
Abstract
Background
Myeloperoxidase (MPO) -containing macrophages and neutrophils have been described at sites of plaque rupture. The presence of these cells in precursor lesions to acute rupture (thin cap atheroma, or vulnerable plaque) and within thrombi adjacent to ruptures has not been described, nor an association with iron-containing macrophages within unstable plaques.
Methods
We studied 61 acute ruptures, 15 organizing ruptures, 31 thin cap fibroatheromas, and 28 fibroatheromas from 72 sudden coronary death victims by immunohistochemical and histochemical techniques. Inflammatory cells were typed with anti-CD68 (macrophages), anti-BP-30 (neutrophil bactericidal glycoprotein), and anti-MPO. Iron was localized by Mallory's Prussian blue stain. In selected plaques alpha smooth muscle actin (DAKO, Carpinteria, CA, clone M0851) was performed.
Results
MPO positive cells were present in 79% of ruptured caps, 28% of thin cap fibroatheroma, and no fibroatheromas; neutrophils were present in 72% of ruptures, 8% of thin cap fibroatheromas, and no fibroatheromas. Iron containing foam cells were present in the caps of 93% of acute ruptures, of 85% of organizing ruptures, 20% of thin cap atheromas, and 10% of fibroatheromas. MPO positive cells were more frequent in occlusive than non-occlusive thrombi adjacent to ruptures (p = .006) and were more numerous in diabetics compared to non-diabetics (p = .002)
Conclusion
Unstable fibrous caps are more likely to contain MPO-positive cells, neutrophils, and iron-containing macrophages than fibrous caps of stable fibroatheromas. MPO-positive cells in thrombi adjacent to disrupted plaques are associated with occlusive thrombi and are more numerous in diabetic patients.
Similar content being viewed by others


Background
Plaque rupture is a major cause of coronary thrombosis, and is morphologically characterized by an interruption in a thin fibrous cap overlying a lipid rich core. [1–4] Often there are numerous macrophages infiltrating the fibrous cap of rupture plaques, suggesting a critical role in inciting plaque rupture. More recently, myeloperoxidase (MPO) expressing monocytes and neutrophils have been described at the site of plaque disruption, suggesting a role of MPO in plaque rupture. [5, 6] However, the precise factor precipitating plaque rupture, as well as the morphologic and cellular differences that identify a vulnerable plaque (thin cap fibroatheroma) prone to rupture are currently not known.
Circulating monocytes produce hypochlorous acid by activation of MPO, increasing oxidative stress. [7, 8] Recent evidence suggests that MPO-producing macrophages may persist in the atherosclerotic plaque and contribute to plaque instability. [9] In addition, serum levels of MPO seem to correlate with adverse risks in acute coronary syndrome patients.[10]
The purpose of this study was threefold: (1) to corroborate Sugiyama et al's and Naruko's finding that MPO positive cells including neutrophils are increased at the site of plaque instability; [5, 6] (2) to quantitate MPO positive cells and neutrophils for the first time in thin-cap fibroatheroma; and (3) to correlate numbers of MPO positive cells in thrombi adjacent to ruptures with thrombus characteristics and risk factors.
Methods
Autopsy sections of coronary artery plaques were selected from patients dying with severe coronary atherosclerosis. Inclusion for study included apparent natural cardiac deaths at the time of autopsy, with drug-related deaths subsequently excluded in cases of positive toxicology. Cases were seen in consultation at the Armed Forces Institute of Pathology from the Office of the Chief Medical Examiner in Baltimore, Maryland, under institutional review board approval. Hearts with severe coronary artery disease (≥ 75% cross section luminal narrowing of ≥ 1 epicardial artery) were included for study. Sections selected for study included 135 sections from 72 patients, namely 28 fibroatheromas, 31 thin-cap fibroatheromas, 61 acute plaque ruptures, 15 healing plaque ruptures. Plaque types were defined as previously described [11–16].
The distribution of lesions per arterial segment is presented on Table 1. Healing rupture denoted areas of acute thrombus overlying plaque disruption, with endothelial cell organization of portion of the fibrin thrombus with persistence of fibrin within the thrombus. [12]. Risk factors were determined by clinical history obtained from investigators and by serum corroboration when necessary. No cases of intra-coronary intervention were included in the study.
