
Abstract
Chimeric antigen receptor T (CAR-T) cell therapy has significantly improved the life expectancy for patients with refractory or relapse B cell lymphoma. As for B cell acute lymphoblastic leukemia (B-ALL), although the primary response rate is promising, the high incidence of early relapse has caused modest long-term survival with CAR-T cell alone. One of the main challenges is the limited persistence of CAR-T cells. To further optimize the clinical effects of CAR-T cells, many studies have focused on modifying the CAR structure and regulating CAR-T cell differentiation. In this review, we focus on CAR-T cell persistence and summarize the latest progress and strategies adopted during the in vitro culture stage to optimize CAR-T immunotherapy by improving long-term persistence. Such strategies include choosing a suitable cell source, improving culture conditions, combining CAR-T cells with conventional drugs, and applying genetic manipulations, all of which may improve the survival of patients with hematologic malignancies by reducing the probability of recurrence after CAR-T cell infusion and provide clues for solid tumor CAR-T cell therapy development.
Similar content being viewed by others

Background
Chimeric antigen receptor T (CAR-T) cell immunotherapy has rapidly impacted the malignant tumor field and has achieved remarkable effects in recent years as the latest promising adoptive cell therapy. The CAR concept was first proposed in 1989, when G Gross et al. designed a chimeric T-cell receptor (TCR) gene consisting of the TCR constant domain and the antibody variable domain and transfected it into cytotoxic T cells, conferring these T cells with antibody-like specificity [1]. Moreover, the chimeric TCR was able to signal effectively upon activation and to execute its effector function [1]. Subsequently, with the emergence and development of transgenic technology, there are new options for adoptive T-cell therapy [2,3,4,5,6]. Up to 2011, CAR-T cells were used to treat patients with lymphoblastic leukemia, with striking results [7,8,9]. Based on multiple clinical trials with laudable results, the FDA approved the first CAR-T cell therapeutic product (Kymriah, CTL-019 from Novartis) for patients with relapsed/refractory B-cell acute lymphoblastic leukemia (r/r B-ALL) in 2017, and multiple similar products have since been approved [10,11,12,13]. Overall, the complete remission (CR) rate in patients treated with CD19 CAR-T is 30–70%, and in some trials, the rate was over 90% [10, 12, 14]. The fact that these products were generated using diverse technical schemes indicates the marked efficiency of CAR-T cells for treatment of lymphoblastic leukemia [15,16,17,18,19]. CAR-T cells have also been used for treating solid tumors, but curative effects are lacking compared to those for lymphoblastic leukemia [20,21,22,23,24]. Hence, there is an urgent need to further optimize CAR-T therapy to resolve existing issues, especially the limited persistence, high recurrence rate, and insufficient cytotoxicity, with regard to solid tumors.
The conventional CAR-T cell production process includes five steps: (1) T-cell collection and activation; (2) CAR structure preparation and transduction; (3) CAR-T cells expansion in vitro; (4) Verification of CAR-T cell phenotype and function; (5) CAR-T cells cryopreservation and storage until administration to the patient [25]. The five steps require approximately one to two weeks, at which point qualified CAR-T cells are ready for delivery to patients. Based on this process, two main areas of focus for optimizing CAR-T therapy have been developed: the design of CAR structure and the intervention during CAR-T cell expansion stage [26]. First, the CAR structure, including the binding domain, extracellular spacer, transmembrane domain, and intracellular signaling domain, is regarded as the core entity of the CAR-T cells [26, 27]. This structure has undergone constant remodeling, encompassing five generations of evolution and optimization since the origin of CAR-T cells [28, 29]. These five generations have been comprehensively studied and applied and thus are not introduced in detail in this review [29,30,31]. The second-generation CAR is the most widely adopted structure, which includes one costimulatory domain within the intracellular domain that is most commonly derived from CD28 or 4-1BB and can enhance TCR signaling [3, 32,33,34,35,36].
In addition to structural design, the intervention during CAR-T cell expansion stage could also contribute to the persistence after infusion. Sophisticated engineering strategies are applied to improve antitumor activity by regulating CAR-T cell development and differentiation. Overall, CAR-T cells exhibit strong proliferation and differentiation abilities in the culture stage [37]. These activities are accompanied by competition between effector function and long persistence, which determines the final quality of the cell therapy after infusion into patients. In this review, we describe the different approaches that have been recently adopted in vitro to improve the persistence and immunophenotype of CAR-T cells, including the choice of suitable cells, the improvements in the in vitro culture conditions, the application of conventional drugs with CAR-T cells, and the use of genetic manipulations. These approaches may improve the persistence of CAR-T cells which have superior immunotherapeutic effects.
Strategies for choosing a suitable source of cells for CAR-T therapy
AutoCAR-T cells or alloCAR-T cells
First, the quality and characteristics of T cells are crucial for the therapeutic efficacy of CAR-T cells [38]. CAR-T cells are classified as autologous or allogeneic (autoCAR-T or alloCAR-T) according to the source of T cells, and both have been evaluated in clinical trials [39,40,41]. Auto- and alloCAR-T cells have unique benefits and challenges that need to be addressed. For autoCAR-T, the T cells are isolated from patients, and thus, immunological rejection does not occur [42]. However, long-term infiltration in the tumor microenvironment (TME) decreases cytotoxicity and induces exhaustion of T cells, which might lead to insufficient therapeutic efficacy of autoCAR-T cells for tumors [40]. Moreover, the whole stage, from cell collection to cell infusion, is time-consuming and is required for each patient to undergo autoCAR-T cell therapy, and this process is overly lengthy for some critically ill patients. Furthermore, the cost of CAR-T cell therapy is too high to wildly applied. AlloCAR-T cells potentially overcome these problems. They can be collected and prepared in advance and then supplied to patients without delay. Healthy donor T cells possess greater cytotoxicity when compared to T cells from patients. Moreover, multiple and systematic production of these cells may reduce the average cost and result in a process more easily carried out. The greatest obstacles for the application of alloCAR-T cells are host versus graft disease (HvGD) and graft versus host disease (GvHD), which need to be overcome by genetic manipulation of TCR and human leukocyte antigen (HLA), and it can be accomplished via the TALEN and CRISPR systems [43].
Beyond the principle and technical factors, recently published clinical results show that autoCAR-T cells have better therapeutic efficacy than alloCAR-T cells [44]. For example, the initial overall response rate for traditional autoCAR-T(CTL-019) was reported to be approximately 90%, with a 5-year response rate reaching 58% [45]. These patients with diffuse large B-cell lymphoma and follicular lymphoma were followed for a median of 60.7 months [45, 46]. For patients with B-ALL who were treated with alloCAR-T, the initial overall response rate was 67%, the 6-month response rate was 55%, and the median survival time was 4.1 months (NCT02808442 and NCT02746952). The quantity and persistence of CAR-T cells are important factors for tumor recurrence. In one study, the CAR transgene was continuously detectable beyond 1 year in approximately 50% of patients who achieved complete remission after autoCAR-T treatment, but only 1 patient treated with alloCAR-T had a detectable CAR transgene beyond 120 days [47]. Furthermore, cytokine-release syndrome (CRS) occurs in 57% of patients administered autoCAR-T cells and in 91% of those administered alloCAR-T cells, with a similar neurotoxicity rate (39% vs. 38%) [44]. In comparison with alloCAR-T cells, autoCAR-T cells demonstrated better performance in a recent clinical trial in almost all aspects, especially in persistence [44]. Regardless, the core advantage of alloCAR-T cells, namely, their “off-the-shelf” nature, cannot be ignored and represents a significant benefit for large-scale systematic production [44]. In addition, the CRISPR genomic editing tool allows for easy manipulation of alloCAR-T cells, which is worth exploring in future tumor immunotherapy studies [48]. Moreover, T-cell-derived induced pluripotent stem cells have been verified as an ideal source of autoCAR-T cells that do not cause GvHD, which may facilitate the large-scale development of potent autoCAR-T cells for a broad range of immunotherapies [49].
Subgroups of T cells
In addition to the source, cell subgroup also affects CAR-T cell persistence and therapeutic efficacy. For conventional CAR-T cell therapies, T cells are collected and isolated using the marker CD3, mainly resulting in two subgroups, CD4+ and CD8+ T cells [38]. The CD4/CD8 ratio is not constant among patients or donors, which has attracted some attention. Several studies have focused on the relationship between the CD4/CD8 ratio and therapeutic efficacy in clinical trials [50]. First, as the bases of T cells, both CD4+ and CD8+ T cell subsets are capable of efficiently killing tumor cells [51]. Compared to the use of individual T cell subsets for CAR-T cells, the combination of CD4+ and CD8+ subsets exhibits synergetic antitumor effects both in vitro and in vivo [51]. Moreover, more uniform potency was obtained with defined T cell subsets compared with unselected T cells. Subsequently, a clinical trial involving 29 adult B-ALL patients with defined CD4+ and CD8+ T subsets (half and half) for CD19 CAR-T cells observed a marked effect (NCT01865617). This result highlights the advantage of consistent and standardized CAR-T cells and response evaluation [52, 53]. Nonetheless, a conflicting result has been reported, whereby CD4+ CAR-T cells showed superior antitumor activity in glioblastoma compared to CD8+ CAR-T cells, especially regarding the long-term antitumor response [54]. Additionally, a recent study that followed two patients for 10 years after CAR-T cell treatment showed a proportion of CD4+ CAR-T cells over 99%, whereas that of CD8+ CAR-T cells was less than 1% [15]. These findings suggest exaggerated persistence of CD4+ T cells in vivo [15]. Such a discrepant result might be caused by differences in tumor cell characteristics, the TME, the receptor, tumor antigens, and even the transduction and expansion methods. In summary, these studies consistently highlight the importance of defined CD4+ and CD8+ CAR-T cell compartments for effective antitumor activity.
