
Abstract
The relentless rise of atmospheric CO2 is causing large and unpredictable impacts on the Earth climate, due to the CO2 significant greenhouse effect, besides being responsible for the ocean acidification, with consequent huge impacts in our daily lives and in all forms of life. To stop spiral of destruction, we must actively reduce the CO2 emissions and develop new and more efficient “CO2 sinks”. We should be focused on the opportunities provided by exploiting this novel and huge carbon feedstock to produce de novo fuels and added-value compounds. The conversion of CO2 into formate offers key advantages for carbon recycling, and formate dehydrogenase (FDH) enzymes are at the centre of intense research, due to the “green” advantages the bioconversion can offer, namely substrate and product selectivity and specificity, in reactions run at ambient temperature and pressure and neutral pH. In this chapter, we describe the remarkable recent progress towards efficient and selective FDH-catalysed CO2 reduction to formate. We focus on the enzymes, discussing their structure and mechanism of action. Selected promising studies and successful proof of concepts of FDH-dependent CO2 reduction to formate and beyond are discussed, to highlight the power of FDHs and the challenges this CO2 bioconversion still faces.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others



Keywords
- Carbon dioxide utilisation
- Formic acid
- Formate dehydrogenase
- Molybdenum
- Tungsten
- Hydride transfer
- Biocatalyst
- Green chemistry
- Energy
- Biotechnology
1 The Relentless Rise of Carbon Dioxide
In 2018 alone, more than 36Gt of CO2 [1] were dumped into the atmosphere as waste material from fossil resources-based energy and chemical industries! In that year, the global atmospheric CO2 concentration reached an annual average value of 407 ppm, an increase of 150% since pre-industrial times (277 ppm in 1750) (Fig. 1) [1]. Yet, new records are being set, and a monthly average of 416 ppm was already observed this March 2020 [2]. This ever-increasing atmospheric CO2 concentration is causing large and unpredictable impacts on the Earth climate, due to the CO2 significant greenhouse effect, besides being responsible for the ocean acidification, with consequent huge impacts in our daily lives and in all forms of life.

Source NOAA Climate.gov, based on EPICA Dome C data provided by NOAA NCEI Paleoclimatology Program (https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide)
Relentless rise of carbon dioxide. Global atmospheric CO2 concentrations in parts per million (ppm) for the past 800,000 years. The peaks and valleys track ice ages (low CO2) and warmer interglacials (higher CO2). During these cycles, CO2 was never higher than 300 ppm. In 2018, it reached 407.4 ppm. On the geologic time scale, the increase (blue dashed line) looks virtually instantaneous
Atmospheric CO2 concentration results from the balance between CO2 emission and uptake [3]. CO2 is emitted from human activities, such as fossil fuel combustion and oxidation from other energy and industrial processes (10.0Gt of carbon in 2018 [1]) and deliberate activities on land, mainly deforestation (1.5Gt of carbon in 2018 [1]), as well as, from natural processes, such as volcanic eruptions and biological emissions. On the other plate of the scale, the small “CO2 sinks” are mainly provided by physical and biological processes in oceans (2.6Gt of carbon in 2018 [1]) and land (3.5Gt of carbon in 2018 [1]). To break this largely unfavourable imbalance (more than 5Gt in 2018), we must actively reduce the CO2 emissions and develop new and more efficient “CO2 sinks”—new individual actions and political decisions are needed, as reviewed by Seixas and Ferreira in this book [3].
Until recently, the debate often focused only on “passive CO2 mitigation”, searching for strategies for CO2 capture and sequestration (CCS). Instead, we should be looking at the opportunities for the energy and chemical industries provided by exploiting this novel and huge carbon feedstock (Fig. 2), such as (a) storage of “intermittent” renewable energy sources (RES) (wind, solar and hydropower energy, which are now rapidly growing and becoming economically viable), (b) conversion of RES-derived electricity into fuels (mainly for mobility and transport sector, in particular aviation and heavy freight over long distances, the major polluters), (c) production of added-value compounds (VC) and feedstock chemicals for making all the modern-world chemical commodities (from bulk chemicals to plastics, fertilisers and even pharmaceuticals). Regarding atmospheric CO2 reduction, points (a) and (b) (energy industry) are of major relevance, as the different scales of energy and chemical industries impede the VC production to function as a quantitative “sink” for the massive fossil fuels-dependent CO2 emissions. Together, these three axes, storage/conversion/production, will certainly provide a straightforward way to actively reduce the CO2 emissions, while actively consuming the CO2 already released—”two-in-one solution”.

Directing CO2 into the axes storage/conversion/production through the formate route. (icons from www.icons8.com)
But, how to direct CO2 into the storage/conversion/production axes? Formic acid/formateFootnote 1 offers key advantages (Fig. 2)!
2 Formic Acid—The Stepping Stone Towards Carbon Dioxide Utilisation
Formic acid was identified in fifteenth century as an acidic vapour in ant hills, from where its name derivates—”formica”, the Latin word for ant. It was first synthesised only in the nineteenth century from hydrocyanic acid by the famous French chemist and physicist Joseph Gay-Lussac and also from carbon monoxide by another French chemist, Marcellin Berthelot. However, formic acid received little industrial attention until the last quarter of twentieth century, when it started to be used as a preservative and antibacterial in livestock feed due to its low toxicity (LD50 of 1.8 g/kg). More recently, formic acid regained a new interest, due to some features that are key to the longed-for “post-fossil era” (Fig. 2).
-
(a)
Formic acid is a stable product that can be formed by the “simple” two-electron reduction of CO2 (Eq. 1), what, noteworthy, resulted in a considerable attention to the electrochemical CO2 reduction in the last decade (see Sect. 3.).
-
(b)
Besides formic acid, also carbon monoxide is a stable product of the two-electron CO2 reduction (Eq. 2). However, its high toxicity, low solubility and low mass transfer rate make the carbon monoxide subsequent utilisation challenging. In contrast, formic acid is a highly soluble and stable liquid, easy to store and transport.
-
(c)
Formic acid is also not explosive, what represents an important advantage relatively to dihydrogen, an ideal “clean” fuel (see below).
-
(d)
Formic acid is already used as a “building block” in chemical industry.
-
(e)
Formic acid can be a substrate for further reduction to a carbon-based fuel, methanol and methane, what might not be the most obvious option regarding CO2 consumption.
-
(f)
Formic acid, formed from CO2 and dihydrogen (Eq. 3), can be used as a “storage form” of dihydrogen, an ideal “clean” fuel (potentially zero contribution to the global carbon cycle, with a high gravimetric energy density) [4,5,6,7,8,9,10,11,12,13]. Although formic acid is not a perfect dihydrogen “storage medium”, due to its relatively small hydrogen content (4.4%(m/m) or 5.3%(m/v)), it is currently still one of the best options to circumvent the technical difficulties associated with dihydrogen handling, storage and transport. For this purpose, formic acid produced by CO2 hydrogenation or any other approach is converted back to dihydrogen when needed.
-
(g)
Moreover, formic acid fuel cells are being the centre of a renewed interest [14,15,16,17,18].
-
(h)
From a biotechnological point of view, formate can be both produced and assimilated by many natural and biotechnologically engineered organisms and, unlike dihydrogen (that is “just” oxidised to form reducing power), act as a carbon source for “formatotrophic” organisms, thus, enabling considerably higher biomass formation and VC and fuels production yields [19].

3 How to Convert Carbon Dioxide to Formic Acid/Formate?—The Chemical Way
CO2 is a kinetically and thermodynamically stable molecule, with a high negative value of the reduction potential of the CO2/HCOOH pair (highly pH dependent), what makes its activation and reduction a difficult task [8]. Hence, perhaps the first answer that comes to mind to reduce CO2 to formate is: electrochemically [20,21,22,23,24,25,26,27,28,29,30,31,32,33]. However, the feasibility—meaning essentially the economic viability—of this process, that is currently the centre of intense research, depends on the Faradaic efficiency and energetic efficiency of CO2 reduction (avoiding high electrochemical overpotentials) and on the rate and selectivity (purity) of formate production. The other “cost” to be considered is obviously the environmental one, and this depends on the use of a RES-derived electricity and on the sustainability of the electrodes (composition and durability).
The second answer is probably going to be photoreduction, which is the most straightforward way to use a RES to convert CO2. Solar energy (photogenerated electrons) can be used to drive chemical reactions, and this solar-to-chemical energy conversion followed by storage in the form of chemical bonds is generally called “artificial photosynthesis” (as it is a mimic of photosynthetic process used by living organism to fix CO2). The progress in this field has been quite remarkable, and several highly efficient and promising systems have been developed for CO2 reduction (as well as water oxidation and hydrogen evolution), and formic acid can be produced with high rates and selectivity [34,35,36,37,38,39,40,41,42,43,44]. However, some problems have yet to be solved. In a very simplified way, artificial photosynthesis needs two fundamental components: an ideal light absorber/photosensitiser (for light harvest, charge separation and charge transfer) and an ideal catalyst (with high intrinsic activity and stability and low overpotential). Therefore, the heterogenisation of the molecular catalysts and engineering of applicable devices are the main challenges towards the development of effective artificial photosynthesis devices (practical problems, such as density and exposure of the catalyst active sites, conductivity, mass transport and stability of the catalyst-derived material or electrode, all make the catalyst intrinsic activity and efficiency quite different from the device performance numbers). In addition, scale-up feasibility and whole device long-term stability (also associated with the costs of using expensive high-purity semiconductors to achieve high efficiency) have to be attained. Nevertheless, artificial photosynthesis devices might become economically viable sooner than many anticipate: considering the energy consumption forecasted for 2050, the future solar energy devices only need a ≈ 10% solar-to-fuel efficiency (already achievable in proof of concept devices!) if 1% of the Earth’s surface is covered [44].
Formic acid can also be produced chemically from CO2 and dihydrogen. This CO2 hydrogenation is just the thermal overall CO2 reduction by dihydrogen using molecular catalysts (Eq. 3), as an alternative to direct electrochemical or photoelectrochemical reduction of CO2 [5, 10, 12, 13, 45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. Hence, here, it is the rational design of the catalytic systems (efficiency and selectivity) that must be attained and the systems that have been developed to date exhibit selectivity and yield lower than desirable, besides requiring a high temperature and/or high pressure. Dihydrogen is a “clean” fuel (potentially zero contribution to the global carbon cycle), but its real environmental impact depends on how it is produce. The industrial production of dihydrogen (primarily from methane) requires harsh temperatures and emits as much CO2 into the atmosphere as natural gas burning [72]. To be environmentally friendly, dihydrogen must be produced by electrolysis of water using a RES and selected heterogeneous or homogeneous catalysts or biological systems [73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93]. As noted above, besides producing formic acid itself, CO2 hydrogenation is thought as a relevant way to storage dihydrogen. Therefore, also the reversible interconversion of formic acid to CO2 and dihydrogen must be carefully considered.
4 How to Convert Carbon Dioxide to Formic Acid/Formate?—Exploiting the Power of Formate Dehydrogenases (Enzymes for Solving Humankind’s Problems)
4.1 The Biochemical Way
In contrast to purely physicochemical, biological processes are substrate and product-specific (life requires a well-defined metabolism) and occur under truly “green”, sustainable conditions, at ambient temperature and pressure and close to neutral pH. Biological catalysts—enzymes—offer selectivity and specificity, coupled with high specific activity (in terms of active sites) and maximal rate (under the respective cellular context). Enzymes have evolved to become perfect catalystsFootnote 2, comprising (a) specific surface patches to establish contact with specific biomolecules (for cell localisation, integration into metabolic complexes, crosstalk or simple electron transfer), (b) channels where only (or mainly) the correct substrates come in and well-determined products come out, as well as, (c) highly defined active sites, assembled to promote the formation of key transition states and intermediates and, thus, lower the reaction energy barriers and energy loss. For that, active sites are built with precise steric features, electrostatic and hydrogen bonding interactions, fine-tuned reduction potentials and pKa values and optimised (and often synchronised) electron and proton transfer paths. The power and efficiency of biological catalysis is such that enzymes cascades are the cornerstone of all metabolic pathways that sustain life on Earth. Hence, there is a growing interest in making use of all the advantages the “biochemical way” can provide. Numerous hybrid systems have been (are being) designed to merge the best of the two worlds—chemical and biochemical—and the CO2 reduction to formate is no exception.
4.2 Formate Dehydrogenases—Enzymatic Machineries
To interconvert CO2 and formate, living organisms use formate dehydrogenase (FDH) enzymes. FDHs are a heterogeneous and broadly distributed group of enzymes that catalyse the reversible two-electron interconversion of formate and CO2 (Eq. 1) [94,95,96,97,98,99,100,101]. These enzymes evolved to take part in diverse metabolic pathways, being used by some prokaryotic organism to fix (reduce) CO2 into formate, while other prokaryotes use FDHs to derive energy, by coupling the formate oxidation (which has a very low reduction potential value, Eº′(CO2/HCOO−) = −0.43 V) to the reduction of several terminal electron acceptors; FDHs are also broadly used by both prokaryotes and eukaryotes in C1 metabolism.
FDHs can be divided into two major classes, based on their cofactor content and the consequent chemical strategy used to carry out the formate/CO2 interconversion. One class comprises FDHs that have no metal ions or other redox-active centres—the metal-independent FDHs class [102,103,104,105,106,107,108,109]. These enzymes are widespread, being found in bacteria, yeasts, fungi and plants, are all (as far as is known) NAD-dependent and belong to the D-specific dehydrogenases of 2-oxyacids family. The other class—the metal-dependent FDHs class Footnote 3—comprises only prokaryotic enzymes that hold different redox-active centres (Table 1) and whose active site harbours one molybdenum or one tungsten centre (molybdenum-containing FDH (Mo-FDH) or tungsten-containing FDH (W-FDH), respectively) [94,95,96,97,98,99,100,101, 110,111,112].
4.2.1 The Metal-Independent Formate Dehydrogenases
Comparatively, the metal-independent FDHs are quite simple enzymes, generally forming homodimers, containing a NAD(H) and a formate-binding pockets in a close vicinity of each other (Fig. 3) [102,103,104,105,106,107,108,109]. The formate-binding site harbours a conserved arginine and asparagine residues, while an aspartate and serine residues make contacts to the nicotinamide ring, with another arginine residue binding the phosphate moiety linker of NAD(H).

C. boidinii formate dehydrogenase. A Three-dimensional structure view of the homodimer. B Arrangement of NAD and azide shown in the same orientation (but not same scale) as in (A). C Enzyme active site, with azide and NAD bound. The structures shown are based on the PDB file 5DN9 [109] (\(\alpha\) helices and \(\beta\) sheets are shown in red and cyan, respectively)
4.2.2 The Metal-Dependent Formate Dehydrogenases
Because the metal-dependent FDHs are involved in diverse metabolic pathways (energy and C1 metabolism), for which different “interfaces” are needed, this class is extraordinarily heterogeneous, comprising enzymes with diverse redox-active centres, such as iron–sulfur centres (Fe/S), haems and flavins, besides the characteristic molybdenum or tungsten active sites, organised in different subunit compositions and quaternary structures (Table 1) [94,95,96,97,98,99,100,101, 110,111,112]. This structural diversity is well exemplified by Escherichia coli, that expresses one “simple” monomeric cytoplasmatic enzyme, containing only the molybdenum centre and one [4Fe–4S] centre (the FDH H; Fig. 4) [113,114,115,116,117], and two “complex” heteromeric ((αβγ)3) membrane-bound respiratory enzymes that harbour seven additional redox-active centres ([4Fe–4S] centres and b-type haems) in addition to the molybdenum centre (the FDH N [118,119,120] (Fig. 5) and FDH O [121,122,123]). Also, the sulfate-reducing bacteria of the Desulfovibrio genus contain diverse Mo-FDHs and W-FDHs [124,125,126,127,128,129], such as the heterodimeric (αβ) periplasmatic W-FDH of D. gigas [130] or D. vulgaris [129, 131, 132], with “only” four [4Fe–4S] centres and one tungsten centre (Fig. 6) [133, 134], or the more “complex” heteromeric (αβγ) Mo-FDH of D. desulfuricans [135,136,137] or D. vulgaris [129, 131] that contains eight redox-active centres ([4Fe–4S] centres and c-type haems) in addition to the molybdenum centre. Remarkably, the overall protein fold of the molybdenum—and tungsten-containing subunits, including the arrangement of Fe/S centre, is highly conservedFootnote 4 [116, 117, 119, 130, 132, 138,139,140].

