
Abstract
Sepsis-associated encephalopathy (SAE) is associated with an increased rate of morbidity and mortality. It is not understood what the exact mechanism is for the brain dysfunction that occurs in septic patients, but brain inflammation and oxidative stress are a possible theory. Such events can occur through the alteration of molecules that perpetuate the inflammatory response. Thus, it is possible to postulate that CD40 may be involved in this process. The aim of this work is to evaluate the role of CD40-CD40L pathway activation in brain dysfunction associated with sepsis in an animal model. Microglia activation induces the upregulation of CD40-CD40L, both in vitro and in vivo. The inhibition of microglia activation decreases levels of CD40-CD40L in the brain and decreases brain inflammation, oxidative damage and blood brain barrier dysfunction. Despite this, anti-CD40 treatment does not improve mortality in this model. However, it is able to improve long-term cognitive impairment in sepsis survivors. In conclusion, there is a major involvement of the CD40-CD40L signaling pathway in long-term brain dysfunction in an animal model of sepsis.
Similar content being viewed by others

Introduction
Sepsis and its consequences are the most common causes of mortality in intensive care units (1,2). Sepsis-associated encephalopathy (SAE) has many different clinical presentations, but one of the most common findings is the altered level of consciousness that ranges from confusion to delirium and coma (3). Furthermore, SAE is associated with an increased rate of morbidity and mortality (4).
The exact mechanism to explain brain dysfunction in septic patients is not fully understood, but it appears to involve different mechanisms that include oxidative stress, cerebral energy dysfunction, blood-brain barrier (BBB) disruption and disturbances in neurotransmission (2,6–8). All these alterations could be related to an inappropriate activation of the immune system (5). In this context, evidence suggests that there is a critical role for the CD40-CD40 ligand (L) signaling pathway in several inflammatory disorders. This pathway was described in different conditions associated with both neuroinflammation and brain oxidative stress (9–12), suggesting that it can be relevant to the development of SAE. Moreover, it has been proven in sepsis that there is a systemic increase of CD40 and its ligand (13,14). CD40-CD40L interaction stimulates many transcription factors, thereby increasing the expression of many mediators such as adhesion molecules, cytokines, growth factors and metalloproteinases (MMPs) (15). The integrity of the BBB appears to play a major role in the progression of brain dysfunction after sepsis. Additionally, the activation of the CD40-CD40L pathway could interfere in inflammatory mediator signaling that may participate in the breakdown of BBB. Thus, we hypothesize that the CD40-CD40L signaling could be of major relevance in brain dysfunction associated with sepsis.
The aim of this work is to evaluate the role of CD40-CD40L pathway activation in the brain dysfunction associated with sepsis in an animal model.
Materials and Methods
Drugs
CD40 and CD40L were purchased from Abcam (Cambridge, MA, USA). Dulbecco modified Eagle medium (DMEM), fetal bovine serum, trypsinethylenediaminetetraacetic acid (Trypsin-EDTA), deoxyribonuclease I, penicillin-streptomycin and minocycline were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Ethics
The experimental procedures involving animals were in accordance with the National Institutes of Health (Bethesda, MD, USA) Guide for the Care and Use of Laboratory Animals, adopted by the National Institutes of Health (NIH) (16) and the approval of our institutional ethics committee.
Primary Microglia Culture
Primary mixed glial cell cultures were prepared from the cerebral cortex of 7-d-old rats (Wistar) as described previously (17). Cells isolated from cerebral hemispheres were dissociated in PBS containing 0.25% trypsin and 0.02 mg/mL deoxyribonuclease I and plated at a density of 0.1 × 106 cells/cm2 in 75-cm2 culture flasks in DMEM supplemented with 10% fetal bovine serum. At confluency (12–14 d), primary glial cultures were used to isolate microglial cells as described previously (17). Mixed glial cultures were shaken briefly to dislodge any microglial cells that were loosely attached to the astrocytes. Microglial cells were purified by preplating the cells for 30 min into culture flasks (75 cm2) at a density of 0.1 × 106 cells/cm2. The contaminated cells were removed by changing the culture medium. After isolation, microglial cultures were treated with lipopolysaccharides (LPS) (100 µg/mL) for 24 h, then treated with anti-CD40 (100 µg/flask) for an additional 24 h. After this period, both medium and cells were collected to different analyses (see below).
