
Abstract
Background
The disability rate associated with rheumatoid arthritis (RA) ranks high among inflammatory joint diseases. However, the cause and potential molecular events are as yet not clear. Here, we aimed to identify differentially expressed genes (DEGs), pathways and immune infiltration involved in RA utilizing integrated bioinformatics analysis and investigating potential molecular mechanisms.
Materials and methods
The expression profiles of GSE55235, GSE55457, GSE55584 and GSE77298 were downloaded from the Gene Expression Omnibus database, which contained 76 synovial membrane samples, including 49 RA samples and 27 normal controls. The microarray datasets were consolidated and DEGs were acquired and further analyzed by bioinformatics techniques. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of DEGs were performed using R (version 3.6.1) software, respectively. The protein-protein interaction (PPI) network of DEGs were developed utilizing the STRING database. Finally, the CIBERSORT was used to evaluate the infiltration of immune cells in RA.
Results
A total of 828 DEGs were recognized, with 758 up-regulated and 70 down-regulated. GO and KEGG pathway analyses demonstrated that these DEGs focused primarily on cytokine receptor activity and relevant signaling pathways. The 30 most firmly related genes among DEGs were identified from the PPI network. The principal component analysis showed that there was a significant difference between the two tissues in infiltration immune.
Conclusion
This study shows that screening for DEGs, pathways and immune infiltration utilizing integrated bioinformatics analyses could aid in the comprehension of the molecular mechanisms involved in RA development. Besides, our study provides valuable data related to DEGs, pathways and immune infiltration of RA and may provide new insight into the understanding of molecular mechanisms.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) occurs in approximately 5 per 1000 people and can inevitably prompt severe joint damage and disability. Significant progress has been made over the past two decades with respect to the disease pathophysiology, optimal outcome measures, and effective treatment strategies, including the understanding of the comprehension in diagnosing and treating RA in the early stage [1]. The disability rate of RA ranks high among the arthritic which occurs in multiple-joint on the human body, and the incidence of this kind of arthritis is increasing year by year. The incidence of RA is occult, early diagnosis is difficult, and imaging manifestations occur comparatively late. At the point when RA is identified, the patients are usually at an advanced stage of this disease. RA would lead to multiple-joint dysfunction, disability, lower quality of life, respiratory illness, cardiovascular disease, and other comorbidities in patients not receiving intervention [2]. The etiology of RA is still ambiguous. All things considered, both genetic factors and environmental factors, contribute to the occurrence and development of RA [3].
At present, the frequently used methods for early detection and diagnosis of RA are magnetic resonance imaging, ultrasound and serological examination (including rheumatoid factor, anti-cyclic citrullinated peptide, etc). Be that as it may, there are confinements of these techniques, so the exactness and accuracy is not high [4, 5]. Computed tomography and X-rays can only detect lesions in its advanced stage, but do not detect early impairment. At the moment, the treatment of RA incorporates drug treatment, immunologic purging, functional training, surgical operation, and complementary and/or alternative medicine, and so forth [6, 7]. Therefore, it is critical to study the potential molecular mechanisms of RA synovial membrane and consequently identify more valid diagnostic techniques and more reliable molecular markers for detecting occurrence and evaluating prognosis, as well as to investigate more valid methods to control and prevent RA. Gene expression microarrays have been generally applied in studying gene expression profiles which provides a moderately new way for exploring genes and offers broad application prospects for drug-based molecular targeting. At present, large amounts of data have been published on Gene Expression Omnibus (GEO) [8]. Furthermore, integrating these databases can permit a more profound study of molecular mechanisms.
In this study, we downloaded four original microarray datasets (including GSE55235, GSE55457, GSE55584 and GSE77298) from the GEO database which incorporated a total of 76 samples, with 27 healthy controls and 49 RA samples. Differentially expressed genes (DEGs) in RA samples and control group (CG) were screened utilizing packages in R (version 3.6.1) software, and gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enrichment analysis of DEGs were additionally performed. After that, the protein-protein interaction (PPI) network was used to analyze the anticipated associations for a particular group of proteins through the STRING online database. Finally, the immune infiltration was analyzed by performing the CIBERSORT algorithm between RA and normal tissues, which was widely used to assess the relationship and the relative content of 22 types of immune cell subsets. Because of the limited samples and selection differences, not all the DEGs, relevant pathways and immune infiltration being screened out could be directly used as biomarkers.
Materials and methods
Search strategy
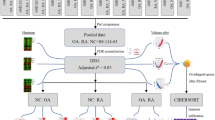
We used the keyword “Rheumatoid arthritis” to search the GEO database (https://www.ncbi.nlm.nih.gov/geo/), and there was a sum of 5543 results for “Rheumatoid arthritis” in the GEO database from their inception up to November 7, 2019. By restricting the entry type (series), study type (expression profiling by array) and tissue sources (Homo sapiens), 5386 pieces of items that were not related to the purpose of this study were excluded. After further selection with title, summary and samples, we discovered the absence of required data in 153 items. Finally, 4 series from 2 platforms were included, and gene expression profiles of GSE55235, GSE55457, GSE55584 and GSE77298 were downloaded. Figure 1 depicted the details of the selection process.