E. coli formate dehydrogenase H. A Three-dimensional structure view. B Arrangement of the redox-active centres shown in the same orientation (but not same scale) as in (A). C Molybdenum catalytic centre of oxidised enzyme. D Molybdenum catalytic centre of reduced enzyme as suggested by Boyington et al. in 1997 [116]. E Molybdenum catalytic centre of reduced enzyme as suggested by Raaijmakers and Romão in 2006 [117]. The structures shown are based on the PDB files 1FDO (A, B, C), 1AA6 (D) [116] and 2IV2 (E) [117] (\(\alpha\) helices and \(\beta\) sheets are shown in red and cyan, respectively)

E. coli formate dehydrogenase N. A Three-dimensional structure view. B Arrangement of the redox-active centres shown in the same orientation (but not same scale) as in (A). C Molybdenum catalytic centre of oxidised enzyme. The structures shown are based on the PDB file 1KQF [119] (\(\alpha\) helices and \(\beta\) sheets are shown in red and cyan, respectively)

D. gigas (A, B, E) and D. vulgaris (C, D, F, G) formate dehydrogenases. A Three-dimensional structure view of D. gigas W-FDH. B Arrangement of the redox-active centres of D. gigas W-FDH, shown in the same orientation (but not same scale) as in (A). C Three-dimensional structure view of D. vulgaris W-FDH. D Arrangement of the redox-active centres of D. vulgaris W-FDH, shown in the same orientation (but not same scale) as in (C). E Tungsten catalytic centre of oxidised D. gigas W-FDH. F Tungsten catalytic centre of oxidised D. vulgaris W-FDH. G Tungsten catalytic centre of formate-reduced D. vulgaris W-FDH. The structures shown are based on the PDB files 1H0H (A, B, E) [130], 6SDR (C, D, F) and 6SDV (G) [132] (\(\alpha\) helices and \(\beta\) sheets are shown in red and cyan, respectively)
The diversity of metal-dependent FDHs is also observed through their “molecular plasticity”. Some FDHs take part in formate-hydrogen lyase systems, as is the case of FDH H from E. coli (Mo-FDH) [141], Pectobacterium atrosepticum (Mo-FDH) [142] or C. carboxidovorans (W-FDH) [143,144,145,146,147]. Physiologically, the E. coli formate-hydrogen lyase is a membrane-bound system involved in formate oxidation and dihydrogen formation under fermentative growth conditions [141, 148,149,150,151]. The system comprises two enzymes, the cytoplasmatic Mo-FDH (described above) and a membrane-bound, cytoplasm-faced nickel/iron-containing hydrogenase (Ni/Fe-Hase); FDH oxidises formate to CO2 and the resulting reducing equivalents are transferred, through three Fe/S proteins, to the Ni/Fe-Hase that reduces protons to dihydrogen (Fig. 7).

Adapted with permission from Ref. [149]
Predicted architecture of the E. coli formate-hydrogen lyase. B, F and G represent three Fe/S proteins. See text for details
A different rearrangement of the same basic features (FDH plus Hase) is found in cytoplasmatic dihydrogen-dependent FDHs (better denominated as CO2 reductases), that physiologically catalyse the reduction of CO2 to formate with the simultaneous and direct oxidation of dihydrogen, that is, without the intervention of an external electron-transfer protein or molecule, as reviewed by Litty and Müller in this Book [152] and also [153,154,155,156,157,158,159]. The dihydrogen-dependent CO2 reductase of the acetogen A. woodii is a tetramer (αβγδ), holding one FDH-like subunit comprising one molybdenum and one [4Fe–4S] centres, where CO2 is reduced; the necessary electrons are transferred intramolecularly from an iron–iron hydrogenase-like (Fe/Fe-Hase) subunit (second active site), via two small electron-transfer subunits (each with four [4Fe–4S] centres) (Fig. 8) [153]. A tungsten-containing homologue enzyme is found in Thermoanaerobacter kivui [156].

Adapted with permission from Ref. [156]. http://creativecommons.org/licenses/by/4.0/
Structural organisation of A. woodii (molybdenum-dependent) and T. kivui (tungsten-dependent) dihydrogen-dependent CO2 reductases. ET represents two small electron-transfer subunits. See text for details
A further example of the “plasticity” of FDH-like proteins is provided by N-formyl-methanofuran dehydrogenases (FMFDH) that also have two physically separated active sites: one catalyses the reduction of CO2 to formate, which is then intramolecularly transferred to the second active site, where it is condensed with methanofuran to form N-formyl-methanofuran (Eq. 4) [140, 160, 161]. The FMFDHs are even more complex than FDHs and the enzyme from the methanogen M. wolfeii is a tetramer of (αβγδεω) units, whose CO2-reducing subunit shares the tungsten and [4Fe–4S] centres, as well as, the protein fold of the W-FDHs and Mo-FDHs (Fig. 9).

M. wolfeii N-formyl-methanofuran dehydrogenase. A Three-dimensional structure view. B Arrangement of the redox-active centres shown in the same orientation (but not same scale) as in (A). C Tungsten catalytic centre of oxidised enzyme. The structures shown are based on the PDB file 5T5I [140] (\(\alpha\) helices and \(\beta\) sheets are shown in red and cyan, respectively)
In contrast to the structural and organisational diversity, the active site of all presently known metal-dependent FDHs and FMFDH is very well conserved [94,95,96,97,98,99,100,101, 110,111,112, 140]. In the oxidised form, the active site harbours one molybdenum ion (in the case of Mo-FDHs and Mo-FMFDHs) or one tungsten ion (in W-FDHs and W-FMFDHs) coordinated by the cis-dithiolene (–S–C = C–S–) group of two pyranopterin cofactor molecules (Fig. 10), as is characteristic of this family of mononuclear molybdenum and tungsten enzymes [97, 110,111,112, 162,163,164,165]. The metal first coordination sphere is completed by one terminal sulfido group (Mo/W = S)Footnote 5 plus one sulfur or selenium atom from a cysteine or selenocysteine residue (Mo/W–S(Cys) or Mo/W–Se(SeCys)) (abbreviated as Cys–Mo–FDH, Cys–W–FDH, SeCys–Mo–FDH and SeCys–W–FDH), in a distorted trigonal prismatic. Noteworthy, there is no apparent relation (as far as is presently known) between the metal and the bound amino acid residue (examples of the four combinations Cys–Mo–FDH, Cys–W–FDH, SeCys–Mo–FDH and SeCys–W–FDH are known for long; Table 1) or the enzyme activity. The active site also comprises two other residues that are strictly conserved to all known FDHs and FMFDHs and are thought to be crucial to the catalytic cycle (as discussed below), one arginine and one histidine (this linked (C-terminal side) to the selenocysteine or cysteine that coordinates the molybdenum or tungsten ion) [116, 117, 119, 130].

Active site of formate dehydrogenase and N-formyl-methanofuran dehydrogenase. Structure of the pyranopterin cofactor (top). The pyranopterin cofactor molecule is formed by pyrano(green)-pterin(blue)-dithiolene(red)-methylphosphate(black) moieties; in all so far characterised FDHs, the cofactor is found esterified with a guanosine monophosphate (dark grey). The dithiolene (–S–C = C–S–) group forms a five-membered ene-1,2-dithiolene chelate ring with the molybdenum or tungsten ion, here indicated as M (from metal). Structure of the molybdenum/tungsten centre in the oxidised state (middle). For simplicity, only the dithiolene moiety of the pyranopterin cofactor is represented. Structure of the molybdenum/tungsten centre in the reduced state (bottom). For simplicity, only the dithiolene moiety of the pyranopterin cofactor is represented. Contrary to the oxidised state (that is consensually accepted), the structure of the reduced state is still under debate, as discussed below, under Sect. 4.3.2b. The two hypotheses under debate are represented, with the cysteine or selenocysteine residue bound to the metal and with the residue dissociated from the metal (Sect. 4.3.2b)
4.3 Formate Dehydrogenases—Mechanism of Action
4.3.1 The Metal-Independent Formate Dehydrogenases
The metal-independent FDHs are NAD-dependent enzymes, whose chemical strategy to interconvert formate and CO2 is surprisingly simple and well established (Fig. 11) [102,103,104,105,106,107,108,109]: the enzyme binds formate and NAD+ in close proximity of each other (1.4 Å distance between H-(formate) and C4-(pyridine ring)) and makes NAD+ acquire a bipolar conformation, which increases its electrophilicity and, thus, facilitates the hydride transfer. The reaction, then, proceeds by straightforward hydride transfer from formate to NAD+. In accordance, the rate-limiting step of the catalytic cycle is the formate C–H bond cleavage, as shown by kinetic studies of the 2H-labelled formate isotopic effect), and the enzyme operate via a ternary complex (FDH-formate-NAD+) kinetic mechanism [107, 170,171,172,173,174,175,176,177,178].

Hydride transfer mechanism proposed for metal-independent NAD-dependent formate dehydrogenases
4.3.2 The Metal-Dependent Formate Dehydrogenases
Several experimental and some computational approaches have been exploited to elucidate how the metal-dependent FDHs carry out the formate/CO2 interconversion and over the years a few mechanistic proposals have been put forward [116, 117, 137, 166, 169, 179,180,181,182,183,184,185]. Presently, several key points are well established, but two remain a matter of debate, and all are discussed below, before the currently accepted mechanistic hypotheses are introduced.
-
(a)
Presently, several key points are well established
-
(i)
The formate/CO2 interconversion occurs at the molybdenum or tungsten centre, in a reaction that is intermediated by the metal, which cycles between the +6 and +4 oxidation states (Eq. 5a–5d), as demonstrated by numerous spectroscopic and kinetic studies. The electrons necessary to carry out CO2 reduction or released from formate oxidation are intramolecularly transferred from the physiological partner (electron donor or electron acceptor), through the different redox centres of each enzyme (Fe/S centres, haems, FAD (see above)) that act like a “wire” to facilitate the fast and effective electron transfer. Therefore, the intramolecular electron transfer is, thus, an integral aspect of the global reaction. Depending on the enzyme (on the biochemical pathway where the enzyme is involved in), the physiological redox partner can be membrane quinols, cytoplasmatic and periplasmatic cytochromes, ferredoxins, NAD(P) or coenzyme F420 [94,95,96,97,98,99,100,101]. For those enzymes like the dihydrogen-dependent CO2 reductase (see above), the electrons are directly provided by the co-substrate oxidation (dihydrogen in this case) that occurs in the enzyme second active site. As a consequence of the physical separation of the oxidation and reduction half-reactions (that occur at different enzyme centres), all these enzymes operate via a ping-pong kinetic mechanism, as observed experimentally.
-
(ii)
Although the formate/CO2 interconversion occurs at the molybdenum or tungsten centre, the reaction is not one of oxygen atom transfer, as is characteristic of many molybdoenzymes and tungstoenzymes (Fig. 12) [94, 97, 110,111,112, 162,163,164,165]: the substrate for “CO2 reduction” is in fact CO2 and not hydrogencarbonate (Eq. 6) [186], and the product of formate oxidation is CO2 and not hydrogencarbonate (Eq. 7), as was clearly demonstrated by the formation of 13C16O2 gas during the oxidation of 13C-labelled formate in 18O-enriched water [166]. Therefore, to catalyse the formate oxidation, FDH has to abstract one proton plus two electrons (Eq. 8) or one hydride (Eq. 9) from the formate molecule (or the reverse for CO2 reduction).
Fig. 12 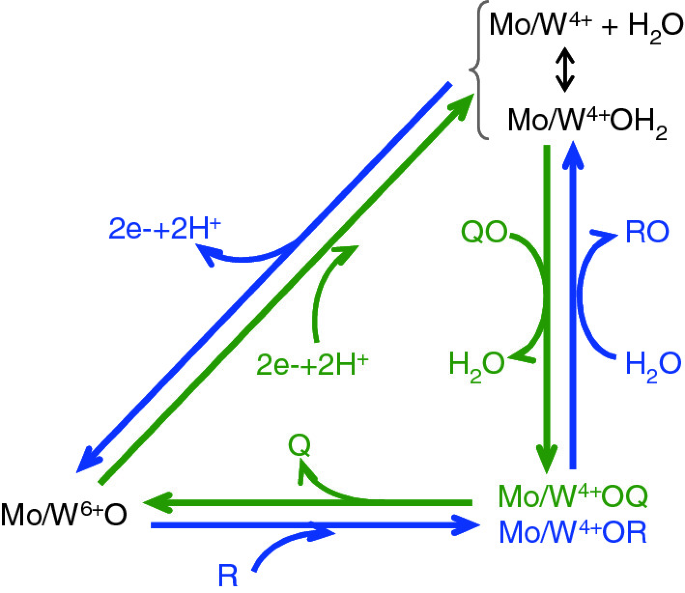
Oxygen atom transfer in molybdo—and tungstoenzymes. Typically, these enzymes catalyse the transfer of an oxygen atom from water to product—oxygen atom insertion (blue arrows)—or from substrate to water—oxygen atom abstraction (green arrows)—in reactions that entail a net exchange of two electrons, in which the molybdenum/tungsten atom cycle between Mo/W6+ and Mo/W4+, and, most importantly, where the metal is the direct oxygen atom acceptor or donor. This feature was coined by Holm and others in the 1980s as the “oxo transfer hypothesis”





-
(iii)
A simple chemical reasoning, based on the pKa values of formic acid (HCOOH/HCOO− = 3.77; HCOO−/CO22− ≫ 14), demonstrates that it is much more difficult to abstract the Cα proton from formate (Eq. 8) than abstract a hydride (Eq. 9), which, in addition, lead to the formation of a stable product (CO2), instead of a carbonanion (CO22−) (Scheme 1). The simple mechanistic strategy followed by the metal-independent FDHs (Sect. 4.3.1.), that is, direct reaction with NAD+, with no enzyme cofactors involved, further confirms that it must be exceptionally facile (thermodynamics) to abstract a hydride from the formate molecule. On its turn, CO2, with an electronic structure O −−δC+2δ− O−δ and a carbon-localised LUMO, is susceptible to attack by nucleophiles and to reduction, being a good hydride acceptor, as supported by the chemistry of several synthetic transition metal-hydride complexes that mimic the FDH catalysis [65, 67, 187,188,189,190,191,192].
Scheme 1 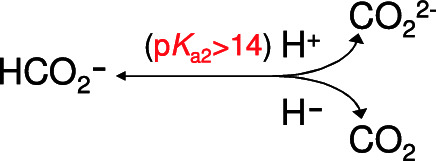
Products formed by proton abstraction (ruled by a pKa2 >> 14) or hydride abstraction from formate
-
(iv)
The terminal sulfido group of the active site (Fig. 10) is well documented as a hydride acceptor/donor. Since the 1970s, the sulfido group is established as the hydride acceptor in the oxidised molybdenum centre (Mo6+=S) of xanthine oxidase and aldehyde oxidaseFootnote 6 [110,111,112, 162,163,164,165, 193,194,195,196,197,198,199,200,201,202], as well as, the hydride donor in the reduced centre (Mo4+–SH) of hydroxybenzoyl-CoA reductaseFootnote 7 [203, 204]. This “twin” behaviour (oxidised/hydride acceptor versus reduced/hydride donor) is supported by a remarkable characteristic of the Mo/W-ligands: the pKa values of the coordinated ligands change dramatically with the oxidation state of the metal and determine that the higher oxidation states should hold deprotonated ligands, that is Mo/W6+=S, while the lower oxidation states should hold protonated ligands, that is Mo/W4+–SH [205,206,207]. This behaviour (Eq. 10) enables the metal-sulfido to act as a hydride acceptor/donor and is supported by the high covalency of the terminal sulfur atom in the metal sulfido group, with an available S π-bond well suited to accept a hydride.

