
Abstract
Background
Ustilago maydis is known for its natural potential to produce a broad range of valuable chemicals, such as itaconate, from both industrial carbon waste streams and renewable biomass. Production of itaconate, and many other secondary metabolites, is induced by nitrogen limitation in U. maydis. The clustered genes responsible for itaconate production have recently been identified, enabling the development of new expression tools that are compatible with biotechnological processes.
Results
Here we report on the investigation of two of the native promoters, P tad1 and P mtt1 , from the itaconate cluster of U. maydis MB215. For both promoters the specific activation upon nitrogen limitation, which is known to be the trigger for itaconate production in Ustilago, could be demonstrated by gfp expression. The promoters cover a broad range of expression levels, especially when combined with the possibility to create single- and multicopy construct integration events. In addition, these reporter constructs enable a functional characterization of gene induction patterns associated with itaconate production.
Conclusions
The promoters are well suited to induce gene expression in response to nitrogen limitation, coupled to the itaconate production phase, which contributes towards the further improvement of organic acid production with Ustilago.
Similar content being viewed by others
Background
The family of Ustilaginaceae has sparked great interest as promising industrial production organisms in recent years. The growing biotechnological attention results from their native ability to utilize a range of bio-based substrates [10, 18, 20, 62] and to produce a broad variety of value–added chemicals, such as glycolipids, polyols, and organic acids [12, 19, 33]. Considerable efforts have been undertaken to optimize fermentation and process conditions in order to increase the yield, titer and rate of glycolipids [42], erythritol [28], malate [61], itaconate [16, 41] and 2-hydroxyparaconate [15, 22]. The biochemical pathways and associated gene clusters of several secondary metabolites have been characterized and engineered, including those of cellobiose lipids [51], mannosyl erythritol lipids [23, 27], malate [60], itaconate [17, 59], and 2-hydroxyparaconate [16]. These efforts have accelerated recently largely thanks to a growing suite of efficient genetic engineering tools, including marker recycling through the FLP/FRT system [32], Golden Gate Cloning [53] and Cas9-based genome engineering [48]. Although originally developed for the model organism U. maydis, these tools can also be adapted for use in other Ustilaginaceae [59, 60]. However, critical limitations remain that are characteristic for fungal biotechnology, including the availability of suitable promoters. Specific sets of promoters have been developed for the production of proteins and chemicals of high industrial relevance in different organisms, such as Aspergillus niger [55, 56], Escherichia coli [37, 38], and Penicillium chrysogenum [44]. Modern online analysis systems have become an attractive means to investigate promoter properties, enabling high-resolution characterization of the specific time of induction, the induction trigger, or the promoter strength, for different conditions or promoter variants [64]. One particularly important trigger for the production of organic acids and glycolipids in Ustilaginaceae is nitrogen limitation. Such a limitation causes a strict temporal separation of the very different cellular objectives associated with biomass growth and product formation [14, 26, 52]. This characteristic of fungal secondary metabolite production may be exploited for metabolic engineering. Constitutive promoters, such as P otef and P oma [13, 46, 49], are mainly active in the growth phase, while inducible promoters, such as Pcrg1, Pnar1 or the tet-system, come with other drawbacks [4, 6, 63]. Pcrg1 relies on arabinose and is efficiently repressed by glucose and xylose, two of the main carbon sources for industrial production processes [4]. Pnar1 is induced by nitrate and repressed by ammonium [6] and is therefore not suitable for nitrogen-limited production. Other inducible promoters are associated with specific phases of the Ustilago life cycle, making them equally problematic for engineering of secondary metabolite production [2]. Hence new promoters, which are specifically activated by nitrogen limitation hold much promise for metabolic engineering.
Recently, the gene cluster responsible for itaconate production in U. maydis was identified [17] and its promoters are promising candidates to overcome current limitations. From the different functions of the genes in the cluster, including catalytic, transport and transcriptional regulation activities, it may be deduced that the individual promoters should possess different inherent properties with respect to induction and strength. This was confirmed by qRT–PCR [17]. Two- to ninefold elevated activity for all seven genes in this cluster, except for rdo1, was shown during the itaconate production phase compared to non-induced conditions in rich medium [17]. Further, the regulation of most genes in the cluster is strongly dependent on the putative transcriptional regulator ria1. By deletion of ria1, the transcription level for all genes within the cluster was lowered by up to ninefold and overexpression triggered expression of most cluster genes [17]. Therefore these promoters show much promise for biotechnological usage as expression tools.
Here we investigate a set of promoters from the genes responsible for itaconate production in U. maydis that are induced during the non-growing production phase as initiated by nitrogen limitation.
Methods
Strains and cultivation conditions
All strains used and constructed within this work are listed in Table 1.
