
Abstract
Despite effective therapeutic and preventive strategies, atherosclerosis and its complications still represent a substantial health burden. Leukocytes and inflammatory mechanisms are increasingly recognized as drivers of atherosclerosis. Neutrophil granulocytes within the circulation were recently shown to undergo neutrophil extracellular trap (NET) formation, linking innate immunity with acute complications of atherosclerosis. In this chapter, we summarize mechanisms of NET formation, evidence for their involvement in atherosclerosis and thrombosis, and potential therapeutic regimens specifically targeting NET components.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Atherosclerosis
- Cl-amidine
- Deoxyribonuclease
- Heparin
- Myocardial infarction
- Neutrophil extracellular traps
- Neutrophils
- Thrombosis
1 Introduction
Atherosclerosis accounts for a substantial global disease burden. In recent decades, significant progress in understanding atherosclerosis was made. The identification of modifiable risk factors (Catapano et al. 2016), e.g. arterial hypertension or hypercholesterolemia, and the advent of percutaneous primary coronary intervention for treating acute complications of atherosclerosis like acute myocardial infarction (MI) (Beran et al. 2002) have led to a further reduction in incidence, cardiovascular morbidity, and markedly improved prognosis (Herrington et al. 2016). Still, due to the growing global population, the absolute number of deaths attributed to atherosclerotic disease is rising (Barquera et al. 2015) and increasing the need for novel therapies.
Besides traditional risk factors, inflammation and leukocytes are increasingly recognized as contributors to vascular disease and its complications. Pathomechanisms are manifold, with T cells (Bullenkamp et al. 2016), B cells (Sage et al. 2019), dendritic cells (Gil-Pulido and Zernecke 2017), monocytes, and macrophages (Moroni et al. 2019) being implicated. Recently, neutrophils were suggested to significantly promote atherosclerosis (Doring et al. 2015), particularly its acute vascular syndromes (Mangold et al. 2015). Upon activation, neutrophils are able to undergo drastic morphological changes, leading to cellular disintegration and release of intracellular content into the extracellular space, in a process called neutrophil extracellular trap (NET) formation (Brinkmann et al. 2004). Besides atherosclerosis (Doring et al. 2017), NETs have emerged as important drivers of disease (Papayannopoulos 2018), including auto-immunity, sepsis, and cancer.
In this chapter, we aim to give a broad overview of the role of vascular NETs in atherosclerotic disease and its specific manifestations. Finally, we discuss potential therapeutic regimens targeting NETs and their components.
2 Neutrophils and Neutrophil Extracellular Traps
Under physiological conditions, neutrophil granulocytes comprise approximately 60% of total leukocytes in humans (Bainton et al. 1971). Neutrophils have a rather short lifespan and are released from the bone marrow into the circulation (Ley et al. 2018). Neutrophils are crucial fighters of the innate immune system, being the first cell population recruited to sites of inflammation and injury (Distelmaier et al. 2014). Neutrophils are critical for host defense (Wang and Arase 2014).
Their abundance and cellular properties enable them to effectively fight pathogens by phagocytosis, degranulation, or cytokine secretion (Witko-Sarsat et al. 2000). Neutrophils are equipped with numerous types of granules (Cowland and Borregaard 2016) containing catalytic enzymes, such as myeloperoxidase (MPO) and serine proteases. Upon phagocytosis of pathogens, neutrophils can fuse their granules with phagolysosomes, resulting in intracellular degradation of pathogens. Alternatively, neutrophils can release their granular content into the extracellular space (Cowland and Borregaard 2016).
Another effector mechanism of neutrophils was identified only 15 years ago, although first evidence had already emerged in 1996: It was reported that phorbol myristate acetate (PMA) could rapidly induce cell death of neutrophils. Thereby, neutrophils underwent substantial morphological changes different from apoptosis or necrosis, including signs of nuclear decondensation (Takei et al. 1996). Extending this finding, the group of Arturo Zychlinsky demonstrated that not only PMA, but also lipopolysaccharide (LPS) or interleukin (IL)-8 led to the appearance of extracellular structures described as fragile fibers of decondensed DNA covered in histones and granule proteins. Gram-positive as well as gram-negative bacteria could be ensnared by these NETs, and presence of neutrophil elastase (NE) or MPO promoted degradation of important bacterial virulence factors (Brinkmann et al. 2004).
Using live-cell imaging, NET formation was found to be an active process, different from apoptosis and necrosis. After stimulation, isolated neutrophils first flattened, forming intracellular vacuoles, followed by loss of the nuclear lobular shape and further expanding in the cytoplasm. In this stage, neutrophils were still viable, containing the vital dye calcein blue but were not positive for Annexin V. This was reversed upon ultimate rupture of the plasma membrane. Furthermore, and in contrast to apoptosis, chromatin was decondensated. Intracellular membranes were fragmented, enabling mixing of nuclear, granular, and cytoplasmic components, which was not a feature of necrosis (Fuchs et al. 2007).
An important pre-requisite for NET formation is the presence of reactive oxygen species (ROS) (Fuchs et al. 2007; Hakkim et al. 2011). Inhibiting ROS production diminished NET formation, and neutrophils isolated from chronic granulomatous disease patients carrying mutations in the phagocyte NADPH oxidase did not produce NETs in response to PMA. However, when hydrogen peroxide was added to the system, the ability to form NETs was restored. These experiments indicated that NETosis is dependent on assembly and activation of the NADPH oxidase (Fuchs et al. 2007) and can be triggered by protein kinase C signaling via the raf-MEK-ERK pathway (Hakkim et al. 2011).
ROS serve as substrate for MPO, but presence of both is an essential stimulus for activation of NE (Metzler et al. 2014). Upon nuclear translocation, NE degrades core histones, facilitating decondensation of chromatin in synergy with histone citrullination by the calcium-dependent enzyme peptidyl arginine deiminase 4 (PAD-4) (Wang et al. 2004). Subsequent disintegration of intracellular membranes enables adsorption of granular proteases and antimicrobials onto chromatin. Rupture of the outer cell membrane finally leads to expulsion of cellular meshwork resulting in formation of NETs (Remijsen et al. 2011). Many of these released NET-associated proteins were shown to be degraded by neutrophil proteases in vitro, probably reducing their capacity to act as autoantigens in vivo (de Bont et al. 2020).
Since their discovery, pathways of NET formation are under thorough debate. Numerous triggers were reported; however, key events essential for NETosis could hardly be connected by signaling molecules to describe defined intracellular cascades. Even the absolute necessity of PAD-4 activity for NET formation is in question. Knockout or inhibition of PAD-4 was, on the one hand, reported to disrupt mouse and human NET formation (Lewis et al. 2015; Li et al. 2010; Martinod et al. 2013) while, on the other hand, other groups still observed NETs in response to the same stimuli independent of PAD-4 activity (Claushuis et al. 2018; Kenny et al. 2017). These conflicting data highlight the problems regarding different methods and also different interpretations of results. Nevertheless, citrullinated histone H3 (citH3) is still considered the most specific marker for NET formation. Differentiation between “vital” and “suicidal” NETosis even questions terminology itself by indicating that NET formation does not have to result in immediate cell death (Desai et al. 2016; Madhusoodanan 2017). Despite uncertainty regarding NET formation, it is increasingly recognized that presence of NETs fundamentally influences disease, including atherosclerosis and thrombosis.
Recently, the exclusivity of extracellular trap (ET) formation to neutrophils came into debate. Among granulocytes, mast cells (Campillo-Navarro et al. 2017) and eosinophils (Mukherjee et al. 2018) were reported capable of forming ETs. Another group even found monocytes to release ETs containing myeloperoxidase and citH3 (Granger et al. 2017). Furthermore, evidence implicated macrophages to equally expel their intracellular content in a process of ET formation (Doster et al. 2018). However, the significance of these non-neutrophil-associated ETs so far remains incompletely understood.
3 NETs in Venous Thrombosis
Deep vein thrombosis (DVT) and its major complications are prevalent in Europe and associated with high morbidity and mortality. Virchow’s triad (Kumar et al. 2010) serves as an excellent framework for understanding risk factors of thrombosis, which are hypercoagulability, vascular dysfunction, and stasis. Recently, however, it was proposed to extend this triad to a tetrad, taking into account the paramount influence of the immune system and its dysregulation on thrombosis (Kapoor et al. 2018). In a rat model of inferior vena cava ligation, pro-inflammatory neutrophils were observed in emerging thrombi and vein walls (Wakefield et al. 1995). In humans, the pro-inflammatory markers interleukin (IL)-6 and C-reactive protein in plasma were increased in DVT and gradually declined after disease onset (Roumen-Klappe et al. 2002), while C-reactive protein predicted post-thrombotic syndrome (Roumen-Klappe et al. 2009).