Serials selection process
Microarray data information
GSE55235, GSE55457 and GSE55584 were three multi-center genome-wide transcriptomic data sets (Affymetrix HG- U133 A) from a total of 79 individuals, including 20 healthy controls, as well as 26 osteoarthritis patients and 33 RA patients. The platform for GSE77298 was GPL570, [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array, which included 7 synovial tissue from healthy joints and 16 synovial tissue from rheumatoid arthritis joints. Platform and series matrix file(s) were downloaded from the GEO and saved as TXT files. R software (version 3.6.1) was used to process the downloaded files.
Integration of microarray data and DEGs
These four raw datasets were incorporated for the analysis. The coordinated microarray datasets were batch-normalized by R software (https://bioconductor.org/biocLite.R) utilizing limma packet (biocLite “limma”) analysis and then saved as a TXT file. The operating instruction codes, utilizing the R software, were processed automatically and the DEGs in CG and RA samples were analyzed by the limma package. Using R software to run the instruction code. Up or down-regulated genes were obtained independently and utilized for further analysis. The downloaded files (including platform and series of the matrix) were converted annotation package utilizing the R software.
Data processing and identification of DEGs
Volcano plot was used to display all of the up-regulated and down-regulated DEGs using the same limma package. Then, using pheatmap package and restricting the items to 100 DEGs, the hierarchical clustering was performed. The results were visualized using a heatmap. The ID associated with the probe name was converted into gene symbol using Perl programming (https://www.perl.org) language (version 5.30.0) as well as a genome-wide annotation library (http://www.bioconductor.org/packages/release/data/annotation/html/org. Hs.eg.db.html) and then saved in a TXT file. Adjusted P-value<0.05 and log fold change (logFC)>2 were considered as DEGs.
GO and KEGG pathway enrichment analyses of DEGs
The functional and pathway enrichment of the proteins encoded by candidate genes were analyzed, and these genes were annotated using the R software. GO and KEGG pathway analysis of DEGs were performed utilizing the appropriate packages (biocLite “DOSE”, “clusterProfiler” and “pathview”). In this study, we analyzed the DEGs that were significantly up and down-regulated as determined from integrated microarray RA data, and an adjusted P-value of<0.05 was considered statistically significant.
PPI network integration
The database STRING (https://string-db.org, version 11) was a precomputed worldwide resource for the exploration and analysis of interactions between known and predicted protein-protein interaction. Concerning a specific group of proteins, the network view analyzed the predicted associations. Each network node represented a different protein and the association between these nodes represented the interaction of biological molecules, which can be used for identifying interactions and associated pathways between these proteins encoded by DEGs in RA. The central nodes which were closely related to other corresponding proteins may be the core or key proteins and exert significant physiological functions.
Immune infiltration by CIBERSORT analysis
We used the CIBERSORT [9] to analyze the normalized data filtered by Perl programming language, and immune cells infiltration matrix were acquired. In this study, twenty-two immune cells included macrophages M2, plasma cells, neutrophils, mast cells activated, T cells CD8, macrophages M1, T cells gamma delta, B cells memory, monocytes, B cells naive, T cells follicular helper, NK cells activated, dendritic cells resting, T cells CD4 memory activated, T cells CD4 naive, NK cells resting, T cells regulatory (Tregs), dendritic cells activated, eosinophils, macrophages M0, T cells CD4 memory resting and mast cells resting. The percentage of immune cells in the gene expression matrix and the relationship between two immune cells by installing “corrplot” package was calculated [10]. After that, using “ggplot2” package, the principal component analysis (PCA) was performed to discover whether there was a difference between RA and normal synovial tissues.
Results
An aggregate of 828 DEGs was acquired, of which 758 were up-regulated and 70 down-regulated respectively (Fig. 2). Top of the 50 up and down-DEGs from the integrated data are shown in Table 1 separately. The smaller its adjusted P-value, the greater possibility of DEG and higher ranking in this experiment. R-heatmap software was utilized to draw a heatmap of the 50 up and 50 down-regulated DEGs, as shown in Fig. 3.