As host for cloning experiments, E. coli DH5α (DSM 6897) was used. All E. coli strains were grown at 37 °C shaking at 200 rpm (shaking diameter 25 mm) in lysogeny broth (LB) medium. For recombinant strains 100 mg L−1 ampicillin were added to the medium.
Ustilago maydis cultures were cultivated in YEPS light medium containing 20 g L−1 D–sucrose, 5 g L−1 yeast extract, and 10 g L−1 peptone at 30 °C shaking at 200 rpm (shaking diameter 25 mm).
For physiological experiments 96 flat-bottom round-well plates or 48-well flower plates were used as described below.
As cultivation medium MTM was used containing 50 g L−1 glucose, 0.2 g L−1 MgSO4 · 7 H2O, 0.01 g L−1 FeSO4 · 7 H2O, 0.5 g L−1 KH2PO4, 1 mL L−1 vitamin solution, 10 mL L−1 trace element solution, 19.5 g L−1 2-(N-morpholino)ethanesulfonic acid (MES) as buffer, and differing NH4Cl concentrations. MES buffer was used as stock solution with pH adjusted to 6.5 with NaOH. The vitamin solution contained (per liter) 0.05 g D-biotin, 1 g D-calcium panthotenate, 1 g nicotinic acid, 25 g myo-inositol, 1 g thiamine hydrochloride, 1 g pyridoxol hydrochloride, and 0.2 g para–aminobenzoic acid. The trace element solution contained (per liter) 1.5 g EDTA, 0.45 g ZnSO4 · 7 H2O, 0.10 g MnCl2 · 4 H2O, 0.03 g CoCl2 · 6 H2O, 0.03 g CuSO4 · 5 H2O, 0.04 g Na2MoO4 · 2 H2O, 0.45 g CaCl2 · 2 H2O, 0.3 g FeSO4 · 7 H2O, 0.10 g H3BO3, and 0.01 g KI [19]. For phosphate limitation 3.2 g L−1 NH4Cl and 0.1 g L−1 KH2PO4 were used.
Precultures for analytical experiments were cultivated for 24 h in 500 mL shake flasks without baffles with 50 mL MTM containing 4 g L−1 NH4Cl, to ensure that no nitrogen limitation occurs prior to inoculation of the main culture.
Analytical methods
All experiments were performed in triplicates, unless stated otherwise. The arithmetic mean of the biological replicates is shown. Error bars and ± values indicate standard error of the mean.
HPLC analysis was performed as described previously [62]. Centrifuged samples (13,000g, 5 min) were filtered through cellulose acetate filters (diameter 0.2 µm, VWR, Germany) and subsequently diluted 1:10 with distilled water. For analysis a Dionex Ultimate 3000 HPLC (Dionex, USA) with an Organic Acid Resin column (CS-Chromatographie, Germany) kept at 75 °C, with a constant flow rate of 0.8 ml min−1 of 5 mM sulfuric acid as eluent was used. For detection, a Shodex RI101 detector at 35 °C and a variable wavelength UV detector (Dionex, USA) at 210 nm were used.
Ammonium concentration was determined by a colorimetric assay according to Willis [57]. For this 50 µL of the sample (maximal 50 mg L−1− NH4 +) were mixed with 1 mL reagent solution and afterwards with 0.25 mL hypochlorite solution. The mixture was incubated for at least 12 min at room temperature before measuring the absorbance at 685 nm. The reagent solution contained 32 g sodium salicylate (anhydrous), 40 g trisodium phosphate (TSP) and 0.5 g sodium nitrosylpentacyanoferrate(III) (sodium nitroprusside) dissolved in 1 L of water. For the hypochlorite solution, 50 mL of commercially available bleach (Clorox) containing 5–5.25% sodium hypochlorite were diluted with water to 1 L with a final concentration of ~ 0.25% hypochlorite.
Initial promoter characterization experiments were performed in 96-well plates using a BioLector (m2p-labs, Baesweiler, Germany) at 30 °C shaking at 1000 rpm (shaking diameter: 3 mm). GFP fluorescence intensity was determined at 488/520 nm (excitation/emission) with the gain set to 70. For biomass determination backscatter intensity in the far red range 620/620 nm with the gain set to 10 was used. Specific promoter activities are expressed as GFP fluorescence over biomass backscatter. Since both these signals are measured in arbitrary units, this specific activity is expressed without dimension. Values are normalized (point-to-point) for the arithmetic mean of the corresponding wildtype. Induction ratios were determined by dividing the maximum specific promoter activity by the average activity from 2–10 h.