Procedures—Sepsis Induction and Treatments
Rats were subjected to cecal ligation and puncture (CLP) as described previously (18). They were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (10 mg/kg). Under aseptic conditions, a 3-cm midline laparotomy was performed to expose the cecum and adjoining intestine. The cecum was ligated with a 3.0 silk suture at its base, below the ileocecal valve, and was perforated once with 14-gauge needle. The cecum was then squeezed gently to extrude a small amount of feces through the perforation site. The cecum was then returned to the peritoneal cavity, and the laparotomy was closed with 4.0 silk sutures. Animals were resuscitated with regular saline (50 mL/kg) subcutaneously (SC) immediately after and 12 h after CLP. All animals received antibiotics (ceftriaxone at 30 mg/kg and clindamycin 25 mg/kg) every 6 h SC for 3 d. In the sham-operated group, the rats were submitted to all surgical procedures but the cecum was neither ligated nor perforated. To minimize variability between different experiments, the CLP procedure was always performed by the same investigator.
For some of the subsets of the experiment, animals were treated with anti-CD40 or isotype IgG as a control. Immediately after the CLP procedure, animals were placed in a stereotactic apparatus and the hippocampus was cannulated and anti-CD40 (1 µg/kg, 10 µg/kg or 100 µg/kg) was administered (19). In addition, some animals were treated with minocycline as described previously (20). In brief, immediately after sepsis induction, minocycline (100 µg/kg) or saline was administered as a single intracerebroventricular injection. Depending on the experiment, some animals were followed to the determination of mortality, subjected to behavioral tasks or killed at different times. The hippocampus was then removed to perform several analyses (see below).
Measures
Survival studies. Forty-four animals were observed at regular (12 h) intervals to the determination of mortality over ten days after sepsis induction (sham-operated animals, CLP plus anti-CD40 (100 µg/kg), CLP plus saline, n = 18 animals each group)
Behavior tests. The animals (n = 10) separately underwent two different behavioral tasks 10 d after surgery. The behavioral procedures were conducted between the hours of 1300 and 1600 in a sound-isolated room, and a single animal performed only one behavioral test at only one point in time after surgery. All behavioral tests were recorded by the same person who was blind to the animal group.
The inhibitory avoidance procedure was described in a previous report (21). The apparatus was an acrylic box (50 × 25 × 25 cm) whose floor consisted of parallel-caliber stainless-steel bars (1-mm diameter) spaced 1 cm apart, and a platform measuring 7 cm wide and 2.5 cm high. Animals were placed on the platform and their latency to step down on the grid with all four paws was measured with an automatic device. Training sessions were performed 10 d after surgery. Immediately after stepping down on the grid, animals received a foot shock of 0.3 mA for 2 s. In test sessions carried out 24 h after training, no foot shock was given and the step-down latency (maximum of 180 s) was used as a measure of retention.
The habituation to the open-field task evaluates motor performance in the training section and nonassociative memory in the retention test session. Habituation to an open field was carried out in a 40 × 60 cm open field surrounded by 50 cm high walls made of brown plywood with a frontal glass wall. The floor of the open field was divided into 12 equal rectangles by black lines. The animals were gently placed on the left rear quadrant and allowed to explore the arena for 5 min (training session). Immediately following this, the animals were taken back to their home cage and 24 h later submitted again to a similar open-field session (test session). Crossing of the black lines and rearing performed in both sessions were counted. The decrease in the number of crossings and rearings between the two sessions was taken as a measure of the retention of habituation (22).
CD40 and CD40L levels. To perform immunoblotting, samples of brain tissue were homogenized in Laemmli buffer (62.5 mmol/L Tris-HCl, pH 6.8, 1% [w/v] sodium dodecyl sulfate [SDS], 10% [v/v] glycerol) and equal amounts of protein (100 µg/well) were fractionated by polyacrylamide gel electrophoresis-sodium dodecyl sulphate (SDS-PAGE) and electrotransferred to nitrocellulose membranes. The efficiency of electrotransfer was verified by Ponceau S staining, and the membrane was blocked in Tris-Tween buffer saline (TTBS) (100 mmol/L Tris-HCl, pH 7.5, containing 0.9% NaCl and 0.1% Tween 20) with 5% milk. The membranes were incubated overnight at 4°C with rabbit polyclonal anti-CD40 (1:1000) (Abcam) or anti-CD40L (1:1000) (Abcam). Secondary anti-rabbit IgG was incubated with the membrane for 2 h (1:10000). The membrane was washed with TTBS and the immunoreactivity was detected by chemiluminescence using enhanced chemiluminescent (ECL) (Bio-Rad, São Paulo, Brazil) substrates for detection of horseradish peroxidase (HRP) enzyme activity. Densitometry analysis was performed with the software ImageJ v.1.34 (NIH, Bethesda, MD, USA). All results are expressed as a relative ratio between CD40 or CD40L and β-actin.
