary tumors and their local control. Metastatic disease may need to be considered separately even though it clearly plays a significant role in survival of the patient. Research to develop predictive assays for tumor RT response should generally measure local control and normal tissue effects [16, 51, 52].
5.7.2.1 Tumor Oxygen Status
Tumor vascular beds differ significantly from those in normal tissues in their structure and physiological characteristics. Tumor-related blood vessels are composed of single-layered endothelium, commonly containing gaps between the endothelium taken up by tumor cells, resulting in immature capillaries. A dysfunctional blood supply through the tumor reduces oxygen delivery, resulting in areas of tumor hypoxia, acidic intra-TME nutritional deprivation, and therewith the tumor response to IR For more information about predictive tests to assess Oxygen Effect to Tumor Hypoxia see Sect. 5.8.
5.7.2.2 Repopulation
Tumor repopulation is a key factor contributing to treatment failure after RT. Alternative fractionation schemes have been proposed as methods for modulating interfraction tumor repopulation. A phase III randomized trial in over 1000 patients with head and neck cancer showed significant improvement in regional control following accelerated and hyperfractionated RT as compared to conventional fractionated RT. Additional evidence, which has shown the importance of proliferation, demonstrated that higher doses are needed to control a tumor when overall time of treatment is prolonged. Clinical evidence that tumor repopulation is an important mechanism for treatment failure is notably apparent in a subset of patients. Therefore, evaluation of tumor repopulation has been a priority for developing predictive tests [52]. Table 5.5 shows several tests that can be performed in vitro and in vivo to measure tumor repopulation.
5.7.2.3 Intrinsic Radiosensitivity
Various types of cell death, such as apoptosis and autophagy, which also result in a loss of colony-forming ability, contribute to tissue reactions caused by IR. Many publications proposed that cellular radiosensitivity could be measured by the clonogenic assay. A technical challenge of dispersing tumor cells ex vivo has interfered with its clinical application, however [52].
For more information about predictive tests to assess radiosensitivity, see Chap. 7.
5.7.3 Modern Factors
Radiation responsiveness was traditionally defined using the 6R’s (see Sect. 5.4) but only three factors proved reliable as prognostic markers: hypoxia, repopulation, and radiosensitivity, and hence RT regimens have been modified according to fraction size, dose per fraction, and overall treatment time as depicted in Fig. 5.16. Individual assessments of these parameters could have predictive value since each of these parameters has a substantial effect on the outcome of the RT. Assays based on measurements of these parameters, however, had mixed success in developing predictive assays for many reasons. Firstly, the lack of success may be explained by the fact that few quantitative differences exist between human tumors and normal tissues, and their heterogeneity overlaps in many ways. Secondly, it was intended that the 6R’s can be used to understand emerging phenomena in radiation biology rather than predicting its outcomes.

Review of modern biomarkers used to obtain information on relevant features of radiobiology
The first molecular techniques were applied to radiobiology about two decades ago and soon revealed the existence of proteins and genes that respond to and influence the cellular outcome of IR [53]. Radiation response of tumors is associated with a complex series of gene and protein alterations some which also are influenced by the underlying genomic alterations, for example, mutations. When cells experience IR-induced damage, it was early on observed that key proteins are induced [57, 58]. An example is the p53 protein which upon exposing cells to a photon beam is induced and control multiple pathways. For instance, a single fraction of 20 Gy X-rays was observed to induce key proteins such as MDM2 and CDKN1A in some cell lines, both which is regulated by p53 [58]. Furthermore, the response to radiation is influenced by polymorphisms in genes encoding proteins that participate in DNA damage repair, as well as by mutations affecting these genes. When cells experience radiation insult, multiple genes undergo a series of up and down regulations interacting through many pathways including p53-regulated genes such as p21 (CIP1/WAF1) and GADD45A. Also, the response to radiation is influenced by methylation, acetylation, ubiquitylation, phosphorylation, and sumoylation of genes and proteins which control the DNA damage repair. For instance, the presence of hyper-methylated promoters means that a gene is becoming actively transcribed; hyper-methylation of promoters attracts proteins that inhibit transcription and turn it off. Non-coding RNAs also contribute to radiation response. There are complementary forms of RNA, namely microRNA (miRNA) that are not translated into protein and play an important role in the initiation and progression, repopulation and programmed cell death (see Chap. 3). Therefore, this mechanism is reflected in terms of sensitivity or resistance to IR [16, 59]. Overall, gene and protein expression are altered in the tumor itself and by radiation which affects cellular outcomes and causes a heterogeneity of RT response in tumors. Accordingly, protein, DNA and RNA analysis can be used to obtain information on relevant features of radiobiology using several types of measurements such as genomics, transcriptomics, epigenomics, or proteomics, which analyze DNA, RNA, DNA–chromatin interactions and proteins, respectively, as described in Fig. 5.17 and Table 5.6.

Schematic view of biomarkers. Proteins, DNA chromatin, DNA, or RNA that are analyzed by proteomics, genomics epigenomics, genomics, or transcriptomics, respectively
5.8 Tumor Hypoxia and Therapeutic Approaches
Hypoxia refers to conditions with low oxygen. The oxygen concentration of most normal tissue in the human body is around 5–7% and tissues with less than 3% oxygen are regarded as hypoxic. Hypoxic cells are known to be more resistant to radiation and chemotherapy. Hypoxia is also a potent microenvironmental factor promoting metastatic progression of cancer [60].
Hypoxia in tumor cells can be of two types—acute hypoxia or chronic hypoxia. Acute hypoxia is a transient perfusion-limited hypoxia due to transiently occluded blood vessels [61]. Chronic hypoxia is a diffusion-limited hypoxia and can lead to necrosis [62].
Oxygen can generally diffuse approximately 150 μm at the arterial end of the capillary and less at the venous end (Fig. 5.18). Therefore, when the radius of the tumor is less than 160 μm, there is no central necrotic region. Between 160 and 200 μm, there may or may not be a hypoxic center. When the radius is more than 200 μm, the central portion consists of anoxic necrotic cells while the next layers consist of cells with different degrees of hypoxia and aerobic actively dividing cells at the outer layer [62]. The central portion becomes necrotic because the cells are deprived of both oxygen and nutrients.

Diffusion of oxygen through tumor. As the distance from the blood supply increases, the oxygen levels available for the cells decreases. As the cells grow more hypoxic, they become more radioresistant
As hypoxic cells are more radioresistant than oxygenated cells (Chap. 3), irradiation of a tumor will predominantly kill the outer layer of cells leaving the hypoxic cells. One way to reduce this problem is to divide the radiation dose into many daily fractions, which will allow the hypoxic cells nearest to the oxygenated cells to be reoxygenated after the oxygenated cells have been killed [63]. In this section, we will give an overview of the most important mechanisms and pathways induced by hypoxia since these are potential targets in connection with treatments.
5.8.1 Oxygen Effect
As described in Chap. 3, oxygen modifies the biological effects of low LET IR. For such radiation, DNA damage predominantly occurs through the indirect effect of radiation-induced water radicals. In the absence of oxygen, DNA radicals can become chemically restituted through donation of hydrogen atoms by SH-compounds (such as glutathione). However, if molecular oxygen is present, RO2· is produced which cannot be restored. Oxygen thus “fixes” the damage produced by free radicals (i.e., makes the radiation damage permanent). Therefore, cells are more sensitive to radiation in the presence of oxygen than in its absence.
The relative radiosensitivity of cells increases dramatically when the oxygen tension increases from 3 mmHg (0.4% O2) to about 30 mmHg (4% O2), which corresponds to the oxygen concentration in venous blood [64]. Beyond this, it reaches a plateau with no further effect of increasing the oxygen tension. In order to compete with restitution, molecular oxygen must be present during or within microseconds after the radiation exposure because the lifetime of the free radicals generated by the radiation is less than about 10−5 s.
The oxygen enhancement ratio (OER) is the ratio of doses under hypoxic to aerated conditions that produce the same biologic effect. In vitro studies have shown OER of X-rays to be around 3.5 for high doses and around 2.5 in low dose regions of the cell survival curves [65].
High LET radiations like alpha particles cause direct and complex damage to DNA with little possibility for restitution. Therefore, there is no enhanced effect with presence of oxygen (i.e., OER is 1). It is intermediate for neutrons and comparatively high for low LET radiations such as X-rays and gamma rays [66, 67]. OER decreases as LET increases. The OER falls slowly until about 60 keV/μm of LET, then falls rapidly and reaches 1, i.e., no oxygen effect, when LET reaches about 200 keV/μm [66]. Thus, one way to target hypoxia is to use high LET radiation (Box 5.9).
Box 5.9 The Oxygen Enhancement Ratio
The oxygen enhancement ratio varies with:
-
Cell cycle phase
-
Type/quality of radiation
-
Radiation dose and dose rate
5.8.2 Hypoxia Response Pathways
When a tumor grows, the existing vasculature will not be able to provide oxygen and nutrients to the more distant cells resulting in regions of hypoxia. During hypoxic conditions, the cells that survive are those that undergo adaptive responses, which are not only critical to survival but also promote malignancy and metastasis. The main purposes of the adaptive responses are to uphold ATP production and save energy as well as nutrients. ATP production is maintained by the formation of new blood vessels (angiogenesis), increased production of red blood cells (erythropoiesis), switch to anaerobic metabolism, and migration to a more favorable environment (metastasis). The processes to save energy include reduction of protein synthesis and recirculation of nutrients (autophagy). The signaling pathways regulating these processes are induced by upregulation of hypoxia-inducible factors (HIFs) and activation of the unfolded protein response (UPR).
5.8.2.1 Hypoxia-Inducible Factor (HIF)
HIF-1 was discovered in 1995. Its importance is emphasized by the award of the Nobel prize in Physiology or Medicine 2019 to William Kaelin Jr., Peter J. Ratcliffe and Gregg L. Semenza for their work in elucidating how HIF senses and adapts cellular response to oxygen availability. The HIF protein family consists of three different alpha subunits (HIF-1α, HIF-2α, HIF-3α), which bind to the same type of β-subunit. In this section, we will only discuss HIF-1, which is the only one which is ubiquitously expressed and the most well studied. In the presence of molecular oxygen, proline-hydroxylase enzymes (PHD) hydroxylate HIF-1α, i.e., PHDs add two OH-groups to proline residues on HIF-1α using oxygen as a cofactor. Hydroxylated HIF-1α is recognized by von-Hippel-Lindau (VHL)-ubiquitin ligase complexes, which then adds ubiquitin-groups that target HIF-1α for degradation. Without oxygen, HIF-1α is stabilized allowing it to bind to the β-subunit and activate gene transcription.
HIF-1 is known to induce transcription of more than 60 genes by binding to a common promoter called hypoxia response element (HRE). Some of these genes are involved in increasing oxygen supply, such as VEGF (which induces angiogenesis), erythropoietin (which stimulate red blood cell production), and iron transport to erythroid tissue. HRE-regulated genes are also involved in cell proliferation, such as insulin-like growth factor-2 (IGF-2) and transforming growth factor-α (TGF-α). HIF-1 also induces transcription of genes involved in switching of metabolic pathways to use glycolysis as a primary mechanism of ATP production. HIF thus regulate transcription of genes involved in glucose transport into the cell (i.e., GLUT1 and GLUT3), and in maintaining intracellular pH during increased lactate production such as carbonic anhydrase 9 (CAIX) and monocarboxylate transporter 4 (MCT4).
In addition to promoting hypoxia tolerance, HIF-1 also upregulates transcription of genes involved in several steps of metastasis: angiogenesis, epithelial-mesenchymal transition (EMT), cell motility, intra/extravasation, and the formation of a premetastatic niche facilitating colonization of metastatic tumor cells.
Stabilization of HIF and activation of its downstream pathways occur at relatively moderate levels of hypoxia (<about 2% O2) as compared to radiation resistance, which occurs below about 0.1% O2 [68].
5.8.2.2 The Unfolded Protein Response (UPR)
Similar to IR resistance, activation of the unfolded protein response (UPR) occurs below about 0.2% O2 and contributes to hypoxia tolerance by reducing protein synthesis and inducing autophagy. UPR is activated by accumulation of unfolded and misfolded proteins in the endoplasmic reticulum (ER). Such accumulation takes place under hypoxia because post-translational disulfide bond formation, which is necessary for correct protein folding, have been shown to be oxygen dependent [69]. There are three ER stress sensors in the ER membrane, PERK (protein kinase RNA-like endoplasmic reticulum kinase), IRE1a (inositol requiring kinase 1a), and ATF6 (activating transcription factor 6). All three are activated by the release of BiP (Binding immunoglobin Protein) from the ER membrane. Activated PERK phosphorylates eIF2α (Eukaryotic translation initiation factor 2 subunit α), which inhibits translation of protein except for some key proteins, which remain translated [70]. One of these is ATF4 (activating transcription factor 4), which induces transcription of genes required for autophagy, amino acid synthesis and import, redox balance, and angiogenesis [71]. However, if the stress becomes excessive, ATF4 can switch from pro-survival to pro-death and induce apoptosis [72]. Active IRE1α (Inositol-requiring transmembrane kinase/endoribonuclease 1α) reduces protein synthesis through the degradation of selected mRNAs. It also activates an inflammatory response and promotes autophagy as well as apoptosis. BIP release allows ATF6 translocation to Golgi, where cleavage of this protein results in release of transcriptionally active ATF6 (DNA-binding cytoplasmic domain of ATF6), which contributes to restoring ER homeostasis [70].
5.8.3 Measurement of Tumor Hypoxia
Hypoxia in tumor cells can be measured using oxygen probes or protein markers for hypoxia. Polarographic electrodes, in particular Eppendorf electrodes, have long been regarded as the gold standard for measuring tumor hypoxia. The limitations of these are that they must be inserted into the tumor damaging the tissue and that they consume oxygen resulting in a high signal-to-noise ratio for low oxygen levels [73]. Fiber-optic probes have also been developed [74], but the disadvantage of both is that this is an invasive method, which can only be used in accessible tumors. Another method is injection of pimonidazole to patients before biopsies or total resection of the tumor. Pimonidazole is a 2-nitroimidazole compound, which binds to thiol-containing proteins specifically in hypoxic cells, and it can be used in immunohistochemical analyses to quantitative assessment of tumor hypoxia [75].
A noninvasive alternative method is hypoxia PET imaging. 2-Nitroimidazoles have been developed as radiosensitizers due to their ability to undergo up to six electron reductions. However, they can also be used as radiotracers carrying a positron emitter. The reduction requires the activity of reductases that are only present in viable hypoxic cells resulting in the radiolabeled molecules being accumulated in hypoxic regions visible to PET imaging. Examples of nitroimidazole or nitroimidazole-derivative tracers are 18F-Fluoromisonidazole (18F-FMISO), 18F-Fluoroazomycin-Arabinofuranoside (18F-FAZA), 18F-labeled fluoroerythronitroimidazole (18F-FETNIM), 18F-fluoroetanidazole (18F-HX4), and 18F-2-(2-Nitro-1H-imidazol-1-yl)-N-(2,2,3,3,3-pentafluoropropyl) acetamide (18F-EF5) [73, 76]. Other redox-sensitive compounds include Copper(II) diacetyl-bis (N4-methylthiosemicarbazone) (60Cu-ATSM) and 123I-iodoazomycinarabinoside (123I-IAZA) [77]. Of these, 18F-MISO is the most extensively used in both preclinical and clinical studies. F-MISO is highly lipophilic, which means it can easily diffuse across cell membranes but also that the clearance of the tracer from normoxic tissue is very slow. This results in a low tumor to blood ratio and the need to wait for typically 2 h between tracer injection and imaging. A general problem in PET imaging is the low spatial resolution.
Magnetic resonance imaging (MRI) is non-ionizing and has a better spatial resolution than PET. Different MRI techniques have been evaluated for quantification of the blood oxygenation level in tissue. Blood oxygen level-dependent MRI (BOLD-MRI) uses the paramagnetic quality of deoxyhemoglobin in contrast to oxygenated hemoglobin. This can be measured as an increase in T2*-relaxation rate. A similar technique, tissue oxygen level-dependent MRI (TOLD-MRI) uses an increase in T1 relaxation caused by the presence of dissolved oxygen. A third technique is dynamic contrast-enhanced MRI (DCE-MRI) in which a paramagnetic contrast agent such as gadolinium is used and imaged over time [78].
HIF and its target proteins such as CA9 and GLUT1 are upregulated under hypoxia (see above). Accordingly, they have been studied as endogenous markers for hypoxia and seem to co-localize with 2-nitroimidazoles. However, their expression can also be regulated by other factors than oxygen, which reduces their reliability in quantifying tumor hypoxia.
5.8.4 Therapeutic Approaches to Tumor Hypoxia
Tumor hypoxia is a major barrier for effectively treating cancer patients. Various approaches have been investigated both in vitro and in vivo to overcome hypoxia in tumor cells. The main clinical strategies to mitigate treatment resistance due to tumor hypoxia are either to improve oxygenation, mimic the effect of oxygen, use drugs that are cytotoxic only in hypoxic cells, apply dose escalation to hypoxic areas, or inhibit pathways that are important for cell survival under hypoxia (Table 5.7).
5.8.4.1 Improving Oxygenation to Tumors
The simplest approach to reduce tumor hypoxia is to let the patient breathe hyperbaric oxygen (HBO) (2–4 atmosphere 100% oxygen). However, this may cause vasoconstriction. To avoid this, a mixture of 5% carbon dioxide and 95% oxygen called carbogen was introduced. Carbogen breathing reduces chronic diffusion limited hypoxia. For acute hypoxia, nicotinamide can be used to mitigate transient fluctuations in tumor blood flow [79]. In the ARCON strategy, a combination of carbogen and nicotinamide was used in association with accelerated hyperfractionated RT to reduce tumor proliferation during treatment and spare normal tissues [80].
5.8.4.2 Hypoxic Cell Radiosensitizers
Hypoxic cell radiosensitizers are chemical or pharmacologic agents that can increase the radiosensitivity of cells, which are deficient in oxygen. These agents are oxygen substitutes that diffuse into poorly vascularized areas of tumors and penetrate to reach hypoxic cells within the tissue. Therefore, when administered with radiation these agents cause fixation of the free radical damage caused by IR and thereby increases its lethal effect. A radiosensitizer should have an increased effect on tumors relative to normal cells to achieve a therapeutic window/gain. An ideal hypoxic cell radiosensitizer should be chemically stable; have low metabolic breakdown; be highly soluble in water and lipids; act at all phases of cell cycle; be active at low doses of radiation; and cause low toxicity to normal cells [81].
The commonly used hypoxic cell radiosensitizers belong to the nitroimidazole group of drugs. In the nitroimidazole ring structure, a nitro group can be present in any of positions 2–5. Based on the position of the nitro group, drugs are named 2-nitroimidazole to 5-nitroimidazole. The presence of the nitro group in position 2 increases electron affinity and IR sensitization capacity [82]. Therefore, 2-nitroimidazole is a more efficient hypoxic cell radiosensitizer. Three widely used nitroimidazole compounds in clinical trials and their characteristics are summarized in Table 5.8.
The enhancement ratio is the ratio of X-ray doses needed to control 50% of the tumors in the absence and presence of the drug. It was found to be around 1.82 for misonidazole in a mouse mammary tumor model for a single-dose treatment but for multifraction regimens, it is usually less [83].
The Danish Head and Neck Cancer 5 (DAHANCA 5) study which comprised 422 patients with pharyngeal and supraglottic carcinomas found a clear benefit of using nimorazole as a radiosensitizer. Thus, no increase in late radiation toxicity and a highly significant benefit in 5-year local control rates and disease-free survival rates. The use of nimorazole as a radiosensitizer in head and neck cancer is standard practice in Denmark [55].
5.8.4.3 Bioreductive Drugs
Bioreductive drugs are drugs that are reduced by cells to form active cytotoxic agents only under hypoxic conditions while being non-reduced in an oxic cell. Thus, the differential action on hypoxic cells (i.e., tumor cells) compared to oxic cells, i.e., normal cells) gives a therapeutic window/gain. Unlike the direct action of hypoxic cell radiosensitizers, which increases the radiosensitivity of cells, bioreductive drugs act synergistic with radiation to improve tumor cell kill [84].
The hypoxic cell cytotoxicity ratio (HCR) is the dose of a drug required to kill a certain fraction of oxic cells compared to the dose of drug required to kill an equal fraction of hypoxic cells. HCR varies for different classes of bioreductive drugs [85]. The main classes of bioreductive compounds are fused ring benzoquinones (e.g., mitomycin C), organic N-oxides (e.g., tirapazamine), and nitroheterocyclic compounds (e.g., RSU-1089), among which tirapazamine is the widely investigated drug.
5.8.4.4 Hypoxia-Targeted Radiotherapy
As described above, imaging techniques such as PET and MRI can be used to identify hypoxic regions within the tumor. They are used in RT treatment planning to define radioresistant sub-volumes that can be targeted with boost dose or dose escalation. Targeting hypoxic sub-volumes in tumors by adaptive RT/dose painting can lead to improved tumor control. There is early clinical evidence from a phase 2 randomized trial that dose escalation to hypoxic volumes using 18F-FMISO PET is feasible without increased normal tissue toxicity in head and neck cancers [86]. The utility of hypoxia-directed RT dose painting is being studied in other cancer subsites such as lung, pancreas, and cervix cancer [87,88,89].
5.8.4.5 Molecular Targeted Drugs
Drugs that target specific molecules to improve radiosensitivity in hypoxic tumors are being investigated in preclinical and clinical studies. Examples are dichloroacetate, electron transport chain inhibitors, and other pathway inhibitors.
Dichloroacetate (DCA) is a specific inhibitor of the pyruvate dehydrogenase kinase (PDK), which has been shown to increase ROS production in hypoxic cancer cells but not in aerobic cells leading to radiosensitization of hypoxic tumor cells [90]. Electron transport chain inhibitors have been developed to reduce oxygen consumption and thereby improve tumor oxygenation. One such is an anti-diabetic drug, Metformin, which has been shown to reduce oxygen consumption through inhibition of mitochondrial complex I and thus improve tumor oxygenation and response to RT [91]. Other approaches include inhibition of pathways that are important for cell survival under hypoxia (e.g., HIF-1α, CAIX, MCT4, VEGF, and lactate dehydrogenase-A (LDHA) [92].
Box 5.10 Tumor Hypoxia in Radiotherapy
-
Tumor hypoxia can be acute (perfusion-limited) or chronic (diffusion-limited).
-
Tumor hypoxia leads to resistance to RT and chemotherapy.
-
Hypoxic tumor cells undergo adaptive responses that are critical to survival but also promote malignancy and metastasis.
-
Strategies to either increase oxygenation or interfere with the hypoxic response pathways can improve the outcome of RT.
-
Drugs that either mimic the effect of oxygen or are toxic under hypoxic conditions can be used to increase the effect of RT.
5.9 Tumor Resistance and Progression
-
RT is an effective cancer treatment, but a large portion of patients subsequently experience radioresistance and recurrence of their cancers.
-
Mechanisms of radioresistance in cancer treatment are related to several factors: Increased DNA damage repair capacity, cell-cycle redistribution, cancer stem cell resilience, signaling pathways, epithelial-mesenchymal transition (EMT), and tumor metabolism.
5.9.1 Introduction
RT is one of the most efficient therapeutic regimens for various tumors. The main factor in determining the therapeutic effect is the tumor radiation response that is correlated to intrinsic or acquired radioresistance after the treatment [93]. Intrinsic radioresistance relies on inherent characteristics of the tumor (for instance, the presence of cancer stem cells (CSCs). The acquired radioresistance is a process of tumor adaptation to the RT-induced changes and develop resistance to the IR. This complex process involves multiple genes, factors, and mechanisms [94, 95]. Radioresistance leads to poor prognosis in cancer patients, and it represents the main reason for RT failure, which can ultimately lead to tumor recurrence and/or progression/metastases [95]. Radioresistance of tumors is influenced by several factors, including increased DNA damage repair capacity, cell-cycle redistribution, CSCs resilience, signaling pathways, epithelial-mesenchymal transition (EMT), and tumor metabolism (Fig. 5.19).

Description of the tumor biological responses to radiation and the mechanisms for its resistance
5.9.2 DNA Damage Repair Ability
RT kills cancer cells mostly by inducing DNA damage. Radiation-induced DNA lesions are multiple including base and sugar modifications, cross-links, single-stranded breaks (SSBs), and double-stranded breaks (DSBs) [96] (see also Chap. 3). DNA DSBs are responsible for most of IR-induced cell killing [97]. To respond to DNA damage, cells initiate signaling pathways named DNA damage response (DDR) signaling. The capacity of tumor cells to trigger a DDR following radiation, through initiation of DNA repair and cell-cycle checkpoints, promotes radiation resistance and tumor cell survival. In radiobiology, it is considered that cells with a high capacity to trigger DSBs repair will be radioresistant, while cells with frail repair ability will be more sensitive in response to radiation [93].
Activation of DDR and proper repair of DNA lesions are of paramount importance for cellular integrity, survival, and genomic stability [98]. There are four main DNA repair pathways in tumor cells: DNA DSB repair, base excision repair (BER), nucleotide excision repair (NER), and mismatch repair (MMR) [99]. BER and SSB repair (SSBR) are important for repairing altered bases and SSBs. Besides DSBs caused directly by IR unrepaired damaged bases and SSBs, can generate DNA DSBs when they face a replication fork. Non-homologous end joining (NHEJ) and homologous recombination (HR) two principal cellular DSB repair pathways (see Chap. 3).
5.9.2.1 Dysfunctional DNA Repair in Cancer
Defects in DNA repair factors are a characteristic of many cancers which promote genomic instability and mutation in cells [98, 100,101,102]. Deficiency of DDR and DNA repair genes caused diseases such as Xeroderma pigmentosum, Ataxia Telangiectasia (ataxia telangiectasia mutated (ATM)), Nijmegen breakage Syndrome (Nibrin (NBS1)), Werner syndrome (WRN), Fanconi anemia (FA), associated breast cancer syndrome (BRCA1 and BRCA2), lead to cancer susceptible disease syndromes (reviewed in [98, 102] and in Chap. 7). Cancers present germ line mutations, somatic mutations or distinctive expression of DDR components that lead to the impairment of DDR and altered checkpoints. In addition, DNA replication stress due to oncogene stimulation also causes initiation of DDR and tumor progression.
The same DNA repair deregulation that commenced carcinogenesis is associated with cancer progression, aggressiveness [102]. There are situations in which the tumor is preserved after the therapy, becoming even more aggressive. It is known that IR can activate stress and adaptive response to DNA lesions causing cancer progression, metastatic behavior [103]. Several dysfunctional DDR signaling genes are found in cancer and correlated with radioresistance (Table 5.9).
5.9.3 Cell-Cycle Redistribution
Activation of cell-cycle checkpoints is important downstream IR-induced DDR signaling. Several molecules in the cell-cycle checkpoints regulate and block cell-cycle progression to allow for DNA repair and prohibit premature entering into mitosis with unrepaired DNA lesions. The same machinery responsible for DNA damage recognition, ATM and ATR, activates the cell-cycle checkpoints kinases CHK1 and CHK2 to regulate the checkpoints and repair DNA damage. Two distinct signaling cascades, the ATM-Chk2 and ATR-Chk1 axes respond to DSBs damages, respectively, to ssDNA. Activated Chk1 and Chk2 inactivate CDC25C by phosphorylation. In the absence of CDC25C phosphatase activity, WEE1 kinase impedes cyclin-dependent kinase 1 and 2 (CDK1/2) activity triggering cell-cycle arrest in the G2/M phase and promoting DNA damage repair (reviewed in [93, 95, 111]). Molecules in the cell-cycle checkpoints were also found to be linked with the development of radioresistance (e.g., ATM, p53, p21, Chk2, Cdc2).
5.9.4 Modification of Extracellular Signaling Pathways and Tumor Suppressors
A considerable amount of research has been done on the modulation of cellular signaling pathways in response to IR and many of them are involved in radioresistance, including extracellular and intracellular signaling cascades. This subsection focuses specifically on the major oncogenic signaling pathways such as Ras and phosphatidylinositol-3′-kinase pathways (PI3K)/AKT that contribute to radioresistance (Fig. 5.20).