The involvement of the sulfido group as the hydride acceptor during FDH catalysis was demonstrated by electron paramagnetic resonance (EPR) spectroscopic studies that showed that, in formate-reduced FDH, the formate Cα hydrogen atom is transferred to an acceptor group located within magnetic contact to the molybdenum atom of FDHs from different sources (E. coli [166], D. desulfuricans [136] or Cupriavidus necator (previously known as Ralstonia eutropha) [184]). The observation of a strongly coupled, solvent-exchangeable and substrate-derived proton, with a hyperfine constant of 20–30 MHz, is consistent with the hydrogen atom being transferred to a ligand in the first coordination sphere of the molybdenum atom upon its reduction [196, 197, 200, 201, 208]. Similar hyperfine constant values were determined in model complexes [209] and also in real enzymes, as in xanthine oxidase, where the strong coupled hydrogen is originated from the xanthine C8 hydrogen atom (the position that is hydroxylated by that enzyme (see Footnote 6) [196,197,198, 200,201,202, 208]. It should be noted that a hyperfine interaction of this magnitude could not arise from the transfer of the formate Cα hydrogen atom to an acceptor in the second coordination sphere of the metal, for example, transfer to the conserved histidine residue, as initially proposed [116, 117, 166, 169, 180,181,182,183], or transfer to the selenocysteine/cysteine residue if it had been dissociated from the molybdenum/tungsten ion [117, 169, 180,181,182,183]—an hypothesis discussed below in point (vi). Photolysis assays with 77Se-enriched FDH, described below in point (vi) and Footnote 8, further confirmed that the selenocysteine residue cannot be the hydrogen atom acceptor [166].
Further evidence that the sulfido group becomes protonated upon reduction was also provided by a recent X-ray absorption spectroscopy (XAS) study with the R. capsulatus Mo-FDH [210].
-
(v)
The terminal sulfido group is essential to both formate oxidation and CO2 reduction. It is well established, by numerous spectroscopic and kinetic studies, that cyanide reacts with the active site sulfido group of different molybdoenzymes, such as xanthine oxidase, and abstracts it in the form of thiocyanate, yielding a desulfo enzyme form that harbours an oxo group in the place of the native sulfido group [110,111,112, 193,194,195, 198, 199, 202]. The sulfido by oxo replacement renders xanthine oxidase and other enzymes inactive, because its active site is no longer able to accept a hydride (see Footnote 6). The same and complete cyanide inhibition is observed in several FDH, such as the ones from Methanobacterium formicicum [211], Alcaligenes eutrophus [212], E. coli [167], R. capsulatus (where the sulfido was observed to be replaced by an oxo group) [210], or D. desulfuricans (where thiocyanate formation accounted to 0.87 per molybdenum atom) [137]. Together with the experimental evidences that support the involvement of the sulfido group as a hydrogen atom acceptor during FDH catalysis (described above), these inhibitory results demonstrate that the sulfido group acts as a hydride acceptor/donor in FDH catalysis.
-
(b)
Two interrelated points are not yet consensual
-
(vi)
Does the active site cysteine or selenocysteine residue dissociate from the metal during catalysis?
If the configuration of the oxidised active site is consensually accepted, the reduced form still finds a few contradictory experimental evidences (Fig. 10).
X-ray crystallography: In a reinterpretation of the crystallographic data of the reduced E. coli SeCys–Mo–FDH H originally obtained by Boyington et al. in 1997 [116], Raaijmakers and Romão in 2006 [117] suggested that the polypeptide loop containing the selenocysteine was not properly traced in the original work and that the selenocysteine residue is not bound to the metal, but, instead, is found dissociated from the molybdenum ion and shifted away (12 Å) (Fig. 4). Therefore, those authors suggested that, while in the oxidised state the selenocysteine residue is coordinated to the metal, the enzyme reduction triggers the residue dissociation, resulting in a square pyramidal penta-coordinated centre, where the molybdenum ion is coordinated by the cis-dithiolene (–S–C = C–S–) group of two pyranopterin cofactor molecules (in the equatorial positions) plus the terminal sulfido group (in the axial position) (Fig. 10). Regardless of this reinterpretation, all other crystallographic structures so far available, for FDH and FMFDH (Figs. 4, 5, 6 and 9, and references herein), show a stable hexa-coordination, with the cysteine or selenocysteine always bound to the molybdenum/tungsten ion. This is also the case of the recently solved structure of the formate-reduced D. vulgaris SeCys–W–FDH [132] and also of the NADH-reduced R. capsulatus Cys–Mo–FDH, whose structure was determined by cryo-electron microscopy [139].
XAS: Two recent XAS studies at the Mo K-edge suggested that, in R. capsulatus Cys–Mo–FDH, the Mo5+ state holds the cysteine residue bound to the metal, as the oxidised Mo6+ one, with a Mo-S(Cys) bond of 2.63 Å, while the Mo4+ state of formate-reduced enzyme has its cysteine displaced form the metal [169, 210]. However, contrary results were obtained with SeCys–Mo–FDHs from E. coli [213] and D. desulfuricans [214], which showed a metal bound residue in all oxidised and reduced states: the E. coli enzyme EXAFS data at both the Mo and Se K-edges were interpreted as indicating the presence of one Mo–Se bond of 2.62 Å, plus one Se–S bond of 2.19 Å (between the sulfido group and the selenocysteine selenium) [213].
EPR spectroscopy: Further experimental evidence for the stable molybdenum/tungsten hexa-coordination came from EPR spectroscopy that clearly showed that the selenocysteine/cysteine must remain bound to the Mo5+ centre of formate-reduced enzyme [208]. When the EPR spectrum is obtained from 77Se-enriched enzyme, a very strong and anisotropic interaction with selenium is observed (A1,2,3(77Se) = 13.2, 75, 240 MHz) [166]. This interaction and the observation of the expected 95,97Mo hyperfine coupling confirms that the selenium atom of the selenocysteine is directly coordinated to the Mo5+ and further suggests that the unpaired electron is delocalised over the selenium (17–27%) and molybdenum atoms (73–83%) [166]. Also, the hydrogen atoms of the β-methylene carbon of the selenocysteine residue are thought to be in the close proximity of the molybdenum atom, being responsible for an interaction with a not solvent-exchangeable protons (A1 = 35.1 MHz) [136]. Photolysis assays additionally confirmed that the selenium/sulfur ligation is retained in the FDH Mo5+ centre (the light beam did not affect the strong selenium–molybdenum EPR interaction observed in 77Se-enriched FDH)Footnote 8 [166]. The Mo5+ hexa-coordination (resulting from having the selenocysteine/cysteine residue bound to the molybdenum ion) was also supported by theoretical calculations on the signals-giving species of FDHs [215].
Inhibition assays: A different type of experimental evidence came from inhibition studies with iodoacetamide, an alkylating agent that reacts with “free” ionised selenocysteine or cysteine residues (carboxamidomethylation). Native E. coli SeCys–Mo–FDH H and its cysteine mutant [216] and native R. capsulatus Cys–Mo–FDH [183] are not inhibited by iodoacetamide treatment. However, when the preliminary iodoacetamide treatment (incubation) is carried out in the presence of formate (not under turnover), both native and cysteine-containing mutant E. coli FDHs are inhibited [216]. Inhibition is also observed in the R. capsulatus FDH, but only when the iodoacetamide treatment (incubation) is carried out in the presence of nitrate; in this case, the cysteine carboxamidomethylation was confirmed by mass spectroscopy [183]. On the other hand, native D. vulgaris SeCys–W–FDH is inhibited by iodoacetamide, but mass spectroscopy clearly showed that the inhibition is not due to the carboxamidomethylation of the active site selenocysteine residue (but of 9 other cysteine residues not present in the active site) [132]. In addition, other FDHs are not at all affected by iodoacetamide [135, 136]. Hence, the inhibition results available were not obtained under formate/CO2 turnover conditions and the inconsistency of the results, once more, do not contribute to provide a definitive answer.
Overall, the majority of experimental evidences points towards a molybdenum/tungsten stable hexa-coordination, with the cysteine/selenocysteine residue always bound to the metal. Regarding the results showing a metal penta-coordination, with unbound cysteine/selenocysteine residue, it is possible that the crystallisation/irradiation had induced some artefacts that are not relevant to the enzyme activity; but it is also possible that the species crystallographically characterised, being catalytically relevant, bear no relation to the species observed by XAS and EPR (with these being not catalytically relevant). Certainly, high-resolution structures are needed to confirm the existence of the two alternating conformations of the selenocysteine/cysteine-containing polypeptide loop and to discuss the catalytic relevance of each conformation.
-
(vii)
Do formate/CO2 bind directly to the molybdenum/tungsten ion during catalysis?
Inspired by the oxotransfer chemistry displayed by several molybdenum—and tungsten-dependent enzymes (Fig. 12) [110,111,112, 162,163,164,165], and in particular by periplasmatic nitrate reductasesFootnote 9, it was suggested that FDH catalysis necessarily involves the formate/CO2 direct binding to the molybdenum/tungsten ion [180, 218].
To begin the discussion of this point, it should be noted that the direct formate/CO2 binding would involve an unprecedented hepta-coordinated molybdenum/tungsten centre or it would depend on the dissociation of the cysteine/selenocysteine residue from the metal, in order to create a vacant position (penta-coordinated centre) for substrate binding. While the hypothesis of the hepta-coordinated metal centre was (so far) never perused, the dissociation of the cysteine/selenocysteine is, as discussed above, controversial.
The two recent XAS studies mentioned above in point (vi) suggested that, in the presence of formate, the cysteine ligation of (active) R. capsulatus Cys–Mo–FDH is replaced by a long Mo–O bond of 2.15 Å, which was interpreted as arising from the Mo–OCO(H) complex [169, 210]. The strong and competitive inhibition of E. coli SeCys–Mo–FDH H-catalysed formate oxidation by azide, cyanate, thiocyanate, nitrite and nitrate (1–00 μM range) was also evoked to support that formate, as well as those inhibitors, bind directly to the molybdenum ion [219]. Yet, competitive inhibition can arise if the inhibitor binds in the active site, but not directly to the metalFootnote 10 [220]; this seems to be the case at least of azide, a well documented inhibitor of both metal-independent [104, 171, 177] and metal-dependent [136, 166] FDHs (as suggested by EPR [136, 215]) and nitrite (as suggested by crystallographyFootnote 11). Regarding nitrate, (once more) several other FDH enzymes are not inhibited or the inhibition constants are 2–3 orders of magnitude higher than Km(formate) [132, 135, 136, 166], or it is though as a substrate (even though a very poor one; see Footnote 9 [182]). Moreover, the same study [219] showed that the inhibition of the E. coli SeCys-Mo-FDH H-catalysed CO2 reduction by those anions is very weak (in the range of 1–50 mM) and not competitive in nature, results that are contradictory to the hypothesis that the reduced active site (the one that reacts with CO2) becomes penta-coordinated, with an unbound selenocysteine residue, and with an available position to bind inhibitors and CO2.
Therefore, except from the abovementioned XAS data, there are no other direct experimental evidences of the direct formate or CO2 binding to the FDH molybdenum/tungsten ion; namely, there are no crystallographic structures showing the formate molecule in the active site and there are no EPR signals showing the presence of formate in the first coordination sphere of molybdenum/tungsten [136, 166, 184].
Theoretically, it can be argued that, since the FDH-catalysed reaction does not involve the transfer of an oxygen atom (as explained above in point (ii)), there is no need to form the otherwise expected Mo/W6+–OCO(H) or Mo/W4+–OCO complexes (follow Mo4+–OR and Mo4+–OQ in Fig. 12). It can also be argued in the opposite way: if formate/CO2 binds directly to the molybdenum/tungsten ion, why there is no oxygen atom transfer to form hydrogencarbonate (Eq. 7)? Overall, in the absence of more definitive experimental evidences, we must continue to ask: Does the direct formate/CO2 binding to the metal occur? Is it necessary or desirable to interconvert formate and CO2? Is the penta-coordinated metal centre with unbound cysteine/selenocysteine catalytically relevant?
-
(c)
Currently accepted mechanistic hypotheses
In accordance with the well-established points (i) to (v) highlighted above, the FDH-catalysed formate oxidation and CO2 reduction are presently recognised to occur through hydride transfer (Eq. 9), with the oxidised and reduced active site sulfido group, Mo/W6+=S and Mo/W4+–SH, acting as the direct hydride acceptor and donor, respectively. Yet, points (vi) and (vii) still raise questions to some authors regarding the coordination of the active site and substrates binding during FDH catalysis.
As originally proposed by Niks et al. [184] for formate oxidation and shortly after also for CO2 reduction [137], we suggest that FDH catalysis proceeds as follows (the reaction mechanism is suggested to be identical in Mo–FDH and W–FDH, as well as in FMFDH):
Formate oxidation (Fig. 13, blue arrows) is initiated with the formate binding to the oxidised active site, but not directly to the molybdenum/tungsten atom. Following the example provided by the metal-independent FDH, where the formate-binding site harbours arginine and asparagine residues [102,103,104,105,106,107,108,109], it is suggested that the conserved arginine residue is essential to drive the formate Cα hydrogen towards the sulfido ligand, by establishing hydrogen bond(s) with its oxygen atom(s). Also, azide (N3−, isoelectronic with CO2) is suggested to bind (tightly) to the same site and not directly to the molybdenum/tungsten ion (as had been previously suggested for the D. desulfuricans FDH inhibition by azide [136, 215]). The binding of azide and formate to the same site, and not to the molybdenum/tungsten atom itself, explains why azide is a powerful inhibitor of both metal-independent (Ki = 40 nM for Candida boidinii) [104, 171, 177] and metal-dependent FDHs [136, 166]. A similar reasoning applies to the inhibitor nitrite (isoelectronic with formate). Formate oxidation, then, proceeds by a straightforward hydride transfer from formate to the sulfido group of the oxidised molybdenum/tungsten centre, Mo/W6+=S, leading to the formation of Mo/W4+–SH and CO2. The re-oxidation of Mo/W4+ to Mo/W6+ (via intramolecular electron transfer to the enzyme other(s) redox centre(s) and, eventually, to the physiological partner) and the release of CO2 close the catalytic cycle. The now oxidised Mo/W6+ favours the sulfido group deprotonation (dictated by the ligand pKa [205,206,207]), and the initial oxidised molybdenum/tungsten centre, Mo/W6+=S, is regenerated. Under non-steady-state catalytic conditions (as the ones created in EPR experiments) the molybdenum/tungsten one-electron oxidation should be favoured (Mo/W4+→Mo/W5+), leading to the formation of the EPR detectable Mo5+–SH species.

Reversible hydride transfer mechanism proposed for metal-dependent formate dehydrogenase and N-formyl-methanofuran dehydrogenase [137, 184]. Reaction mechanism proposed for formate oxidation (blue arrows) and CO2 reduction (green arrows). For simplicity, the mechanism is represented only for a molybdenum, selenocysteine-containing enzyme, but it should be similar for tungsten and cysteine-containing enzymes. See text for details. A similar hydride transfer mechanism can also take place with a penta-coordinated reduced metal centre, with a dissociated selenocysteine/cysteine residue (see text for details)
CO2 reduction is suggested to follow the reverse reaction mechanism (Fig. 13, green arrows). First, CO2 binds to the reduced active site, not directly to the molybdenum/tungsten ion, but at the same site as formate (and azide), with the conserved arginine anchoring its oxygen atom(s) through hydrogen bond(s) and orienting its carbon atom towards the protonated sulfido ligand. In an approximated way, based on the inhibition and Michaelis–Menten constants for the D. desulfuricans FDH, the “binding strength” is suggested to follow the order CO2 (Km ≈ 15 μM [137]) > azide (Ki ≈ 30 μM [136]) > formate (Km ≈ 60 μM [137]). Then, the reaction proceeds through straightforward hydride transfer from the protonated sulfido group of the reduced molybdenum/tungsten centre, Mo/W4+–SH, to the CO2 carbon, whose LUMO have predominant C–π orbital character, prone to nucleophile attack and reduction. This yields a formate moiety and Mo/W6+=S. The subsequent re-reduction of Mo/W6+ to Mo/W4+ (via intramolecular electron transfer from the enzyme physiological partner, through its redox centre(s)) and formate release closes the catalytic cycle. The now reduced Mo/W4+ favours the sulfido group protonation and the initial reduced molybdenum/tungsten centre, Mo/W4+–SH, is regenerated.
The FDH-catalysed reaction is reversible and the equilibrium between formate oxidation versus CO2 reduction is determined by the availability of formate versus CO2 and the ability to maintain the active site oxidised (Mo/W6+) versus reduced (Mo/W4+), which, in its turn, determines the protonation state of the metal sulfido group in a concerted and straightforward way.
Overall, the chemical strategy herein suggested is exactly the same as the one proposed for the metal-independent FDHs: both bind formate in a close proximity to an oxidised, electrophilic, hydride acceptor, which in metal-independent enzymes is a NAD+ molecule and in metal-dependent enzymes is the M6+=S group; both bind CO2 in a close proximity of a reduced, nucleophilic, hydride donor, a NADH molecule or the M4+–SH group.
As expected, this mechanistic proposal faces some criticism and the most relevant one concerns the role of the active site selenocysteine/cysteine residue. In fact, although the mechanism is suggested to operate in a hexa-coordinated metal centre (Fig. 13), it can also take place in a penta-coordinated centre (Fig. 10), with an unbound selenocysteine/cysteine—the sixth ligand does not seem to interfere with the hydride transferFootnote 12. Even though there are experimental evidences (as discussed above) and mechanistic arguments can be envisaged to support the necessity of having a bound selenocysteine/cysteine (as discussed in [96, 98]), in the absence of clear and definitive experimental evidences, both scenarios—dissociated and bound selenocysteine/cysteine—seem to be possible and this is an aspect that will remain in open for now. Certainly, future research will shed light in these aspects of the FDH reaction, allowing a critic evaluation of this mechanistic proposal.
4.4 Formate Dehydrogenases in the Context of Carbon Dioxide Utilisation
The majority of currently known FDHs function in vivo to oxidise formate, with only a few participating in metabolic pathways to reduce (fix) CO2—the reaction direction that is interesting to “solve humankind’s problem” with atmospheric CO2. However, the CO2/formate interconversion is thermodynamically reversible (E º′(CO2/HCOO−) = −0.43 V) and in vitro there is no a priori reason for a FDH to be unable to catalyse the CO2 reduction, as long as there is sufficient reducing power availableFootnote 13. Regarding the metal-independent FDHs, this simply means the adequate NADH/NAD+ ratio (Eq. 11); in what concerns the metal-dependent FDHs, this means that the molybdenum/tungsten active site centre must be kept reduced at the proper reduction potential (Eq. 12). Although at first sight obvious, the necessity to keep the enzyme active site reduced is most often overlooked, what may explain why there are so many reports in the literature of FDHs unable to reduce CO2. This is particularly true for metal-dependent FDHs: if the reduction potential of one (or more) of the FDH redox centres is (are) relatively high, it could be difficult to “push” the electrons into the active site (the centre with the higher reduction potential could stay reduced, “blocking” the electron transfer to the other(s) centre(s) with lower reduction potentials and the active site in particular). In addition to thermodynamics, also the kinetics has to be taken into account to evaluate if the CO2 reduction is going to be efficient, or too slow relatively to the formate oxidation to be relevant (rate of formate oxidation versus CO2 reduction, Eqs. 13, 14). The key point here is that FDH kinetics is determined by four parameters, the Km and kcat for the two substrates (CO2 and formate), and the reaction can be run under different regimes (mainly forward, mainly reverse and equilibrium, as determined by kcat/Km and imposed conditions). However, except for protein engineering (a difficult task on its own), there is not much that can be done to modify the kinetic parameters to further favour the CO2 reduction.
Also, the enzymes stability and potential interfering compounds must be well thoughtout. The lifetime of a CO2 converter device is a critical issue, and it would greatly depend on the time the enzyme maintains its full activity. In this respect, it should be emphasised that the purifying processes often decrease the enzymes stability and even make them unstable (while taken out of their biological environment), thus hampering their usage in a sustained (“real life”) way or just making the scale up process unviable. The inhibition and inactivation by compounds that might be present in the “substrate reaction mixture”; for example, dioxygen or carbon monoxide, are pitfalls that must be considered to avoid the need (additional cost) of using purified CO2. The inhibition or inactivation (or, on the contrary, improved stability) by the materials used to build the device cannot be overlook also.
The enzyme–material “communication” is another major challenge in hybrid systems. It is necessary to properly orient and “link” the enzyme to the material (for example, electrode or light absorber), via electrostatic or covalent interactions to maximise the charge transfer. In this respect, features as the enzyme size (in the nanoscale) and local-specific surface charge/hydrophilicity/hydrophobicity must be taken into consideration when choosing the materials and its functionalisations.
Having these general points tackled, any FDH could be used to build a device to promote the CO2 reduction. The same would be true for whole-cell devices, but considering in that case the organism whole metabolism (carbon and energy needs).
4.5 Formate Dehydrogenases in Action
The use of enzymes and whole-cells systems to convert CO2 into VC is growing exponentially due to the “green” advantages the “biochemical way” can offer, namely substrate and product specificity (ability to discriminate the substrate in a complex mixture and to produce only the product of interest) in reactions at ambient temperature and pressure and neutral pH. Numerous hybrid systems are currently being exploited to convert CO2 into formate, following the same master lines as described in Sect. 3 (Fig. 2). Like electrochemistry, bioelectrochemistry is currently under intense research, as is reviewed in [221,222,223,224,225,226,227] and references herein (below). Most interest is also being focused on the biophotoreduction of CO2, as solar light represents the most straightforward way to use a RES to convert CO2. Semi-artificial photosynthesis systems have been devised, where enzymes and also entire metabolic pathways within cells are interfaced with synthetic materials to develop new solar-to-VC and solar-to-fuel devices, which would not be feasible with natural or artificial systems alone [228 and references herein (below)]. The direct CO2 hydrogenation is also getting enormous attention, mimicking metabolic pathways, where the formate-hydrogen lyase systems are the most explored examples, but using also whole-cells systems. Most important are the breakthroughs achieved by exploiting the recently identified metabolic pathways of acetogens and its dihydrogen-dependent CO2 reductase enzymes (Sect. 4.2.2.), as is reviewed by Litty and Müller in this Book [152] and also [153,154,155,156,157,158,159] and references herein (below).
Herein (below), a few promising studies and successful proof of concepts of FDH-dependent CO2 reduction to formate and beyond are discussed, to highlight the power of FDHs and the challenges this CO2 bioconversion still faces.
One of the most efficient CO2 reducers so far described (along with the T. kivui enzyme described below) is a SeCys–W–FDH from the Synthrobacter fumaroxidans that displays an impressive rate of CO2 reduction of ≈ 2.5 × 103s−1 (reported as 900Umg−1; K CO2m not determined, assays with 10 mM hydrogencarbonate), with a slightly lower formate oxidation rate (≈ 1.9 × 103s−1 (reported as 700Umg−1); K HCOO−m of 40 μM) [229,230,231]. This enzyme is also a good electrocatalyst to carry out the electrochemical reduction of CO2 to formate, using mild conditions and applying small overpotentials, with a maximum current density of ≈ 80 μAcm−2 that corresponds to ≈ 110 s−1 (from a monolayer of enzyme) [232]. Intriguingly, while in homogeneous catalysis in solution the CO2 reduction is slightly faster than the formate oxidation, in the electrochemical-assisted reduction/oxidation is the formate oxidation that is more than 2 times faster (with a current density of ≈ 200 μAcm−2 [232]). S. fumaroxidans expresses another very fast CO2 reducer SeCys-W-FDH, with a rate of ≈ 200 s−1 (reported as 90Umg−1) [229,230,231], but its CO2 reduction activity cannot kinetically compete with its highly efficient formate oxidation, rate of ≈ 5.6 × 103s−1 (value reported as 2700Umg−1) and K HCOO−m of 10 μM. Unfortunately, these enzymes are extremely oxygen-sensitive, and no further studies towards a biotechnological application were pursuit, as far as we know.
Several other FDHs have been described to be able to reduce CO2, but at considerably lower rates. Numerous studies have been conducted with metal-independent FDHs, many of which relying on sacrificial electron donors [233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248]. This is the case of the C. boidinii NAD-dependent metal-independent FDH, that, in spite of its considerably low k HCO3−cat value of only 0.009 s−1 (K HCO3−m ≈ 27.3 mM [240]; kHCO3− ≈ 0.3 M−1s−1; k HCOO−cat ≈ 5.0 s−1; K HCOO−m ≈ 5.0 mM; kHCOO− ≈ 1.0 × 103M-1s-1 [109]), has been largely exploited for its ability to reduce CO2. To push the reaction in the desired, but thermodynamically unfavourable, directionFootnote 14 is important to remove NAD+/regenerate NADH (also essential for the process to become cost-effective, since NADH is a very expensive reducing agent). Four selected examples of different strategies to force the reaction towards the CO2 reduction are: (a) an electroenzymatic cell where NADH is electrochemically regenerated through a rhodium complex, with which a formate formation rate of ≈ 3.2 × 10−4 μmolmin−1mg−1 was achieved [240]; (b) electrochemical NADH regeneration, but with an electropolymerised mediator-regenerator (neutral red) in a novel cathode with immobilised FDH, which is able to produce formate at a rate of ≈ 60 μMmin−1) [249]; (c) photocatalytical NADH regeneration using a rhodium complex and a visible light-active photocatalyst) that enabled a formate formation rate of ≈ 1 μmolmin−1 [250]; (d) and enzymatic regeneration, using glutamate dehydrogenase with NAD(H) being covalently attached to micro-particles, to be easily recovered and reused, in an approach that allowed to improve the reaction yield from 0.12 to 1.27 methanol formed/NADH consumed (in this study, formate was further reduced to methanol) [251]. The Thiobacillus sp KNK65MA NAD-dependent metal-independent FDH exhibit an as well low kcat value (k HCO3−cat ≈ 0.32 s−1; K HCO3−m ≈ 9.2 mM; kHCO3− ≈ 35 M−1s−1), but its specificity for formate is only 3 times superior (k HCOO−cat ≈ 1.8 s−1; K HCOO−m ≈ 16 mM; kHCOO− ≈ 110 M.1s.1) [252]. This Thiobacillus enzyme was successfully used to reduce CO2 by coupling it with a NADH photoelectrochemical regeneration system (Fig. 14), with a formate production rate of 2 μMmin−1 (current density ≈ 3.5mAcm−2) [245].

