
Abstract
Background
The TWNK gene encodes the twinkle protein, which is a mitochondrial helicase for DNA replication. The dominant TWNK variants cause progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant 3, while the recessive variants cause mitochondrial DNA depletion syndrome 7 and Perrault syndrome 5. Perrault syndrome is characterized by sensorineural hearing loss in both males and females and gonadal dysfunction in females. Patients with Perrault syndrome may present early-onset cerebellar ataxia, whereas middle-age-onset cerebellar ataxia caused by TWNK variants is rare.
Case presentation
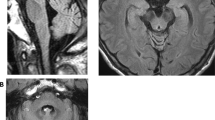
A Japanese female born to consanguineous parents presented hearing loss at age 48, a staggering gait at age 53, and numbness in her distal extremities at age 57. Neurological examination revealed sensorineural hearing loss, cerebellar ataxia, decreased deep tendon reflexes, and sensory disturbance in the distal extremities. Laboratory tests showed no abnormal findings other than a moderate elevation of pyruvate concentration levels. Brain magnetic resonance imaging revealed mild cerebellar atrophy. Using exome sequencing, we identified a homozygous TWNK variant (NM_021830: c.1358G>A, p.R453Q).
Conclusions
TWNK variants could cause middle-age-onset cerebellar ataxia. Screening for TWNK variants should be considered in cases of cerebellar ataxia associated with deafness and/or peripheral neuropathy, even if the onset is not early.
Similar content being viewed by others

Background
The Twinkle protein, encoded by the TWNK gene, is a mitochondrial helicase for DNA replication. The dominant TWNK variants cause progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant 3 (PEOA3), while the recessive variants cause mitochondrial DNA depletion syndrome 7 (MTDPS7) and Perrault syndrome 5 (PRLTS5) [1]. PRLTS is characterized by sensorineural hearing loss in both males and females and gonadal dysfunction in females. Early-onset cerebellar ataxia and other neurological symptoms, including sensory neuropathy, muscle weakness, ophthalmoplegia, nystagmus, and intellectual disability, may appear in patients with PRLTS. Only two reports have described patients with PRLTS presenting with cerebellar ataxia at their middle ages [2, 3]. In MTDPS7, cerebellar ataxia is characterized by infantile onset [4]. PEOA3 rarely causes cerebellar ataxia [5]. We report a homozygous TWNK variant in a patient with middle-age-onset cerebellar ataxia associated with deafness and sensory neuropathy.
Case presentation
A Japanese female presented hearing loss at age 48, a staggering gait at age 53, and numbness in her distal extremities at age 57. Her parents were consanguineous, and she had three siblings without neurological symptoms and no children (Fig. 1a). Neurological examination at age 58 revealed gaze-directional nystagmus, ataxic dysarthria, severe sensorineural hearing loss, decreased deep tendon reflexes, cerebellar incoordination in the limbs, sensory disturbance in the distal extremities, positive Romberg’s sign, and inability to walk in tandem. Laboratory tests at age 58 showed no abnormal findings, including in CK, lactic acid, and pyruvate levels. A moderate elevation of pyruvate concentration levels was observed at age 69 (0.98 mg/dL, normal range: 0.30–0.94 mg/dL). Magnetic resonance imaging showed mild cerebellar atrophy. We performed exome sequencing using SureSelect Human All Exon V6 and a HiSeq 2500 sequencer. We used BWA (http://bio-bwa.sourceforge.net) for mapping, SAMtools (http://samtools.sourceforge.net) and Picard (http://www.htslib.org) for SAM/BAM handling, GATK (https://gatk.broadinstitute.org), SAMtools, and Pindel (http://gmt.genome.wustl.edu/packages/pindel/) for variant calling, and ANNOVAR (http://annovar.openbioinformatics.org) for variant annotation. We detected 109,876 variants and used filtering criteria consisting of zygosity, function, allele frequencies in open databases, regions of runs of homozygosity, and Combined Annotation Dependent Depletion (CADD) score (http://cadd.gs.washington.edu/home). After filtering, we identified two homozygous variants: TWNK (NM_021830):c.1358G>A,p.R453Q (rs760988188); and TMBIM4 (NM_016056):c.713delA,p.K238Sfs*3 (rs767782535). We determined that the TWNK variant was causative, as it has been reported that TWNK variants cause cerebellar ataxia, sensorineural deafness, and sensory neuropathy, which our patient presented. We validated this variant using Sanger sequencing (Fig. 1b). A segregation study was not conducted because we were unable to contact family members. The heterozygous variant has been reported as a very rare variant in gnomAD (https://gnomad.broadinstitute.org/; allele frequency in East Asian: 0, total: 0.000003976), whereas the homozygous variant was not found in the database. The variant pathogenicity was confirmed by three prediction tools: CADD score, 23.4; PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), possibly damaging; and MutationTaster (http://www.mutationtaster.org/), disease causing. The variant was classified as likely pathogenic according to the guidelines of the American College of Medical Genetics and Genomic and the Association for Molecular Pathology [6].