Immunohistochemical inflammatory markers studied were CD68 (KP1 clone, Dako, Carpinteria, CA, dilution 1:50, pan-macrophage marker), BP 30/Cathepsin G (R & D Systems, Minneapolis, 1:80, neutrophil marker), and MPO (Biodesign, Kennebunk ME, dilution 1:250). Iron stain was performed using standard Perls modification of Mallory Prussian blue iron histochemical stain. In selected plaques alpha smooth muscle actin (DAKO, Carpinteria, CA, clone M0851) was performed at 1:400 dilution. Quantitation of cellular elements was performed in the fibrous cap region by computerized morphometric measurements (IPLab SpectrumTM image processing software, Signal Analytics Corporation, Vienna, VA). Comparisons of two means was performed using Student's T test, and of multiple categories was performed using ANOVA means table with Fisher's post hoc testing. Non-parametric comparison was performed using Mann-Whitney comparison means testing. Statistical analysis was performed using SAS software (Cary, NC).
Results
The patient age ranged from 37 to 82 (mean, 52 years), and there were 66 male and 6 females patients. Seventy-three percent were white and 26% black. The heart weights ranged from 204 to 875 (mean 511 g). Risk factors included 10 diabetic patients, 22 hypertensive, 37 hypercholesterolemic and 36 smokers. There were no statistical differences between the three groups (acute rupture, healing rupture, stable plaque) in regards to sex, age, gender, or heart weight (Table 2). Risk factors and demographic data are presented in Table 2, as well as distribution of sampled plaque types by culprit plaque morphology. Of the 72 patients, culprit plaques were 51 acute ruptures (Figures 1 and 2), 5 healing ruptures, and 16 stable plaques without plaque disruption. The plaques sampled were 61 ruptures, 15 healing ruptures, 31 thin cap fibroatheromas, and 28 fibroatheromas with thick caps.

Acute plaque rupture with iron-containing macrophages at the rupture site. A. Low magnification of plaque rupture with occlusive thrombus. Movat pentachrome. B. Higher magnification of A showing rupture site demonstrating fragmented cap (top) and thrombus below. C. Perl's iron stain of the same area as B, demonstrating a large number of hemosiderin-laden macrophages at the rupture site.

Acute plaque rupture with iron-containing macrophages at the rupture site. A. Low magnification of plaque rupture with occlusive thrombus. Movat pentachrome. B. Higher magnification of A showing rupture site with prominent cholesterol crystals. C. Perl's iron stain, low magnification demonstrating hemosiderin-laden macrophages at the rupture site (arrow), seen at higher magnification in D.
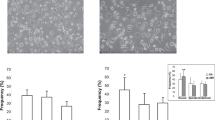
Table 3 demonstrates the density of macrophages (identified by CD 68 immunostaining), neutrophils (identified by cathepsin G immunostaining), MPO containing macrophages/neutrophils (Figures 3 and 4), and iron-containing macrophages within the fibrous caps of fibroatheromas, thin cap fibroatheromas, and acute and healing ruptures.

MPO positive cells and neutrophils proximal to and at acute rupture sites. A. 3 mm proximal to an acute rupture site there were only scattered MPO-positive macrophages at the luminal surface. B. The rupture site demonstrates numerous MPO positive cells. C and D are corresponding images of the sites of A and B, respectively, demonstrating that many of the MPO positive cells express cathepsin G, a neutrophil marker.

MPO positive cells in the rupture site thrombus. A demonstrates a low magnification of rupture of a large necrotic core. The arrow is the rupture site shown at higher magnification at B, which shows the fibrous cap (fc) above a thrombus rich in MPO-positive cells (immunohistochemical stain for myeloperoxidase). C is a slightly higher magnification demonstrating the thrombus (thr) just adjacent to the torn fibrous cap.