Review of tumor suppressors and the molecular signal pathways that contribute to radioresistance
Radiation causes many behavioral changes in the extracellular domain of EGFR as well as to its kinase domain, including truncations and common mutations of an extracellular domain of EGFR [16, 116, 117]. Photon radiation can cause EGFR aberrations that over-activate downstream pro-oncogenic signaling pathways, including the RAS-RAF-MEK-ERK MAPK and AKT-PI3K-mTOR pathways that modulate cell processes, including cell resistance to RT, angiogenesis, migration, invasion, and apoptosis [118,119,120].
When EGFR is activated by photon radiation, this activation, triggers the RAS-RAF-MEK-ERK MAPK pathways. Radiation resistance is associated with this pathway targets due to its pro-survival nature, and mutated RAS has been associated with resistance to photon-irradiation in cancer cells [121,122,123,124].
Also, when EGFR is activated by photon radiation, this triggers the AKT-PI3K-mTOR pathways and activated EGFR induces the phosphorylation of PI3K and downstream AKT phosphorylation [124]. Radiation resistance is also linked to activation of this target with tumor cells being protected by decreased autophagy and apoptosis, as well as increased DNA repair capacity [125].
Overall resistance to photon irradiation appears to be modulated by the transmembrane protein EGFR but also repression of tumor suppressors. In this circumstance, the tumor suppressors LKB1 and PTEN has been reported to correlate to resistance.
5.9.5 Activation of Epithelial-to-Mesenchymal Transition (EMT)
EMT is a series of changes involving epithelial cells that phenotypically transform into mesenchymal cells [126]. To acquire local invasiveness, the first step involves a profound phenotypic shift within the primary tumor of cancer cells. The progression of cancer is dependent on tumor cells undergoing the EMT and invading blood vessels [127, 128]. This results in invasion and distant metastasis, the main cause of cancer-related death. Normal epithelial cell layers cannot accommodate the motility and invasiveness of malignant carcinoma cells, yet this epithelial organization plan continues to be recognized in many primary carcinomas. Despite their overall topology differing quite a bit from that of comparable normal epithelia, these tumors contain well-organized sheets of epithelial cells. Although it’s not confirmed, cancer cells require a partial or complete EMT to become invasive as in the following steps [129]:
-
1.
Factors, including growth factor (GF), initiate EMT by promoting E-cadherin replacement by N-cadherin.
-
2.
Due to its ability to allow these tumor cells to form homotypic interactions with a variety of types of mesenchymal cells, N-cadherin increases the affinity of cancer cells for stromal cells composed of fibroblasts, which support tumor cell invasion.
-
3.
N-cadherin forms a weaker intermolecular band than E-cadherin, so molecules whose intermolecular bands are weaker favor motility.
When normal cells transform into cancerous cells, E-cadherin turns into N-cadherin and such cancer cells can escape their keratinocyte neighbors. Thus, the switch from E- to N-cadherin expression promotes invasion of the stroma. N-cadherin allows cancer cells to form homotypic interactions with mesenchymal cells, including endothelium, fibroblasts, and others in the stroma of the organ (Fig. 5.21).

An overview of a typical EMT program that causes cadherin shifts in the cell and invasion of cancers
In vitro and in vivo, EMT is induced by a variety of factors, including radiation, ROS, hypoxia, and transforming growth factor [130,131,132]. Radiation causes many changes in a cell, which are directly affiliated with epithelial cancer. These include aberrant multicellular organization, genomic instability, dysregulation of differentiation, and phenotypic transition, i.e., EMT. Multiple evidence suggests that Notch1 signaling is essential for EMT [133]. Unlike normal tissue, when cancerous tissues experience radiation insult, the irradiated cells show overexpression of an active Notch-1 protein in both cytoplasm and nucleus. This results in decreased expression of E-cadherin and increased expression of vimentin and fibronectin, which consequently alleviate radiation-induced EMT as reported both in in vitro and in vivo models [134, 135]. Furthermore, low dose IR promotes NF-κB p65 signaling in cervical cancer cells. The transcription factor NF-κB regulates tumorigenesis as well as apoptosis and is essential for the initiation and maintenance of EMT. This signaling subsequently induces EMT that downregulates of expression of the epithelial markers E-cadherin and Cytokeratin 18 (CK-18), which is a hepatocyte-secreted protein that is released into the blood during apoptosis and necrosis while it upregulates vimentin and the mesenchymal markers N-cadherin. Therefore, this leads to improving the progression of cancer cells [136]. Additionally, a variety of evidence indicates that Granulocyte Colony-Stimulating Factor (G-CSF) (formerly believed to promote granulocytes) is strongly associated with lung cancer progression [137]. Moreover, it has been reported that IR activates Granulocyte Colony-Stimulating Factor Receptor (G-CSFR)/JAK/STAT3 signaling pathways via secreted G-CSF, resulting in EMT in non-small cell lung cancer cells (NSCLCs) [137]. The matrix metalloproteinase (MMP) family proteins are essential for EMT, as well. The γ-IR treatment of nodular NSCLC cells was associated with increased invasion via EMT induction, which is characterized by a highly active MMPs system [138]. Following irradiation, an EMT program is induced, causing breast cancer cells to develop a resistance and progression behavior. The proto-oncogene tyrosine-protein kinase (SRC), being a non-receptor kinase, integrates diverse signal transduction pathways that contribute to a variety of cellular processes including survival, proliferation, angiogenesis, differentiation, migration, and angiogenesis. Upon irradiation, SRC is activated, which promotes EMT through AKT, p38, and PI3K activation. Similarly, fractionated IR inhibits PTEN-related expression, which is associated with activation of the Drosophila embryonic protein SNAI1 (Snail) protein accumulation and Akt/GSK-3β signaling and provoking EMT [139]. As a result of radiation, GSK-3β is phosphorylated at serine 9 residue, and silencing the tank-binding kinase-1 (TBK1) tones down the phosphorylation, indicating that TBK1 inhibition is associated with activating GSK-3β. Indeed, GSK-3β functions in several key signaling pathways that can contribute to EMT, such as the Wnt, TGF-β, Hedgehog, Notch, and PI3K pathways. Upon phosphorylation at its N-terminus, GSK-3β can be inactivated and Snail is stabilized and succumbs to EMT. In this way, it is suggested that TBK1 inhibition has the potential to reduce radiation-induced Zinc Finger E-Box Binding Homeobox 1 (ZEB1) expression via activation of GSK-3β. This hypothesis is supported by the observation that pre-treatment of cells with the GSK-3β inhibitor SB216763 increases TBK1 inhibition, increases E-cadherin expression, and reduces ZEB1. In conclusion, it was seen that TBK1 signaling is influenced by GSK-3β in radiation-induced EMT [140]. RT can cause both lessening of tumor cells and killing of them as it can also lead to damage to normal tissues, radioresistance, invasion, and distant metastases. Several studies have suggested that EMT generates malignant characteristics such as a propensity to recurrence, resistance to treatment, and dissemination of metastatic cells. Radiation is generally considered a key initiating factor for EMT, as it activates signal pathways that initiate the process.
5.9.6 Changing Tumor Metabolism
Radiation resistance has in multiple studies been linked to tumor metabolism alterations as a result of radiation-induced changes in mitochondrial metabolism or glycolysis [141, 142]. The development of radioresistance is closely related to glucose- as well as mitochondrial metabolism. The radiosensitivity of the tumors can be explained by radiation-induced metabolic changes in glucose and mitochondrial functions or by tumor-induced metabolic changes (Table 5.10).
After the tumor cells have been exposed to radiation for an extended period of time, these cells experience alteration in the glucose metabolic pathway. As an important kinase, AKT regulates multiple biological processes, including cellular metabolism. As described in the previous section AKT is phosphorylated in cells after IR exposure to radiation which results in radioresistance of tumor cells through the AKT-mediated alteration of cellular glucose metabolism. A disruption in glucose metabolism also occurs after radiation exposure of tumor cells, resulting in an accumulation of lactic acid. This is the primary product of glycolysis and contributes to the development of malignant tumors. Moreover, glycolysis may cause tumor metastasis, recurrences, and resistance to RT.
Radiation exposure of tumor cells likewise disrupts mitochondrial metabolism. In the post-RT setting, manganese superoxide dismutase (MnSOD) activity increases significantly in pancreatic cancer cells irradiation. MnSOD is the enzyme which catalyzes the formation of superoxide anion radicals, fighting the effects of oxidative stress ROS-induced damage. As a consequence, G2 checkpoint block is activated and cells are protected against mitochondrial oxidative stress by mitochondrial antioxidant systems, promoting radioresistance development. Radiation exposure to Burkitt lymphoma similarly disrupts mitochondrial metabolism by inducing the proteins (mitochondrial proteomes) that prompt radioresistance. Radiation of tumor cells targets Mitochondrial MAPK phosphatase (MKP1). Radiation increases MKP1 expression in breast cancer cells, where it translocates into the mitochondria and inhibits apoptosis by phosphorylating the c-Jun N-terminal kinase (JNK).
5.10 Palliative Radiotherapy
Box 5.11 Palliative Radiotherapy
-
Palliative RT may be used to slow down tumor growth, prevent or treat symptoms, and improve quality of life.
-
Common applications include prevention of bleeding and pain and treatment of metastases.
-
Brain and spinal cord metastases are often treated to relieve neurological symptoms.
-
Palliative RT is often hypofractionated with potentially unique radiobiology.
-
The radiobiological mechanisms of palliative RT are currently not well understood.
While radical RT aims to eliminate cancer cells with the intention of cure, palliative RT is used to slow down tumor growth, to prevent or treat symptoms and improve quality of life. It is estimated that more than 50% of RT patients receive palliative RT. This is particularly relevant in populations in less well-resourced countries where the burden of advanced disease is high, and palliation is commonplace (Box 5.11).
Typically, palliative RT is used to treat or prevent bleeding [143,144,145,146] or obstruction [147]. In addition, palliative RT is commonly used to treat pain, particularly bone pain, as well as brain and spinal cord metastases to address neurological symptoms [148]. Sites frequently treated palliatively include bone, brain, pelvis, and lung.
Despite the widespread use of RT with palliative intent, the radiobiology of palliative RT is not well understood. Radiation can reduce tumor burden by targeting proliferating tumor cells, and thus reduce tumor growth and result in relief of obstructions and pressure. However, mechanistic effects of how RT can stop bleeding and pain are still somewhat obscure. It has been speculated that cytokine modulation may alter pain levels and targeting of vascular components may prevent bleeding [149]. Palliative regimens are also typically shorter and frequently involve hypofractionated approaches or single (7–8 Gy) fraction schedules, which may induce differential biological effects to those induced by typical conventionally fractionated radical treatments. Enhanced vascular targeting and immune modulation have been implicated [150].
5.11 Tumor Microenvironment Changes Tumor Sensitivity
Box 5.12 The Tumor Microenvironment and Radiotherapy
-
Tumor microenvironments (TME) can recourse the tumor origination, expansion, invasion, metastasis, and its response to therapies like RT and chemotherapy.
-
RT immunomodulates the TME to induce a local anti-tumor immune response leading to tumor regression.
The tumor microenvironment (TME) is an “ecological niche” to facilitate the promotion and the succession of cancer cells. TME intricacy is coupled with tumor expansion, metastasis, and reaction to therapy. Potent alteration stirring in the TME is the basis of tumor cell variant selection, which may endorse genomic instability [31]. Cancer is an exceptionally assorted disease as the cells within the tumor have diverse mutations at different locations within the primary tumor and the metastatic site. Tumor growth and development are not only by actions relating tumor cells, but also by the milieu they inhabit, the TME. In 1889, Stephen Paget introduced “seed and soil” proposition which recommended that metastasis does not occur due to accidental events, but some tumor cells (the “seeds”) nurture ideally in particular organs (the “soil”) and metastases only emerges when the appropriate seed and soil combination takes place [151] (Box 5.12).
5.11.1 Components of the TME
Tumors are normally greatly heterogeneous and intricate in genetics. Different cell types, such as adipocytes, fibroblasts, immune cells, endothelial cells, and neuroendocrine (NE) cells, possess unique purpose in TME (Table 5.11). Acellular constituents of the TME like the extracellular matrix (ECM), extracellular vesicles (EVs), and cytokines remain adjacent to these cells. Physical and chemical uniqueness of the TME that are considered as vital the microenvironmental components comprise of the high interstitial pressure, hypoxia, fibrosis, and low pH. Further, communications involving cells and stromal elements also play an important role in cancer evolution and development [152].
5.11.2 Effect of Radiation on TME
IR can modify exchanges among tumor cells and their microenvironment. The TME does not only persuade tumor cell proliferation, invasion and metastasis of cancer cells, angiogenesis, modulation of immune cell infiltration and the immune response, but also has a brunt on the therapeutic response [151]. The effects of radiation on the TME diverge with dose and fractionation plan. Radiation stimulates the cellular and DNA damage that can lead to both production as well as release of tumor-associated neo-antigens and secretion of cytokines from tumor and the stromal cells. These proceedings can budge the equilibrium towards an immuno-reactive microenvironment, as contrasting to immunosuppressive microenvironment, causing the recruitment and activation of cytotoxic T cells. Figure 5.22 gives an overview as to how radiation affects the TME.

Overview of effects of radiation on the tumor microenvironment. (Figure adapted from Barker et al. [153], with permission)
The damage induced by IR affects many cells in the TME. The sensitivity of tumor cells is higher to radiation and their death commences the inflammatory signaling cascade. Also, the levels of intercellular adhesion molecule 1 (ICAM) and vascular cell adhesion molecule 1 (VCAM) expression go high as that of the cells of the innate immune system. Interestingly, the upregulation of the integrins present on the endothelial cells is associated with better survival as it acts as a system for radioresistance. Vascular diminution heightened the effect of hypoxia inducing the HIF-1α signaling. It also leads to the pro-angiogenic and pro-vasculogenic stimuli through VEGF and chemokine (C-X-C motif) ligand 12 (CXCL12). Following radiation cancer-associated fibroblast (CAF) activation stimulated the altered growth factor secretion and secret several ECM and cytokine modulators. Also, the transforming growth factor-β (TGF-B) signaling pathway is not only complex but also pleiotropic. It directly affects the tumor cells and CAFs, driving the HIF-1 signaling and reducing the activation of T cells and the dendritic cells (DC). In the immune compartment, due to high levels of mTOR there is not only an increase in the accessibility of tumor cell antigen but also a boost in the processing of the antigen in combination with damage-associated molecular pattern (DAMP) related Toll-like receptor (TLR) responses. Also, there is an increased pro-inflammatory cytokine signaling to activate the DCs and as a consequence T cells. The activated DCs move to the proximal lymph nodes. In the TME, such signaling is frequently obstructed by high Treg cytotoxic T lymphocyte antigen 4 (CTLA-4) suppression of co-stimulation. Radiation upregulates the NKG2D signals on the tumor cells that enforce direct cytotoxic effects by the CD8+ T cells and NK cells. However, there are other tumor cells which getaway with the PD-L1 signaling but the MDSC derived IL-10 immunosuppression remains intact.
Hence, depending on the organ, there are organ-specific interstitial cells such as the presence of astrocytes in the central nervous system or the osteoblasts in bone tissues. Such cells are collectively called as the stroma of the tumor and along with other essential factors like the surrounding pH, oxygen levels, extracellular matrix make up the microenvironment of the tumor [139].
5.11.2.1 Effect on Stroma and Cancer-Associated Fibroblasts (CAFs)
The tumor cells are known to interact with the stroma or the adjacent milieu. The tumor stroma comprises of a range of diverse cells, and extracellular matrix (ECM), that are known to advocate in limiting the host immune reaction against tumor cells. The tumor stroma puts forward a versatile complex network that maintains the tumor propagation by secreting immunosuppressive cytokine, diverse cellular processes and metabolic alterations [154]. Stromal cells involving the endothelial cells and adipocytes can modify the radiosensitivity by their roles in angiogenesis and vasculogenesis, and their secreted adipokines, respectively. The extracellular matrix can control radiation sensitivity by manipulating the oxygen proximity and managing the equilibrium and bioavailability of growth factors and cytokines [155]. Even though several RT-mediated stromal transformations like the renewal of or polarization towards tumor-suppressing immunity are valuable, RT can operate as a double-edged sword in tumors.
Chronic inflammation is one of the radiation effects on the stroma which acts as a chief driver of fibrosis during which there is occurrence of persistent immune responses in addition to tissue remodeling and repair processes. Some of the radiation-induced alteration on the TME is by altering the way CAFs manage their collagen assembly. In addition, CAFs that receive RT-undergo variation and modification in terms of their variety, secretome, and phenotype. Moreover, RT augments the stimulation of the proliferating machinery linking the RAS and mitogen-activated protein kinase (MAPK) signaling pathway; the invasion pathway that is associated with tumor evolution, resistance, and metastasis. When the stroma receives RT, there is augmentation in the invasiveness of the tumor because of the augmented hepatocyte growth factor (HGF)/c-Met (HGF receptor) signaling and MAPK activity. This aids the improvement in the tumor movability which can prove to be fatal. RT can also stimulate the stromal traits that can potentially impart resistance to therapy. Studies are still ongoing in understanding as to how fractionated RT alters the TMEs stromal machinery and how all the modifications can affect after the cancer cells response to RT [155].
5.11.2.2 Effect on Vasculature
An important feature of solid tumors is the vascular network that develops from the vasculogenesis, angiogenesis, and fibroblasts. A characteristic of these vessels is that they lack their supporting pericytes or basement membrane, which makes them vulnerable to radiation [156,157,158]. When tumor tissue is exposed to radiation, blood vessels at the level of the microvessel network are firstly destructed and the degree of this destruction depends on the radiation dose. The vessel’s thickness increases, which raises the risk of atherosclerotic changes and intimal proliferation, since the destructed microvessel network reduces distance between functional vessels and vascular density, resulting in hypoperfusion. Irradiation not only modulates the vessel structure, but it also has long-term effects, including medial necrosis and fibrosis, but these effects are affected by radiation dose. When a radiation dose is high, for instance, blood flow is permanently reduced; necrosis and fibrosis are produced, and the tumor is revascularized by vasculogenesis, which is a disorganized process of vessel growth in comparison to angiogenesis. Secondly, radiation to tumor tissue induces the chemokine receptor CXCR4 which induces bone marrow of matrix metalloprotease 9 that is responsible for radiation vasculogenesis and subsequent post irradiation [159, 160].
5.11.2.3 Effect on Immune System
IR kills cancer cells directly (atomic ionization) and indirectly (radiolysis of water) [16, 20, 158]. IR also induces a host immune response to tumor cells and facilitates tumor recognition and healing. These effects occur in a variety of ways, including immune response signaling. When cells are dying, they translocate calreticulin (ERp57) and their endoplasmic reticulum (ER) protein complex to the cell membrane which functions as a “eat me” signal. Secondly, upon IR of a tumor, inflammatory molecules, such as ATP and high mobility group protein B1 (HMGB1), are released, promoting a T cell and DC response. Signaling of this type uses three types of signals in anti-tumor immune responses, which are:
-
1.
DCs take captive of released danger signals and process them.
-
2.
Danger signals are presented into T cells by MHC molecules. As a result, naïve tumor-specific T cells are activated and become effector T cells.
-
3.
Finally, macrophages and DCs may be recognized by cytotoxic T cells, and they destroy remaining cells of tumor tissues through their circulation through the bloodstream [161,162,163,164,165].
However, not all irradiated cells sustain damage, and some survive. These IR surviving cells may expose surface molecules like antigen-presenting molecules of major histocompatibility complex class I (MHCI), death receptor Fas, and ICAMs which lead to an improvement in the recognition and killing of tumors by immune cells that are reactivated by tumor antigens.
To cure tumors, radiation needs to cause cell death. There are five different types of cell death depending on the pathways in the response system of DNA damage, namely, apoptosis, senescence, autophagy, mitotic catastrophe, and necrosis [13, 157, 158]. Via apoptosis pathways macrophages may be triggered to clean out these dying cells which causes polarized M1 macrophages thereby driving an anti-tumor immune response and indirectly killing of tumors. However, this process may also contribute to carcinogenesis and inflammation [163].
5.11.3 Tumor Radiosensitivity and Underlying Mechanisms
The most well-known tumors in declining order of radiosensitivity as follows: (1) lymphoma, (2) embryonal tumors, (3) cellular anaplastic tumors, (4) basal cell carcinoma, (5) adenoma and adenocarcinoma, (6) desmoplastic tumors, such as squamous carcinoma, (7) fibroblastic sarcoma; osteosarcoma; neurosarcoma. Tumors composed of fast-growing cells like the embryonal tumors are sensitive to radiotherapy.
Lymphoid cells are predominantly vulnerable to radiation. On the other hand, the radioresistant category comprises of the neurosarcomas, gliomas, and melanomas. Tumor sensitivity to radiation is affected by the variation on the radiosensitivity exhibited by the subgroups of major cancer forms. There are numerous factors affecting the cancer radiosensitivity and at times they are even obscure. The most important factor that plays the most vital role in influencing the tumor behavior under RT is the general condition of the patient. Also, there is an explicit association between radiosensitivity and the grade of malignancy which might not be uniform always. Hence, all these factors should be carefully evaluated before imparting radiotherapy and predicting its outcomes.
5.11.3.1 Factors Affecting Tumor Radiosensitivity
One of the most prominent factors imparting the tumor radiosensitivity and which was recognized very early on was the high metabolism of the tumor cells. This is similar to the fast growth and also understood as the tumor growth rate imparting radiosensitivity. Augmented or uneven vascularity also goes with fast growth. These factors combined provide the rapidly growing tumor cells with sensitivity to radiation. Especially when tumors are bulky hypersensitivity can be encountered in the treatment of such tumors. Therefore, to avoid accidents like bulky necrosis or interstitial hemorrhage appropriate precaution should be taken. Other factors like the large quantity of autolytic cell ferments and susceptibility of mitotic nuclei are also crucial in imparting tumor sensitivity to rapidly growing tumor cells. It is seen that the tumors exhibiting the embryonic trait in the origin cells provide the complete group of embryonal tumors a very high radiosensitivity. Hence, their response to radiation treatment is probably quicker and complete compared to other tumors. Therefore, such tumors should be identified as especially favorable.
5.12 Systemic Anti-tumor Immune Responses and Abscopal Effects
5.12.1 Introduction
While radiation effects classically result from direct damage to cells and tissues within the target, effects outside the field also known as abscopal effects have been observed in both tumors and normal tissue. For example, irradiation often leads to systemic fatigue [166], and bilateral pneumonitis sometimes occurs after unilateral lung irradiation [167]. Occasionally, metastatic tumors have been observed to regress dramatically outside the primary irradiation field, which is a manifestation of the systemic anti-tumor effect [168].
An illustrative example is the case of a man diagnosed with ulcerated malignant melanoma in his left arm [169]. The arm was resected, and no further treatment was indicated. Twenty months later, the patient had a painful skin lesion on the head and an asymptomatic metastatic lesion in the lung. The skin lesion was irradiated with 3 Gy in 10 sessions, after which the lesion and pain disappeared. Unexpectedly, the lung lesion also disappeared although it had never been treated. Reports of cases of abscopal effects in melanoma are relatively common. Another example is the case of a woman with squamous carcinoma of the anal canal with metastases to pelvic lymph nodes, liver, and bone. After palliative RT limited to the pelvis with sensitizing chemotherapy, complete regressions were observed 4 months after treatment not only in the primary tumor but also in the bone and liver metastases, an effect that persisted 4 years later [170]. Figure 5.23 gives an overview as to how radiation induces Abscopal Effects.