Identification of the TWNK variant. a Family tree in this study. The filled circles indicate affected individuals, and the open circles and boxes indicate non-affected individuals. The proband is indicated by an arrow. b Sanger sequences of the TWNK variant in the patient and a control subject. c Domain architecture of Twinkle and previously reported variants for three phenotypes: dominant progressive external ophthalmoplegia type 3 (PEOA3); recessive mitochondrial DNA depletion syndrome 7 (MTDPS7), and Perrault syndrome 5 (PRLTS5). The variant in our case is marked in red. c Conservation of protein sequence at the nine residues with variants. Mutated residues are marked in red. The variant in our case is marked in bold
Discussion and conclusions
We reported a case of middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant. Because we could not confirm gonadal dysfunction, we were not able to diagnose PRLTS. However, cerebellar ataxia associated with hearing loss and sensory neuropathy in our patient was consistent with the neurological symptoms of PRLTS. PRLTS is classified as PRLTS1 to PRLTS6, caused by genes HSD17B4, HARS2, CLPP, LARS2, TWNK, and ERAL1 [1, 7]. We first reported that TWNK was a causative gene for PRLTS5 in two families of Japanese and European ancestry [1]. According to our original report and subsequent studies [3, 8,9,10,11,12], cerebellar ataxia in patients with PRLTS5 starts between ages 3 and 43, and hearing loss develops between ages 3 and 13 (Table 1). In contrast, our patient presented cerebellar ataxia at age 53 and hearing loss at age 48. Of the previously reported TWNK variants, eight are located in the helicase domain and three in the primase-related domain (Fig. 1c). The three families with the primase-related domain variants (families III, IV, and VII) tended to have a younger age onset than those with the helicase domain variants (Table 1). On the other hand, among the six families with helicase domain variants, families other than family VI and our patients had at least one variant located at a fully conserved amino acid in vertebrates (Fig. 1d). Patients in family VI presented cerebellar ataxia at older ages, although they had a nonsense variant in another allele (Table 1). Because the mutated amino acid in family VI and in our patient is less conserved in vertebrates, dysfunction of helicase activity may not be as severe as in other families. Therefore, our patient may have developed at an older age due to a less damaging variant located in the helicase domain of Twinkle.
R453, where the variant in our patient is located, may be important for Twinkle functions. It lies in conserved helicase motif H1a, which is involved in ATP binding and hydrolysis. In addition, it interacts with L381, where the variant causing PEOA3 is located. Functional analysis revealed that the L381P variant reduces the ATPase and helicase activities of Twinkle [13]. Collectively, the R453Q variant may lead to Twinkle ATPase and helicase dysfunction.
In conclusion, TWNK variants may cause middle-age-onset cerebellar ataxia. Therefore, screening for TWNK variants should be considered in cases of cerebellar ataxia with hearing loss and/or sensory neuropathy, even if the onset is not early.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. The sequence datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised.