The potential role of NETs in venous thrombosis was indicated by a flow chamber experiment, where NETs provided a fibrous scaffold for fibrin, von Willebrand factor (vWF) and platelets. NETs were then observed in thrombi of baboons subjected to experimental DVT (Fuchs et al. 2010). In a mouse model of DVT, large amounts of DNA were observed in thrombi, forming NET-like structures (von Bruhl et al. 2012). These observations were complemented by another group showing that fresh parts of thrombi were rich in the NET-specific marker citH3, which co-localized with vWF (Brill et al. 2012).
The first observation of NETs in human venous thrombosis was presented in a case report of a patient suffering from microscopic polyangiitis and DVT: both in kidney and thrombus samples, NETs were abundantly present (Nakazawa et al. 2012). Characterizing thrombi based on histological methods, DNA webs and citH3 were concentrated in organizing sections of thrombi, but not in already organized parts (Savchenko et al. 2014). Extending these findings, plasma DNA levels were found increased, diagnosing DVT with a sensitivity of 81%. Also, thrombus DNA positively correlated with D-dimer, vWF activity, the clinical Wells score, and neutrophil-derived MPO (Diaz et al. 2013). Similar results were obtained with concentrations of nucleosomes, which were elevated in DVT patients and positively correlated with neutrophil activation (van Montfoort et al. 2013).
Venous thromboembolism, mainly presenting as pulmonary embolism (PE), is a major complication of DVT (Di Nisio et al. 2016). Levels of nuclear DNA were shown to be elevated in PE (Arnalich et al. 2013) and independently predictive of mortality (Jimenez-Alcazar et al. 2018). In chronic thromboembolic pulmonary hypertension, a long-term sequela of PE (Lang 2004) characterized by the apposition of non-resolving, organized clots (Galie et al. 2016), neutrophils were shown to be hyperresponsive (Rose et al. 2003) and present in superficial areas of thrombi (Quarck et al. 2015), while soluble NET surrogates were increased compared to healthy controls (Aldabbous et al. 2016).
4 NETs in Atherosclerosis and Arterial Thrombosis
4.1 Atherosclerosis
First evidence for the importance of neutrophils in human atherosclerosis arose indirectly, when increased MPO levels predicted risk of coronary artery disease independently of traditional risk factors (Zhang et al. 2001). High numbers of circulating neutrophils as important source of MPO were linked to both formation and severity of atherosclerotic lesions (Huang et al. 2001) and chronic stable angina pectoris (Avanzas et al. 2004). Direct assessment of human atherosclerotic lesions revealed presence of MPO (Daugherty et al. 1994) and neutrophils (Tavora et al. 2009) producing pro-inflammatory IL-8 (Marino et al. 2015). The extent of neutrophil infiltration was associated with a pro-inflammatory state and rupture-prone lesions (Ionita et al. 2010). Furthermore, levels of the NET surrogate markers dsDNA and chromatin were independently associated with the severity of coronary atherosclerosis and occurrence of adverse cardiovascular events (Borissoff et al. 2013).
Experimental models to identify mechanistic pathways of atherosclerosis typically rely on mice deficient for Apo E or low-density lipoprotein receptor, which are fed with a high-fat diet to develop atherosclerotic lesions. This led to the identification of a plethora of contributors underlying lesion formation and progression and confirmed a significant role for neutrophils in plaque development.
MPO-positive neutrophils were predominantly found in lesional caps of plaques (van Leeuwen et al. 2008) and plaque regions with already high inflammatory activity outnumbering present macrophages (Rotzius et al. 2010). Depletion of neutrophils was shown to attenuate lesion formation (Zernecke et al. 2008); however, protective effects were only apparent if depletion was performed within the first weeks, confining the effect of neutrophil activity to early stages of plaque development (Drechsler et al. 2010).
In Apo E knockout mice, neutrophils adhered to the luminal site of carotid atherosclerotic lesions and released DNA, indicative of NET formation, an interpretation which was supported by visualization of NETs in human endarterectomy samples (Megens et al. 2012). Presence of NETs was further verified in atherosclerotic lesions of mice in conjunction with an increase of the pro-inflammatory markers IL-1α, IL-1β, and IL-6 (Warnatsch et al. 2015). NETs, via histones, induced lytic cell death of smooth muscle cells in atherosclerotic lesions, leading to decreased plaque stability (Silvestre-Roig et al. 2019).
Recently, atherosclerotic lesions were classified into rupture-prone or erosion-prone phenotypes (Quillard et al. 2017). A growing body of evidence suggests that pathomechanisms are profoundly different in these two entities. Rupture-prone lesions typically contain many macrophages, harbor large lipid pools but have low interstitial collagen and few smooth muscle cells covered by thin fibrous caps. Disruption of fibrous caps makes up about two thirds of coronary events (Prati et al. 2013; Virmani et al. 2000). Conversely, eroded plaques typically present with a thick or even intact fibrous cap with a discontinuous endothelial layer (Libby 2017). Neutrophil infiltration appears to be critical for erosion as shown by an optical coherence tomography study to distinguish between plaque rupture and erosion in acute coronary syndrome. Of 25 included patients, seven exhibited erosion, and levels of systemic MPO were strikingly increased compared to patients with plaque rupture (Ferrante et al. 2010). Characterization of human endarterectomy samples demonstrated that presence of NETs was positively correlated with endothelial cell apoptosis, a hallmark feature of eroded plaques. Yet, the extent of NET burden was not different in both lesion types, emphasizing the power of neutrophil effector function (Quillard et al. 2015).
4.2 Arterial Thrombosis
The role of NETs in arterial thrombosis was mostly studied in two major conditions, acute myocardial infarction and ischemic stroke.
4.2.1 Acute Myocardial Infarction
Apart from atherosclerosis itself, neutrophils are critically involved in myocardial infarction, an acute manifestation of stable disease. Naruko et al. discovered that neutrophils were abundantly present in both ruptured and eroded plaques of patients who have died from MI (Naruko et al. 2002). The emergence of catheter-based thrombus aspiration in MI (Beran et al. 2002) made a detailed examination of thrombi in a high number of patients possible, and enabled a new view on atherosclerosis outside of autopsy specimens. This provided crucial insights into pathomechanisms underlying coronary thrombosis. Analyses revealed that leukocytes are a major component of fresh thrombi (Rittersma et al. 2005). The majority of thrombus leukocytes were neutrophils which co-localized with large quantities of endothelin-1 (Adlbrecht et al. 2007), a potent vasoconstrictor and pro-inflammatory mediator associated with left ventricular dysfunction after MI (Taylor et al. 2004). Neutrophil accumulation at the culprit site was associated with a local increase in pro-thrombotic complement factors and infarct size (Distelmaier et al. 2009). This was corroborated by the observation that thrombus neutrophil count was associated with impaired coronary microcirculation and reduced left ventricular function at six-month follow-up (Arakawa et al. 2009).
Ultimately, the presence of NETs was demonstrated in culprit site thrombi (de Boer et al. 2013). NETs were decorated with pro-inflammatory interleukin-17, which drives neutrophil accumulation (Liao et al. 2012) and is suggested to be important in the pathogenesis of MI (Mora-Ruiz et al. 2019). NETs in culprit site thrombi were confirmed by another group, which demonstrated NET formation to be induced by high mobility group box 1, an important danger-associated molecular pattern (Maugeri et al. 2014). In comparison with venous thrombi, NET burden was significantly higher in coronary thrombi and positively correlated with infarct size (Mangold et al. 2015). Recently, levels of dsDNA measured one day after MI were also associated with microvascular obstruction, myocardial salvage index, and left ventricular ejection fraction at four months (Helseth et al. 2019). Furthermore, neutrophils isolated from the culprit lesion site were more prone to undergo NETosis ex vivo in comparison with neutrophils harvested from a non-infarct related coronary artery (Stakos et al. 2015). The same group also found NETs to be decorated with tissue factor, an important mediator of coagulation. NETs were also shown to contribute to myocardial fibrosis by leading to increased activation and differentiation of fibrocytes at the culprit site (Hofbauer et al. 2019).