Due to the enormous success of CAR-T immunotherapy, interest in engineering innovative CARs using other types of immune cells has emerged in recent years. Although CAR-NK cells are well-known immune cell therapy that are undergoing comprehensive, meticulous preclinical and clinical trials, they are not reviewed here [55,56,57,58,59,60]. Natural killer T cells (NKTs), γδTs, regulatory T cells (Tregs), and a series of infrequent T cell subsets are also potential candidate for immune cell therapy. NKT cells are a group of T cells with both T cell receptors and NK cell markers and thus possess features of both cell types. These features include the nonspecific killing function of NK cells and the specific killing function of T cells, endowing NKT cells with powerful surveillance and killing capabilities [61]. Since their discovery and exploration, NKT cells have been used for cellular immunotherapy beginning in 2010 for patients with liver carcinoma, lung carcinoma, and gastric carcinoma [62]. In 2019, NKT cells were loaded with a GD2 CAR structure to specifically recognize and kill neuroblastoma cells, and clinical trials have shown a positive therapeutic effect [63]. More CAR-NKT cells are advancing in clinical trials [63, 64].
T cells can be defined as two main subsets according to the difference in TCR molecules: αβT and γδT. The proportion of αβT cells exceeds 60%, and that of γδT cells is less than 5% [65]. Regarding anticancer efficacy, γδT cells can kill target cells through the perforin-granzyme-B pathway, Fas-FasL pathway, and antibody-dependent cell-mediated cytotoxicity (ADCC) pathway [66, 67]. Such multiple modes of action suggest a predominant anticancer effect [68]. In addition, γδT cells can recognize targets directly without major histocompatibility complex (MHC) restriction, which is their core advantage compared to αβT cells for use as off-the-shelf alloCAR-T cells without the complication of GvHD [65]. Based on these characteristics, several preclinical trials have demonstrated positive results using CAR-γδT cells, with favorable persistence in vivo, and the relevant clinical products have been described [69, 70]. A recent preclinical study in 2022 reported that CD20 CAR-γδT cells have powerful tumor inhibition and low sensitivity between grafts and hosts, highlighting the potential advantages of CAR-γδT cells [69, 71].
As a type of immunosuppressive cell, Tregs are critical for immune system tolerance and hyperimmune response avoidance [72, 73]. Therefore, unlike other CAR cells for anticancer treatment, CAR-Tregs maintain the Treg phenotype and function. These cells are guided to the target tissue by the CAR structure and show enhanced suppression of autoimmune disease in susceptible organisms [74]. Furthermore, CAR-Tregs avoid MHC restriction, and compared to polyclonal Tregs, fewer cells are required. Preclinical studies demonstrate that CAR-Tregs protect vascularized grafts in fully immunocompetent recipients [75]. These cells have been modified to treat GvHD, type 1 diabetes, and Alzheimer’s disease, and it is worth noting that CAR-Tregs markedly expand the applications of CAR engineering [76]. However, CAR-Tregs are only in the initial stages of development and application, and further optimization is urgently needed to improve their efficiency and stability for clinical application.
In summary, as the main component of CAR-T cells, the source and characteristics of T cells significantly determine the persistence and efficiency. Therefore, choosing a suitable source is the first and important step for successful CAR-T cell therapy (Table 1).
Strategies for optimizing CAR-T cell culture conditions in vitro
Differentiation of T cells influences CAR-T cell efficiency
In addition to the original subsets and proportion of T cells, their differentiation plays a significant role in regulating CAR-T cell anticancer activity. Similar to the components, the differentiation state of T cells also varies among individuals and culture platforms. T cells can be subdivided into five principal and typical subgroups according to the expression of cell surface markers, as shown in Fig. 1 [77]. For every T cell subpopulation, these different phenotypes suggest functional characteristics and diversity, which are vital to developing and understanding new strategies for CAR-T cell therapy [78, 79].

The T cell differentiation process and related changes in T cell metabolism. TN, TSCM, TCM, TEM, and TEFF are divided according to the surface markers (CCR7, CD45RO, CD45RA, CD62L, CD95). Different subsets show diverse functions and dynamic metabolic characteristics
Naïve T cells Before interacting with their cognate antigen or pathogen, immature T cells are considered naïve T (TN) cells. These cells exhibit high expression of the CD45RA isoform, L-selectin (CD62L), and CXC chemokine receptor 7 (CCR7). The latter two representative markers indicate the ability of these cells to home towards lymph nodes [80]. TN cells possess high reproductive and survival abilities, but a sharp decline in the TN proportion during the expansion stage of CD3/CD28 antibody stimulation and activation cannot be avoided [37]. After stimulation and activation by CD3/CD28 antibodies and cytokines, TN cells experience rapid differentiation into memory T cells and effector T cells [37]. Stem cell memory T cells As the most recently discovered T cell subset, stem cell memory T (TSCM) cells have attracted much attention, as they display two properties of TN and TM cells and express the associated surface markers, including CD62L, CCR7, CD45RA and CD95 [79, 80]. It is worth noting that the gene expression pattern of TSCM cells indicates that they are at an earlier differentiated stage than memory T cells. Studies have shown that TSCM cells can quickly respond to antigenic stimulation and maintain strong self-renewal capacity [80, 81]. These findings suggest a tremendous potential of these cells for adoptive T cell therapy, despite their extremely low cell proportion [79, 82]. Central memory T cells Similar to TSCM cells, central memory T (TCM) cells express CD62L, CCR7, and CD95, but they express the CD45RO isoform instead of the CD45RA isoform [83]. As TCM cells have been previously exposed to an antigen, a second instance of antigen exposure causes rapid proliferation and an effective immune response. Compared to effector T cells, TCM cells are less cytotoxic, but their characteristics of rapid proliferation and long persistence enable superior performance [84, 85]. Both TSCM and TCM cells maintain the ability to home towards lymph nodes, which is important for long persistence [85]. Thus, the ratio of TCM and TSCM cells has become a key performance indicator for evaluating CAR-T therapeutic effects (NCT01318317 and NCT01815749) [86]. Further comparison of the two memory T subsets reveals that TSCM cells can retain a higher proportion of naïve phenotypes than TCM cells, indicating stronger self-renewal ability [83, 87]. Effector memory T cells Another type of memory T cell is the effector memory T (TEM) cell, which lacks CCR7 and CD62L expression, in contrast to TSCM and TCM cells. This lack of expression prevents these cells from homing towards lymph nodes, but they can access target tissues. Once the cell encounters antigens, effector functions rapidly ensue as well as secretion of a series of cytokines, such as TNF-α, IFN-γ, and perforin [88, 89]. Terminal effector T cells Continuous antigen exposure and stimulation induce TEM cells to further differentiate into terminal effector T (TEFF) cells, the terminus of effector T cells. These cells do not express CD62L, CCR7, or CD45RO, but they do express CD45RA. TEFF cells are the primary T cell subset of anticancer cells. However, TEFF cells have a short lifespan and hardly any self-renewal ability. After addressing a chronic infection or killing cancer cells, TEFF cells inevitably lose their effector function and become exhausted [79, 89].
These five differentiated T cell subsets have diverse features in terms of proliferation ability and antitumor effect that substantially contribute to the clinical efficacy of CAR-T cells [79]. Several preclinical and clinical trials have revealed that compared to traditional CAR-T cells, CD19 CAR-T cells derived from TSCM cells or from a higher ratio of memory T cells show long-term persistence and responses [90, 91]. Therefore, a consensus has been reached that TSCM and TCM cells have better persistence and antitumor activity in vivo than TEM and TEFF cells. Accordingly, researchers have focused on strategies to alter T cell differentiation to induce more TSCM and TCM cells in vitro [37, 92].
Culture conditions influence CAR-T cell differentiation and function
In addition to differences in surface markers and functions, metabolic requirements among the five subsets of T cells are diverse [93, 94]. It has gradually become clear that T cell metabolic reprogramming occurs at the same time as T cell activation and is another critical component for adoptive therapy [94]. For TN cells, the main metabolic pathway and energy acquisition methods are oxidative phosphorylation (OXPHOS) and fatty acid oxidation (FAO) in mitochondria [37]. In response to a CD3/CD28 antigen stimulation signal, TN cells accelerate metabolism to satisfy the increased biosynthetic demands [37]. The PI3K-AKT-mTOR pathway is activated and subsequently promotes aerobic glycolysis (Warburg effect) [95]. This metabolic reprogramming also results in the transformation of TN cells to TEFF cells [80]. For TSCM and TEM cells, the metabolic pattern is similar to that of TN cells and mainly depends on OXPHOS and low-level glycolysis [93].