The abscopal effect of RT. On the left side of this schematic case, the patient suffers from a tumor in the shoulder (e.g., a melanoma) that has metastasized to the lung. The treatment protocol consisted of surgery of the primary tumor and the metastatic nodules were treated with a combination of RT and immune checkpoint blockade, which promotes abscopal anti-tumor immunity. On the right side of this schematic case, RT triggers the release of tumor antigens, neoantigens, and DAMPs, which are recognized and processed by macrophages and DCs. Subsequently, these antigen-presenting cells present these tumor antigens to T cells in a draining lymph node of the tumor. The antigen-presenting cells are then taken up by cytotoxic T cells, which subsequently destroy the remaining cells of the cancerous tissue
The previous clinical cases showing unexpected regression of tumors outside the irradiated field are examples of a phenomenon first described by Mole as the “abscopal effect” [171]. The word abscopal comes from the Latin ab (position away from) and Scopus (target). However, there is now clear evidence that the abscopal effects associated with RT are due to systemic anti-tumor immune responses triggered by the RT. However, despite millions of patients receiving RT, abscopal effects are too unpredictable to be a therapeutic target, as only a limited number of cases have been identified. In a recent study, only 46 cases were identified between 1969 and 2014 [172]. With the advent of immune checkpoint blockade therapy, clinical interest has soared. Since the introduction of the anti-CTLA-4 antibody ipilimumab into clinical practice in 2011, abscopal effects have been observed in several patients who received RT while taking ipilimumab [173, 174]. Other immune checkpoint therapies have also shown promise. There are reports of abscopal effects following treatment with RT and anti-PD1 therapies such as pembrolizumab or nivolumab [175]. In this section, we will cover the biological events in cancer cells that precede the occurrence of radiation-induced abscopal effects.
5.12.2 Radiation-Induced Immunogenicity
Radiation can alter the immunogenicity of tumors by promoting tumor antigens, neoantigens, and danger signals.
5.12.2.1 Tumor Antigens
Tumor antigens result from protein degradation due to damage to cellular proteins caused by radiation-induced free radicals. Radicals such ROS are generated during radiation-induced radiolysis of water. RT then causes cellular damage that is independent of DNA and leads to an increase in the pool of peptides, i.e., tumor antigens that bind to the major histocompatibility complex (MHC) [176].
Tumor neoantigens are tumor-specific antigens resulting from the expression of a large number of mutations which may be a result of tumor genetic instability but also due to severe DNA damage. The degrees of DNA damage can strongly influence the immunogenicity of the tumor. In this context, RT leads to DNA SSBs or DSBs or other DNA alterations (see Chap. 3) [177]. DSBs are the most lethal DNA lesions if not repaired. Repair depends on the type of radiation causing the damage. Low LET radiation causes simple DSBs, which are repaired by NHEJ pathway. High LET radiation causes clustered DNA lesions (or multiple damaged sites) that are usually repaired by HR. Non-DSB lesions at clustered DNA damage sites are corrected with Base Excision Repair. If the DNA repair mechanisms are not sufficient to repair the damage, or if the RT itself causes defects in the DNA repair mechanisms, mutations occur that can lead to the expression of tumor neoantigens. These neoantigens are then processed by tumor cells and presented by MHCs [178].
5.12.2.2 Damage-Associated Molecular Pattern (DAMP)
Damage signals properly known as Damage-Associated Molecular Patterns (DAMPs) are endogenous molecular signals released by irradiated cells after damage or when dying [176]. These signals enable enhanced recognition and killing of the tumor by macrophages and DCs resulting in antigen presentation to T cells [161, 164]. Some of the most frequently found radiation-induced DAMPs in tumor cells are:
-
Fragmented double-stranded DNA and micronuclei. These self-DNA fragments and micronuclei appear as cytosolic DNA in tumor cells, which are later recognized by immune cells.
-
High mobility group box 1 (HMGB1) is a nuclear protein associated with chromatin that is released extracellularly by dying tumor cells, normally necrotic cells [179]. HMGB1 binds to Toll-like receptor 4 (TLR4) expressed by DCs and activates them.
-
Nucleotide release by apoptotic cells function as a chemotactic signal for phagocytic myeloid cells including DCs by stimulating the Purinergic receptor 2 (P2RY2) [180].
-
Extracellular ATP can also function through the activation of Purinergic receptor 7 (P2RX7) to initiate inflammasome activation and subsequent Interleukin 1β production. This mechanism was shown to be required for the induction of tumor antigen-specific CD8+ T cells following challenge with dying tumor cells [181].
-
Calreticulin is an ER protein that gets translocated to the cellular membrane after RT. Calreticulin is essential for efficient uptake of dying tumor cells by antigen-presenting cells [182].
-
Chemokines are a frequent part of DAMPs. Chemokines, such as C-X-C motif (CXC) ligand 16, 10, and 19 (CXCL16, CXCL10, and CXCL9), are known for being released after radiation exposure; they trigger differentiation of immune cells, and stimulate immune modulators such as Interferons, Tumor necrosis factor α, and Interleukin 1β [162, 183,184,185].
In summary, radiation increases the immunogenicity of tumors by simultaneously promoting the release of tumor antigens, neoantigens, and DAMPs. This radiation-induced immunogenicity may counteract the progression of tumors that have escaped immune surveillance, especially if an abscopal effect can be triggered.
5.12.2.3 Anti-tumor Immune Responses: Abscopal Effects
To reproduce the radiation-induced abscopal effect has been challenging as its underlying biological mechanisms are not fully understood. However, some crucial cellular signaling pathways have been identified alongside the IR doses that appear to be fundamental for their occurrence.
-
The accumulation of cytosolic double-stranded DNA fragments (dsDNA) is usually a sign of microbial infection and alerts the host’s innate immune system to initiate a defense response. Interestingly, the accumulation of dsDNA after RT has been shown to be an essential step in the abscopal response. Mitochondrial DNA and dsDNA are recognized by cyclic GMP-AMP synthase (cGAS), which produces messengers that bind and activate the transmembrane protein Stimulator of interferon genes (STING), triggering a robust innate immune response [186]. In particular, activation of the type I interferon signaling pathway (IFN-1) and production of Interferon β cytokines by cancer cells trigger the accumulation of basic leucine zipper ATF-like transcription factor 3 (Batf3)-dependent DCs, which are required for the maturation of anti-tumor CD8+ T cells.
-
The irradiation dose and fractionation schemes are crucial for the induction of abscopal effects. With increasing radiation dose, more dsDNA fragments accumulate in the cytoplasm of cancer cells. However, the increased radiation dose of over 12 Gy also triggers the activation of three prime repair exonuclease 1 (Trex1), which degrades dsDNA and prevents an immune response. Interestingly, Interferon β production can be enhanced with repeated radiation doses that do not trigger Trex1 [186]. Radiation regimens that have been shown to be effective in preclinical and clinical studies are 3 × 8 and 5 × 6 Gy.
-
Transforming growth factor beta (TGF-ß) is a potent immunosuppressive cytokine in established tumors that impairs the antigen presentation function of DCs, prevents T cell priming and thus inhibit the abscopal effect [164].
-
TGF-β expression increases in irradiated tissues. However, the abscopal effect can be restored if TGF-β neutralizing antibodies are administered during RT. Current research is investigating if the use of dual-function drugs that simultaneously target TGF-β and PD -L1 in combination with RT can be a good strategy to overcome tumor immune escape [187].
5.13 Normal Tissue Damage and Response to Radiotherapy
5.13.1 Introduction
The tolerance of normal tissues to RT is the limiting factor in delivering a radiation dose high enough to eradicate tumor cells [188, 189]. The dose delivered to the target structure is limited by the dose constraints to the organs at risk (OAR), which are in close proximity to the tumor. In the case of external RT, standard recommendations for OAR doses have been issued based on the volumes irradiated and are regularly updated to reflect new practices.
Despite ongoing technical and technological advances, the planning target volume (PTV) necessarily contains a volume of normal tissue for several reasons. First, the target volume, which receives the prescribed dose, is always larger than the actual tumor volume. Indeed, this volume must take into account the visible tumor volume (gross tumor volume, GTV), the possible microscopic extension of the tumor (clinical target volume, CTV), the movements of the different volumes (internal target volume, ITV), small errors in patient set-up and technical precision, all together making up the PTV. Secondly, the tumor contains normal tissues such as soft tissues and blood vessels, which receive the entire prescribed dose. Finally, especially for external beam RT, the radiation beam inevitably passes through normal tissue, depositing doses that may be clinically relevant. Effective RT is therefore necessarily associated with a risk of adverse effects. Side effects may occur during RT or a few weeks after treatment (acute effects). In the long term, late effects, which may occur months or years after RT, are the most critical because they are chronic, disabling, painful, and most often irreversible.
Limiting adverse events to reduce morbidity and optimize the therapeutic index of RT remains a priority in the fight against cancer. To this end, NTCP modeling is widely used in treatment planning as a tool to differentiate treatment plans [11]. In future, cellular and animal models of radiotoxicity aimed at understanding the sequence of molecular and cellular events that drive the pathogenesis of early and late normal tissue radiation injury should enable the development of tools to reduce the impact of RT on normal tissues [190].
5.13.2 Acute Tissue Response
Box 5.13 Radiation-Induced Acute Normal Tissue Response
-
Acute effects of radiation are observed in days to weeks after exposure.
-
The time scale of clinical manifestation of most acute responses is independent of the radiation dose and related to the proliferative rate of injured cells.
-
Acute effects are transient and reversible.
-
Typical acute responding normal tissues are: intestine, mucosa, skin, hair follicles, bone marrow.
Acute responses are primarily observed in tissues with rapid cell renewal where cell division is required to maintain the function of the organ. In these renewing tissues, physiological cell loss occurs constantly from the post-mitotic tissue compartments while actively cycling cell populations in the germinal parts of the tissue proliferate to replenish them. The radiation response is related to death of critical cell populations such as the stem cells in the crypts of the small intestines, in the bone marrow, or in the basal layer of the skin. When sufficiently large numbers of critical cell populations are affected, cell production capacity is no longer able to compensate for the physiologically occurring cell loss, leading to hypoplasia and cell depletion. For this reason, the time scale of clinical manifestation of most acute responses is independent of dose and instead reflects the rate of loss of functional cells and the demand of proliferation of the supporting stem cells of the different tissues (Box 5.13).
In general, acute responses are transient, but sensitive to the overall radiation treatment time. Recovery may occur by rapid repopulation from the surviving stem cell compartment or by recruitment of stem cells from neighboring sites (non-irradiated/damaged areas). Hence, the latent period of manifestation of tissue reactions is specific, depending on the cell type and its proliferation rate (Table 5.12) as well as on its intrinsic sensitivity to radiation and capacity to repair damaged DNA.
Apart from the number of sterilized cells, several other non-lethal mechanisms like proliferative impairment and disturbances in molecular cell signaling play a role in the acute tissue reaction to radiation. For example, the release of 5-hydroxytryptamine by mast cells have been shown to be responsible for several early clinical reactions such as erythema following irradiation of the skin and nausea/vomiting following irradiation of the intestines. Immunological reactions associated with local and systemic release of cytokines have also been demonstrated. In the hematopoietic system, apoptosis plays an important role in acute normal tissue response. Apoptotic death directly following irradiation, particularly of the lymphoid lineage, causes additional cell loss on top of the physiological cellular loss rate of circulating cells and explains earlier onset particularly for the lymphoid lineage. Furthermore, the typical tissue or organ architecture has a principal role in the response to irradiation. Threshold irradiation doses (Table 5.13) are often dependent on the irradiated normal tissue volume.
The tissue architecture is generally organized in the so-called functional subunits (FSUs). Some tissues are built of anatomically demarcated FSUs, like nephrons in the kidney, liver- and lung lobules. These types of organs—with a parallel arrangement of FSUs show large reserve capacity. Non-radiation exposed volumes of the organ can take over the function of the damaged tissue. Other tissue types like the spinal cord and mucosa do not show clear anatomical demarcation in FSUs. In such serial arranged FSUs tissues, radiation injury to a small tissue volume can result in function loss of a larger volume or even the whole organ. The threshold radiation dose can be defined as the safe, tolerated, dose below which no tissue-specific reaction occurs. This particular dose is difficult to determine [191]. In general, it is the estimated dose that is required to cause a typical tissue reaction in 1% of the exposed individuals (ED1) relative to non-irradiated controls. To be noticed is that radiation doses <ED1 could also induce biological effects above baseline levels in non-irradiated, age-matched individuals, i.e., above natural incidence. Table 5.13 lists threshold doses and latency times for tissue reactions to a single radiation exposure for a selected number of healthy tissues and organs.
5.13.3 Late Tissue Response
Box 5.14 Radiation-Induced Late Normal Tissue Response
-
Late effects of radiation occur months, years, or decades after radiation therapy.
-
Late effects are progressive and irreversible.
-
The latent phase of chronic reactions is inversely proportional to the radiation dose.
-
The late effects are based on an interactive response of parenchymal cells, vascular endothelium and fibroblasts, with a contribution from immune cells, especially macrophages, as well as other cells.
-
Tissues and organs are affected by atrophy, fibrosis, and/or necrosis, which can severely impair their functions and lead to a loss of function.
The classification that distinguishes between early and late effects is based exclusively on the time of first diagnosis of pathological changes, with a threshold arbitrarily set at 3 months after the start of RT [5, 193]. Unfortunately, there is no general biological rationale for this threshold, whereas a classification of early and late effects is clinically relevant and essential for comparing studies. Nevertheless, there are specific biological characteristics of early and late effects that distinguish them (Tables 5.13 and 5.14). As discussed in Sect. 5.13.2, the early symptoms most often appear in highly proliferative tissues, such as the intestinal mucosa, epidermis, and bone marrow. In these tissues, irradiation causes a progressive but potentially reversible depletion of cells by preventing their physiological renewal, associated with a direct or indirect radiation-induced inflammatory reaction. In contrast, late side effects can affect all tissues. The pathogenesis leads to fibrosis, atrophy, vascular damage as well as other detrimental side effects on normal tissues such as hormonal deficiencies, infertility, and second malignancies (discussed in Chap. 7). Late side effects are chronic, progressive, in most cases irreversible and their severity tends to increase over time (Fig. 5.24 and Box 5.14).

Radiation-induced chronic damage to healthy tissues. Late damage develops within months to decades post RT and may concern all normal tissues. Successive cycles of tissue remodeling and repair, together with chronic inflammation induce vascular and parenchymal damage leading to tissue atrophy/fibrosis/necrosis compromising organ function
The mechanisms that lead to these effects are more complex than for acute effects. They involve organ-specific changes in parenchymal cells, including cell death and alterations in cellular metabolism. Consequential late effects from severe acute reactions may contribute to chronic damage due to unresolved regeneration of rapidly proliferating tissues which contributes additional damage to connective tissue and endothelium [194].
The pathogenesis leading to radiation-induced fibrosis is a result of fibroblast differentiation into myofibroblasts, proliferation of surviving fibroblasts and extracellular matrix and collagen deposition. Seen as wound healing that goes wrong, this pathological process plays a key role in the development and expression of most late effects. Atrophy is caused by the loss of fibroblasts and collagen reabsorption. Examples of fibrotic-atrophic response include hardening and shrinkage of an irradiated breast, or strictures and malabsorption of irradiated small intestine. Vascular damage is caused by either small vessel dilation or constriction as well as losses of the vascular endothelial cells from small blood vessels and capillaries. Vascular damage results, for instance, in skin telangiectasias, bleeding, ischemia with intestinal perforation and fistula formation. The immune system also contributes significantly to the tissue response through the involvement of macrophages and mast cells, which interact with other cells in the irradiated tissue and other organs through the release of cytokines and growth factors [195,196,197,198]. More generally, the response of a tissue is mediated by different types of cells such as inflammatory, stromal, endothelial, and parenchymal cells that actively communicate through the release of cytokines, chemokines and growth factors, and/or the activation of molecular pathways downstream of these messengers. Altogether, these effects lead progressively to parenchymal damage and potentially to loss of organ function in the irradiated volume.
The tissues of cancer survivors treated with RT still bear the traces of RT to varying degrees. Although patients are usually asymptomatic, all irradiated patients, especially those with late effects, have a common histological feature of radiation-induced fibrosis and atrophy. Fibrosis predominates in the breast, skin, small intestine, lungs, kidneys, and liver, while atrophy and necrosis predominate in the later stages after RT alone or in combination with surgery and local trauma to bone (osteoradionecrosis of the mandible, ORN), nerve, or brain. The clinical severity correlates with the extent of the underlying pathophysiological process, which is usually invisible and often depends on the level of parenchymal cell loss.
It is often mentioned that 5–10% of patients, and sometimes up to 20% for the treatment of pelvic malignancies, including prostate, rectal and cervical cancer, develop late side effects. However, some authors believe that the rates of patients with late side effects may be greatly underestimated [193]. For example, in the case of abdominal or pelvic cancers, more than half of patients would suffer from some form of chronic bowel dysfunction [199]. Most of the effects observed today were caused several years ago by RT techniques that are less used today (2D- and 3D-CRT), and which are progressively being replaced by more precise and more efficient techniques and technologies (IMRT, SBRT and their derivatives, hadrontherapy, and maybe FLASH RT in the future). It is therefore likely that the landscape of side effects will be completely different in a few years.
As cancer detection and management continue to improve, there is an increase in the number of long-term cancer survivors in the more economically developed countries. For example, in the USA, the 5-year survival rate has increased from 49% in the period 1975–1977 to 67% in the period 2010–2016 (American Cancer Society, Cancer Facts and Figures, Atlanta, Georgia, 2021). On the other hand, given that about half of patients are treated with RT, it can be estimated that several tens of thousands of patients will develop side effects each year in this country. Worldwide, with more than 19 million new cancer cases in 2020 (and about 30 million expected in 2040), and an estimated 5-year prevalence of more than 50 million [200], the number of patients developing side effects affecting their quality of life is expected to be in the millions each year. Beyond the fact that the late side effects of RT still limit the effectiveness of this treatment, these figures reveal a real public health concern facing the public authorities and the medical profession.
5.13.4 Radionecrosis
Box 5.15 Radiation-Induced Fibrosis and Necrosis
-
10% survivors of RT will suffer from fibrosis/radionecrosis.
Radionecrosis is a late toxicity phenomenon with the occurrence depending on radiation dose, the tissue affected and a number of site-specific risk factors; as treatment options are scant, preventive measures should be facilitated by providing the treatment team with the forecasted 3D RT isodose curves (Box 5.15).
Radiation accidents and therapy have shown that, in principle, all human tissues can suffer from necrosis as a late toxicity as result of progressive ischemia of irradiated tissues in the context of chronic inflammation. Pathologic samples show necrosis with fibrinous exudates and dystrophic changes of the vessels in the exposed tissues. In daily practice, radionecrosis most commonly involves bone (head and neck (jaws-mastoïd-temporal bone-larynx-cartilage)/femoral head), breast, CNS, bowel, skin and rarely ribs or sclera (Sect. 5.14.3). Even when standard dosage schedules are followed, serious radiation complications would occur in 5–10% of long-term survivors. Yet, general incidence rates on most tissues are difficult to present as most studies have specific settings and constraints resulting in large heterogeneity of data. In the following section, osteonecrosis of the jaws and brain radionecrosis will be discussed in greater detail.
5.13.4.1 Osteoradionecrosis (ORN) of the Jaw
Osteoradionecrosis (ORN) of the jaw is a late complication of RT in the treatment of head and neck cancer. It is defined as exposed irradiated bone that fails to heal over a period of 3 months without any evidence of persisting, recurrent tumor or metastatic disease. ORN occurs in bone that was exposed to a radiation total dose exceeding 60 Gy. However, in the presence of concomitant risk factors, lesions can develop in bone exposed to a lower dose, usually above 50 Gy. The overall susceptibility ratio between mandible and maxilla is for the development of ORN 24/1, with the posterior areas of the mandible most at risk and the upper jaw rarely affected.
The overall incidence of ORN in IMRT patients is reported to vary between 5.1% and 12.4% [201] with excess figures (up to 25.5%) [202] in the presence of risk factors and higher figures with longer follow-up. ORN usually develops during the first 3 to 24 months after RT; however, the real risk for ORN lasts a lifetime and can occur at any time following RT.
The pathophysiology of ORN is still uncovered. In essence, the viability of the irradiated bone is lost due to ischemic necrosis in the irradiated atrophic tissue without sufficient capability of repair, leading to secondary soft tissue breakdown and exposure of bone. Pathological fractures following ORN typically form no callus formation [203], illustrating the absence of periosteal healing. The presence of Actinomyces in necrotic bone is best detected with a PCR-based method and its role needs further investigation.
ORN may remain asymptomatic for a prolonged period but signs and symptoms may also occur before the development of bony exposure. Presenting clinical features include pain, tooth mobility, mucosal swelling, erythema, ulceration, malocclusion, dysphagia, trismus, paresthesia, or even anesthesia of the associated branch of the trigeminal nerve.
Different classifications of ORN exist, usually based on following criteria: extent of the lesion [204], symptoms [204, 205] and response to hyperbaric oxygen therapy. The extent of lesions can vary and range from a non-healing extraction site to exposure and necrosis of large sections of the jaw. Late stage ORN often present with fistula from the oral mucosa or skin, complete devitalization of bone, pathological fractures, and even life-threatening complications.
Panoramic radiographs are mostly used for diagnosis, follow-up, and monitoring patients who are at risk of osteonecrosis. However, only at a loss of 30–50% in bone density injury will be visible on X-ray. CBCT, CT, and MRI allow to analyze the jaws more extensively and to better assess the extent of injuries and are also very helpful in differentiating osteonecrosis from other causes of osteolysis.
Although ORN may occur spontaneously [206], most ORN develop after dental surgery (extractions of teeth, dento-alveolar surgery, dental implant placement).
The most important risk factors for ORN are dose >50–60 Gy and post-RT dento-alveolar surgery in the high-risk zone. Other factors are tumor size, proximity of the tumor to bone, age >60 years, diabetes mellitus, poor oral hygiene, concomitant chemotherapy, active smoking, excessive alcohol consumption, and chronic use of corticosteroids [207]. Many of these published risk factors still need confirmation with robust data and study designs.
Pre-treatment dental screening aims to reduce the risk of developing ORN after RT by eliminating all teeth with an elevated risk in an area of bone that will get exposed to a high dose of IR. It is therefore mandatory to provide the dentist and/or oral surgical team with the forecasted 3D RT isodose curves (Fig. 5.25) to allow a differential approach for teeth within and outside the high ORN risk perimeter [208].

3D RT isodose curves
In the areas with a high risk of developing ORN (>50 Gy), an extraction is done whenever teeth represent a risk for future need for extraction of a risk for future infection. Teeth which will be extracted as part of the surgical resection approach can be left in situ. In other areas of the jaws, the extraction therapy will depend on regular extraction guidelines, the clinical experience of the supervising surgeon considering the level of oral hygiene of the patient and the expected future limitation of mouth opening. In the upper jaw, due to the far lower incidence of ORN, most clinicians opt for regular extraction guidelines.
Depending on the extent of the affected area (both soft tissues and bone), the symptoms, the existence of a pathological fracture the treatment will vary from a conservative to a surgical approach. In early stages, conservative measures such as antibiotics, debridement, and irrigation will be preferred while surgical resection and reconstruction (reconstruction plates, free vascularized osteomyocutaneous flaps) are reserved for more advanced cases. Whenever resection of ORN is needed, 3D RT isodose curves should be allowed to be included in the virtual planning of the procedure.
HBO as adjunctive therapy to conventional treatment has not been proven to yield consistently significantly favorable results compared to conventional treatment alone. Therapeutic regimens composed of Pentoxifylline and Tocopherol combined have been shown to have a synergistic effect in treating small areas of ORN with visual and symptomatic resolution of the condition. Clonodrate, a first-generation non-aminobisphosphonate, has been described as effective when combined with pentoxifylline and tocopherol for refractory ORN (PENTOCLO-protocol).
Lifestyle changes should accompany both conservative and surgical procedures: proper oral hygiene, smoking and alcohol cessation, healthy and adequate nutrition intake, well-fitting dentures [209]. Dento-alveolar surgical procedures in a highly radiated mandible should be avoided if possible and whenever needed following principles should be kept in mind: minimal periosteal degloving, antibiotic coverage, local anesthesia without epinephrine.
5.13.4.2 Brain Radionecrosis
Brain radionecrosis (RN) is an irreversible late radiation-induced tissue complication that can occur after irradiation of brain parenchyma inducing a vascular lesion of the white matter, developing in the irradiation field, secondary to chronic inflammation of the brain parenchyma, with a tendency for spontaneous extension [210]. Its pathophysiology is not yet clear. Brain RN induces hypocellular zones of necrosis and fibrinous exudates with degenerative or dystrophic changes in the vasculature, with telangiectasia, hyaline thickening of vessels, and fibrinoid necrosis including intravascular thrombosis responsible for an increase of vascular permeability. The occurrence and severity are correlated with dose-volume parameters [211]. An actuarial incidence of brain RN up to 34% two years after stereotactic radiotherapy (SRT) was recently reported—symptomatic and sometimes lethal or severely debilitating in 10–17% of the patients [212]. Approximately 80% of cases occur within 3 years from the completion of RT.
The symptoms of RN are those of a non-specific intracerebral expansive process. A seizure is inaugural in half of the cases, signs of intracranial hypertension and a progressive deficit syndrome (sensory, motor, or aphasia) are frequently present. The semiology often reproduces the initial signs of the primary tumor. In pituitary tumors, lesions preferentially affect the chiasma and the optic nerves causing severe visual disturbances; damage to the temporal, frontal, and hypothalamus lobes is often associated, causing cognitive impairment.
The main differential diagnosis is tumor progression due to very similar clinical and radiological characteristics.
The gold standard for the diagnosis with certainty is the pathological analysis. On histological analysis, 50% of lesions are pure RN, the remaining 50% associated with radionecrosis and tumor cells without predicting their viability.
There is not yet a validated imaging technique that distinguishes the two entities though advanced imaging techniques such as DTI (ADC and fractional anisotropy ratios), perfusion MR imaging (CBV, rPH, and relative PSR), MR spectroscopy, and amino acid PET hold promise [213]. The MRI shows a persistent central hypointense and an enlargement of a pre-existing enhancement in T1 gadolinium associated with a hypersignal in T2 with an appearance of “Swiss cheese” or “soap bubble.” Perfusion MRI, spectro-MRI and PET amino acid imaging may provide additional arguments. Other avenues are showing interest in the differential diagnostic strategy—notably radiomics.
When this documentation is not possible, the decision-making process is guided by clinical and imaging criteria collected over a significant period of follow-up. Such criteria were proposed by the Association of Neuro-Oncologists of French Expression (ANOCEF) [214]. The treatment options of brain RN include steroids, bevacizumab, surgical resection, and hyperbaric oxygen.
5.13.5 Pathogenesis of Early and Late Normal Tissue Radiation Injury
Box 5.16 Normal Tissue Radiation Pathology
-
Cellular depletion by radiation-induced death is not the only one responsible for initiation and progression of lesions.
-
Radiation-induced effects on the vascular endothelium drive the propagation of the inflammatory response and chronic effects.
-
Molecular and cellular damage after exposure to IR impacts cellular homeostasis and potentially leads to chronic organ dysfunction.
-
The notion of a continuum of effects, orchestrated by all the compartments and chronic cytokine cascades, opens up fields of therapeutic approaches.
Improving the quality of life of patients by reducing sequelae of cancer treatment is one of the main future challenges. Beyond the dose itself to the organs at risk, the probability of occurrence of side effects is related to a multitude of factors: the nature of the radiation (photons, electrons, charged particles), the volume irradiated, the fractionation, the spread, the dose rate, but also the nature of the exposed tissue (hierarchical versus flexible tissue, in parallel or in series organization) or the individual susceptibility of the patient. Furthermore, in a simplified manner, acute toxicity is mainly observed in rapidly proliferating tissues (skin, gastrointestinal tract, and hematopoietic system) and late effects are observed in slower proliferating tissues (central nervous system, kidney, heart) [215]. Historically, tissue response to radiation has long been explained by the target cell concept which suggests that the severity of tissue effects is mainly due to the depletion of cells in a target compartment by radiation-induced death resulting in a functional impairment of the organ. This hypothesis can be considered for early effects but is more questionable for late effects. Cellular depletion by radiation-induced death is an important element of the tissue response, but it is not the only one responsible for the initiation and progression of lesions. Molecular and cellular damage after exposure to IR will disrupt cellular homeostasis and potentially lead to chronic organ dysfunction. It is now agreed that the tissue response to IR is the result of the activation and integrated involvement of all the compartments that make up the tissue (Fig. 5.26 and Box 5.16).

Irradiation and progression of radiation-induced normal tissues damage. Tissue damage results from several acute events such as cell loss and endothelial cells activation. Damage progression includes a continuum of effects orchestrated in time and space leading to tissue fibrosis/necrosis and organ dysfunction
The notion of a continuum of effects, orchestrated by all the compartments and chronic cytokine cascades, opens up fields of investigation into various therapeutic approaches [190]. The contemporary view involves several cell types and molecular mechanisms, which together form an orchestrated response, and contribute to the initiation, progression, and chronicity of radiation-induced injury. A better understanding of these event kinetics should allow the identification of molecular and cellular targets, associated functions, and relevant times for therapeutic action. Radiation-induced effects on the vascular endothelium and epithelial barriers are important for the propagation of the inflammatory response and the recruitment of immune cells. The concept that the microvasculature plays a central role in the radiation toxicity of many tissues is emerging and demonstrated now. Irradiation leads to endothelial cell apoptosis, increased vascular permeability, and acquisition of a pro-inflammatory and pro-coagulant phenotype. Moreover, tissue-specific deletion in the endothelium of key molecular actors impacts the severity of acute and normal tissue injury [216].
Rapidly after exposure to IR, damage to the endothelium and epithelial cells leads to the release of damager signals (such as DAMPS) and the activation of adhesion molecules. This reaction allows the recruitment of a large panel of immune cells to the damaged site, which are able to repair the tissue but which, in the case of chronic inflammation, can strongly also participate in the installation of fibrosis [196]. For example, macrophages are rapidly recruited after irradiation and are a heterogeneous immune cell population with multiple pro- or anti-inflammatory as well as pro- or anti-fibrosis functions. The recruitment dynamics of macrophages, as well as their phenotypic orientation impacted by their microenvironment over time, are increasingly shown to play an essential role in the evolution of radiation-induced injury [195]. Radiation-induced immune effects are propagated by a large panel of cytokines including interferon-γ (IFNγ), Interleukin-1β (IL-1β), Interleukin-6 (IL-6), CC-chemokine ligand 2 (CCL2), tumor necrosis factor (TNF), and transforming growth factor-β (TGFβ). Interestingly, beyond their roles in the inflammatory response, some of these cytokines also play essential roles in several other processes contributing to the evolution of radiation-induced lesions. TGFβ induces the differentiation of fibroblasts into myo-fibroblasts with a consequent increase in the extracellular matrix. In addition, in association with other cytokines such as IL-1β, TGFβ promotes endothelial-mesenchymal (endoMT) [217] and EMT, two key processes also demonstrated in radiation-induced lesions to healthy tissues [218]. Finally, it has recently been shown in several studies that senescence also contributes to the pathogenesis of radiation-induced injury to healthy tissue. Senescence is a durable cell-cycle arrest with a persistent pro-inflammatory Senescence-Associated Secretory Phenotype (SASP) characterized by the secretion of multiple growth factors and cytokines, the senescence-messaging secretome (SMS) [219]. Premature senescence can be produced by a large panel of DNA-damaging agents and genotoxic stress including IR. In several preclinical models of radiation-induced lung injury, it has been shown that many types of cells bear senescence marks such as pneumocytes, macrophages, and endothelial cells [220, 221]. Interestingly, senolytic agents that selectively can kill senescent cells limit radiation-induced lung injury provided the evidence that senescence participates to the pathogenesis and that senolytic drugs could be a good strategy to reduced late normal tissue damages [222].
Recent research clearly shows that normal tissue injury is a dynamic and progressive process. The main challenge in the future will be to perfectly decipher this dynamic of events for each organ and its own characteristics. This will allow to propose new molecular and functional tools to predict, prevent, and treat damage to healthy tissues after irradiation.
5.13.6 Dose-Volume Effects and Constraints (QUANTEC, PENTEC, and HyTEC)
The evaluation of treatment plans in the treatment planning system is based on dose-volume histogram analysis for PTV and critical organs. Final plan quality evaluation should be based on plan complexity, plan robustness, and dose distribution analysis including dose-volume control. Constraints for any dose-volume relationship should be connected to the radiobiological outcome.
In 2010, a series of articles were published in the International Journal of Radiation Oncology Biology Physics as a meta-analysis of published dose–response observations for different critical organs. The project called Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC) aimed to review meaningful data published in the previous 18 years for common critical organs in terms of dose/volume values connected to radiobiological effects [223]. This endeavor was a challenge because it involved the amalgamation of different analytic methodologies, calculation methods, endpoints, and grading schemes, which were used in different studies to address the relationship between dosimetric parameters and the clinical outcomes of normal tissues.
QUANTEC consists of two introductory papers about the overview and history with some scientific issues related to the QUANTEC effort and about the suggestions on how to rationally incorporate the QUANTEC metrics/models into clinical practice. The core of the QUANTEC project is described in 16 articles for different organs at risk or complications:
-
Bladder
-
Brain
-
Brainstem
-
Esophagus
-
Hearing loss
-
Heart
-
Kidney
-
Larynx and pharynx
-
Liver
-
Lung
-
Optic nerves and chiasm
-
Penile bulb
-
Rectum
-
Salivary gland
-
Spinal cord
-
Stomach and small bowel
For each organ, there are associated sections describing: clinical significance, endpoints, challenges in defining volumes, review of dose/volume data, factors affecting risk, mathematical/biological models, special situations, recommended dose/volume limits, future toxicity studies, and toxicity scoring.
The QUANTEC reviews provide focused summaries of the dose/volume/outcome information for many organs, but these were usually obtained for 3D conformal RT or other techniques that have in many cases already been replaced by more modern techniques, such as VMAT or SRT. It should be emphasized that dose/volume constraints and other information in QUANTEC are expected to be updated in the future for relevant techniques. The data is not intended to be extrapolated to pediatric patients. Pediatric Normal Tissue Effects in the Clinic (PENTEC) is a recent initiative to review tolerance constraints for children, who may have different tolerance to that of adults [224].
For hypofractionated RT such as SRS or SBRT, relatively small target volumes receive hypofractionated RT schedules, typically in 1–5 fractions. As an extension to QUANTEC and other previous guides to tissue tolerance, high dose per fraction, Hypofractionated Treatment Effects in the Clinic (HyTEC) was published as a series of articles [225]. This project served to provide guidance on dose/volume constraints for hypofractionated regimens for 7 normal tissues as well as 9 disease sites (TCP). Interestingly, the possibility of so-called “new radiobiology” of hypofractionation is also alluded to—the possibility that large fractions may induce enhanced radiobiological effects in tumors by additional vascular targeting and anti-tumor immune responses [150]. Radiobiological aspects of hypofractionation are discussed in detail in Chap. 5.
Overall, the recommendations of QUANTEC, PENTEC, HyTEC, and other constraint guidelines should be used judiciously as a guide and should not replace clinical judgment.
5.13.7 Radiobiology Models for Normal Tissue Toxicity
Box 5.17 Normal Tissue Toxicity Modeling
-
Normal tissue complication probability (NTCP) describes the probability of organ/structure complication related to radiation treatment specified by physical and clinical factors in radiation oncology.
-
There are various approaches to NTCP modeling which usually are based on different statistical distributions.
-
The predictive power of the NTCP model is dependent on the parameters of models, which can include dosimetry as well as other clinical and treatment conditions.
The normal tissue complication probability (NTCP) is the probability that for a given dose distribution organ or structure complication can be expected. These complications can be multiple for one organ or structure and usually are called as endpoints in the models. NTCP is aimed at quantification of dependence of tolerance dose for a certain radiation effect on the size of treated volume. The NTCP models are supposed to be predictive and to be used to estimate the complication risk for organs at risk (OARs) after RT. OARs can be used to individualize the tumor dose for a given acceptable NTCP (Box 5.17).
NCTP models are used to describe dose–response curve shape for particular endpoint for an organ at risk, which is usually sigmoidal. These models are usually connected to Dose-Volume Histogram (DVH) of the applied treatment plan; therefore the models are sometimes called DVH-reduction models. More complex approach [226] moves towards spatial dose distribution in the patient and not dose-volume reduction only. When voxel-based evidence on organ radiosensitivity was acknowledged and attempts were made to develop a probabilistic atlas for NTCP in radiation oncology. However, there are other clinical factors that influence complications, such as chemotherapy, fraction size, pre-existing medical conditions, and comorbidities. The predictive strength for models can be enhanced with considering other important clinical and medical features for the patient. This information is expected to provide a boost for further deployment of biological models in the clinical treatment planning process.
Common NTCP models as described by [227] are:
-
1.
Lyman-Kutcher-Burman (LKB) model (Gaussian)
-
2.
Parallel architecture model (Logit)
-
3.
Weibull model (Weibull)
-
4.
Critical element model (Poisson)
-
5.
Relative seriality model (Poisson)
-
6.
Critical volume model (Binomial)
-
7.
Inverse tumor model (Poisson)
Models are based on different statistical distributions (in parentheses). The first four models are using cell-survival-based response, while others are phenomenological. However, each model may be expressed in terms of the parameters D50 (dose that is associated with the 50% response probability) and γ50 (gradient of the dose-response curve at the level of the 50% response probability). The steepness of the NTCP curve can be expressed in the models by parameter m. It is inversely proportional to the steepness of the dose response.
Commonly used model is the Lyman-Kutcher-Burman (LKB) model. This model assumes that the tolerance dose increases inversely as power of n of the partial volume irradiated. Examples of NTCP curves obtained for the LKB model are presented in Fig. 5.27.