Volcano plot of the differentially expressed genes between RA and normal synovial tissues. Black points represent the adjusted P-value>0.05. Green points represent adjusted P-value<0.05 and down-regulated genes. Red points represent adjusted P-value<0.05 and the up-regulated genes

Heatmap of the top 100 DEGs according to the adjusted P-value and logFC. Red indicates higher gene expression and green indicates lower gene expression
GO term and KEGG pathway enrichment analysis of DEGs
The GO term and KEGG pathway enrichment analyses of up and down-regulated genes with an adjusted P-value of<0.05 were obtained respectively. The results of the GO term in RA were shown in Table 2 and Fig. 4 (a and b). The visual analysis results of the KEGG enrichment of DEGs in RA were shown in Table 3 and Fig. 4 (c and d). The up-regulated genes were mainly enriched in cytokine receptor activity, G protein-coupled chemoattractant receptor activity, chemokine receptor activity, MHC protein complex binding, chemokine binding, chemokine receptor binding and cytokine activity. The down-regulated genes were mostly amassed in peroxidase activity and oxidoreductase activity, acting on peroxide as acceptor. In the KEGG analysis, the up-regulated genes were mainly enriched in the chemokine signaling pathway, hematopoietic cell lineage, cytokine-cytokine receptor interaction, viral protein interaction with cytokine and cytokine receptor, primary immunodeficiency, leishmaniasis, osteoclast differentiation, rheumatoid arthritis, cell adhesion molecules (CAMs). The down-regulated enriched KEGG pathways of DEGs included PPAR signaling pathway, regulation of lipolysis in adipocytes, adipocytokine signaling pathway, glucagon signaling pathway, AMPK signaling pathway, calcium signaling pathway, thyroid hormone synthesis, apelin signaling, cGMP-PKG signaling pathway. Besides, the pathway map for targeted RA (Fig. 5) was described using in KEGG pathway enrichment. The significantly enriched terms and pathways may enlighten our minds and assist us in further study of the role of DEGs in RA.

GO and KEGG pathway enrichment analysis of DEGs in GSE55235, GSE55457, GSE55584 and GSE77298. a GO terms in the enrichment analysis of the up-regulated genes. b GO terms in the enrichment analysis of the down-regulated genes. c KEGG terms in the enrichment analysis of the up-regulated genes. d KEGG terms in the enrichment analysis of the down-regulated genes

KEGG pathway enrichment analysis and pathway map for RA
Analyzing DEGs in RA using a PPI network
The DEG expression products in RA were constructed by way of the STRING database to construct PPI networks (minimum required interaction score: 0.990). After deleting all isolated and partially disconnected nodes, an integrated network was built, as shown in Fig. 6a. The 30 most significant genes (Fig. 6b) which had been displaying statistical significant interaction were CDK1, KIF11, CDC20, CCNB1, CCNB2, MAD2L1, BUB1B, NDC80, AURKA, CCNA2, ISG15, NCAPG, TTK, DLGAP5, LCP2, TPX2, CD247, CKS2, LCK, VAV1, CCL5, CD3E, FOXM1, KIF20A, MX1, NUSAP1, SYK, ZAP70, ZWINT and ASPM. Among these genes, CDK1, KIF11, and CDC20 possessed the highest node degree.

a PPI network (828 DEGs filtered into the PPI network that contained 103 nodes and 168 edges). b The predicted association rank (from low to high) of the top 30 genes in the PPI network
Immune infiltration analyses
PCA depicted no overlap of these two elliptical clusters and showed that there was a significant difference (P<0.05) in immune cell infiltration between the RA group and the healthy control group (Fig. 7a). The corheatmap (Fig. 7b) result showed that mast cells activated and eosinophils had a positive correlation (value = 0.68). T cells CD8 had a significant negative correlation with T cells CD4 memory resting (value = − 0.64). Correlation heatmap (Fig. 7c) summarized the results obtained from 69 filtered gene expression matrix and the relative percent of the 22 immune cells was shown in Fig. 8a. Compared with normal tissue, the violin plot of the immune cell showed that, plasma cells, T cells CD8, T cells CD4 memory activated, T cells follicular helper, T cells gamma delta and macrophages M1 infiltrated statistically more, while T cells CD4 memory resting, NK cells activated, monocytes, macrophages M2, dendritic cells resting, mast cells activated and eosinophils infiltrated statistically less in RA tissue (Fig. 8b).

Results of CIBERSORT analysis of Gene Expression Omnibus database. a Principal component analysis (PCA) was performed on two groups. Red points and ellipse indicate RA sample, and green points and ellipse indicate normal samples. b Correlation matrix of infiltration degree of immune cells in RA samples. Red indicates trends consistent with the positive correlation and blue indicates trends consistent with the negative correlation between two immune cells. The bigger size of the numbers statistics data represents the more positive or negative correlation. c Landscape of immune cell infiltration

The landscape of immune infiltration between RA and normal controls. a The distribution of 22 immune cells in 69 filtered gene matrix. Red indicates higher immune infiltration expression and green indicates lower expression. b Violin diagram of immune cell proportions in two groups. The blue fusiform fractions on the left represent the normal group and the red fusiform fractions on the right represent the RA group.