To mediate T cell differentiation and improve the TSCM and TCM ratio during the CAR-T cell expansion phase, which impact the persistence and clinical performance, culture conditions and operating steps have been explored in depth. Combinations of multiple cytokines IL-2 is a widely used cytokine that not only induces rapid T cell proliferation in vitro but also evokes a switch from OXPHOS to glycolysis. It induces formation of more effector T cells and reduces memory T cell population [96]. Other cytokines, including IL-7, IL-15, and IL-21, mediate metabolic adaptation by boosting OXPHOS and inhibiting glycolysis [97, 98]. The combination of IL-7 and IL-15 has been reported to intervene CAR-T cells with higher proportions of TSCM and TCM cells and superior anticancer activity in vivo compared with CAR-T cells cultured with only IL-2 [99]. This strategy has been applied for clinical trials in lymphoma, sarcoma, and osteosarcoma [9, 100]. Metabolite adjustments In addition to cytokines, glucose is the direct target mediating glycolysis [101, 102]. Many studies have explored and validated the abilities of several drugs and molecules to restrict or interfere with glycolysis metabolism. For example, 2-deoxy-D-glucose (2-DG), a synthetic glucose analogue, enters cells through glucose transporters (GLUTs) and is subsequently phosphorylated by hexokinase, acting as a competitive glucose inhibitor [101]. In the presence of 2-DG during expansion in vitro, CD8+ T cells alter their differentiation trajectory for more memory cells, leading to persistent antitumor functionality [101]. Metabolites have also been verified to influence T cell metabolic adaptability and differentiation fate. Metabolic analysis has shown that intracellular L-arginine is markedly depleted after TN cells become activated [101, 103]. Additional L-arginine supplementation in the culture medium regulates T cell metabolic alternation from glycolysis to OXPHOS and increases the ratio of TSCM cells, leading to greater antitumor activity [103]. Glutamine is a substrate of the TCA cycle, and glutamine metabolism is augmented once TN cells are activated [104]. Glutamine is required for effector T differentiation and function, and its depletion increases the percentage of TCM cells [104, 105]. Furthermore, glutamine-deficient condition caused by the antagonist 6-diazo-5-oxo-L-norleucine (DON) in the culture medium reportedly increases OXPHOS and reduces glycolysis in CD19 CAR-T cells [106]. Ultimately, these changes induce stronger lysis in vitro and more robust elimination of tumor cells in vivo, indicating a promising approach to optimize CAR-T immunotherapy [106]. Moreover, another study found that carnosine limits extracellular acidification to shift the metabolic profile from an acidic, stressed state towards an oxidative, energetic state [107]. Therefore, addition of carnosine enhances lentiviral gene delivery in activated T cells [107]. This finding provides the potential to improve the overall quality of the cell culture medium for CAR-T cell therapies. In addition to these specific metabolites, several growth factors originating from the immune microenvironment might participate in T cell proliferation, differentiation, or function. Recent studies have shown that adding human platelet lysate to the CAR-T cell culture medium enriches the TCM cell subset and ultimately improvs the antitumor effect in a mouse xenograft model [107].
Hence, it can be concluded that the core mechanism by which these strategies optimize culture conditions is related to the regulation of T cell metabolism through the increase in OXPHOS and appropriate inhibition of glycolysis in vitro. These processes lead to a high percentage of TSCM and TCM cells, ultimately resulting in superior longevity and antitumor potential for CAR-T immunotherapy.
Strategies for novel application of conventional drugs in the CAR-T cell expansion phase
In addition to improvements in the culture medium, several activators and inhibitors that target metabolic pathways and even conventional drugs used to treat other diseases have been used with CAR‑T cells to achieve superior cancer immunotherapy. In this section, we focus on the application of these clinical molecules and drugs that potentially modulate metabolism and T cell differentiation as novel CAR-T adjuvants (Table 2).
PI3K-AKT-mTOR inhibitors
Hyperactivation of PI3K has been verified to induce tumorigenesis in hematologic malignancies and in many solid tumors, such as melanoma, lung cancer, and breast cancer, which indicates its potential as a therapeutic target [118]. However, development of pan-PI3K inhibitors has advanced slowly for many years because of serious toxicity and low tolerance [119]. Several PI3K subunit inhibitors have shown remarkable antitumor effects in lymphoid cancer and have been approved by the FDA based on the results of clinical trials [120]. Moreover, in normal T cells, activation of PI3K family members, especially PI3K-δ and -γ, is pivotal for transmitting signals from the TCR/CD3 complex to activate downstream transcription factors (e.g., HIF-1α and c-Myc), promoting glycolysis and differentiation [121]. The effects of several PI3K inhibitors on T/CAR-T cells have recently been studied. In 2016, Rasha Abu Eid et al. demonstrated that the pan-PI3K inhibitor GDC-0941 (GDC) enhances the proliferative ability and survival of CD8+ T cells by delaying terminal differentiation and preserving the memory phenotype, which significantly slows tumor growth in B16 tumor bearing mice [108, 122]. Subsequently, pretreatment with a series of PI3K inhibitors (e.g., Idelalisib, Duvelisib, and LY294002) during the expansion phase improved the CD19/CD33 CAR-T cells, as indicated by a higher frequency of CCR7+CD62L+ T cells and better antitumor therapeutic capacity in vivo [109, 110, 112, 123]. Inhibitors of components downstream of the PI3K pathway, such as AKT and mTOR, may have a similar effect. For example, as a traditional AKT inhibitor for treatment of chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), Ibrutinib supplementation during CAR-T cell expansion phase might enrich the less-differentiated T cells and reduce expression of exhaustion markers [111, 124, 125]. Thus, this is an option to further improve the clinical outcomes of CLL patients [111]. Rapamycin (RAPA) has traditionally been administered to patients undergoing allogeneic hematopoietic stem cell transplantation (allo-HSCT) and kidney transplant to attenuate GvHD in clinical trials [126]. It has also been reported that pretreatment with RAPA enhances the infiltration capacity of EpCAM CAR-T cells into the bone marrow and increases the elimination of AML in mice by inhibiting mTOR activity [113].
Epigenetic drugs
Studies have gradually clarified that epigenetic factors, including DNA methylation, histone modification, and noncoding RNA, play vital roles not only in tumorigenesis and progression but also in T cell differentiation, metabolism, and function [127, 128]. Knowledge of epigenetic remodeling suggests that epigenetic-targeted drugs may serve as an adjuvant for CAR-T cell immunotherapy. DNA methylation inhibitors DNA methyltransferases are gradually become activated during T cell differentiation [129]. Additionally, de novo DNA methylation was shown to promote T cell exhaustion and limit immunotherapy [129]. Therefore, inhibition of DNA methylation is capable of inducing T cell rejuvenation and restricting exhaustion. A recent study revealed the effect of Decitabine, a clinical DNA methylation inhibitor, on CAR-T cells [37]. Addition of this drug to CAR-T cell medium increased the expression of memory-related genes, strengthened the proliferative potential, increased cytokine production, and strengthened tumor cell lytic capacity, even at a very low effector/target ratio, compared to untreated CAR-T cells [37]. Therefore, adding demethylating drugs represents a convenient and economical option to improve the persistence and anticancer properties of CAR-T cells. HDAC inhibitors As significant posttranscriptional modifications of histone proteins, acetylation and deacetylation have been analyzed in detail [130]. Histone acetyltransferases (HATs) use acetyl-CoA, a critical intermediate metabolite and important signal transducer, as the primary substrate for histone acetylation, and acetyl group addition to a histone reduces the positive charge, leading to a relaxed DNA state suitable for active transcription. In contrast, histone deacetylases (HDACs) perform the opposite function [131]. HATs and HDACs have been considered important targets for various diseases [132]. Several HDAC inhibitors have received FDA approval for tumor therapy and are particularly utilized in clinical studies in combination with anti-PD-L1 mAb to restore the host immune response [133]. A recent study demonstrated that SAHA, a clinical HDAC inhibitor, reduced the expression of immunosuppressive molecules (e.g., CTLA-4 and TET2) in B7-H3 CAR-T cells and sharply increased therapeutic efficacy at a low dose [114]. These findings support an unexpected application and potential clinical translation of HDAC inhibitors. BET bromodomain inhibitors Through defined comprehensive epigenetic target screening of chemical probes, JQ1, an inhibitor of bromodomain and extraterminal motif (BET) proteins, was selected because it is able to maintain the functional properties of TSCM cells [115]. Mechanistically, JQ1 inhibits the BET protein BRD4 and directly regulates the expression of the transcription factor BATF in CD8+ T cells, with an improved memory phenotype [115]. CAR-T cells pretreated with JQ1 exhibit enhanced persistence and antitumor effects in mice [115].
Several other conventional drugs with potential applications in CAR-T cell therapy
Metformin For more than 100 years, metformin has been known to reduce glucose levels, and it has been approved by the FDA since 1995 as a first-time treatment for type II diabetes [134]. Many studies have shown that metformin also has many other functions, such as reversing ageing and inhibiting tumor growth [135]. Recently, immunoregulatory potential has been revealed for metformin. Specifically, metformin reprograms T cell differentiation and maintains the phenotype and function of TSCM and TCM cells through the AMPK-miRNA-EOMES-PD1 pathway [92]. These activities lead to increased cytotoxicity in vivo, which may benefit cancer patients [92]. Moreover, a recent identification of targets and mechanisms may lead to a wider and a more accurate application of metformin [136]. Sulforaphane Sulforaphane (SFN) is a naturally occurring antioxidant enriched in cruciferous vegetables that has been regarded as one of the most promising treatments for autism spectrum disorder [137]. SFN can arrest the cell cycle and inhibit tumor progression, moreover, SFN has also been found to modulate immune cell differentiation and function [137]. Based on this, Chunyi Shen et al. evaluated the effect of SFN on CAR-T cells and reported that SFN pretreatment downregulates PD-1 expression in meso CAR-T cells by inhibiting the PI3K-AKT pathway and improves antitumor efficiency both in vivo and in vitro [116]. Auranofin Auranofin is a gold-containing phosphine compound that has been approved for treating patients with rheumatoid arthritis [138]. It can significantly reduce accumulation of intracellular ROS, and pretreatment of CD19 CAR-T cells or TILs with auranofin increases the elimination of CD19+ tumor cells and autologous tumor spheroids [117]. These data suggest a potential strategy involving use of Auranofin to improve adoptive cellular immunotherapy [117].
In addition to single-drug studies, integrated drug screening and profiling have been implemented to identify small-molecule drugs that modulate CAR T-cell performance among a library of more than 500 approved or investigational compounds [139]. Taken together, these studies show that combining CAR-T cells with traditional drugs can elicit unexpected effects, indicating potential new uses of these drugs as immunomodulatory agents. This offers a new approach for CAR-T cells optimization.
Strategies for enhancing CAR-T cell function by genetic manipulation
Genetic manipulation has been extensively applied across the bioscience and medical field. In particular, the clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9 (CRISPR/Cas9) system is considered the next generation of genomic editing technology and it has been widely applied [140]. This system represents a revolutionary innovation for research on the functional genome of various diseases, including monogenic disorders, polygenic disorders, and cancer, which can be used in immunotherapy [141, 142]. In this section, we provide an overview of recent approaches and applications of CRISPR/Cas9 to enhance CAR-T cell functions.
