
Abstract
Background
We present the clinical, MRI and CT findings in a case of a new mitochondrial genome mutation (tRNA arginine gene), characterized by brain calcifications which are indicative of Kearns–Sayre syndrome (KSS). Some radiological features resembled those of Fahr’s disease (affecting the PDGFRB gene).
Case presentation
A 36-year-old male presented some typical clinical features of KSS, including onset before 20 years of age, pigmentary retinopathy, progressive external ophthalmoplegia and ptosis. However, the hallmark radiological finding of diffuse calcifications in the nuclear ganglia resembles some cases related to the PDGRFB mutation. Genetic investigation revealed a new mutation in the mitochondrial tRNA-arginine gene.
Conclusions
Brain calcifications are a common feature of mitochondrial diseases, but little is known about their pathophysiology. Here, we describe radiological similarities between a new mitochondrial DNA mutation and other genetic conditions, which are related to Fahr’s disease. These similarities could provide new insights into putative genotype–phenotype correlations.
Similar content being viewed by others


Background
Mitochondrial dysfunction is emerging as a common contributor to neurodegeneration and spongiotic brain diseases and presents with a wide range of signs and severity (Ignatenko et al. 2020). The relationships between genotype and phenotype, as well as the exceptional variability of non-overlapping diseases, remain poorly understood (Chung et al. 2021).
Kearns–Sayre syndrome (KSS) is a rare, sporadic mitochondrial oculo-cranial-somatic neuromuscular disorder with an estimated prevalence of around 1/125,000 (Leal et al. 2016; Pasquini et al. 2020). KSS is usually associated with a single large-scale mitochondrial DNA (mtDNA) deletion, which can range from 1,000 to 10,000 base pairs in length (Khambatta et al. 2014). Over 150 deletions have been identified, the most common being a deletion of a 4.9 Kb fragment, which extends from base 8,469 to 13,147 and causes ~ 20% of cases (Alemi et al. 2007; Sequiera et al. 2021). In rare cases, KSS has also been associated with point mutations, frequently involving mitochondrial transfer RNA (tRNA) (Seneca et al. 2001; Emmanuele et al. 2011). These mutations are generally not inherited but instead arise spontaneously during early embryonic development (Ainslie et al. 2016). Normal mitochondrial function depends on the expression of the mitochondrial genome. Human mtDNA is a 16,569-kb circular molecule which contains 13 genes for the mitochondrial respiratory chain complexes subunits and 24 genes for mitochondrial protein biosynthesis, including 22 tRNA genes (Scaglia 2008).
KSS classically presents before 20 years of age with a clinical triad of pigmentary retinopathy, progressive external ophthalmoplegia and cardiac conduction anomalies. Many additional manifestations have been described, including weakness, fatigue, elevated levels of lactate and pyruvate, mental retardation, short stature, ataxia, hearing loss, and certain endocrinopathies (e.g. diabetes, hypoparathyroidism, dysthyroidism and suprarenal alterations) (Khambatta et al. 2014; Zavatta and Clarke 2021; Sharma et al. 2016). High protein levels have also been identified in the cerebrospinal fluid (CSF) (Khambatta et al. 2014).
KSS is usually confirmed by the presence of “ragged red fibres (RRF)” on muscle biopsy and cytochrome c oxidase (COX) deficiency (Goldstein and Falk 2003; Challa et al. 2004). Ragged red fibres correspond to clumps of altered mitochondria in the subsarcolemmal region of the muscle fibre and can usually be identified when muscle is stained with Gömöri trichrome stain (Reichmann et al. 1996). Cytochrome c oxidase is an essential enzyme that is active in mitochondria and its deficiency can result from mutation in the mitochondrial genome since three of its 13 structural subunits are encoded by mtDNA (Shoubridge 2001). COX/succinate dehydrogenase is identified by double-labelling histochemistry staining (Ross 2011).
