
Abstract
Background
Fecal bacterial densities are proxy indicators of beach water quality, and beach posting decisions are made based on Beach Action Value (BAV) exceedances for a beach. However, these traditional beach monitoring methods do not reflect the full extent of microbial water quality changes associated with BAV exceedances at recreational beaches (including harmful cyanobacteria). This proof of concept study evaluates the potential of metagenomics for comprehensively assessing bacterial community changes associated with BAV exceedances compared to non-exceedances for two urban beaches and their adjacent river water sources.
Results
Compared to non-exceedance samples, BAV exceedance samples exhibited higher alpha diversity (diversity within the sample) that could be further differentiated into separate clusters (Beta-diversity). For Beach A, Cyanobacterial sequences (resolved as Microcystis and Pseudanabaena at genus level) were significantly more abundant in BAV non-exceedance samples. qPCR validation supported the Cyanobacterial abundance results from metagenomic analysis and also identified saxitoxin genes in 50% of the non-exceedance samples. Microcystis sp and saxitoxin gene sequences were more abundant on non-exceedance beach days (when fecal indicator data indicated the beach should be open for water recreational purposes). For BAV exceedance days, Fibrobacteres, Pseudomonas, Acinetobacter, and Clostridium sequences were significantly more abundant (and positively correlated with fecal indicator densities) for Beach A. For Beach B, Spirochaetes (resolved as Leptospira on genus level) Burkholderia and Vibrio sequences were significantly more abundant in BAV exceedance samples. Similar bacterial diversity and abundance trends were observed for river water sources compared to their associated beaches. Antibiotic Resistance Genes (ARGs) were also consistently detected at both beaches. However, we did not observe a significant difference or correlation in ARGs abundance between BAV exceedance and non-exceedance samples.
Conclusion
This study provides a more comprehensive analysis of bacterial community changes associated with BAV exceedances for recreational freshwater beaches. While there were increases in bacterial diversity and some taxa of potential human health concern associated with increased fecal indicator densities and BAV exceedances (e.g. Pseudomonas), metagenomics analyses also identified other taxa of potential human health concern (e.g. Microcystis) associated with lower fecal indicator densities and BAV non-exceedances days. This study can help develop more targeted beach monitoring strategies and beach-specific risk management approaches.
Similar content being viewed by others
Introduction:
Recreational water ecosystems, such as freshwater beaches, are subject to fecal contamination, resulting in beach postings deeming beaches unsuitable for public recreational activities [1, 2]. Deterioration of water quality can cause gastrointestinal illnesses among beachgoers and may be caused by fecal contamination sources, including wastewater treatment plants, septic tank systems, combined sewer overflows and animal/bird feces [3, 4]. Fecal indicator bacteria, including E. coli and Enterococcus, are correlated with gastrointestinal illness and are thus proxy indicators of water quality for recreational water ecosystems [2, 5]. Despite their common use, fecal indicator bacteria (FIB) are limited by a lack of host specificity and sensitivity [6, 7] and may not necessarily be correlated with the broad spectrum of enteric and non-enteric pathogens of health concern [8]. Additionally, FIB density can be biased due to environmental and physicochemical factors, including water temperature [9], the persistence of FIB for an extended period of time outside the host cell [10], and potential to regrow in beach sediments [11].
Traditional methods for fecal indicator testing in recreational water ecosystems, such as beaches, include culture-based enumeration [12]. E. coli and Enterococcus are commonly used fecal indicators for beach quality monitoring, and beaches are posted/closed for public visits based on exceedance above Beach Action Value (BAV) thresholds recommended by the United States Environmental Protection Agency [13] and Health Canada [2]. These Beach Action Values are determined based on epidemiological studies (mainly from US Beaches) on the correlation between gastrointestinal diseases among beachgoers/swimmers and fecal indicator densities [1, 14, 15]. However, the occurrence and abundance of fecal indicator bacteria can be variable from different fecal pollution sources [16,17,18], and may not be indicative of many pathogens of human health concern from non-fecal sources (e.g. toxigenic cyanobacteria).
Molecular methods such as quantitative PCR and digital PCR have been tested as alternatives to augment culturing-based methods for monitoring beach water quality [19, 20]. However, these PCR methods typically rely on a limited number of microbial markers such as Enterococcus for FIB monitoring and HF183 for human fecal source tracking, which may not provide a comprehensive overview of the microbial communities in recreational waters. Additionally, obtaining broad microbial information from PCR-based methods requires multiple assays, which can be laborious and costly for complex microbial environments such as water ecosystems.In comparison, a DNA sequencing-based metagenomic approach can simultaneously characterize most of the taxa in a beach water sample and provide gene profiles of the identified organisms [21, 22]. Therefore, a broader range of information obtained from the metagenomics-based approach can potentially provide a valuable screen of microbial changes and improve beach monitoring and management strategies.
The current study evaluates the potential of a metagenomics-based approach to augment beach monitoring strategies by obtaining a comprehensive overview of bacterial community changes associated with Beach Action Value exceedances for recreational freshwater beaches. The questions we focused on are: (1) Do the beach water microbial communities differ significantly between Beach Action Value Exceedance and Non-Exceedance Beach Days? (2) Do the two different beaches show similar patterns of bacterial community changes in response to fecal indicator exceedances? (3) Can the adjacent river and creek water sources account for bacterial changes observed at the beaches? (4) Are there any bacterial taxa of human health concern that do not correlate with fecal indicator densities? (5) Do Antibiotic Resistance Genes (ARGs) abundance correlate with changes in fecal indicator densities?