Reproduced (from Ref. [245]) by permission of the Royal Society of Chemistry. All rights reserved. https://doi.org/10.1039/c6gc02110g
Schematic diagram of enzymatic photosynthesis of formic acid using Thiobacillus FDH coupled with photoelectrochemical regeneration of nicotinamide cofactors. Co-Pi, cobalt phosphate. See text and Ref. [245] for details.
The metal-dependent FDHs display a wide range of CO2 reduction rates. The Clostridium carboxidivorans NAD-dependent SeCys-W-FDH exhibits a considerably low k COcat 2 value (only 0.08 s−1; K HCO3−m ≈ 50 μM) [144, 145, 147]. Nevertheless, it enabled the photoelectrochemical CO2 reduction to formate within an enzyme cascade that led to the methanol production at ≈ 4 μMmin−1) [253]. The Methylobacterium extorquens AM1 NAD-dependent Cys-W-FDH has also been exploited with different approaches to drive the electrochemical CO2 reduction [242, 254,255,256,257,258]. Using mediated enzymatic bioelectrocatalysis with gas diffusion electrodesFootnote 15, current densities of 15–20mAcm−2 were attained [254, 255]; in a whole-cell catalyst, M. extorquens was able to electrochemically produced formate concentrations of up to 60 mM [242].
The R. capsulatus [182] and Cupriavidus oxalaticus [259] NAD-dependent Cys–Mo–FDH enzymes, on the other hand, have k COcat 2 values, of 1.5 s−1 and ≈ 3 s−1, respectively, but ≈ 25 and ≈ 30 times (respectively) lower than the one for formate oxidation (R. capsulatus K HCOO−m ≈ 280 μM and K COm 2 not determined, assays with 100 mM hydrogencarbonate; C. oxalaticus K HCOO−m ≈ 100 μM and K HCO3−m ≈ 40 mM). The C. necator NAD-dependent Cys–Mo–FDH, on the contrary, catalyses the reduction of CO2 with a k COcat 2 ≈ 11 s−1 (K COm 2 ≈ 2.7 mM; K NADHm ≈ 45 μM) [260, 261]. To fulfil the potential industry application of this oxygen-tolerant and robust enzyme, it is necessary to implement a NADH regenerating system that pushes the reaction towards CO2 reduction (as discussed above; see Footnote 14) [262]. With the C. necator FDH, this was successfully achieved with the inclusion of glucose dehydrogenase in the system (Fig. 15), which, while catalysing the re-reduction of NAD+ to NADH, enabled the continuous electron delivery to drive the CO2 reduction and, therefore, improved the reaction yield from 0.2 to 1.8 formate formed/NADH consumed [262].

Schematic diagram of the enzymatic cascade reaction C. necator FDH and glucose dehydrogenase. See text and Ref. [262] for details
The E. coli SeCys-Mo-FDH H was also shown to be able to reduce CO2 [263], but at rates considerably lower than the ones of formate oxidation, < 1 versus 160 s−1 [263]. Interestingly, when the reaction is driven electrochemically (protein film voltammetry), the formate oxidation was only two times higher than the CO2 reduction, with current densities of 180 versus 80 μAcm−2, respectively [263]. This E. coli enzyme feature has been exploited in fuel cell devices (FDH immobilised in redox mediators-functionalised redox polymers) [15, 16], where CO2 could be reduced with a very high Faradaic efficiency (99%) and a current density of ≈ 60 μAcm−2 (K HCO3−m ≈ 2.5 mM) [16]. Thanks to the FDH H-containing formate-hydrogen lyase system (Sect. 4.2.2.)Footnote 16, engineered E. coli whole cells were also used as a “cell factory” to very efficiently produce formate from a gaseous mixture of CO2 and dihydrogen (56:44; up to 10 bar) (Fig. 16); an 100% of CO2 conversion was achieved, with formate (more than 500 mM) being accumulated outside the bacterial cells [264]. Intact E. coli cells were also used in a microbial electrolysis system using an iron-modified carbon cathode, with which a formate production rate of ≈ 10 μMmin−1, with a Faradaic efficiency of ≈ 60%, was attained [265].

Adapted with permission from Ref. [264]. http://creativecommons.org/licenses/by/4.0/
Schematic diagram of a “cell factory” to produce formate using E. coli whole-cells. FHL, formate-hydrogen lyase. See text and Ref. [264] for details
FDHs from sulfate-reducing bacteria constitute other very interesting systems to exploit, exhibiting high rates of CO2 reduction. The D. desulfuricans SeCys–Mo–FDH is a strikingly efficient CO2 reducer. With a \(k_{\text{cat}}^{{{\text{CO}}_{2} }}\) ≈ 50 s−1 and particularly low \(K_{m}^{{{\text{CO}}_{2} }}\) ≈ 15 μM, this enzyme has a superior specificity for CO2 (\(k^{{{\text{CO}}_{2} }}\) ≈ 3.3 × 106M−1s−1) [137]. The high Km value for formate (K HCOO−m ≈ 55 μM; k HCOO−cat ≈ 550 s−1; kHCOO− ≈ 10 × 106M−1s−1) enables D. desulfuricans SeCys–Mo–FDH to be a powerful CO2 reducer, as long as the formate concentration is kept low (is removed from the system). In addition, once the catalysis is initiated (occurring at steady-state rates), this enzyme robustness allows the reaction to fully proceed even in the presence of dioxygen [139]. Moreover, the D. desulfuricans SeCys–Mo–FDH is also a good electrocatalyst (unmediated electrochemistry) to carry out the electrochemical reduction of CO2 with good catalytic currents being attained [266]. The ability of D. desulfuricans to produce formate was also demonstrated in whole-cells catalysis, where the continuous formate production exhibited a maximum specific formate production rate of 14 mM formate/gdcwh, and more than 45 mM of formate were obtained with a production rate of 0.40mMh−1 [267].
The D. vulgaris contains also several interesting FDHs. One SeCys–Mo–FDH is able to catalyse the CO2 reduction at a rate of ≈ 3.4 s−1 (reported as 1Umg−1) [129, 131, 268]. However, its extremely low Km value for formate (K HCOO−m of 8 μM) and higher rate of formate oxidation (k HCOO−cat ≈ 260 s−1) makes this enzyme a very interesting biocatalyst to oxidise formate instead, namely to be coupled to dihydrogen production. The proof of concept that D. vulgaris is able to produce dihydrogen at high volumetric and specific rates (0.125 dm3 H2/dm3h1 and 2.5 dm3 H2/gdcwh) was obtained recently, with the demonstration that whole cells are able to grow by catalysing the oxidation of formate to hydrogencarbonate and dihydrogen, in the absence of sulfate or a syntrophic partner [268, 269].
The D. vulgaris SeCys–W–FDH, on the other hand, is better suited for CO2 reduction, with a k COcat 2 ≈ 315 s−1 (\(K_{m}^{{{\text{CO}}_{2} }}\) ≈ 420 μM; \(k^{{{\text{CO}}_{2} }}\) ≈ 0.75 × 106M−1s−1) [132]. Even though the CO2 specificity of this enzyme is considerably lower (100 times) than that for formate (k HCOO−cat ≈ 1310 s−1; K HCOO−m ≈ 17 μM; kHCOO− ≈ 77.5 × 106M−1s−1 [132]), this D. vulgaris W-FDH is at the base of several very well succeeded proof-of-principle devices for semi-artificial photosynthesis and production/storage of dihydrogen. A D. vulgaris W-FDH-containing cathode wired to a T. elongatus photosystem II-containing photoanode with a synthetic dye with complementary light absorption was successfully employed to drive light-dependent CO2 conversion to formate, using water as an electron donor (Fig. 17) [270]. In this photoelectrochemical tandem device, electrons are photogenerated in the photosystem II, which oxidises water to dioxygen, and transferred to the FDH cathode (this biocathode catalyses the formate formation with a current density of 240 μAcm−2 (at—0.6 V versus SHE) and a Faradaic efficiency of ≈ 80%). The whole system is able to efficiently produce formate at 0.185 μmolcm−2, with Faradaic efficiency of ≈ 70%, but progressive photosystem II photodegradation (due to prolonged irradiation) resulted in an irreversible decrease in the CO2 photoreduction. A different D. vulgaris W-FDH-material configuration was recently devised, based on a ruthenium dye [271]. The employment of this dye-sensitised TiO2-adsorped FDH enable the visible light-driven CO2 reduction to formate with a turnover frequency of 11 s−1, in the absence of a soluble redox mediator (Fig. 18) [271] (comparatively, this bioelectrode reached a current density of 100 μAcm−2 (at −0.6 V versus SHE), with a Faradaic efficiency of 92.5%). Furthermore, the D. vulgaris FDH-mediated electroenzymatic CO2 reduction to formate was attained using a redox viologen-based polymer/enzyme-modified gas diffusion electrode [272]. The D. vulgaris W-FDH has also been exploited to drive the dihydrogen formation/storage in a system that mimics the natural formate-hydrogen lyase systems (see Sect. 4.2.2.) [273]. The semi-artificial formate-hydrogen lyase system consists of the D. vulgaris W-FDH and D. vulgaris Ni/Fe-Hase immobilised on a conductive scaffold of indium tin oxide that acts as an electron relay. This configuration enables the overall reaction to proceed reversibly towards formate conversion into CO2 plus dihydrogen or towards formate formation, with minimal bias in either direction (Fig. 19), thus allowing the longed-for dihydrogen storage and release on demand. The system is able to produce dihydrogen (upon formate addition) at a rate of 4nmolmin−1 (turnover number of 23 × 103 and turnover frequency of 6.4 s−1 for the Hase) or to produce formate (in the presence of dihydrogen) at a rate of 22nmolmin−1 (turnover number of 16 × 103 and turnover frequency of e.4 s−1 for the FDH) for 8 h (this bioelectrode system reached current densities of 185 and 450 μAcm−2, for CO2 and H+ reduction, respectively (at −0.6 V versus SHE) and of 300 and 440 μAcm−2 for formate and H2 oxidation, respectively (at −0.2 V versus SHE), with Faradaic efficiencies for H2 and formate production of 77 and 76%, respectively). Moreover, this semi-artificial formate-hydrogen lyase concept can be deployed in either an electrochemical cell or a self-assembled colloidal suspension, thus providing versatility for applications in different contexts.