Discussion
The characteristics of RA is synovitis, systemic inflammation, and the arrival of autoantibodies [2]. As a result, the synovial membrane breaks down the body’s immune system, causing chronic inflammation, destruction of cartilage and bone, and dysfunction to other essential organs [11,12,13]. It is reported that 50% of the risk for occurrence and development of RA is related to genetic factors. At the same time, smoking is an environmental risk factor for RA. The early onset of RA is not easy to identify, and in the meantime, cartilage and bone disintegration are frequently found in the end stages of this disease. RA occurrence and development can occur at any age, gender, and nationality for complex biological processes, and the positive rate of serum examination is low as well as non-specific. Consequently, it is miles critical to observe and study the mechanisms and development of RA at the molecular level. Based on this, differentially expressed genes (DEGs) have been efficaciously used to predict the response of therapeutic approaches for RA patients. As an example, the capability of certain genes (type I interferon-responsive) to predict the nonresponder of rituximab [14] and anti-tumor necrosis factor [15].
In this study, we integrated gene expression profile datasets from four specific groups (GSE55235, GSE55457, GSE55584 and GSE77298) and used R (version 3.6.1) to analyze these datasets. A total of 828 DEGs were identified using the limma package, consisting of 758 up-regulated genes and 70 down-regulated genes. The pinnacle 20 most significantly up-regulated genes were ADAMDEC1, IGHM, IGJ, IGKC, IGLL3, IGLV1–44, IGLC1, TNFRSF17, IGLL5, CRTAM, CXCL9, IGLJ3, TRAT1, SDC1, TPD52, IGK, CD27, CXCL10, IL21R and IGHG1. The pinnacle 20 most significantly down-regulated genes were SLC19A2, PLIN1, KLF9, ADCY2, PPAP2B, EBF2, ADH1B, KLF4, PPARGC1A, GABARAPL1, TRHDE, PHKA1, FBXW12, TCEAL2, PCK1, PCDH9, MAFF, LEP, RERGL and SGCA. Constructing a PPI network (minimum required interaction score = 0.990) of DEG-encoding proteins from the STRING database and screening the 30 most significant related genes. The enriched GO of DEGs in RA were analyzed by R software, and correlation analysis confirmed that the up-regulated genes have been specifically involved in cytokine receptor activity, G protein-coupled chemoattractant receptor activity, chemokine receptor activity, MHC protein complex binding, chemokine binding, chemokine receptor binding and cytokine activity, and that the down-regulated DEGs were mainly involved in peroxidase activity and oxidoreductase activity, acting on peroxide as acceptor. This finding is consistent with the knowledge that cytokine, chemokine and peroxidase activity play crucial roles in the RA occurrence and progression.
The detection of autoantibodies (including RA, A-CCP, CRP) in RA patients is an identification that distinguishes the disease from other inflammatory arthritis, such as psoriatic arthritis, reactive arthritis and osteoarthritis. In addition to the clinical symptoms and signs arising from arthritis processes in the joints, muscle weakness around joints is also commonly reported by RA patients [16,17,18]. Takashi Yamada et al. [18] discovered that altered Ca2+ and free radical signaling (such as reactive oxygen and reactive nitrogen species) can result in RA-based muscle weakness. In a general way, RA with CCP + RF+ subjects had excessively high citrulline-specific IgG binding, and CCP + RF- and CCP-RF+ subjects had modest binding to array peptides [19]. As a systemic autoimmune disease, RA is characterized by inflammation and angiogenesis in the synovium. Many cytokines and inflammatory medium are observed in synovial tissues and synovial fluids, whose function is to display angiogenic properties. Inhibitor of DNA binding 1, one of the transcription factors, is a marker of cellular self-renewal. This factor within the bone marrow causes a significant reduction of endothelial progenitor cell association with tumor-related vasculogenesis [20, 21]. Amélie Simon et al. [22] observed that microscopic polyangiitis is vasculitides typical of necrotizing inflammation for small-sized vessels and is usually connected with serum positivity for those anti-neutrophil cytoplasmic antibodies. In most conditions, anti-neutrophil cytoplasmic antibodies are directed against two constituents of neutrophil primary granules as well as monocyte lysosomes: myeloperoxidase or proteinase 3.
Furthermore, the up-regulated enriched Kyoto Encyclopedia of Genes and (KEGG) pathways of DEGs included the chemokine signaling pathway, hematopoietic cell lineage, cytokine-cytokine receptor interaction, viral protein interaction with cytokine and cytokine receptor, primary immunodeficiency, leishmaniasis, osteoclast differentiation, rheumatoid arthritis, cell adhesion molecules (CAMs). The down-regulated enriched KEGG pathways of DEGs included PPAR signaling pathway, regulation of lipolysis in adipocytes, adipocytokine signaling pathway, glucagon signaling pathway, AMPK signaling pathway, calcium signaling pathway, thyroid hormone synthesis, apelin signaling, cGMP-PKG signaling pathway. Relative studies have demonstrated that fibroblast-like synoviocytes play a crucial role by producing cytokines in all stages of RA. Once fibroblast-like synoviocytes are activated during RA, a series of inflammatory factors and proteases will be produced involved in the inflammatory response, causing progressive destruction of bone and cartilage [23].