Levels of cytokines. Concentrations of TNF-α, IL-1β and IL-6 were determined by enzyme-linked immunosorbent assay (ELISA) on microplate reader using commercial kits (Peprotech, São Paulo, Brazil).
Blood-Brain Barrier Permeability. The integrity of the BBB was investigated in animals using Evans blue dye extravasation (23). The dye was administered (2% wt/vol in phosphate-buffered saline [PBS]) intravenously (3 mL/kg) through the femoral vein of the rats. The animals were perfused 1.5 h after using normal saline (250 mL) through the left ventricle at 110 mmHg pressure until colorless perfusion fluid was obtained from the right atrium. Quantitative evaluation of BBB permeability was achieved by measuring the content of Evans blue in the hippocampus by its fluorescence intensity (nanogram per milligram of brain tissue) (Spectramax M2 microplate reader, Molecular Devices, Sunnyvale, CA, USA).
Thiobarbituric Acid Reactive Species. The formation of thiobarbituric acid reactive substances (TBARS) during an acid-heating reaction was measured as an index of oxidative stress as described previously (24). The samples were mixed with 1 mL of trichloroacetic acid 10% and 1 mL of thiobarbituric acid 0.67% (Sigma-Aldrich) and then heated in a boiling water bath for 15 min. Malondialdehyde (MDA) equivalents were determined by the absorbance at 535 nm using 1,1,3,3- tetramethoxypropane (Sigma-Aldrich) as an external standard. Results were expressed as MDA equivalents per mg of protein.
Measurement of Nitrite-Nitrate Concentration. Nitrite-nitrate concentration was assayed spectrophotometrically using Griess reagents (1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-naphthylethylenediamine dihydrochloride in bidistilled H2O [NED solution]) and vanadium (III) chloride as described previously (25). A standard curve was run simultaneously with each set of samples and the optical density at 550 nm (OD550) was measured using ELISA microplate reader (25).
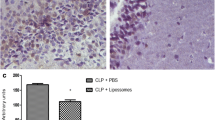
Myeloperoxidase activity. The tissue was homogenized (50 mg/mL) in 0.5% of hexadecyltrimethylammonium bromide (Sigma-Aldrich) and centrifuged (8765g). The suspension was sonicated and an aliquot of supernatant was mixed with a solution of 1.6 mmol/L 3,3′,5,5′-tetramethylbenzidine (TMB) and 1 mmol/L H2O2. The myeloperoxidase (MPO) activity was measured spectrophotometrically at 650 nm at 37°C. The results were expressed as mU/mg protein. (26).
Protein determination. The Lowry protein assay was used to measure the total protein concentration (27).
Statistical Analysis
Data collected were analyzed with oneway analysis of variance (ANOVA) followed by the Tukey post hoc method and expressed as mean ± standard deviation. Data from the open-field task were analyzed with ANOVA followed by Tukey post hoc method and expressed as mean ± SEM. Data from the inhibitory avoidance task is reported as median and interquartile ranges and comparisons among groups were performed using the Mann-Whitney U tests. For behavioral analyses, individual groups were analyzed by the Wilcoxon tests. Mortality was analyzed by the Kaplan-Meier survival curves and log-rank test. For all comparisons, p < 0.05 indicated statistical significance.
Results
Microglia Activation Induces the Upregulation of CD40-CD40L
The in vitro activation of microglia by LPS was able to increase the cellular expression of CD40 and the secretion of CD40L (Figure 1). The treatment with anti-CD40 was able to decrease CD40 expression in microglia, causing a positive feedback loop (see Figure 1). This also was observed in vivo. By using the CLP model of sepsis, it was demonstrated that from 12 to 48 h sepsis induced and increased the hippocampal content of both CD40 and CD40L (see Figure 1). The upregulation of CD40-CD40L seems to be dependent of microglia activation in vivo, since the inhibition of microglia by minocycline was able to abrogate sepsis-induced upregulation of CD40-CD40L 24 h after CLP (see Figure 1).