Adapted with permission from Ref. [270]
Schematic diagram of a semi-artificial photosynthetic tandem PEC cell coupling water oxidation to CO2 reduction by D. vulgaris FDH. dpp, phosphonated diketopyrrolopyrrole dye, POs, [poly(1-vinylimidazole-coallylamine)-[Os(bipy)2Cl]Cl redox polymer], PS II, photosystem II. See text and Ref. [270] for details

Adapted with permission from Ref. [271]
Schematic diagram of a photocatalyst system for CO2 conversion using a dye-semiconductor-D. vulgaris FDH arrangement. ATR-IR, attenuated total reflection infrared, PFV, protein film voltammetry, QCM, quartz crystal microbalance, TEOA, triethanolamine. See text and Ref. [271] for details

Adapted with permission from Ref. [273]
Schematic diagram of a semi-artificial formate-hydrogen lyase system for the reversible and selective interconversion of dihydrogen and CO2 into formate using D. vulgaris FDH. The concept can be deployed in either an electrochemical cell (top) or a self-assembled colloidal suspension (bottom). H2ase, hydrogenase, ITO. indium tin oxide, NP, nanoparticle. See text and Ref. [273] for details
Acetogens and methanogens are organisms that reduce (fix) CO2 in vivo [274, 275], and, as such, they have been the focus of intense research to develop new CO2 converter devices, enzymatic and whole-cell systems, as is reviewed by Litty and Müller in this Book [152] and also [153,154,155,156,157,158,159]. Herein, we only highlight the dihydrogen-dependent CO2 reductases (Sect. 4.2.2.) from Acetobacterium woodii and T. kivui: the former is a SeCys–Mo–FDH that catalyses the CO2 hydrogenation with a kcat of 28 s−1 (reported as 10Umg−1; K HCO3−m ≈ 37 mM) and displays slightly higher rates of formate oxidation (CO2 plus dihydrogen formation with a kcat ≈ 39 s−1, reported as 14Umg−1; K HCOO−m ≈ 1 mM) [153, 154]; the second is a outstanding Cys–W–FDH that catalyses the CO2 hydrogenation with a kcat of 2.5 × 103s−1 (900 μmol formate min−1mg−1; K H2m ≈ 130 μM—one of the fastest CO2 reducers so far described), with the reverse reaction being catalysed with a kcat of 2.7 × 103s−1 (930 μmol dihydrogen min−1mg−1; K HCOO−m ≈ 550 μM) [156]. The CO2 hydrogenation equilibrium constant close to one (∆G º′ = 3.5KJmol−1) makes these systems ideal biocatalysts for dihydrogen storage and production. A. woodii was successfully used as a whole-cell biocatalyst to produce dihydrogen from formate, reaching a specific dihydrogen formation rate of ≈ 70 mmol g −1protein 1h−1 (≈ 30 mmol g −1cdw h−1) and a volumetric dihydrogen evolution rate of ≈ 80 mMh−1, with yields up to 1 mol dihydrogen per mol formate [155]. T. kivui was successfully exploited in a whole-cell system to convert dihydrogen plus CO2 (hydrogencarbonate) into formate, achieving a specific formate formation rate of ≈ 235 mmol g −1protein 1h−1 (≈ 150 mmol g −1cdw h−1) and a volumetric formate production rate of ≈ 270mMh−1; high titres up to 130 mM of formate were reached, with the key advantage of having the unwanted acetate formation abolished [159].
5 Outlook
The global energy demand and the present high dependence on fossil fuels have caused the increase in the atmospheric CO2 concentration for the highest values since records began. Due to its significant greenhouse effect, CO2 rise is responsible for large and unpredictable impacts on the Earth climate, besides being responsible for ocean acidification (its major sink). While some authors defend that these alterations are no longer reversible, the CO2 emissions must be greatly decelerate and new and more efficient “CO2 sinks” must be developed to avoid worsen this (already huge) “carbon crisis”. The three axes, storage/conversion/production, are envisaged by many authors as the best strategy to actively reduce the CO2 emissions, while actively consuming the CO2 already released—”two-in-one solution”. Along with chemical strategies, the “biochemical way” is proving is high value in different hybrid and biological systems to convert CO2 into fuels and VC. FDHs are efficient catalysts to reduce CO2 to formate and are in the right way to became key partners in the longed-for safe energy/stable climate solution.
Notes
- 1.
pKa1 (formic acid (methanoic acid, HCOOH)/formate) = 3.77.
- 2.
It should be kept in mind that enzymes did not evolve to maximise “our” VC production. Enzymes and all cellular components evolved to achieve sustained life. The statement of “perfect catalysts” must be taken within the respective context.
- 3.
It should be noted that the difference between the two FDHs classes is the absence or presence of redox-active centres. All (so far known) metal-independent FDHs are NAD(P)-dependent. In contrast, there are some metal-dependent FDHs that use NAD(P)+/NAD(P)H as a co-substrate, while many other use other physiological redox partners (such as membrane quinols, cytoplasmatic and periplasmatic cytochromes, ferredoxins or coenzyme F420).
- 4.
Presently, only five FDHs have been structurally characterised: the E. coli Mo-FDHs FDH H [116, 117] and FDH N [119], the D. gigas W-FDH [130], the D. vulgaris W-FDH [132] and the Rhodobacter capsulatus Mo-FDH [138] were crystallographically characterised; the R. capsulatus enzyme structure was also determined by cryo-electron microscopy [139]. In addition, also the crystallographic structure of the tungsten-containing Methanothermobacter wolfeii N-formyl-methanofuran dehydrogenase, a structurally related enzyme (see below), was solved [140].
- 5.
Although initially thought to be an oxygen [116], it is now unambiguously established that this terminal atom it is a sulfur, in both Mo-FDHs and W-FDHs, as well as in FMFDH, as established by X-ray crystallography and XAS [117, 140, 166]. In addition, it was already identified the sulfotransferase that, in conjunction with the IscS cysteine desulfurase, catalyses the insertion of this ligand in the active site [167,168,169].
- 6.
Xanthine oxidase catalyses the hydroxylation of xanthine to urate. To carry out this reaction, xanthine oxidase promotes the cleavage of the C8–H bond of xanthine, with the hydride being transferred from the xanthine moiety to the active site sulfido group (Mo6+=S → Mo4+–SH); simultaneously, the active site catalyses the insertion of an oxygen atom in the xanthine moiety to produce urate (Mo6+–O− → Mo4+). Aldehyde oxidase catalyses the conversion of aldehydes into the respective carboxylates, following the same chemical strategy: cleavage of the C–H bond, with transfer of hydride to the sulfido group, and subsequent insertion of an oxygen atom.
- 7.
Hydroxybenzoyl-CoA reductase catalyses the reverse reaction of the xanthine oxidase one, with insertion of a hydride and abstraction of an oxygen atom.
- 8.
These photolysis assays also demonstrate that the selenocysteine residue is not the formate Cα hydrogen acceptor [166]: while the light beam did not affect the 77Se interaction, it induced the photolysis of the solvent-exchangeable formate-derived proton, showing that the selenocysteine residue does not bind the strongly coupled proton mentioned above.
- 9.
The C. necator periplasmatic nitrate reductase catalyses the reduction of nitrate to nitrite (ONO2− + 2e−+ 2H+ → NO2− + H2O), and it was described to share with FDHs the same molybdenum coordination sphere, with the cis-dithiolene (–S–C = C–S–) group of two pyranopterin cofactor molecules, one terminal sulfido group and one sulfur atom from a cysteine residue [217]. The similarity in the active site metal centre led some authors to suggest similar mechanistic strategies for nitrate reductases and FDHs—leading to the so-called sulfur-shift mechanism [180, 218].
To give further support to the hypothesis of a similar mechanistic strategy, the nitrate reductase activity of different FDHs was investigated. The R. capsulatus Cys-Mo-FDH was described to be able to catalyse the nitrate reduction to nitrite, even though at an extremely low catalytic constant (kcat(nitrate) = 0.21 min−1 [183] that compares very poorly with kcat(formate) = 2124 min−1 and kcat(CO2) = 89 min−1 [182]), and the catalysis was suggested by XAS to involve the dissociation of the cysteine residue from the molybdenum ion, with the subsequent nitrate binding [183]. However, other FDHs failed to reduce nitrate (such as the ones from D. desulfuricans [135, 136], D. vulgaris [132]).
- 10.
The active site conserved arginine residue, below suggested to be key to “anchor” formate and CO2 during turnover, could also be involved in the binding of these inhibitory anions through electrostatic interactions—to have a strong and competitive inhibition of the formate oxidation, it is not necessary that those anions bind directly to the molybdenum/tungsten ion it self.
The observed very weak inhibition of the CO2 reduction by those anions (mentioned below in this point (vii)) could be explained by subtle conformational changes involving the conserved histidine residue upon reduction of the active site. Such conformational changes were described for D. vulgaris FDH [132] and could explain why those inhibitory anions would not be stabilised at the arginine spot within the reduced active site. Therefore, both the formate oxidation (strong inhibition) and the CO2 reduction (weak inhibition) could be inhibited without evoking the direct binding of the inhibitory anions to the metal or the dissociation of the selenocysteine residue.
- 11.
Boyington et al. [116] described the structure of E. coli SeCys-Mo-FDH treated with the inhibitor nitrite as showing the selenocysteine bound to the molybdenum ion and the nitrite molecule with one of its oxygen atoms at 2.58Å from the molybdenum.
- 12.
It should be noted that, xanthine oxidase, for example, that also uses a terminal sulfido group as the hydride acceptor in the conversion of xanthine to urate, has a molybdenum penta-coordinated active site, with no amino acid residues bound to it (the molybdenum ion is coordinated by the cis-dithiolene (–S–C = C–S–) group of one pyranopterin cofactor molecule, the terminal sulfido group plus two oxo groups (see previous Footnotes and references in the text).
- 13.
In vivo, in the great majority of the cases, the reaction is tuned to operate only in one direction; this is determined by the reduction potential of the enzyme redox centres, by the available physiological electron partners and substrates and by the redox status of the subcellular location where the reaction takes place. The few exceptions are mainly related with regulation points of the metabolism.
- 14.
The reduction potential values of the NAD(P)+/NAD(P)H (−0.32 V) and CO2/HCOO− (−0.43 V) pairs indicate that the NADH-dependent CO2 reduction (Eq. 11) is thermodynamically highly unfavourable. To force the reaction towards the CO2 reduction is important to remove the product (NAD+) and maintain (regenerate) the substrate (NADH) concentration.
- 15.
Gas diffusion electrodes (GDE) promote electrochemical reactions between the liquid and the gaseous phase, thus eliminating the limitations arising from slow mass transport when hydrogencarbonate/carbonate is used as CO2 source.
- 16.
Under physiological conditions the E. coli formate-hydrogen lyase system catalyses the formate oxidation to CO2 coupled to the reduction of protons to dihydrogen (Sect. 4.2.2.); yet, under a pressurised (up to 10 bar) atmosphere of CO2 and dihydrogen, engineered E. coli (with abolished “respiratory” FDH, pyruvate-formate lyase and all major hydrogenases) whole cells efficiently catalyse the reverse reaction of formate formation.
Abbreviations
- CCS:
-
Carbon dioxide capture and sequestration
- Cys-Mo-FDH:
-
Cysteine, molybdenum-containing formate dehydrogenase
- Cys-W-FDH:
-
Cysteine, tungsten-containing formate dehydrogenase
- EPR:
-
Electron paramagnetic resonance spectroscopic
- Fe/Fe-Hase:
-
Iron–iron hydrogenase
- Fe/S:
-
Iron–sulfur centres
- FDH:
-
Formate dehydrogenase
- FMFDH:
-
N-formyl-methanofuran dehydrogenase
- GDE:
-
Gas diffusion electrode
- Mo-FDH:
-
Molybdenum-containing formate dehydrogenase
- Ni/Fe-Hase:
-
Nickel/iron-containing hydrogenase
- RES:
-
Renewable energy sources
- SeCys-Mo-FDH:
-
Selenocysteine, molybdenum-containing formate dehydrogenase
- SeCys-W-FDH:
-
Selenocysteine, tungsten-containing formate dehydrogenase
- XAS:
-
X-ray absorption spectroscopy
- VC:
-
Added-value compounds or valuable compounds
- W-FDH:
-
Tungsten-containing formate dehydrogenase
References
Friedlingstein P, Jones MW, O’Sullivan M et al (2019) Global carbon budget 2019. Earth Syst Sci Data 11:1783–1838
CO2 Earth (2020). https://www.co2.earth/monthly-co2. Accessed 15 Apr 2020
Seixas J, Ferreira F (2020) Carbon economy and carbon footprint. In: Moura JJG, Moura I, Maia L (ed) Enzymes for solving humankind’s problems, Springer Nature Switzerland AG (in press
Loges B, Boddien A, Junge H, Beller M (2008) Controlled generation of hydrogen from formic acid amine adducts at room temperature and application in H2/O2 fuel cells. Angew Chem 47:3962–3965
Enthaler S, Langermann J, Schmidt T (2010) Carbon dioxide and formic acid-the couple for environmental-friendly hydrogen storage? Energy Environ Sci 3:1207–1217
Jiang HL, Singh SK, Yan JM, Zhang XB, Xu Q (2010) Liquid-phase chemical hydrogen storage: catalytic hydrogen generation under ambient conditions. Chemsuschem 3:541–549
Yadav M, Xu Q (2012) Liquid-phase chemical hydrogen storage materials. Energy Environ Sci 5:9698–9725
Appel AM, Bercaw JE, Bocarsly AB, Dobbek H, DuBois DL, Dupuis M, Ferry JG, Fujita E, Hille R, Kenis PJA, Kerfeld CA, Morris RH, Peden CHF, Portis AR, Ragsdale SW, Rauchfuss TB, Reek JNH, Seefeldt LC, Thauer RK, Waldrop GL (2013) Frontiers, opportunities, and challenges in biochemical and chemical catalysis of CO2 fixation. Chem Rev 113:6621–6658
Kuehnel MF, Wakerley DW, Orchard KL, Reisner E (2015) Photocatalytic formic acid conversion on cds nanocrystals with controllable selectivity for H2 or CO. Angew Chem 54:9627–9631
Wang W, Himeda Y, Muckerman JT, Manbeck G, Fujita E (2015) CO2 Hydrogenation to formate and methanol as an alternative to photo—and electrochemical CO2 reduction. Chem Rev 115:12936–12973
Preuster P, Papp C, Wasserscheid P (2017) Liquid organic hydrogen carriers (LOHCs): toward a hydrogen-free hydrogen economy. Acc Chem Res 50:74–85
Sordakis K, Tang CH, Vogt LK, Junge H, Dyson PJ, Beller M, Laurenczy G (2018) Homogeneous catalysis for sustainable hydrogen storage in formic acid and alcohols. Chem Rev 118:372–433
Zhong H, Iguchi M, Chatterjee M, Himeda Y, Xu Q, Kawanami H (2018) Formic acid-based liquid organic hydrogen carrier system with heterogeneous catalysts. Adv Sustain Syst 2:1700161
Yu X, Pickup PG (2008) Recent advances in direct formic acid fuel cells (DFAFC). J Power Sources 182:124–132
Sahin S, Cai R, Milton RD, Abdellaoui S, Macazo FC, Minteer SD (2018) Molybdenum-dependent formate dehydrogenase for formate bioelectrocatalysis in a formate/O2 enzymatic fuel cell. J Electrochem Soc 165:109–113
Yuan M, Sahin S, Cai R, Abdellaoui S, Hickey DP, Minteer SD, Milton RD (2018) Creating a low-potential redox polymer for efficient electroenzymatic CO2 reduction. Angew Chem 57:6582–6586
EPFL News (2020). https://actu.epfl.ch/news/the-world-s-first-formic-acid-based-fuel-cell/. Accessed 15 Apr 2020
BBC News (2020). https://www.bbc.com/news/business-40403351. Accessed 15 Apr 2020
Bar-Even A, Noor E, Flamholz A, Milo R (2013) Design and analysis of metabolic pathways supporting formatotrophic growth for electricity-dependent cultivation of microbes. Biochim Biophys Acta 1827:1039–1047
Benson EE, Kubiak CP, Sathrum AJ, Smieja JM (2009) Electrocatalytic and homogeneous approaches to conversion of CO2 to liquid fuels. Chem Soc Rev 38:89–99
DuBois MR, DuBois DL (2009) Development of molecular electrocatalysts for CO2 reduction and H2 production/oxidation. Acc Chem Res 42:1974–1982
Whipple DT, Kenis PJA (2010) Prospects of CO2 utilization via direct heterogeneous electrochemical reduction. J Phys Chem Lett 1:3451–3458