RA is associated with an increase in mortality. Previous research displayed that the occurrence rate of the malignancies in RA patients has been reported to be high [6]. A review of scientific studies compiled in Romania demonstrated findings that anemia and other chronic disease manifestations are relatively common in approximately 6–10% of RA patients, and are all related to worse outcomes in particular functional impairment and mortality [24, 25]. The adaptive immune system is closely connected with the generation of the anti-tumor immune response. For that reason, RA patients with gastrointestinal cancer history must be carefully monitored while receiving the treatment of disease-modifying antirheumatic drugs [26]. However, in many factors, tumor necrosis factor-α is recognized as performing biological functions associated with the pathogenesis of RA [27]. Its capabilities include chemokine amplification, endothelial cell activation, leukocyte accumulation [28], experiencing cardiovascular comorbidity [29], acceleration destruction of osteoclast and chondrocyte, and demonstrating metabolic syndrome [30]. Related studies have reported that peroxisome proliferator-activated receptor (PPAR)-γ may additionally induce activation Wnt/β-catenin signaling [31]. Numerous studies have indicated that decreased expression of adipocyte genes such as nuclear receptors PPARg in the RA synovial tissue [32, 33], and PPARg mediates mesenchymal stem cells as well as fibroblast-like synovial cells differentiation into adipocytes [34]. PPAR-γ activators performed significantly anti-inflammatory and anti-degeneration roles in rheumatoid arthritis [35]. According to a study of adjuvant-induced arthritis in rats’ synovium, inhibition PPAR-γ expression by T0070907 or PPAR-γ siRNA could significantly promote the proliferation of fibroblast-like synoviocytes and expressions of c-Myc, Cyclin D1, MMP-1, and MMP-9, except for TIPM-1 [31, 36]. Meanwhile, compared with normal tissues, PPAR-γ was obviously reduced whether immunohistochemical technique or protein detection by western blot [36]. A lot of experimental results were commendably consistent with the result in this integrated study and suggested that PPAR-γ might play a pivotal role during RA synovial tissue activation. As for the gene expression of AMPK in those newly diagnosed RA patients, a master regulator of the metabolic process was decreased in the peripheral blood leukocytes and elevated levels of TGF-β1 in plasma accounts for the occurrence of RA pathogenesis [37]. Recent data evidence suggested that S100A8/A9 was a member of the Ca2+ binding S100 protein family and had become a hot topic as a critical alarmin modulating the inflammatory response. Using small-molecule inhibitors that block off S100A8/A9 activity can exhibit beneficial functions on disease relative activities in animal models of autoimmune diseases such as RA [38, 39].
We constructed a PPI network of the protein encoded by DEGs and identified the subsequent top 10 closely related genes: CDK1, KIF11, CDC20, CCNB1, CCNB2, MAD2L1, BUB1B, NDC80, AURKA and CCNA2. These genes are key nodes for the construction a PPI network and play a distinct role in the pathogenesis of RA. In accordance with the proinflammatory CDK signaling, p16INK4A protein as a Cyclin-Dependent Kinases inhibitor in synovial fibroblasts also demonstrated an inhibitory action in the development of RA [40]. Ectopic expression of p16INK4A protein can also suppress LPS-induced IL-6 expression in macrophages [41], and simultaneously enhance the observations that CDK inhibitory proteins relative features to counteract inflammation [42]. Interleukin-6 (IL-6) signaling was a critical target in inflammatory pathways [43]. In patients with RA, the high level of IL-6 and IL-6R are found in both serum and synovial fluid of related joints affected by the disease. IL-6 is a cytokine serving several biological and biochemical functions that affect the immune and vasculature system. Generally speaking, conventional IL-6 signaling is in charge of the anti-inflammatory capabilities of IL-6, conversely, trans-signaling is in charge of the pro-inflammatory properties of IL-6. Consequently, disorders of the IL-6 axis can result in the onset or progression of disease states, especially in autoimmune and inflammatory dysregulation [44]. Activation of epidermal growth factor receptor (EGFR) signaling leads to the propagation and metabolism of synovial fibroblasts in RA. Beyond that, in addition to its function in propagation and metabolism, EGFR can generate cytokine in synovial tissues during the pathogenesis of RA. Some animal experiments have yielded potentially prospective results aiming at target EGFR involving RA. As a result, pharmacologic modulations or its ligands targeting EGFR may reveal undiscovered methods for the treatment of RA [45]. EGF receptor is a tyrosine kinase. At present, only NEK6 and CDK1 kinases can phosphorylate KIF11 at Ser1033 and Thr926 respectively, causing the combination of microtubules and KIF11 in the process of mitotic spindle assembly [46,47,48]. Some inflammatory cytokines are controlled by the expression of the c-Fos. Both IL-1β and c-Fos are interacted with each other, including its gene expression and activities, and causing a cross-linking effect that is a vital mechanism to arthritic joint destruction. As a result, the blockade of IL-1β, c-Fos, or link between both can be effective therapeutically as a treatment method for RA patients’ joint destruction [49]. Researchers at the University of Chicago found through mice experiments that the inhibition of c-Myc or c-Raf-1 can significantly decrease the invasiveness of RA synovial fibroblasts. Besides, dominant-negative mutants c-Raf-1 reduced the expression of phosphorylated c-Jun in vivo as well as the expression of disease-relevant MMPs [50].