Circadian rhythms and neutrophil aging were recently proposed to modulate neutrophil migratory properties into tissues with substantial influence on vascular health and thrombo-inflammatory reactions in ischemia reperfusion (Adrover et al. 2019; Steffens et al. 2017).
4.2.2 Acute Ischemic Stroke
NETs were also shown to be associated with acute ischemic stroke. Concentrations of plasma DNA identified patients at risk of death at follow-up (Rainer et al. 2003), while nucleosomes were correlated with neurological dysfunction and infarction volume (Geiger et al. 2006). Another group demonstrated that DNA was increased after stroke (Tsai et al. 2011). Correspondingly, immunohistological analysis of thrombectomy samples revealed large numbers of neutrophils positive for citH3 (Laridan et al. 2017). NET burden in ischemic stroke thrombi retrieved via endovascular therapy was associated with the complexity of intervention, measured as duration of procedure and number of required wire passes (Ducroux et al. 2018). With respect to prognosis and outcome, soluble NET markers were associated with a higher NIHSS score, an indicator of stroke severity. Increased all-cause mortality was reported in patients with citH3 levels ranging in the upper quartile (Valles et al. 2017). Mechanistically, a mouse model of cerebral artery ischemia/reperfusion revealed significant exacerbation of brain injury after infusion of exogenous histones, highlighting the cytotoxic properties of NET components and their devastating influence on vascular tissues (De Meyer et al. 2012).
4.2.3 Other Conditions Associated with Arterial Thrombosis
In other diseases associated with arterial thrombosis, most evidence is available in abdominal aortic aneurysm (AAA). AAA, characterized by vessel dilation and formation of multilayered intraluminal thrombi (Delbosc et al. 2011), is increasingly being recognized as an inflammatory condition (Piechota-Polanczyk et al. 2015). Neutrophils appear to be crucial, as their depletion using a specific, cytotoxic antibody resulted in drastically reduced AAA formation (Eliason et al. 2005). NETs were shown to be abundantly present in the luminal part of human AAA thrombi and adventitia, and to be induced by periodontal pathogens (Delbosc et al. 2011) associated with AAA progression (Nakano et al. 2011). Finally, NETs co-localized with IL-1β in AAA thrombi (Meher et al. 2018), a pro-inflammatory mediator that drives AAA (Johnston et al. 2013) and NET (Keshari et al. 2012) formation.
In other pathologies associated with arterial thrombosis, evidence for the influence of NETs is scarce. Peripheral artery disease thrombi contain NETs to a similar extent as coronary and stroke thrombi (Farkas et al. 2019). Comparing plasma samples of DVT and PAD patients, neutrophil elastase alpha1 anti-trypsin complex, a specific marker for NETs, was increased in PAD (Kremers et al. 2019). Increased NET markers were also observed in thrombotic microangiopathies like thrombotic thrombocytopenic purpura or hemolytic uremic syndrome, with plasma levels being reflective of disease activity (Fuchs et al. 2012).
5 Neutrophil Extracellular Traps as a Therapeutic Target
Given their importance in the pathogenesis of atherosclerotic vascular disease, there is an interest in finding therapeutic compounds to inhibit NETs and block their detrimental effects. Several pathways have been suggested, targeting various NET components.
5.1 PAD-4 Inhibitors: Cl-Amidine
Since activation of PAD-4 is regarded critical for efficient uncoiling of chromatin in NETosis (Wang et al. 2009), inhibition of this key enzyme was suggested as a potential therapeutic regimen. Initially, synthetic PAD-4 inhibitors such as Cl-amidine were envisioned for treatment of rheumatoid arthritis (Kearney et al. 2005; Luo et al. 2006), a disease exacerbated by PAD-mediated excessive formation of citrullinated proteins that promote auto-immunity (Turunen et al. 2016). As Cl-amidine was characterized to irreversibly block PAD4, it critically interferes with NET formation (Wang et al. 2009). Cl-amidine was shown to attenuate disease severity in mouse models of sepsis (Biron et al. 2017), collagen-induced arthritis (Willis et al. 2011) and systemic lupus erythematosus, where it decreased NET formation and reduced deposition of inflammatory immunoglobulin and complement factors in the kidney (Knight et al. 2013). Daily subcutaneous treatment of ApoE knockout mice with Cl-amidine could alleviate atherosclerotic lesions under high-fat diet, while accumulation of neutrophils and macrophages into lesions was reduced (Knight et al. 2014). Thrombus formation induced by photochemical injury of the carotid artery could be significantly delayed by pre-treatment with Cl-amidine (Knight et al. 2013). It was demonstrated that Cl-amidine treatment reduces infarct size in a mouse model of coronary artery ligation and was associated with improved cardiac function (Novotny et al. 2018). In a model of ischemic stroke, administration of Cl-amidine prevented thrombotic occlusions (Pena-Martinez et al. 2019). These observations emphasize a certain dependency of NETosis on enzymatic PAD4 activity despite conflicting results and render pharmaceutical PAD-4 inhibition, a potentially promising target for treatment of human atherosclerotic and thrombotic disease.
5.2 Deoxyribonuclease
Deoxyribonuclease (DNase) degrades NETs by hydrolysis of the DNA backbone (Fuchs et al. 2010). Two isoforms of different cellular origins target DNA strands in vivo. DNase 1, secreted by the non-hematopoietic compartment, preferentially degrades protein-free DNA, while leukocyte-derived DNase 1 like 3 (DNase 1L3) cleaves DNA:protein complexes (Napirei et al. 2009). Adequate DNase activity was suggested to be crucial for a homeostatic balance between NET formation and degradation. Indeed, neutrophilic mice deficient in both plasmatic DNases developed severe disseminated thrombosis. However, reconstitution with or presence of just one functional DNase type was sufficient to protect mice from vascular occlusion (Jimenez-Alcazar et al. 2017). Targeting chromatin and NETs by DNase 1 was shown to be beneficial in experimental DVT (Brill et al. 2012; von Bruhl et al. 2012) and ischemic injury, including intestinal ischemia (Boettcher et al. 2017a), testicular torsion (Boettcher et al. 2017b), and ischemic stroke (Pena-Martinez et al. 2019).
In atherosclerotic mice, injection of DNase reduced lesion size and attenuated lesional NET burden as well as pro-inflammatory cytokines (Warnatsch et al. 2015). Furthermore, DNase 1 was used in rodent models of cardiac ischemia. Although one study reports reduction of neutrophil infiltration by DNase 1 alone in a model of ischemia/reperfusion, the authors could only show improved cardiac function by co-administration of tissue plasminogen activator (tPA) (Ge et al. 2015). In contrast, DNase 1 treatment improved cardiac function in mice after coronary artery ligation, without any reduction in neutrophil infiltration to the ischemic myocardium (Vogel et al. 2015). These apparently contradictory results may be due to variations in methodology of ligation, duration of ischemia, and timing of DNase application. In vitro, DNase 1 was shown to accelerate tPA-mediated thrombolysis of human coronary (Mangold et al. 2015) and cerebral (Laridan et al. 2017) thrombi in comparison with tPA alone. Importantly, in MI patients, low DNase activity was associated with increased infarct size (Mangold et al. 2015).
Considering the mounting evidence on the benefits of DNase application in different well-established disease models, these data raise the possibility of DNase 1 treatment of patients in neutrophil-driven disease settings in which NET formation plays a pathogenic role.
5.3 Heparin
Due to their anticoagulant properties, unfractionated heparin and low molecular weight heparins are long recognized therapeutic cornerstones for deep vein thrombosis (Mazzolai et al. 2018) and MI (Neumann et al. 2019). Given the inflammatory component of thrombotic diseases, it is intriguing that heparins have a variety of anti-inflammatory effects (Mulloy et al. 2016; Rao et al. 2010) which seem to be unrelated to their anticoagulant activity (Rao et al. 2010). In a model of acute inflammation, diminished accumulation of neutrophils was at least in part ascribed to the ability of heparin oligosaccharides to block L- and P-selectin (Nelson et al. 1993). Likewise, CD11b-dependent adhesion could be attenuated by interaction with heparin (Salas et al. 2000; Wang et al. 2002). Furthermore, heparins can not only limit NET formation itself as shown by treatment in vitro and in vivo (Manfredi et al. 2017), but can also target existing NETs in various ways: heparin was shown to inhibit enzymatic activity of neutrophil elastase and Cathepsin G (Fryer et al. 1997), two major enzymes present in NETs (Folco et al. 2018). Binding and displacement of histones by heparin promotes NETs disassembly, limiting their pro-thrombotic properties in vitro (Fuchs et al. 2010) and in vivo (von Bruhl et al. 2012). At the same time, excessive release of histones during cell death and NETosis was suggested to counteract the anti-thrombotic function of heparin, potentially explaining non-responsiveness to heparin (Longstaff et al. 2016).