NTCP curves calculated from Lyman-Kutcher-Burman model for two parameters combinations. Parameter m is inversely proportional to the steepness of the curve. (a) NTCP curve calculated by LKB model for D50 = 50 Gy and m = 0.50. For a dose of 50 Gy, the value of NTCP is 0.50. (b) NTCP curve calculated by LKB model for D50 = 60 Gy and m = 1. For a dose of 50 Gy, the value of NTCP is 0.43
Serial (critical element models) and parallel (critical volume models) are also common models. These mechanistic models are based on tissue architecture. It is assumed that organs consist of functional subunits, which can be organized in chains for serial organs or independently for parallel organs. Damage of one functional subunit impairs the function of the whole organ, while the function of a parallel organ is more dependent on the irradiated volume.
NTCP models are usually incorporated into in-house developed software in RT centers. Currently, they are also available in commercial treatment planning systems. Parameters for different NTCP models adopted from literature must be used with caution when the probability estimation is applied as a decision criterion for the treatment plan. NTCP can be used also for comparisons between different treatment plans or RT modalities. In these cases, NTCP is used as a relative value in the plan evaluation process and this approach is safer. However, the software should always allow the user to update model parameters. There should be detailed documentation for the models available. It is obvious that the value of NTCP is strongly dependent on the parameters of the model, and therefore should be used with caution.
5.14 Stem Cells in Radiotherapy
5.14.1 Introduction
Stem cells have been described as undifferentiated cells which are found in most adult mammalian tissues. Stem cells are divided into two principal groups: embryonic and adult stem cells. Embryonic cells, which have a pluripotency phenotype have the blastocyst inner cell mass as their origin. It means that they can be differentiated into all cells from the three main germ layers (endo-, meso-, and ectoderm). On the other hand, adult stem cells can be differentiated into cell types according to their origin tissue, thus they are multipotent. Under physiological conditions, adult stem cells are slow growing with a long G0 cell-cycle phase. The main function of such stem cells is to maintain tissue homeostasis including continuous regeneration and associated constant number of cells. The way they are divided is as follows: from the origin stem cell arises one daughter cell with stem cell properties and one progenitor cell with a higher proliferative capacity [228]. Below the normal stem cells in different tissues are described alongside cancer stem cells and their IR response or resistance together underlying molecular mechanisms.
5.14.2 Normal Stem Cells in Different Tissues
In the healthy human tissue, there are multiple cell populations with different stem cell phenotypic characteristics and radiation sensitivity. These different stem cell niches are discussed below based on the tissue localization.
5.14.2.1 Bone Marrow Stem Cells
Stem cells of the bone marrow are divided into two groups: hematopoietic and mesenchymal stem cells. From the hematopoietic stem cells arise leukocytes, erythrocytes, and thrombocytes and from the mesenchymal stem cells adipocytes, chondrocytes, myocytes, and osteocytes are generated [229]. One function of mesenchymal stem cells is to establish the hematopoietic stem cell niche [230]. When it comes to IR toxicity, the progenitors from the hematopoietic stem cells are more sensitive than the origin, more primitive, stem cells. Such a difference is linearly dose dependent in the progenitor cells and is one of the factors causing the development of one of the early radiation effects, the hematopoietic syndrome [231]. Importantly, both of these stem cell populations are responsible for the repopulating of the damaged bone-marrow homeostasis after IR exposure.
5.14.2.2 Neural Stem Cells
The neural stem cell pool can be divided according to the localization. Hence, we recognize the subventricular and the subgranular group [232]. Even within these two subtypes are heterogenous. Thus, one can distinguish four main types of cells from the subventricular niche: activated neural stem cells, dormant neural stem cells, progenitor cells, and quiescent neural stem cells [233]. Generally, neural stem cells can differentiate into multiple neuronal- and glial cell types. The most IR sensitive populations are activated neural stem cells and progenitor cells because IR induces their cell death, for example, apoptosis. Such effects lead to a reduced population of new neurons. To prevent the negative effects of IR on the neural stem cells different protective strategies have been tested, for example, administration of lithium [234, 235] or the natural polyphenol resveratrol [236]. It has been shown that lithium pre-treatment can reduce DNA damage and increase microglial activation [234, 235]. Resveratrol, on the other hand, has a neuroprotective effect, because it can reduce oxidative stress [236].
5.14.2.3 Skin Stem Cells
Several types of the stem cells exist in the skin which can differentiate into more than two dozen cell types, including epidermal-, keratinocyte-, and melanocyte stem cells [237]. The keratinocytes progenitors are the most IR sensitive ones, if damaged they are eliminated thereby contributing to the high epidermal sensitivity to IR. In contrast, more primitive keratinocyte stem cells possess active repair mechanisms and increased cell survival, but their rapid and faultier repair contribute to the genomic instability. Interestingly, while the keratinocyte stem cells favor repair of DNA damage, the melanocyte stem cells are not involved in tissue regeneration after IR damage [231].
5.14.2.4 Intestinal Stem Cells
Gastrointestinal syndrome, a known acute toxicity response to IR, promoted the first exploration of intestinal stem cells in radiation biology studies, which included the exposure of mice to doses greater than 14 Gy, inducing death after 7 to 12 days due to small intestine damage. This high sensitivity has been attributed to the fast cell turnover in the intestinal mucosa which, in mice, completely renews the epithelium every 5 days [231]. These studies allowed the characterization of intestinal regeneration, revealing the presence of a stem cell population near the bottom of the intestinal crypt. These actively cycling cells are highly sensitive to IR, undergoing apoptosis in response to doses as low as 1 Gy although this sensitivity seems to be dependent upon their position within the crypt [238]. Another stem cell subpopulation, known as crypt base columnar cells and characterized by the expression of Lgr5 (Leucine-Rich Repeat Containing G Protein-Coupled Receptor 5), are less radiosensitive than the previously described cells yet more sensitive than small intestine progenitor cells. Radiation toxicity can occur at low doses; however, crypt loss is only observed after exposure to higher radiation doses. This may be because crypts only disappear after total loss of the stem cell population, which only happens at doses greater than 8 Gy. This radiosensitivity could be caused by the accumulation of DNA damage or pro-apoptotic proteins after genotoxic stress, i.e., p53, ATM, and PUMA. The difference in radiosensitivity between the small intestine and the colon could be due to a more efficient p53 signaling, DNA repair and G2-phase checkpoint delay in the latter. The high expression level of the anti-apoptotic protein Bcl-2 in colon progenitors could be another reason. Paradoxically, the risk of developing cancer after exposure to IR is lower in the small intestine, suggesting the interplay between cell resistance and lower genomic stability. It should be noted that most of this knowledge is based on studies in mice; therefore, human models are still required in order to understand the intestinal stem cell radiation biology [231] (Box 5.18).
Box 5.18 Normal Tissue Stem Cells and Radiosensitivity
-
Stem cells are divided into two principal groups, embryonic and adult stem cells, respectively. Some examples are bone marrow-, neural, skin, and intestinal stem cells.
-
The main function of stem cells is to maintain the tissue homeostasis.
-
In the normal human tissue, there are multiple cell populations with different stem cell phenotypic characteristics and different IR sensitivity.
5.14.3 Cancer Stem Cells: Their Role in Radiation Therapy Sensitivity and Resistance
Tumor heterogeneity is found among patients with the same histological diagnosis as well as within each patient’s tumor as a result of genetic or phenotypic variations [31]. Further, the tissue of origin also influences the inter-tumoral heterogeneity because some of the driving signaling networks (e.g., those that maintain genomic integrity) may vary. Moreover, tumor progression, treatment sensitivity including towards RT and tumour aggressiveness are largely influenced by the origin of the carcinogenic transformation as well as the TME. Tumor heterogeneity plays a significant role in cancer cell survival, thus setting a significant challenge in the development of effective cancer treatment, or in the prevention of tumor progression and metastasis [239].
Recent studies describe two models, i.e., clonal evolution and cancer stem cells (CSCs) which in part can explain tumor heterogeneity as well as alterations during progression of malignancy. The clonal evolution model shares the idea that all cells can accumulate genetic mutations; therefore, any cell has tumorigenic potential [240, 241]. On the other hand, the CSCs’ model describes a hierarchy system in which tumor growth and progression can be maintained with a small proportion of cancer cells displaying stem-like characteristics, such as self-renewal. These stem-like cancer cells can drive tumorigenesis and differentiation, which can to some extent explain tumor cell heterogeneity [240, 242, 243]. It is believed that CSCs originate from the malignant transformation of normal stem cells or progenitor cells. Thus, CSCs possess key properties such as self-renewing and differentiation capacity, thereby being able to produce a phenotypically variable progeny [244]. Due to these characteristics, CSCs are thought to be important for tumor formation, recurrence, and resistance. Indeed, experimental data from xenograft studies in mice where different tumor cells with diverse CSCs characteristics have been engrafted and formed tumors, have demonstrated that CSCs are involved in tumor growth and metastasis and that they are resistant to a multitude of cancer treatments including RT [245,246,247,248,249,250,251]. It has been noted that only a few surviving CSCs in a heterogeneous tumor are enough to cause local tumor relapse after RT but also to promote metastasis [243, 247]. It is difficult to calculate the frequency of CSCs within a tumor as it is dependent on the type of malignancy. Further, the identification of CSCs is challenging as specific markers are not entirely clear. This is in part due to the high intra- and inter-tumor heterogeneity as well as by tumor plasticity, and variable genotypes and phenotypes. Regardless, a few markers, i.e., CD44, CD98, CD90, CD44+/CD24−, and CD133, are robust enough to be used in the identification of CSCs (Table 5.15) in breast cancer, small-cell lung cancer (SCLC), esophageal cancer, larynx, head and neck cancer, non-small cell lung cancer (NSCLC), etc. Interestingly, these markers are also associated with response to RT as illustrated in NSCLC and glioblastoma [245, 246, 248,249,250,251].
The variable phenotype of CSCs has a strong association with cells of origin thus making the comparison between different tumors complicated. In addition, the number of CSCs within a tumor is relatively small; therefore, the use of CSC markers is a poor predictor for treatment response [258]. Regardless, the characterization of CSCs remains important, as it may provide essential information in the development of more efficient treatment strategies and the prevention of tumor relapse as well as metastasis [240, 247,248,249, 251, 259].
A successful RT treatment largely depends on the elimination of cells with tumorigenic capacity, i.e., the number of clonogenic cells, which in part are CSCs, seeking to inactivate these permanently and to take control of tumor growth. Should a single CSC survive, the possibility of tumor relapse is tangible, with the consequent concern that this new tumor may now be RT resistant. In part RT resistance of tumors depends on the number of CSCs within the tumor mass, with greater numbers often being responsible for the failure of therapy [240, 260]. Some of the signaling networks involved are shown in Fig. 5.28.

CSCSs are also reported to have activated DDR signaling and increased DNA repair capacity, for example, ATM, Chk1/2, and NHEJ [245, 250], increased cell death resistance as well as upregulation of the signaling pathways involved in cell survival and proliferation, such as HIF-1α, WNT (Wingless-related integration site), NOTCH, or Hedgehog [247]. Moreover, it was recently shown that tumors with a higher count of CSCs had an impaired local control of the tumor and lower effect of RT than tumors with less CSCs [240, 261]. Several studies have suggested that RT sensitization is linked to the same signaling pathways involved in the preservation of CSCs cells, for example, WNT, NOTCH, or Hedgehog signaling pathways; therefore, these pathways have been indicated as possible targets for CSC-targeted therapies [247, 253]. Other strategies propose the inhibition of the DNA damage response (ATM, Chk1/2), the promotion of apoptosis, and the inhibition of epigenetic-related proteins, for example, histone deacetylase (HDAC), Enhancer of zeste homolog (EZH2). [247]. However, at present very few of these strategies have been clinically tested, due to complex cellular characteristics of CSCs. Nevertheless, further CSCs exploration and their signaling networks could reveal new potential therapeutic targets (Box 5.19).
Box 5.19 Cancer Stem Cells and Radiosensitivity
-
Tumor heterogeneity is explained by theory of clonal evolution and/or existence of CSCs.
-
Clonal evolution presumes accumulation of genetic mutation(s) while CSCs model describes a hierarchy system of tumor growth and progression maintained with only by a small subpopulation of CSCs.
-
Few specific markers are at hand for characterization of CSCs due to high inter- and intra-tumor heterogeneity, tumor plasticity, and variable genotypes and phenotypes.
-
Tumors with a higher count of CSCs show lower efficacy of RT and impaired local tumor control.
-
Multiple signaling cascades controlling DDR signaling, cell death, EMT, and hypoxia are reported to be altered in CSCs of tumors offering a putative way for RT sensitization for the future.
5.15 Radiotherapy and the Human Microbiota (Box 5.20)
Box 5.20 The Human Microbiota and Radiotherapy
-
The microbiota is composed of many microorganisms such as bacteria (most represented and studied), viruses, fungi, and archaea.
-
The microbiota plays a key physiological role in maintaining the gut health and well-being of the host.
-
Composition and abundance of the microbiota can be modified by various stresses and a stable alternative state of the microbiota can lead to pathologies.
-
Reduction of the fecal microbiota diversity and composition after RT are consistently associated with intestinal toxicity.
-
The microbiota can modify the tumor response effectiveness of RT.
-
The microbiota can be a therapeutic target for personalized medicine that might be used to increase patient’s quality of life during or after RT.
5.15.1 What Is the Human Microbiota?
The human body has around 500 billion cells including microorganisms such as bacteria, viruses, fungi, and archaea, on the surface of organs in contact with the outside. All of these microorganisms, hosted by the body, represent the human microbiota. Several microbiotas exist in an organism: in the digestive tract system (from mouth to anus), in the respiratory system, in the urogenital tract, and on the skin. Nevertheless, the larger community of microorganisms resides in the digestive system. The intestinal microbiota can be considered as an organ, given that it has specific functions of its own. Indeed, the intestinal microbiota makes it possible to maintain intestinal homeostasis by transforming nutrients that cannot be digested by the intestine into essential metabolites, by maintaining an effective epithelial barrier avoiding intestinal colonization by pathogens and also by participating to the development as well as the function of the immune system.
Among microorganisms, the most prevalent and studied ones are bacteria. The human GI tract is colonized by more than 2000 different individual bacteria species. Proportional representation and genus level distribution are dependent on the organ localization and vary with diet, age, and geographical localization of the host. As other microorganisms within the microbiota, bacteria have an important role in maintaining the health and well-being of the host. In the healthy gastrointestinal tract, Firmicutes and Bacteroidetes represent the predominant phylum in the microbiota (up to 80%). Other phylums which are less represented in the microbiota are Actinobacteria (3%), Proteobacteria (1%), Verrucomicrobia, and Fusobacteria (less than 1%).
In a physiological environment, the microbiota is in a state considered stable and healthy, also called eubiosis, where very little modifications take place in its composition. The relationship between host and microbiota is therefore beneficial for both entities. However, multifactorial events can cause transitory disturbances in the microbiota state and therefore population reorganization. Because of the resilience ability of the microbiota, such modifications are often transient. The microbiota then has the capability to return to its basic stable healthy state. However, the microbiota can also tend towards another stable state, called alternated state or dysbiosis, which becomes deleterious for the host [262]. Dysbiosis is defined as a condition where there is an excessive presence of pathogenic microorganisms, a defect in the communities of beneficial microorganisms and a loss of ecosystem structure, i.e., decrease in richness and diversity of microorganism species and increase of the low-grade inflammation, the intestinal permeability, and the oxidative stress. The dysbiotic state of the microbiota questions the scientific and medical community about its involvement in the development of certain pathologies like inflammatory bowel disease (IBD), metabolic disease (obesity or diabetes), neurological pathology (Alzheimer, autism, or Parkinson) but also cancers. Recently, dysbiosis was also reported in patients treated by RT (Fig. 5.29).