Induction profiles of U. maydis MB215 P tad1 -gfp under different cultivation conditions were analyzed in 48-well flower plates (M2P-labs, Baesweiler, Germany) with an in-house constructed screening system based on the established BioLector setup [45, 54] with a fluorospectrometer, which features excitation and emission monochromators for free wavelength selection in the UV/Vis range (Fluoromax-4, HORIBA Jobin–Yvon). GFP fluorescence intensity was determined at 495/507 nm (bandpass: 8 nm, integration time: 600 ms) and biomass was measured via backscatter intensity at 650/650 nm (bandpass: 4 nm, integration time: 1200 ms).
Cloning procedures
The genome of U. maydis 521 was taken as reference sequence [29].
Plasmids P tad1 -gfp, P itp1 -gfp, P adi1 -gfp, and P mtt1 -gfp were cloned by amplifying the promoter regions of genes UMAG_05076 (tad1), UMAG_05077 (itp1), UMAG_05078 (adi1), and UMAG_05079 (mtt1) with the oligonucleotide primers listed in Table 2 and exchanging P otef on the vector pUMa43 [34]. For UMAG_05077 (itp1), UMAG_05078 (adi1), and UMAG_05079 (mtt1) 2000 bp in front of the respective start codon were used and for UMAG_05076 (tad1) 1500 bp were used (Fig. 1).

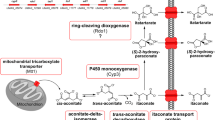
Clustered itaconate genes in U. maydis including trans-aconitate decarboxylase (tad1), a major facilitator superfamily transporter (itp1), an aconitate–Δ–isomerase (adi1), a mitochondrial tricarboxylate transporter (mtt1), and a transcriptional regulator (ria1) [17]. Red arrows indicate the promoter regions investigated in this study
The resulting plasmids were linearized with the restriction enzyme SspI prior to U. maydis transformation and constructs were integrated into the ip-locus of U. maydis strain MB215 by homologous recombination [40]. Transformation of U. maydis was performed using standard protocols [47]. For selection of transformants, PDA plates with 2 μg ml−1 carboxin were used. Correct integration of constructs and number of integration events was verified by Southern blot analysis using enzyme EcoRV for genome restriction and the Cbx-cassette from vector pMF1–C [5] as a probe for the detection of specific DNA fragments.
Results and discussion
Expression strengths of promoters from the itaconate cluster of U. maydis
The recently characterized genes encoding core catalytic enzymes and transporters, which are required for itaconate production (tad1, itp1, adi1, mtt1) were chosen as primary targets for the investigation of promoter activities. From previous studies it was known that their native activity is strongly influenced by the activity of the transcriptional regulator Ria1 which is itself also encoded in the itaconate gene cluster [17].
For analysis of induction conditions and promoter strength we fused each of the four promoters to gfp. GFP has been shown to be a suitable tool for expression analysis in several organisms including Escherichia coli [11, 43], Saccharomyces cerevisiae [1] and Pseudomonas putida [64]. Also in U. maydis GFP was used before to investigate host–pathogen interactions [49], to identify and localize proteins, for example a motor protein [39], and to investigate promoter activities [3, 49].
The plasmids containing the promoter-gfp fusions were integrated into the ip-locus of U. maydis MB215 [9, 31]. The resulting mutants were screened for ip–locus and single (s) or multiple (m) integration events by PCR and Southern Blotting. This targeted in-locus integration avoids undesired polar effects, such as gene disruption, and ensures optimal comparability between different strains by avoiding locus- and copy number-dependent expression differences [49]. For all promoters, one clone with single construct integration was chosen for further characterization (Fig. 2).

Fluorescence signals for U. maydis single integration mutants cultivated in MTM containing 0.8 g L−1 NH4Cl and 50 g L−1 glucose expressing gfp under the control of four promoters from the itaconate gene cluster. GFP fluorescence intensities (normalized against the wildtype) are shown for single integration mutants under control of P otef (red), P tad1 (green), P itp1 (orange), P adi1 (purple), and P mtt1 (blue) after 24 h of cultivation. Error bars indicate standard error of the mean (n = 3)
Fluorescence increased over time for all strains (data not shown), however, with great differences in the absolute intensity range. While P itp1 and P adi1 showed very low fluorescence intensities after 24 h of cultivation, about two-to-three orders of magnitude below that of the strong P otef , P tad1 showed 48-fold and P mtt1 fivefold lower intensities. These data indicate pronounced differences of promoter activity within the itaconate cluster from U. maydis, with P adi1 and P itp1 as relatively weak promoters and P mtt1 as a strong promoter. The differences are in line with the physiological roles of the regulated genes. The mitochondrial transporter Mtt1 is the limiting step for itaconate biosynthesis in U. maydis [17], requiring relatively high expression. In contrast, high expression of Adi1 would result in a surplus of trans–aconitate, which is a potent inhibitor for vital metabolic reactions, such as the conversion of citrate to isocitrate by aconitase in the TCA–cycle [21]. The in-trans expression of P adi1 and P itp1 outside of their original genomic context might influence their activity, since the orientation in the genome suggests a possible bidirectional promoter (Fig. 1), which might be influenced by additional upstream elements in their native context. Based on those results, P tad1 and P mtt1 were chosen as the two strongest promoters for further characterization and for the development as expression tools.