These observations stimulated the development of heparinoids that lack anticoagulant properties while keeping their anti-inflammatory effects (Rao et al. 2010). Indeed, sevuparin, a low-anticoagulant heparin analog, inhibited NE and histone H4, proteins associated with NETs (Rasmuson et al. 2019). In another study, non-anticoagulant heparin prevented histone-mediated cytotoxicity and improved survival in a murine sepsis model (Wildhagen et al. 2014). Thus, it might be a crucial therapeutic add-on to reduce NET burden without increasing risk of bleeding complications.
6 Summary
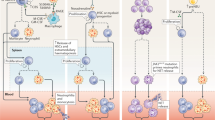
A summary of the content of this chapter is provided in the central Fig. 1. Taken together, NETs are important for the initiation and progression of thrombosis in the setting of atherosclerosis, including VTE, MI, and stroke. For these reasons, NETs have emerged as potential targets for treatment. Despite promising in vitro and experimental in vivo data, adequately powered clinical trials are required to assess clinical benefits and safety of anti-NETotic regimens.

The central figure summarizes mechanisms of NET formation, its pathways and consequences in atherosclerosis and thrombosis. The figure was constructed using Biorender and Microsoft PowerPoint
Abbreviations
- AAA:
-
Abdominal aortic aneurysm
- citH3:
-
Citrullinated histone H3
- DNase:
-
Deoxyribonuclease
- dsDNA:
-
Double-stranded DNA
- DVT:
-
Deep vein thrombosis
- ET:
-
Extracellular traps
- HIT:
-
Heparin-induced thrombocytopenia
- IL:
-
Interleukin
- MI:
-
Myocardial infarction
- MPO:
-
Myeloperoxidase
- NE:
-
Neutrophil elastase
- NETs:
-
Neutrophil extracellular traps
- PAD:
-
Peripheral artery disease
- PAD-4:
-
Peptidylarginine deiminase 4
- PE:
-
Pulmonary embolism
- PMA:
-
Phorbol myristate acetate
- ROS:
-
Reactive oxygen species
- tPA:
-
Tissue plasminogen activator
- VTE:
-
Venous thromboembolism
References
Adlbrecht C, Bonderman D, Plass C, Jakowitsch J, Beran G, Sperker W, Siostrzonek P, Glogar D, Maurer G, Lang IM (2007) Active endothelin is an important vasoconstrictor in acute coronary thrombi. Thromb Haemost 97:642–649
Adrover JM, Del Fresno C, Crainiciuc G, Cuartero MI, Casanova-Acebes M, Weiss LA, Huerga-Encabo H, Silvestre-Roig C, Rossaint J, Cossio I, Lechuga-Vieco AV, Garcia-Prieto J, Gomez-Parrizas M, Quintana JA, Ballesteros I, Martin-Salamanca S, Aroca-Crevillen A, Chong SZ, Evrard M, Balabanian K, Lopez J, Bidzhekov K, Bachelerie F, Abad-Santos F, Munoz-Calleja C, Zarbock A, Soehnlein O, Weber C, Ng LG, Lopez-Rodriguez C, Sancho D, Moro MA, Ibanez B, Hidalgo A (2019) A neutrophil timer coordinates immune defense and vascular protection. Immunity 50:390–402.e10. https://doi.org/10.1016/j.immuni.2019.01.002
Aldabbous L, Abdul-Salam V, McKinnon T, Duluc L, Pepke-Zaba J, Southwood M, Ainscough AJ, Hadinnapola C, Wilkins MR, Toshner M, Wojciak-Stothard B (2016) Neutrophil extracellular traps promote angiogenesis: evidence from vascular pathology in pulmonary hypertension. Arterioscler Thromb Vasc Biol 36:2078–2087. https://doi.org/10.1161/atvbaha.116.307634
Arakawa K, Yasuda S, Hao H, Kataoka Y, Morii I, Kasahara Y, Kawamura A, Ishibashi-Ueda H, Miyazaki S (2009) Significant association between neutrophil aggregation in aspirated thrombus and myocardial damage in patients with ST-segment elevation acute myocardial infarction. Circ J 73:139–144
Arnalich F, Maldifassi MC, Ciria E, Codoceo R, Renart J, Fernandez-Capitan C, Herruzo R, Garcia-Rio F, Lopez-Collazo E, Montiel C (2013) Plasma levels of mitochondrial and nuclear DNA in patients with massive pulmonary embolism in the emergency department: a prospective cohort study. Crit Care 17:R90. https://doi.org/10.1186/cc12735
Avanzas P, Arroyo-Espliguero R, Cosin-Sales J, Quiles J, Zouridakis E, Kaski JC (2004) Multiple complex stenoses, high neutrophil count and C-reactive protein levels in patients with chronic stable angina. Atherosclerosis 175:151–157. https://doi.org/10.1016/j.atherosclerosis.2004.03.013
Bainton DF, Ullyot JL, Farquhar MG (1971) The development of neutrophilic polymorphonuclear leukocytes in human bone marrow. J Exp Med 134:907–934
Barquera S, Pedroza-Tobias A, Medina C, Hernandez-Barrera L, Bibbins-Domingo K, Lozano R, Moran AE (2015) Global overview of the epidemiology of atherosclerotic cardiovascular disease. Arch Med Res 46:328–338. https://doi.org/10.1016/j.arcmed.2015.06.006
Beran G, Lang I, Schreiber W, Denk S, Stefenelli T, Syeda B, Maurer G, Glogar D, Siostrzonek P (2002) Intracoronary thrombectomy with the X-sizer catheter system improves epicardial flow and accelerates ST-segment resolution in patients with acute coronary syndrome: a prospective, randomized, controlled study. Circulation 105:2355–2360
Biron BM, Chung CS, O'Brien XM, Chen Y, Reichner JS, Ayala A (2017) Cl-amidine prevents histone 3 citrullination and neutrophil extracellular trap formation, and improves survival in a murine sepsis model. J Innate Immun 9:22–32. https://doi.org/10.1159/000448808
Boettcher M, Eschenburg G, Mietzsch S, Jimenez-Alcazar M, Klinke M, Vincent D, Tiemann B, Bergholz R, Reinshagen K, Fuchs TA (2017a) Therapeutic targeting of extracellular DNA improves the outcome of intestinal ischemic reperfusion injury in neonatal rats. Sci Rep 7:15377. https://doi.org/10.1038/s41598-017-15807-6
Boettcher M, Meier D, Jimenez-Alcazar M, Eschenburg G, Mietzsch S, Vincent D, Klinke M, Trochimiuk M, Appl B, Tiemann B, Bergholz R, Reinshagen K, Fuchs TA (2017b) Degradation of extracellular DNA by DNase1 significantly reduces testicular damage after testicular torsion in rats. Urology 109:223.e1–223.e7. https://doi.org/10.1016/j.urology.2017.07.031
Borissoff JI, Joosen IA, Versteylen MO, Brill A, Fuchs TA, Savchenko AS, Gallant M, Martinod K, Ten Cate H, Hofstra L, Crijns HJ, Wagner DD, Kietselaer B (2013) Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler Thromb Vasc Biol 33:2032–2040. https://doi.org/10.1161/atvbaha.113.301627
Brill A, Fuchs TA, Savchenko AS, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD (2012) Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost 10:136–144. https://doi.org/10.1111/j.1538-7836.2011.04544.x
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303:1532–1535. https://doi.org/10.1126/science.1092385
Bullenkamp J, Dinkla S, Kaski JC, Dumitriu IE (2016) Targeting T cells to treat atherosclerosis: odyssey from bench to bedside. Eur Heart J Cardiovasc Pharmacother 2:194–199. https://doi.org/10.1093/ehjcvp/pvw001
Campillo-Navarro M, Leyva-Paredes K, Donis-Maturano L, Gonzalez-Jimenez M, Paredes-Vivas Y, Cerbulo-Vazquez A, Serafin-Lopez J, Garcia-Perez B, Ullrich SE, Flores-Romo L, Perez-Tapia SM, Estrada-Parra S, Estrada-Garcia I, Chacon-Salinas R (2017) Listeria monocytogenes induces mast cell extracellular traps. Immunobiology 222:432–439. https://doi.org/10.1016/j.imbio.2016.08.006
Catapano AL, Graham I, De Backer G, Wiklund O, Chapman MJ, Drexel H, Hoes AW, Jennings CS, Landmesser U, Pedersen TR, Reiner Z, Riccardi G, Taskinen MR, Tokgozoglu L, Verschuren WMM, Vlachopoulos C, Wood DA, Zamorano JL, Cooney MT (2016) 2016 ESC/EAS guidelines for the management of dyslipidaemias. Eur Heart J 37:2999–3058. https://doi.org/10.1093/eurheartj/ehw272