Agarwal AS, Zhai Y, Hill D, Sridhar N (2011) The electrochemical reduction of carbon dioxide to formate/formic acid: engineering and economic feasibility. Chemsuschem 4:1301–1310
Schneider J, Jia HF, Muckerman JT, Fujita E (2012) Thermodynamics and kinetics of CO2, CO, and H + binding to the metal centre of CO2 reduction catalysts. Chem Soc Rev 41:2036–2051
Costentin C, Robert M, Saveant JM (2013) Catalysis of the electrochemical reduction of carbon dioxide. Chem Soc Rev 42:2423–2436
Jhong HR, Ma S, Kenis PJA (2013) Electrochemical conversion of CO2 to useful chemicals: current status, remaining challenges, and future opportunities. Curr Opin Chem Eng 2:191–199
Clark ML, Grice KA, Moore CE, Rheingold AL, Kubiak CP (2014) Electrocatalytic CO2 reduction by M(bpy–R) (CO)4 (M = Mo, W; R = H, tBu) complexes. electrochemical, spectroscopic and computational studies and comparison with group 7 catalysts. Chem Sci 5:1894–1900
Kopljar D, Inan A, Vindayer P, Wagner N, Klemm E (2014) Electrochemical reduction of CO2 to formate at high current density using gas diffusion electrodes. J Appl Electrochem 44:1107–1116
Lu X, Leung DY, Wang H, Leung MK, Xuan J (2014) Electrochemical reduction of carbon dioxide to formic acid. ChemElectroChem 1:836–849
Qiao J, Liu Y, Hong F, Zhang J (2014) A review of catalysts for the electroreduction of carbon dioxide to produce low carbon fuels. Chem Soc Rev 43(2):631–675
Martín AJ, Larrazábal GO, Pérez-Ramírez J (2015) Towards sustainable fuels and chemicals through the electrochemical reduction of CO2: lessons from water electrolysis. Green Chem 17:5114–5130
Pletcher D (2015) The cathodic reduction of carbon dioxide—what can it realistically achieve? a mini review. Electrochem Commun 61:97–101
Yoo JS, Christensen R, Vegge T, Nørskov JK, Studt F (2016) Theoretical Insight into the trends that guide the electrochemical reduction of carbon dioxide to formic acid. Chemsuschem 9:358–363
Morris AJ, Meyer GJ, Fujita E (2009) Molecular approaches to the photocatalytic reduction of carbon dioxide for solar fuels. Acc Chem Res 42:1983–1994
Doherty MD, Grills DC, Muckerman JT, Polyansky DE (2010) Toward more efficient photochemical CO2 reduction: Use of scCO2 or photogenerated hydrides. Coord Chem Rev 254:2472–2482
Takeda H, Ishitani O (2010) Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord Chem Rev 254:346–354
Tamaki Y, Morimoto T, Koike K, Ishitani O (2012) Photocatalytic CO2 reduction with high turnover frequency and selectivity of formic acid formation using Ru(II) multinuclear complexes. Proc Natl Acad Sci U S A 109:15673–15678
Izumi Y (2013) Recent advances in the photocatalytic conversion of carbon dioxide to fuels with water and/or hydrogen using solar energy and beyond. Coord Chem Rev 257:171–186
Navalon S, Dhakshinamoorthy A, Garcia AMH (2013) Photocatalytic CO2 reduction using non-titanium metal oxides and sulfides. Chemsuschem 6:562–577
Oh Y, Hu X (2013) Organic molecules as mediators and catalysts for photocatalytic and electrocatalytic CO2 reduction. Chem Soc Rev 42:2253–2261
Das S, Daud WMAW (2014) Photocatalytic CO2 transformation into fuel: a review on advances in photocatalyst and photoreactor. Renew Sustain Energy Rev 39:765–805
Zhou X, Liu R, Sun K, Chen Y, Verlage E, Francis SA, Lewis NS, Xiang C (2016) Solar-driven reduction of 1 atm of CO2 to formate at 10% energy-conversion efficiency by use of a TiO2-protected III-V tandem photoanode in conjunction with a bipolar membrane and a Pd/C cathode. ACS Energy Lett 1:764–770
Nocera DG (2017) Solar fuels and solar chemicals industry. Acc Chem Res 50:616–619
Zhang B, Sun L (2019) Artificial photosynthesis: opportunities and challenges of molecular catalysts. Chem Soc Rev 48:2216–2264
Graf E, Leitner W (1992) Direct formation of formic-acid from carbon-dioxide and dihydrogen using the [(Rh(Cod)Cl)2]Ph2P-(CH2)4PPh2 catalyst system. J Chem Soc Chem Commun 1992:623–624
Jessop PG, Ikariya T, Noyori R (1994) Homogeneous catalytic-hydrogenation of supercritical carbon-dioxide. Nature 368:231–233
Gassner F, Leitner W (1993) Hydrogenation of carbon-dioxide to formic-acid using water-soluble rhodium catalysts. J Chem Soc Chem Commun 1993:1465–1466
Jessop PG, Ikariya T, Noyori R (1995) Homogeneous hydrogenation of carbon-dioxide. Chem Rev 95:259–272
Leitner W (1995) Carbon dioxide as a raw material: synthesis of formic acid and its derivatives from CO2. Angew Chem 34:2207–2221
Munshi P, Main AD, Linehan JC, Tai CC, Jessop PG (2002) Hydrogenation of carbon dioxide catalyzed by ruthenium trimethylphosphine complexes: the accelerating effect of certain alcohols and amines. J Am Chem Soc 124:7963–7971
Jessop PG, Joó F, Tai CC (2004) Recent advances in the homogeneous hydrogenation of carbon dioxide. Coord Chem Rev 248:2425–2442
Enthaler S (2008) Carbon dioxide—the hydrogen-storage material of the future? Chemsuschem 1:801–804
Fukuzumi S (2008) Bioinspired energy conversion systems for hydrogen production and storage. Eur J Inorg Chem 2008:1351–1362
Federsel C, Jackstell R, Boddien A, Laurenczy G, Beller M (2010) Ruthenium-catalyzed hydrogenation of bicarbonate in water. Chemsuschem 3:1048–1050
Loges B, Boddien A, Gartner F, Junge H, Beller M (2010) Catalytic generation of hydrogen from formic acid and its derivatives: useful hydrogen storage materials. Top Catal 53:902–914
Himeda Y, Miyazawa S, Hirose T (2011) Interconversion between formic acid and H2/CO2 using rhodium and ruthenium catalysts for CO2 fixation and H2 storage. Chemsuschem 4:487–493
Langer R, Diskin-Posner Y, Leitus G, Shimon LJW, Ben-David Y, Milstein D (2011) Low-pressure hydrogenation of carbon dioxide catalyzed by an iron pincer complex exhibiting noble metal activity. Angew Chem 50:9948–9952
Schmeier TJ, Dobereiner GE, Crabtree RH, Hazari N (2011) Secondary coordination sphere interactions facilitate the insertion step in an iridium(iii) CO2 reduction catalyst. J Am Chem Soc 133:9274–9277
Wang W, Wang S, Ma X, Gong J (2011) Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 40:3703–3727
Hull JF, Himeda Y, Wang W-H, Hashiguchi B, Periana R, Szalda DJ, Muckerman JT, Fujita E (2012) Reversible hydrogen storage using CO2 and a proton-switchable iridium catalyst in aqueous media under mild temperatures and pressures. Nat Chem 4:383–388
Maenaka Y, Suenobu T, Fukuzumi S (2012) Catalytic interconversion between hydrogen and formic acid at ambient temperature and pressure. Energy Environ Sci 5:7360–7367
Yadav M, Xu Q (2012) Liquid-phase chemical hydrogen storage materials Energy. Environ Sci 5:9698–9725
Wesselbaum S, Hintermair U, Leitner W (2012) Continuous-flow hydrogenation of carbon dioxide to pure formic acid using an integrated scCO2 process with immobilized catalyst and base. Angew Chem 51:8585–8588
Huff CA, Sanford MS (2013) Catalytic CO2 hydrogenation to formate by a ruthenium pincer complex. ACS Catal 3:2412–2416
Jeletic MS, Mock MT, Appel AM, Linehan JCA (2013) Cobalt-based catalyst for the hydrogenation of CO2 under ambient conditions. J Am Chem Soc 135:11533–11536
Muller K, Sun Y, Thiel WR (2013) Ruthenium(II) phosphite complexes as catalysts for the hydrogenation of carbon dioxide. ChemCatChem 5:1340–1343
Wang W-H, Muckerman JT, Fujita E, Himeda Y (2013) Mechanistic insight through factors controlling effective hydrogenation of CO2 catalyzed by bioinspired proton-responsive iridium(iii) complexes. ACS Catal 3:856–860
Bays JT, Priyadarshani N, Jeletic MS, Hulley EB, Miller DL, Linehan JC, Shaw WJ (2014) The influence of the second and outer coordination spheres on Rh(diphosphine)2 CO2 hydrogenation catalysts. ACS Catal 4:3663–3670
Filonenko GA, van Putten R, Schulpen EN, Hensen EJM, Pidko E (2014) A highly efficient reversible hydrogenation of carbon dioxide to formates using a ruthenium PNP-Pincer catalyst. ChemCatChem 6:1526–1530
Hsu S-F, Rommel S, Eversfield P, Muller K, Klemm E, Thiel WR, Plietker BA (2014) Rechargeable hydrogen battery based on Ru catalysis. Angew Chem 53:7074–7078
Moret S, Dyson PJ, Laurenczy G (2014) Direct synthesis of formic acid from carbon dioxide by hydrogenation in acidic media. Nat Commun 5:5017
Barelli L, Bidini G, Gallorini F, Servili S (2008) Hydrogen production through sorption-enhanced steam methane reforming and membrane technology: a review. Energy 33:554–570
Fihri A, Artero V, Razavet M, Baffert C, Leibl W, Fontecave M (2008) Cobaloxime-based photocatalytic devices for hydrogen production. Angew Chem 47:564–567
Gloaguen F, Rauchfuss TB (2009) Small molecule mimics of hydrogenases: hydrides and redox. Chem Soc Rev 38:100–108
Losse S, Vos JG, Rau S (2010) Catalytic hydrogen production at cobalt centres. Coord Chem Rev 254:2492–2504
Artero V, Chavarot-Kerlidou M, Fontecave M (2011) Splitting water with cobalt. Angew Chem 50:7238–7266
Kilgore UJ, Roberts JAS, Pool DH, Appel AM, Stewart MP, DuBois MR, Dougherty WG, Kassel WS, Bullock RM, DuBois DL (2011) [Ni(PPh2NC6H4X2)2]2 + complexes as electrocatalysts for H2 production: effect of substituents, acids, and water on catalytic rates. J Am Chem Soc 133:5861–5872
Du P, Eisenberg R (2012) Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: recent progress and future challenges. Energy Environ Sci 5:6012–6021
Tran PD, Barber J (2012) Proton reduction to hydrogen in biological and chemical systems. Phys Chem Chem Phys 14:13772–13784
Wang M, Chen L, Sun LC (2012) Recent progress in electrochemical hydrogen production with earth-abundant metal complexes as catalysts. Energy Environ Sci 5:6763–6778
Chen WF, Iyer S, Sasaki K, Wang CH, Zhu YM, Muckerman JT, Fujita E (2013) Biomass-derived electrocatalytic composites for hydrogen evolution. Energy Environ Sci 6:1818–1826
Eckenhoff WT, McNamara WR, Du PW, Eisenberg R (2013) Cobalt complexes as artificial hydrogenases for the reductive side of water splitting. Biochim Biophys Acta—Bioenerg 1827:958–973
Thoi VS, Sun YJ, Long JR, Chang CJ (2013) Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem Soc Rev 42:2388–2400
Faber MS, Jin S (2014) Earth-abundant inorganic electrocatalysts and their nanostructures for energy conversion applications. Energy Environ Sci 7:3519–3542
Liu YM, Yu HT, Quan X, Chen S, Zhao HM, Zhang YB (2014) Efficient and durable hydrogen evolution electrocatalyst based on nonmetallic nitrogen doped hexagonal carbon. Sci Rep 4:6843
McKone JR, Marinescu SC, Brunschwig BS, Winkler JR, Gray HB (2014) Earth-abundant hydrogen evolution electrocatalysts. Chem Sci 5:865–878
Pan LF, Li YH, Yang S, Liu PF, Yu MQ, Yang HG (2014) Molybdenum carbide stabilized on graphene with high electrocatalytic activity for hydrogen evolution reaction. Chem Commun 50:13135–13137
Xiao P, Sk MA, Thia L, Ge XM, Lim RJ, Wang JY, Lim KH, Wang X (2014) Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ Sci 7:2624–2629
Clough AJ, Yoo JW, Mecklenburg MH, Marinescu SC (2015) Two-dimensional metal-organic surfaces for efficient hydrogen evolution from water. J Am Chem Soc 137:118–121
Cui W, Liu Q, Xing ZC, Asiri AM, Alamry KA, Sun X (2015) MoP nanosheets supported on biomass-derived carbon flake: One-step facile preparation and application as a novel high-active electrocatalyst toward hydrogen evolution reaction. Appl Catal B 164:144–150
Morozan A, Goellner V, Zitolo A, Fonda E, Donnadieu B, Jones D, Jaouen F (2015) Synergy between molybdenum nitride and gold leading to platinum-like activity for hydrogen evolution. Phys Chem Chem Phys 17:4047–4053
Darensbourg MY, Eduaran EL, Ding S, Lunsford AL, Pathirana KD, Ghosh P, Yang X (2020) Hydrides, carbonyls, and metal—metal bonds: organometallic chemistry control of hydrogenases. In: Moura JJG, Moura I, Maia L (eds) Enzymes for solving humankind’s problems, Springer Nature Switzerland AG (in press)
Martins M, Pereira IAC, Pita M, De Lacey AL (2020) Biological production of hydrogen. In: Moura JJG, Moura I, Maia L (eds) Enzymes for solving humankind’s problems, Springer Nature Switzerland AG (in press)
Grimaldi S, Schoepp-Cothenet B, Ceccaldi P, Guigliarelli B, Magalon A (2013) The prokaryotic Mo/W-bisPGD enzymes family: a catalytic workhorse in bioenergetic. Biochim Biophys Acta 1827:1048–1085
Hartmann T, Schwanhold N, Leimkühler S (2015) Assembly and catalysis of molybdenum or tungsten-containing formate dehydrogenases from bacteria. Biochim Biophys Acta 1854:1090–1100
Maia LB, Moura JJG, Moura I (2015) Molybdenum and tungsten-dependent formate dehydrogenases. J Biol Inorg Chem 20:287–309
Magalon A, Ceccaldi P, Schoepp-Cothenet B (2017) The Prokaryotic Mo/W-bisPGD Enzymes Family. In: Hille R, Schulzke C, Kirk M (eds) Molybdenum and tungsten enzymes: biochemistry, RSC Metallobiology Series No. 5, The Royal Society of Chemistry, Cambridge, Chap. 5, p 143–191
Maia LB, Moura I, Moura JJG (2017) Molybdenum and tungsten-containing formate dehydrogenases: aiming to inspire a catalyst for carbon dioxide utilization. Inorg Chim Acta 455:350–363
Niks D, Hille R (2018) Reductive activation of CO2 by formate dehydrogenases. Methods Enzymol 613:277–295
Niks D, Hille R (2019) Molybdenum—and tungsten-containing formate dehydrogenases and formylmethanofuran dehydrogenases: structure mechanism, and cofactor insertion. Ptot Sci 28:111–122
Nielsen CF, Lange L, Meyer AS (2019) Classification and enzyme kinetics of formate dehydrogenases for biomanufacturing via CO2 utilization. Biotechnol Adv 37:107408
Kato N (1990) Formate dehydrogenase from methylotrophic yeasts. Methods Enzymol 188:459–462
Vinals C, Depiereux E, Feytmans E (1993) Prediction of structurally conserved regions of D-specific hydroxy acid dehydrogenases by multiple alignment with formate dehydrogenase. Biochem Biophys Res Commun 192:182–188
Popov VO, Lamzin VS (1994) NAD + -dependent formate dehydrogenase. Biochem J 301:625–643
Filippova EV, Polyakov KM, Tikhonova TV, Stekhanova TN, Boiko KM, Popov KO (2005) Structure of a new crystal modification of the bacterial NAD-dependent formate dehydrogenase with a resolution of 2.1 Å. Crystallogr Rep 50:796–800
Schirwitz K, Schmidt A, Lamzin VS (2007) High resolution structures of formate dehydrogenase from Candida boidinii. Protein Sci 16:1146–1156
Shabalin IG, Polyakov KM, Tishkov VI, Popov VO (2009) Atomic resolution crystal structure of NAD(+)-dependent formate dehydrogenase from bacterium moraxella Sp. C-1. Acta Nat 1:89–93
Alekseeva AA, Savin SS, Tishkov VI (2011) NAD (+) -dependent formate dehydrogenase from plants. Acta Nat 3:38–54
Guo Q, Gakhar L, Wickersham K, Francis K, Vardi-Kilshtain A, Major DT, Cheatum CM, Kohen A (2016) Structural and kinetic studies of formate dehydrogenase from candida boidinii. Biochemistry 55:2760–2771
Hille R, Hall J, Basu P (2014) The mononuclear molybdenum enzymes. Chem Rev 114:3963–4038
Maia L, Moura I, Moura JJG (2017) Molybdenum and tungsten-containing enzymes: an overview. In: Hille R, Schulzke C, Kirk M (eds) Molybdenum and Tungsten Enzymes: Biochemistry, RSC Metallobiology Series No. 5, The Royal Society of Chemistry, Cambridge, Chap. 1, p 1–80
Maia L, Moura JJG (2018) Mononuclear molybdenum-containing enzymes. Reference module in chemistry, molecular sciences and chemical engineering, pp 1–19. https://doi.org/10.1016/B978-0-12-409547-2.13932-0
Zinoni F, Birkmann A, Stadtman TC, Böck A (1986) Nucleotide sequence and expression of the selenocysteine-containing polypeptide of formate dehydrogenase (Formate-Hydrogen-Lyase-Linked) from Escherichia coli. Proc Natl Acad Sci USA 83:4650–4654
Axley MJ, Grahame DA, Stadtman TC (1990) Escherichia coli formate-hydrogen Lyase. J Biol Chem 265:18213–18218
Gladyshev VN, Boyington JC, Khangulov SV, Grahame DA, Stadtman TC, Sun PD (1996) Characterization of crystalline formate dehydrogenase h from Escherichia coli. J Biol Chem 271:8095–8100
Boyington JC, Gladyshev VN, Khangulov SV, Stadtman T, Sun PD (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine and an Fe4S4 cluster. Science 275:1305–1308
Raaijmakers HCA, Romão MJ (2006) Formate-reduced E coli formate dehydrogenase H: the reinterpretation of the crystal structure suggests a new reaction mechanism. J Biol Inorg Chem 11:849–854