In the current study, using CIBERSORT analytical tool, the relative percent and content of 22 immune cell subsets, what we have performed, were the most comprehensive analysis in RA synovial tissues to date. We found that the proportion of mast cells activated, eosinophils and T cells CD4 memory resting was high in the normal synovial tissue, while T cells CD8 accounted for RA synovial high expression. A study has found that T cells, particularly CD8 infiltration in the synovial tissues, were considered as predictors of RA development and the presence of antibodies against citrullinated peptides [51]. In the correlation analysis of the infiltration degree of immune cells, CD8+ T cells were negatively correlated with the infiltration degree of resting CD4+ memory T cells, which meant the binding of CD4 + T cells and RA synovial antigen in the microenvironment was decreased [52].
In the last few years, DEGs identification and further bioinformatics analysis was performed and these results may provide new perspective for the study of RA. However, a limitation of this study was not experimentally validated, which may need to perform in the future studies.
Conclusions
It is beneficial for the research community to study this network to further examine and understand the interaction between RA relevant DEGs. These findings may help us to enhance our general understanding of the molecular mechanism of RA. However, further relevant molecular biological experiments are required to affirm the function of the identified genes associated with RA.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- DEGs:
-
Differentially expressed genes
- RA:
-
Rheumatoid arthritis
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- logFC:
-
Log fold change
- PPI:
-
Protein-protein interaction
- PCA:
-
Principal component analysis
- CG:
-
Control group
- PPAR:
-
Peroxisome proliferator-activated receptor
- IL-6:
-
Interleukin-6
- EGFR:
-
Epidermal growth factor receptor
References
Myasoedova E, Crowson CS, Kremers HM, Therneau TM, Gabriel SE. Is the incidence of rheumatoid arthritis rising?: results from olmsted county, Minnesota. Arthritis Rheum. 2010;62(6):1576–82.
Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet. 2010;376(9746):1094–108.
Viatte S, Barton A. Genetics of rheumatoid arthritis susceptibility, severity, and treatment response. Semin Immunopathol. 2017;39(4):395–408.
Groves C, Chandramohan M, Chew NS, Aslam T, Helliwell PS. Clinical examination, ultrasound and mri imaging of the painful elbow in psoriatic arthritis and rheumatoid arthritis: which is better, ultrasound or mr, for imaging enthesitis? Rheumatol Ther. 2017;4(1):71–84.
Kourilovitch M, Galarza-Maldonado C, Ortiz-Prado E. Diagnosis and classification of rheumatoid arthritis. J Autoimmun. 2014;48-49:26–30.
De Cock D, Hyrich K. Malignancy and rheumatoid arthritis: epidemiology, risk factors and management. Best Pract Res Clin Rheumatol. 2018;32(6):869–86.
Misiuniene N, Baranauskaite A. diagnosis and treatment of rheumatoid arthritis. Medicina (Kaunas). 2002;38(1):110–6.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. Ncbi geo: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–82.
Harrison WT, Yeates RM, Phillips ML, Nenoff TM. New framework connectivity patterns in templated networks: the creatinine zinc phosphites c4n3oh7.Znhpo3, c4n3oh7.Zn(h2o)hpo3, and (c4n3oh7)2.Znhpo3.H2o. Inorg Chem. 2003;42(5):1493–8.
Choy E. Understanding the dynamics: pathways involved in the pathogenesis of rheumatoid arthritis. Rheumatology (Oxford). 2012;51(Suppl 5):v3–11.
Liu L, Wong CW, Han M, Farhoodi HP, Liu G, Liu Y, et al. Meta-analysis of preclinical studies of mesenchymal stromal cells to treat rheumatoid arthritis. EBioMedicine. 2019;47:563–77.
Smolen JS, Aletaha D, Redlich K. The pathogenesis of rheumatoid arthritis: new insights from old clinical data? Nat Rev Rheumatol. 2012;8(4):235–43.