KSS-related neuroradiological findings are unspecific. Spongiosis, myelin loss and gliosis are the main pathological findings (Finsterer 2018; Alston et al. 2017), explained by increased T2-spin echo signal on MRI examination. In a recent study (Yu et al. 2016), MRI showed symmetric high T2 signals in cerebral and cerebellar white matter, as well as in the brainstem. Although both grey and white matter can be affected, patients most often show symmetric high T2-weighted MRI signal in subcortical U-fibres and in the periventricular white matter. Subcortical involvement is suggestive of KSS and is differentiated from the prevalent deep white matter lesions seen in other mitochondrial disorders and leukodystrophies. The subcortical abnormalities can extend into the deep cerebral white matter (internal capsule, splenium, cerebral and cerebellar peduncles), basal ganglia (46.7% of patients), thalamus (53.3%) and thalamocortical connections. The tegmentum of the brainstem, efferent cerebellar fibres to the thalamus and the dentate nuclei are also frequently involved (Yu et al. 2016; Demange et al.1989; Chu et al. 1999; Pasquini et al. 2020). It is quite common to have restricted diffusion (evidenced by diffusion-weighted imaging (DWI)) at the involved sites due to the status spongiosus of the tissue (Sacher et al. 2005). Occasionally, T2 prolongation areas also show siderocalcific deposits, especially in basal ganglia and subcortical regions (Robertson et al. 1979). Finally, cerebral, cerebellar and brainstem atrophy may be reported (Chu et al. 1999).
This report presents a case of KSS in a 36-year-old male with white matter abnormalities and diffuse calcifications in the nuclear ganglia. These were explained by a newly identified single mutation in the mitochondrial tRNA arginine gene. The radiological and clinical features may be explained by KSS, but some aspects resemble Fahr’s disease, a related genetic condition.
Case presentation
A 36-year-old male presented with some typical clinical findings of KSS, including onset before 20 years of age, pigmentary retinopathy, progressive external ophthalmoplegia (CPEO) and ptosis. The patient had a history of complex childhood-onset neurological disease with fatigue, muscle weakness, hearing loss, cognitive impairment, headaches, and ataxia. There was no evidence of alterations in cardiac conduction on electrocardiogram (ECG), as recently confirmed in our department. A muscle biopsy showed mitochondrial aggregates which altered the muscle fibre contour and created a “ragged” appearance (intracellular RRF; Gomori Trichrome staining; Vogel 2001). This was due to the accumulation of abnormal mitochondria underneath the plasma membrane. 35–40% of fibres were also COX-deficient using combined COX/SDH staining (Ross 2011), which confirmed mitochondrial disease.
Genetic investigation did not highlight any chromosomal alterations (karyotyping, chromosome microarray analysis) or mutations using whole exome sequencing (Ambry Genetics; Ploski 2016). MtDNA analysis (sequencing of mtDNA using Illumina MiSeq; McElhoe et al. 2014) did not find any known pathogenic point mutations associated with mitochondrial disease, but a new mutation (mtDNA; heteroplasmic variant, m10466C > T) was detected, which corresponded to the mitochondrial tRNA-arginine gene.
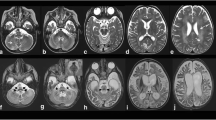
MRI and CT investigations showed mild involvement of the periventricular white matter (high T2 signal), without clear effects on subcortical tissue (Fig. 1). No local calcifications were evident at these sites. Ventricles and sulci were within normal size limits.

Magnetic resonance axial sections T2 spin-echo sequence showing hyperintensities in the white matter
At the level of the basal ganglia, we found symmetric T1-shortening involving the head and body of the caudate nuclei, pallidus, putamen, and posterior-medial thalami (Fig. 2). The T2*GE sequence and a CT scan confirmed the presence of diffuse calcifications, particularly in the bilateral globus pallidus (Figs. 3, 4), where the T1-SE and T2-SE signal was hypointense. Some thin linear calcifications extended from the caudate into the semioval centre and towards the cortex, following vessels and cortical projections. The brainstem and the cerebellar white matter were not affected.

a Magnetic resonance axial sections T1 spin-echo sequence showing areas of hyperintense signal in thalami and hypointense signal in pars medialis of the globus pallidus, b axial T2 spin echo hyposignal in pars medialis of the globus pallidus

T2* gradient recalled echo (GRE) sequence MRI axial sections showing extensive brain calcification

Non-enhanced axial CT sections demonstrating bilateral, nearly symmetric calcifications in the basal ganglia and posterior thalamus. Less intense and punctuated calcifications present in the caudate nuclei
This case of genetic mitochondriopathy is characterized by symmetric basal ganglia calcifications (BGCs) and T2 hyperintensities in the white matter. Mitochondrial disorders (MDs) primarily affect muscles and the brain, where focal lesions, such as white and grey matter stroke-like lesions, atrophy, BGCs and subcortical calcifications, can be found (Finsterer and Kopsa 2005; Carafoli 2010; Finsterer and Torres de Carvalho 2017).