Generation of alloCAR-T cells by CRISPR/Cas9
As discussed above, alloCAR-T cells have enormous advantages for immunotherapy, including a short production cycle, low cost, and consistent curative effects. However, given differences in MHC and TCR on the T cell surface between patients and donors, the issues of GvHD and HvGD must be completely resolved. Before the CRISPR system was developed, the first two generations of genomic editing tools, zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs), were applied to disrupt endogenous TCRs in alloCAR-T cell [29]. Compared to ZFN and TALEN, the CRISPR system has the highest editing effectiveness and simplest flexibility. Therefore, it has been widely adopted for alloCAR-T cells. Justin Eyquem et al. first targeted a CAR to the TRAC locus with CRISPR/Cas9 and enhanced its tumor elimination ability compared to traditional CAR-T [143, 144]. Subsequently, Xiaojuan Liu et al. verified that CRISPR/Cas9 mediated editing of TRAC and B2M is readily applicable to alloCAR-T cells [145]. A series of relevant clinical trials have also been initiated [43, 146]. And it has been verified that a one-step CRISPR process can yield alloCAR-T cells with up to four gene deficiencies, highlighting the enormous potential for simultaneous CAR-T cell remodeling [145, 147]. Nonetheless, it is worth noting that coexpression of endogenous TCR plus CAR leads to superior persistence of T cells and significantly prolongs leukemia control in vivo compared to TCR knockout CAR-T cells, which suggests the significance of the TCR structure for T cells [148].
Improving CAR-T cell efficiency by CRISPR/Cas9
Compared to its significant curative effects in hematological malignancies, there are multiple obstacles for CAR-T immunotherapy application in solid tumors, primarily due to immunosuppressive TMEs, which disable and exhaust T cells. Knockout of several typical cell-surface immune checkpoint molecules, including PD-1, TIM-3, LAG-3 and CTLA-4, has been implemented in CAR-T cells and corresponding clinical trials are ongoing [149,150,151,152,153]. In addition to these coinhibitory molecules, a series of intracellular negative regulators of TCR signaling, such as tyrosine phosphatase 1B (PTP1B) and Cbl Proto-Oncogene B (CBLB), have been disrupted to enhance CAR-T cell function [154, 155]. PTP1B attenuates cytokine induced JAK/STAT signaling by dephosphorylating and deactivating JAK2 and TYK2 [154]. PTP1B is upregulated in tumor infiltrating T cells and it limits T cell expansion and cytotoxicity. PTP1B-specific deletion in CD8+ T cells enhances antigen-induced expansion and cytotoxicity to suppress tumor growth by activating JAK/STAT5 signaling [154]. Furthermore, the pharmacologic inhibition or genetic deletion of PTP1B enhances the efficacy of CAR-T cells against solid tumors [154]. CBLB is an E3 ubiquitin ligase that plays a crucial role in TIL dysfunction [155]. Inhibition or deletion of CBLB could restore the effector function of TILs and reduce PD-1 and TIM-3 levels. In CAR-T cells, this deletion has a similar effect on preventing CAR-T cell exhaustion with superior antitumor capacity [155]. Considering the immunosuppressive effect of several chemokines in TME especially in solid tumors, their receptors are gradually drawing more attention. Disruption of TGF-β receptor II (TGFBRII) in CAR-T cells could decrease exhaustion and improve the killing efficiency to solid tumors [156, 157]. As a key immunosuppressive metabolite in TME, adenosine could impair CAR-T cell function and induce sluggishness. Additionally, knockout of its receptor, A2AR, increases the capacity to inhibit solid tumor growth and development, indicating a potential target to promote CAR T-cell therapy [158, 159]. Collectively, these data indicated that targeting several immunosuppressive factors in TME and their receptors in CAR-T cells was a favorable option that could significantly improve the use of CAR-T cell therapy in solid tumors.
Improving CAR-T cell efficiency via activator coexpression
The above studies show the remarkable results of strengthening CAR-T abilities by disrupting negative immune regulators with the CRISPR system. Correspondingly, several attempts to improve CAR-T therapy by coexpression of positive immune factors have acquired distinguished effects. Interleukins Addition of IL-2, IL-7, and IL-15 to culture medium stimulates T cell proliferation and preserves the TSCM and TCM proportions (reviewed in Sect. 2). Furthermore, the coexpression of interleukin-encoding genes with the CAR framework has led to promising results in preclinical and clinical studies. IL-7 or IL-15 coexpression in CAR-T cells has been assessed in clinical trials (NCT04381741, NCT03932565, and NCT03721068). Furthermore, recent research has shown the superior antitumor activity of IL-7 receptor (IL7R) coexpression in CAR-T cells targeting neuroblastoma and glioblastoma [160]. Related clinical trials have begun to assess the persistence and efficacy of this approach [160]. Further studies have revealed that coexpression of IL-12, IL-18, IL-21, or IL-23 in CAR-T cells significantly enhances the therapeutic effect against different malignancies, suggesting the significance of interleukins for CAR-T cell optimization [161]. Therefore, IL-12 not only enhances the ability to directly kill tumor cells, but also recruits host immune cells to enhance the anticancer immune response [162]. IL-18-secreting CAR-T cells were shown to modulate the TME and to evoke an effective endogenous antitumor immune response [163]. IL-21 was verified to enhance expansion of CAR-T cells after antigenic stimulation and reduce the apoptosis rate [164]. IL-23 overexpression could regulate CAR-T cells with increased granzyme B expression and decreased PD-1 expression [165]. Chemokine Receptors In the context of solid tumors, the first challenge is the weak infiltration of CAR-T cells into tumors due to the lack of sufficient and appropriate migratory signals. Chemokine systems offer promise as an approach to overcome this obstacle, and such systems have been confirmed to modulate the migration and function of immune cells through interactions between chemokines and specific chemokine receptors (CCRs). Further studies have revealed a significant correlation between chemokine genes and T cell migration. Hence, some studies have focused on rescuing the chemokine and CCR axes to improve the activity of CAR-T cells [166]. Linchun Jin et al. demonstrated that the coexpression of CXCR1 or CXCR2, the CXCL8 receptor, increased the migration and persistence of CAR-T cells in the TME. These increases led to superior tumor regression and long-term immunologic memory in several aggressive tumors, such as glioblastoma, ovarian cancer, and pancreatic cancer [167]. Synergy with CXCR2 coexpression was reported in an additional study [168]. CCL2-CCR2 signaling is another signaling pathway that has been confirmed to enhance the intratumor infiltration of immune cells. In the context of malignant pleural mesotheliomas that secrete high levels of CCL2, CCR2 transduction into meso CAR-T cells increases CCL2-induced calcium flux, cell migration and cell death [168]. Compared with control meso CAR T cells, those with CCR2 coexpression showed a 12.5-fold increase in tumor infiltration in a mouse xenograft tumor model [168]. Moreover, expression levels of proinflammatory cytokines, including IL-2, IFN-γ, and TNF-α, were increased by CCR2 in the treatment of non-small cell lung carcinoma [169]. Another study showed that adoptive T cell intratumoral trafficking is improved by CXCL16-CXCR6 (significantly higher IFN-γ production compared to conventional CAR-T) [170], CXCL12-CXCR4 (enhanced CAR-T cells recruitment into CXCL12-rich bone marrow in an acute myeloid leukemia mouse model) [171], and CCL22-CCR4 (enhanced antitumor efficacy against a subcutaneous xenograft model of human Hodgkin’s lymphoma) [166, 172]. Taken together, the results from abundant studies have highlighted the potential of combining the chemokine and receptor systems with CAR-T cells to markedly enhance immunotherapy, especially against solid tumors. Given that the concentration and variety of chemokines differ among tumors, it is important to select the optimum chemokine and receptor pathway for combination with CAR-T cells to optimize immunotherapy. Other Active Regulators Several active regulators of TCR signaling or downstream pathways have been gradually found to enhance CAR-T cell efficacy, such as CD80, 41BBL, OX40, and CD40L [173,174,175,176]. Moreover, IL-7 induces T cell polyfunctionality by activating signal transducer and activator of transcription 5 (STAT5) [177, 178]. Beyond IL-7 supplementation in the culture medium during CAR-T cell expansion phase, persistent STAT5 activation by genetic manipulation has shown great results. Ectopic expression of a constitutively active form of STAT5 (CASTAT5) modulates CD4+ T cells with robust proliferation and vigorous infiltration abilities and enhances the antitumor response [179]. Further analysis suggested that CASTAT5 enables the remodeling of the genome-wide chromatin structure in CD4+ T cells and establishes a distinct epigenetic and transcriptional landscape [179]. When CASTAT5 was coexpressed in CD19 CAR-T cells, an optimal therapeutic outcome was achieved in a B-cell lymphoma model [179]. Activator protein 1 (AP1) is an important transcriptional regulator that participates in several cellular processes, including immune regulation [180]. Importantly, AP1 dysregulation in CD8+ T cells induces CAR-T cell exhaustion, involving downregulation of cytokines, reduction in expansion, increase in checkpoints, and exaggerated effector differentiation [181]. Further research has shown that c-Jun, a member of the AP1 family, is sufficiently dominant and its overexpression enabled an increase in the proportion of TSCM and TCM cells, long-term proliferation, cytokine secretion and T cells exhaustion [181]. In the context of c-Jun coexpression, CAR-T cells show superior self-renewal ability, resistance to exhaustion, and antitumor ability in lymphoma and solid tumors [182]. C3aR, the receptor of complement fragment C3a, has been verified to enhance T cell responses and its cooperation in CAR-T cells tended to induce memory T cell phenotype with superior therapeutic potential in extramedullary leukemia [183]. Moreover, the introduction of Toll/interleukin-1 receptor domain of Toll-like receptor 2 in CD19 CAR-T cells showed improved expansion, persistency, and effector function and it has been adopted for relapse or refractory B-ALL patients [184, 185]. Therefore, overexpression of several core positive cytokines and regulators might address the major barriers (weak infiltration and poor persistence) to CAR-T efficiency, especially for solid tumors [182].