Materials and methods
Study design
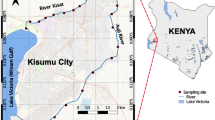
This study focused on microbial changes associated with fecal contamination at two Toronto recreational freshwater beaches, including Marie Curtis Park East Beach (referred to as Beach A) and Sunnyside Beach (referred to as Beach B), along with their adjacent river and creek water sources as assessed by shotgun DNA sequencing. The water sampling was performed 3 days a week for the summer season 2021 (June 1–August 26). On each sampling day, eight samples were collected, including one each from Etobicoke Creek (referred to as River A and source water for MCPEB) and Humber River (referred to as River B and source water for Sunnyside Beach), and three from each beach transect 30W (43.585610–79.540054), 30W replicate, and 32W (43.585110–79.540560) for Marie Curtis Park East Beach, and transect 18W (43.636612–79.452670), 21W (43.637110–79.457530) and 21W replicate for Sunnyside Beach. These sampling sites and the names used (30W, 32W, 18W, and 21W) were selected according to Toronto Public Health's Beach Monitoring Program (Fig. 1). A total of 309 samples corresponding to 38 beach days were collected. The samples were collected between 5:30 and 7 am and were transported (on ice) to the lab within 1 h for further processing.

Sampling sites for Marie Curtis Park East Beach, Etobicoke Creek, Sunnyside Beach and Humber River
Water sample collection, filtration and DNA extraction
Water samples were collected 30 cm below the water surface in sterile screw-capped polyethylene terephthalate (PET) bottles (1000 mL). For E. coli culturing and Enterococcus qPCR, a 100 mL sample was passed through a 0.45 µm mixed cellulose esters (MCE) filter (Millipore Corp., Bedford, MA), while for metagenomic DNA extraction, 100 mL of water sample was filtered through 0.22 µm nitrocellulose filters (Millipore Corp., Bedford, MA). For Enterococcus qPCR, we used the DNA extraction protocol as described previously [23]. Briefly, the membrane filters were placed in a 2 mL microcentrifuge tube with 0.3 gm glass beads (600 µL of 0.2 µg/mL Salmon sperm DNA as extraction buffer) and bead beaten for 60 s, followed by centrifuging for 60 s at 12, 000 g. The supernatant (400 µL) was collected in a 1.5 mL tube and centrifuged for 5 min at 12, 000 g to collect DNA extract (~ 350 µL) for qPCR. For DNA Sequencing, DNA extraction was performed by using the Norgen Soil Plus DNA Extraction kit (Norgen Biotek Corp., Canada) as described previously [24], followed by DNA quantification using Qubit fluorometer (dsDNA High-Sensitivity Assay kit, Thermo Fisher Scientific, USA).
E. coli enumeration by culturing
E. coli enumeration was performed for all samples using Differential Coliform Agar (OxoidTM) with cefsulodin as described previously [23]. Beach water samples were directly processed (100 mL), while Creek/River samples were diluted 1:10 for E. coli culturing. The membrane filters were placed on 47 mm agar plates and incubated for 24 h at 44.5 °C [25]. The phosphate buffer saline (passed through the membrane filters) was used alongside the water sample as a negative control. Each sample was tested in triplicate, and enumeration counts for each sampling site were recorded as the mean.
Enterococcus qPCR
Enterococcus qPCR was performed as described previously [23, 26] using Method 1609.1: Enterococci in Water by TaqMan Quantitative Polymerase Chain Reaction (qPCR) with Internal Amplification Control (IAC) Assay [27]. In brief, the stock cultures (109 CFUs) of Enterococcus faecalis (ATCC 2921) were used to prepare calibrator-positive controls (104 CFUs) and for DNA extraction to prepare Standard Curves. For standard curves, DNA was extracted from the stock cultures using the Norgen Soil Plus Extraction kit described in the previous section. The DNA quantification was done using a Qubit fluorometer (dsDNA High-Sensitivity Assay kit, Thermo Fisher Scientific, USA), followed by the calculation of Target Sequence Copies. Ten-fold dilutions (10–40,000 Target Sequence Copies) were prepared from Stock Culture DNA, and four individual standard curves were used to create a composite standard curve. Alongside each batch of samples, two calibrator positive controls, two method blanks (phosphate buffer saline passed through filters), and two non-template controls (DNA extract replaced by nuclease-free water) were analyzed to test for DNA extraction efficiency and contamination. Each PCR reaction was carried out in duplicate and comprised DNA Recovery Control (Salmon DNA qPCR) and a PCR inhibition control (Internal Amplification Control). Each qPCR reaction (25 µL) comprised of 12.5 µL TaqMan Environmental Master mix (Thermo Fisher Scientific, USA), 3.0 µL of primer–probe working solution (primers and probes concentration was 1.0 µM and 80.0 nm, respectively), 2.0 µL of internal amplification control and 2.5 µL Bovine Serum Albumin (2 mg/mL). All the reactions were performed on Bio-Rad CFX96 Touch Real-Time PCR (Bio-Rad Inc. USA). The Enterococcus Calibrator Cell Equivalents were calculated using an Excel sheet (https://www.epa.gov/cwa-methods/other-clean-water-act-test-methods-microbiological#file-183743) provided by USEPA.
Sample selection criteria for shotgun DNA sequencing
The following considerations were used for sample selection: (1) samples having E. coli > 235 CFU/100 mL by plate counting or having Enterococcus > 1000 calibrator cell equivalents by qPCR as determined previously [23, 26] were defined as BAV exceedances, (2) Sampling Days showing exceedance or non-exceedance for all the sampling sites for a beach were prioritized, (3) an equal number of beach samples were selected for BAV Exceedance and Non-exceedance groups, (4) for a single Beach Day, DNA from all the sampling sites for a beach was pooled and, (5) For creek/river water sources (Etobicoke Creek and Humber River), sampling dates matching to the selected beach samples were sequenced for comparison of bacterial trends with associated beaches. In total, 48 pooled water samples were selected for shotgun sequencing (Additional file 1: Table 1).