Activation of the P tad1 and P mtt1 occurs in response to nitrogen limitation
For expression of genes coupled to itaconate production the induction time can be critical, depending on the expressed product, since some products are toxic during the growth phase, and the production of itaconate poses a drain on the primary metabolite cis-aconitate. Consequently we investigated the induction time and conditions (Fig. 3) for P tad1 and P mtt1 using P otef as a control. Additionally, we investigated the differences resulting from clones with single construct integration compared to multiple integration, hypothesizing that multiple insertions may allow for a wider range of activities. It has to be noted, however, that the copy number for multiple integration events could not be quantified.

Induction profiles of gfp under control of P tad1 (a), P mtt1 (b), and P otef (c). Itaconate concentration (red) and NH4Cl concentration (black) correlated to the specific promoter activity (GFP over biomass, normalized against the WT) for U. maydis strains containing single (light green) and multiple (dark green) insertions. Error bars indicate standard error of the mean (n = 3)
Promoter activities are expressed as GFP fluorescence intensity over biomass (measured as scattered light). The activity of both P tad1 and P mtt1 increased strongly after 14 h of cultivation, corresponding to the time point where ammonium was depleted from the culture medium (Fig. 3). Production of itaconate started approximately two hours later. This correlates well with the known induction of itaconate production for U. maydis [14]. In contrast, the strong constitutive P otef promoter gave a high and relatively stable signal during the growth- and production phases. The low fluorescence intensity of the P tad1 construct during the initial growth phase hints at a weak, but tightly controlled activity with an induction ratio of 72 (single integration) and 122 (multiple integrations). P mtt1 in contrast is a strong, but leaky promoter, with a basal activity during the growth phase and a lower induction ratio of 13 (single integration) and 11 (multiple integrations). The basal activity of P mtt1 in the growth phase may result from the generally high promoter activity, or from an activation due to expression out of the original chromosomal context.
We next investigated the impact of multiple integration on the expression activity. With values of 0.03 (P tad1 , single, 23–32 h), 0.40 (P tad1 , multi, 28–29 h), 0.33 (P mtt1 , single, 27–32 h), 1.32 (P mtt1 , multi, 29–32 h), 1.38 (P otef , single, 25–26 h), and 3.00 (P otef , multi, 25–26 h), the maximum expression level increased by about twofold (P otef ), fourfold (P mtt1 ), and 13-fold (P tad1 ) for multiple construct integrations compared to single integration. Taking into consideration the promoter activities of single-copy P tad1 and P mtt1 constructs, and multiple integration, expression levels covering a nearly 50-fold dynamic range with high resolution appears realistic. This value may even be increased by selecting higher integration numbers for P mtt1 .
The impact of growth limiting nutrients on P tad1
Itaconate production by U. maydis is generally induced upon nitrogen limitation. However, the amount of nitrogen source as a growth-limiting nutrient has a strong impact on the efficiency of itaconate production [16, 41]. In other itaconate producers, such as Aspergillus terreus, phosphate-limiting media are generally used [36, 58], although a P-limitation is not strictly needed for efficient induction [25, 35]. In order to further investigate the effect of different growth limitations on the activation of P tad1 , we cultivated U. maydis P tad1 -gfp with different combinations of NH4Cl and KH2PO4. Keeping the initial concentration of KH2PO4 at 0.5 g L−1 we increased the initial NH4Cl concentrations from 0.8 to 1.6 and 3.2 g L−1, resulting in higher biomass concentrations and nitrogen limitation at a later time-point during cultivation. To ensure a phosphate-limited culture, we lowered the initial concentration of KH2PO4 to 0.1 g L−1 combined with an initial NH4Cl concentration of 3.2 g L−1 (Fig. 4).