Newly discovered targets for CAR-T cells with longer lifetimes and better efficiency
Numerous studies have focused on improving CAR-T immunotherapy using CRISPR mediated knockout or ectopic overexpression of genes. The majority of these genes have been verified to perform some function in the antitumor immune response. Prominent results have been achieved with this strategy, and there are several ongoing related clinical trials. To further optimize adoptive T cell therapy, especially for solid tumors, exploration of novel and undetected targets is urgently needed. The CRISPR mediated functional genome-wide screening platform has become an excellent tool for meticulously discovering new gene functions and molecular mechanisms [186, 187]. Indeed, this screening strategy has been utilized to identify potential regulators of tumor-immune interactions as new immunotherapy targets in cancer treatment with impacts on processes such as T cell activation, effector function, exhaustion, and cytokine secretion and signaling (Table S1).
Screening for T cell activation
To comprehensively search for genes that regulate T cell activation, Wanjing Shang et al. first developed a single-cell-based readout of T cell activation by measuring the expression of CD69, a well-defined early marker of T cell activation, using flow cytometry in Jurkat cells derived from T cells [188]. An unbiased genome-wide screening using a lentiviral-infected Jurkat cell library was performed. Cell was collected and analyzed according to the expression level of CD69, and the screening result confirmed the abundance of well-known regulators involved in T cell activation and identified several previously unexplored genes. For example, family with sequence similarity 49-member B (FAM49B), which was highly ranked in the CD69high subset, is considered a negative regulator. Further study validated that FAM49B regulates cytoskeletal remodeling via the Rac-PAK axis during T cell activation and that the negative regulatory effect was reversed by FAM49B deficiency [188].
Screening for T cell effector function
In addition to high-throughput loss-of-function screening, gain-of-function screening using a modified CRISPR system has been applied to identify functional boosters in primary T cells. Lupeng Ye et al. developed a dead-guide RNA-based genome-wide gain-of-function CRISPR activation screening system [189]. During the screening process, CD107a was chosen as the marker to reflect the ability of cytotoxic CD8+ T lymphocytes to kill tumor cells [189]. CD8+ T cells expressing CD107a at high levels (top 5%) were sorted and sequenced. Further analysis revealed several uncharacterized genes enriched in CD107ahigh cells, among which proline dehydrogenase 2 (PRODH2) was verified to reprogram proline metabolism and promote proliferation in CD8+ T cells [189]. Moreover, PRODH2 engineering by genomic knockin or lentiviral overexpression increases CD22 CAR-T cytotoxicity towards cancer cells [189].
Screening for T cell exhaustion
For a more comprehensive understanding of the molecular events regulating CAR T-cell exhaustion, Dongrui Wang et al. developed a robust method for whole genome CRISPR-KO screening of human IL13Rα2 CAR-T cells targeting glioblastoma cells [190]. After transduction with sgRNA library lentivirus, the CAR-T cells were recursively incubated with excess GBM stem cells. The CAR-T cells were then sorted from the coculture system and grouped based on expression of the inhibitory receptor PD-1, a typical marker of T cell exhaustion [190]. A potential target for repressing CAR-T antitumor activity was detected in the PD-1low subset. Knockout of the top hits (TLE4, IKZF2, TMEM184B, and EIF5A) repressed PD-1 expression, finally inhibiting exhaustion and improving CAR-T cell cytotoxicity in vitro [190].
Screening for T cell persistence
CD62L was selected as a pivotal marker to further improve persistence of CAR-T cells [81]. Devikala Gurusamy et al. performed CRISPR screening of CD8+ T cells in mice [191]. Primary CD8+ T cells were stimulated twice and then classified by CD62, ROS, γH2AX, and proliferation signaling. The sgRNA distributions showed that p38 kinase can regulate the desired phenotypes of T cells. Further genetic and pharmacological studies suggested that p38 kinase blockade improves T cell fitness via metabolic and transcriptional alterations. Finally, p38 inhibition improved the persistence and antitumor efficacy of CAR-based adoptive immunotherapies [191]. Similar screening studies have also been related to T cell proliferation, cytokine regulation, and tumor cell resistance, but these studies differed in biosample types, library sources, sgRNA numbers, and screening protocols (Table S1) [192,193,194].
Taken together, increased application of whole-genome screening in immuno-oncology networks based on the CRISPR system has gradually revealed the astonishing ability to identify potential immune regulators. Thus, such studies may ultimately uncover novel opportunities and strategies for improving the efficiency of current immunotherapy agents.
Conclusion
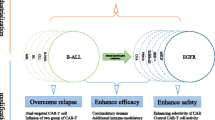
CAR-T cell immunotherapy has emerged and rapidly developed into a promising treatment option for various types of cancer, especially hematologic malignancies. However, several barriers exist that need to be overcome to achieve superior clinical results. In recent years, numerous studies have concentrated on identifying strategies to optimize CAR-T cell efficiency. The aims are to regulate the T cell phenotype and drive T cell differentiation for superior effector function and longer persistence [93]. Therefore, we systematically examined the optimizating strategies that aim to improve the long-term persistence and antitumor performance of CAR-T cells (Fig. 2).

Summary of strategies to improve CAR-T persistence. Optimization approaches to improve CAR-T cell persistence and antitumor performance, including choosing a suitable cell source, improving culture conditions with metabolites, combining CAR-T cells with conventional drugs, and applying genetic manipulations
As the T cell source and components influence the applicable targets of CAR-T cells, they should be prudently chosen. According to existing clinical data, CD4+ T cell-based CAR-T cells show better persistence than CD8+ T cells, and autoCAR-T cells exist in vivo longer than alloCAR-T cells [15, 44]. Culture conditions also influence CAR-T cell proliferation and differentiation by regulating cellular metabolism. Several metabolites, such as glutamine inhibitors, L-arginine, and 2-DG, have been found to alter the T cell development trajectory by enhancing OXPHOS and restricting glycolysis [101, 103, 106]. Interestingly, some conventional drugs improve the antitumor performance of CAR-T cells, relying on their ability to regulate metabolism and differentiation and thus prolong persistence. These drugs might have clinical applications as CAR-T adjuvants. In fact, Idelalisib, Rapamycin, Duvelisib, Decitabine, and Ibrutinib have been deemed safe in clinical trials (Table 2). Moreover, genetic manipulation has been quickly applied to CAR-T cells, especially with the discovery and development of the CRISPR system [195]. This approach not only targets traditional checkpoints but also several newfound targets. It is expected that the CRISPR system will increase CAR-T cell applications and lead to improvements. It is worth noting that combining these methods might result in superior in vivo persistence of CAR-T cells, particularly in the context of solid tumors surrounded by a hostile microenvironment [195]. To take advantage of these strategies for CAR-T cells to treat cancer, there is an urgent need to better understand the underlying mechanisms and to quickly carry out preclinical and clinical trials. It’s worth noting that several factors in specific clinical setting also influenced the CAR-T persistence, such as the choice and effect of previous lymphodepleting chemotherapy which could influence the state of TME, the distribution and density of antigen target which induces CAR-T cells exhaustion [196,197,198,199,200]. Based on this, the clinical setting should not be ignored for improving CAR-T cells persistence in vivo.
Notably, several recent studies have revolutionized the in vitro CAR-T cell expansion process by shortening the time required [201,202,203]. Compared to the traditional 1- to 2-week waiting period, the new process only requires 1 to 3 days [201]. The CAR-T cells generated using this shorter in vitro culture process exhibited better proliferation, longer persistence, and better tumor killing ability than conventional CAR-T cells. A recent clinical study in which r/r B-ALL patients were treated with FasTCAR-T cells reported a complete CR at Day 28 and a CR rate of 83.3% at the 3-month assessment [204]. Further attention should be given to comparison between conventional and short-term CAR-T cells. Interestingly, several other strategies about vaccination and viruses application to improve CAR-T cells persistence and clinical efficacy have successively emerged. For example, when CAR-T cells were generated by donor Epstein–Barr virus-specific T-cells, they showed superior expansion/persistence ability and limited CRS, neurotoxicity, and GvHD in clinical trials [205]. Another virus, oncolytic virus, was capable to enter into tumor cells and delivers target protein which was recognized and killed by CAR-T cells, and this combination strategy showed promising results for multiple solid tumors [206]. Similarly, a nanoparticulate RNA vaccine has also been applied to deliver a protein into solid tumors as CAR-T target, which could selectively enhance the expansion of CAR-T cells in vivo [207]. These remarkable studies broadened the application and efficacy especially for solid tumors.
In this review, we summarize several strategies to regulate CAR-T cell metabolism and differentiation to improve CAR-T persistence, which aims at providing methods or clues for recurrence and refractory problem after CAR-T cell therapy. Also, the strategies could be applied in expanding immune cell therapy to more solid tumor, auto-immune diseases and cost reduction to benefit more patients.
Availability of data and materials
Not applicable.