From microbiota healthy state to dysbiosis and pathologies: case of RT effects
5.15.2 Pelvic Radiotherapy and the Human Fecal Dysbiosis: Prospective Clinical Trials
Currently, at least eight prospective clinical studies assessed the effect of RT combined [263,264,265,266] or not [267,268,269,270] with other anti-tumors treatments, like chemotherapy, on the gut microbiota dysbiosis. The results of these studies have recently been presented in Byeongsang Oh’s review in 2021 [271]. Fecal microbiota changes by pelvic cancers (gynecological, colorectal/rectal, prostate, lymph node, and anal cancers) and/or after RT are briefly summarized below. Prior to RT, patients suffering from pelvic cancers have a loss of their fecal microbial diversity [266, 269]. The clinical studies also performed taxonomic analyses at the phyla level in feces from cancer patients. Results highlight variations of the relative bacteria abundance with an increase of the Firmicutes [269] and the Actinobacteria [266] and decrease of the Bacteroidetes [269] and the Fusobacteria [266]. Low fecal bacterial diversity has also been described in patients during and after pelvic RT [266, 267, 269]. Pelvic RT gradually reshapes microbiota bacterial composition in such cancer patients. Indeed, the prospective clinical studies demonstrate that during pelvic RT, the fecal relative abundance of the phylum Bacteroidetes tends to decrease and conversely that of the Fusobacteria significantly increases [266]. After the completion of pelvic RT, fecal relative abundance of the phyla Firmicutes is reduced [266, 269] and that of the phylum Bacteroidetes, Fusobacteria [266], Proteobacteria [270], and Actinobacteria [267] are enhanced.
5.15.3 Consequences of the Human Fecal Dysbiosis in Pelvic RT-Induced Digestive Toxicity
The prospective clinical studies described above, also show that the reduction of the fecal microbial diversity during and after pelvic RT, are consistently associated with radiation toxicity and therefore with intestinal complications, i.e., enteropathy, enteritis, and diarrhea [266,267,268,269]. Indeed, as suggested by a personal view of Andreyev’s team published in The Lancet Oncology in 2014 [272], clinical data seem to indicate that microbiota through its composition change might be an actor involved in radiation-induced intestinal toxicity. In 2018, Gerassy-Vainberg et al. [273] published preclinical data supporting this assumption. Indeed, results show that, rectal irradiation by brachytherapy leads to microbial dysbiosis in chronic post-exposure phase (6 weeks). Irradiated microbiota transplantation in germ-free mice transmits susceptibility to radiation damages at least in part through the production by epithelial cells of Interleukin-1 (IL-1). Nevertheless, in 2020, data published in Science by Guo Hua et al. show that gut microbiome-metabolome network plays a crucial role in substantial protection against radiation-induced toxicity though host defense regulation [274]. Indeed, a small proportion of mice named “elite-survivors” can survive a high dose of total body irradiation and that these individuals also reduce their susceptibility to radiation-induced digestive toxicity and damages. The families Lachnospiraceae and Enterococcaceae, together with downstream metabolites represented by propionate and tryptophan pathway members, contribute substantially to radioprotection. Even if a role of the gut microbiota is suggested on RT-induced tissue toxicity or protection, very little data exists concerning its involvement and the underlying mechanisms. Wang et al. showed that the reduction of the fecal microbial diversity is even more pronounced in patients with pelvic cancers who later progressed to RT-induced side effects (fatigue, diarrhea) [269]. Mitra et al. propose that compositional characteristics of microbiota in cancer patients could also be relevant to be predictive of end-of-anti-cancer treatment bowel toxicity [265].
5.15.4 Consequences of the Human Fecal Dysbiosis in Radiotherapy Efficiency
RT efficiency, regarding anti-tumor effects, passes in part through the induction of immunogenic cell death in which CD8+ cytotoxic T cell, CD11b+ myeloid cells and dendritic cells all have been described as major actors [275]. There is growing evidence of the existence of bidirectional effects of RT and microbiome composition. Indeed, RT-induced reduction of gut microbiota diversity, richness, and composition could be followed by the host immune response alteration which in turn could lead to an effectiveness change of the anticancer treatment themselves. In 2021, data published by Shiao et al. in the Cancer Cell, robustly demonstrated that, within the gut microbiota, commensal bacteria and fungi differentially regulate tumor response to RT [276]. Indeed, commensal bacteria are required for efficient immune anti-tumor effect (activation of T cells) of RT. Currently, no study has identified bacterial subjects involved in RT efficiency. By contrast, Shiao et al. demonstrated that commensal fungi regulate the immunosuppressive microenvironment of tumor (with combined effects on T cells and macrophages) after RT leading to a reduction of treatment efficiency. They highlighted a role of Saccharomycetales orders and specific Candida Albicans genera in fungal effect after RT.
5.15.5 Conclusions
Fecal microbial signature in patients with pelvic cancers may be a tool for RT risk assessment and/or efficiency. In order to robustly demonstrate this assumption, further experiments and clinical trials should be performed. The use of high-throughput data generation by multi-omics approaches (e.g., microbiota shotgun sequencing or metabolomics analyses) and mathematical models will give an added value compared to previous studies. After RT, a better understanding of individual states of different microorganism populations within the microbiota or more largely within the intestinal ecosystem could help to guide personalized medicine. Indeed, prophylactic or curative treatments like rich fiber diet, probiotic or fecal microbiota transplantation could prevent or reduce RT-induced toxicity and/or improve radiotherapy efficiency on tumor control.
5.16 Radiomics, Data Science, and Artificial Intelligence in Radiation Oncology
5.16.1 Basic Methods of Data Analysis
In radiation research, different techniques are used to measure complete molecular- or genomic profiles of organisms, resulting in various collected data types. Omics, a general term for specified measurements and studied biological fields, include genomics, proteomics, transcriptomics, metabolomics, phenomics, lipidomics, and many more. The suffix “omics” indicates interest in all molecules or genes of a specified type and their interactions rather than individual observations. For example, in genomics studies the genome, a set of all genes expressed in the cell, tissue, or organism, and their relationships with each other and with the environment is analyzed. So other omics-based platforms such as proteomics and transcriptomics study proteome (all proteins) and transcriptome (all types of transcripts like mRNAs and miRNAs), respectively. Omics data analysis helps to understand the influence of molecules and genes on a phenotype. The measurements can be obtained with techniques like microarrays, high-throughput sequencing technologies (Illumina, Oxford Nanopore, etc.), mass spectrometry (MS) (including MS imaging), flow-, and mass cytometry. The amount of collected data may vary from few records to millions, and the measurements may be taken for one point in time or more. With increased throughput, a large amount of data is generated, which requires advanced methods to perform a comprehensive analysis. By combining the domain expertise and knowledge of mathematics and statistics, data science extracts insights from big data.
Different statistical approaches are applied depending on the experimental design, type of data, sample size, number of replicates, number of time points, and so on. The first step of any analysis is data preprocessing that includes data cleaning and normalization. Omics data may have missing values, duplicated observations or outliers which need to be handled. Outliers are observations that deviate from other observations due to equipment failure or recording errors. An outlier can be corrected or removed from the analysis. There are many outlier detection methods that can detect one or more anomalies, like Chauvenet’s criterion, Grubbs’ criterion, Dixon’s procedure, Tukey’s or Huberta’s method. Which one to choose depends mainly on the data distribution and sample size. Observations with missing values can be ignored, removed or the values may be imputed with mean, median, mode, or constant value. Missing data imputation can also be carried out with, for example, the Nearest Neighbor algorithm, which finds k nearest observations to the observation with missing values and the aggregate of these measurements, as mean/median value, is used to impute the missing one. Before the analysis, data should be normalized to guarantee their numerical scale similarity across different experiments. The standardization techniques, asz-, t-score transformation, or local re-scaling, are often applied to partially correct the batch effect or reference instability.
For a comparative study, depending on the data distribution, a variety of statistical tests can be applied. Usually, if data contain one or two experimental groups, one-step testing is performed. Three or more groups or measurements collected from several time points (time series) require a two-step procedure to determine the difference profile—the omnibus type test (from ANOVA family, for example) followed by pairwise comparisons. The test hypothesis is verified with a p-value, the probability that the test statistics would take a value at least as extreme as observed, assuming the null hypothesis is true. The lower the p-value, the stronger the evidence against the null hypothesis, and if the value is equal or smaller than the assumed significance level α, it confirmed the presence of the effect studied.
Due to the number of data collected, the omics analyses usually require more than one hypothesis to be verified, which leads to the problem of multiple testing. In the case of a single test performed, the first type of error is controlled by significance level, but in the case of multiple testing, the number of false-positive results has to be maintained for the whole test family. This can be done with the use of Bonferroni correction, Simes-Hochberg procedure, Dunn-Šidák, Holm, or Hommel methods to control family-wise-error (FWER) or Benjamini and Hochberg procedure or Storey’s algorithm when focusing on false-discovery-rate (FDR). However, the p-value depends on sample size, and if the sample is sufficiently large, the statistical test will almost always indicate a significant difference. Therefore, in big data, it is recommended to calculate effect size together with the p-value. Effect size is a quantitative measure of the strength of a phenomenon calculated based on data and is independent of the sample size. A lot of different measures of effect size exist, and they can be divided into two categories: for indicating differences between groups (e.g., risk difference, risk ratio, odds ratio, Cohen’s d, Glass’s delta, Hedges’ g, the probability of superiority, ω2) and estimating measure of similarity between variables (e.g., the correlation coefficient r, R2, Spearman’s ρ, Kendall’s τ, φ coefficient, Cramer’s V, Cohen’s f, η2).
It is possible to integrate data and results from different experiments to get a unified view to them, in situations when the same experiment is performed on a different set of data or the same data is used in a different experiment (different method) but concerning the same characteristic (null hypothesis). The p-value integration can be carried out, among others, with Fisher product, Lancaster, Stouffer method, or weighted z-transformation. The adaptive rank truncated product method can also be applied at the pathway analysis level.
If measurements were taken for multiple characteristics to estimate the relationship and its strength between them (between a dependent variable and independent variables), a regression analysis can be conducted. Regression is a statistical method that tries to fit a model (function) to the data. The model has different forms, the most common one is a linear function, which is a line that closely fits the data according to a specific criterion. In other words, the dependent variable is a linear combination of the model parameters.
5.16.2 Artificial Intelligence and Machine Learning Methods for Knowledge Discovery
In radiation oncology, data science (incorporating the disciplines of computer science and statistics) attempts to provide clinical insight and clinical decision support using structured or unstructured clinical data that incorporates multiple variables descriptive of patient cohorts [277]. Daily clinical workflows produce a vast array of data comprising of electronic health records, treatment information, genomic data, multimodal imaging, and patient outcomes [278]. Inconsistencies in annotations of medical records presents a problem when utilizing unstructured data [279]. Here machine learning and artificial intelligence are key to detecting patterns within these vast data matrices [280, 281], providing opportunities for the development of diagnostic and prognostic tools. Increasingly clinical data and -omics data on the intrinsic biological characteristics of the patient are being integrated to derive models predictive of outcome metrics such as cancer survival and treatment response [282]. As the volume of clinical data available increases, innovative methods to process and interpret the data is required, translating the information into useable knowledge.
Machine learning (ML) and artificial intelligence (AI) approaches are capable of both identifying intrinsic patterns within data (termed unsupervised techniques) and developing models linking matrices of clinical data to identifying factors such as diagnostic or prognostic criteria (termed supervised techniques) [283]. For the latter type of model, the standard approach is to randomly separate the available data from patients into a training set which is presented to a machine learning algorithm capable of identifying important variables that link the patient data to the target variable (which is itself representative of the diagnostic or prognostic criterion). Subsequently, the generalizability of the learnt algorithm to new data is interrogated by presenting the algorithm with the unseen test data which remains from the available dataset after separation of the training data.
Typical applications of these approaches include prediction of toxicity to radiation therapy with dosimetric factors in head and neck cancer [284] to prediction of survival in pancreatic cancer [282]. A key advantage of machine learning in predicting therapy outcomes over conventional models such as NTCP and TCP which use dosimetric data [285] is the application of additional clinical and biochemical data [284, 286]. Many models demonstrate high performance owing to validation taking place using data which bears a close relationship to the training dataset. This is a particular concern when translating algorithms to a clinical setting, as models may not be evaluated on an external dataset [287, 288], and as such their generalizability is in question. In general, clinical translation of AI-based technologies requires generalizable, robust models which are validated in prospective, randomized clinical trials, and this represents a key challenge to their adoption [289].
Recently, deep learning-based algorithms have been employed, which perform automated image segmentation [290, 291] without the requirement for feature engineering, though the potential for overfitting and a lack of generalizability can persist with such approaches. Deep learning (DL) is a widely researched area in radiobiology and radiation oncology with models being developed for tasks such as modeling outcomes using dose-volume metrics, radiomic feature discovery, image and tumor segmentation, and treatment outcome prediction.
One area where machine learning approaches have seen substantial application is in the development of methodologies utilizing medical imaging data such as CT, MRI, and PET for predictive modeling, particularly of radiotherapeutic outcome. In this instance, the field has been termed radiomics [292, 293]. Here the lack of standardized image acquisition protocols (e.g., slice thickness and tube current in CT) can affect the quality and reliability of the radiomic features extracted by automated algorithms [248, 249, 294, 295]. Often manual clinical delineation of regions of interest of prognostic value prior to modeling (which is termed “feature engineering”) is utilized though this can introduce interobserver variability [296].
The success of radiomics in this context is in the development of “interpretable” machine learning or AI algorithms for a range of applications. Features may be extracted from 2D and 3D imaging data, reducing them to multiple features describing tumor intensity, shape, and texture [297]. These may subsequently be utilized to quantify tumor heterogeneity, where tumors with high heterogeneity have been shown to demonstrate resistance to treatment [298]. Once the features have been extracted machine learning or statistical learning can be applied to match feature patterns to “ground truth” data. Radiomic approaches have been applied to multiple imaging formats including CT, MRI, PET, and ultrasound [299,300,301,302]. These approaches have been successfully applied to segmentation and detection problems such as the differentiation of prostate cancers with Gleeson grade 6 and 7 [303], the discrimination of breast cancer subtypes [304] and in TNM staging [305]. Potential clinical applications of radiomics-based classification extend to the detection of lung nodules providing a prognostic and diagnostic aid to the clinician [306]. In terms of personalization of treatment, these approaches have been used to identify prostate cancer patients at risk of biochemical recurrence post-treatment from MRI-based imaging [307], to distinguish between HPV-positive and HPV-negative head and neck cancers [308] and to monitor the response to RT [309]. Similarly, ML models are also being examined in radiation genomics, or radiogenomics, which explores tumor and normal tissue response to radiation at a genomic level [310]. Linking imaging characteristics with genomic data has the potential to aid clinical cancer research and improve decision-making capabilities and personalize therapy [310,311,312]. Various deep learning methodologies are currently used to generate new knowledge from radiogenomic data including convolutional neural networks (CNNs) [313] and deep neural networks (DNNs) [314].
However, as highlighted earlier, significant challenges remain regarding the clinical interpretability of radiomic and radiogenomic analyses and models, image acquisition standardization and data storage in the era of “big data” [292, 312, 315, 316]. Randomized controlled trials will remain the gold standard in evaluating diagnostic and prognostic interventions that are AI or ML based will be evaluated in oncology [317, 318], where data science approaches can also provide complementary information [319, 320].
5.16.3 Radiomics in Radiobiology and Clinical Oncology
5.16.3.1 Techniques in Medical Imaging
Several imaging modalities exist in radiology and nuclear medicine with different physical, acquisition, and reconstruction principles which have strengths and weaknesses but all of them are indispensable for differential diagnosis.
-
Ultrasound sonography (USG) uses high-frequency mechanical waves to differentiate tissues based on various reflectivity on the tissue edges. USG is an affordable and inexpensive modality with minimal burden for the patient.
-
Roentgenography (X-ray imaging, RX) uses electromagnetic waves with energy in the X-ray range most often between 40 and 120 keV. RX is a fast and inexpensive modality with small radiation exposure. If one collects thousands of 2D RX images from different angles, one can use computer algorithms for creating 3D images and one is talking about CT. Advantages of CT are in special resolution, high contrast, and 3D information; however, at the cost of high radiation exposure and higher price than RX.
-
MRI also uses also electromagnetic waves as well as RX of CT but with an energy in the radio range most often between 240 and 500 neV which cannot ionize biological tissue (no radiation exposure) which is together with high tissue contrast advantage of MRI. The disadvantages are longer examination time (15–45 min), price, availability, and several contraindications.
Imaging methods in nuclear medicine are characterized by lower spatial resolution than the radiological methods mentioned above, but they contain very specific functional information. Small amount of a specific radiopharmaceutical is injected into the patient and then emitted gamma photons are detected. Depending on the isotope used, one speaks of positron emission tomography (PET, beta+ tracer) or single photon emission computed tomography (SPECT, gamma tracer). Both methods can be combined into hybrid modalities mostly with CT for obtaining anatomical information.
5.16.3.2 Main Steps in Medical Image Analysis
In the first step, it is necessary to find the pathological area(s) and make a basic description. The process that identifies such areas or, in general, region of interest (ROI) is named segmentation or delineation and can be done manually or with the support of ML algorithms. Manual segmentation is a time consuming and demanding task, with relatively low level of reproducibility so it is beneficial to use semi- or fully automatic methods.
Sometimes it is helpful to segment the pathology based on different histological tissue types, for example, necrotic part, active tumor, or edema but also precise anatomical localization, diameter or volume, shape, intensity, and changes compared to the previous examination.
After all variables are collected, the radiologist must decide which kinds of pathologies it could be. Occasionally, the radiological diagnoses are not unequivocal, for example, the pathology looks like high grade gliomas but with non-negligible probability it could be metastasis of some other primary tumor and the treatment of such different entities are completely different. Each hospital produces thousands of images per day, and it is obvious that usage of the same kind of computer algorithms or AI can save time, increase reproducibility and precision of diagnosis.
5.16.3.3 Radiomics: Definition, Features, and Examples
The concept of radiomics appeared in 2012 [321]. While the traditional analysis of imaging is based on the visual interpretation of simple features—such as tumor size, general shape, contrast uptake, or signal intensity—radiomics processes any type of imaging computationally and translates into complex quantitative data. Radiomics is based on qualitative and quantitative analyses, combining numerical data from medical imaging with clinical and biological characteristics to obtain predictive and/or prognostic information about patients. Indeed, the study of cellular interaction within tissues and intrinsic characteristics of medical imaging reflect the physiology and pathophysiology of the affected organ. The radiomics approach is: (1) Noninvasive; (2) Allows an evaluation of the studied tissues in their globality, thus characterizing their spatial heterogeneity; (3) Represents an easy way to follow the patient over time, allowing understanding the changes throughout the history of the disease and the therapeutic sequence. A typical radiomics workflow follows five steps as illustrated in Fig. 5.30.