(A) Western blotting analysis on activated microglial culture with LPS and treated with antiCD40 and CD40L. (B) Expression CD40 and CD40L for Western blotting 24 h in the hippocampus of rats submitted for sham and CLP or CLP and CLP plus minocycline. Levels of CD40 and CD40L (C) 12 h (D) 24 h and (E) 48 h by Western blotting. Data are expressed as protein levels/β-actin. For five independent experiments performed in duplicate (mean ± SD). Representative bands. *Different from sham (p < 0.05); #different from CLP plus control (p < 0.05).
The Upregulation of CD40-CD40L Induces Brain Inflammation
Microglia increased the secretion of TNF-α, IL-1β and IL-6 (Figures 2A-C) in response to the in vitro LPS treatment. Blocking the activation of CD40-CD40L by anti-CD40 decreased the in vitro secretion of the cytokines mentioned previously (see Figures 2A-C). To confirm these results, we determined hippocampal levels of TNF-α, IL-1β and IL-6, 24 h after CLP. As demonstrated previously, sepsis is associated with an increase in hippocampal levels of TNF-α, IL-1β and IL-6, and the intrahippocampal administration of anti-CD40 decreases cytokine levels in a way that resembles a dose-response shape (Figures 2D-F). This also was observed in the hippocampal activity of MPO as a neutrophil accumulation marker (Figure 2G).

Cytokine levels (A) TNF-α, (B) IL-1 β and (C) IL-6 in microglial culture activated with LPS and treated or not treated with anti-CD40; cytokines (D) TNFα, (E) IL-1 β and (F) IL-6 in the hippocampus of rats submitted to sepsis by CLP treated or not with anti-CD40 (1, 10 or 100 µg/kg) and MPO activity in the hippocampus of rats submitted to sepsis by CLP treated or not treated with anti-CD40 (1, 10 or 100 µg/kg). Data are expressed as pg/mg protein and nmol/mg protein, for independent experiments performed in duplicate (mean ± SD). *Different from sham (p < 0.05); #different from CLP plus saline (p < 0.05); &different from CLP plus anti-CD401 µg/kg (p < 0.05).
Anti-CD40 Treatment Decreases Markers of Brain Injury after Sepsis
After sepsis, brain inflammation could induce oxidative and nitrosative stress as well as cause dysfunction of the BBB. Hippocampal levels of nitrite and TBARS were increased 24 h after CLP (Figures 3A, B). Treatment with anti-CD40 was able to consistently decrease hippocampal levels of nitrite in all studied doses (see Figure 3A). Additionally, the high anti-CD40 dose was able to decrease hippocampal levels of TBARS (see Figure 3B). Dysfunction of the BBB also occurs in the CLP model 24 h after surgery as described previously (28) and improvement was seen with anti-CD40 treatment at 10 and 100 µg/kg (Figure 3C).

Nitrate concentration (A) thiobarbituric acid reactive substances (TBARS) (B) and permeability of blood brain barrier (BBB) (C) in the hippocampus of rats submitted to sepsis by CLP treated or not treated with anti-CD40 (1, 10 or 100 µg/kg). Data are expressed as nmol/mg protein and ng/mL, for independent experiments performed in duplicate (mean ± SD). *Different from sham (p < 0.05); #different from CLP plus saline (p < 0.05); &different from CLP plus anti-CD401 µg/kg (p < 0.05).
Anti-CD40 Treatment Does Not Improve Mortality after Sepsis
Since brain dysfunction in the clinical setting is associated with higher mortality rates, we aimed to determine if the protective effects of anti-CD40 on brain inflammation were able to improve mortality as an adjunctive treatment to antibiotics and fluid administration. Despite this, anti-CD40 at the higher dose (100 µg/kg) was not able to improve mortality in this model (Figure 4).

Kaplan-Meier curve of survival times in sham, CLP and CLP plus anti-CD40 (100 µg/kg) groups. *Different from sham (p < 0.05). Data are analyzed to log-rank.