Basal ganglia calcifications. BGCs are not an unusual finding on CT scans (3–6 per 1,000) (Brannan et al. 1980) or on autopsy and are generally unrelated to calcium abnormalities or neurological disease (Johnson et al. 2013; Verulashvili et al. 2006). CT and MRI scans are important for identifying calcification patterns and to establish possible differential diagnoses. The most appropriate tool is CT, since MRIs can underestimate calcium load (calcium deposits reduce the T1 relaxation time, on T2-images the lesions appear hypointense and on T2* gradient recalled echo (GRE), the signal is very low). BGCs, which consist of hydroxyapatite, zinc, iron and magnesium in a protein rich stroma, most commonly involve the globus pallidus, caudate and dentate nucleus. Other common intracranial calcifications include the pineal gland, falx, arachnoid granulations, and choroid plexus. The specific pathogenesis of BGCs is unknown, but alterations in alkaline phosphatase activity, calcium metabolism and vascular supply have been proposed as possible mechanisms. Neuronal cell loss and gliosis are usually absent at the site of basal ganglia calcification, but when present, may determine neurological dysfunction. Patients with bilateral BGCs most commonly experience movement abnormalities, cognitive impairment and psychiatric symptoms. These symptoms may be due to neuronal degeneration and mineralization, affecting mainly dopaminergic transmission in the basal ganglia, which leads to fronto-subcortical loop dysfunction (including the motor, oculomotor, prefrontal, and anterior cingulate circuits). Congruently, PET and SPECT studies demonstrate decreased metabolism in the basal ganglia and frontal lobe of patients with BGCs (Oliveira and Oliveira 2013).
Symmetric BGCs can be idiopathic (Fahr’s disease) (Tai and Batla 2015; Nicolas et al. 2013) or secondary to a long list of causes (Table 1). Fahr’s disease is a genetic condition which can affect the SLC20A2, PDGFB, PDGFRB genes, where secondary (non-genetic) causes are not recognizable. Non-idiopathic Fahr’s disease is commonly associated with parathyroid gland diseases.
In Fahr’s disease, there is commonly a pattern of symmetric calcifications in the pallidus, thalamus and dentate nucleus. Other areas of calcification include the brainstem, cerebellum and cortical/subcortical white matter, but these are less consistently reported. Hypoparathyroidism is often associated with tissue calcification in the basal ganglia as consequence of altered calcium/phosphate homeostasis. The pattern of intracranial calcification in some PDGFRB patients (Nicolas et al. 2013), which primary involves the caudate nuclei, thalami and lenticular nuclei, is quite similar to that found in our patient.
BGCs have also been described in mitochondrial diseases, such as mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leigh syndrome and KSS. However, these are uncommon (Finsterer and Kopsa 2005; Valanne et al. 1998) and calcium/phosphate metabolism is usually normal. Progressive and symmetric calcifications in the globus pallidus, putamen, caudate, thalamus and corona radiata are the most common radiological findings in MELAS, together with parietal stroke-like lesions and severe cortical atrophy (Valanne et al. 1998; Sue et al. 1998; Harrington et al. 1981).
Bilateral BGCs have been described in some cases of Leigh syndrome, mainly involving the putamen. The syndrome is characterized by symmetrical T2 hyperintensities in periventricular white matter, which is suggestive of hypomyelination (Valanne et al. 1998; Angural et al. 2018).
In some patients, KSS has been associated with cerebellar and brainstem atrophy, as well as calcification in the basal ganglia (Chu et al. 1999; Valanne et al. 1998). In particular, some patients show punctate symmetric T2-hyperintensities in the globus pallidi or in the medial thalamic nuclei and tegmentum of the brainstem. In certain cases, CT scans show diffuse calcifications in the globus pallidi and caudate nuclei (Robertson et al. 1979; Valanne et al. 1998).