Claushuis TAM, van der Donk LEH, Luitse AL, van Veen HA, van der Wel NN, van Vught LA, Roelofs J, de Boer OJ, Lankelma JM, Boon L, de Vos AF, van ‘t Veer C, van der Poll T (2018) Role of peptidylarginine deiminase 4 in neutrophil extracellular trap formation and host defense during Klebsiella pneumoniae-induced pneumonia-derived sepsis. J Immunol 201:1241–1252. https://doi.org/10.4049/jimmunol.1800314
Cowland JB, Borregaard N (2016) Granulopoiesis and granules of human neutrophils. Immunol Rev 273:11–28. https://doi.org/10.1111/imr.12440
Daugherty A, Dunn JL, Rateri DL, Heinecke JW (1994) Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest 94:437–444. https://doi.org/10.1172/jci117342
de Boer OJ, Li X, Teeling P, Mackaay C, Ploegmakers HJ, van der Loos CM, Daemen MJ, de Winter RJ, van der Wal AC (2013) Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost 109:290–297. https://doi.org/10.1160/th12-06-0425
de Bont CM, Eerden N, Boelens WC, Pruijn GJM (2020) Neutrophil proteases degrade autoepitopes of NET-associated proteins. Clin Exp Immunol 199:1–8. https://doi.org/10.1111/cei.13392
De Meyer SF, Suidan GL, Fuchs TA, Monestier M, Wagner DD (2012) Extracellular chromatin is an important mediator of ischemic stroke in mice. Arterioscler Thromb Vasc Biol 32:1884–1891. https://doi.org/10.1161/atvbaha.112.250993
Delbosc S, Alsac JM, Journe C, Louedec L, Castier Y, Bonnaure-Mallet M, Ruimy R, Rossignol P, Bouchard P, Michel JB, Meilhac O (2011) Porphyromonas gingivalis participates in pathogenesis of human abdominal aortic aneurysm by neutrophil activation. Proof of concept in rats. PLoS One 6:e18679. https://doi.org/10.1371/journal.pone.0018679
Desai J, Mulay SR, Nakazawa D, Anders HJ (2016) Matters of life and death. How neutrophils die or survive along NET release and is “NETosis” = necroptosis? Cell Mol Life Sci 73:2211–2219. https://doi.org/10.1007/s00018-016-2195-0
Di Nisio M, van Es N, Buller HR (2016) Deep vein thrombosis and pulmonary embolism. Lancet 388:3060–3073. https://doi.org/10.1016/s0140-6736(16)30514-1
Diaz JA, Fuchs TA, Jackson TO, Kremer Hovinga JA, Lammle B, Henke PK, Myers DD Jr, Wagner DD, Wakefield TW (2013) Plasma DNA is elevated in patients with deep vein thrombosis. J Vasc Surg Venous Lymphat Disord:1. https://doi.org/10.1016/j.jvsv.2012.12.002
Distelmaier K, Adlbrecht C, Jakowitsch J, Winkler S, Dunkler D, Gerner C, Wagner O, Lang IM, Kubicek M (2009) Local complement activation triggers neutrophil recruitment to the site of thrombus formation in acute myocardial infarction. Thromb Haemost 102:564–572. https://doi.org/10.1160/th09-02-0103
Distelmaier K, Winter MP, Dragschitz F, Redwan B, Mangold A, Gleiss A, Perkmann T, Maurer G, Adlbrecht C, Lang IM (2014) Prognostic value of culprit site neutrophils in acute coronary syndrome. Eur J Clin Investig 44:257–265. https://doi.org/10.1111/eci.12228
Doring Y, Drechsler M, Soehnlein O, Weber C (2015) Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol 35:288–295. https://doi.org/10.1161/atvbaha.114.303564
Doring Y, Soehnlein O, Weber C (2017) Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res 120:736–743. https://doi.org/10.1161/circresaha.116.309692
Doster RS, Rogers LM, Gaddy JA, Aronoff DM (2018) Macrophage extracellular traps: a scoping review. J Innate Immun 10:3–13. https://doi.org/10.1159/000480373
Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O (2010) Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 122:1837–1845. https://doi.org/10.1161/circulationaha.110.961714
Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, Ben Maacha M, Blanc R, Redjem H, Ciccio G, Smajda S, Fahed R, Michel JB, Piotin M, Salomon L, Mazighi M, Ho-Tin-Noe B, Desilles JP (2018) Thrombus neutrophil extracellular traps content impair tPA-induced thrombolysis in acute ischemic stroke. Stroke 49:754–757. https://doi.org/10.1161/strokeaha.117.019896
Eliason JL, Hannawa KK, Ailawadi G, Sinha I, Ford JW, Deogracias MP, Roelofs KJ, Woodrum DT, Ennis TL, Henke PK, Stanley JC, Thompson RW, Upchurch GR Jr (2005) Neutrophil depletion inhibits experimental abdominal aortic aneurysm formation. Circulation 112:232–240. https://doi.org/10.1161/circulationaha.104.517391
Farkas AZ, Farkas VJ, Gubucz I, Szabo L, Balint K, Tenekedjiev K, Nagy AI, Sotonyi P, Hidi L, Nagy Z, Szikora I, Merkely B, Kolev K (2019) Neutrophil extracellular traps in thrombi retrieved during interventional treatment of ischemic arterial diseases. Thromb Res 175:46–52. https://doi.org/10.1016/j.thromres.2019.01.006
Ferrante G, Nakano M, Prati F, Niccoli G, Mallus MT, Ramazzotti V, Montone RA, Kolodgie FD, Virmani R, Crea F (2010) High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 122:2505–2513. https://doi.org/10.1161/circulationaha.110.955302
Folco EJ, Mawson TL, Vromman A, Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G, Luscinskas FW, Libby P (2018) Neutrophil extracellular traps induce endothelial cell activation and tissue factor production through interleukin-1alpha and cathepsin G. Arterioscler Thromb Vasc Biol 38:1901–1912. https://doi.org/10.1161/atvbaha.118.311150
Fryer A, Huang YC, Rao G, Jacoby D, Mancilla E, Whorton R, Piantadosi CA, Kennedy T, Hoidal J (1997) Selective O-desulfation produces nonanticoagulant heparin that retains pharmacological activity in the lung. J Pharmacol Exp Ther 282:208–219
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A (2007) Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 176:231–241. https://doi.org/10.1083/jcb.200606027
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD (2010) Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A 107:15880–15885. https://doi.org/10.1073/pnas.1005743107
Fuchs TA, Kremer Hovinga JA, Schatzberg D, Wagner DD, Lammle B (2012) Circulating DNA and myeloperoxidase indicate disease activity in patients with thrombotic microangiopathies. Blood 120:1157–1164. https://doi.org/10.1182/blood-2012-02-412197
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M (2016) 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 37:67–119. https://doi.org/10.1093/eurheartj/ehv317
Ge L, Zhou X, Ji WJ, Lu RY, Zhang Y, Zhang YD, Ma YQ, Zhao JH, Li YM (2015) Neutrophil extracellular traps in ischemia-reperfusion injury-induced myocardial no-reflow: therapeutic potential of DNase-based reperfusion strategy. Am J Physiol Heart Circ Physiol 308:H500–H509. https://doi.org/10.1152/ajpheart.00381.2014
Geiger S, Holdenrieder S, Stieber P, Hamann GF, Bruening R, Ma J, Nagel D, Seidel D (2006) Nucleosomes in serum of patients with early cerebral stroke. Cerebrovasc Dis 21:32–37. https://doi.org/10.1159/000089591
Gil-Pulido J, Zernecke A (2017) Antigen-presenting dendritic cells in atherosclerosis. Eur J Pharmacol 816:25–31. https://doi.org/10.1016/j.ejphar.2017.08.016
Granger V, Faille D, Marani V, Noel B, Gallais Y, Szely N, Flament H, Pallardy M, Chollet-Martin S, de Chaisemartin L (2017) Human blood monocytes are able to form extracellular traps. J Leukoc Biol 102:775–781. https://doi.org/10.1189/jlb.3MA0916-411R
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, Waldmann H (2011) Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7:75–77. https://doi.org/10.1038/nchembio.496
Helseth R, Shetelig C, Andersen GO, Langseth MS, Limalanathan S, Opstad TB, Arnesen H, Hoffmann P, Eritsland J, Seljeflot I (2019) Neutrophil extracellular trap components associate with infarct size, ventricular function, and clinical outcome in STEMI. Mediat Inflamm 2019:7816491. https://doi.org/10.1155/2019/7816491
Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S (2016) Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res 118:535–546. https://doi.org/10.1161/circresaha.115.307611
Hofbauer TM, Mangold A, Scherz T, Seidl V, Panzenbock A, Ondracek AS, Muller J, Schneider M, Binder T, Hell L, Lang IM (2019) Neutrophil extracellular traps and fibrocytes in ST-segment elevation myocardial infarction. Basic Res Cardiol 114:33. https://doi.org/10.1007/s00395-019-0740-3
Huang ZS, Jeng JS, Wang CH, Yip PK, Wu TH, Lee TK (2001) Correlations between peripheral differential leukocyte counts and carotid atherosclerosis in non-smokers. Atherosclerosis 158:431–436
Ionita MG, van den Borne P, Catanzariti LM, Moll FL, de Vries JP, Pasterkamp G, Vink A, de Kleijn DP (2010) High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler Thromb Vasc Biol 30:1842–1848. https://doi.org/10.1161/atvbaha.110.209296
Jimenez-Alcazar M, Rangaswamy C, Panda R, Bitterling J, Simsek YJ, Long AT, Bilyy R, Krenn V, Renne C, Renne T, Kluge S, Panzer U, Mizuta R, Mannherz HG, Kitamura D, Herrmann M, Napirei M, Fuchs TA (2017) Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358:1202–1206. https://doi.org/10.1126/science.aam8897
Jimenez-Alcazar M, Limacher A, Panda R, Mean M, Bitterling J, Peine S, Renne T, Beer JH, Aujesky D, Lammle B, Fuchs TA (2018) Circulating extracellular DNA is an independent predictor of mortality in elderly patients with venous thromboembolism. PLoS One 13:e0191150. https://doi.org/10.1371/journal.pone.0191150
Johnston WF, Salmon M, Su G, Lu G, Stone ML, Zhao Y, Owens GK, Upchurch GR Jr, Ailawadi G (2013) Genetic and pharmacologic disruption of interleukin-1beta signaling inhibits experimental aortic aneurysm formation. Arterioscler Thromb Vasc Biol 33:294–304. https://doi.org/10.1161/atvbaha.112.300432
Kapoor S, Opneja A, Nayak L (2018) The role of neutrophils in thrombosis. Thromb Res 170:87–96. https://doi.org/10.1016/j.thromres.2018.08.005
Kearney PL, Bhatia M, Jones NG, Yuan L, Glascock MC, Catchings KL, Yamada M, Thompson PR (2005) Kinetic characterization of protein arginine deiminase 4: a transcriptional corepressor implicated in the onset and progression of rheumatoid arthritis. Biochemistry 44:10570–10582. https://doi.org/10.1021/bi050292m
Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, Brinkmann V, Bernuth HV, Zychlinsky A (2017) Diverse stimuli engage different neutrophil extracellular trap pathways. elife 6. https://doi.org/10.7554/eLife.24437
Keshari RS, Jyoti A, Dubey M, Kothari N, Kohli M, Bogra J, Barthwal MK, Dikshit M (2012) Cytokines induced neutrophil extracellular traps formation: implication for the inflammatory disease condition. PLoS One 7:e48111. https://doi.org/10.1371/journal.pone.0048111
Knight JS, Zhao W, Luo W, Subramanian V, O'Dell AA, Yalavarthi S, Hodgin JB, Eitzman DT, Thompson PR, Kaplan MJ (2013) Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest 123:2981–2993. https://doi.org/10.1172/jci67390
Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, Guo C, Grenn RC, Thompson PR, Eitzman DT, Kaplan MJ (2014) Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res 114:947–956. https://doi.org/10.1161/circresaha.114.303312
Kremers BMM, Birocchi S, van Oerle R, Zeerleder S, Spronk HMH, Mees BME, Luken BM, Ten Cate H, Ten Cate-Hoek AJ (2019) Searching for a common thrombo-inflammatory basis in patients with deep vein thrombosis or peripheral artery disease. Front Cardiovasc Med 6:33. https://doi.org/10.3389/fcvm.2019.00033
Kumar DR, Hanlin E, Glurich I, Mazza JJ, Yale SH (2010) Virchow's contribution to the understanding of thrombosis and cellular biology. Clin Med Res 8:168–172. https://doi.org/10.3121/cmr.2009.866
Lang IM (2004) Chronic thromboembolic pulmonary hypertension--not so rare after all. N Engl J Med 350:2236–2238. https://doi.org/10.1056/NEJMp048088
Laridan E, Denorme F, Desender L, Francois O, Andersson T, Deckmyn H, Vanhoorelbeke K, De Meyer SF (2017) Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol 82:223–232. https://doi.org/10.1002/ana.24993
Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, Bicker KL, Bingham RP, Campbell M, Chen YH, Chung CW, Craggs PD, Davis RP, Eberhard D, Joberty G, Lind KE, Locke K, Maller C, Martinod K, Patten C, Polyakova O, Rise CE, Rudiger M, Sheppard RJ, Slade DJ, Thomas P, Thorpe J, Yao G, Drewes G, Wagner DD, Thompson PR, Prinjha RK, Wilson DM (2015) Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol 11:189–191. https://doi.org/10.1038/nchembio.1735
Ley K, Hoffman HM, Kubes P, Cassatella MA, Zychlinsky A, Hedrick CC, Catz SD (2018) Neutrophils: new insights and open questions. Sci Immunol 3. https://doi.org/10.1126/sciimmunol.aat4579
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862. https://doi.org/10.1084/jem.20100239
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX, Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, Yuan J, Jevallee H, Wei F, Shi GP, Cheng X (2012) Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol 59:420–429. https://doi.org/10.1016/j.jacc.2011.10.863
Libby P (2017) Superficial erosion and the precision management of acute coronary syndromes: not one-size-fits-all. Eur Heart J 38:801–803. https://doi.org/10.1093/eurheartj/ehw599
Longstaff C, Hogwood J, Gray E, Komorowicz E, Varju I, Varga Z, Kolev K (2016) Neutralisation of the anti-coagulant effects of heparin by histones in blood plasma and purified systems. Thromb Haemost 115:591–599. https://doi.org/10.1160/th15-03-0214
Luo Y, Knuckley B, Lee YH, Stallcup MR, Thompson PR (2006) A fluoroacetamidine-based inactivator of protein arginine deiminase 4: design, synthesis, and in vitro and in vivo evaluation. J Am Chem Soc 128:1092–1093. https://doi.org/10.1021/ja0576233
Madhusoodanan J (2017) Core concept: role player or cellular rubbish? Biologists debate the function of neutrophil extracellular traps. Proc Natl Acad Sci U S A 114:13309–13311. https://doi.org/10.1073/pnas.1719978115
Manfredi AA, Rovere-Querini P, D'Angelo A, Maugeri N (2017) Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol Res 123:146–156. https://doi.org/10.1016/j.phrs.2016.08.008
Mangold A, Alias S, Scherz T, Hofbauer T, Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C, Kammerlander A, Mascherbauer J, Winter MP, Distelmaier K, Adlbrecht C, Preissner KT, Lang IM (2015) Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res 116:1182–1192. https://doi.org/10.1161/circresaha.116.304944
Marino F, Tozzi M, Schembri L, Ferraro S, Tarallo A, Scanzano A, Legnaro M, Castelli P, Cosentino M (2015) Production of IL-8, VEGF and elastase by circulating and intraplaque neutrophils in patients with carotid atherosclerosis. PLoS One 10:e0124565. https://doi.org/10.1371/journal.pone.0124565
Martinod K, Demers M, Fuchs TA, Wong SL, Brill A, Gallant M, Hu J, Wang Y, Wagner DD (2013) Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci U S A 110:8674–8679. https://doi.org/10.1073/pnas.1301059110
Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D'Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA (2014) Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost 12:2074–2088. https://doi.org/10.1111/jth.12710