Impact of growth limiting nutrients on P tad1 activity. Specific promoter activity (GFP over biomass, normalized against the wildtype) for U. maydis P tad1- gfp cultivated in MTM containing 0.5 g L−1 KH2PO4 with 0.8 g L−1 (green), 1.6 g L−1 (blue), and 3.2 g L−1 (black) NH4Cl, and 0.1 g L−1 KH2PO4 with 3.2 g L−1 NH4Cl (red). Single values are shown for two biological replicates (n = 2; diamonds) and the mean value (line)
Higher NH4Cl concentrations resulted in a delay of P tad1 induction, correlated with a later depletion of nitrogen. However, specific P tad1 promoter activity was about 2.5-fold (1.6 g L−1) and 7.5-fold (3.2 g L−1) lower for the cultures containing more NH4Cl, compared to the culture containing 0.8 g L−1 NH4Cl, and for the phosphate-limited culture the specific promoter activity was negligible (Fig. 4). These data further support the role of nitrogen limitation as major trigger for the production of itaconate, as opposed to general growth limitation. They also explain previous observations, where the use of 4 and 0.8 g L−1 NH4Cl resulted in similar volumetric production rates [16], even though much more biomass was formed at 4 g L−1 NH4Cl. Apparently, lower nitrogen concentrations result in a stronger induction of itaconate production genes, leading to more efficient production. This may be related to pH in the employed system of batch cultures with a soluble 100 mM MES buffer starting at pH 6.5. Higher nitrogen concentrations may lead to a stronger pH drop during growth, and itaconate formation might be triggered at a sub-optimal pH level with preference over other secondary metabolites. Indeed, U. maydis favors the production of glycolipids over organic acids at a low pH [24, 27, 50], and the regulatory interplay between different secondary metabolites is complex [7, 8, 30]. A strong relationship between pH and induction of itaconate production has also been observed for A. terreus [35].
In summary, we were able to show that the activation of the itaconate cluster in U. maydis is induced specifically in response to nitrogen limitation, and the level of induction was strongly dependent on the initial nitrogen concentration. Although the depletion of nitrogen triggers the activation, different other factors, such as pH and the concentration of other nutrients, can be optimized for fine tuning of the promoters activities.
Conclusions
The potential of Ustilaginaceae as production organisms for different industrially-relevant compounds has been convincingly demonstrated in several instances. The presented investigation of the promoters from Ustilago’s itaconate cluster provides a new set of genetic tools that will enable heterologous gene expression under nitrogen limitation. Activity of these promoters is clearly coupled to the production phase, with a broad range of activities that reach up to the level of the commonly used P otef . The investigation of these promoters opens new doors for future metabolic engineering strategies. These strategies aim for an improved match with the different cellular objectives during growth- and production phases compared to P otef and P oma , which are mainly active during growth phase or e.g. Pcrg1 and Pnar1, which are activated or repressed under conditions incompatible with Ustilago’s production phase. In addition, the GFP fusions enable further detailed investigations into the mechanism of induction of secondary metabolite production in U. maydis, and specifically give further insight into the regulation of the itaconate cluster of Ustilago and of the itaconate production pathway.
References
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA. 2005;102:12678–83.
Basse CW. Dissecting defense-related and developmental transcriptional responses of maize during Ustilago maydis infection and subsequent tumor formation. Plant Physiol. 2005;138:1774–84.
Basse CW, Stumpferl S, Kahmann R. Characterization of a Ustilago maydis gene specifically induced during the biotrophic phase: evidence for negative as well as positive regulation. Mol Cell Biol. 2000;20:329–39.
Bottin A, Kamper J, Kahmann R. Isolation of a carbon source-regulated gene from Ustilago maydis. Mol Gen Genet. 1996;253:342–52.
Brachmann A, König J, Julius C, Feldbrügge M. A reverse genetic approach for generating gene replacement mutants in Ustilago maydis. Mol Genet Genom. 2004;272:216–26.
Brachmann A, Weinzierl G, Kamper J, Kahmann R. Identification of genes in the bW/bE regulatory cascade in Ustilago maydis. Mol Microbiol. 2001;42:1047–63.
Brakhage AA. Regulation of fungal secondary metabolism. Nat Rev Microbiol. 2013;11:21–32.
Brakhage AA, Schroeckh V. Fungal secondary metabolites—strategies to activate silent gene clusters. Fungal Genet Biol. 2011;48:15–22.
Broomfield PL, Hargreaves JA. A single amino-acid change in the iron-sulphur protein subunit of succinate dehydrogenase confers resistance to carboxin in Ustilago maydis. Curr Genet. 1992;22:117–21.
Couturier M, Navarro D, Olive C, Chevret D, Haon M, Favel A, Lesage-Meessen L, Henrissat B, Coutinho PM, Berrin JG. Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genom. 2012;13:57.
De Mey M, Maertens J, Lequeux GJ, Soetaert WK, Vandamme EJ. Construction and model-based analysis of a promoter library for E. coli: an indispensable tool for metabolic engineering. BMC Biotechnol. 2007;7:34.
Feldbrügge M, Kellner R, Schipper K. The biotechnological use and potential of plant pathogenic smut fungi. Appl Microbiol Biotechnol. 2013;97:3253–65.
Flor-Parra I, Vranes M, Kamper J, Perez-Martin J. Biz1, a zinc finger protein required for plant invasion by Ustilago maydis, regulates the levels of a mitotic cyclin. Plant Cell. 2006;18:2369–87.
Geiser E. Itaconic acid production by Ustilago maydis. Aachen: Apprimus; 2015.