Shotgun DNA sequencing and quality control analysis
To avoid DNA concentration bias during library pooling, input DNA from each sample was normalized to 200 ng. The library was prepared using NEBNext® Ultra™ II DNA library preparation kit with TruSeq3 paired-end adapters. The fragment size and read length were 500 bp and 150 bp, respectively. DNA sequencing was performed on Illumina NextSeq 2000 (2 × 150) at the Farncombe Sequencing Institute at McMaster University. The quality of raw reads was analyzed using FASTQC [28]. Adapter trimming, decontamination (removal of reads mapping to human), quality filtration (Quality Score > 30, by the sliding window algorithm, widowsize = 4 bases), Length Filtration (> 75 bp) and removal of tandem repeats were performed using the KneadData pipeline (Available at: http://huttenhower.sph.harvard.edu/kneaddata).
Cyanobacteria and cyanotoxin gene qPCR
The number of Cyanobacterial and Cyanotoxin (Microcystin, Saxitoxin and Cylindrospermopsin) gene copies from metagenomics sequencing were validated using CyanoDTec Total Cyanobacteria and Toxin Kit (Phytoxigene™) according to the manufacturer's instructions. In total, 17 (8 for non-exceedance and 9 for exceedance beach days) pooled DNA samples from Marie Curtis Park East Beach were used for Cyanobacteria/toxin qPCR. Four standard curves were run separately for each assay (Total Cyanobacteria and Toxin), followed by preparing a composite standard curve. The standard curve range for each assay was 10–100,000 gene copies. Each reaction comprised 20 µL of mastermix/primer–probe solution and 5 µL of DNA from pooled samples (the same DNA used for shotgun sequencing). The gene copy numbers for each sample were calculated using the Slope-Intercept equation from the composite standard curve and normalized to gene copies per nanogram of DNA. A gene was only considered present in a sample if the gene copies/Threshold-cycle values were within the range of the standard curve, and the results were only accepted if the internal amplification control threshold-cycle (Ct) value for a sample was not offset more than 1.5 compared to non-template control.
Bioinformatics and data analysis
Clean reads were aligned against NCBI RefSeq protein Database (Accessed on February 24 2023) using DIAMOND BLASTx on sensitive mode [29], followed by annotation using MEGAN6 (Weighted Lowest Common Ancestor (LCA) method: Minimum-Score = 50, Top-percent filter = 10%, Minimum-Support = 50) [30, 31]. For lateral comparison, annotated reads were rarefied (normalized) to the sample of the smallest size (~ 4.8 million reads) to neutralize bias associated with sequencing depth [31,32,33]. For the Core Microbiome Analysis, bacterial genera present in ≥ 50% of the samples with a relative abundance of ≥ 0.1% were selected [34, 35]. For the Alpha diversity analysis, Shannon–Weaver and Simpson's Reciprocal diversity indices measurements were calculated [36, 37]. Two alpha diversity matrices were used for cross-validation of in-sample diversity. For Antibiotic resistance genes (ARGS) analysis, quality-filtered sequences were assembled into contigs using the MEGAHIT (Metagenome Assembler: k_min + 1 = 2, Min = kmer Size = 21, Max kmer Size = 99, k-step = 20, and minimum-contig size = 200) [38], followed by ARGs annotation using Pathofact pipeline (Combines DeepARG and Resistance gene identifier results for cross-validation) [39]. Statistical analysis was performed using STAMP metagenomic data statistical analysis software [40]. Analysis of Similarity (ANOSIM) was used for the Beta-Diversity assessment [24]. Shapiro–Wilk's normality testing (Stats v3.6.2 R package) was used to determine the normal distribution of the tested microbial variables, followed by either Welch's (and one-way ANOVA) or Wilcoxon-Mann–Whitney t-test for comparison between the groups [41] and Spearman's rank test (Log-transformed data) for correlation analysis between the variables [42, 43].
Results
Quality control analytics
A total of 48 pooled DNA samples from recreational beaches and associated creek/river water were sequenced (Additional file 1: Table 1). Next-generation DNA sequencing provided 12.9 ± 3.2 and 13.7 ± 3.5 million raw reads for Beach A and B, respectively (Additional file 1: Table 2). A high proportion (83%) of raw reads passed the quality control criteria and were processed for downstream taxonomic/functional analysis. The R2 value for the qPCR composite standard curves (Enterococcus, Total Cyanobacteria and Toxin Assays) was within 0.992–0.999, while slope and intercept values were between − 3.25 to − 3.46 and 38.66 to 39.13, respectively (Additional file 1: Table 2). Additional file 1: Figure 2 shows the taxonomic identification after annotation/Least Common Ancestor (LCA) analysis. Bacterial sequences dominated all the samples (93–96%), followed by eukaryotes (3–4%), viruses (1–2%) and archaea (< 1%). The results below highlight bacterial diversity/composition changes associated with the Beach Action Value exceedance and non-exceedance of fecal indicator bacteria.
Diversity between beach action value (BAV) exceedance and non-exceedance beach days
Alpha diversity (diversity within the samples) was approximately 10% higher in Beach Action Value (BAV) Exceedance day samples than the non-exceedance samples for both Beach A (4.4 ± 0.5 versus 4.0 ± 0.3) and Beach B (4.1 ± 0.1 versus 3.7 ± 0.7) (Table 1). Interestingly, exceedance samples from Beach A showed a ~ 9% higher alpha diversity than associated river source. Additionally, when concatenated at the genus level, exceedance samples were segregated into separate clusters from non-exceedance samples for both beaches on principal component analysis plots (Fig. 2 and Additional file 1: Figure 3). Analysis of core microbiome differences identified 9 (24%) and 3 (10%) bacterial genera exclusive to BAV Exceedance beach days from Beach A and Beach B, respectively (Additional file 1: Figure 4).