Mazzolai L, Aboyans V, Ageno W, Agnelli G, Alatri A, Bauersachs R, Brekelmans MPA, Buller HR, Elias A, Farge D, Konstantinides S, Palareti G, Prandoni P, Righini M, Torbicki A, Vlachopoulos C, Brodmann M (2018) Diagnosis and management of acute deep vein thrombosis: a joint consensus document from the European Society of Cardiology working groups of aorta and peripheral vascular diseases and pulmonary circulation and right ventricular function. Eur Heart J 39:4208–4218. https://doi.org/10.1093/eurheartj/ehx003
Megens RT, Vijayan S, Lievens D, Doring Y, van Zandvoort MA, Grommes J, Weber C, Soehnlein O (2012) Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb Haemost 107:597–598. https://doi.org/10.1160/th11-09-0650
Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, Serbulea V, Leitinger N, Ailawadi G, Upchurch GR Jr (2018) Novel role of IL (Interleukin)-1beta in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 38:843–853. https://doi.org/10.1161/atvbaha.117.309897
Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V (2014) A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep 8:883–896. https://doi.org/10.1016/j.celrep.2014.06.044
Mora-Ruiz MD, Blanco-Favela F, Chavez Rueda AK, Legorreta-Haquet MV, Chavez-Sanchez L (2019) Role of interleukin-17 in acute myocardial infarction. Mol Immunol 107:71–78. https://doi.org/10.1016/j.molimm.2019.01.008
Moroni F, Ammirati E, Norata GD, Magnoni M, Camici PG (2019) The role of monocytes and macrophages in human atherosclerosis, plaque neoangiogenesis, and atherothrombosis. Mediat Inflamm 2019:7434376. https://doi.org/10.1155/2019/7434376
Mukherjee M, Lacy P, Ueki S (2018) Eosinophil extracellular traps and inflammatory pathologies-untangling the web! Front Immunol 9:2763. https://doi.org/10.3389/fimmu.2018.02763
Mulloy B, Hogwood J, Gray E, Lever R, Page CP (2016) Pharmacology of heparin and related drugs. Pharmacol Rev 68:76–141. https://doi.org/10.1124/pr.115.011247
Nakano K, Wada K, Nomura R, Nemoto H, Inaba H, Kojima A, Naka S, Hokamura K, Mukai T, Nakajima A, Umemura K, Kamisaki Y, Yoshioka H, Taniguchi K, Amano A, Ooshima T (2011) Characterization of aortic aneurysms in cardiovascular disease patients harboring Porphyromonas gingivalis. Oral Dis 17:370–378. https://doi.org/10.1111/j.1601-0825.2010.01759.x
Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A (2012) Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front Immunol 3:333. https://doi.org/10.3389/fimmu.2012.00333
Napirei M, Ludwig S, Mezrhab J, Klockl T, Mannherz HG (2009) Murine serum nucleases--contrasting effects of plasmin and heparin on the activities of DNase1 and DNase1-like 3 (DNase1l3). FEBS J 276:1059–1073. https://doi.org/10.1111/j.1742-4658.2008.06849.x
Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, Ehara S, Yoshiyama M, Takeuchi K, Yoshikawa J, Becker AE (2002) Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 106:2894–2900
Nelson RM, Cecconi O, Roberts WG, Aruffo A, Linhardt RJ, Bevilacqua MP (1993) Heparin oligosaccharides bind L- and P-selectin and inhibit acute inflammation. Blood 82:3253–3258
Neumann FJ, Sousa-Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, Byrne RA, Collet JP, Falk V, Head SJ, Juni P, Kastrati A, Koller A, Kristensen SD, Niebauer J, Richter DJ, Seferovic PM, Sibbing D, Stefanini GG, Windecker S, Yadav R, Zembala MO (2019) 2018 ESC/EACTS guidelines on myocardial revascularization. Eur Heart J 40:87–165. https://doi.org/10.1093/eurheartj/ehy394
Novotny J, Chandraratne S, Weinberger T, Philippi V, Stark K, Ehrlich A, Pircher J, Konrad I, Oberdieck P, Titova A, Hoti Q, Schubert I, Legate KR, Urtz N, Lorenz M, Pelisek J, Massberg S, von Bruhl ML, Schulz C (2018) Histological comparison of arterial thrombi in mice and men and the influence of Cl-amidine on thrombus formation. PLoS One 13:e0190728. https://doi.org/10.1371/journal.pone.0190728
Papayannopoulos V (2018) Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol 18:134–147. https://doi.org/10.1038/nri.2017.105
Pena-Martinez C, Duran-Laforet V, Garcia-Culebras A, Ostos F, Hernandez-Jimenez M, Bravo-Ferrer I, Perez-Ruiz A, Ballenilla F, Diaz-Guzman J, Pradillo JM, Lizasoain I, Moro MA (2019) Pharmacological modulation of neutrophil extracellular traps reverses thrombotic stroke tPA (tissue-type plasminogen activator) resistance. Stroke 50:3228–3237. https://doi.org/10.1161/strokeaha.119.026848
Piechota-Polanczyk A, Jozkowicz A, Nowak W, Eilenberg W, Neumayer C, Malinski T, Huk I, Brostjan C (2015) The abdominal aortic aneurysm and intraluminal thrombus: current concepts of development and treatment. Front Cardiovasc Med 2:19. https://doi.org/10.3389/fcvm.2015.00019
Prati F, Uemura S, Souteyrand G, Virmani R, Motreff P, Di Vito L, Biondi-Zoccai G, Halperin J, Fuster V, Ozaki Y, Narula J (2013) OCT-based diagnosis and management of STEMI associated with intact fibrous cap. JACC Cardiovasc Imaging 6:283–287. https://doi.org/10.1016/j.jcmg.2012.12.007
Quarck R, Wynants M, Verbeken E, Meyns B, Delcroix M (2015) Contribution of inflammation and impaired angiogenesis to the pathobiology of chronic thromboembolic pulmonary hypertension. Eur Respir J 46:431–443. https://doi.org/10.1183/09031936.00009914
Quillard T, Araujo HA, Franck G, Shvartz E, Sukhova G, Libby P (2015) TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: implications for superficial erosion. Eur Heart J 36:1394–1404. https://doi.org/10.1093/eurheartj/ehv044
Quillard T, Franck G, Mawson T, Folco E, Libby P (2017) Mechanisms of erosion of atherosclerotic plaques. Curr Opin Lipidol. https://doi.org/10.1097/mol.0000000000000440
Rainer TH, Wong LK, Lam W, Yuen E, Lam NY, Metreweli C, Lo YM (2003) Prognostic use of circulating plasma nucleic acid concentrations in patients with acute stroke. Clin Chem 49:562–569
Rao NV, Argyle B, Xu X, Reynolds PR, Walenga JM, Prechel M, Prestwich GD, MacArthur RB, Walters BB, Hoidal JR, Kennedy TP (2010) Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol 299:C97–C110. https://doi.org/10.1152/ajpcell.00009.2010
Rasmuson J, Kenne E, Wahlgren M, Soehnlein O, Lindbom L (2019) Heparinoid sevuparin inhibits streptococcus-induced vascular leak through neutralizing neutrophil-derived proteins. FASEB J:fj201900627R. https://doi.org/10.1096/fj.201900627R
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R, Noppen S, Delforge M, Willems J, Vandenabeele P (2011) Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 21:290–304. https://doi.org/10.1038/cr.2010.150
Rittersma SZ, van der Wal AC, Koch KT, Piek JJ, Henriques JP, Mulder KJ, Ploegmakers JP, Meesterman M, de Winter RJ (2005) Plaque instability frequently occurs days or weeks before occlusive coronary thrombosis: a pathological thrombectomy study in primary percutaneous coronary intervention. Circulation 111:1160–1165. https://doi.org/10.1161/01.cir.0000157141.00778.ac
Rose F, Hattar K, Gakisch S, Grimminger F, Olschewski H, Seeger W, Tschuschner A, Schermuly RT, Weissmann N, Hanze J, Sibelius U, Ghofrani HA (2003) Increased neutrophil mediator release in patients with pulmonary hypertension--suppression by inhaled iloprost. Thromb Haemost 90:1141–1149. https://doi.org/10.1160/th03-03-0173
Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng CN, Bjorkstrom NK, Malmberg KJ, Lindbom L, Eriksson EE (2010) Distinct infiltration of neutrophils in lesion shoulders in ApoE−/− mice. Am J Pathol 177:493–500. https://doi.org/10.2353/ajpath.2010.090480
Roumen-Klappe EM, den Heijer M, van Uum SH, van der Ven-Jongekrijg J, van der Graaf F, Wollersheim H (2002) Inflammatory response in the acute phase of deep vein thrombosis. J Vasc Surg 35:701–706