Abbreviations
- CAR-T:
-
Chimeric antigen receptor T
- TME:
-
Tumor microenvironment
- TCR:
-
T-cell receptor
- r/r B-ALL:
-
Relapsed/refractory B-cell acute lymphoblastic leukemia
- CR:
-
Complete remission
- autoCAR-T:
-
Autologous CAR-T
- alloCAR-T:
-
Allogeneic CAR-T
- TIL:
-
Tumor infiltrating lymphocyte
- HvGD:
-
Host versus graft disease
- GvHD:
-
Graft versus host disease
- HLA:
-
Human leukocyte antigen
- CRS:
-
Cytokine-release syndrome
- NKTs:
-
Natural killer T cells
- Tregs:
-
Regulatory T cells
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- MHC:
-
Major histocompatibility complex
- CCR7:
-
CXC chemokine receptor 7
- TN :
-
Naïve T
- TSCM :
-
Stem cell memory T
- TCM :
-
Central memory T
- TEM :
-
Effector memory T
- TEFF :
-
Terminal effector T
- OXPHOS:
-
Oxidative phosphorylation
- FAO:
-
Fatty acid oxidation
- CLL/SLL:
-
Chronic lymphocytic leukemia/small lymphocytic lymphoma
- allo-HSCT:
-
Allogeneic hematopoietic stem cell transplantation
- HATs:
-
Histone acetyltransferases
- HDACs:
-
Histone deacetylases
- BET:
-
Bromodomain and extraterminal motif
- CRISPR/Cas9:
-
Clustered regularly interspaced short palindromic repeat (CRISPR)-associated protein 9
- ZFNs:
-
Zinc finger nucleases
- TALENs:
-
Transcription activator-like effector nucleases
- PTP1B:
-
Tyrosine phosphatase 1B
- CBLB:
-
Cbl proto-oncogene B
- TGFBRII:
-
TGF-β receptor II
- CCRs:
-
Chemokine receptors
- FAM49B:
-
Family with sequence similarity 49-member B
- PRODH2:
-
Proline dehydrogenase 2
- GLUTs:
-
Glucose transporters
References
Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024–8.
Kochenderfer JN, Yu Z, Frasheri D, Restifo NP, Rosenberg SA. Adoptive transfer of syngeneic T cells transduced with a chimeric antigen receptor that recognizes murine CD19 can eradicate lymphoma and normal B cells. Blood. 2010;116(19):3875–86.
Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–64.
Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–28.
Gandhi M, Jones K. Optimizing tumor-targeting chimeric antigen receptor T cells in B-cell lymphoma patients. Immunotherapy. 2011;3(12):1441–3.
Micklethwaite KP, Savoldo B, Hanley PJ, Leen AM, Demmler-Harrison GJ, Cooper LJN, et al. Derivation of human T lymphocytes from cord blood and peripheral blood with antiviral and antileukemic specificity from a single culture as protection against infection and relapse after stem cell transplantation. Blood. 2010;115(13):2695–703.
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73.
Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70.
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–9.
Liu Y, Chen X, Han W, Zhang Y. Tisagenlecleucel, an approved anti-CD19 chimeric antigen receptor T-cell therapy for the treatment of leukemia. Drugs Today (Barc). 2017;53(11):597–608.
Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol. 2019;10:2040620719841581.
Mullard A. FDA approves fourth CAR-T cell therapy. Nat Rev Drug Discov. 2021;20(3):166.
First CAR-T. therapy to target BCMA gets FDA nod. Nat Biotechnol. 2021;39(5):531.
Dai H, Wu Z, Jia H, Tong C, Guo Y, Ti D, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13(1):30.
Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4 CAR T cells. Nature. 2022;602(7897):503–9.
Zhang C, Wang X, Zhang R, Liu F, Wang Y, Yan Z, et al. Donor-derived CD19 CAR-T cell therapy of relapse of CD19-positive B-ALL post allotransplant. Leukemia. 2021;35(6):1563–70.
O’Leary MC, Lu X, Huang Y, Lin X, Mahmood I, Przepiorka D, et al. FDA Approval Summary: Tisagenlecleucel for Treatment of Patients with Relapsed or Refractory B-cell Precursor Acute Lymphoblastic Leukemia. Clin Cancer Res. 2019;25(4):1142–6.
Liu J, Zhong JF, Zhang X, Zhang C. Allogeneic CD19-CAR-T cell infusion after allogeneic hematopoietic stem cell transplantation in B cell malignancies. J Hematol Oncol. 2017;10(1):35.
Roex G, Timmers M, Wouters K, Campillo-Davo D, Flumens D, Schroyens W, et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J Hematol Oncol. 2020;13(1):164.
Newick K, O’Brien S, Moon E, Albelda SM. CAR T Cell Therapy for Solid Tumors. Annu Rev Med. 2017;68:139–52.
Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther. 2020;28(11):2320–39.
Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. 2021;18(5):1085–95.
Gao TA, Chen YY. Engineering Next-Generation CAR-T, Cells. Overcoming Tumor Hypoxia and Metabolism. Annu Rev Chem Biomol Eng. 2022;13:193–216.
Edeline J, Houot R, Marabelle A, Alcantara M. CAR-T cells and BiTEs in solid tumors: challenges and perspectives. J Hematol Oncol. 2021;14(1):65.
Levine BL, Miskin J, Wonnacott K, Keir C. Global Manufacturing of CAR T Cell Therapy. Mol Ther Methods Clin Dev. 2017;4:92–101.
Huang R, Li X, He Y, Zhu W, Gao L, Liu Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86.
Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136(14):1632–44.
Jayaraman J, Mellody MP, Hou AJ, Desai RP, Fung AW, Pham AHT, et al. CAR-T design: Elements and their synergistic function. EBioMedicine. 2020;58:102931.
Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1):132.
Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer. 2019;120(1):26–37.
Mazinani M, Rahbarizadeh F. CAR-T cell potency: from structural elements to vector backbone components. Biomark Res. 2022;10(1):70.
Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–27.
Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang C-H, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–9.
Textor A, Grunewald L, Anders K, Klaus A, Schwiebert S, Winkler A, et al. CD28 Co-Stimulus Achieves Superior CAR T Cell Effector Function against Solid Tumors Than 4–1BB Co-Stimulus. Cancers (Basel). 2021;13(5):1050.
Vinanica N, Yong A, Wong D, Png YT, Seow SV, Imamura M, et al. Specific stimulation of T lymphocytes with erythropoietin for adoptive immunotherapy. Blood. 2020;135(9):668–79.
Muliaditan T, Halim L, Whilding LM, Draper B, Achkova DY, Kausar F, et al. Synergistic T cell signaling by 41BB and CD28 is optimally achieved by membrane proximal positioning within parallel chimeric antigen receptors. Cell Rep Med. 2021;2(12):100457.
Wang Y, Tong C, Dai H, Wu Z, Han X, Guo Y, et al. Low-dose decitabine priming endows CAR T cells with enhanced and persistent antitumour potential via epigenetic reprogramming. Nat Commun. 2021;12(1):409.
Wang X, Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015.
Hu Y, Wang J, Wei G, Yu J, Luo Y, Shi J, et al. A retrospective comparison of allogenic and autologous chimeric antigen receptor T cell therapy targeting CD19 in patients with relapsed/refractory acute lymphoblastic leukemia. Bone Marrow Transplant. 2019;54(8):1208–17.
Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–99.
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013.
Bartoló-Ibars A, Uribe-Herranz M, Muñoz-Sánchez G, Arnaldos-Pérez C, Ortiz-Maldonado V, Urbano-Ispizua Á, et al. CAR-T after Stem Cell Transplantation in B-Cell Lymphoproliferative Disorders: Are They Really Autologous or Allogenic Cell Therapies? Cancers (Basel). 2021;13(18):4664.
Hu Y, Zhou Y, Zhang M, Ge W, Li Y, Yang L, et al. CRISPR/Cas9-Engineered Universal CD19/CD22 Dual-Targeted CAR-T Cell Therapy for Relapsed/Refractory B-cell Acute Lymphoblastic Leukemia. Clin Cancer Res. 2021;27(10):2764–72.
DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19 malignancies take autologous CAR T cells ‘off the shelf’? Nat Rev Clin Oncol. 2021;18(4):195–6.
Chong EA, Ruella M, Schuster SJ. Five-Year Outcomes for Refractory B-Cell Lymphomas with CAR T-Cell Therapy. N Engl J Med. 2021;384(7):673–4.
Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017;377(26):2545–54.
Benjamin R, Graham C, Yallop D, Jozwik A, Mirci-Danicar OC, Lucchini G, et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: results of two phase 1 studies. Lancet. 2020;396(10266):1885–94.
Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in Universal CAR-T Cell Therapy. Front Immunol. 2021;12:744823.
van der Stegen SJC, Lindenbergh PL, Petrovic RM, Xie H, Diop MP, Alexeeva V, et al. Generation of T-cell-receptor-negative CD8αβ-positive CAR T cells from T-cell-derived induced pluripotent stem cells. Nat Biomed Eng. 2022;6(11):1284–97.
Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6(261):261ra151.
Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8 + and CD4 + subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492–500.
Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8 + and CD4 + CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116.
Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8 + composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–38.
Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4 + CAR T cells mediate superior antitumor activity. JCI Insight. 2018;3(10):e99048.
Franks SE, Wolfson B, Hodge JW. Natural Born Killers: NK Cells in Cancer Therapy. Cancers (Basel). 2020;12(8):2131.
Baghery Saghchy Khorasani A, Yousefi A-M, Bashash D. CAR NK cell therapy in hematologic malignancies and solid tumors; obstacles and strategies to overcome the challenges. Int Immunopharmacol. 2022;110:109041.
Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14(1):7.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168.
Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14(1):73.
Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. Biomark Res. 2022;10(1):12.
Delfanti G, Dellabona P, Casorati G, Fedeli M. Adoptive Immunotherapy With Engineered iNKT Cells to Target Cancer Cells and the Suppressive Microenvironment. Front Med (Lausanne). 2022;9:897750.
Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. 2014;124(18):2824–33.
Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. 2020;26(11):1686–90.
Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced Persistence and Antitumor Activity against Neuroblastoma. Clin Cancer Res. 2019;25(23):7126–38.
Silva-Santos B, Mensurado S, Coffelt SB. γδ T cells: pleiotropic immune effectors with therapeutic potential in cancer. Nat Rev Cancer. 2019;19(7):392–404.
Silva-Santos B, Serre K, Norell H. γδ T cells in cancer. Nat Rev Immunol. 2015;15(11):683–91.