Differential abundance principal component analysis plots for beach action value (BAV) exceedance and non-exceedance samples from Beach A and Beach B
Bacterial community changes associated with fecal indicator exceedances/non-exceedances
Bacterial community composition on the phylum level was similar for both beaches (Fig. 3). Proteobacteria (38–75%) were the most abundant, followed by Bacteroidetes (20–40%), Actinobacteria (10–30%), Verrucomicrobia (5–7%), Cyanobacteria (2–4%) and Firmicutes (1–2%). For Beach A, at the phylum level, Proteobacteria (p = 1.04e−5) and Fibrobacteres (p = 0.049) were significantly more abundant on Beach Action Value Exceedance days, while Actinobacteria (p = 7.64e−4) and Cyanobacteria (p = 0.021) were significantly more abundant on non-exceedance beach days (Additional file 1: Figure 5). Compared to corresponding sampling dates from beach and river, Cyanobacterial sequences were more abundant for non-exceedance beach samples (Fig. 4). Fibrobacteres sequences were more abundant for exceedance samples and showed a similar abundance pattern between river A and beach A samples (Additional file 1: Figure 6). Additional file 1: Figures 7 and 8 show that Cyanobacterial sequences classified into Microcystis and Pseudanabaena genera. Metagenomics analyses indicated that Microcystis sequences were absent in exceedance samples but detected in 6/9 = 67% of non-exceedance samples (and 100% of samples from late July through August). Pseudanabaena sequences were detected in both exceedance and non-exceedance samples. However, for Pseudanabaena, we did not observe any statistically significant (p = 0.36) difference between BAV exceedance and non-exceedance samples.

Relative abundance profile for six top most abundant phyla identified in beach action value exceedance and non-exceedance beach day samples

Bar plot of cyanobacterial abundance for samples from Marie Curtis Park East Beach and Etobicoke Creek. The horizontal line represents the average number of normalized sequences for each group
We further validated the Cyanobacterial findings using 16S rRNA (Cyanobacterial-specific) qPCR (Fig. 5). Cyanobacterial-specific qPCR results confirmed the metagenomic analysis. qPCR results validated that BAV-non-exceedance beach days had significantly higher (p = 9.99e−4) cyanobacterial gene copies compared to BAV exceedance days for Beach A. Although qPCR did not detect Cyanobacteria-associated microcystin and cylindrospermopsin genes (Table 2), 50% (4/8) of the qPCR tested BAV non-exceedance samples (corresponding to samples with higher Cyanobacterial gene copies) showed the presence of saxitoxin genes.

Bar plot of Microcystis abundance measured by qPCR for Beach A samples. The horizontal line represents the average number of gene copies for each group
For Beach B, at the phylum level, only Spirochaetes sequences were significantly (p = 0.016) more abundant in BAV exceedance samples than non-exceedance samples, and corresponding sampling dates from River B also showed higher mean abundance for sampling dates corresponding to BAV exceedance beach samples (Additional file 1: Figure 9). At the genus level, Spirochaetes sequences were mainly classified into Leptospira and showed comparatively higher mean abundance in BAV exceedance samples (Additional file 1: Figure 10), though the difference was not significant (p = 0.25).
For Beach A, at the genus level, abundances of Pseudomonas (p = 9.41e−3), Acinetobacter (p = 5.09e−3), and Clostridium (p = 0.038) were significantly higher for BAV exceedance days than non-exceedance day samples (Fig. 6). Compared with non-exceedance beach samples, the abundances of Pseudomonas and Acinetobacter were higher in both Etobicoke Creek and Marie Curtis Park East Beach for the sampling dates corresponding to Beach Action Value Exceedances (Additional file 1: Figures 11 and 12). Table 3 shows the correlation between fecal indicator densities and differentially abundant genera for Beach A and River A. For the Beach A samples, the fecal indicator densities (E. coli by culture and Enterococcus by qPCR) showed a significant positive correlation with Pseudomonas (rs = 0.7, p < 0.01), Acinetobacter (rs = 0.6 and 0.5, p ≤ 0.01), and Clostridium (rs = 0.5, p ≤ 0.05). River A samples also showed a significant positive correlation between fecal indicator densities with Pseudomonas (rs = 0.7 and 0.8, p < 0.01) and Acinetobacter (rs = 0.5, p = 0.01).

Differential abundance of statistically significant bacterial genera between beach action value exceedance and non-exceedance beach days from Beach A
For Beach B, at the genus level, 14 genera, including Burkholderia (p = 0.034) and Vibrio (p = 0.05), were significantly more abundant on BAV exceedance days than non-exceedance day samples (Fig. 7). The mean abundance of Burkholderia and Vibrio was also higher in River B on the sampling dates corresponding to BAV exceedance at Sunnyside Beach (Additional file 1: Figures 13 and 14). For correlation with fecal indicator densities, only the Burkholderia counts showed a significant positive correlation with E. coli density for Beach A (Table 4).

Differential abundance of statistically significant bacterial genera between beach action value exceedance and non-exceedance beach days from Beach B
Escherichia sequences were detected in more samples than Enterococcus (Additional file 1: Figures 15 and 16). E. coli sequences were detected in 90% of the samples for both beaches, while Enterococcus sequences were detected in only one sample for each beach. Additionally, the difference in mean proportions between BAV exceedance and non-exceedance samples for Escherichia was not significant (p > 0.05) for both beaches.
Relationship between antibiotics resistance genes and fecal indicator exceedances/non-exceedances
Antibiotic resistance analysis revealed that for both Marie Curtis Park East and Sunnyside Beaches, the five most abundant resistant gene groups were those involved in Beta-lactam antibiotic resistance, Multidrug resistance efflux pumps, Aminoglycoside resistance, Macrolides-Lincosamides resistance and Tetracycline resistance (Fig. 8). However, the difference in mean proportions for the antibiotic-resistance genes was not statistically significant (p > 0.05) between the Beach Action Value Exceedance and Non-exceedance days for both beaches. Additionally, there was no significant correlation between fecal indicator densities and antibiotic resistance gene abundances (Additional file 1: Table 3).