T2-hyperintensities in the white matter. Mitochondrial encephalopathies are associated with structurally and/or functionally abnormal mitochondria, which affects the brain because of its high dependence on oxidative metabolism. Cerebral and cerebellar white matter involvement is increasingly being recognized as a feature of mitochondrial disorders, consisting mainly of cyst-like lesions, neuroinflammation and demyelinating conditions. Clinically, patients with leukoencephalopathy most often present with recurrent episodes of neurological regression. Onset is often in infancy and progresses with a neurodegenerative course. In KSS, status spongiosus of white and grey matter is the most typical lesion and involves the cerebrum, cerebellum (white matter) and brainstem (grey matter). The disorder is also characterized by neuronal degeneration, astrocytosis and demyelination. In particular, the distinguishing feature of KSS is the involvement of subcortical white matter (areas of T2 prolongation, sometimes with subcortical calcifications), as opposed to the deep white matter pathology seen in other mitochondrial disorders. KSS-related lesions involve the frontal and temporal regions (subcortical U fibres), as well as the periventricular and deep white matter but spare the immediate periventricular layer. Leigh’s syndrome is characterized by the presence of microcystic cavitation following necrosis of the temporal and frontal white matter. Transient infarct-like lesions are the hallmark of MELAS and mainly involve the temporo-parieto-occipital white and grey matter, with slight mass effect (Valanne et al. 1998; Sue et al. 1998; Bindu et al. 2018).
Conclusions
This case of a 36-year-old male presents a complex clinical and radiological condition with features resembling both KSS and Fahr’s disease. A new mutation in the mitochondrial tRNA-arginine gene was identified. The correlation between tRNA mitochondrial genes and brain calcification has not been clarified and warrants further study. Studying this putative relationship could provide new insight into the pathophysiological pathways which lead to similar phenotypes and related genetic conditions. The interactions between mtDNA mutations and energy homeostasis could create new avenues for therapeutic intervention.
Availability of data and materials
All the data are available for further investigations.
Abbreviations
- KSS:
-
Kearns–Sayre syndrome
- mtDNA:
-
Mitochondrial DNA
- CSF:
-
Cerebrospinal fluid
- RRF:
-
Ragged red fibres
- COX:
-
Cytochrome c oxidase
- BGCs:
-
Basal ganglia calcifications
- MD:
-
Mitochondrial disorders
- TORCH:
-
Toxoplasma gondii, others, rosolia, cytomegalovirus, herpes simplex virus
- MRI:
-
Magnetic resonance imaging
- MELAS:
-
Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes
References
Ainslie GR, Gibson KM, Vogel KR (2016) mTOR, autophagy, aminoacidopathies, and human genetic disorders. In: Molecules to medicine with mTOR, chapter 9. Academic Press, pp 143–166
Alemi M, Prigione A, Wong A, Schoenfeld R, DiMauro S, Hirano M, Taroni F, Cortopassi G (2007) Mitochondrial DNA deletions inhibit proteasomal activity and stimulate an autophagic transcript. Free Radic Biol Med 42(1):32–43
Alston CL, Rocha MC, Lax NZ, Turnbull DM, Taylor RW (2017) The genetics and pathology of mitochondrial disease. J Pathol 241:236–250
Angural A, Sharma I, Pandoh P, Sharma V, Spolia A, Rai E, Singh V, Razdan S, Pandita KK, Sharma S (2018) A case report on a novel MT-ATP6 gene variation in atypical mitochondrial Leigh syndrome associated with bilateral basal ganglia calcifications. Mitochondrion 46:209–213. https://doi.org/10.1016/j.mito.2018.06.005
Bindu PS, Sonam K, Chiplunkar S, Govindaraj P, Nagappa M, Vekhande CC, Aravinda HR, Ponmalar JJ, Mahadevan A, Gayathri N, Bharath MS, Sinha S, Taly AB (2018) Mitochondrial leukoencephalopathies: a border zone between acquired and inherited white matter disorders in children? Multiple Sclerosis Related Disorders 20:84–92. https://doi.org/10.1016/j.msard.2018.01.003
Brannan TS, Burger AA, Chaudhary MY (1980) Bilateral basal ganglia calcifications visualized. J Neurol Neurosurg Psychiatry 43:403–406