Raterman HG, Vosslamber S, de Ridder S, Nurmohamed MT, Lems WF, Boers M, et al. The interferon type i signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res Ther. 2012;14(2):R95.
Stuhlmuller B, Haupl T, Hernandez MM, Grutzkau A, Kuban RJ, Tandon N, et al. Cd11c as a transcriptional biomarker to predict response to anti-tnf monotherapy with adalimumab in patients with rheumatoid arthritis. Clin Pharmacol Ther. 2010;87(3):311–21.
Fraser A, Vallow J, Preston A, Cooper RG. Predicting 'normal' grip strength for rheumatoid arthritis patients. Rheumatology (Oxford). 1999;38(6):521–8.
Stenstrom CH, Minor MA. Evidence for the benefit of aerobic and strengthening exercise in rheumatoid arthritis. Arthritis Rheum. 2003;49(3):428–34.
Yamada T, Steinz MM, Kenne E, Lanner JT. Muscle weakness in rheumatoid arthritis: the role of ca (2+) and free radical signaling. EBioMedicine. 2017;23:12–9.
Zheng Z, Mergaert AM, Fahmy LM, Bawadekar M, Holmes CL, Ong IM, et al. Disordered antigens and epitope overlap between anti-citrullinated protein antibodies and rheumatoid factor in rheumatoid arthritis. Arthritis Rheumatol. 2020;72(2):262–72.
Edhayan G, Ohara RA, Stinson WA, Amin MA, Isozaki T, Ha CM, et al. Inflammatory properties of inhibitor of DNA binding 1 secreted by synovial fibroblasts in rheumatoid arthritis. Arthritis Res Ther. 2016;18:87.
Gao D, Nolan DJ, Mellick AS, Bambino K, McDonnell K, Mittal V. Endothelial progenitor cells control the angiogenic switch in mouse lung metastasis. Science. 2008;319(5860):195–8.
Simon A, Subra JF, Guilpain P, Jeannin P, Pignon P, Blanchard S, et al. Detection of anti-pentraxin-3 autoantibodies in anca-associated vasculitis. PLoS One. 2016;11(1):e0147091–e91.
Bartok B, Firestein GS. Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev. 2010;233(1):233–55.
Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD, Tanasescu R. Extra-articular manifestations in rheumatoid arthritis. Maedica. 2010;5(4):286–91.
Firestein GS, McInnes IB. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46(2):183–96.
Kleinert S, Waldner M, Wendler J, Kunzmann V. Disease-modifying antirheumatic drugs in rheumatoid arthritis patients with a history of colorectal cancer. Z Rheumatol. 2016;75(1):41–6.
Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118(11):3537–45.
Feldmann M, Maini RN. Anti-tnf alpha therapy of rheumatoid arthritis: what have we learned? Annu Rev Immunol. 2001;19:163–96.
Dixon WG, Symmons DP. What effects might anti-tnfalpha treatment be expected to have on cardiovascular morbidity and mortality in rheumatoid arthritis? A review of the role of tnfalpha in cardiovascular pathophysiology. Ann Rheum Dis. 2007;66(9):1132–6.
Sattar N, McCarey DW, Capell H, McInnes IB. Explaining how "high-grade" systemic inflammation accelerates vascular risk in rheumatoid arthritis. Circulation. 2003;108(24):2957–63.
Li XF, Sun YY, Bao J, Chen X, Li YH, Yang Y, et al. Functional role of ppar-γ on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci Rep. 2017;7(1):12671.
Brenner M, Linge CP, Li W, Gulko PS. Increased synovial expression of nuclear receptors correlates with protection in pristane-induced arthritis: a possible novel genetically regulated homeostatic mechanism. Arthritis Rheum. 2011;63(10):2918–29.
Brenner M, Laragione T, Gulko PS. Short-term low-magnesium diet reduces autoimmune arthritis severity and synovial tissue gene expression. Physiol Genomics. 2017;49(4):238–42.
Yamasaki S, Nakashima T, Kawakami A, Miyashita T, Tanaka F, Ida H, et al. Cytokines regulate fibroblast-like synovial cell differentiation to adipocyte-like cells. Rheumatology. 2004;43(4):448–52.
Liu Y, Qu Y, Liu L, Zhao H, Ma H, Si M, et al. Ppar-γ agonist pioglitazone protects against il-17 induced intervertebral disc inflammation and degeneration via suppression of nf-κb signaling pathway. Int Immunopharmacol. 2019;72:138–47.
Marder W, Khalatbari S, Myles JD, Hench R, Lustig S, Yalavarthi S, et al. The peroxisome proliferator activated receptor-γ pioglitazone improves vascular function and decreases disease activity in patients with rheumatoid arthritis. J Am Heart Assoc. 2013;2(6):e000441.