Geiser E, Przybilla SK, Engel M, Kleineberg W, Buttner L, Sarikaya E, Hartog TD, Klankermayer J, Leitner W, Bolker M. Genetic and biochemical insights into the itaconate pathway of Ustilago maydis enable enhanced production. Metab Eng. 2016;38:427–35.
Geiser E, Przybilla SK, Engel M, Kleineberg W, Buttner L, Sarikaya E, Hartog TD, Klankermayer J, Leitner W, Bolker M, Blank LM, Wierckx N. Genetic and biochemical insights into the itaconate pathway of Ustilago maydis enable enhanced production. Metab Eng. 2016;38:427–35.
Geiser E, Przybilla SK, Friedrich A, Buckel W, Wierckx N, Blank LM, Bölker M. Ustilago maydis produces itaconic acid via the unusual intermediate trans-aconitate. Microb Biotechnol. 2016;9:116–26.
Geiser E, Reindl M, Blank LM, Feldbrugge M, Wierckx N, Schipper K. Activating intrinsic carbohydrate-active enzymes of the smut fungus Ustilago maydis for the degradation of plant cell wall components. Appl Environ Microbiol. 2016;82:5174–85.
Geiser E, Wiebach V, Wierckx N, Blank LM. Prospecting the biodiversity of the fungal family Ustilaginaceae for the production of value-added chemicals. BMC Fungal Biol Biotechnol. 2014;1:2.
Geiser E, Wierckx N, Zimmermann M, Blank LM. Identification of an endo-1,4-beta-xylanase of Ustilago maydis. BMC Biotechnol. 2013;13:59.
Glusker J. Aconitase. In: Boyer P, editor. The enzymes. 3rd ed. New York: Academic Press; 1971. p. 413–39.
Guevarra ED, Tabuchi T. Production of 2-hydroxyparaconic and itatartaric acids by Ustilago cynodontis and simple recovery process of the acids. Agric Biol Chem. 1990;54:2359–65.
Gunther M, Grumaz C, Lorenz S, Stevens P, Lindemann E, Hirth T, Sohn K, Zibek S, Rupp S. The transcriptomic profile of Pseudozyma aphidis during production of mannosylerythritol lipids. Appl Microbiol Biotechnol. 2015;99:1375–88.
Günther M, Hirth T, Zibek S, Rupp S. Produktion von Biotensiden mit Pseudozyma-Stämmen. BIOspektrum. 2013;19:813–5.
Hevekerl A, Kuenz A, Vorlop KD. Filamentous fungi in microtiter plates-an easy way to optimize itaconic acid production with Aspergillus terreus. Appl Microbiol Biotechnol. 2014;98:6983–9.
Hewald S, Josephs K, Bölker M. Genetic analysis of biosurfactant production in Ustilago maydis. Appl Environ Microbiol. 2005;71:3033–40.
Hewald S, Linne U, Scherer M, Marahiel MA, Kämper J, Bölker M. Identification of a gene cluster for biosynthesis of mannosylerythritol lipids in the basidiomycetous fungus Ustilago maydis. Appl Environ Microbiol. 2006;72:5469–77.
Jeya M, Lee KM, Tiwari MK, Kim JS, Gunasekaran P, Kim SY, Kim IW, Lee JK. Isolation of a novel high erythritol-producing Pseudozyma tsukubaensis and scale-up of erythritol fermentation to industrial level. Appl Microbiol Biotechnol. 2009;83:225–31.
Kämper J, Kahmann R, Bölker M, Ma LJ, Brefort T, Saville BJ, Banuett F, Kronstad JW, Gold SE, Muller O, Perlin MH, Wosten HAB, de Vries R, Ruiz-Herrera J, Reynaga-Pena CG, Snetselaar K, McCann M, Perez-Martin J, Feldbrügge M, Basse CW, Steinberg G, Ibeas JI, Holloman W, Guzman P, Farman M, Stajich JE, Sentandreu R, Gonzalez-Prieto JM, Kennell JC, Molina L, Schirawski J, Mendoza-Mendoza A, Greilinger D, Munch K, Rossel N, Scherer M, Vranes M, Ladendorf O, Vincon V, Fuchs U, Sandrock B, Meng S, Ho ECH, Cahill MJ, Boyce KJ, Klose J, Klosterman SJ, Deelstra HJ, Ortiz-Castellanos L, Li WX, Sanchez-Alonso P, Schreier PH, Hauser-Hahn I, Vaupel M, Koopmann E, Friedrich G, Voss H, Schluter T, Margolis J, Platt D, Swimmer C, Gnirke A, Chen F, Vysotskaia V, Mannhaupt G, Guldener U, Munsterkotter M, Haase D, Oesterheld M, Mewes HW, Mauceli EW, DeCaprio D, Wade CM, Butler J, Young S, Jaffe DB, Calvo S, Nusbaum C, Galagan J, Birren BW. Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature. 2006;444:97–101.