Jormakka M, Tornroth S, Abramson J, Byrne B, Iwata S (2002) Purification and crystallization of the respiratory complex formate dehydrogenase-N from Escherichia coli. Acta Crystallogr D Biol Crystallogr 58:160–162
Jormakka M, Törnroth S, Byrne B, Iwata S (2002) Molecular basis of proton motive force generation: structure of formate dehydrogenase-N. Science 2295:1863–1868
Wang H, Gunsalus RP (2003) Coordinate regulation of the Escherichia coli formate dehydrogenase fdnGHI and fdhF genes in response to nitrate, nitrite and formate: roles fro NarL and NarP. J Bacteriol 185:5076–5085
Pommier J, Mandrand MA, Holt SE, Boxer DH, Giodano G (1992) A second phenazine methosulphate-linked formate dehydrogenase isoenzyme in Escherichia coli. Biochim Biophys Acta 1107:305–313
Plunkett G, Burland V, Daniels DL, Blattner FR (1993) Analysis of the Escherichia coli genome. Nucleic Acids Res 21:3391–3398
Abaibou H, Pommier J, Benoit JP, Giordano G, Mandrand M (1995) Expression and characterization of the Escherichia coli fdo locus and a possible physiological role for aerobic formate dehydrogenase. J Bacteriol 177:7141–7149
Bursakov S, Liu M-Y, Payne WJ, LeGall J, Moura I, Moura JJG (1995) Isolation and preliminary characterization of a soluble nitrate reductase from the sulfate reducing organism Desulfovibrio desulfuricans ATCC 27774. Anaerobe 1:55–60
Sebban C, Blanchard L, Bruschi M, Guerlesquin F (1995) Purification and characterization of the formate dehydrogenase from Desulfovibrio vulgaris hildenborough. FEMS Microbiol Lett 133:143–149
Brondino CD, Passeggi MCG, Caldeira J, Almendra MJ, Feio MJ, Moura JJG, Moura I (2004) Incorporation of either molybdenum or tungsten into formate dehydrogenase from Desulfovibrio alaskensis NCIMB 13491. J Biol Inorg Chem 9:145–151
Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, Eisen JA, Ward N, Methe B, Brinkac LM, Daugherty SC, Deboy RT, Dodson RJ, Durkin AS, Madupu R, Nelson WC, Sullivan SA, Fouts D, Haft DH, Selengut J, Peterson JD, Davidsen TM, Zafar N, Zhou L, Radune D, Dimitrov G, Hance M, Tran K, Khouri H, Gill J, Utterback TR, Feldblyum TV, Wall JD, Voordouw G, Fraser CM (2004) The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris hildenborough. Nat Biotechnol 22:554–559
Mota CS, Valette O, Gonzalez PJ, Brondino CD, Moura JJG, Moura I, Dolla A, Rivas MG (2011) Effects of molybdate and tungstate on expression levels and biochemical characteristics of formate dehydrogenases produced by Desulfovibrio alaskensis NCIMB 13491. J Bacteriol 193:2917–2923
Silva SM, Pimentel C, Valente FMA, Rodrigues-Pousada C, Pereira IAC (2011) Tungsten and molybdenum regulation of formate dehydrogenase expression in Desulfovibrio vulgaris hildenborough. J Bacteriol 193:2908–2916
Raaijmakers H, Macieira S, Dias JM, Texierira S, Bursakov S, Huber R, Moura JJG, Moura I, Romão MJ (2002) Gene sequence and the 18Å crystal structure of the tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Structure 10:1261–1272
da Silva SM, Voordouw J, Leitão C, Martins M, Voordouw G, Pereira IAC (2013) Function of formate dehydrogenases in Desulfovibrio vulgaris hildenborough energy metabolism. Microbiology 159:1760–1769
Oliveira AR, Mota C, Mourato C, Domingos RM, Santos MFA, Gesto D, Guigliarelli B, Santos-Silva T, Romão MJ, Pereira IAC (2020) Toward the mechanistic understanding of enzymatic CO2 reduction. ACS Catal 10:3844–3856
Almendra MJ, Brondino CD, Gavel O, Pereira AS, Tavares P, Bursakov S, Duarte R, Caldeira J, Moura JJ, Moura I (1999) Purification and characterization of a tungsten-containing formate dehydrogenase from Desulfovibrio gigas. Biochemistry 38:16366–16372
Raaijmakers H, Teixeira S, Dias JM, Almendra MJ, Brondino CD, Moura I, Moura JJ, Romão MJ (2001) Tungsten-containing formate dehydrogenase from Desulfovibrio gigas: metal identification and preliminary structural data by multi-wavelength crystallography. J Biol Inorg Chem 6:398–404
Costa C, Teixeira M, LeGall J, Moura JJG, Moura I (1997) Formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774: isolation and spectroscopic characterization of the active sites. J Biol Inorg Chem 2:198–208
Rivas M, Gonzalez P, Brondino CD, Moura JJG, Moura I (2007) EPR characterization of the molybdenum(V) forms of formate dehydrogenase from Desulfovibrio desulfuricans ATCC 27774 upon formate reduction. J Inorg Biol 101:1617–1622
Maia L, Fonseca L, Moura I, Moura JJG (2016) Reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase: a kinetic and mechanistic study. J Am Chem Soc 138:8834–8846
Young T, Niks D, Hakopian S, Tam TK, Yu X, Hille R, Blaha GM (2020) Crystallographic and kinetic analyses of the FdsBG subcomplex of the cytosolic formate dehydrogenase FdsABG from Cupriavidus necator. J Biol Chem 295:6570–6585
Radon C, Mittelstädt G, Duffus BR, Bürger J, Hartmann T, Mielke T, Teutloff C, Leimkühler S, Wendler P (2020) Cryo-EM structures reveal intricate Fe-S cluster arrangement and charging in Rhodobacter capsulatus formate dehydrogenase. Nat Commun 11:1912
Wagner T, Ermler U, Shima S (2016) The methanogenic CO2 reducing-and-fixing enzyme is bifunctional and contains 46 [4Fe-4S] clusters. Science 354:114–117
Trchounian K, Poladyan A, Vassilian A, Trchounian A (2012) Multiple and reversible hydrogenases for hydrogen production by Escherichia coli. Crit Rev Biochem Mol Biol 47:236–249
Babujee L, Apodaca J, Balakrishnan V, Liss P, Kiley PJ, Charkowski AO, Glasner JD, Perna NT (2012) Evolution of the metabolic and regulatory networks associated with oxygen availability in two phytopathogenic enterobacteria. BMC Genom 13:110
Liou JS-C, Balkwill DL, Drake GR, Tanner RS (2005) Clostridium carboxidivorans Sp. Nov., a solvent-producing clostridium isolated from an agricultural settling lagoon, and reclassification of the acetogen clostridium scatologenes strain SL1 as clostridium drakei Sp Nov. Int J Syst Evol Microbiol 55:2085–2091
Ragsdale SW, Pierce E (2008) Acetogenesis and the wood-Ljungdahl pathway of CO2 fixation. Biochim Biophys Acta 1784:1873–1898
Bruant G, Levesque M-J, Peter C, Guiot SR, Masson L (2010) Genomic analysis of carbon monoxide utilization and butanol production by clostridium carboxidivorans strain P7T. PLoS One 5:e13033
Paul D, Austin FW, Arick T, Bridges SM, Burgess SC, Dandass YS, Lawrence ML (2010) Genome sequence of the solvent-producing bacterium clostridium carboxidivorans strain P7T. J Bacteriol 192:5554–5555
Alissandratos A, Kim HK, Matthews HK, Hennessey JE, Philbrook A, Easton CJ (2013) Clostridium carboxidovorans strain P7T recombinant formate dehydrogenase catalyzes reduction of CO2 to formate. Appl Environ Micorobiol 79:741–744
Bagramyan K, Trchounian A (2003) Structural and functional features of formate hydrogen lyase, an enzyme of mixed-acid fermentation from Escherichia coli. Biochem Moscow 68:1159–1170
McDowall JS, Murphy BJ, Haumann M, Palmer T, Armstrong FA, Sargent F (2014) Bacterial formate hydrogenlyase complex. Proc Natl Acad Sci U S A 111:E3948–E3956
Mcdowall JS, Hjersing MC, Palmer T, Sargent F (2015) Dissection and engineering of the Escherichia coli formate hydrogenlyase complex. FEBS Lett 589:3141–3147
Pinske C, Sargent F (2016) Exploring the directionality of Escherichia coli formate hydrogenlyase: a membrane-bound enzyme capable of fixing carbon dioxide to organic acid. MicrobiologyOpen 5:721–737
Litty D, Müller V (2020) Acetogenic bacteria for biotechnological applications. In: Moura JJG, Moura I, Maia L (eds) Enzymes for solving humankind’s problems, Springer Nature Switzerland AG (in press)
Schuchmann K, Müller V (2013) Direct and reversible hydrogenation of CO2 to formate by a bacterial carbon dioxide reductase. Science 342:1382–1385
Schuchmann K, Vonck J, Müller V (2016) A bacterial hydrogen-dependent CO2 reductase forms filamentous structures. FEBS J 283:1311–1322
Kottenhahn P, Schuchmann K, Müller V (2018) Efficient whole cell biocatalyst for formate-based hydrogen production. Biotechnol Biofuels 11:93
Schwarz FM, Schuchmann K, Müller V (2018) Hydrogenation of CO2 at ambient pressure catalyzed by a highly active thermostable biocatalyst. Biotechnol Biofuels 11:237
Müller V (2019) New horizons in acetogenic conversion of one-carbon substrates and biological hydrogen storage. Trends Biotechnol 37:1344–1354
Schoelmerich MC, Müller V (2019) Energy conservation by a hydrogenase-dependent chemiosmotic mechanism in an ancient metabolic pathway. Proc Natl Acad Sci USA 116:6329–6334
Schwarz FM, Müller V (2020) Whole-cell biocatalysis for hydrogen storage and syngas conversion to formate using a thermophilic acetogen. Biotechnol Biofuels 13:32
Bertram PA, Karrasch M, Schmitz RA, Böcher R, Albracht SPJ, Thauer RK (1994) Formylmethanofuran dehydrogenases from methanogenic Archaea Substrate specificity, EPR properties and reversible inactivation by cyanide of the molybdenum or tungsten iron-sulfur proteins. Eur J Biochem 220:477–484
Hochheimer A, Hedderich R, Thauer RK (1998) The formylmethanofuran dehydrogenase isozymes in Methanobacterium wolfeii and Methanobacterium terhmoautotrophicum: induction of the molybdenum isozyme by molybdate and constitutive synthesis of the tungsten isozyme. Arch Microbiol 170:389–393
Hille R (1996) The mononuclear molybdenum enzymes. Chem Rev 96:27575–27816
Johnson MK, Rees DC, Adams MWW (1996) Tungstoenzymes Chem Rev 96:2817–2839
Bevers LE, Hagedoorn PL, Hagen WR (2009) The bioinorganic chemistry of tungsten. Coord Chem Rev 253:269–290
Hagen WR (2017) Tungsten-containing enzymes. In: Hille R, Schulzke C, Kirk M (eds) Molybdenum and tungsten enzymes: biochemistry, RSC Metallobiology Series No. 5, The Royal Society of Chemistry, Cambridge, Chap 10, pp 313–342
Khangulov SVV, Gladyshev VN, Dismukes GC, Stadtman TC (1998) Selenium-containing formate dehydrogenase H from Escherichia coli: a molybdenum enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry 37:3518–3528
Thome R, Gust A, Toci R, Mendel R, Bittner F, Magalon A, Walburger A (2012) A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. J Biol Chem 287:4671–4678
Arnoux P, Ruppelt C, Oudouhou F, Lavergne J, Siponen MI, Toci R, Mendel RR, Bittner F, Pignol D, Magalon A, Walburger A (2015) Sulphur shuttling across a chaperone during molybdenum cofactor maturation. Nat Commun 6:6148
Schrapers P, Hartmann T, Kositzki R, Dau H, Reshke S, Schulzke C, Leimkühler S, Haumann M (2015) Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from Rhodobacter capsulatus. Inorg Chem 54:3260–3271
Blanchard JS, Cleland WW (1980) Kinetic and chemical mechanisms of yeast formate dehydrogenase. Biochemistry 19:3543–3550
Rotberg NS, Cleland WW (1991) Secondary 15 N isotope effects on the reactions catalyzed by alcohol and formate dehydrogenases. Biochemistry 30:4068–4071
Lamzin VS, Dauter Z, Popov VO, Harutyunyan EH, Wilson KS (1994) High resolution structures of holo and apo formate dehydrogenase. J Mol Biol 236:759–785
Tishkov VI, Matorin AD, Rojkova AM, Fedorchuk VV, Savitsky PA, Dementieva LA, Lamzin VS, Mezentzev AV, Popov VO (1996) Site-directed mutagenesis of the formate dehydrogenase active centre: role of the His332-Gln313 pair in enzyme catalysis. FEBS Lett 390:104–108
Mesentsev AV, Lamzin VS, Tishkov VI, Ustinnikova TB, Popov VO (1997) Effect of pH on kinetic parameters of NAD + -dependent formate dehydrogenase. Biochem J 321:475–480
Tishkov VI, Popov VO (2004) Catalytic mechanism and application of formate dehydrogenase. Biochemistry (Mosc) 69:1252–1267
Castillo R, Oliva M, Marti S, Moliner V (2008) A theoretical study of the catalytic mechanism of formate dehydrogenase. J Phys Chem B 112:10012–10022
Bandaria JN, Cheatum CM, Kohen A (2009) Examination of enzymatic H-tunneling through kinetics and dynamics. J Am Chem Soc 131:10151–10155
Nilov DK, Shabalin IG, Popov VO, Svedas VK (2012) Molecular modeling of formate dehydrogenase: the formation of the Michaelis complex. J Biomol Struct Dyn 30:170–199
Leopoldini M, Chiodo SG, Toscano M, Russo N (2008) Reaction mechanism of molybdoenzyme formate dehydrogenase. Chemistry 14:8674–8681
Mota CS, Rivas MG, Brondino CD, Moura I, Moura JJG, Gonzalez PG, Cerqueira NMFSA (2011) The mechanism of formate oxidation by metal-dependent formate dehydrogenases. J Biol Inorg Chem 16:1255–1268
Tiberti M, Papaleo E, Russo N, Gioia L, Zampella G (2012) Evidence for the formation of a Mo-H intermediate in the catalytic cycle of formate dehydrogenase. Inorg Chem 51:8331–8339
Hartmann T, Leimkuhler S (2013) The oxygen-tolerant and NAD + -dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J 280:6083–6096
Hartmann T, Schrapers P, Utesch T, Nimtz M, Rippers Y, Dau H, Mroginski MA, Haumann M, Leimkühler S (2016) The molybdenum active site of formate dehydrogenase is capable of catalyzing C-H bond cleavage and oxygen atom transfer reactions. Biochemistry 55:2381–2389
Niks D, Duvvuru J, Escalona M, Hille R (2016) Spectroscopic and kinetic properties of the molybdenum-containing, NAD + -dependent formate dehydrogenase from Ralstonia eutropha. J Biol Chem 291:1162–1174
Dong G, Ryde U (2018) Reaction mechanism of formate dehydrogenase studied by computational methods. J Biol Inorg Chem 23:1243–1255
Thauer RK, Kaufer B, Fuchs G (1975) The active species of ‘CO2’ utilized by reduced ferredoxin: CO2 oxidoreductase from clostridium pasteurianum. Eur J Biochem 55:111–117
Arnoux P, Sabaty M, Alric J, Frangioni B, Guigliarelli B, Adriano JM, Pignol D (2003) Structural and redox plasticity in the heterodimeric periplasmic nitrate reductase. Nat Struct Biol 10:928–934
Tanaka R, Yamashita M, Nozaki K (2009) Catalytic hydrogenation of carbon dioxide using Ir(III)-pincer complexes. J Am Chem Soc 131:14168–14169
Ziebart C, Federsel C, Anbarasan P, Jackstell R, Baumann W, Spannenberg A, Beller M (2012) Well-defined iron catalyst for improved hydrogenation of carbon dioxide and bicarbonate. J Am Chem Soc 134:20701–20704
Filonenko GA, Hensen EJM, Pidko EA (2014) Mechanism of CO2 hydrogenation to formates by homogeneous Ru-PNP pincer catalyst: from a theoretical description to performance optimization. Catal Sci Technol 4:3474–3485
Maiti BK, Maia LB, Pal K, Pakhira B, Avilés T, Moura I, Pauleta SR, Nuñez JL, Rizzi AC, Brondino CD, Sarkar S, Moura JJG (2014) One electron reduced square planar bis(benzene-1,2-dithiolato) copper dianionic complex and redox switch by O2/HO−. Inorg Chem 53:12799–12808
Lothrop AP, Snider GW, Flemer S, Ruggles EL, Davidson RS, Lamb AL, Hondal RJ (2014) Compensating for the absence of selenocysteine in high-molecular weight thioredoxin reductases: the electrophilic activation hypothesis. Biochemistry 53:664–674
Massey V, Edmondson D (1970) On the mechanism of inactivation of xanthine oxidase by cyanide. J Biol Chem 245:6595–6598
Edmondson DE, Ballou D, Vanheuvelen A, Palmer G, Massey V (1973) Kinetic studies on the substrate reduction of xanthine oxidase. J Biol Chem 248:6135–6144
Olson JS, Ballou DP, Palmer G, Massey V (1974) The mechanism of action of xanthine oxidase. J Biol Chem 249:4363–4382
Gutteridge S, Tanner SJ, Bray RC (1978) The molybdenum centre of native xanthine oxidase. evidence for proton transfer from substrates to the centre and for existence of an anion-binding site. Biochem J 175:869–878
Gutteridge S, Tanner SJ, Bray RC (1978) Comparison of the molybdenum centres of native and desulpho xanthine oxidase. the nature of the cyanide-labile sulphur atom and the nature of the proton-accepting group. Biochem J 175:887–897
Bray RC, Gutteridge S, Stotter DA, Tanner SJ (1979) The mechanism of xanthine oxidase The relationship between the rapid and very rapid electron-paramagnetic-resonance signals. Biochem J 177:357–360
Coughlan MP, Johnson JL, Rajagopalan KV (1980) Mechanisms of inactivation of molybdoenzymes by cyanide. J Biol Chem 255:2694–2699
Gutteridg S, Bray RC (1980) Oxygen-17 splitting of the very rapid molybdenum(V) e.p.r. signal from xanthine oxidase. rate of exchange with water of the coupled oxygen atom. Biochem J 189:615–623