Legut M, Cole DK, Sewell AK. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell Mol Immunol. 2015;12(6):656–68.
Ou L, Wang H, Huang H, Zhou Z, Lin Q, Guo Y, et al. Preclinical platforms to study therapeutic efficacy of human γδ T cells. Clin Transl Med. 2022;12(6):e814.
Lee D, Rosenthal CJ, Penn NE, Dunn ZS, Zhou Y, Yang L. Human γδ T Cell Subsets and Their Clinical Applications for Cancer Immunotherapy. Cancers (Basel). 2022;14(12):3005.
Rozenbaum M, Meir A, Aharony Y, Itzhaki O, Schachter J, Bank I, et al. Gamma-Delta CAR-T Cells Show CAR-Directed and Independent Activity Against Leukemia. Front Immunol. 2020;11:1347.
Nishimoto KP, Barca T, Azameera A, Makkouk A, Romero JM, Bai L, et al. Allogeneic CD20-targeted γδ T cells exhibit innate and adaptive antitumor activities in preclinical B-cell lymphoma models. Clin Transl Immunology. 2022;11(2):e1373.
Jin K, Parreau S, Warrington KJ, Koster MJ, Berry GJ, Goronzy JJ, et al. Regulatory T Cells in Autoimmune Vasculitis. Front Immunol. 2022;13:844300.
Richardson N, Wootton GE, Bozward AG, Oo YH. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin Immunopathol. 2022;44(4):461–74.
Arjomandnejad M, Kopec AL, Keeler AM. CAR-T Regulatory (CAR-Treg) Cells: Engineering and Applications. Biomedicines. 2022;10(2):287.
Ellis GI, Coker KE, Winn DW, Deng MZ, Shukla D, Bhoj V, et al. Trafficking and persistence of alloantigen-specific chimeric antigen receptor regulatory T cells in Cynomolgus macaque. Cell Rep Med. 2022;3(5):100614.
Qu G, Chen J, Li Y, Yuan Y, Liang R, Li B. Current status and perspectives of regulatory T cell-based therapy. J Genet Genomics. 2022;49(7):599–611.
van den Broek T, Borghans JAM, van Wijk F. The full spectrum of human naive T cells. Nat Rev Immunol. 2018;18(6):363–73.
Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel). 2016;8(3):36.
Tantalo DG, Oliver AJ, von Scheidt B, Harrison AJ, Mueller SN, Kershaw MH, et al. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J Immunother Cancer. 2021;9(5):e002555.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–7.
Lugli E, Dominguez MH, Gattinoni L, Chattopadhyay PK, Bolton DL, Song K, et al. Superior T memory stem cell persistence supports long-lived T cell memory. J Clin Investig. 2013;123(2):594–9.
Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. 2020;43(1):1–9.
Lugli E, Galletti G, Boi SK, Youngblood BA. Stem, Effector, and Hybrid States of Memory CD8 T Cells. Trends Immunol. 2020;41(1):17–28.
Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012;35(9):651–60.
Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8 + T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102(27):9571–6.
Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127(24):2980–90.
López-Cantillo G, Urueña C, Camacho BA, Ramírez-Segura C. CAR-T Cell Performance: How to Improve Their Persistence? Front Immunol. 2022;13:878209.
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–84.
Brummelman J, Pilipow K, Lugli E. The Single-Cell Phenotypic Identity of Human CD8 and CD4 T Cells. Int Rev Cell Mol Biol. 2018;341:63–124.
Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8 + memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–28.
Arcangeli S, Falcone L, Camisa B, De Girardi F, Biondi M, Giglio F, et al. Next-Generation Manufacturing Protocols Enriching T CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front Immunol. 2020;11:1217.
Zhang Z, Li F, Tian Y, Cao L, Gao Q, Zhang C, et al. Metformin Enhances the Antitumor Activity of CD8 T Lymphocytes via the AMPK-miR-107-Eomes-PD-1 Pathway. J Immunol. 2020;204(9):2575–88.
Zhang M, Jin X, Sun R, Xiong X, Wang J, Xie D, et al. Optimization of metabolism to improve efficacy during CAR-T cell manufacturing. J Transl Med. 2021;19(1):499.
Kouidhi S, Elgaaied AB, Chouaib S. Impact of Metabolism on T-Cell Differentiation and Function and Cross Talk with Tumor Microenvironment. Front Immunol. 2017;8:270.
Patel CH, Powell JD. Warburg meets epigenetics. Science (New York, NY). 2016;354(6311):419–20.
Bishop EL, Gudgeon N, Dimeloe S. Control of T Cell Metabolism by Cytokines and Hormones. Front Immunol. 2021;12:653605.
Loschinski R, Böttcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget. 2018;9(17):13125–38.
Huang Y, Si X, Shao M, Teng X, Xiao G, Huang H. Rewiring mitochondrial metabolism to counteract exhaustion of CAR-T cells. J Hematol Oncol. 2022;15(1):38.
Gong W, Hoffmann J-M, Stock S, Wang L, Liu Y, Schubert M-L, et al. Comparison of IL-2 vs IL-7/IL-15 for the generation of NY-ESO-1-specific T cells. Cancer Immunol Immunother. 2019;68(7):1195–209.
Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo C-F, et al. IL15 Enhances CAR-T Cell Antitumor Activity by Reducing mTORC1 Activity and Preserving Their Stem Cell Memory Phenotype. Cancer Immunol Res. 2019;7(5):759–72.
Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8 + T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479–88.
Marchesi F, Vignali D, Manini B, Rigamonti A, Monti P. Manipulation of Glucose Availability to Boost Cancer Immunotherapies. Cancers (Basel). 2020;12(10):2940.
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell. 2016;167(3):829-842.e13.
Nabe S, Yamada T, Suzuki J, Toriyama K, Yasuoka T, Kuwahara M, et al. Reinforce the antitumor activity of CD8 T cells via glutamine restriction. Cancer Sci. 2018;109(12):3737–50.
Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013–21.
Shen L, Xiao Y, Zhang C, Li S, Teng X, Cui L, et al. Metabolic reprogramming by ex vivo glutamine inhibition endows CAR-T cells with less-differentiated phenotype and persistent antitumor activity. Cancer Lett. 2022;538:215710.
Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, et al. Enhancing Chimeric Antigen Receptor T Cell Anti-tumor Function through Advanced Media Design. Mol Ther Methods Clin Dev. 2020;18:595–606.
Schöffski P, Cresta S, Mayer IA, Wildiers H, Damian S, Gendreau S, et al. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res. 2018;20(1):109.
Stock S, Übelhart R, Schubert M-L, Fan F, He B, Hoffmann J-M, et al. Idelalisib for optimized CD19-specific chimeric antigen receptor T cells in chronic lymphocytic leukemia patients. Int J Cancer. 2019;145(5):1312–24.
Funk CR, Wang S, Chen KZ, Waller A, Sharma A, Edgar CL, et al. PI3Kδ/γ inhibition promotes human CART cell epigenetic and metabolic reprogramming to enhance antitumor cytotoxicity. Blood. 2022;139(4):523–37.
Fan F, Yoo HJ, Stock S, Wang L, Liu Y, Schubert M-L, et al. Ibrutinib for improved chimeric antigen receptor T-cell production for chronic lymphocytic leukemia patients. Int J Cancer. 2021;148(2):419–28.
Zheng W, O’Hear CE, Alli R, Basham JH, Abdelsamed HA, Palmer LE, et al. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia. 2018;32(5):1157–67.
Nian Z, Zheng X, Dou Y, Du X, Zhou L, Fu B, et al. Rapamycin Pretreatment Rescues the Bone Marrow AML Cell Elimination Capacity of CAR-T Cells. Clin Cancer Res. 2021;27(21):6026–38.
Lei X, Ou Z, Yang Z, Zhong J, Zhu Y, Tian J, et al. A Pan-Histone Deacetylase Inhibitor Enhances the Antitumor Activity of B7-H3-Specific CAR T Cells in Solid Tumors. Clin Cancer Res. 2021;27(13):3757–71.
Kong W, Dimitri A, Wang W, Jung I-Y, Ott CJ, Fasolino M, et al. BET bromodomain protein inhibition reverses chimeric antigen receptor extinction and reinvigorates exhausted T cells in chronic lymphocytic leukemia. J Clin Invest. 2021;131(16):e145459.
Shen C, Zhang Z, Tian Y, Li F, Zhou L, Jiang W, et al. Sulforaphane enhances the antitumor response of chimeric antigen receptor T cells by regulating PD-1/PD-L1 pathway. BMC Med. 2021;19(1):283.
Renken S, Nakajima T, Magalhaes I, Mattsson J, Lundqvist A, Arnér ESJ, et al. Targeting of Nrf2 improves antitumoral responses by human NK cells, TIL and CAR T cells during oxidative stress. J Immunother Cancer. 2022;10(6):e004458.
Yang Q, Jiang W, Hou P. Emerging role of PI3K/AKT in tumor-related epigenetic regulation. Semin Cancer Biol. 2019;59:112–24.
Wang X, Ding J, Meng L-h. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36(10):1170–6.
Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72(12):2337–47.
Saravia J, Raynor JL, Chapman NM, Lim SA, Chi H. Signaling networks in immunometabolism. Cell Res. 2020;30(4):328–42.
Hasslacher S, Schneele L, Stroh S, Langhans J, Zeiler K, Kattner P, et al. Inhibition of PI3K signalling increases the efficiency of radiotherapy in glioblastoma cells. Int J Oncol. 2018;53(5):1881–96.
Zhang H, Hu Y, Shao M, Teng X, Jiang P, Wang X, et al. Dasatinib enhances anti-leukemia efficacy of chimeric antigen receptor T cells by inhibiting cell differentiation and exhaustion. J Hematol Oncol. 2021;14(1):113.