Differential abundance of antibiotic resistance genes in beach action value exceedance and non-exceedance beach days from Beach A and Beach B
Discussion
Freshwater Beach monitoring programs use densities of fecal indicators, including E. coli and Enterococcus, as a reference for evaluating beach water quality. For each fecal indicator and analysis method (enumeration by culturing or quantitative PCR), a specific Beach Action Value (BAV) is recommended by the Public Health Authorities, and beaches are posted for recreational uses if the levels of fecal indicator densities are above the BAV [2, 5]. However, limited experimental data is available for microbial community changes associated with Beach Action Value exceedances, and a single fecal indicator may not be indicative of diverse potential health risks from enteric and non-enteric pathogens, toxigenic cyanobacteria and aspects such as antimicrobial resistance (Ferguson et al., 2012; Li et al., 2021). Compared to conventional analysis methods (culturing and PCR), a metagenomics-based approach provides a robust and comprehensive taxonomic and functional screening profile for water ecosystems. Additionally, metagenomic analysis can provide a foundation for targetted water quality monitoring by identifying region/site-specific microbial/functional differences [37, 44]. This study aimed to provide a comprehensive profile of bacterial community changes associated with fecal indicator Beach Action Value Exceedances and Non-exceedances across two urban recreational freshwater beaches.
Alpha diversity for water samples from Great Lakes beaches has been found to range from 3.5 [45, 46] to 7 [47], and our results for Marie Curtis Park East and Sunnyside Beaches were within this range. Beach Action Value Exceedance samples from both beaches showed comparatively higher alpha diversity than non-exceedance and associated creek/river water sources, indicating other bacterial groups may be present in exceedance day samples. Aside from creeks or rivers, sand/sediment resuspension and other fecal pollution sources can impact bacterial diversity in recreational waters, including birds and mammals defecating nearby around the beach ecosystem [3, 48, 49]. Beach Action Value Exceedance and non-exceedance samples from both beaches separated into independent clusters on principle component analysis plots, which indicates bacterial abundance and diversity differences. Bacterial genera that differed between Beach Action Value Exceedance days and Non-exceedance days differed between the two beaches, suggesting localized influences around each beach rather than regional processes drive microbial community changes. Differences in diversity and the core microbiome between BAV exceedance and non-exceedance beach day samples could be due to diverse environmental factors, including rain events that can increase bacterial diversity on BAV exceedance beach days by increasing the bacterial load from urban runoff, creek/river plumes entering lakes, or increased flows dislodging soil and sediment-attached microbial communities [50, 51].
At the phylum level, Proteobacteria, Bacteroidetes, Actinobacteria, Verrucomicrobia, and Firmicutes were abundant in all tested samples, consistent with other findings (surface water and sediment) for the Great Lakes region [24, 52, 53]. Interestingly, for Beach A, Cyanobacteria sequences were significantly more abundant in BAV non-exceedance samples than exceedance days or associated with the adjacent creek water source. Additionally, the increased abundance of Cyanobacteria and Saxitoxin genes was more notable in the later summer weeks (July–August 2021), corresponding to BAV non-exceedance beach days. Thus, we detected Microcystis, Pseudanabaena, and saxitoxin gene sequences on many non-exceedance beach days when E. coli data indicated Beach A should be open for water recreation. Both Beach A and B are not routinely monitored for cyanobacteria or harmful algal species and would typically be tested in response to visual complaints of bloom formations. Cyanobacterial genera, including Microcystis, can lead to deteriorating (eutrophication and toxin production) water quality for recreational purposes [54, 55]. Similar to our findings, a study on recreational waters [56] identified decreased fecal indicator densities associated with higher Cyanobacterial (specifically Microcystis) levels. Therefore, relying solely on the fecal indicator densities for recreational water quality may provide an incomplete perception of human health risks at beaches. Fibrobacteres species (cellulose-degrading bacteria) are specific to the rumen microbiome of ruminant animals [57], while Spirochaetes are found in farm animals (cows and pigs) but not human fecal material [58]. Leptospira contamination can be from domestic and wild animals [59], which may indicate that fecal contamination for Beach B can be from both water sources (Humber River) and localized (wild animals). The increased abundance of Fibrobacteres and Spirochaetes, along with increased E. coli levels, on BAV exceedance days at our two beaches may indicate fecal contamination from livestock or other ruminants in runoff to river sources that subsequently impacts the beaches.
On the genus level, Pseudomonas, Acinetobacter and Clostridium were significantly more abundant in BAV exceedance samples from Beach A (and River A samples for the same dates), while Bulkholderia and Vibrio were more abundant for BAV exceedance samples from Beach B (and River B samples for the same dates). Studies in the Great Lakes region [24, 50] have identified Pseudomonas, Clostridium and Acinetobacter as common genera in stormwater, which may explain the influx of these genera into Beach A from the adjacent creek water source. Both River A and River B are significantly impacted by stormwater systems at times, contributing to increased fecal contamination at Marie Curtis Park East and Sunnyside Beaches [60, 61]. Burkholderia and Vibrio sequences have been identified to be associated with both human and animal fecal contamination in urban recreational waters [62], which is in agreement with our findings for Beach B and may indicate fecal contamination from both associated waters (Humber River) and localized (wild animals) sources.
The metagenomics method also provided a screen for detecting a diverse range of antibiotic resistance genes. We detected numerous Antibiotic Resistance Genes (ARGs) of clinical concern in water samples from both Beaches. Similar to our beaches, other studies have also identified ARGs at beaches [63] and across ~ 350 lakes in Canada [64]. We found no significant association between numbers of antibiotic resistance genes (ARGs) and fecal indicators, with exceedances based on E. coli or Enterococcus numbers, this suggests the occurrence of AMR genes was not solely driven by AMR genes associated with these two FIBs. AMR genes are likely associated with diverse other bacteria, including those unrelated to fecal pollution, which is another limitation of using traditional culture (E. coli) or qPCR (Enterococcus) methods to predict overall AMR gene occurrence. However, ARG pools were consistently present at our two beaches, with the potential for horizontal gene transfer to bacterial species of human health concern. While water can play a role in routes of ARG exposure, quantifying that role and its associated human health risks requires further research [65].