There was significant increase in macrophages in thin cap fibroatheromas, ruptures, and healing ruptures vs. fibroatheromas (p < .0001). No fibroatheroma had intracap neutrophils, vs. 8% of thin cap fibroatheromas, 79% of acute ruptures, and 35% of healing ruptures (p < .0001). There was no difference in intracap MPO positive macrophages or neutrophils between thin cap fibroatheromas with culprit plaque acute ruptures as compared to stable plaque. MPO cells were absent in fibroatheromas, but present in 28% of thin cap fibroatheromas, 79% of ruptures, and 60% of healing ruptures (p < .0001). Iron containing macrophages were present similarly more frequently in acute and healing ruptures than fibroatheromas and thin cap fibroatheromas (p < .0001).
Occlusive thrombi were longer than non-occlusive (Table 4), although there was no difference in length of necrotic core. Density of cap MPO macrophages/neutrophils did not differ between occlusive and non-occlusive thrombi; however, adjacent thrombus had a higher concentration of MPO and CD68 macrophages than non-occlusive thrombi (p < .01, Table 4). Density of intracap macrophages overlying disrupted plaques (acute or organizing ruptures) did not vary by risk factor; however, the density of MPO positive cells was greater in diabetics with borderline significance for smokers (Table 5). Acute ruptures characterized by thin caps without actin positive cells, variable numbers of actin positive smooth muscle cells at the base of the plaque towards the internal elastic laminae (IEL). Healing ruptures, the rupture site characterized by fibrin, granulation tissue and none to few actin positive cells, again variable numbers of actin positive smooth muscle cells at the base of the plaque towards the IEL. In thin cap fibroatheromas, the cap itself contained no smooth muscle cells, but there were variable numbers of actin positive smooth muscle cells at the base of the plaque towards the IEL."
Discussion
The current study corroborates work by others, showing increased MPO-containing cells and neutrophils in plaque rupture sites. [5, 6] It reveals new information regarding MPO content comparing plaque subtype, especially small numbers in thin cap atheroma. MPO co-localizes with iron containing macrophages on the surface and its density in thrombotic lesions correlates well with thrombus size. The numbers of MPO containing cells are also significantly higher in patients with diabetes and smoking history, creating a link between risk factors and histomorphological findings.
MPO has emerged as a potential participant in the promotion of atherosclerosis. [17] It is stored and secreted from activated neutrophils and monocytes, and is an important component in degranulation material of leukocytes, critical in human innate host defenses. [5, 6] The MPO role in atherosclerosis initiation and propagation is related to its potential to activate lipid peroxidation and promoting post-translational modification of target proteins.[18, 19] MPO catalyzes LDL oxidation and releases HOCl, degrading extracellular matrix. Recent studies have found that MPO is capable of promoting oxidation of lipoproteins which could lead to the increase in cholesterol deposit and formation of foam cells in fatty streaks.[18, 20]
Tissue localization of MPO has been described in a variety of inflammatory conditions,[19] MPO was found to be increased in human atheromas more than a decade ago.[21] More recently, studies have focused on the presence of MPO not only in early lesions, but also in acute complications of atherosclerosis and plaque vulnerability.[9] The link between MPO levels and CAD is strongly supported by epidemiological studies. Individuals who have high MPO levels are more likely to demonstrate abnormal coronary angiograms compared to controls.[22] Individuals with MPO deficiency have some protection against CAD and others harboring a polymorphism that decreases MPO expression have markedly reduced rates of CAD, myocardial infarction and cardiac death. [23–25]
The current study corroborates the findings of Naruko et al who analyzed 126 coronary sections using immunohistochemical studies for macrophages (CD66b), neutrophils (CD11b) and myeloperoxidase. [5] The population in their study was composed of material from autopsies and atherectomy procedures. In the control of patients dying form non-cardiac deaths, MPO was not indentified in any case, whereas ruptured or eroded plaques had MPO positivity mostly in neutrophils, and only occasionally in macrophages. A similar approach was used by Sugiyama et al, who found increased numbers of MPO-containing macrophages in eroded and ruptured plaques, with little to no MPO in fatty streaks. The expression of MPO was also variably dependent on the stage of atherosclerotic lesions, with special high expression with fibrous caps and activated plaques. [6] Our finding of higher MPO expression in patients with diabetes substantiates clinical studies that measured MPO in plasma of diabetic patients versus controls. [26, 27]
It is known that free iron in the plasma catalyzes lipid peroxidation and this reaction has been involved in the development of atherosclerosis in the way of hydroxyl radical formation by neutrophils requiring exogenous iron.[28] Iron is stored as either ferritin or hemosiderin. Ferritin consists of an outer protein shell with iron complexed within the core. It has been postulated that either MPO or lactoferrin could inhibit the formation of the hydroxyl radical upon neutrophil degranulation. Since it can be seen by light microscopy as gold-brown granules and is demonstrated by the Prussian blue stain, we assessed positivity of iron containing cells in culprit lesions and found that these were more frequently present in acute and healing ruptures than fibroatheromas and thin cap fibroatheromas. Although the molecular relationship between iron and myeloperoxidase is unknown, MPO contains an iron porphyrin prosthetic group. In other inflammatory conditions, such as asthma, there seems to be an association between levels of MPO and iron, and it is suggested they may act in a concerted manner in the pathogenesis of the disease. [29] While our findings of elevated iron and MPO do not elucidate its possible mechanistic relationship, it brings new data to the discussion of the pathogenesis of atherosclerosis progression.