Typical radiomics workflow. The different steps are: (1) Data selection: choosing the image to analyze, the imaging protocol to use and the correlated outcome. (2) Imaging and segmentation with (semi-) automatic methods to improve reproducibility. (3) Feature extraction and selection with appropriate algorithms. (4) Modeling using available machine learning models. (5) Reporting results. (Adopted from Keek et al. [322] with permission)
Combining radiomics features with deep learning features or semantic features may further improve prognostic performance. The changes over time may also be integrated (delta-radiomics). Several studies have proven the effectiveness of using these features independently in predictive modeling. As was mentioned above, radiologists make basic descriptions of pathology like volume, shape, etc. but the number of these descriptors are limited by the radiologist’s time and his/her eyes. But there exist tens or hundreds of different descriptors which can describe pathology and a method which analyses all these descriptive parameters is called Radiomics and these parameters are called features.
One can divide radiomics features into several classes like: (1) First order; (2) Two and three (2D/3D) Shape and; (3) Grey level class (e.g., Size zone, Neighboring tone, Run length, Co-occurrence, Dependency).
The first order features (more than 15) characterized distribution of voxels intensities so they can be commonly known histogram parameters like median, mean, or several quartiles. But they also include mathematically sophisticated parameters like energy, entropy, mean absolute deviation, root mean squared, skewness, or kurtosis (“Sharpness of the peak”).
Shape features (2D or 3D, more than 20) are intensity-independent parameters which are extracted from segmented binary mask image or triangle mash. For example, 2D features can be mesh surface, perimeter, sphericity, maximum 2D diameter or elongation. As 3D shape features, one can mention, for example, mesh volume, surface area to volume ratio, compactness, or flatness.
The biggest features class (which can be subdivided) with more than 50 features is grey level class. For example, grey level size zone features are trying to quantify connected voxels in an image which share the same intensity and one can extract features like grey level non-uniformity, size-zone non-uniformity, grey level variance or zone entropy, etc. Neighboring grey tone features quantify differences between intensity of voxel and average intensity of neighbors’ voxels within defined distance and one can extract features like coarseness, contrast, complexity, or strength.
Radiomics create a model to predict clinical outcomes based on extracted features. Not all features have to be used, selection of features are done before modeling because lots of them are correlated to each other or can be unstable across a dataset. Clinical outcome which radiomics model can be diagnosis (benign or malignant, subtype or stage), treatment evaluation, or prognosis (survival coefficients).
5.16.3.4 Clinical Applications
These days there exist hundreds of papers which evaluate the usefulness of radiomics in clinical practice mostly on CT data, but MRI and PET are becoming more common. Radiomics can be used in diagnosis as well as treatment evaluation of different oncological diseases like brain tumors, breast, lung-, prostate-, or colorectal cancer. Radiomics are being applied in the field of oncology in different settings to help decision-making such as:
-
Differentiation between human papillomavirus-positive and human papillomavirus-negative oropharyngeal tumors on contrast-enhanced CT [323].
-
Prediction of tumor aggressiveness in prostate cancer [324].
-
Assistance to automatic segmentation and sub-target volume definition in prostate cancer [325].
-
Prediction of treatment response and outcome in head and neck and lung cancer (with combination of genomic features) [326,327,328], rectum [329], esophageal [330], or prostate cancer [331].
-
Prediction of toxicity in head and neck (xerostomia) [332] and lung (pneumonitis) [333] cancers.
-
Differential diagnosis between recurrence and RT-induced radionecrosis in brain [334].
Although promising, the predictive power sometimes evidenced in the pilot studies need to be externally validated in independent datasets with numerous methodological pitfalls including imaging technique standardization.
5.17 Exercises and Self-Assessment
-
Q1.
What is meant by the “therapeutic window”? Mention several methods to widen the therapeutic window.
-
Q2.
The tumor volume doubling time (VDT) is heterogeneous among tumors and influences RT response. Discuss and reflect on different parameters that control VDT of tumors.
-
Q3.
It is important to estimate the growth fraction (GF) of tumors and several methods may be used in vitro and in vivo to assess this. Give some examples of methods and in what context they are applied.
-
Q4.
Discuss the link between the Hallmarks of Radiobiology and the Hallmarks of Cancer.
-
Q5.
Which of the below statements is wrong?
-
(a)
Lowering the dose rate leads to greater sparing of late responding normal tissues than of tumors.
-
(b)
The process of redistribution might push cells from a radioresistant to a radiosensitive cell-cycle phase.
-
(c)
During chronic low dose rate exposure, cells with long repair half times will be spared relative to their counterparts with rapid DNA damage repair.
-
(d)
Low dose rate irradiation can be considered as a form of extreme fractionation.
-
(a)
-
Q6.
How can dose rate affect be explained in terms of linear-quadratic model?
-
Q7.
What are the classical factors that are used to predict RT response in a tumor?
-
Q8.
List four techniques which are used to measure biomarkers to predict RT response.
-
Q9.
Oxygen enhancement ratio (OER) is seen with some but not all IR qualities. Please indicate which type (a–d) that doesn’t have OER.
-
(a)
X-rays
-
(b)
Gamma-rays
-
(c)
Neutrons
-
(d)
α-particles
-
(a)
-
Q10.
All of the following statements about hypoxic cell radiosensitizers are true except one, please indicate and explain.
-
(a)
Increases radiosensitivity of hypoxic cells
-
(b)
Nitroimidazole groups of drugs are commonly used
-
(c)
Presence of nitro group in second position, decreases electron affinity and sensitization
-
(d)
Dose-limiting toxicity of Misonidazole is neurotoxicity
-
(a)
-
Q11.
Give some examples how photon radiation can induce modification extracellular signaling pathways.
-
Q12.
Two principal mechanisms of tumor metabolism participate in radiation resistance. Give their names.
-
Q13.
What is radiation-induced abscopal effect in oncology?
-
Q14.
Describe the typical acute and late effects following exposure of the skin to radiation. Hints: target cells at risk, latent period, volume effect, pathology, recovery.
-
Q15.
Define and describe the late adverse effects of RT.
-
Q16.
Can radionecrosis be avoided by choosing a more appropriate radiation modality?
-
Q17.
How can access to the forecasted 3D RT isodose curves allow for a better prevention of osteoradionecrosis of the mandible?
-
Q18.
What is the main function of the stem cells?
-
Q19.
What is the most radiosensitive group of stem cells?
-
Q20.
Describe some different characteristics of cancer stem cells which may contribute to RT resistance?
-
Q21.
Describe potential role of the intestinal microbiota in RT-induced adverse side effects (gut toxicity) or in RT efficiency concerning anti-tumor effects.
5.18 Exercise Solutions
-
SQ1.
Therapeutic window: The difference between tumor control probability and normal tissue complication probability at identical irradiation dose. Methods to widen the therapeutic window: Dose fractionation, reduction of the normal tissue/organ at risk exposed volume, combination therapy.
-
SQ2.
VDT is influenced by localization of the tumor, i.e., tumor site. It is also influenced if the tumor is a primary or a metastatic lesion where the latter often have reduced VDT as a result of limited nutrition and oxygen levels. VDT is also influenced by histology of the tumor, i.e., the inherited growth capacity of the cells. Finally, VDT is influenced by tumor heterogeneity in proliferative signaling cascades which is a consequence of different genomic- and signaling make ups of the individual tumors.
-
SQ3.
In vitro tumor cell progression through S-phase can be monitored by BrdUrd or IdUrd-labeling of cells. These tracers are incorporated into DNA as tumor pass through S-phase and by using an antibody against BrdUrd or IdUrd, cells in S-phase can be determined using flowcytometry. Another method utilizes 3H-thymidine to assess DNA-synthesis by flow cytometry. The third method is based on PET-analyses of tumors in vivo which have been pulsed with radio-labeled 18F-fluoro-3′-deoxy-3′-l-fluorothymidine (FLT). FLT is phosphorylated by Thymidine Kinase 1 which has an S-phase activity. Hence, FLT tracer levels are a surrogate for S-phase cells which can be evaluated by PET scanning. Finally, the proliferation rate in a tumor biopsy can be analyzed by immunohistochemical staining for the nuclear Ki-67 antigen, reflecting S-phase proportion of cells.
-
SQ4.
Likely links are shown below.
Hallmark of radiobiology
Hallmark of cancer
Repair
Genomic instability and mutations, enabling replicative immortality
Redistribution
Sustaining proliferative signaling
Repopulation
Evading growth suppressors, sustaining proliferative signaling, tumor-promoting activation
Radiosensitivity
Resisting cell death, deregulating cellular energetics
Reoxygenation
Inducing angiogenesis
Reactivation of the immune response
Avoiding immune destruction, tumor-promoting activation, activation invasion and metastasis
-
SQ5.
Alternative (c) is the wrong answer. Accumulation of DNA damage is larger in cells with long repair half times than for cells which show rapid repair of their DNA damages.
-
SQ6.
Single-track and double-track actions can both induce DNA double strand breaks. There is no correlation between dose rate and single-track X-ray lesion (αcontribution in the LQ model). In a double-track action, different X-ray photon tracks produce the two interactions of single strand DNA lesions, and therefore the formation of double strand lesions is proportional to the radiation dose squared (β in the LQ model).
-
SQ7.
Tumor oxygen status, the degree of repopulation or proliferation rate and intrinsic radiosensitivity.
-
SQ8.
Proteomics, genomics, epigenomics, genomics, or transcriptomics, used for measuring proteins, DNA/chromatin, DNA, or RNA and transcription, respectively.
-
SQ9.
Alternative (d). OER is 1 for high LET radiation like α- particles.
-
SQ10.
Alternative (c). The presence of the nitro group in second position, increases electron affinity and radiosensitization.
-
SQ11.
Photon beam activates major oncogenic signaling pathways such as Ras, MAPK/ERK, and PI3K/AKT in part via the epidermal growth factor receptor (EGFR) cascade. Radiation resistance is associated with these signaling cascades due to their pro-survival nature. For example, when AKT is phosphorylated, tumor cells are protected by decreased autophagy and apoptosis, as well as increased DNA repair capacity. Mutated RAS has also been associated with resistance to photons in cancer cells.
-
SQ12.
The mitochondrial and or glucose metabolism, respectively.
-
SQ13.
Abscopal effects are radiation-induced systemic anti-tumor immune responses in which irradiation of a primary tumor or large metastasis causes remission of distant, non-irradiated lesions.
-
SQ14.
Acute effects: Dry skin (impairment of cell production), epilation (injury to hair follicles), erythema (vascular leakage). Latency time: Few weeks. Large volume effect: The smaller the volume, the higher the tolerance to radiation. Transient effect: Reversible injury. Late effects: Gangrene, ulcer, telangiectasia (vascular damage), fibrosis (increase in collagen fibers). Latency: Months-years. Large volume effect: The smaller the volume, the higher the tolerance to radiation. Chronic, irreversible injury.
-
SQ15.
Late effects of radiation are progressive, irreversible and occur months, years, or decades after radiation therapy. They are based on an interactive response of parenchymal cells, vascular endothelium and fibroblasts, with a contribution from immune cells, especially macrophages. Tissues and organs are affected by atrophy, fibrosis, or necrosis, which can severely impair their functions and lead to a loss of function.
-
SQ16.
No. Since the risk of radionecrosis remains life-long the affected tissues are at danger with the total radiation dose being the primary risk factor for the tissue involved, combined with other risk factors.
-
SQ17.
Adapting the preventive extraction of teeth to the risk zone of >50–60 Gy would allow for a more appropriate dental management: more aggressive in the >60 Gy zone and far less aggressive in the other areas of the jaw, improving the quality of life of these patients. The fewer extractions in highly irradiated areas, the lesser the risk for ORN.
-
SQ18.
The principal function of stem cells is to maintain tissue homeostasis including continuous regeneration and associated constant number of cells.
-
SQ19.
Bone marrow stem cells.
-
SQ20.
The answer is displayed in Fig. 5.29. In brief, CSC may have (1) Increased DNA repair capacity which allows them to handle IR-induced DNA DSBs; (2) Increased signaling networks that block IR-induced cell death including deficient pro-apoptotic signaling and increased anti-apoptotic signaling; (3) CSCs have slow proliferation and may therefore not be so sensitivity to IR-induced DNA damage.
-
SQ21.
Studies showed evidence of the existence of bidirectional effects of RT on the tumor and on the intestinal microbiota. In prospective clinical studies, a reduction of the fecal microbial diversity during and after pelvic RT was measured in patients suffering from intestinal complications. Also, RT-induced modification of microbiota diversity and composition can modify the host immune response and in turn the effectiveness of the anticancer treatment themselves including RT.
References
Borras JM, Lievens Y, Dunscombe P, Coffey M, Malicki J, Corral J, Gasparotto C, Defourny N, Barton M, Verhoeven R, van Eycken L, Primic-Zakelj M, Trojanowski M, Strojan P, Grau C. The optimal utilization proportion of external beam radiotherapy in European countries: an ESTRO-HERO analysis. Radiother Oncol. 2015;116(1):38–44. https://doi.org/10.1016/j.radonc.2015.04.018.
Nikjoo H, Uehara S, Wilson WE, Hoshi M, Goodhead DT. Track structure in radiation biology: theory and applications. Int J Radiat Biol. 1998;73(4):355–64. https://doi.org/10.1080/095530098142176.
Fowler JF. The linear-quadratic formula and progress in fractionated radiotherapy. Br J Radiol. 1989;62(740):679–94. https://doi.org/10.1259/0007-1285-62-740-679.
Moulder JE, Seymour C. Radiation fractionation: the search for isoeffect relationships and mechanisms. Int J Radiat Biol. 2018;94(8):743–51. https://doi.org/10.1080/09553002.2017.1376764.
Dörr W. Pathogenesis of normal-tissue side-effects. In: Joiner MC, van der Kogel AJ, editors. Basic clinical radiobiology. 4th ed. London: Hodder Arnold; 2009. p. 169–89.
Tinganelli W, Durante M. Carbon ion radiobiology. Cancers (Basel). 2020;12(10):3022. https://doi.org/10.3390/cancers12103022.
Vanderwaeren L, Dok R, Verstrepen K, Nuyts S. Clinical progress in proton radiotherapy: biological unknowns. Cancers (Basel). 2021;13(4):604. https://doi.org/10.3390/cancers13040604.
Grellier N, Belkacemi Y. Effets biologiques des hautes doses par fraction [Biologic effects of high doses per fraction]. Cancer Radiother. 2020;24(2):153–8. https://doi.org/10.1016/j.canrad.2019.06.017.
Lin B, Gao F, Yang Y, Wu D, Zhang Y, Feng G, Dai T, Du X. FLASH radiotherapy: history and future. Front Oncol. 2021;11:644400. https://doi.org/10.3389/fonc.2021.644400.
McBride WH, Schaue D. Radiation-induced tissue damage and response. J Pathol. 2020;250(5):647–55. https://doi.org/10.1002/path.5389.
Marks LB, Yorke ED, Jackson A, Ten Haken RK, Constine LS, Eisbruch A, Bentzen SM, Nam J, Deasy JO. Use of normal tissue complication probability models in the clinic. Int J Radiat Oncol Biol Phys. 2010;76(3 Suppl):S10–9. https://doi.org/10.1016/j.ijrobp.2009.07.1754.
Higgins GS, O’Cathail SM, Muschel RJ, McKenna WG. Drug radiotherapy combinations: review of previous failures and reasons for future optimism. Cancer Treat Rev. 2015;41(2):105–13. https://doi.org/10.1016/j.ctrv.2014.12.012.
Hall EJ, Giaccia AJ, editors. Radiobiology for the radiologist. 8th ed. Philadelphia, PA: Wolters Kluwer; 2019.
Khaled S, Held K. Radiation biology: a handbook for teachers and students. 1st ed. International Atomic Energy Agency; 2012.
Podgorsak EB. Radiation oncology physics. 1st ed. Vienna: IAEA; 2005.
Joiner MC, Van der Kogel AJ. Basic clinical radiobiology. 5th ed. CRC Press; 2018.
Shrieve DC, Loeffler JS, editors. Human radiation injury. 1st ed. Lippincott Williams & Wilkins; 2011.
Khan FM, Sperduto PW, Gibbons JP. Khan’s treatment planning in radiation oncology. Lippincott Williams & Wilkins; 2016.
Chang DS, Lasley FD, Das IJ, Mendonca MS, Dynlacht JR. Basic radiotherapy physics and biology. 1st ed. Springer International Publishing; 2014.
Mayles P, Nahum A, Rosenwald J. Handbook of radiotherapy physics: theory and practice. 1st ed. CRC Press; 2007.
Alpen E. Radiation biophysics. 2nd ed. Academic Press; 1997.
Halperin E, Brady L, Wazer D, Perez C. Perez & Brady’s principles and practice of radiation oncology. 7th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2019.
Miller I, Min M, Yang C, Tian C, Gookin S, Carter D, Spencer SL. Ki67 is a graded rather than a binary marker of proliferation versus quiescence. Cell Rep. 2018;24(5):1105–1112.e5. https://doi.org/10.1016/j.celrep.2018.06.110.
Hall EJ, Giaccia AJ. Radiobiology for the radiologist. Philadelphia: Lippincott Wilkins & Williams; 2006.
Withers HR. The four R’s of radiotherapy. In: Advances in radiation biology, vol. 5. Elsevier; 1975. p. 241–71.
Steel GG, McMillan TJ, Peacock JH. The 5Rs of radiobiology. Int J Radiat Biol. 1989;56(6):1045–8. https://doi.org/10.1080/09553008914552491.
Boustani J, Grapin M, Laurent PA, Apetoh L, Mirjolet C. The 6th R of radiobiology: reactivation of anti-tumor immune response. Cancers (Basel). 2019;11(6):860. https://doi.org/10.3390/cancers11060860.
Lehnert S. Biomolecular action of ionizing radiation. 1st ed. CRC Press; 2007.
Shuryak I, Hall EJ, Brenner DJ. Dose dependence of accelerated repopulation in head and neck cancer: supporting evidence and clinical implications. Radiother Oncol. 2018;127(1):20–6. https://doi.org/10.1016/j.radonc.2018.02.015.
Daguenet E, Khalifa J, Tolédano A, Borchiellini D, Pointreau Y, Rodriguez-Lafrasse C, Chargari C, Magné N. To exploit the 5 ‘R’ of radiobiology and unleash the 3 ‘E’ of immunoediting: ‘RE’-inventing the radiotherapy-immunotherapy combination. Ther Adv Med Oncol. 2020;12:1758835920913445. https://doi.org/10.1177/1758835920913445.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Bristow RG, Alexander B, Baumann M, Bratman SV, Brown JM, Camphausen K, Choyke P, Citrin D, Contessa JN, Dicker A, Kirsch DG, Krause M, Le QT, Milosevic M, Morris ZS, Sarkaria JN, Sondel PM, Tran PT, Wilson GD, Willers H, Wong RKS, Harari PM. Combining precision radiotherapy with molecular targeting and immunomodulatory agents: a guideline by the American Society for Radiation Oncology. Lancet Oncol. 2018;19(5):e240–51. https://doi.org/10.1016/s1470-2045(18)30096-2.
Harrington K, Jankowska P, Hingorani M. Molecular biology for the radiation oncologist: the 5Rs of radiobiology meet the hallmarks of cancer. Clin Oncol (R Coll Radiol). 2007;19(8):561–71. https://doi.org/10.1016/j.clon.2007.04.009.
Withers HR, Taylor JM, Maciejewski B. The hazard of accelerated tumor clonogen repopulation during radiotherapy. Acta Oncol. 1988;27:131–146.
Mitchell JB, Bedford JS, Bailey SM. Dose-rate effects in mammalian cells in culture III. Comparison of cell killing and cell proliferation during continuous irradiation for six different cell lines. Radiat Res. 1979;79:537–51.
Friedl AA, Prise KM, Butterworth KT, Montay-Gruel P, Favaudon V. Radiobiology of the FLASH effect. Med Phys. 2021; https://doi.org/10.1002/mp.15184. Epub ahead of print.
Sabloff M, Tisseverasinghe S, Babadagli ME, Samant R. Total body irradiation for hematopoietic stem cell transplantation: what can we agree on? Curr Oncol. 2021;28(1):903–17.
Gore EM, Lawton CA, Ash RC, Lipchik RJ. Pulmonary function changes in long-term survivors of bone marrow transplantation. Int J Radiat Oncol Biol Phys. 1996;36(1):67–75. https://doi.org/10.1016/S0360-3016(96)00123-X.
Soule BP, Simone NL, Savani BN, Ning H, Albert PS, Barrett AJ, Singh AK. Pulmonary function following total body irradiation (with or without lung shielding) and allogeneic peripheral blood stem cell transplant. Bone Marrow Transplant. 2007;40(6):573–8. https://doi.org/10.1038/sj.bmt.1705771.
Wheldon TE. The radiobiological basis of total body irradiation. Br J Radiol. 1997;70(840):1204–7. https://doi.org/10.1259/bjr.70.840.9505837.
Spencer K, Parrish R, Barton R, Henry A. Palliative radiotherapy. BMJ. 2018;23(360):k821. https://doi.org/10.1136/bmj.k821.
Sakamoto K. Radiobiological basis for cancer therapy by total or half-body irradiation. Nonlinearity Biol Toxicol Med. 2004;2(4):293–316. https://doi.org/10.1080/15401420490900254.
Hoeben BAW, Pazos M, Albert MH, Seravalli E, Bosman ME, Losert C, et al. Towards homogenization of total body irradiation practices in pediatric patients across SIOPE affiliated centers. A survey by the SIOPE radiation oncology working group. Radiother Oncol. 2021;155:113–9. https://doi.org/10.1016/j.radonc.2020.10.032.
Cosset JM, Girinsky T, Malaise E, Chaillet MP, Dutreix J. Clinical basis for TBI fractionation. Radiother Oncol. 1990;18(Suppl 1):60–7. https://doi.org/10.1016/0167-8140(90)90179-Z.
Thomas ED, Clift RA, Hersman J, Sanders JE, Stewart P, Buckner CD, et al. Marrow transplantation for acute nonlymphoblastic leukemia in first remission using fractionated or single-dose irradiation. Int J Radiat Oncol Biol Phys. 1982;8(5):817–21. https://doi.org/10.1016/0360-3016(82)90083-9.
Cheng J, Schultheiss T, Wong J. (2008). Impact of drug therapy, radiation dose, and dose rate on renal toxicity following bone marrow transplantation. Int J Radiat Oncol Biol Phys. 2008;71(5):1436–43. https://doi.org/10.1016/j.ijrobp.2007.12.009.
Gogna N, Morgan G, Downs K, Atkinson K, Biggs J. Lung dose rate and interstitial pneumonitis in total body irradiation for bone marrow transplantation. Australas Radiol. 1992;36(4):317–20. https://doi.org/10.1111/j.1440-1673.1992.tb03208.x.
Kim TH, Rybka WB, Lehnert S, Podgorsak EB, Freeman CR. Interstitial pneumonitis following total body irradiation for bone marrow transplantation using two different dose rates. Int J Radiat Oncol Biol Phys. 1985;11(7):1285–91. https://doi.org/10.1016/0360-3016(85)90243-3.
Graves SS, Storer BE, Butts TM, Storb R. Comparing high and low total body irradiation dose rates for minimum-intensity conditioning of dogs for dog leukocyte antigen-identical bone marrow grafts. Biol Blood Marrow Transplant. 2013;19(11):1650–4. https://doi.org/10.1016/j.bbmt.2013.08.007.
Storb R, Raff RF, Appelbaum FR, Deeg HJ, Graham TC, Schuening FG, Sale G, Bryant E, Seidel K. Fractionated versus single-dose total body irradiation at low and high dose rates to condition canine littermates for DLA-identical marrow grafts. Blood. 1994;83(11):3384–9. https://doi.org/10.1182/blood.V83.11.3384.3384.
Begg AC. Predicting response to radiotherapy: evolutions and revolutions. Int J Radiat Biol. 2009;85(10):825–36.
Torres-Roca JF, Stevens CW. Predicting response to clinical radiotherapy: past, present, and future directions. Cancer Control. 2008;15(2):151–6. https://doi.org/10.1177/107327480801500207.
Yaromina A, Krause M, Baumann M. Individualization of cancer treatment from radiotherapy perspective. Mol Oncol. 2012;6(2):211–21. https://doi.org/10.1016/j.molonc.2012.01.007.
Hirst DG, Robson T. Molecular biology: the key to personalised treatment in radiation oncology? Br J Radiol. 2010;83(993):723–8. https://doi.org/10.1259/bjr/91488645.
Overgaard J, Hansen HS, Overgaard M, Bastholt L, Berthelsen A, Specht L, Lindeløv B, Jørgensen K. A randomized double-blind phase III study of nimorazole as a hypoxic radiosensitizer of primary radiotherapy in supraglottic larynx and pharynx carcinoma. Results of the Danish Head and Neck Cancer Study (DAHANCA) Protocol 5-85. Radiother Oncol. 1998;46(2):135–46. https://doi.org/10.1016/S0167-8140(97)00220-X.
West C, Davidson S, Roberts S, Hunter R. Intrinsic radiosensitivity and prediction of patient response to radiotherapy for carcinoma of the cervix. British journal of cancer. 1993;68(4):819–23. https://doi.org/10.1038/bjc.1993.434.
Lee JM, Bernstein A. p53 mutations increase resistance to ionizing radiation. Proc Natl Acad Sci U S A. 1993;90(12):5742–6.
Papathanasiou MA, Kerr NC, Robbins JH, McBride OW, Alamo I Jr, Barrett SF, Hickson ID, Fornace AJ Jr. Induction by ionizing radiation of the gadd45 gene in cultured human cells: lack of mediation by protein kinase C. Mol Cell Biol. 1991;11(2):1009–16.
Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9(2):102–14. https://doi.org/10.1038/nrg2290.
Brown JM. Tumor hypoxia, drug resistance, and metastases. J Natl Cancer Inst. 1990;82:338–9.
Chaplin DJ, Olive PL, Durand RE. Intermittent blood flow in a murine tumor: radiobiological effects. Cancer Res. 1987;47(2):597–601.
Thomlinson RH, Gray LH. The histological structure of some human lung cancers and the possible implications for radiotherapy. Br J Cancer. 1955;9(4):539–49. https://doi.org/10.1038/bjc.1955.55.
van Putten LM. Tumour reoxygenation during fractionated radiotherapy; studies with a transplantable mouse osteosarcoma. Eur J Cancer. 1968;4(2):172–82. https://doi.org/10.1016/0014-2964(68)90015-7.
Wright EA, Howard-Flanders P. The influence of oxygen on the radiosensitivity of mammalian tissues. Acta Radiol. 1957;48(1):26–32. https://doi.org/10.3109/00016925709170930.
Palcic B, Skarsgard LD. Reduced oxygen enhancement ratio at low doses of ionizing radiation. Radiat Res. 1984;100(2):328–39.
Barendsen GW, Koot CJ, Van Kersen GR, Bewley DK, Field SB, Parnell CJ. The effect of oxygen on impairment of the proliferative capacity of human cells in culture by ionizing radiations of different LET. Int J Radiat Biol Relat Stud Phys Chem Med. 1966;10(4):317–27. https://doi.org/10.1080/09553006614550421.
Broerse JJ, Barendsen GW, van Kersen GR. Survival of cultured human cells after irradiation with fast neutrons of different energies in hypoxic and oxygenated conditions. Int J Radiat Biol Relat Stud Phys Chem Med. 1968;13(6):559–72. https://doi.org/10.1080/09553006814550621.
Koumenis C, Wouters BG. “Translating” tumor hypoxia: unfolded protein response (UPR)-dependent and UPR-independent pathways. Mol Cancer Res. 2006;4(7):423–36. https://doi.org/10.1158/1541-7786.MCR-06-0150.
Koritzinsky M, Levitin F, van den Beucken T, Rumantir RA, Harding NJ, Chu KC, Boutros PC, Braakman I, Wouters BG. Two phases of disulfide bond formation have differing requirements for oxygen. J Cell Biol. 2013;203(4):615–27. https://doi.org/10.1083/jcb.201307185.
Bartoszewska S, Collawn JF. Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol Biol Lett. 2020;25:18. https://doi.org/10.1186/s11658-020-00212-1.
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–33. https://doi.org/10.1016/S1097-2765(03)00105-9.
Singleton DC, Harris AL. Targeting the ATF4 pathway in cancer therapy. Expert OpinTher Targets. 2012;16(12):1189–202. https://doi.org/10.1517/14728222.2012.728207.
Colliez F, Gallez B, Jordan BF. Assessing tumor oxygenation for predicting outcome in radiation oncology: a review of studies correlating tumor hypoxic status and outcome in the preclinical and clinical settings. Front Oncol. 2017;7:10. https://doi.org/10.3389/fonc.2017.00010.
Griffiths JR, Robinson SP. TheOxyLite: a fibre-optic oxygen sensor. Br J Radiol. 1999;72(859):627–30. https://doi.org/10.1259/bjr.72.859.10624317.
Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC Jr, Raleigh JA. Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol. 1998;71(2):270–7. https://doi.org/10.1006/gyno.1998.5163.
Komar G, Seppänen M, Eskola O, Lindholm P, Grönroos TJ, Forsback S, Sipilä H, Evans SM, Solin O, Minn H. 18F-EF5: a new PET tracer for imaging hypoxia in head and neck cancer. J Nucl Med. 2008;49(12):1944–51. https://doi.org/10.2967/jnumed.108.053785.
Al-Arafaj A, Ryan EA, Hutchison K, Mannan RH, Mercer J, Wiebe LI, AJ ME. An evaluation of iodine-123 iodoazomycinarabinoside as a marker of localized tissue hypoxia in patients with diabetes mellitus. Eur J Nucl Med. 1994;21(12):1338–42. https://doi.org/10.1007/BF02426699.
Lyng H, Malinen E. Hypoxia in cervical cancer: from biology to imaging. Clin Transl Imaging. 2017;5(4):373–88. https://doi.org/10.1007/s40336-017-0238-7.
Kjellen E, Joiner MC, Collier JM, Johns H, Rojas A. A therapeutic benefit from combining normobaric carbogen or oxygen with nicotinamide in fractionated X-ray treatments. Radiother Oncol. 1991;22(2):81–91. https://doi.org/10.1016/0167-8140(91)90002-X.
Tharmalingham H, Hoskin P. Clinical trials targeting hypoxia. Br J Radiol. 2019 Jan;92(1093):20170966.
Adams GE, Flockhart IR, Smithen CE, Stratford IJ, Wardman P, Watts ME. Electron-affinic sensitization. VII. A correlation between structures, one-electron reduction potentials, and efficiencies of nitroimidazoles as hypoxic cell radiosensitizers. Radiat Res. 1976;67(1):9–20.
Hall EJ, Giaccia AJ. The biology and exploitation of tumor hypoxia. In: Hall EJ, Giaccia AJ, editors. Radiobiology for the radiologist. 8th ed. Philadelphia, PA: Wolters Kluwer; 2019. p. 825–46.
Sheldon PW, Foster JL, Fowler JF. Radiosensitization of C3H mouse mammary tumours by a 2-nitroimidazole drug. Br J Cancer. 1974;30(6):560–5. https://doi.org/10.1038/bjc.1974.235.
Zeman EM, Hirst VK, Lemmon MJ, Brown JM. Enhancement of radiation-induced tumor cell killing by the hypoxic cell toxin SR 4233. Radiother Oncol. 1988;12(3):209–18. https://doi.org/10.1016/0167-8140(88)90263-0.
Stratford IJ, Stephens MA. The differential hypoxic cytotoxicity of bioreductive agents determined in vitro by the MTT assay. Int J Radiat Oncol Biol Phys. 1989;16(4):973–6. https://doi.org/10.1016/0360-3016(89)90898-5.