Anti-CD40 Treatment Improves Long-term Cognitive Impairment in Sepsis Survivor Animals
Another clinically relevant outcome after brain dysfunction is long-term cognitive impairment. We consistently demonstrated that survivors from this model had several different cognitive deficits 10 d after CLP (29). Thus, it was determined that anti-CD40 was able to improve long-term cognitive impairment. Animals subjected to sepsis presented with impairment in both open-field and inhibitory avoidance tasks (Figures 5A, B). Anti-CD40 treatment (100 µg/kg) improved both aversive and nonaversive memory deficits induced by sepsis (see Figures 5A, B).

Habituation on (A) the open field and (B) inhibitory avoidance. Animals were submitted to sham-operated and CLP treated or not treated with anti-CD40 (100 µg/kg). Data are presented as mean ± SEM to open field and as median and interquartile ranges to inhibitory avoidance, n = 10 rats per group. *Different training (p < 0.05). sec, Seconds.
Discussion
We have demonstrated that microglia activation is followed by upregulation of CD40 and secretion of CD40L. This seems to be of importance in the development of brain inflammation associated with sepsis. However, anti-CD40 treatment does not improve mortality, but improves long-term cognitive impairment observed in sepsis survivors in this model.
The CD40-CD40L pathway seems to be involved in inflammatory responses implicated in the pathophysiology and progression of various autoimmune and inflammatory disorders (30–33). When microglial cells are exposed to proinflammatory mediators, the expression of CD40 is rapidly upregulated (11,34,35). LPS is a strong inducer of CD40 expression in microglia, possibly by the endogenous production of interferon-beta, which contributes to CD40 expression by the activation of STAT-1 (36). Additionally, patients with severe sepsis have higher circulating levels of soluble CD40L than healthy controls, and these levels are associated with the outcome (37). Thus, there is a biological plausibility that CD40 and its ligand could be upregulated in the brain of animals submitted to sepsis, as we have demonstrated here. To date, there is no evidence to suggest that sepsis could activate the CD40-CD40L pathway in the brain during sepsis development.
Activated microglia are known for playing a key role in mediating inflammatory processes associated with various central nervous system disorders (38,39). Activated microglia release compounds such as glutamate, free radicals, proteases, cytokines, leukotrienes and nitric oxide (NO) that could contribute to brain dysfunction (31,40). This can be further aggravated by the dysfunction of the BBB that is implicated in the pathogenesis of SAE (41). Microglia activation is an early event after systemic LPS injection (42), and can be maintained for longer periods of time after sepsis induction (43). We had previously demonstrated that microglial inhibition by minocycline was able to decrease brain inflammation and improve long-term cognitive dysfunction after sepsis (20). In this model, we demonstrated that a major player in microglia activation after sepsis is the activation of CD40-CD40L pathway. Furthermore, the inhibition of CD40-CD40L is able to decrease brain inflammation and oxidative damage as well as BBB dysfunction. Thus, all these alterations seem to be relevant to the development of SAE (44). Despite the positive results related to anti-CD40 treatment on brain inflammation, this was not associated with higher survivor rates.
Since the control of brain inflammation was not able to improve mortality in our model, we aimed to determine if anti-CD40 treatment could improve long-term brain dysfunction in sepsis survivors. We had demonstrated previously that the control of acute inflammation was associated with improved long-term outcome in the CLP model of sepsis (45–47). Here we demonstrated that the downregulation of CD40 signaling was able to improve long-term cognitive function in sepsis survivor rats. In neurodegenerative disorders such as Alzheimer’s disease, microglial activation occurs leading to an increase in CD40 signaling pathway (10,11,34). It was demonstrated that activation of the CD40-CD40L pathway modulates amyloid β-induced innate immune responses in microglia, including decreased microglia phagocytosis of exogenous amyloid β 1–42 (48). These data provide a mechanistic explanation for our previous work showing that long-term cognitive impairment is associated with increased brain content of amyloid-β and decreased levels of synaptophysin (49).
Conclusion
There is a major involvement of the CD40-CD40L signaling pathway in brain dysfunction in an animal model of sepsis. By blocking its activation, it is possible to decrease brain inflammation and oxidative damage, as well as long-term cognitive impairment.
Disclosures
The authors declare they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
References
Warren HS. (1997) Strategies for the treatment of sepsis. New Engl. J. Med. 336:952–3.
Hotchkiss RS, Karl IE. (2003) The pathophysiology and treatment of sepsis. New Engl. J. Med. 348:138–50.