Carafoli E (2010) The fateful encounter of mitochondria with calcium: how did it happen? Biochem Biophys Acta 1797:595–606
Challa S, Kanikannan MA, Jagarlapudi MMK, Bhoompally VR, Surath M (2004) Diagnosis of mitochondrial diseases: clinical and histological study of sixty patients with ragged red fibers. Neurol India 52(3):353–358
Chu BC, Terae S, Takahashi C, Kikuchi Y, Miyasaka K, Abe S, Minowa K, Sawamura T (1999) MRI of the brain in the Hearns-Sayre syndrome: report of four cases and a review. Neuroradiology 41:759–764
Chung CY, Valdebenito GE, Ar C, Duchen MR (2021) Rewiring cell signalling pathways in pathogenic mtDNA mutations. Trends Cell Biol. https://doi.org/10.1016/j.tcb.2021.10.005
Demange P, Gia HP, Kalifa G, Sellier N (1989) MR of Kearns-Sayre syndrome. Am J Neuroradiol 10(Suppl):S91
Emmanuele V, Silvers DS, Sotiriou E, Tanji K, DiMauro S, Hirano M (2011) MERRF and Kearns-Sayre overlap syndrome due to the mtDNA m.3291T>C mutation. Muscle Nerve 44(3):448–451
Finsterer J, Kopsa W (2005) Basal ganglia calcifications in mitochondrial disorders. Metabolic Brain Disease 219–226
Finsterer J, Torres de Carvalho EH (2017) Cerebral manifestations of mitochondrial disorders. Can J Neurol Sci 44:654–663
Finsterer J, Zarrouk-Mahjoub S (2018) Involvement of the spinal cord in mitochondrial disorders. J Neurosci Rural Pract 9:245–251
Goldstein A, Falk MJ. Mitochondrial DNA deletion syndromes. 2003 Dec 17 [Updated 2019 Jan 31]. In: Adam MP, Ardinger HH, Pagon RA et al (eds) GeneReviews® [Internet]. University of Washington, Seattle, pp 1993–2022. https://www.ncbi.nlm.nih.gov/books/NBK1203/
Harrington MG, Macpherson P, McIntosh WB, Allam BF, Bone I (1981) The significance of the incidental finding of basal ganglia calcification on computed tomography. J Neurol Psychiatry 44:1168–1170
Ignatenko O, Nikkanen J, Kononov A, Zamboni N, Ince-Dunn G, Suomalainen A (2020) Mitochondrial spongiotic brain disease: astrocytic stress and harmful rapamycin and ketosis effect. Life-Science-Alliance Org. https://doi.org/10.26508/lsa.202000797
Johnson JM, Legesse B, Camprodon JA, Murray E, Price BH (2013) The clinical significance of bilateral basal ganglia calcification presenting with mania and delusions. J Neuropsychiatry Clin Neurosci 25:1. https://doi.org/10.1176/appi.neuropsych.12090222
Khambatta S, Nguyen DL, Beckman TJ, Wittich CM (2014) Kearns-Sayre syndrome: a case series of 35 adults and children. Int J General Med 7:325–332
Leal M, Dhoble C, Lee J, Lopez D, Menéndez LS (2016) A rare case of Kearns-Sayre syndrome in a 17-year-old Venezuelan male with bilateral ptosis as the initial presentation. Oxf Med Case Rep 2016(3):34–36
McElhoe JA, Holland MM, Makova KD, Shu-Wei SuM, Paul IM, Baker CH, Faith SA, Youngd B (2014) Development and assessment of an optimized next-generation DNA sequencing approach for the mtgenome using the Illumina MiSeq. Forensic Sci Int Genet 13:20–29. https://doi.org/10.1016/j.fsigen.2014.05.007
Nicolas G, Pottier C, Charbonnier C, Guyant-Marechal L, Le Ber I, Pariente J, Labauge P, Ayrignac X, Defebvre L, Maltete D, Martinaud O, Lefaucheur R, Guillin O, Wallon D, Chaumette B, Rondepierre P, Derache N, Fromager G, Schaeffer S, Krystkowiak P, Verny C, Jurici S, Sauvee M, Verin M, Lebouvier T, Rouaud O, Thauvin-Robinet C, Rousseau A, Rovelet-Lecrux A, Frebourg T, Campion D, Hannequin D (2013) Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136:3395–3407
Oliveira JRM, Oliveira MF (2013) Basal ganglia calcification as a putative cause for cognitive decline. Dementia Neuropsychol 7(2):151–154. https://doi.org/10.1590/S1980-57642013DN70200003
Pasquini L, Guarnera A, Rossi-Espagnet MC, Napolitano A, Martinelli D, Deodato F, Diodato D, Carrozzo R, Dionisi-Vici C, Longo D (2020) Spinal cord involvement in Kearns-Sayre syndrome: a neuroimaging study. Neuroradiology 62(10):1315–1321
Ploski R (2016) Next generation sequencing—general information about the technology, possibilities, and limitations. Clinical applications for next-generation sequencing, pp 1–18, https://www.sciencedirect.com/science/article/pii/B9780128017395000015