Welz S, Mönnich D, Pfannenberg C, Nikolaou K, Reimold M, La Fougère C, Reischl G, Mauz PS, Paulsen F, Alber M, Belka C, Zips D, Thorwarth D. Prognostic value of dynamic hypoxia PET in head and neck cancer: results from a planned interim analysis of a randomized phase II hypoxia-image guided dose escalation trial. Radiother Oncol. 2017;124(3):526–32. https://doi.org/10.1016/j.radonc.2017.04.004.
Daniel M, Andrzejewski P, Sturdza A, Majercakova K, Baltzer P, Pinker K, Wadsak W, Mitterhauser M, Pötter R, Georg P, Helbich T, Georg D. Impact of hybrid PET/MR technology on multiparametric imaging and treatment response assessment of cervix cancer. Radiother Oncol. 2017;125(3):420–5. https://doi.org/10.1016/j.radonc.2017.10.036.
Elamir AM, Stanescu T, Shessel A, Tadic T, Yeung I, Letourneau D, Kim J, Lukovic J, Dawson LA, Wong R, Barry A, Brierley J, Gallinger S, Knox J, O’Kane G, Dhani N, Hosni A, Taylor E. Simulated dose painting of hypoxic sub-volumes in pancreatic cancer stereotactic body radiotherapy. Phys Med Biol. 2021;66(18) https://doi.org/10.1088/1361-6560/ac215c.
Even AJ, van der Stoep J, Zegers CM, Reymen B, Troost EG, Lambin P, van Elmpt W. PET-based dose painting in non-small cell lung cancer: comparing uniform dose escalation with boosting hypoxic and metabolically active sub-volumes. Radiother Oncol. 2015;116(2):281–6. https://doi.org/10.1016/j.radonc.2015.07.013.
de Mey S, Dufait I, Jiang H, Corbet C, Wang H, Van De Gucht M, Kerkhove L, Law KL, Vandenplas H, Gevaert T, Feron O, De Ridder M. Dichloroacetate radiosensitizes hypoxic breast cancer cells. Int J Mol Sci. 2020;21(24):9367. https://doi.org/10.3390/ijms21249367.
Zannella VE, Dal Pra A, Muaddi H, McKee TD, Stapleton S, Sykes J, Glicksman R, Chaib S, Zamiara P, Milosevic M, Wouters BG, Bristow RG, Koritzinsky M. Reprogramming metabolism with metformin improves tumor oxygenation and radiotherapy response. Clin Cancer Res. 2013;19(24):6741–50. https://doi.org/10.1158/1078-0432.CCR-13-1787.
Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W, Zhao F, You L, Zheng L, Zhang T, Zhao Y. Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol. 2021;14(1):14. https://doi.org/10.1186/s13045-020-01030-w.
Liu YP, Zheng CC, Huang YN, He ML, Xu WW, Li B. Molecular mechanisms of chemo- and radiotherapy resistance and the potential implications for cancer treatment. MedComm. 2021;2(3):315–40. https://doi.org/10.1002/mco2.55.
Galeaz C, Totis C, Bisio A. Radiation resistance: a matter of transcription factors. Front Oncol. 2021;11:662840. https://doi.org/10.3389/fonc.2021.662840.
Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F, Gong Z, Guo C, Li X, Deng H, Cao K, Zhou M, Xiang B, Li X, Li Y, Li G, Xiong W, Zeng Z. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37(1):87. https://doi.org/10.1186/s13046-018-0758-7.
Chen HHW, Kuo MT. Improving radiotherapy in cancer treatment: Promises and challenges. Oncotarget. 2017;8(37):62742–58. https://doi.org/10.18632/oncotarget.18409.
Morgan MA, Lawrence TS. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin Cancer Res. 2015;21(13):2898–904. https://doi.org/10.1158/1078-0432.CCR-13-3229.
Bhattacharya S, Asaithamby A. Repurposing DNA repair factors to eradicate tumor cells upon radiotherapy. Transl Cancer Res. 2017;6(Suppl 5):S822–39. https://doi.org/10.21037/tcr.2017.05.22.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. https://doi.org/10.1038/nrc2342.
Huang R, Zhou PK. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct Target Ther. 2021;6(1):254. https://doi.org/10.1038/s41392-021-00648-7.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. https://doi.org/10.1038/nature08467.
Kiwerska K, Szyfter K. DNA repair in cancer initiation, progression, and therapy-a double-edged sword. J Appl Genet. 2019;60(3–4):329–34. https://doi.org/10.1007/s13353-019-00516-9.
Al-Dimassi S, Abou-Antoun T, El-Sibai M. Cancer cell resistance mechanisms: a mini review. Clin Transl Oncol. 2014;16(6):511–6. https://doi.org/10.1007/s12094-014-1162-1.
Squatrito M, Brennan CW, Helmy K, Huse JT, Petrini JH, Holland EC. Loss of ATM/Chk2/p53 pathway components accelerates tumor development and contributes to radiation resistance in gliomas. Cancer Cell. 2010;18(6):619–29. https://doi.org/10.1016/j.ccr.2010.10.034.
Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J, Yuan J, Wang M, Chen D, Sun Y, Woodward WA, Liu Y, Dean DC, Liang H, Hu Y, Ang KK, Hung MC, Chen J, Ma L. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16(9):864–75. https://doi.org/10.1038/ncb3013.
Kotula E, Berthault N, Agrario C, Lienafa MC, Simon A, Dingli F, Loew D, Sibut V, Saule S, Dutreix M. DNA-PKcs plays role in cancer metastasis through regulation of secreted proteins involved in migration and invasion. Cell Cycle. 2015;14(12):1961–72. https://doi.org/10.1080/15384101.2015.1026522.
Srivastava M, Nambiar M, Sharma S, Karki SS, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh RK, Ray P, Natarajan R, Kelkar M, De A, Choudhary B, Raghavan SC. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell. 2012;151(7):1474–87. https://doi.org/10.1016/j.cell.2012.11.054.
Kan C, Zhang J. BRCA1 mutation: a predictive marker for radiation therapy? Int J Radiat Oncol Biol Phys. 2015;93(2):281–93. https://doi.org/10.1016/j.ijrobp.2015.05.037.
Kinzel L, Ernst A, Orth M, Albrecht V, Hennel R, Brix N, Frey B, Gaipl US, Zuchtriegel G, Reichel CA, Blutke A, Schilling D, Multhoff G, Li M, Niyazi M, Friedl AA, Winssinger N, Belka C, Lauber K. A novel HSP90 inhibitor with reduced hepatotoxicity synergizes with radiotherapy to induce apoptosis, abrogate clonogenic survival, and improve tumor control in models of colorectal cancer. Oncotarget. 2016;7(28):43199–219. https://doi.org/10.18632/oncotarget.9774.
Madhusudan S. Evolving drug targets in DNA base excision repair for cancer therapy. Curr Mol Pharmacol. 2012;5(1):1–2. https://doi.org/10.2174/1874467211205010001.
Li LY, Guan YD, Chen XS, Yang JM, Cheng Y. DNA repair pathways in cancer therapy and resistance. Front Pharmacol. 2021;11:629266. https://doi.org/10.3389/fphar.2020.629266.
Baptistella AR, Landemberger MC, Dias MVS, Giudice FS, Rodrigues BR, da Silva PPCE, Cassinela EK, Lacerda TC, Marchi FA, Leme AFP, Begnami MD, Aguiar S Jr, Martins VR. Rab5C enhances resistance to ionizing radiation in rectal cancer. J Mol Med (Berl). 2019;97(6):855–69. https://doi.org/10.1007/s00109-019-01760-6.
Uraki S, Ariyasu H, Doi A, Kawai S, Takeshima K, Morita S, Fukai J, Fujita K, Furuta H, Nishi M, Sugano K, Inoshita N, Nakao N, Yamada S, Akamizu T. Reduced expression of mismatch repair genes MSH6/MSH2 directly promotes pituitary tumor growth via the ATR-Chk1 pathway. J Clin Endocrinol Metab. 2018;103(3):1171–9. https://doi.org/10.1210/jc.2017-02332.
Peng H, Yao S, Dong Q, Zhang Y, Gong W, Jia Z, Yan L. Excision repair cross-complementing group 1 (ERCC1) overexpression inhibits cell apoptosis and is associated with unfavorable prognosis of esophageal squamous cell carcinoma. Medicine (Baltimore). 2018;97(31):e11697. https://doi.org/10.1097/MD.0000000000011697.
Li Q, Ma R, Zhang M. XRCC1 rs1799782 (C194T) polymorphism correlated with tumor metastasis and molecular subtypes in breast cancer. Onco Targets Ther. 2018;11:8435–44. https://doi.org/10.2147/OTT.S154746.
Brewer MR, Yun CH, Lai D, Lemmon MA, Eck MJ, Pao W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc Natl Acad Sci. 2013;110(38):E3595–604. https://doi.org/10.1073/pnas.1220050110.
Endres NF, Barros T, Cantor AJ, Kuriyan J. Emerging concepts in the regulation of the EGF receptor and other receptor tyrosine kinases. Trends Biochem Sci. 2014;39(10):437–46. https://doi.org/10.1016/j.tibs.2014.08.001.
Fidler IJ, Kim SJ, Langley RR. The role of the organ microenvironment in the biology and therapy of cancer metastasis. J Cell Biochem. 2007;101(4):927–36. https://doi.org/10.1002/jcb.21148.
Rodemann HP, Dittmann K, Toulany M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int J Radiat Biol. 2007;83(11–12):781–91. https://doi.org/10.1080/09553000701769970.
Schmidt-Ullrich RK, Mikkelsen RB, Dent PE, Todd DG, Valerie K, Kavanagh BD, Chen PB. Radiation-induced proliferation of the human A431 squamous carcinoma cells is dependent on EGFR tyrosine phosphorylation. Oncogene. 1997;15(10):1191–7. https://doi.org/10.1038/sj.onc.1201275.
Affolter A, Fruth K, Brochhausen C, Schmidtmann I, Mann WJ, Brieger J. Activation of mitogen-activated protein kinase extracellular signal-related kinase in head and neck squamous cell carcinomas after irradiation as part of a rescue mechanism. Head Neck. 2011;33(10):1448–57. https://doi.org/10.1002/hed.21623. Epub 2010 Nov 10.
Grana TM, Rusyn EV, Zhou H, Sartor CI, Cox AD. Ras mediates radioresistance through both phosphatidylinositol 3-kinase-dependent and Raf-dependent but mitogen-activated protein kinase/extracellular signal-regulated kinase kinase-independent signaling pathways. Cancer Res. 2002;62(14):4142–50.
Konings K, Vandevoorde C, Baselet B, Baatout S, Moreels M. Combination therapy with charged particles and molecular targeting: a promising avenue to overcome radioresistance. Front Oncol. 2020:128. https://doi.org/10.3389/fonc.2020.00128.
Toulany M, Kasten-Pisula U, Brammer I, Wang S, Chen J, Dittmann K, Baumann M, Dikomey E, Rodemann HP. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clin Cancer Res. 2006;12(13):4119–26. https://doi.org/10.1158/1078-0432.CCR-05-2454.
Kim TJ, Lee JW, Song SY, Choi JJ, Choi CH, Kim BG, et al. Increased expression of pAKT is associated with radiation resistance in cervical cancer. Br J Cancer. 2006;94(11):1678–82.
Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–42. https://doi.org/10.1038/nrm1835.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–8. https://doi.org/10.1172/JCI39104.
Weinberg RA. The biology of cancer. Garland Science; 2013.
Weinberg RA. The biology of cancer, vol. 544. 1st ed. New York: Garland Science, Taylor & Francis Group, LLC; 2007. p. 560–1.
Kawamoto A, Yokoe T, Tanaka K, Saigusa S, Toiyama Y, Yasuda H, et al. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol Rep. 2012;27(1):51–7. https://doi.org/10.3892/or.2011.1485.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105(17):6392–7. https://doi.org/10.1073/pnas.0802047105.
Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST, Lee HB. Role of reactive oxygen species in TGF-1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–75.
Leong KG, Niessen K, Kulic I, Raouf A, Eaves C, Pollet I, Karsan A. Jagged1-mediated Notch activation induces epithelial-to-mesenchymal transition through Slug-induced repression of E-cadherin. J Exp Med. 2007;204(12):2935–48. https://doi.org/10.1084/jem.20071082.
Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK. A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002;21(8):1948–56. https://doi.org/10.1093/emboj/21.8.1948.
Zavadil J, Cermak L, Soto-Nieves N, Böttinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. EMBO J. 2004;23(5):1155–65. https://doi.org/10.1038/sj.emboj.7600069.
Yan S, Wang Y, Yang Q, Li X, Kong X, Zhang N, Yuan C, Yang N, Kong B. Low-dose radiation-induced epithelial-mesenchymal transition through NF-κB in cervical cancer cells. Int J Oncol. 2013;42(5):1801–6. https://doi.org/10.3892/ijo.2013.1852.
Cui YH, Suh Y, Lee HJ, Yoo KC, Uddin N, Jeong YJ, Lee JS, Hwang SG, Nam SY, Kim MJ, Lee SJ. Radiation promotes invasiveness of non-small-cell lung cancer cells through granulocyte-colony-stimulating factor. Oncogene. 2015;34(42):5372–82. https://doi.org/10.1038/onc.2014.466.
Park JK, Jang SJ, Kang SW, Park S, Hwang SG, Kim WJ, Kang JH, Um HD. Establishment of animal model for the analysis of cancer cell metastasis during radiotherapy. Radiat Oncol. 2012;7:153. https://doi.org/10.1186/1748-717X-7-153.
He E, Pan F, Li G, Li J. Fractionated ionizing radiation promotes epithelial-mesenchymal transition in human esophageal cancer cells through PTEN deficiency-mediated Akt activation. PLoS One. 2015;10(5):e0126149. https://doi.org/10.1371/journal.pone.0126149.
Liu W, Huang YJ, Liu C, Yang YY, Liu H, Cui JG, Cheng Y, Gao F, Cai JM, Li BL. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3β and repression of ZEB1. Lab Invest. 2014;94(4):362–70. https://doi.org/10.1038/labinvest.2013.153.
Bhatt AN, Chauhan A, Khanna S, Rai Y, Singh S, Soni R, et al. Transient elevation of glycolysis confers radio-resistance by facilitating DNA repair in cells. BMC Cancer. 2015;15(1):1–12. https://doi.org/10.1186/s12885-015-1368-9.
Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci U S A. 2004;101(6):1714–9.
Abratt RP, Shepherd LJ, Salton DG. Palliative radiation for stage 3 non-small cell lung cancer—a prospective study of two moderately high dose regimens. Lung Cancer. 1995;13(2):137–43. https://doi.org/10.1016/0169-5002(95)00487-4.
Crane CH, Janjan NA, Abbruzzese JL, Curley S, Vauthey J, Sawaf HB, Dubrow R, Allen P, Ellis LM, Hoff P, Wolff RA, Lenzi R, Brown TD, Lynch P, Cleary K, Rich TA, Skibber J. Effective pelvic symptom control using initial chemoradiation without colostomy in metastatic rectal cancer. Int J Radiat Oncol Biol Phys. 2001;49(1):107–16. https://doi.org/10.1016/S0360-3016(00)00777-X.
Dirix P, Vingerhoedt S, Joniau S, Van Cleynenbreugel B, Haustermans K. Hypofractionated palliative radiotherapy for bladder cancer. Support Care Cancer. 2016;24(1):181–6. https://doi.org/10.1007/s00520-015-2765-y.
Yan J, Milosevic M, Fyles A, Manchul L, Kelly V, Levin W. A hypofractionated radiotherapy regimen (0–7-21) for advanced gynaecological cancer patients. Clin Oncol (R Coll Radiol). 2011;23(7):476–81. https://doi.org/10.1016/j.clon.2011.01.001.
Choi HS, Jeong BK, Jeong H, Ha IB, Kang KM. Role of radiotherapy in the management of malignant airway obstruction. Thorac Cancer. 2020;11(8):2163–9. https://doi.org/10.1111/1759-7714.13523.
Bezjak A, Adam J, Barton R, Panzarella T, Laperriere N, Wong CS, Mason W, Buckley C, Levin W, McLean M, Wu JS, Sia M, Kirkbride P. Symptom response after palliative radiotherapy for patients with brain metastases. Eur J Cancer. 2002;38(4):487–96. https://doi.org/10.1016/S0959-8049(01)00150-2.
Jones B, Dale RG. Further radiobiologic modeling of palliative radiotherapy: use of virtual trials. Int J Radiat Oncol Biol Phys. 2007;69(1):221–9.
Song CW, Glatstein E, Marks LB, Emami B, Grimm J, Sperduto PW, Kim MS, Hui S, Dusenbery KE, Cho LC. Biological principles of stereotactic body radiation therapy (SBRT) and stereotactic radiation surgery (SRS): indirect cell death. Int J Radiat Oncol Biol Phys. 2021;110(1):21–34. https://doi.org/10.1016/j.ijrobp.2019.02.047.
Ansems M, Span PN. The tumor microenvironment and radiotherapy response; a central role for cancer-associated fibroblasts. Clin Transl Radiat Oncol. 2020;22:90–7. https://doi.org/10.1016/j.ctro.2020.04.001.
Wei R, Liu S, Zhang S, Min L, Zhu S. Cellular and extracellular components in tumor microenvironment and their application in early diagnosis of cancers. Anal Cell Pathol. 2020;6283796:2020. https://doi.org/10.1155/2020/6283796.
Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15(7):409–25. https://doi.org/10.1038/nrc3958.
Menon H, Ramapriyan R, Cushman TR, Verma V, Kim HH, Schoenhals JE, Atalar C, Selek U, Chun SG, Chang JY, Barsoumian HB, Nguyen QN, Altan M, Cortez MA, Hahn SM, Welsh JW. Role of radiation therapy in modulation of the tumor stroma and microenvironment. Front Immunol. 2019;10:193. https://doi.org/10.3389/fimmu.2019.00193.
Krisnawan VE, Stanley JA, Schwarz JK, DeNardo DG. Tumor microenvironment as a regulator of radiation therapy: new insights into stromal-mediated radioresistance. Cancers (Basel). 2020;12(10):2916. https://doi.org/10.3390/cancers12102916.
Baker DG, Krochak RJ. The response of the microvascular system to radiation: a review. Cancer Investig. 1989;7(3):287–94. https://doi.org/10.3109/07357908909039849.
Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9(6):685–93. https://doi.org/10.1038/nm0603-685.
Khan FM, Gibbons JP. Khan’s the physics of radiation therapy. 5th ed. Lippincott Williams & Wilkins; 2014.
Levitt S, Purdy J, Perez C, Vijayakumar S. Technical basis of radiation therapy. 4th ed. Berlin, Heidelberg: Springer; 2012.
Donlon NE, Power R, Hayes C, Reynolds JV, Lysaght J. Radiotherapy, immunotherapy, and the tumour microenvironment: turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021;502:84–96. https://doi.org/10.1016/j.canlet.2020.12.045.
Rodriguez-Ruiz ME, Rodriguez I, Garasa S, Barbes B, Solorzano JL, Perez-Gracia JL, Labiano S, Sanmamed MF, Azpilikueta A, Bolaños E, Sanchez-Paulete AR, Aznar MA, Rouzaut A, Schalper KA, Jure-Kunkel M, Melero I. Abscopal effects of radiotherapy are enhanced by combined immunostimulatory mAbs and are dependent on CD8 T cells and crosspriming. Cancer Res. 2016;76(20):5994–6005. https://doi.org/10.1158/0008-5472.CAN-16-0549.
Siva S, MacManus MP, Martin RF, Martin OA. Abscopal effects of radiation therapy: a clinical review for the radiobiologist. Cancer Lett. 2015;356(1):82–90. https://doi.org/10.1016/j.canlet.2013.09.018.
Sprung CN, Forrester HB, Siva S, Martin OA. Immunological markers that predict radiation toxicity. Cancer Lett. 2015;368(2):191–7. https://doi.org/10.1016/j.canlet.2015.01.045.
Vanpouille-Box C, Diamond JM, Pilones KA, Zavadil J, Babb JS, Formenti SC, Barcellos-Hoff MH, Demaria S. TGFβ is a master regulator of radiation therapy-induced antitumor immunity. Cancer Res. 2015;75(11):2232–42. https://doi.org/10.1158/0008-5472.CAN-14-3511.
Sureka CS, Armpilia C. Radiation biology for medical physicists. 1st ed. CRC Press; 2017.
Bower JE. Cancer-related fatigue—mechanisms, risk factors, and treatments. Nat Rev Clin Oncol. 2014;11(10):597–609. https://doi.org/10.1038/nrclinonc.2014.127.
Morgan GW, Breit SN. Radiation and the lung: a reevaluation of the mechanisms mediating pulmonary injury. Int J Radiat Oncol Biol Phys. 1995;31(2):361–9. https://doi.org/10.1016/0360-3016(94)00477-3.
Formenti SC, Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718–26. https://doi.org/10.1016/S1470-2045(09)70082-8.
De la Cruz V, Sanz Á, Torrego JC, Fiorini AB. The strange abscopal effect. Rev Clin Esp. 2014;214(3):170–1. https://doi.org/10.1016/j.rce.2013.12.005.
Joe MB, Lum JJ, Watson PH, Tonseth RP, McGhie JP, Truong PT. Radiation generates an abscopal response and complete resolution of metastatic squamous cell carcinoma of the anal canal: a case report. J Gastrointest Oncol. 2017;8(6):E84–9. https://doi.org/10.21037/jgo.2017.06.15.
Mole RH. Whole body irradiation; radiobiology or medicine? Br J Radiol. 1953;26(305):234–41. https://doi.org/10.1259/0007-1285-26-305-234.
Abuodeh Y, Venkat P, Kim S. Systematic review of case reports on the abscopal effect. Curr Probl Cancer. 2016;40(1):25–37. https://doi.org/10.1016/j.currproblcancer.2015.10.001.
Grimaldi AM, Simeone E, Giannarelli D, Muto P, Falivene S, Borzillo V, Giugliano FM, Sandomenico F, Petrillo A, Curvietto M, Esposito A, Paone M, Palla M, Palmieri G, Caracò C, Ciliberto G, Mozzillo N, Ascierto PA. Abscopal effects of radiotherapy on advanced melanoma patients who progressed after ipilimumab immunotherapy. Onco Targets Ther. 2014;3:e28780. https://doi.org/10.4161/onci.28780.
Hiniker SM, Chen DS, Reddy S, Chang DT, Jones JC, Mollick JA, Swetter SM, Knox SJ. A systemic complete response of metastatic melanoma to local radiation and immunotherapy. Transl Oncol. 2012;5(6):404–7. https://doi.org/10.1593/tlo.12280.
Qin Q, Nan X, Miller T, Fisher R, Teh B, Pandita S, Farach AM, Pingali SR, Pandita RK, Butler EB, Pandita TK, Iyer SP. Complete local and abscopal responses from a combination of radiation and nivolumab in refractory Hodgkin’s lymphoma. Radiat Res. 2018;190(3):322–9. https://doi.org/10.1667/RR15048.1.
Burnette B, Fu YX, Weichselbaum RR. The confluence of radiotherapy and immunotherapy. Front Oncol. 2012;2:143. https://doi.org/10.3389/fonc.2012.00143.
Harrison L, Hatahet Z, Wallace SS. In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J Mol Biol. 1999;290(3):667–84. https://doi.org/10.1006/jmbi.1999.2892.
Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, Guo C, Wu X, Li Y, Li X, Li G, Xiong W, Zeng Z. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer. 2019;18(1):128. https://doi.org/10.1186/s12943-019-1055-6.
Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–9. https://doi.org/10.1038/nm1622.
Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M, Sharma P, Lysiak JJ, Harden TK, Leitinger N, Ravichandran KS. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461(7261):282–6. https://doi.org/10.1038/nature08296.
Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, Vermaelen K, Panaretakis T, Mignot G, Ullrich E, Perfettini JL, Schlemmer F, Tasdemir E, Uhl M, Génin P, Civas A, Ryffel B, Kanellopoulos J, Tschopp J, André F, Lidereau R, McLaughlin NM, Haynes NM, Smyth MJ, Kroemer G, Zitvogel L. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–8. https://doi.org/10.1038/nm.2028.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Métivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54–61. https://doi.org/10.1038/nm1523.
Craig DJ, Nanavaty NS, Devanaboyina M, Stanbery L, Hamouda D, Edelman G, Dworkin L, Nemunaitis JJ. The abscopal effect of radiation therapy. Future Oncol. 2021;17(13):1683–94. https://doi.org/10.2217/fon-2020-0994.
Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29(3):372–83. https://doi.org/10.1016/j.immuni.2008.08.004.
Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, van Endert P, Zitvogel L, Kroemer G. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14(10):1848–50. https://doi.org/10.1038/sj.cdd.4402201.
Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ, Inghirami G, Coleman CN, Formenti SC, Demaria S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat Commun. 2017;8:15618. https://doi.org/10.1038/ncomms15618.
Lan Y, Moustafa M, Knoll M, Xu C, Furkel J, Lazorchak A, Yeung TL, Hasheminasab SM, Jenkins MH, Meister S, Yu H, Schlegel J, Marelli B, Tang Z, Qin G, Klein C, Qi J, Zhou C, Locke G, Krunic D, Derner MG, Schwager C, Fontana RE, Kriegsmann K, Jiang F, Rein K, Kriegsmann M, Debus J, Lo KM, Abdollahi A. Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell. 2021;39(10):1388–1403.e10. https://doi.org/10.1016/j.ccell.2021.08.008.
Emami B, Lyman J, Brown A, Coia L, Goitein M, Munzenrider JE, Shank B, Solin LJ, Wesson M. Tolerance of normal tissue to therapeutic irradiation. Int J Radiat Oncol Biol Phys. 1991;21(1):109–22. https://doi.org/10.1016/0360-3016(91)90171-Y.
Milano MT, Constine LS, Okunieff P. Normal tissue tolerance dose metrics for radiation therapy of major organs. Semin Radiat Oncol. 2007;17(2):131–40. https://doi.org/10.1016/j.semradonc.2006.11.009.
Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nat Rev Cancer. 2006;6(9):702–13. https://doi.org/10.1038/nrc1950.
ICRP. ICRP statement on tissue reactions/early and late effects of radiation in normal tissues and organs—threshold doses for tissue reactions in a radiation protection context. ICRP Publication 118. Ann ICRP. 2012;41(1/2).
ICRP. Statement on tissue reactions, ref 4825-3093-1464. 2011.
Bentzen SM, Dörr W, Anscher MS, Denham JW, Hauer-Jensen M, Marks LB, Williams J. Normal tissue effects: reporting and analysis. Semin Radiat Oncol. 2003;13(3):189–202. https://doi.org/10.1016/S1053-4296(03)00036-5.
Dörr W, Hendry JH. Consequential late effects in normal tissues. Radiother Oncol. 2001;61(3):223–31. https://doi.org/10.1016/S0167-8140(01)00429-7.
Cytlak UM, Dyer DP, Honeychurch J, Williams KJ, Travis MA, Illidge TM. Immunomodulation by radiotherapy in tumour control and normal tissue toxicity. Nat Rev Immunol. 2022;22(2):124–38. https://doi.org/10.1038/s41577-021-00568-1. Epub 2021 Jul 1.
François A, Milliat F, Guipaud O, Benderitter M. Inflammation and immunity in radiation damage to the gut mucosa. Biomed Res Int. 2013;2013:123241. https://doi.org/10.1155/2013/123241.
Meziani L, Deutsch E, Mondini M. Macrophages in radiation injury: a new therapeutic target. Oncoimmunology. 2018;7(10):e1494488. https://doi.org/10.1080/2162402X.2018.1494488.
Najafi M, Motevaseli E, Shirazi A, Geraily G, Rezaeyan A, Norouzi F, Rezapoor S, Abdollahi H. Mechanisms of inflammatory responses to radiation and normal tissues toxicity: clinical implications. Int J Radiat Biol. 2018;94(4):335–56. https://doi.org/10.1080/09553002.2018.1440092.
Hauer-Jensen M, Denham JW, Andreyev HJ. Radiation enteropathy—pathogenesis, treatment and prevention. Nat Rev Gastroenterol Hepatol. 2014;11(8):470–9. https://doi.org/10.1038/nrgastro.2014.46.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Willaert R, Nevens D, Laenen A, Batstone M, Politis C, Nuyts S. Does intensity-modulated radiation therapy lower the risk of osteoradionecrosis of the jaw? A long-term comparative analysis. Int J Oral Maxillofac Surg. 2019;48(11):1387–93. https://doi.org/10.1016/j.ijom.2019.04.018.
Raguse JD, Hossamo J, Tinhofer I, Hoffmeister B, Budach V, Jamil B, Jöhrens K, Thieme N, Doll C, Nahles S, Hartwig ST, Stromberger C. Patient and treatment-related risk factors for osteoradionecrosis of the jaw in patients with head and neck cancer. Oral Surg Oral Med Oral Pathol Oral Radiol. 2016;121(3):215–21.e1. https://doi.org/10.1016/j.oooo.2015.10.006.
Van Camp N, Verhelst PJ, Nicot R, Ferri J, Politis C. Impaired callus formation in pathological mandibular fractures in medication-related osteonecrosis of the jaw and osteoradionecrosis. J Oral Maxillofac Surg. 2021;79(9):1892–901. https://doi.org/10.1016/j.joms.2021.04.024.
Notani K, Yamazaki Y, Kitada H, Sakakibara N, Fukuda H, Omori K, Nakamura M. Management of mandibular osteoradionecrosis corresponding to the severity of osteoradionecrosis and the method of radiotherapy. Head Neck. 2003;25(3):181–6. https://doi.org/10.1002/hed.10171.
Lyons A, Osher J, Warner E, Kumar R, Brennan PA. Osteoradionecrosis—a review of current concepts in defining the extent of the disease and a new classification proposal. Br J Oral Maxillofac Surg. 2014;52(5):392–5. https://doi.org/10.1016/j.bjoms.2014.02.017.
Spijkervet FKL, Brennan MT, Peterson DE, Witjes MJH, Vissink A. Research frontiers in oral toxicities of cancer therapies: osteoradionecrosis of the jaws. J Natl Cancer Inst Monogr. 2019;53:lgz006. https://doi.org/10.1093/jncimonographs/lgz006.
Grisar K, Schol M, Schoenaers J, Dormaar T, Coropciuc R, Vander Poorten V, Politis C. Osteoradionecrosis and medication-related osteonecrosis of the jaw: similarities and differences. Int J Oral Maxillofac Surg. 2016;45(12):1592–9. https://doi.org/10.1016/j.ijom.2016.06.016.
van Baar GJC, Leeuwrik L, Lodders JN, Liberton NPTJ, Karagozoglu KH, Forouzanfar T, Leusink FKJ. A novel treatment concept for advanced stage mandibular osteoradionecrosis combining isodose curve visualization and nerve preservation: a prospective pilot study. Front Oncol. 2021;11:630123. https://doi.org/10.3389/fonc.2021.630123.
Schuurhuis JM, Stokman MA, Witjes MJ, Dijkstra PU, Vissink A, Spijkervet FK. Evidence supporting pre-radiation elimination of oral foci of infection in head and neck cancer patients to prevent oral sequelae. A systematic review. Oral Oncol. 2015;51(3):212–20. https://doi.org/10.1016/j.oraloncology.2014.11.017.
Walker AJ, Ruzevick J, Malayeri AA, Rigamonti D, Lim M, Redmond KJ, Kleinberg L. Postradiation imaging changes in the CNS: how can we differentiate between treatment effect and disease progression? Future Oncol. 2014;10(7):1277–97. https://doi.org/10.2217/fon.13.271.
Ruben JD, Dally M, Bailey M, Smith R, McLean CA, Fedele P. Cerebral radiation necrosis: incidence, outcomes, and risk factors with emphasis on radiation parameters and chemotherapy. Int J Radiat Oncol Biol Phys. 2006;65(2):499–508. https://doi.org/10.1016/j.ijrobp.2005.12.002.
Kohutek ZA, Yamada Y, Chan TA, Brennan CW, Tabar V, Gutin PH, Yang TJ, Rosenblum MK, Ballangrud Å, Young RJ, Zhang Z, Beal K. Long-term risk of radionecrosis and imaging changes after stereotactic radiosurgery for brain metastases. J Neurooncol. 2015;125(1):149–56. https://doi.org/10.1007/s11060-015-1881-3.
Shah R, Vattoth S, Jacob R, Manzil FF, O’Malley JP, Borghei P, Patel BN, Curé JK. Radiation necrosis in the brain: imaging features and differentiation from tumor recurrence. Radiographics. 2012;32(5):1343–59. https://doi.org/10.1148/rg.325125002.
Le Rhun E, Dhermain F, Vogin G, Reyns N, Metellus P. Radionecrosis after stereotactic radiotherapy for brain metastases. Expert Rev Neurother. 2016;16(8):903–14. https://doi.org/10.1080/14737175.2016.1184572.
De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5(1):13. https://doi.org/10.1038/s41572-019-0064-5.
Guipaud O, Jaillet C, Clément-Colmou K, François A, Supiot S, Milliat F. The importance of the vascular endothelial barrier in the immune-inflammatory response induced by radiotherapy. Br J Radiol. 2018;91(1089):20170762. https://doi.org/10.1259/bjr.20170762.
Mintet E, Rannou E, Buard V, West G, Guipaud O, Tarlet G, Sabourin JC, Benderitter M, Fiocchi C, Milliat F, François A. Identification of endothelial-to-mesenchymal transition as a potential participant in radiation proctitis. Am J Pathol. 2015;185(9):2550–62. https://doi.org/10.1016/j.ajpath.2015.04.028.
Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. https://doi.org/10.1002/path.2277.
Muñoz-Espín D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15(7):482–96. https://doi.org/10.1038/nrm3823.
Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, Meng A. Inhibition of Bcl-2/xl with ABT-263 selectively kills senescent type II pneumocytes and reverses persistent pulmonary fibrosis induced by ionizing radiation in mice. Int J Radiat Oncol Biol Phys. 2017;99(2):353–61. https://doi.org/10.1016/j.ijrobp.2017.02.216.
Soysouvanh F, Benadjaoud MA, Dos Santos M, Mondini M, Lavigne J, Bertho A, Buard V, Tarlet G, Adnot S, Deutsch E, Guipaud O, Paget V, François A, Milliat F. Stereotactic lung irradiation in mice promotes long-term senescence and lung injury. Int J Radiat Oncol Biol Phys. 2020;106(5):1017–27. https://doi.org/10.1016/j.ijrobp.2019.12.039.
Prasanna PG, Citrin DE, Hildesheim J, Ahmed MM, Venkatachalam S, Riscuta G, Xi D, Zheng G, Deursen JV, Goronzy J, Kron SJ, Anscher MS, Sharpless NE, Campisi J, Brown SL, Niedernhofer LJ, O’Loghlen A, Georgakilas AG, Paris F, Gius D, Gewirtz DA, Schmitt CA, Abazeed ME, Kirkland JL, Richmond A, Romesser PB, Lowe SW, Gil J, Mendonca MS, Burma S, Zhou D, Coleman CN. Therapy-induced senescence: opportunities to improve anticancer therapy. J Natl Cancer Inst. 2021;113(10):1285–98. https://doi.org/10.1093/jnci/djab064.