Roumen-Klappe EM, Janssen MC, Van Rossum J, Holewijn S, Van Bokhoven MM, Kaasjager K, Wollersheim H, Den Heijer M (2009) Inflammation in deep vein thrombosis and the development of post-thrombotic syndrome: a prospective study. J Thromb Haemost 7:582–587. https://doi.org/10.1111/j.1538-7836.2009.03286.x
Sage AP, Tsiantoulas D, Binder CJ, Mallat Z (2019) The role of B cells in atherosclerosis. Nat Rev Cardiol 16:180–196. https://doi.org/10.1038/s41569-018-0106-9
Salas A, Sans M, Soriano A, Reverter JC, Anderson DC, Pique JM, Panes J (2000) Heparin attenuates TNF-alpha induced inflammatory response through a CD11b dependent mechanism. Gut 47:88–96. https://doi.org/10.1136/gut.47.1.88
Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, Libby P, Goldhaber SZ, Mitchell RN, Wagner DD (2014) Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost 12:860–870. https://doi.org/10.1111/jth.12571
Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, Winter J, Adrover JM, Santos GS, Froese A, Lemnitzer P, Ortega-Gomez A, Chevre R, Marschner J, Schumski A, Winter C, Perez-Olivares L, Pan C, Paulin N, Schoufour T, Hartwig H, Gonzalez-Ramos S, Kamp F, Megens RTA, Mowen KA, Gunzer M, Maegdefessel L, Hackeng T, Lutgens E, Daemen M, von Blume J, Anders HJ, Nikolaev VO, Pellequer JL, Weber C, Hidalgo A, Nicolaes GAF, Wong GCL, Soehnlein O (2019) Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 569:236–240. https://doi.org/10.1038/s41586-019-1167-6
Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, Ritis K (2015) Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J 36:1405–1414. https://doi.org/10.1093/eurheartj/ehv007
Steffens S, Winter C, Schloss MJ, Hidalgo A, Weber C, Soehnlein O (2017) Circadian control of inflammatory processes in atherosclerosis and its complications. Arterioscler Thromb Vasc Biol 37:1022–1028. https://doi.org/10.1161/atvbaha.117.309374
Takei H, Araki A, Watanabe H, Ichinose A, Sendo F (1996) Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol 59:229–240
Tavora FR, Ripple M, Li L, Burke AP (2009) Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc Disord 9:27. https://doi.org/10.1186/1471-2261-9-27
Taylor AJ, Bobik A, Richards M, Kaye D, Raines G, Gould P, Jennings G (2004) Myocardial endothelin-1 release and indices of inflammation during angioplasty for acute myocardial infarction and stable coronary artery disease. Am Heart J 148:e10. https://doi.org/10.1016/j.ahj.2004.03.018
Tsai NW, Lin TK, Chen SD, Chang WN, Wang HC, Yang TM, Lin YJ, Jan CR, Huang CR, Liou CW, Lu CH (2011) The value of serial plasma nuclear and mitochondrial DNA levels in patients with acute ischemic stroke. Clin Chim Acta 412:476–479. https://doi.org/10.1016/j.cca.2010.11.036
Turunen S, Huhtakangas J, Nousiainen T, Valkealahti M, Melkko J, Risteli J, Lehenkari P (2016) Rheumatoid arthritis antigens homocitrulline and citrulline are generated by local myeloperoxidase and peptidyl arginine deiminases 2, 3 and 4 in rheumatoid nodule and synovial tissue. Arthritis Res Ther 18:239. https://doi.org/10.1186/s13075-016-1140-9
Valles J, Lago A, Santos MT, Latorre AM, Tembl JI, Salom JB, Nieves C, Moscardo A (2017) Neutrophil extracellular traps are increased in patients with acute ischemic stroke: prognostic significance. Thromb Haemost 117:1919–1929. https://doi.org/10.1160/th17-02-0130
van Leeuwen M, Gijbels MJ, Duijvestijn A, Smook M, van de Gaar MJ, Heeringa P, de Winther MP, Tervaert JW (2008) Accumulation of myeloperoxidase-positive neutrophils in atherosclerotic lesions in LDLR−/− mice. Arterioscler Thromb Vasc Biol 28:84–89. https://doi.org/10.1161/atvbaha.107.154807
van Montfoort ML, Stephan F, Lauw MN, Hutten BA, Van Mierlo GJ, Solati S, Middeldorp S, Meijers JC, Zeerleder S (2013) Circulating nucleosomes and neutrophil activation as risk factors for deep vein thrombosis. Arterioscler Thromb Vasc Biol 33:147–151. https://doi.org/10.1161/atvbaha.112.300498
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM (2000) Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 20:1262–1275
Vogel B, Shinagawa H, Hofmann U, Ertl G, Frantz S (2015) Acute DNase1 treatment improves left ventricular remodeling after myocardial infarction by disruption of free chromatin. Basic Res Cardiol 110:15. https://doi.org/10.1007/s00395-015-0472-y
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S (2012) Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med 209:819–835. https://doi.org/10.1084/jem.20112322
Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, Schmidt R, Kunkel SL, Greenfield LJ (1995) Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol 15:258–268
Wang J, Arase H (2014) Regulation of immune responses by neutrophils. Ann N Y Acad Sci 1319:66–81. https://doi.org/10.1111/nyas.12445
Wang J-G, Mu J-S, Zhu H-S, Geng J-G (2002) N-desulfated non-anticoagulant heparin inhibits leukocyte adhesion and transmigration in vitro and attenuates acute peritonitis and ischemia and reperfusion injury in vivo. Inflam Res 51:435–443. https://doi.org/10.1007/pl00012403
Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, Sonbuchner LS, McDonald CH, Cook RG, Dou Y, Roeder RG, Clarke S, Stallcup MR, Allis CD, Coonrod SA (2004) Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306:279–283. https://doi.org/10.1126/science.1101400
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA (2009) Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 184:205–213. https://doi.org/10.1083/jcb.200806072
Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V (2015) Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 349:316–320. https://doi.org/10.1126/science.aaa8064
Wildhagen KC, Garcia de Frutos P, Reutelingsperger CP, Schrijver R, Areste C, Ortega-Gomez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GA (2014) Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood 123:1098–1101. https://doi.org/10.1182/blood-2013-07-514984
Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, Luo Y, Levitt B, Glogowska M, Chandra P, Kulik L, Robinson WH, Arend WP, Thompson PR, Holers VM (2011) N-alpha-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol 186:4396–4404. https://doi.org/10.4049/jimmunol.1001620
Witko-Sarsat V, Rieu P, Descamps-Latscha B, Lesavre P, Halbwachs-Mecarelli L (2000) Neutrophils: molecules, functions and pathophysiological aspects. Lab Investig 80:617–653
Zernecke A, Bot I, Djalali-Talab Y, Shagdarsuren E, Bidzhekov K, Meiler S, Krohn R, Schober A, Sperandio M, Soehnlein O, Bornemann J, Tacke F, Biessen EA, Weber C (2008) Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ Res 102:209–217. https://doi.org/10.1161/circresaha.107.160697
Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL (2001) Association between myeloperoxidase levels and risk of coronary artery disease. JAMA 286:2136–2142
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Hofbauer, T.M., Ondracek, A.S., Lang, I.M. (2020). Neutrophil Extracellular Traps in Atherosclerosis and Thrombosis. In: von Eckardstein, A., Binder, C.J. (eds) Prevention and Treatment of Atherosclerosis . Handbook of Experimental Pharmacology, vol 270. Springer, Cham. https://doi.org/10.1007/164_2020_409
Download citation
DOI: https://doi.org/10.1007/164_2020_409
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-86075-2
Online ISBN: 978-3-030-86076-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)