Gofton TE, Young GB. (2012) Sepsis-associated encephalopathy. Nat. Rev. Neurol. 8:557–66.
Sprung CL, et al. (1990) Impact of encephalopathy on mortality in the sepsis syndrome. The Veterans Administration Systemic Sepsis Cooperative Study Group. Crit. Care Med. 18:801–6.
Dal-Pizzol F, Tomasi CD, Ritter C. (2014) Septic encephalopathy: does inflammation drive the brain crazy? Rev. Bras. Psiquiatr. 36:251–8.
Pine RW, et al. (1983) Determinants of organ malfunction or death in patients with intra-abdominal sepsis: A discriminant analysis. Arch. Surg. 118:242–9.
van den Boogaard M, et al. (2010) Endotoxemia-induced inflammation and the effect on the human brain. Crit. Care. 14:R81.
Zampieri FG, Park M, Machado FS, Azevedo LCP. (2011) Sepsis-associated encephalopathy: not just delirium. Clinics. 66:1825–31.
Semmler A, et al. (2008) Sepsis causes neuroinflammation and concomitant decrease of cerebral metabolism. J. Inflammation. 5:38.
Flierl MA, Rittirsch D, Huber-lang MS, Stahel PF. (2010) Pathophysiology of septic encephalopathy—an unsolved puzzle. Crit. Care. 14:165.
Peters AL, Alura L, Stunz GAB. (2010) CD40 and autoimmunity: The dark side of a great activator. Semin. Immunol 21:293–300.
Giunta B, Figueroa KP, Town T, Tan J. (2009) Soluble CD40 ligand in dementia. Drugs Future. 34:333–40.
Lorente L, et al. (2011) Association between serum soluble CD40 ligand levels and mortality in patients with severe sepsis. Crit. Care. 15:R97.
Sinistro A, et al. (2008) Downregulation of CD40 ligand response in monocytes from sepsis patients. Clin. Vaccine Immunol. 15:1851–8.
Chen K, et al. (2006). CD40/CD40L Dyad in the inflammatory and immune responses in the central nervous system. Cell. Mol. Immunol. 3:163–9.
Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Institute for Laboratory Animal Research, Division on Earth and Life Studies, National Research Council of the National Academies. (2011) Guide for the Care and Use of Laboratory Animals. 8th edition. Washington (DC): National Academies Press.
Gordon R, et al. (2011) A simple magnetic separation method for high-yield isolation of pure primary microglia. J. Neurosci. Methods. 194:287–96.
Fink MP, Heard SO. (1990) Laboratory models of sepsis and septic shock. J. Surg. Res. 49:186–96.
Paxinos G, Watson C. (1986) The Rat Brain: Stereotaxic Coordinates. 2nd edition. Academic Press, Sydney, Australia.
Michels M, et al. (2014) The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav. Immun. 43:54–9.
Roesler R, Vianna MR, de-Paris F, Quevedo J. (1999) Memory-enhancing treatments do not reverse the impairment of inhibitory avoidance retention induced by NMDA receptor blockade. Neurobiol. Learn. Mem. 72:252–8.
Vianna MR, Alonso M, Viola H, Izquierdo I. (2000) Role of hippocampal signaling pathways in long-term memory formation of a nonassociative learning task in the rat. Learn. Mem. 7:333–40.
Belayev L, Busto R, Zhao W, Ginsberg MD. (1996) Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res. 739:88–96.
Esterbauer H, Cheeseman KH. (1990) Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol. 186:407–21.
Miranda KM, Espey MG, Wink DA. (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide. 5:62–71.
De Young LM, Kheifets JB, Ballaron SJ, Young JM. (1989) Edema and cell infiltration in the phorbol ester-treated mouse ear are temporally separate and can be differentially modulated by pharmacologic agents. Agents Actions. 26:335–41.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–75.
Comim CM, et al. (2011) Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Int. Care Med 2011; 37:711–8.
Steckert AV, et al. (2013) Late brain alterations in sepsis-survivor rats. Synapse. 67:786–93.
Yi L, Chandrasekaran P, Venkatesan S. (2012) TLR signaling paralyzes monocyte chemotaxis through synergized effects of p38 MAPK and global Rap-1 activation. PloS One. 7:e30404.