Abbreviations
- CADD:
-
Combined Annotation Dependent Depletion
- MTDPS7:
-
Mitochondrial DNA depletion syndrome 7
- PEOA3:
-
Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant 3
- PRLTS:
-
Perrault syndrome
References
Morino H, Pierce SB, Matsuda Y, Walsh T, Ohsawa R, Newby M, et al. Mutations in twinkle primase-helicase cause Perrault syndrome with neurologic features. Neurology. 2014;83:2054–61.
Van Der Knaap MS, Bugiani M, Mendes MI, Riley LG, Smith DEC, Rudinger-Thirion J, et al. Biallelic variants in LARS2 and KARS cause deafness and (ovario)leukodystrophy. Neurology. 2019;92:E1225–37.
Domínguez-Ruiz M, García-Martínez A, Corral-Juan M, Pérez-Álvarez ÁI, Plasencia AM, Villamar M, et al. Perrault syndrome with neurological features in a compound heterozygote for two TWNK mutations: overlap of TWNK-related recessive disorders. J Transl Med. 2019;17:1–10. https://doi.org/10.1186/s12967-019-2041-x.
Pierce SB, Gulsuner S, Stapleton GA, Walsh T, Lee MK, Mandell JB, et al. Infantile onset spinocerebellar ataxia caused by compound heterozygosity for twinkle mutations and modeling of twinkle mutations causing recessive disease. Mol Case Stud. 2016;2:a001107. https://doi.org/10.1101/mcs.a001107.
Schicks J, Synofzik M, Schulte C, Schöls L. POLG, but not PEO1, is a frequent cause of cerebellar ataxia in Central Europe. Mov Disord. 2010;25:2678–82. https://doi.org/10.1002/mds.23286.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Chatzispyrou IA, Alders M, Guerrero-Castillo S, Zapata Perez R, Haagmans MA, Mouchiroud L, et al. A homozygous missense mutation in ERAL1, encoding a mitochondrial rRNA chaperone, causes Perrault syndrome. Hum Mol Genet. 2017;26:2541–50. https://doi.org/10.1093/hmg/ddx152.
Demain LAM, Urquhart JE, O’Sullivan J, Williams SG, Bhaskar SS, Jenkinson EM, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet. 2017;91:302–12.
Lerat J, Jonard L, Loundon N, Christin-Maitre S, Lacombe D, Goizet C, et al. An application of NGS for molecular investigations in Perrault syndrome: study of 14 families and review of the literature. Hum Mutat. 2016;37:1354–62.
Ołdak M, Oziebło D, Pollak A, Stepniak I, Lazniewski M, Lechowicz U, et al. Novel neuro-audiological findings and further evidence for TWNK involvement in Perrault syndrome. J Transl Med. 2017;15:1–13.
Jamali F, Ghaedi H, Tafakhori A, Alehabib E, Chapi M, Daftarian N, et al. Homozygous mutation in TWNK cases ataxia, sensorineural hearing loss and optic nerve atrophy. Arch Iran Med. 2019;22:728–30.
Fekete B, Pentelényi K, Rudas G, Gál A, Grosz Z, Illés A, et al. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med Genet. 2019;20:4–11.
Korhonen JA, Pande V, Holmlund T, Farge G, Pham XH, Nilsson L, et al. Structure-function defects of the TWINKLE linker region in progressive external ophthalmoplegia. J Mol Biol. 2008;377:691–705.
Acknowledgements
Not applicable.
Funding
This study was not supported by any funding.
Author information
Authors and Affiliations
Contributions
KK, HM, and HK designed the study. KK and HM performed the genetic analysis. RM obtained the clinical data. KK and HM drafted the manuscript. KK, HM, RM, YM, RO, YK, YT, TK, and HK revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Human Subjects Committees of Hiroshima University and was performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. Informed written consent was obtained from the participants in this study.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kume, K., Morino, H., Miyamoto, R. et al. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: a case report. BMC Med Genet 21, 68 (2020). https://doi.org/10.1186/s12881-020-01002-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01002-4