Conclusion
MPO localizes in macrophages and neutrophils at the site of plaque rupture, as has been previously demonstrated, and only occasionally in precursor thin cap atheroma. A causative association between MPO and rupture has not been established but remains a possibility that requires further study. Localization of iron at rupture sites is a novel finding that may have implications for imaging of unstable plaques. Whether there is a link between iron deposits at rupture sites and MPO likewise remains an area for investigation.
References
Falk E: Morphologic features of unstable atherothrombotic plaques underlying acute coronary syndromes. Am J Cardiol. 1989, 63 (10): 114E-120E. 10.1016/0002-9149(89)90242-7.
Falk E: Coronary thrombosis: pathogenesis and clinical manifestations. Am J Cardiol. 1991, 68 (7): 28B-35B. 10.1016/0002-9149(91)90382-U.
Farb A, Tang AL, Burke AP, Sessums L, Liang Y, Virmani R: Sudden coronary death. Frequency of active coronary lesions, inactive coronary lesions, and myocardial infarction. Circulation. 1995, 92 (7): 1701-1709.
Wal van der AC, Becker AE, Loos van der CM, Das PK: Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology. Circulation. 1994, 89 (1): 36-44.
Naruko T, Ueda M, Haze K, Wal van der AC, Loos van der CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, et al: Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002, 106 (23): 2894-2900. 10.1161/01.CIR.0000042674.89762.20.
Sugiyama S, Okada Y, Sukhova GK, Virmani R, Heinecke JW, Libby P: Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am J Pathol. 2001, 158 (3): 879-891.
Loria V, Dato I, Graziani F, Biasucci LM: Myeloperoxidase: a new biomarker of inflammation in ischemic heart disease and acute coronary syndromes. Mediators Inflamm. 2008, 2008: 135625-10.1155/2008/135625.
Meuwese MC, Stroes ES, Hazen SL, van Miert JN, Kuivenhoven JA, Schaub RG, Wareham NJ, Luben R, Kastelein JJ, Khaw KT, et al: Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007, 50 (2): 159-165. 10.1016/j.jacc.2007.03.033.
Hazen SL: Myeloperoxidase and plaque vulnerability. Arterioscler Thromb Vasc Biol. 2004, 24 (7): 1143-1146. 10.1161/01.ATV.0000135267.82813.52.
Brennan ML, Hazen SL: Emerging role of myeloperoxidase and oxidant stress markers in cardiovascular risk assessment. Curr Opin Lipidol. 2003, 14 (4): 353-359. 10.1097/00041433-200308000-00003.
Burke AP, Farb A, Malcom GT, Liang Y, Smialek J, Virmani R: Effect of risk factors on the mechanism of acute thrombosis and sudden coronary death in women. Circulation. 1998, 97 (21): 2110-2116.
Burke AP, Farb A, Malcom GT, Liang YH, Smialek J, Virmani R: Coronary risk factors and plaque morphology in men with coronary disease who died suddenly. N Engl J Med. 1997, 336 (18): 1276-1282. 10.1056/NEJM199705013361802.