Overall, this proof of concept study demonstrates the potential value of metagenomics for enabling a more comprehensive screen of bacterial community changes associated with fecal indicator Beach Action Value Exceedance and Non-exceedance conditions at freshwater beaches. The differences in bacterial diversity and abundance in response to BAV exceedances for Beach A were more pronounced (supported by correlation test) compared to Beach B. This may be due to Beach A sampling locations being closer to the mouth of River A than the proximity of Beach B sampling locations to the mouth of River B. In addition, a break wall limits the direct influx from River B into Beach B, while Beach A is on an open coastline that receives unhindered water flow directly from River A under the right wind and current conditions. One limitation of this study is the localization of both tested beaches in the same geographical location or close proximity. However, we identified site-specific microbial differences between the two tested beaches, and future studies can build on our results/methodology to include a larger study area. Additionally, Our results provide insight into localized processes influencing bacterial community changes at freshwater beaches and further identify limitations of existing culture-based and single-gene PCR assay approaches for assessing recreational water quality. The results provide a foundation to guide more comprehensive screening for harmful microorganisms, as well as toxin and antimicrobial resistance genes, in order to improve recreational water quality monitoring and enable more targeted and site-specific risk management strategies.
Conclusion
-
1.
Cyanobacterial sequences (Microcystis and saxitoxin genes in particular) were significantly more abundant in Beach Action Value Non-exceedance samples from Beach A, demonstrating that fecal indicator bacteria densities may not indicate health risks associated with harmful algal blooms and the eutrophication of recreational waters.
-
2.
The increase of Fibrobacteres sequences in BAV exceedance days of Beach A may represent an influx of fecal contamination from livestock or other ruminant animals.
-
3.
Pseudomonas, Acinetobacter, and Clostridium sequences were significantly more abundant on the BAV exceedance days and positively correlated with fecal indicator densities at Beach A.
-
4.
The increase of Spirochaetes (specifically Leptospira), Burkholderia, and Vibrio was significantly associated with Beach Action Value Exceedance samples from Beach B.
-
5.
Similar bacterial diversity and abundance trends between beach and river samples suggest the Creek and River are primary sources of bacterial contamination at the beaches.
-
6.
Pools of Antibiotic Resistance Genes (ARGs) were consistently detected at both beaches, indicating potential for transfer to potentially pathogenic genera by horizontal gene transfer.
-
7.
The metagenomics approach provided the capability of extending beyond E. coli and single gene PCR testing to provide a comprehensive screen of beach water samples for bacterial community composition and toxin and antimicrobial resistance genes associated with changing beach water conditions.
Availability of data and materials
The sequencing data from this study is available from the corresponding author upon reasonable request. All the analytical data supporting the findings in this study is provided in the figures and tables in the main manuscript and supplementary data files.
References:
USEPA. National epidemiologic and environmental assessment of recreational water epidemiology studies. EPA/600/R-10/168. 2009. http://www.epa.gov/neear/files/Report2009v5_508comp.pdf.
Health Canada. Canadian recreational water quality guidelines-indicators of fecal contamination: overview. 2023. https://www.canada.ca/en/health-canada/services/publications/healthy-living/recreational-water-quality-guidelines-indicators-fecal-contamination.html.
Edge TA, Hill S. Multiple lines of evidence to identify the sources of fecal pollution at a freshwater beach in Hamilton Harbour, Lake Ontario. Water Res. 2007;41(16):3585–94.
Edge TA, Hill S, Seto P, Marsalek J. Library-dependent and library-independent microbial source tracking to identify spatial variation in faecal contamination sources along a Lake Ontario beach (Ontario, Canada). Water Sci Technol. 2010;62(3):719–27.
USEPA. Recreational water quality criteria. United States (US) Environmental Protection Agency (EPA). EPA-820-F-12–058. 2012. p. 42. https://www.epa.gov/sites/default/files/2015-10/documents/rwqc2012.pdf.
Ferguson DM, Griffith JF, McGee CD, Weisberg SB, Hagedorn C. Comparison of Enterococcus species diversity in marine water and wastewater using Enterolert and EPA Method 1600. J Environ Public Health. 2013. 2013.
Odonkor ST, Ampofo JK. Escherichia coli as an indicator of bacteriological quality of water: an overview. Microbiol Res. 2013;4(1): e2.
Brumfield KD, Cotruvo JA, Shanks OC, Sivaganesan M, Hey J, Hasan NA, Huq A, Colwell RR, Leddy MB. Metagenomic sequencing and quantitative real-time PCR for fecal pollution assessment in an urban watershed. Front Water. 2021;3: 626849.
Ballesté E, García-Aljaro C, Blanch AR. Assessment of the decay rates of microbial source tracking molecular markers and fecal indicator bacteria from different sources. J Appl Microbiol. 2018;125(6):1938–49.
Ahmed W, Toze S, Veal C, Fisher P, Zhang Q, Zhu Z, Staley C, Sadowsky MJ. Comparative decay of culturable fecal indicator bacteria, microbial source tracking marker genes, and enteric pathogens in laboratory microcosms that mimic a sub-tropical environment. Sci Total Environ. 2021;751: 141475.
Zimmer-Faust AG, Thulsiraj V, Marambio-Jones C, Cao Y, Griffith JF, Holden PA, Jay JA. Effect of freshwater sediment characteristics on the persistence of fecal indicator bacteria and genetic markers within a Southern California watershed. Water Res. 2017;119:1–11.
Hannides A, Elko N, Briggs TR, Kim S-C, Mercer A, Park K, Rosov B, Searcy R, Walther M. US beach water quality monitoring. Shore Beach. 2021;89(3):26.
USEPA. Recreational water quality criteria. United States (US) Environmental Protection Agency (EPA). p. 42. EPA-820-F-12-058. 2012. https://www.epa.gov/sites/default/files/2015-10/documents/rwqc2012.pdf.