Samimi Z, Kardideh B, Zafari P, Bahrehmand F, Roghani SA, Taghadosi M. The impaired gene expression of adenosine monophosphate-activated kinase (AMPK), a key metabolic enzyme in leukocytes of newly diagnosed rheumatoid arthritis patients. Mol Biol Rep. 2019;46(6):6353–60.
Baillet A, Trocmé C, Berthier S, Arlotto M, Grange L, Chenau J, et al. Synovial fluid proteomic fingerprint: S100a8, s100a9 and s100a12 proteins discriminate rheumatoid arthritis from other inflammatory joint diseases. Rheumatology. 2010;49(4):671–82.
Hurnakova J, Hulejova H, Zavada J, Hanova P, Komarc M, Mann H, et al. Relationship between serum calprotectin (s100a8/9) and clinical, laboratory and ultrasound parameters of disease activity in rheumatoid arthritis: a large cohort study. PLoS One. 2017;12(8):e0183420–e20.
Taniguchi K, Kohsaka H, Inoue N, Terada Y, Ito H, Hirokawa K, et al. Induction of the p16ink4a senescence gene as a new therapeutic strategy for the treatment of rheumatoid arthritis. Nat Med. 1999;5(7):760–7.
Murakami Y, Mizoguchi F, Saito T, Miyasaka N, Kohsaka H. P16ink4a exerts an anti-inflammatory effect through accelerated irak1 degradation in macrophages. J Immunol. 2012;189(10):5066.
Schmitz ML, Kracht M. Cyclin-dependent kinases as coregulators of inflammatory gene expression. Trends Pharmacol Sci. 2016;37(2):101–13.
Avci AB, Feist E, Burmester GR. Targeting il-6 or il-6 receptor in rheumatoid arthritis: What's the difference? BioDrugs. 2018;32(6):531–46.
Schett G. Physiological effects of modulating the interleukin-6 axis. Rheumatology (Oxford). 2018;57(suppl_2):ii43–50.
Yuan FL, Li X, Lu WG, Sun JM, Jiang DL, Xu RS. Epidermal growth factor receptor (egfr) as a therapeutic target in rheumatoid arthritis. Clin Rheumatol. 2013;32(3):289–92.
Rapley J, Nicolàs M, Groen A, Regué L, Bertran MT, Caelles C, et al. The nima-family kinase nek6 phosphorylates the kinesin eg5 at a novel site necessary for mitotic spindle formation. J Cell Sci. 2008;121(23):3912.
Cahu J, Olichon A, Hentrich C, Schek H, Drinjakovic J, Zhang C, et al. Phosphorylation by cdk1 increases the binding of eg5 to microtubules in vitro and in xenopus egg extract spindles. PLoS One. 2008;3(12):e3936.
Bertran MT, Sdelci S, Regué L, Avruch J, Caelles C, Roig J. Nek9 is a plk1-activated kinase that controls early centrosome separation through nek6/7 and eg5. EMBO J. 2011;30(13):2634–47.
Shiozawa S, Tsumiyama K. Pathogenesis of rheumatoid arthritis and c-fos/ap-1. Cell Cycle. 2009;8(10):1539–43.
Pap T, Nawrath M, Heinrich J, Bosse M, Baier A, Hummel KM, et al. Cooperation of ras- and c-myc-dependent pathways in regulating the growth and invasiveness of synovial fibroblasts in rheumatoid arthritis. Arthritis Rheum. 2004;50(9):2794–802.
de Hair MJ, van de Sande MG, Ramwadhdoebe TH, Hansson M, Landewé R, van der Leij C, et al. Features of the synovium of individuals at risk of developing rheumatoid arthritis: implications for understanding preclinical rheumatoid arthritis. Arthritis Rheum. 2014;66(3):513–22.
Huang R, Wu J, Zheng Z, Wang G, Song D, Yan P, et al. The construction and analysis of cerna network and patterns of immune infiltration in mesothelioma with bone metastasis. Front Bioeng Biotechnol. 2019;7:257.
Acknowledgments
We thank Dr. Peijian Tong and Dr. Letian Shan for giving us rational advice during the study.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: National Natural Science Foundation of China (Grant No. 81774331 and 82074464). The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Yanzhi Ge and Li Zhou analyzed and extracted the data, contributed analysis tools. Yanzhi Ge, Zuxiang Chen and Ting Li prepared figures and tables. Yanzhi Ge and Letian shan wrote the main protocol and prepared the manuscript. Peijian Tong conceived and designed the study, approved the final draft. The author(s) read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ge, Y., Zhou, L., Chen, Z. et al. Identification of differentially expressed genes, signaling pathways and immune infiltration in rheumatoid arthritis by integrated bioinformatics analysis. Hereditas 158, 5 (2021). https://doi.org/10.1186/s41065-020-00169-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s41065-020-00169-3