Burke AP, Kolodgie FD, Farb A, Weber D, Virmani R: Morphological predictors of arterial remodeling in coronary atherosclerosis. Circulation. 2002, 105 (3): 297-303. 10.1161/hc0302.102610.
Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R: Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001, 103 (7): 934-940.
Farb A, Burke AP, Tang AL, Liang TY, Mannan P, Smialek J, Virmani R: Coronary plaque erosion without rupture into a lipid core. A frequent cause of coronary thrombosis in sudden coronary death. Circulation. 1996, 93 (7): 1354-1363.
Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM: Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000, 20 (5): 1262-1275.
Nicholls SJ, Hazen SL: Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2005, 25 (6): 1102-1111. 10.1161/01.ATV.0000163262.83456.6d.
Podrez EA, Abu-Soud HM, Hazen SL: Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic Biol Med. 2000, 28 (12): 1717-1725. 10.1016/S0891-5849(00)00229-X.
Zhang R, Brennan ML, Shen Z, MacPherson JC, Schmitt D, Molenda CE, Hazen SL: Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J Biol Chem. 2002, 277 (48): 46116-46122. 10.1074/jbc.M209124200.
Roman RM, Wendland AE, Polanczyk CA: Myeloperoxidase and coronary arterial disease: from research to clinical practice. Arq Bras Cardiol. 2008, 91 (1): e11-19. 10.1590/S0066-782X2008001300015.
Daugherty A, Dunn JL, Rateri DL, Heinecke JW: Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994, 94 (1): 437-444. 10.1172/JCI117342.
Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, Topol EJ, Sprecher DL, Hazen SL: Association between myeloperoxidase levels and risk of coronary artery disease. Jama. 2001, 286 (17): 2136-2142. 10.1001/jama.286.17.2136.
Asselbergs FW, Reynolds WF, Cohen-Tervaert JW, Jessurun GA, Tio RA: Myeloperoxidase polymorphism related to cardiovascular events in coronary artery disease. Am J Med. 2004, 116 (6): 429-430. 10.1016/j.amjmed.2003.10.025.
Kutter D, Devaquet P, Vanderstocken G, Paulus JM, Marchal V, Gothot A: Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit?. Acta Haematol. 2000, 104 (1): 10-15. 10.1159/000041062.
Nikpoor B, Turecki G, Fournier C, Theroux P, Rouleau GA: A functional myeloperoxidase polymorphic variant is associated with coronary artery disease in French-Canadians. Am Heart J. 2001, 142 (2): 336-339. 10.1067/mhj.2001.116769.
Ahmed FN, Naqvi FN, Shafiq F: Lipid peroxidation and serum antioxidant enzymes in patients with type 2 diabetes mellitus. Ann N Y Acad Sci. 2006, 1084: 481-489. 10.1196/annals.1372.022.
Wiersma JJ, Meuwese MC, van Miert JN, Kastelein A, Tijssen JG, Piek JJ, Trip MD: Diabetes mellitus type 2 is associated with higher levels of myeloperoxidase. Med Sci Monit. 2008, 14 (8): CR406-410.
Britigan BE, Hassett DJ, Rosen GM, Hamill DR, Cohen MS: Neutrophil degranulation inhibits potential hydroxyl-radical formation. Relative impact of myeloperoxidase and lactoferrin release on hydroxyl-radical production by iron-supplemented neutrophils assessed by spin-trapping techniques. Biochem J. 1989, 264 (2): 447-455.
Ekmekci OB, Donma O, Sardogan E, Yildirim N, Uysal O, Demirel H, Demir T: Iron, nitric oxide, and myeloperoxidase in asthmatic patients. Biochemistry (Mosc). 2004, 69 (4): 462-467. 10.1023/B:BIRY.0000026205.89894.25.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2261/9/27/prepub
Acknowledgements
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LL and MR collected all cases and participated on data analysis. LL and AB participated in the design of the study. FT and AB performed the statistical analysis. AB and FT conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tavora, F.R., Ripple, M., Li, L. et al. Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovasc Disord 9, 27 (2009). https://doi.org/10.1186/1471-2261-9-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2261-9-27