Wade TJ, Calderon RL, Brenner KP, Sams E, Beach M, Haugland R, Wymer L, Dufour AP. High sensitivity of children to swimming-associated gastrointestinal illness: results using a rapid assay of recreational water quality. Epidemiology. 2008;215:375–83.
Wade TJ, Sams E, Brenner KP, Haugland R, Chern E, Beach M, Wymer L, Rankin CC, Love D, Li Q. Rapidly measured indicators of recreational water quality and swimming-associated illness at marine beaches: a prospective cohort study. Environ Health. 2010;9:1–14.
Rochelle-Newall E, Nguyen TMH, Le TPQ, Sengtaheuanghoung O, Ribolzi O. A short review of fecal indicator bacteria in tropical aquatic ecosystems: knowledge gaps and future directions. Front Microbiol. 2015;6:308.
Islam MM, Hofstra N, Islam MA. The impact of environmental variables on faecal indicator bacteria in the Betna river basin, Bangladesh. Environ Processes. 2017;4:319–32.
Harwood VJ, Staley C, Badgley BD, Borges K, Korajkic A. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol Rev. 2014;38(1):1–40.
Nappier SP, Ichida A, Jaglo K, Haugland R, Jones KR. Advancements in mitigating interference in quantitative polymerase chain reaction (qPCR) for microbial water quality monitoring. Sci Total Environ. 2019;671:732–40.
Cao Y, Raith MR, Griffith JF. Droplet digital PCR for simultaneous quantification of general and human-associated fecal indicators for water quality assessment. Water Res. 2015;70:337–49.
Fresia P, Antelo V, Salazar C, Giménez M, D’Alessandro B, Afshinnekoo E, Mason C, Gonnet GH, Iraola G. Urban metagenomics uncover antibiotic resistance reservoirs in coastal beach and sewage waters. Microbiome. 2019;7(1):1–9.
Edge TA, Baird DJ, Bilodeau G, Gagné N, Greer C, Konkin D, Newton G, et al. The Ecobiomics project: advancing metagenomics assessment of soil health and freshwater quality in Canada. Sci Total Environ. 2020;710:135906.
Saleem F, Edge TA, Schellhorn HE. Validation of qPCR method for enterococci quantification at Toronto beaches: application for rapid recreational water monitoring. J Great Lakes Res. 2022;48(3):707–16.
Mohiuddin MM, Botts SR, Paschos A, Schellhorn HE. Temporal and spatial changes in bacterial diversity in mixed use watersheds of the Great Lakes region. J Great Lakes Res. 2019;45(1):109–18.
APHA. Standard methods for the examination of water and wastewater. American Public Health Association. American Water Works Association, Water Environment Federation. 2012.
Saleem F, Schellhorn HE, Simhon A, Edge TA. Same-day Enterococcus qPCR results of recreational water quality at two Toronto beaches provide added public health protection and reduced beach days lost. Can J Public Health. 2023;471:1–12.
USEPA. Method 1609.1: enterococci in water by TaqMan® quantitative polymerase chain reaction (qPCR) with internal amplification control (IAC) assay. EPA-820-R-15-099. 2015. https://www.epa.gov/sites/default/files/2015-08/documents/method_1609-1-enterococcus-iac_2015_3.pdf.
Andrews S. FastQC: a quality control tool for high throughput sequence data. 2010.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60.
Bağcı C, Beier S, Górska A, Huson DH. Introduction to the analysis of environmental sequences: metagenomics with MEGAN. Evolut Genom Stat Comput Methods. 2019;521:591–604.
Bağcı C, Patz S, Huson DH. DIAMOND+ MEGAN: fast and easy taxonomic and functional analysis of short and long microbiome sequences. Curr Protoc. 2021;1(3): e59.
Huson DH, Richter DC, Mitra S, Auch AF, Schuster SC. Methods for comparative metagenomics. BMC Bioinform. 2009;10:1–10.
Pereira MB, Wallroth M, Jonsson V, Kristiansson E. Comparison of normalization methods for the analysis of metagenomic gene abundance data. BMC Genom. 2018;19:1–17.
De Brito LCN, Doolittle-Hall J, Lee C-T, Moss K, Júnior WB, Tavares WLF, Sobrinho APR, Teles FRF. The apical root canal system microbial communities determined by next-generation sequencing. Sci Rep. 2020;10(1):10932.
Astudillo-García C, Bell JJ, Webster NS, Glasl B, Jompa J, Montoya JM, Taylor MW. Evaluating the core microbiota in complex communities: a systematic investigation. Environ Microbiol. 2017;19(4):1450–62.
Chernov TI, Tkhakakhova AK, Kutovaya OV. Assessment of diversity indices for the characterization of the soil prokaryotic community by metagenomic analysis. Eurasian Soil Sci. 2015;48:410–5.
Bae S, Lyons C, Onstad N. A culture-dependent and metagenomic approach of household drinking water from the source to point of use in a developing country. Water Res X. 2019;2: 100026.
Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6.
De Nies L, Lopes S, Busi SB, Galata V, Heintz-Buschart A, Laczny CC, May P, Wilmes P. PathoFact: a pipeline for the prediction of virulence factors and antimicrobial resistance genes in metagenomic data. Microbiome. 2021;9:1–14.
Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–4.
West RM. Best practice in statistics: Use the Welch t-test when testing the difference between two groups. Ann Clin Biochem. 2021;58(4):267–9.
Hu W, Liang J, Ju F, Wang Q, Liu R, Bai Y, Liu H, Qu J. Metagenomics unravels differential microbiome composition and metabolic potential in rapid sand filters purifying surface water versus groundwater. Environ Sci Technol. 2020;54(8):5197–206.
Sun F, Wang C, Yang H. Physicochemical factors drive bacterial communities in an aquaculture environment. Front Environ Sci. 2021;9: 709541.