Reichmann H, Vogler L, Seibel P (1996) Ragged red or ragged blue fibers. Eur J Neurol 36(2):98–102. https://doi.org/10.1159/000117217
Robertson WC Jr, Viseskul C, Lee YE, Lloyd RV (1979) Basal ganglia calcification in Kearns-Sayre syndrome. Arch Neurol 36(11):711–713. https://doi.org/10.1001/archneur.1979.00500470081017
Ross JM (2011) Visualization of mitochondrial respiratory function using cytochrome C oxidase/succinate dehydrogenase (COX/SDH) double-labeling histochemistry. J vis Exp 57:3266. https://doi.org/10.3791/3266
Sacher M, Fatterpekar GM, Edelstein S, Sansaricq C, Naidich TP (2005) MRI findings in an atypical case of Kearns-Sayre syndrome: a case report. Neuroradiology 47:241–244
Scaglia F, Wong LJC (2008) Human mitochondrial transfer RNAs: role of pathogenic mutation in disease. Muscle Nerve 37:150–171
Seneca S, Verhelst H, De Meirleir L, Meire F, Ceuterick-De Groote C, Lissens W, Van Coster R (2001) A new mitochondrial point mutation in the transfer RNA(Leu) gene in a patient with a clinical phenotype resembling Kearns-Sayre syndrome. Arch Neurol 58(7):1113–1118. https://doi.org/10.1001/archneur.58.7.1113
Sequiera GL, Srivastava A, Alagarsamy KN, Rockman-Greenberg C, Dhingra S (2021) Generation and evaluation of isogenic iPSC as a source of cell replacement therapies in patients with Kearns Sayre syndrome. Cells 10:568. https://doi.org/10.3390/cells10030568
Sharma AK, Jain N, Kharwar RB, Narain VS (2016) Classical triad of Kearns-Sayre syndrome. BMJ Case Rep 2016:bcr2016216500. https://doi.org/10.1136/bcr-2016-216500
Shoubridge EA (2001) Cytochrome c oxidase deficiency. Am J Med Genetics Spring 106(1):46–52. https://doi.org/10.1002/ajmg.1378
Sue CM, Crimmins DS, Soo YS, Pamphlett R, Presgrave CM, Kotsimbos N, Jean-Francois MJB, Byrne E, Morris JGL (1998) Neuroradiological features of six kindreds with MELAS tRNALeu A3243G point mutation: implications for pathogenesis. J Neurol Neurosurg Psychiatry 65:233–240
Tai XY, Batla A (2015) Fahr’s disease: current perspectives. Orphan Drugs Res Rev 5:43–49
Valanne L, Ketonen L, Majander A, Suomalainen A, Pihko H (1998) Neuroradiologic findings in children with mitochondrial disorders. Am J Neurordiol 19:369–377
Verulashvili V, Glonti LS, Miminoshvili DK, Maniia MN, Mdivani KS (2006) Basal ganglia calcification: clinical manifestations and diagnostic evaluation. Georgian Med News 140:39–43
Vogel H (2001) Mitochondrial myopathies and the role of the pathologist in the molecular era. J Neuropathol Exp Neurol 60(3):217–227
Yu M, Zhang Z, Wang QQ, Liu J, Zuo YH, Yu L, Xiao JX, Zhang W, Yuan Y, Wang ZX (2016) Clinical and brain magnetic resonance imaging features in a cohort of Chinese patients with Kearns-Sayre syndrome. Chin Med J 129(12):1419–1424
Zavatta G, Clarke BL (2021) Basal ganglia calcification in hypoparathyroidism and psudohypoparathyroidism: local and systemic metabolic mechanisms. J Endocrinol Invest 44:245–253
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation was performed by Dr. PLM, Dr. AA, Dr. RC, Dr. CF, Dr. CR, Dr. AR, MS, and Dr. GV. The first draft of the manuscript was written by Dr. PLM, and all authors commented on subsequent versions of the manuscript. MS edited the final version of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Consent to participate was obtained according to national and European regulations (Italian D.Lgs. 196/2003 and European Declaration 679/2016).
Consent for publication
This case report involves a human participant investigated in our Department for ordinary radiological follow-up. We confirm that the patient expressed his consent to share these images and this information with the scientific and medical communities. Their anonymity has not been compromised.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
La Montanara, P., Albergo, A., Castellana, R. et al. Neuroradiological findings in a young patient bearing a new single mitochondrial gene mutation (case report). Bull Natl Res Cent 46, 265 (2022). https://doi.org/10.1186/s42269-022-00914-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42269-022-00914-w