Malthouse JPG, Gutteridge S, Bray RC (1980) Rapid type 2 molybdenum(V) electron-paramagnetic resonance signals from xanthine oxidase and the structure of the active centre of the enzyme. Biochem J 185:767–770
Xia M, Dempski R, Hille R (1999) The reductive half-reaction of xanthine oxidase Reaction with aldehydes and identification of the catalytically labile oxygen. J Biol Chem 274:3323–3330
Boll M (2005) Key enzymes in the anaerobic aromatic metabolism catalysing birch-like reductions. Biochim Biophys Acta 1707:34–50
Johannes J, Unciuleac M, Friedrich T, Warkentin E, Ermler U, Boll M (2008) Inhibitors of the molybdenum cofactor containing 4-hydroxybenzoyl-CoA reductase. Biochemistry 47:4964–4972
Stiefel EI (1973) Proposed molecular mechanism for the action of molybedenum in enzymes: coupled proton and electron transfer. Proc Natl Acad Sci USA 70:988–992
Stiefel EI (1977) The coordination and bioinorganic chemistry of molybdenum Prog. Inorg Chem 22:1–223
Rajapakshe A, Snyder RA, Astashkin AV, Bernardson P, Evans DJ, Young CG, Evans DH, Enemark JH (2009) Insights into the nature of Mo(V) species in solution: modeling catalytic cycles for molybdenum enzymes. Inorg Chim Acta 362:4603–4608
Maia L, Moura I, Moura JJG (2017) EPR spectroscopy on mononuclear molybdenum-containing enzymes. In: Hanson G, Berliner LJ (eds) Future directions in metalloprotein and metalloenzyme research, Biological Magnetic Resonance, vol 33. Springer International Publishing, Cham, Chap 4, pp 55–101
Wilson GL, Greenwood RJ, Pilbrow JR, Spence JT, Wedd AG (1991) Molybdenum(V) sites in xanthine oxidase and relevant analogue complexes: comparison of molybdenum-95 and sulfur-33 coupling. J Am Chem Soc 113:6803–6812
Duffus BR, Schrapers P, Schuth N, Mebs S, Dau H, Leimkühler S, Haumann M (2020) Anion binding and oxidative modification at the molybdenum cofactor of formate dehydrogenase from Rhodobacter capsulatus studied by X-ray absorption spectroscopy. Inorg Chem 59:214–225
Barber MJ, May HD, Ferry JG (1986) Inactivation of formate dehydrogenase from methanobacterium formicicum by cyanide? Biochemistry 25(1986):8150–8155
Friedebold J, Bowien B (1993) Physiological and biochemical characterization of the soluble formate dehydrogenase, a molybdoenzyme from Alcaligenes eutrophus. J Bacteriol 175:4719–4728
George GN, Colangelo CM, Dong J, Scott RA, Khangulov SV, Gladyshev VN, Stadtman TC (1998) X-ray absorption spectroscopy of the molybdenum site of Escherichia coli formate dehydrogenase. J Am Chem Soc 120:1267–1273
George GN, Costa C, Moura JJG, Moura I (1999) Observation of ligand-based redox chemistry at the active site of a molybdenum enzyme. J Am Chem Soc 121:2625–2626
Mota CS (2011) Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Portugal, PhD Thesis. https://hdl.handle.net/10362/6688
Axley MJ, Böck A, Stadtman TC (1991) Catalytic properties of an Escherichia coli formate dehydrogenase in which sulfur replaces selenium. Proc Natl Acad Sci USA 88:8450–8454
Coelho C, Gonzalez PJ, Moura JG, Moura I, Trincao J, Joao Romao M (2011) The crystal structure of Cupriavidus necator nitrate reductase in oxidized and partially reduced states. J Mol Biol 408:932–948
Cerqueira NMFSA, Fernandes PA, Gonsalez PJ, Moura JJG, Ramos MJ (2013) The sulfur shift: an activation mechanism for periplasmic nitrate reductase and formate dehydrogenase. Inorg Chem 52:10766–10772
Robinson WE, Bassegoda A, Reisner E, Hirst J (2017) Oxidation-state-dependent binding properties of the active site in a mo-containing formate dehydrogenase. J Am Chem Soc 139:9927–9936
Cornish-Bowden A (1995) Fundamentals of enzyme kinetics. Portland Press, London
Silveira CM, Almeida MG (2013) Small electron-transfer proteins as mediators in enzymatic electrochemical biosensors. Anal Bioanal Chem 405:3619–3635
Armstrong FA, Hirst J (2011) Reversibility and efficiency in electrocatalytic energy conversion and lessons from enzymes. Proc Natl Acad Sci U S A 108:14049–14054
Fourmond V, Léger C (2016) Protein electrochemistry: questions and answers. In: Jeuken L (ed) Biophotoelectrochemistry: from bioelectrochemistry to biophotovoltaics. Advances in biochemical engineering/biotechnology, vol 158. Springer, Cham
Fourmond V, Léger C (2017) Modelling the voltammetry of adsorbed enzymes and molecular catalysts. Curr Opin Electrochem 1:110–120
Fourmond V (2017) Direct Electrochemistry of molybdenum and tungsten enzymes. Reference module in chemistry, molecular sciences and chemical engineering, pp 477–488 https://doi.org/10.1016/B978-0-12-409547-2.13354-2
Li H, Buesen D, Dementin S, Léger C, Fourmond V, Plumeré N (2019) Complete protection of O2-sensitive catalysts in thin films. J Am Chem Soc 141:16734–16742
Fourmond V, Wiedner ES, Shaw WJ, Léger C (2019) Understanding and design of bidirectional and reversible catalysts of multielectron, multistep reactions. J Am ChemSoc 141:11269–11285
Kornienko N, Zhang JZ, Sakimoto KK, Yang P, Reisner E (2018) Semi-artificial photosynthesis: interfacing nature’s catalytic machinery with synthetic materials. Nat Nanotechnol 13:890
Bok FA, Hagedoorn PL, Silva PJ, Hagen WR, Schiltz E, Fritsche K, Stams AJ (2003) Two W-containing Formate Dehydrogenases (CO2-reductases) involved in syntrophic propionate oxidation by syntrophobacter fumaroxidans. Eur J Biochem 270:2476–2485
Bok FA, Hagedoorn PL, Silva PJ, Hagen WR, Schiltz E, Fritsche K, Stams AJ (2003) Two W-containing formate dehydrogenases (CO2-reductases) involved in syntrophic propionate oxidation by syntrophobacter fumaroxidans. FEBS J 270:2476–2485
Worm P, Stams AJ, Cheng X, Plugge CM (2011) Growth- and substrate-dependent transcription of formate dehydrogenase and hydrogenase coding genes in Syntrophobacter fumaroxidans and Methanospirillum hungatei. Microbiology 157:280–289
Reda T, Plugge CM, Abram NJ, Hirst J (2008) Reversible interconversion of carbon dioxide and formate by an electroactive enzyme. Proc Natl Acad Sci USA 105:10654–10658
Kuwabata S, Tsuda R, Nishida K, Yoneyama H (1993) Electrochemical conversion of carbon dioxide to methanol with use of enzymes as biocatalysts. Chem Lett 22:1631–1634
Kuwabata S, Tsuda R, Yoneyama H (1994) Electrochemical conversion of carbon dioxide to methanol with the assistance of formate dehydrogenase and methanol dehydrogenase as biocatalysts. J Am Chem Soc 116:5437–5443
Miyatani R, Amao Y (2002) Bio-CO2 fixation with formate dehydrogenase from saccharomyces cerevisiae and water-soluble zinc porphyrin by visible light. Biotechnol Lett 24:1931–1934
Tsujisho I, Toyoda M, Amao Y (2006) Photochemical and enzymatic synthesis of formic acid from CO2 with chlorophyll and dehydrogenase. System Catal Commun 7:173–176
Ihara M, Kawano Y, Urano M, Okabe A (2013) Light driven CO2 fixation by using cyanobacterial photosystem I and NADPH-dependent formate dehydrogenase. PLoS ONE 8:e71581
Amao Y, Shuto N (2014) Formate dehydrogenase-viologen-Immobilized Electrode for CO2 conversion, for development of an artificial photosynthesis system. Res Chem Intermed 40:3267–3276
Amao Y, Takahara S, Sakai Y (2014) Visible-light induced hydrogen and formic acid production from biomass and carbon dioxide with enzymatic and artificial photosynthesis system. Int J Hydrogen Energy 39:20771–20776
Kim S, Kim MK, Lee SH, Yoon S, Jung KD (2014) Conversion of CO2 to formate in an electroenzymatic cell using candida boidinii formate dehydrogenase. J Mol Catal B Enzym 102:9–15
Srikanth S, Maesen M, Dominguez-Benetton X, Vanbroekhoven K, Pant D (2014) Enzymatic electrosynthesis of formate through CO2 sequestration/reduction in a bioelectrochemical system (BES). Bioresour Technol 165:350–354
Hwang H, Yeon YJ, Lee S, Choe H, Jang MG, Cho DH, Park S, Kim YH (2015) Electro-biocatalytic production of formate from carbon dioxide using an oxygen-stable whole cell biocatalyst. Bioresour Technol 185:35–39
Kim SH, Chung GY, Kim SH, Vinothkumar G, Yoon SH, Jung KD (2016) Electrochemical NADH regeneration and electroenzymatic CO2 reduction on cu nanorods/glassy carbon electrode prepared by cyclic deposition. Electrochim Acta 210:837–845
Lee SY, Lim SY, Seo D, Lee JY, Chung TD (2016) Light-driven highly selective conversion of CO2 to formate by electrosynthesized enzyme/cofactor thin film electrode. Adv Energy Mater 6:1502207
Nam DH, Kuk SK, Choe H, Lee S, Ko JW, Son EJ, Choi EG, Kim YH, Park CB (2016) Enzymatic photosynthesis of formate from carbon dioxide coupled with highly efficient photoelectrochemical regeneration of nicotinamide cofactors. Green Chem 18:5989–5993
Schlager S, Dumitru LM, Haberbauer M, Fuchsbauer A, Neugebauer H, Hiemetsberger D, Wagner A, Portenkirchner E, Sariciftci NS (2016) Electrochemical reduction of carbon dioxide to methanol by direct injection of electrons into immobilized enzymes on a modified electrode. Chemsuschem 9:631–635
Ikeyama S, Amao Y (2017) A Novel electron carrier molecule based on a viologen derivative for visible light-driven CO2 reduction to formic acid with the system of zinc porphyrin and formate dehydrogenase. Sustain Energy Fuels 1:1730–1733
Noji T, Jin T, Nango M, Kamiya N, Amao Y (2017) CO2 photoreduction by formate dehydrogenase and a Ru-complex in a nanoporous glass reactor. ACS Appl Mater Interfaces 9:3260–3265
Zhang L, Liu J, Ong J, Li SFY (2016) Specific and sustainable bioelectro-reduction of carbon dioxide to formate on a novel enzymatic cathode. Chemosphere 162:228–234
Yadav RK, Baeg J-O, Oh GH, Park N-J, Kong K-J, Kim J, Hwang DW, Biswas SK (2012) A photocatalyst-enzyme coupled artificial photosynthesis system for solar energy in production of formic acid from CO2. J Am Chem Soc 134:11455–11461
El-Zahab B, Donnelly D, Wang P (2008) Particle-tethered NADH for production of methanol from CO2 catalyzed by coimmobilized enzymes. Biotechnol Bioeng 99:508–514
Choe H, Joo JC, Cho DH, Kim MH, Lee SH, Jung KD, Kim YH (2014) Efficient CO2-reducing activity of NAD-dependent formate dehydrogenase from thiobacillus sp KNK65MA for formate production from CO2 gas. PLoS ONE 9:e103111
Kuk SK, Singh RK, Nam DH, Singh R, Lee JK, Park CB (2017) Photoelectrochemical reduction of carbon dioxide to methanol through a highly efficient enzyme cascade. Angew Chem 56:3827–3832
Sakai K, Kitazumi Y, Shirai O, Kano K (2016) Bioelectrocatalytic formate oxidation and carbon dioxide reduction at high current density and low overpotential with tungsten-containing formate dehydrogenase and mediators. Electrochem Commun 65:31–34
Sakai K, Kitazumi Y, Shirai O, Takagi K, Kano K (2016) Efficient bioelectrocatalytic CO2 reduction on gas-diffusion-type biocathode with tungsten-containing formate dehydrogenase. Electrochem Commun 73:85–88
Sakai K, Kitazumi Y, Shirai O, Takagi K, Kano K (2017) direct electron transfer-type four-way bioelectrocatalysis of CO2/ formate and NAD +/NADH redox couples by tungsten-containing formate dehydrogenase adsorbed on gold nanoparticle-embedded mesoporous carbon electrodes modified with 4-Mercaptopyridine. Electrochem Commun 84:75–79
Sakai K, Sugimoto Y, Kitazumi Y, Shirai O, Takagi K, Kano K (2017) Direct electron transfer-type bioelectrocatalytic interconversion of carbon dioxide/formate and NAD +/NADH redox couples with tungsten-containing formate dehydrogenase. Electrochim Acta 228:537–544
Sakai K, Xia H, Kitazumi Y, Shirai O, Kano K (2018) Assembly of direct-electron-transfer-type bioelectrodes with high performance. Electrochim Acta 271:305–311
Muller U, Willnow P, Ruschig U, Hopner T (1978) Formate dehydrogenase from Pseudomonas oxalaticus. Eur J Biochem 83:485–498
Yu X, Niks D, Mulchandani A, Hille R (2017) Efficient reduction of CO2 by the molybdenum-containing formate dehydrogenase from Cupriavidus necator (Ralstonia eutropha). J Biol Chem 292:16872–16879
Walker LM, Li V, Niks D, Hille R, Elliott SJ (2019) Deconvolution of reduction potentials of formate dehydrogenase from Cupriavidus necator. J Biol Inorg Chem 24:889–898
Yu X, Niks D, Ge X, Liu H, Hille R, Mulchandani A (2019) Synthesis of formate from CO2 gas catalyzed by an O2-tolerant NAD-dependent formate dehydrogenase and glucose dehydrogenase. Biochemistry 58:1861–1868
Bassegoda A, Madden C, Wakerley DW, Reisner E, Hirst J (2014) Reversible interconversion of CO2 and formate by a molybdenum-containing formate dehydrogenase. J Am Chem Soc 136:15473–15476
Roger M, Brown F, Gabrielli W, Sargent F (2018) Efficient hydrogen-dependent carbon dioxide reduction by Escherichia coli. Curr Biol 28:140–145
Singh S, Noori MT, Verm N (2020) Efficient bio-electroreduction of CO2 to formate on a iron phthalocyanine-dispersed CDC in microbial electrolysis system. ElectrochimActa 338:135887
Cordas CM, Campaniço M, Baptista R, Maia LB, Moura I, Moura JJG (2019) Direct electrochemical reduction of carbon dioxide by a molybdenum-containing formate dehydrogenase. J Inorg Biochem 196:110694
Mourato C, Martins M, da Silva SM, Pereira IA (2017) A continuous system for biocatalytic hydrogenation of CO2 to Formate. Bioresour Technol 235:149–156
Martins M, Mourato C, Pereira IAC (2015) Desulfovibrio vulgaris growth coupled to formate-driven H2 production. Environ Sci Technol 49:14655–14662
Martins M, Mourato C, Morais-Silva FO, Rodrigues- Pousada C, Voordouw G, Wall JD, Pereira IAC (2016) Electron transfer pathways of formate-driven H2 production in Desulfovibrio. Appl Microbiol Biotechnol 100:8135–8146
Sokol KP, Robinson WE, Oliveira AO, Warnan J, Nowaczyk MM, Ruff A, Pereira IAC, Reisner E (2018) Photoreduction of CO2 with a formate dehydrogenase driven by photosystem II using a semi-artificial Z-scheme architecture. J Am Chem Soc 140:16418–16422
Miller M, Robinson WR, Oliveira AR, Heidary N, Kornienko N, Warnan J, Pereira IAC, Reisner E (2019) Interfacing formate dehydrogenase with metal oxides for the reversible electrocatalysis and solar-driven reduction of carbon dioxide. Angew Chem 58:4601–4605
Szczesny J, Ruff A, Oliveira AR, Pita M, Pereira IAC, De Lacey AL, Schuhmann W (2020) Electroenzymatic CO2 fixation using redox polymer/enzyme-modified gas diffusion electrodes. ACS Energy Lett 5:321–327
Sokol KP, Robinson WE, Oliveira AR, Zacarias S, Lee C-Y, Madden C, Bassegoda A, Hirst J, Pereira IAC, Reisner E (2019) Reversible and selective interconversion of hydrogen and carbon dioxide into formate by a semiartificial formate hydrogenlyase mimic. J Chem Am Soc 141:17498–17502
Berg IA, Kockelkorn D, Ramos-Vera WH, Say RF, Zarzycki J, Hügler M (2010) Autotrophic carbon fixation in archaea. Nat Rev Microbiol 8:447–460
Martin WF, Thauer RK (2017) Energy in ancient metabolism. Cell 168:953–955
Acknowledgements
This work was supported by the Associate Laboratory for Green Chemistry—LAQV, which is financed by national funds from Fundacão para a Ciência e a Tecnologia, MCTES (FCT/MCTES; UIDB/50006/2020). LBM thanks to FCT/MCTES for the CEEC-Individual 2017 Program Contract.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, duplication, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, a link is provided to the Creative Commons license and any changes made are indicated.
The images or other third party material in this chapter are included in the work's Creative Commons license, unless indicated otherwise in the credit line; if such material is not included in the work's Creative Commons license and the respective action is not permitted by statutory regulation, users will need to obtain permission from the license holder to duplicate, adapt or reproduce the material.
Copyright information
© 2021 The Author(s)
About this chapter

Cite this chapter
Maia, L.B., Moura, I., Moura, J.J.G. (2021). Carbon Dioxide Utilisation—The Formate Route. In: Moura, J.J.G., Moura, I., Maia, L.B. (eds) Enzymes for Solving Humankind's Problems. Springer, Cham. https://doi.org/10.1007/978-3-030-58315-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-030-58315-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-58314-9
Online ISBN: 978-3-030-58315-6
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)