Miller RR, Montoya V, Gardy JL, Patrick DM, Tang P. Metagenomics for pathogen detection in public health. Genome Med. 2013;5:1–14.
Lee CS, Kim M, Lee C, Yu Z, Lee J. The microbiota of recreational freshwaters and the implications for environmental and public health. Front Microbiol. 2016;7:1826.
Hengy MH, Horton DJ, Uzarski DG, Learman DR. Microbial community diversity patterns are related to physical and chemical differences among temperate lakes near Beaver Island. MI PeerJ. 2017;5: e3937.
Nevers MB, Byappanahalli MN, Nakatsu CH, Kinzelman JL, Phanikumar MS, Shively DA, Spoljaric AM. Interaction of bacterial communities and indicators of water quality in shoreline sand, sediment, and water of Lake Michigan. Water Res. 2020;178: 115671.
Rothenheber D, Jones S. Enterococcal concentrations in a coastal ecosystem are a function of fecal source input, environmental conditions, and environmental sources. Appl Environ Microbiol. 2018;84(17):e01038-e1118.
Nakatsu CH, Byappanahalli MN, Nevers MB. Bacterial community 16S rRNA gene sequencing characterizes riverine microbial impact on Lake Michigan. Front Microbiol. 2019;10:996.
Fisher JC, Newton RJ, Dila DK, McLellan SL. Urban microbial ecology of a freshwater estuary of Lake Michigan. Elementa. 2015;3: 000064.
Powers NC, Wallgren HR, Marbach S, Turner JW. Relationship between rainfall, fecal pollution, antimicrobial resistance, and microbial diversity in an urbanized subtropical bay. Appl Environ Microbiol. 2020;86(19):e01229-e1320.
Winters AD, Marsh TL, Brenden TO, Faisal M. Molecular characterization of bacterial communities associated with sediments in the Laurentian Great Lakes. J Great Lakes Res. 2014;40(3):640–5.
Crevecoeur S, Edge TA, Watson LC, Watson SB, Greer CW, Ciborowski JJH, Diep N, et al. Spatio-temporal connectivity of the aquatic microbiome associated with cyanobacterial blooms along a Great Lake riverine-lacustrine continuum. Front Microbiol. 2023;14:1073753.
Paerl HW, Otten TG. Harmful cyanobacterial blooms: causes, consequences, and controls. Microb Ecol. 2013;65:995–1010.
Coffey R, Paul MJ, Stamp J, Hamilton A, Johnson T. A review of water quality responses to air temperature and precipitation changes 2: nutrients, algal blooms, sediment, pathogens. JAWRA J Am Water Resour Assoc. 2019;55(4):844–68.
Halac SR, del Valle Bazán R, Larrosa NB, Nadal AF, Ruibal-Conti AL, Rodriguez MI, Ruiz MA, López AG. First report on negative association between cyanobacteria and fecal indicator bacteria at San Roque reservoir (Argentina): impact of environmental factors. J Freshw Ecol. 2019;34(1):273–91.
Ransom-Jones E, Jones DL, McCarthy AJ, McDonald JE. The Fibrobacteres: an important phylum of cellulose-degrading bacteria. Microb Ecol. 2012;63:267–81.
Jeong JY, Park HD, Lee KH, Weon HY, Ka JO. Microbial community analysis and identification of alternative host-specific fecal indicators in fecal and river water samples using pyrosequencing. J Microbiol. 2011;49:585–94.
Muñoz-Zanzi C, Mason MR, Encina C, Astroza A, Romero A. Leptospira contamination in household and environmental water in rural communities in southern Chile. Int J Environ Res Public Health. 2014;11(7):6666–80.
Staley Z, Grabuski J, Sverko E, Edge TA. Comparison of microbial and chemical source tracking markers to identify fecal contamination sources in the Humber River and associated stormwater outfalls. Appl Environ Microbiol. 2016;82:6357–66.
Staley ZR, Chuong JD, Hill SJ, Grabuski J, Shokralla S, Hajibabaei M, Edge TA. Fecal source tracking and eDNA profiling in an urban creek following an extreme rain event. Sci Rep. 2018;8(1):14390.
Paulino GVB, Félix CR, Silvan CG, Andersen GL, Landell MF. Bacterial community and environmental factors associated to rivers runoff and their possible impacts on coral reef conservation. Mar Pollut Bull. 2020;156: 111233.
Carney RL, Labbate M, Siboni N, Tagg KA, Mitrovic SM, Seymour JR. Urban beaches are environmental hotspots for antibiotic resistance following rainfall. Water Res. 2019;167: 115081.
Kraemer SA, da Costa NB, Oliva A, Huot Y, Walsh DA. A resistome survey across hundreds of freshwater bacterial communities reveals the impacts of veterinary and human antibiotics use. Front Microbiol. 2022;13:995418.
Liguori K, Keenum I, Davis BC, Calarco J, Milligan E, Harwood VJ, Pruden A. Antimicrobial resistance monitoring of water environments: a framework for standardized methods and quality control. Environ Sci Technol. 2022;56(13):9149–60.
Funding
This project was supported with funds provided by the Government of Ontario (Canada-Ontario Agreement grant agreement #2607) and NSERC (Canada) (Alliance grant ALLRP 554507-2020). Such support does not indicate endorsement by the Government of Ontario of the contents or conclusions of this contribution.
Author information
Authors and Affiliations
Contributions
Conceptualization, HES; methodology HES, FS, and TAE; formal analysis and Experimentation, FS, and KLT; writing—original draft preparation, FS; writing—review and editing HES, FS, and TAE, supervision, HES. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary Tables 1–3 and supplementary Figures 1–16.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Saleem, F., Li, E., Edge, T.A. et al. Identification of potential microbial risk factors associated with fecal indicator exceedances at recreational beaches. Environmental Microbiome 19, 4 (2024). https://doi.org/10.1186/s40793-024-00547-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40793-024-00547-8