Keller NP, Turner G, Bennett JW. Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol. 2005;3:937–47.
Keon JP, White GA, Hargreaves JA. Isolation, characterization and sequence of a gene conferring resistance to the systemic fungicide carboxin from the maize smut pathogen, Ustilago maydis. Curr Genet. 1991;19:475–81.
Khrunyk Y, Munch K, Schipper K, Lupas AN, Kahmann R. The use of FLP-mediated recombination for the functional analysis of an effector gene family in the biotrophic smut fungus Ustilago maydis. New Phytol. 2010;187:957–68.
Klement T, Milker S, Jäger G, Grande PM, de Maria PD, Büchs J. Biomass pretreatment affects Ustilago maydis in producing itaconic acid. Microb Cell Fact. 2012;11:43.
König J. Die Identifikation von Ziel-Transkripten des RNA bindenden Proteins Rrm4 aus Ustilago maydis. Germany: Philipps-Universität Marburg; 2008.
Krull S, Hevekerl A, Kuenz A, Prusse U. Process development of itaconic acid production by a natural wild type strain of Aspergillus terreus to reach industrially relevant final titers. Appl Microbiol Biotechnol. 2017;101:4063–72.
Kubicek CP, Punt P, Visser J. Production of organic acids by filamentous fungi. In: Hofrichter M, editor. Industrial applications. Berlin: Springer; 2011. p. 215–34.
Ladner T, Mühlmann M, Schulte A, Wandrey G, Büchs J. Prediction of Escherichia coli expression performance in microtiter plates by analyzing only the temporal development of scattered light during culture. Biotechnol J. 2017;11:20.
Lara AR, Jaén KE, Sigala J-C, Mühlmann M, Regestein L, Büchs J. Characterization of endogenous and reduced promoters for oxygen-limited processes using Escherichia coli. ACS Synth Biol. 2017;6:344–56.
Lehmler C, Steinberg G, Snetselaar KM, Schliwa M, Kahmann R, Bölker M. Identification of a motor protein required for filamentous growth in Ustilago maydis. EMBO J. 1997;16:3464–73.
Loubradou G, Brachmann A, Feldbrugge M, Kahmann R. A homologue of the transcriptional repressor Ssn6p antagonizes cAMP signalling in Ustilago maydis. Mol Microbiol. 2001;40:719–30.
Maassen N, Panakova M, Wierckx N, Geiser E, Zimmermann M, Bölker M, Klinner U, Blank LM. Influence of carbon and nitrogen concentration on itaconic acid production by the smut fungus Ustilago maydis. Eng Life Sci. 2013;14:129–34.
Morita T, Fukuoka T, Imura T, Kitamoto D. Production of glycolipid biosurfactants by basidiomycetous yeasts. Biotechnol Appl Biochem. 2009;53:39–49.
Mutalik VK, Guimaraes JC, Cambray G, Lam C, Christoffersen MJ, Mai QA, Tran AB, Paull M, Keasling JD, Arkin AP, Endy D. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods. 2013;10:354–60.
Polli F, Meijrink B, Bovenberg RAL, Driessen AJM. New promoters for strain engineering of Penicillium chrysogenum. Fungal Genet Biol. 2016;89:62–71.
Samorski M, Müller-Newen G, Büchs J. Quasi-continuous combined scattered light and fluorescence measurements: a novel measurement technique for shaken microtiter plates. Biotechnol Bioeng. 2005;92:61–8.
Sarkari P, Reindl M, Stock J, Muller O, Kahmann R, Feldbrugge M, Schipper K. Improved expression of single-chain antibodies in Ustilago maydis. J Biotechnol. 2014;191:165–75.
Schulz B, Banuett F, Dahl M, Schlesinger R, Schafer W, Martin T, Herskowitz I, Kahmann R. The b alleles of U. maydis, whose combinations program pathogenic development, code for polypeptides containing a homeodomain-related motif. Cell. 1990;60:295–306.
Schuster M, Schweizer G, Reissmann S, Kahmann R. Genome editing in Ustilago maydis using the CRISPR-Cas system. Fungal Genet Biol. 2016;89:3–9.
Spellig T, Bottin A, Kahmann R. Green fluorescent protein (GFP) as a new vital marker in the phytopathogenic fungus Ustilago maydis. Mol Gen Genet. 1996;252:503–9.
Teichmann B. The cellobiose lipid ustilagic acid from Ustilago maydis: biosynthesis and transcriptional regulation. Marburg: Philipps University Marburg; 2009.
Teichmann B, Labbe C, Lefebvre F, Bolker M, Linne U, Belanger RR. Identification of a biosynthesis gene cluster for flocculosin a cellobiose lipid produced by the biocontrol agent Pseudozyma flocculosa. Mol Microbiol. 2011;79:1483–95.