Sayyah M, Najafabadi IT, Beheshti S, Majzoob S. (2003) Lipopolysaccharide retards development of amygdala kindling but does not affect fully-kindled seizures in rats. Epilepsy Res. 57:175–80.
Kotowicz K, Dixon GL, Klein NJ, Peters MJ, Callard RE. (2000) Biological function of CD40 on human endothelial cells: costimulation with CD40 ligand and interleukin-4 selectively induces expression of vascular cell adhesion molecule-1 and P-selectin resulting in preferential adhesion of lymphocytes. Immunology. 100:441–8.
O’Keefe GM, Nguyen VT, Benveniste EN. (2002) Regulation and function of class II major histocompatibility complex, CD40, and B7 expression in macrophages and microglia: Implications in neurological diseases. J. Neurovirol. 8:496–512.
Wesemann DR, Dong Y, O’Keefe GM, Nguyen VT, Benveniste EN. (2002) Suppressor of cytokine signaling 1 inhibits cytokine induction of CD40 expression in macrophages. J. Immunol. 169:2354–60.
Kacimi R, Giffard RG, Yenari M. (2011) Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB, JAK-STAT and JNK stress kinase pathways. J. Inflammation. 8:7.
Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN. (2005) LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood. 106:3114–22.
Ritter C, et al. (2003) Oxidative parameters and mortality in sepsis induced by cecal ligation and perforation. Intensive Care Med. 29:1782–9.
Dehmer T, Lindenau J, Haid S, Dichgans J, Schulz JB. (2000) Deficiency of inducible nitric oxide synthase protects against MPTP toxicity in vivo. J. Neurochem. 74:2213–6.
Jamin N, Junier MP, Granned TG, Cadussaeu J. (2001) Two temporal stages of oligodendroglial response to excitotoxic lesion in the gray matter of the adult rat brain. Exp. Neurol. 172:17–28.
Kremlev SG, Roberts RL, Palmer C. (2004) Differential expression of chemokines and chemokine receptors during microglial activation and inhibition. J. Neuroimmunol. 149:1–9.
Deng YY, Fang M, Zhu GF, Zhou Y, Zeng HK. (2013) Role of microglia in the pathogenesis of sepsis-associated encephalopathy. CNS Neurol. Disord. Drug Targets. 12:720–5.
Henry CJ, Huang Y, Wynne AM, Godbout JP. (2009) Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav. Immun. 23:309–17.
Hernandes MS, et al. (2014) The role of Nox2-derived ROS in the development of cognitive impairment after sepsis. J. Neuroinflammation. 11:36.
Tolenaar JL, et al. (2014) Predicting in-hospital mortality in acute type B aortic dissection: evidence from international registry of acute aortic dissection. Circulation. 130:S45–50.
Barichello T, et al. (2007) Antioxidant treatment prevented late memory impairment in an animal model of sepsis. Crit. Care Med. 35:2186–90.
Biff D, et al. (2013) Correlation of acute phase inflammatory and oxidative markers with long-term cognitive impairment in sepsis survivors rats. Shock. 40:45–8.
Dal-Pizzol F, et al. (2013) Matrix metalloproteinase-2 and metalloproteinase-9 activities are associated with blood-brain barrier dysfunction in an animal model of severe sepsis. Mol. Neurobiol. 48:62–70.
Townsend KP, et al. (2005) CD40 signaling regulates innate and adaptive activation of microglia in response to amyloid beta-peptide. Eur. J. Immunol. 35:901–10.
Schwalm MT, et al. (2014) Acute brain inflammation and oxidative damage are related to long-term cognitive deficits and markers of neurodegeneration in sepsis-survivor rats. Mol. Neurobiol. 49:380–5.
Acknowledgments
This work was supported by CNPq (grant number 476859/2012-3), and NENASC project (PRONEX program from CNPq/FAPESC).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, and provide a link to the Creative Commons license. You do not have permission under this license to share adapted material derived from this article or parts of it.
The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this license, visit (https://doi.org/creativecommons.org/licenses/by-nc-nd/4.0/)
About this article

Cite this article
Michels, M., Danieslki, L.G., Vieira, A. et al. CD40-CD40 Ligand Pathway Is a Major Component of Acute Neuroinflammation and Contributes to Long-term Cognitive Dysfunction after Sepsis. Mol Med 21, 219–226 (2015). https://doi.org/10.2119/molmed.2015.00070
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2119/molmed.2015.00070