Bentzen SM, Constine LS, Deasy JO, Eisbruch A, Jackson A, Marks LB, Ten Haken RK, Yorke ED. Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC): an introduction to the scientific issues. Int J Radiat Oncol Biol Phys. 2010;76(3 Suppl):S3–9. https://doi.org/10.1016/j.ijrobp.2009.09.040.
Constine LS, Ronckers CM, Hua CH, Olch A, Kremer LCM, Jackson A, Bentzen SM. Pediatric normal tissue effects in the clinic (PENTEC): an international collaboration to analyse normal tissue radiation dose-volume response relationships for paediatric cancer patients. Clin Oncol (R Coll Radiol). 2019;31(3):199–207. https://doi.org/10.1016/j.clon.2019.01.002.
Grimm J, Marks LB, Jackson A, Kavanagh BD, Xue J, Yorke E. High dose per fraction, hypofractionated treatment effects in the clinic (HyTEC): an overview. Int J Radiat Oncol Biol Phys. 2021;110(1):1–10. https://doi.org/10.1016/j.ijrobp.2020.10.039.
Palma G, Monti S, Buonanno A, Pacelli R, Cella L. PACE: a probabilistic atlas for normal tissue complication estimation in radiation oncology. Front Oncol. 2019;9:130. https://doi.org/10.3389/fonc.2019.00130.
Adamus-Górka M, Mavroidis P, Lind BK, Brahme A. Comparison of dose response models for predicting normal tissue complications from cancer radiotherapy: application in rat spinal cord. Cancers (Basel). 2011;3(2):2421–43. https://doi.org/10.3390/cancers3022421.
Knoblich JA. Asymmetric cell division during animal development. Nat Rev Mol Cell Biol. 2001;2(1):11–20. https://doi.org/10.1038/35048085.
Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453(7193):306–13. https://doi.org/10.1038/nature07038.
Pontikoglou C, Delorme B, Charbord P. Human bone marrow native mesenchymal stem cells. Regen Med. 2008;3(5):731–41. https://doi.org/10.2217/17460751.3.5.731.
Harfouche G, Martin MT. Response of normal stem cells to ionizing radiation: a balance between homeostasis and genomic stability. Mutat Res. 2010;704(1–3):167–74. https://doi.org/10.1016/j.mrrev.2010.01.007.
Galli R, Gritti A, Bonfanti L, Vescovi AL. Neural stem cells: an overview. Circ Res. 2003;92(6):598–608. https://doi.org/10.1161/01.RES.0000065580.02404.F4.
Chaker Z, Codega P, Doetsch F. A mosaic world: puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip Rev Dev Biol. 2016;5(6):640–58. https://doi.org/10.1002/wdev.248.
Huo K, Sun Y, Li H, Du X, Wang X, Karlsson N, Zhu C, Blomgren K. Lithium reduced neural progenitor apoptosis in the hippocampus and ameliorated functional deficits after irradiation to the immature mouse brain. Mol Cell Neurosci. 2012;51(1–2):32–42. https://doi.org/10.1016/j.mcn.2012.07.002.
Prager I, Patties I, Himmelbach K, Kendzia E, Merz F, Müller K, Kortmann RD, Glasow A. Dose-dependent short- and long-term effects of ionizing irradiation on neural stem cells in murine hippocampal tissue cultures: neuroprotective potential of resveratrol. Brain Behav. 2016;6(10):e00548. https://doi.org/10.1002/brb3.548.
Fukui M, Choi HJ, Zhu BT. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic Biol Med. 2010;49(5):800–13. https://doi.org/10.1016/j.freeradbiomed.2010.06.002.
Brouard M, Barrandon Y. Controlling skin morphogenesis: hope and despair. Curr Opin Biotechnol. 2003;14(5):520–5. https://doi.org/10.1016/j.copbio.2003.09.005.
Potten CS, Grant HK. The relationship between ionizing radiation-induced apoptosis and stem cells in the small and large intestine. Br J Cancer. 1998;78:993–1003. https://doi.org/10.1038/bjc.1998.618.
Konrad CV, Murali R, Varghese BA, Nair R. The role of cancer stem cells in tumor heterogeneity and resistance to therapy. Can J Physiol Pharmacol. 2017;95(1):1–15. https://doi.org/10.1139/cjpp-2016-0079.
Arnold CR, Mangesius J, Skvortsova II, Ganswindt U. The role of cancer stem cells in radiation resistance. Front Oncol. 2020;10:164. https://doi.org/10.3389/fonc.2020.00164.
Laplane L. Cancer stem cells modulate patterns and processes of evolution in cancers. Biol Philos. 2018;33:18. https://doi.org/10.1007/s10539-018-9629-z.
Filip S, Mokry J, Horacek J, English D. Stem cells and the phenomena of plasticity and diversity: a limiting property of carcinogenesis. Stem Cells Dev. 2008;17(6):1031–8. https://doi.org/10.1089/scd.2007.0234.
Filipova A, Seifrtova M, Mokry J, Dvorak J, Rezacova M, Filip S, Diaz-Garcia D. Breast cancer and cancer stem cells: a mini-review. Tumori. 2014;100(4):363–9. https://doi.org/10.1700/1636.17886.
Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer stem cells. Int J Biochem Cell Biol. 2012;44(12):2144–51. https://doi.org/10.1016/j.biocel.2012.08.022.
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–60. https://doi.org/10.1038/nature05236.
Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, Camerini T, Mariani L, Delia D, Calabrò E, Pastorino U, Sozzi G. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106(38):16281–6. https://doi.org/10.1073/pnas.0905653106.
Coppes RP, Dubrovska A. Targeting stem cells in radiation oncology. Clin Oncol (R Coll Radiol). 2017;29(6):329–34. https://doi.org/10.1016/j.clon.2017.03.005.
Lee PJ, Ho CC, Ho H, Chen WJ, Lin CH, Lai YH, Juan YC, Chu WC, Lee JH, Su SF, Chen HY, Chen JJW, Chang GC, Li KC, Yang PC, Chen HW. Tumor microenvironment-based screening repurposes drugs targeting cancer stem cells and cancer-associated fibroblasts. Theranostics. 2021;11(19):9667–86. https://doi.org/10.7150/thno.62676.
Lee J, Steinmann A, Ding Y, Lee H, Owens C, Wang J, Yang J, Followill D, Ger R, MacKin D, Court LE. Radiomics feature robustness as measured using an MRI phantom. Sci Rep. 2021;11(1):3973. https://doi.org/10.1038/s41598-021-83593-3.
Lundholm L, Hååg P, Zong D, Juntti T, Mörk B, Lewensohn R, Viktorsson K. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 2013;4(1):e478. https://doi.org/10.1038/cddis.2012.211.
Moro M, Bertolini G, Pastorino U, Roz L, Sozzi G. Combination treatment with all-trans retinoic acid prevents cisplatin-induced enrichment of CD133+ tumor-initiating cells and reveals heterogeneity of cancer stem cell compartment in lung cancer. J Thorac Oncol. 2015;10(7):1027–36. https://doi.org/10.1097/JTO.0000000000000563.
Smit JK, Faber H, Niemantsverdriet M, Baanstra M, Bussink J, Hollema H, van Os RP, Plukker JT, Coppes RP. Prediction of response to radiotherapy in the treatment of esophageal cancer using stem cell markers. Radiother Oncol. 2013;107(3):434–41. https://doi.org/10.1016/j.radonc.2013.03.027.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F, Cui H. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8. https://doi.org/10.1038/s41392-020-0110-5.
Digomann D, Kurth I, Tyutyunnykova A, Chen O, Löck S, Gorodetska I, Peitzsch C, Skvortsova II, Negro G, Aschenbrenner B, Eisenhofer G, Richter S, Heiden S, Porrmann J, Klink B, Schwager C, Dowle AA, Hein L, Kunz-Schughart LA, Abdollahi A, Lohaus F, Krause M, Baumann M, Linge A, Dubrovska A. The CD98 heavy chain is a marker and regulator of head and neck squamous cell carcinoma radiosensitivity. Clin Cancer Res. 2019;25(10):3152–63. https://doi.org/10.1158/1078-0432.CCR-18-2951.
Martens-de Kemp SR, Brink A, Stigter-van Walsum M, Damen JM, Rustenburg F, Wu T, van Wieringen WN, Schuurhuis GJ, Braakhuis BJ, Slijper M, Brakenhoff RH. CD98 marks a subpopulation of head and neck squamous cell carcinoma cells with stem cell properties. Stem Cell Res. 2013;10(3):477–88. https://doi.org/10.1016/j.scr.2013.02.004.
Sarvi S, Mackinnon AC, Avlonitis N, Bradley M, Rintoul RC, Rassl DM, Wang W, Forbes SJ, Gregory CD, Sethi T. CD133+ cancer stem-like cells in small cell lung cancer are highly tumorigenic and chemoresistant but sensitive to a novel neuropeptide antagonist. Cancer Res. 2014;74(5):1554–65. https://doi.org/10.1158/0008-5472.CAN-13-1541.
Shien K, Toyooka S, Ichimura K, Soh J, Furukawa M, Maki Y, Muraoka T, Tanaka N, Ueno T, Asano H, Tsukuda K, Yamane M, Oto T, Kiura K, Miyoshi S. Prognostic impact of cancer stem cell-related markers in non-small cell lung cancer patients treated with induction chemoradiotherapy. Lung Cancer. 2012;77(1):162–7. https://doi.org/10.1016/j.lungcan.2012.02.006.
Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, Blaudszun AR, Yevsa T, Fricke S, Kossatz-Boehlert U. Cancer stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 2020;11:1280. https://doi.org/10.3389/fimmu.2020.01280.
Liu Y, Yang M, Luo J, Zhou H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. Int J Oral Sci. 2020;12(1):19. https://doi.org/10.1038/s41368-020-00087-0.
Li F, Zhou K, Gao L, Zhang B, Li W, Yan W, Song X, Yu H, Wang S, Yu N, Jiang Q. Radiation induces the generation of cancer stem cells: a novel mechanism for cancer radioresistance. Oncol Lett. 2016;12(5):3059–65. https://doi.org/10.3892/ol.2016.5124.
Krause M, Dubrovska A, Linge A, Baumann M. Cancer stem cells: radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv Drug Deliv Rev. 2017;109:63–73. https://doi.org/10.1016/j.addr.2016.02.002.
Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P. The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol. 2017;15(10):630–8. https://doi.org/10.1038/nrmicro.2017.58.
González-Mercado VJ, Pérez-Santiago J, Lyon D, Dilán-Pantojas I, Henderson W, McMillan S, Groer M, Kane B, Marrero S, Pedro E, Saligan LN. The role of gut microbiome perturbation in fatigue induced by repeated stress from chemoradiotherapy: a proof of concept study. Adv Med. 2020;2020:6375876. https://doi.org/10.1155/2020/6375876.
Jang BS, Chang JH, Chie EK, Kim K, Park JW, Kim MJ, Song EJ, Nam YD, Kang SW, Jeong SY, Kim HJ. Gut microbiome composition is associated with a pathologic response after preoperative chemoradiation in patients with rectal cancer. Int J Radiat Oncol Biol Phys. 2020;107(4):736–46. https://doi.org/10.1016/j.ijrobp.2020.04.015.
Mitra A, Grossman Biegert GW, Delgado AY, Karpinets TV, Solley TN, Mezzari MP, Yoshida-Court K, Petrosino JF, Mikkelson MD, Lin L, Eifel P, Zhang J, Ramondetta LM, Jhingran A, Sims TT, Schmeler K, Okhuysen P, Colbert LE, Klopp AH. Microbial diversity and composition is associated with patient-reported toxicity during chemoradiation therapy for cervical cancer. Int J Radiat Oncol Biol Phys. 2020;107(1):163–71. https://doi.org/10.1016/j.ijrobp.2019.12.040.
Nam YD, Kim HJ, Seo JG, Kang SW, Bae JW. Impact of pelvic radiotherapy on gut microbiota of gynecological cancer patients revealed by massive pyrosequencing. PLoS One. 2013;8(12):e82659. https://doi.org/10.1371/journal.pone.0082659.
Manichanh C, Varela E, Martinez C, Antolin M, Llopis M, Doré J, Giralt J, Guarner F, Malagelada JR. The gut microbiota predispose to the pathophysiology of acute postradiotherapy diarrhea. Am J Gastroenterol. 2008;103(7):1754–61. https://doi.org/10.1111/j.1572-0241.2008.01868.x.
Reis Ferreira M, Andreyev HJN, Mohammed K, Truelove L, Gowan SM, Li J, Gulliford SL, Marchesi JR, Dearnaley DP. Microbiota- and radiotherapy-induced gastrointestinal side-effects (MARS) study: a large pilot study of the microbiome in acute and late-radiation enteropathy. Clin Cancer Res. 2019;25(21):6487–500. https://doi.org/10.1158/1078-0432.CCR-19-0960.
Wang A, Ling Z, Yang Z, Kiela PR, Wang T, Wang C, Cao L, Geng F, Shen M, Ran X, Su Y, Cheng T, Wang J. Gut microbial dysbiosis may predict diarrhea and fatigue in patients undergoing pelvic cancer radiotherapy: a pilot study. PLoS One. 2015;10(5):e0126312. https://doi.org/10.1371/journal.pone.0126312.
Wang Z, Wang Q, Wang X, Zhu L, Chen J, Zhang B, Chen Y, Yuan Z. Gut microbial dysbiosis is associated with development and progression of radiation enteritis during pelvic radiotherapy. J Cell Mol Med. 2019;23(5):3747–56. https://doi.org/10.1111/jcmm.14289.
Oh B, Eade T, Lamoury G, Carroll S, Morgia M, Kneebone A, Hruby G, Stevens M, Boyle F, Clarke S, Corless B, Molloy M, Rosenthal D, Back M. The gut microbiome and gastrointestinal toxicities in pelvic radiation therapy: a clinical review. Cancers (Basel). 2021;13(10):2353. https://doi.org/10.3390/cancers13102353.
Ferreira MR, Muls A, Dearnaley DP, Andreyev HJ. Microbiota and radiation-induced bowel toxicity: lessons from inflammatory bowel disease for the radiation oncologist. Lancet Oncol. 2014;15(3):e139–47. https://doi.org/10.1016/S1470-2045(13)70504-7.
Gerassy-Vainberg S, Blatt A, Danin-Poleg Y, Gershovich K, Sabo E, Nevelsky A, Daniel S, Dahan A, Ziv O, Dheer R, Abreu MT, Koren O, Kashi Y, Chowers Y. Radiation induces proinflammatory dysbiosis: transmission of inflammatory susceptibility by host cytokine induction. Gut. 2018;67(1):97–107. https://doi.org/10.1136/gutjnl-2017-313789.
Guo H, Chou WC, Lai Y, Liang K, Tam JW, Brickey WJ, Chen L, Montgomery ND, Li X, Bohannon LM, Sung AD, Chao NJ, Peled JU, Gomes ALC, van den Brink MRM, French MJ, Macintyre AN, Sempowski GD, Tan X, Sartor RB, Lu K, Ting JPY. Multi-omics analyses of radiation survivors identify radioprotective microbes and metabolites. Science. 2020;370(6516):eaay9097. https://doi.org/10.1126/science.aay9097.
Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97–111. https://doi.org/10.1038/nri.2016.107.
Shiao SL, Kershaw KM, Limon JJ, You S, Yoon J, Ko EY, Guarnerio J, Potdar AA, McGovern DPB, Bose S, Dar TB, Noe P, Lee J, Kubota Y, Maymi VI, Davis MJ, Henson RM, Choi RY, Yang W, Tang J, Gargus M, Prince AD, Zumsteg ZS, Underhill DM. Commensal bacteria and fungi differentially regulate tumor responses to radiation therapy. Cancer Cell. 2021;39(9):1202–1213.e6. https://doi.org/10.1016/j.ccell.2021.07.002.
Rajkomar A, Dean J, Kohane I. Machine learning in medicine. N Engl J Med. 2019;380(14):1347–58. https://doi.org/10.1056/NEJMra1814259.
Kazmierska J, Hope A, Spezi E, Beddar S, Nailon WH, Osong B, Ankolekar A, Choudhury A, Dekker A, Redalen KR, Traverso A. From multisource data to clinical decision aids in radiation oncology: the need for a clinical data science community. Radiothe Oncol. 2020;153:43–54. https://doi.org/10.1016/J.RADONC.2020.09.054.
Sun W, Cai Z, Li Y, Liu F, Fang S, Wang G. Data processing and text mining technologies on electronic medical records: a review. J Healthcare Eng. 2018;2018 https://doi.org/10.1155/2018/4302425.
Fröhlich H, Balling R, Beerenwinkel N, Kohlbacher O, Kumar S, Lengauer T, Maathuis MH, Moreau Y, Murphy SA, Przytycka TM, Rebhan M, Röst H, Schuppert A, Schwab M, Spang R, Stekhoven D, Sun J, Weber A, Ziemek D, Zupan B. From hype to reality: data science enabling personalized medicine. BMC Med. 2018;16(1):150. https://doi.org/10.1186/s12916-018-1122-7.
Willems SM, Abeln S, Feenstra KA, de Bree R, van der Poel EF, Baatenburg de Jong RJ, Heringa J, van den Brekel MWM. The potential use of big data in oncology. Oral Oncol. 2019;98:8–12. https://doi.org/10.1016/j.oraloncology.2019.09.003.
Baek B, Lee H. Prediction of survival and recurrence in patients with pancreatic cancer by integrating multi-omics data. Sci Rep. 2020;10(1):18951. https://doi.org/10.1038/s41598-020-76025-1.
Kelleher JD, Namee BM, D’Arcy A. Machine learning for predictive data analytics. MIT Press; 2015.
El Naqa I, Bradley J, Blanco AI, Lindsay PE, Vicic M, Hope A, Deasy JO. Multivariable modeling of radiotherapy outcomes, including dose-volume and clinical factors. Int J Radiat Oncol Biol Phys. 2006;64(4):1275–86. https://doi.org/10.1016/j.ijrobp.2005.11.022.
Burman C, Kutcher GJ, Emami B, Goitein M. Fitting of normal tissue tolerance data to an analytic function. Int J Radiat Oncol Biol Phys. 1991;21(1):123–35. https://doi.org/10.1016/0360-3016(91)90172-Z.
Gulliford SL, Webb S, Rowbottom CG, Corne DW, Dearnaley DP. Use of artificial neural networks to predict biological outcomes for patients receiving radical radiotherapy of the prostate. Radiother Oncol. 2004;71(1):3–12. https://doi.org/10.1016/J.RADONC.2003.03.001.
Kleppe A, Skrede OJ, De Raedt S, Liestøl K, Kerr DJ, Danielsen HE. Designing deep learning studies in cancer diagnostics. Nat Rev Cancer. 2021;21(3):199–211. https://doi.org/10.1038/s41568-020-00327-9.
Sahiner B, Pezeshk A, Hadjiiski LM, Wang X, Drukker K, Cha KH, Summers RM, Giger ML. Deep learning in medical imaging and radiation therapy. Med Phys. 2019;46(1):e1–e36. https://doi.org/10.1002/MP.13264.
Kelly CJ, Karthikesalingam A, Suleyman M, Corrado G, King D. Key challenges for delivering clinical impact with artificial intelligence. BMC Med. 2019;17(1):195. https://doi.org/10.1186/s12916-019-1426-2.
Ibtehaz N, Rahman MS. MultiResUNet: rethinking the U-Net architecture for multimodal biomedical image segmentation. Neural Netw. 2020;121:74–87. https://doi.org/10.1016/j.neunet.2019.08.025.
Ronneberger O, Fischer P, Brox T. U-Net: convolutional networks for biomedical image segmentation. In: Lecture notes in computer science (including subseries Lecture notes in artificial intelligence and Lecture notes in bioinformatics), vol. 9351; 2015. p. 234–41. https://doi.org/10.1007/978-3-319-24574-4_28.
Gillies RJ, Kinahan PE, Hricak H. Radiomics: images are more than pictures, they are data. Radiology. 2016;278(2):563–77. https://doi.org/10.1148/radiol.2015151169.
van Timmeren JE, Cester D, Tanadini-Lang S, Alkadhi H, Baessler B. Radiomics in medical imaging “how-to” guide and critical reflection. Insights Imaging. 2020;11(1):91. https://doi.org/10.1186/s13244-020-00887-2.
Lu L, Liang Y, Schwartz LH, Zhao B. Reliability of radiomic features across multiple abdominal CT image acquisition settings: a pilot study using ACR CT phantom. Tomography. 2019;5(1):226–31. https://doi.org/10.18383/j.tom.2019.00005.
Park BW, Kim JK, Heo C, Park KJ. Reliability of CT radiomic features reflecting tumour heterogeneity according to image quality and image processing parameters. Sci Rep. 2020;10(1):3852. https://doi.org/10.1038/s41598-020-60868-9.
Fiorino C, Reni M, Bolognesi A, Cattaneo GM, Calandrino R. Intra- and inter-observer variability in contouring prostate and seminal vesicles: implications for conformal treatment planning. Radiother Oncol. 1998;47(3):285–92. https://doi.org/10.1016/S0167-8140(98)00021-8.
Lohmann P, Bousabarah K, Hoevels M, Treuer H. Radiomics in radiation oncology—basics, methods, and limitations. Strahlentherap Onkol. 2020;196(10):848. https://doi.org/10.1007/S00066-020-01663-3.
Emblem KE, Nedregaard B, Nome T, Due-Tonnessen P, Hald JK, Scheie D, Borota OC, Cvancarova M, Bjornerud A. Glioma grading by using histogram analysis of blood volume heterogeneity from MR-derived cerebral blood volume maps. Radiology. 2008;247(3):808–17. https://doi.org/10.1148/radiol.2473070571.
Abdollahi H, Mahdavi SR, Shiri I, Mofid B, Bakhshandeh M, Rahmani K. Magnetic resonance imaging radiomic feature analysis of radiation-induced femoral head changes in prostate cancer radiotherapy. J Cancer Res Ther. 2019;15(8):11. https://doi.org/10.4103/JCRT.JCRT_172_18.
Desideri I, Loi M, Francolini G, Becherini C, Livi L, Bonomo P. Application of radiomics for the prediction of radiation-induced toxicity in the IMRT era: current state-of-the-art. Front Oncol. 2020;10 https://doi.org/10.3389/FONC.2020.01708/FULL.
Qin H, Wu Y-Q, Lin P, Gao R-Z, Li X, Wang X-R, Chen G, He Y, Yang H. Ultrasound image-based radiomics. J Ultrasound Med. 2021;40(6):1229–44. https://doi.org/10.1002/JUM.15506.
Stefano A, Comelli A, Bravatà V, Barone S, Daskalovski I, Savoca G, Sabini MG, Ippolito M, Russo G. A preliminary PET radiomics study of brain metastases using a fully automatic segmentation method. BMC Bioinformatics. 2020;21(8):1–14. https://doi.org/10.1186/S12859-020-03647-7/TABLES/3.
Fehr D, Veeraraghavan H, Wibmer A, Gondo T, Matsumoto K, Vargas HA, Sala E, Hricak H, Deasy JO. Automatic classification of prostate cancer Gleason scores from multiparametric magnetic resonance images. Proc Natl Acad Sci U S A. 2015;112(46):E6265–73. https://doi.org/10.1073/pnas.1505935112.
Monti S, Aiello M, Incoronato M, Grimaldi AM, Moscarino M, Mirabelli P, Ferbo U, Cavaliere C, Salvatore M. DCE-MRI pharmacokinetic-based phenotyping of invasive ductal carcinoma: a radiomic study for prediction of histological outcomes. Contrast Media Mol Imaging. 2018;2018 https://doi.org/10.1155/2018/5076269.
Parmar C, Leijenaar RT, Grossmann P, Rios Velazquez E, Bussink J, Rietveld D, Rietbergen MM, Haibe-Kains B, Lambin P, Aerts HJ. Radiomic feature clusters and prognostic signatures specific for lung and head & neck cancer. Sci Rep. 2015;5:11044. https://doi.org/10.1038/srep11044.
Khawaja A, Bartholmai BJ, Rajagopalan S, Karwoski RA, Varghese C, Maldonado F, Peikert T. Do we need to see to believe? Radiomics for lung nodule classification and lung cancer risk stratification. J Thorac Dis. 2020;12(6):3303–16. https://doi.org/10.21037/JTD.2020.03.105.
Shiradkar R, Ghose S, Jambor I, Taimen P, Ettala O, Purysko AS, Madabhushi A. Radiomic features from pretreatment biparametric MRI predict prostate cancer biochemical recurrence: Preliminary findings. J Magn Reson Imaging. 2018;48(6):1626–36. https://doi.org/10.1002/jmri.26178.
Fujita A, Buch K, Li B, Kawashima Y, Qureshi MM, Sakai O. Difference between HPV-positive and HPV-negative non-oropharyngeal head and neck cancer: texture analysis features on CT. J Comput Assist Tomogr. 2016;40(1):43–7. https://doi.org/10.1097/RCT.0000000000000320.
Fave X, Zhang L, Yang J, Mackin D, Balter P, Gomez D, Followill D, Jones AK, Stingo F, Liao Z, Mohan R, Court L. Delta-radiomics features for the prediction of patient outcomes in non-small cell lung cancer. Sci Rep. 2017;7(1):588. https://doi.org/10.1038/s41598-017-00665-z.
Story MD, Durante M. Radiogenomics. Med Phys. 2018;45(11):e1111–22. https://doi.org/10.1002/MP.13064.
Incoronato M, Aiello M, Infante T, Cavaliere C, Grimaldi AM, Mirabelli P, Monti S, Salvatore M. Radiogenomic analysis of oncological data: a technical survey. Int J Mol Sci. 2017;2017(18):805. https://doi.org/10.3390/IJMS18040805.
Mazurowski MA. Radiogenomics: what it is and why it is important. J Am Coll Radiol. 2015;12(8):862–6. https://doi.org/10.1016/J.JACR.2015.04.019.
Chang XP, Grinband XJ, Weinberg XBD, Bardis XM, Khy XM, Cadena XG, Su M-Y, Cha XS, Filippi XCG, Bota XD, Baldi XP, Poisson XLM, Jain XR, Chow XD. Deep-learning convolutional neural networks accurately classify genetic mutations in gliomas. Am J Neuroradiol. 2018;39(7):1201–7. https://doi.org/10.3174/ajnr.A5667.
Bibault JE, Giraud P, Durdux C, Taieb J, Berger A, Coriat R, Chaussade S, Dousset B, Nordlinger B, Burgun A. Deep learning and radiomics predict complete response after neo-adjuvant chemoradiation for locally advanced rectal cancer. Sci Rep. 2018;8(1):1–8. https://doi.org/10.1038/s41598-018-30657-6.
Chaudhary K, Poirion OB, Lu L, Garmire LX. Deep learning–based multi-omics integration robustly predicts survival in liver cancer. Clin Cancer Res. 2018;24(6):1248–59. https://doi.org/10.1158/1078-0432.CCR-17-0853.
West CM, Barnett GC. Genetics and genomics of radiotherapy toxicity: towards prediction. Genome Med. 2011;3(8):1–15. https://doi.org/10.1186/GM268/TABLES/5.
Sibbald B, Roland M. Understanding controlled trials: why are randomised controlled trials important? BMJ. 1998;316(7126):201. https://doi.org/10.1136/BMJ.316.7126.201.
Van Poucke S, Thomeer M, Heath J, Vukicevic M. Are randomized controlled trials the (g)old standard? From clinical intelligence to prescriptive analytics. J Med Internet Res. 2016;18(7):E185. https://doi.org/10.2196/JMIR.5549. https://www.jmir.org/2016/7/E185
Mayo CS, Matuszak MM, Schipper MJ, Jolly S, Hayman JA, Ten Haken RK. Big data in designing clinical trials: opportunities and challenges. Front Oncol. 2017;7(AUG):187. https://doi.org/10.3389/FONC.2017.00187/BIBTEX.
Weissler EH, Naumann T, Andersson T, Ranganath R, Elemento O, Luo Y, Freitag DF, Benoit J, Hughes MC, Khan F, Slater P, Shameer K, Roe M, Hutchison E, Kollins SH, Broedl U, Meng Z, Wong JL, Curtis L, et al. The role of machine learning in clinical research: transforming the future of evidence generation. Trials. 2021;22(1):1–15. https://doi.org/10.1186/S13063-021-05489-X/FIGURES/3.
Lambin P, Rios-Velazquez E, Leijenaar R, Carvalho S, van Stiphout RG, Granton P, Zegers CM, Gillies R, Boellard R, Dekker A, Aerts HJ. Radiomics: extracting more information from medical images using advanced feature analysis. Eur J Cancer. 2012;48(4):441–6. https://doi.org/10.1016/j.ejca.2011.11.036.
Keek SA, Leijenaar RT, Jochems A, Woodruff HC. A review on radiomics and the future of theranostics for patient selection in precision medicine. Br J Radiol. 2018;91(1091):20170926. https://doi.org/10.1259/bjr.20170926.
Buch K, Fujita A, Li B, Kawashima Y, Qureshi MM, Sakai O. Using texture analysis to determine human papillomavirus status of oropharyngeal squamous cell carcinomas on CT. AJNR Am J Neuroradiol. 2015;36(7):1343–8. https://doi.org/10.3174/ajnr.A4285.
Gugliandolo SG, Pepa M, Isaksson LJ, Marvaso G, Raimondi S, Botta F, Gandini S, Ciardo D, Volpe S, Riva G, Rojas DP, Zerini D, Pricolo P, Alessi S, Petralia G, Summers PE, Mistretta FA, Luzzago S, Cattani F, De Cobelli O, Cassano E, Cremonesi M, Bellomi M, Orecchia R, Jereczek-Fossa BA. MRI-based radiomics signature for localized prostate cancer: a new clinical tool for cancer aggressiveness prediction? Sub-study of prospective phase II trial on ultra-hypofractionated radiotherapy (AIRC IG-13218). Eur Radiol. 2021;31(2):716–28. https://doi.org/10.1007/s00330-020-07105-z.
Shiradkar R, Podder TK, Algohary A, Viswanath S, Ellis RJ, Madabhushi A. Radiomics based targeted radiotherapy planning (Rad-TRaP): a computational framework for prostate cancer treatment planning with MRI. Radiat Oncol. 2016;11(1):148. https://doi.org/10.1186/s13014-016-0718-3.
Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, Bussink J, Monshouwer R, Haibe-Kains B, Rietveld D, Hoebers F, Rietbergen MM, Leemans CR, Dekker A, Quackenbush J, Gillies RJ, Lambin P. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun. 2014;5:4006. Erratum in: Nat Commun. 2014;5:4644. Cavalho, Sara [corrected to Carvalho, Sara]. https://doi.org/10.1038/ncomms5006.
Carvalho S, Leijenaar RTH, Troost EGC, van Timmeren JE, Oberije C, van Elmpt W, de Geus-Oei LF, Bussink J, Lambin P. 18F-fluorodeoxyglucose positron-emission tomography (FDG-PET)-Radiomics of metastatic lymph nodes and primary tumor in non-small cell lung cancer (NSCLC)—a prospective externally validated study. PLoS One. 2018;13(3):e0192859. https://doi.org/10.1371/journal.pone.0192859.
Sörensen A, Carles M, Bunea H, Majerus L, Stoykow C, Nicolay NH, Wiedenmann NE, Vaupel P, Meyer PT, Grosu AL, Mix M. Textural features of hypoxia PET predict survival in head and neck cancer during chemoradiotherapy. Eur J Nucl Med Mol Imaging. 2020;47(5):1056–64. https://doi.org/10.1007/s00259-019-04609-9.
Giannini V, Mazzetti S, Bertotto I, Chiarenza C, Cauda S, Delmastro E, Bracco C, Di Dia A, Leone F, Medico E, Pisacane A, Ribero D, Stasi M, Regge D. Predicting locally advanced rectal cancer response to neoadjuvant therapy with 18F-FDG PET and MRI radiomics features. Eur J Nucl Med Mol Imaging. 2019;46(4):878–88. https://doi.org/10.1007/s00259-018-4250-6.
Xiong J, Yu W, Ma J, Ren Y, Fu X, Zhao J. The role of PET-based radiomic features in predicting local control of esophageal cancer treated with concurrent chemoradiotherapy. Sci Rep. 2018;8(1):9902. https://doi.org/10.1038/s41598-018-28243-x.
Gnep K, Fargeas A, Gutiérrez-Carvajal RE, Commandeur F, Mathieu R, Ospina JD, Rolland Y, Rohou T, Vincendeau S, Hatt M, Acosta O, de Crevoisier R. Haralick textural features on T2 -weighted MRI are associated with biochemical recurrence following radiotherapy for peripheral zone prostate cancer. J Magn Reson Imaging. 2017;45(1):103–17. https://doi.org/10.1002/jmri.25335.
van Dijk LV, Langendijk JA, Zhai TT, Vedelaar TA, Noordzij W, Steenbakkers RJHM, Sijtsema NM. Delta-radiomics features during radiotherapy improve the prediction of late xerostomia. Sci Rep. 2019;9(1):12483. https://doi.org/10.1038/s41598-019-48184-3.
Cunliffe A, Armato SG 3rd, Castillo R, Pham N, Guerrero T, Al-Hallaq HA. Lung texture in serial thoracic computed tomography scans: correlation of radiomics-based features with radiation therapy dose and radiation pneumonitis development. Int J Radiat Oncol Biol Phys. 2015;91(5):1048–56. https://doi.org/10.1016/j.ijrobp.2014.11.030.
Hettal L, Stefani A, Salleron J, Courrech F, Behm-Ansmant I, Constans JM, Gauchotte G, Vogin G. Radiomics method for the differential diagnosis of radionecrosis versus progression after fractionated stereotactic body radiotherapy for brain oligometastasis. Radiat Res. 2020;193(5):471–80. https://doi.org/10.1667/RR15517.1.
Further Reading
Coppes RP, Dubrovska A. Targeting stem cells in radiation oncology. Clin Oncol (R Coll Radiol). 2017;29(6):329–34. https://doi.org/10.1016/j.clon.2017.03.00.
Donlon NE, Power R, Hayes C, Reynolds JV, Lysaght J. Radiotherapy, immunotherapy, and the tumour microenvironment: turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021;502:84–96. https://doi.org/10.1016/j.canlet.2020.12.045.
Horsman MR, Overgaard J. The impact of hypoxia and its modification of the outcome of radiotherapy. J Radiat Res. 2016;57(Suppl 1):i90–8. https://doi.org/10.1093/jrr/rrw007.
Huang Y, Fan J, Li Y, Fu S, Chen Y, Wu J. Imaging of tumor hypoxia with radionuclide-labeled tracers for PET. Front Oncol. 2021;11:731503. https://doi.org/10.3389/fonc.2021.731503.
Joiner MC, Van der Kogel AJ, editors. Basic clinical radiobiology. 5th ed. CRC Press; 2018.
McBride WH, Schaue D. Radiation-induced tissue damage and response. J Pathol. 2020;250(5):647–55. https://doi.org/10.1002/path.5389.
Reda M, Bagley AF, Zaidan HY, Yantasee W. Augmenting the therapeutic window of radiotherapy: a perspective on molecularly targeted therapies and nanomaterials. Radiother Oncol. 2020;150:225–35. https://doi.org/10.1016/j.radonc.2020.06.041.
Sebestyén A, Kopper L, Dankó T, Tímár J. Hypoxia signaling in cancer: from basics to clinical practice. Pathol Oncol Res. 2021;27:1609802. https://doi.org/10.3389/pore.2021.1609802.
Van de Guchte M, Mondot S, Doré J. Dynamic properties of the intestinal ecosystem call for combination therapies, targeting inflammation and microbiota, in ulcerative colitis. Gastroenterology. 2021;161(6):1969–1981.e12. https://doi.org/10.1053/j.gastro.2021.08.057.
Vogin G. Description and management of radiotherapy-induced long-term effects. In: Rauh S, editor. Survivorship care for cancer patients. Cham: Springer; 2021. https://doi.org/10.1007/978-3-030-78648-9_13.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2023 The Author(s)
About this chapter

Cite this chapter
Sminia, P. et al. (2023). Clinical Radiobiology for Radiation Oncology. In: Baatout, S. (eds) Radiobiology Textbook. Springer, Cham. https://doi.org/10.1007/978-3-031-18810-7_5
Download citation
DOI: https://doi.org/10.1007/978-3-031-18810-7_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-18809-1
Online ISBN: 978-3-031-18810-7
eBook Packages: MedicineMedicine (R0)