Teichmann B, Liu LD, Schink KO, Bölker M. Activation of the ustilagic acid biosynthesis gene cluster in Ustilago maydis by the C2H2 zinc finger transcription factor Rua1. Appl Environ Microbiol. 2010;76:2633–40.
Terfrüchte M, Joehnk B, Fajardo-Somera R, Braus GH, Riquelme M, Schipper K, Feldbrügge M. Establishing a versatile Golden Gate cloning system for genetic engineering in fungi. Fungal Genet Biol. 2014;62:1–10.
Wandrey G, Bier C, Binder D, Hoffmann K, Jaeger K-E, Pietruszka J, Drepper T, Büchs J. Light-induced gene expression with photocaged IPTG for induction profiling in a high-throughput screening system. Microb Cell Fact. 2016;15:63.
Wanka F, Arentshorst M, Cairns TC, Jorgensen T, Ram AF, Meyer V. Highly active promoters and native secretion signals for protein production during extremely low growth rates in Aspergillus niger. Microb Cell Fact. 2016;15:145.
Wanka F, Cairns T, Boecker S, Berens C, Happel A, Zheng X, Sun J, Krappmann S, Meyer V. Tet-on, or Tet-off, that is the question: Advanced conditional gene expression in Aspergillus. Fungal Genet Biol. 2016;89:72–83.
Willis RB, Montgomery ME, Allen PR. Improved method for manual, colorimetry determination of total Kjeldahl nitrogen using salicylate. J Agric Food Chem. 1996;44:1804–7.
Willke T, Vorlop KD. Biotechnological production of itaconic acid. Appl Microbiol Biotechnol. 2001;56:289–95.
Zambanini T, Hosseinpour Tehrani H, Geiser E, Merker D, Schleese S, Krabbe J, Buescher JM, Meurer G, Wierckx N, Blank LM. Efficient itaconic acid production from glycerol with Ustilago vetiveriae TZ1. Biotechnol Biofuels. 2017;10:131.
Zambanini T, Hosseinpour Tehrani H, Geiser E, Sonntag CK, Buescher JM, Meurer G, Wierckx N, Blank LM. Metabolic engineering of Ustilago trichophora TZ1 for improved malic acid production. Metab Eng Commun. 2017;4:12–21.
Zambanini T, Kleineberg W, Sarikaya E, Buescher JM, Meurer G, Wierckx N, Blank LM. Enhanced malic acid production from glycerol with high-cell-density Ustilago trichophora TZ1 cultivations. Biotechnol Biofuels. 2016;9:135.
Zambanini T, Sarikaya E, Kleineberg W, Buescher JM, Meurer G, Wierckx N, Blank LM. Efficient malic acid production from glycerol with Ustilago trichophora TZ1. Biotechnol Biofuels. 2016;9:67.
Zarnack K, Maurer S, Kaffarnik F, Ladendorf O, Brachmann A, Kämper J, Feldbrügge M. Tetracycline-regulated gene expression in the pathogen Ustilago maydis. Fungal Genet Biol. 2006;43:727–38.
Zobel S, Benedetti I, Eisenbach L, de Lorenzo V, Wierckx N, Blank LM. Tn7-based device for calibrated heterologous gene expression in Pseudomonas putida. ACS Synth Biol. 2015;4:1341–51.
Authors’ contributions
NW, MS, LMB, and TZ conceived and designed the project. NW, TZ, SKH, GW, JB, and LMB designed experiments and analyzed results. SKH, LMS, LB, HHT, EG, MB, DV, GW, and TZ constructed the strains and performed screening experiments. TZ and NW wrote the manuscript with the help of LMB. All authors read and approved the final manuscript.
Acknowledgements
We gratefully acknowledge Dr. Kerstin Schipper and Prof. Michael Feldbrügge (Heinrich Heine University Düsseldorf) and Prof. Michael Bölker (Philipps University of Marburg) for providing Ustilago vectors and advice.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Consent for publication
Not applicable.
Ethics of approval and consent to participate
Not applicable.
Funding
This work was performed as a part of the Cluster of Excellence “Tailor-Made Fuels from Biomass”, which is funded by the Excellence Initiative of the German federal and state governments to promote science and research at German universities. The scientific activities of the Bioeconomy Science Center were supported financially by the Ministry of Innovation, Science and Research within the framework of the NRW Strategieprojekt BioSC (No. 578 313/323-400-002 13).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zambanini, T., Hartmann, S.K., Schmitz, L.M. et al. Promoters from the itaconate cluster of Ustilago maydis are induced by nitrogen depletion. Fungal Biol Biotechnol 4, 11 (2017). https://doi.org/10.1186/s40694-017-0040-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40694-017-0040-3