Alu A, Lei H, Han X, Wei Y, Wei X. BTK inhibitors in the treatment of hematological malignancies and inflammatory diseases: mechanisms and clinical studies. J Hematol Oncol. 2022;15(1):138.
Wang H, Guo H, Yang J, Liu Y, Liu X, Zhang Q, et al. Bruton tyrosine kinase inhibitors in B-cell lymphoma: beyond the antitumour effect. Exp Hematol Oncol. 2022;11(1):60.
Ghez D, Rubio MT, Maillard N, Suarez F, Chandesris M-O, Delarue R, et al. Rapamycin for refractory acute graft-versus-host disease. Transplantation. 2009;88(9):1081–7.
Akbari B, Ghahri-Saremi N, Soltantoyeh T, Hadjati J, Ghassemi S, Mirzaei HR. Epigenetic strategies to boost CAR T cell therapy. Mol Ther. 2021;29(9):2640–59.
Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol. 2022;23(6):848–60.
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, et al. The epigenetic landscape of T cell exhaustion. Science (New York). 2016; 354(6316):pp. 1165–9.
Akimova T, Beier UH, Liu Y, Wang L, Hancock WW. Histone/protein deacetylases and T-cell immune responses. Blood. 2012;119(11):2443–51.
Gallinari P, Di Marco S, Jones P, Pallaoro M, Steinkühler C. HDACs, histone deacetylation and gene transcription: from molecular biology to cancer therapeutics. Cell Res. 2007;17(3):195–211.
Li Y, Seto E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med. 2016;6(10):a026831.
Que Y, Zhang X-L, Liu Z-X, Zhao J-J, Pan Q-Z, Wen X-Z, et al. Frequent amplification of HDAC genes and efficacy of HDAC inhibitor chidamide and PD-1 blockade combination in soft tissue sarcoma. J Immunother Cancer. 2021;9(2):e001696.
Sanchez-Rangel E, Inzucchi SE. Metformin: clinical use in type 2 diabetes. Diabetologia. 2017;60(9):1586–93.
Cioce M, Pulito C, Strano S, Blandino G, Fazio VM. Metformin: Metabolic Rewiring Faces Tumor Heterogeneity. Cells. 2020;9(11):2439.
Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. 2022;603(7899):159–65.
Singh K, Connors SL, Macklin EA, Smith KD, Fahey JW, Talalay P, et al. Sulforaphane treatment of autism spectrum disorder (ASD). Proc Natl Acad Sci U S A. 2014;111(43):15550–5.
Liu Y, Lu Y, Xu Z, Ma X, Chen X, Liu W. Repurposing of the gold drug auranofin and a review of its derivatives as antibacterial therapeutics. Drug Discov Today. 2022;27(7):1961–73.
Dufva O, Koski J, Maliniemi P, Ianevski A, Klievink J, Leitner J, et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood. 2020;135(9):597–609.
Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096.
Chen M, Mao A, Xu M, Weng Q, Mao J, Ji J. CRISPR-Cas9 for cancer therapy: Opportunities and challenges. Cancer Lett. 2019;447:48–55.
Sharma G, Sharma AR, Bhattacharya M, Lee S-S, Chakraborty C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol Ther. 2021;29(2):571–86.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7.
Tang TCY, Xu N, Nordon R, Haber M, Micklethwaite K, Dolnikov A. Donor T cells for CAR T cell therapy. Biomark Res. 2022;10(1):14.
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27(1):154–7.
Guo Y, Tong C, Su L, Zhang W, Jia H, Liu Y, et al. CRISPR/Cas9 genome-edited universal CAR T cells in patients with relapsed/refractory lymphoma. Blood Adv. 2022;6(8):2695–9.
Dimitri A, Herbst F, Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. 2022;21(1):78.
Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136(12):1407–18.
Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017;11(4):554–62.
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8(10):17002–11.
Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol. 2020;13(1):54.
Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7(1):737.
Zhang H, Zhao P, Huang H. Engineering better chimeric antigen receptor T cells. Exp Hematol Oncol. 2020;9(1):34.
Wiede F, Lu K-H, Du X, Zeissig MN, Xu R, Goh PK, et al. PTP1B Is an Intracellular Checkpoint that Limits T-cell and CAR T-cell Antitumor Immunity. Cancer Discov. 2022;12(3):752–73.
Kumar J, Kumar R, Kumar Singh A, Tsakem EL, Kathania M, Riese MJ, et al. Deletion of Cbl-b inhibits CD8 T-cell exhaustion and promotes CAR T-cell function. J Immunother Cancer. 2021;9(1):e001688.
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5(4):e133977.
Li K, Xu J, Wang J, Lu C, Dai Y, Dai Q, et al. Dominant-negative transforming growth factor-β receptor-armoured mesothelin-targeted chimeric antigen receptor T cells slow tumour growth in a mouse model of ovarian cancer. Cancer Immunol Immunother. 2022. PMID: 36166071. https://doi.org/10.1007/s00262-022-03290-6.
Li N, Tang N, Cheng C, Hu T, Wei X, Han W, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. Oncoimmunology. 2020;9(1):1824643.
Liu G, Zhang Q, Liu G, Li D, Zhang L, Gu Z, et al. Disruption of adenosine 2A receptor improves the anti-tumor function of anti-mesothelin CAR T cells both in vitro and in vivo. Exp Cell Res. 2021;409(1):112886.
Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–51.
Zhang Z, Miao L, Ren Z, Tang F, Li Y. Gene-Edited Interleukin CAR-T Cells Therapy in the Treatment of Malignancies: Present and Future. Front Immunol. 2021;12:718686.
Kueberuwa G, Kalaitsidou M, Cheadle E, Hawkins RE, Gilham DE. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol Ther Oncolytics. 2018;8:41–51.
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018;23(7):2130–41.
Štach M, Ptáčková P, Mucha M, Musil J, Klener P, Otáhal P. Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy. 2020;22(12):744–54.
Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat Biotechnol. 2020;38(4):448–59.
Foeng J, Comerford I, McColl SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med. 2022;3(3):100543.
Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016.
Moon EK, Carpenito C, Sun J, Wang L-CS, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. 2011;17(14):4719–30.
Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine Receptor CCR2b Enhanced Anti-tumor Function of Chimeric Antigen Receptor T Cells Targeting Mesothelin in a Non-small-cell Lung Carcinoma Model. Front Immunol. 2021;12:628906.
Lesch S, Blumenberg V, Stoiber S, Gottschlich A, Ogonek J, Cadilha BL, et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat Biomed Eng. 2021;5(11):1246–60.
Itoh-Nakadai A, Saito Y, Murasawa-Tomizawa M, Kajita H, Ishikawa F. CXCR4-Expressing Anti-CD25 CAR T-Cells Effectively Eliminate Human AML Cells In Vivo. Blood. 2020;136(Supplement 1):35–6.
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402.
Curran KJ, Seinstra BA, Nikhamin Y, Yeh R, Usachenko Y, van Leeuwen DG, et al. Enhancing antitumor efficacy of chimeric antigen receptor T cells through constitutive CD40L expression. Mol Ther. 2015;23(4):769–78.
Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28(4):415–28.
Kuhn NF, Purdon TJ, van Leeuwen DG, Lopez AV, Curran KJ, Daniyan AF, et al. CD40 Ligand-Modified Chimeric Antigen Receptor T Cells Enhance Antitumor Function by Eliciting an Endogenous Antitumor Response. Cancer Cell. 2019;35(3):473-488.e6.
Tan J, Jia Y, Zhou M, Fu C, Tuhin IJ, Ye J, et al. Chimeric antigen receptors containing the OX40 signalling domain enhance the persistence of T cells even under repeated stimulation with multiple myeloma target cells. J Hematol Oncol. 2022;15(1):39.
Li H, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548(7667):338–42.
Onishi M, Nosaka T, Misawa K, Mui AL, Gorman D, McMahon M, et al. Identification and characterization of a constitutively active STAT5 mutant that promotes cell proliferation. Mol Cell Biol. 1998;18(7):3871–9.
Ding Z-C, Shi H, Aboelella NS, Fesenkova K, Park E-J, Liu Z, et al. Persistent STAT5 activation reprograms the epigenetic landscape in CD4 T cells to drive polyfunctionality and antitumor immunity. Sci Immunol. 2020;5(52):eaba5962.
Macián F, García-Cózar F, Im S-H, Horton HF, Byrne MC, Rao A. Transcriptional mechanisms underlying lymphocyte tolerance. Cell. 2002;109(6):719–31.
Seo H, González-Avalos E, Zhang W, Ramchandani P, Yang C, Lio C-WJ, et al. BATF and IRF4 cooperate to counter exhaustion in tumor-infiltrating CAR T cells. Nat Immunol. 2021;22(8):983–95.
Lynn RC, Weber EW, Sotillo E, Gennert D, Xu P, Good Z, et al. c-Jun overexpression in CAR T cells induces exhaustion resistance. Nature. 2019;576(7786):293–300.
Lai P, Chen X, Wang Y, Wang J, Zhang Y, Geng S, et al. C3aR costimulation enhances the antitumor efficacy of CAR-T cell therapy through Th17 expansion and memory T cell induction. J Hematol Oncol. 2022;15(1):68.
Weng J, Lai P, Qin L, Lai Y, Jiang Z, Luo C, et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J Hematol Oncol. 2018;11(1):25.
Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. 2018;32(3):801–8.
Chow RD, Chen S. Cancer CRISPR Screens In Vivo. Trends Cancer. 2018;4(5):349–58.
Liu D, Zhao X, Tang A, Xu X, Liu S, Zha L, et al. CRISPR screen in mechanism and target discovery for cancer immunotherapy. Biochim Biophys Acta Rev Cancer. 2020;1874(1):188378.
Shang W, Jiang Y, Boettcher M, Ding K, Mollenauer M, Liu Z, et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc Natl Acad Sci U S A. 2018;115(17):E4051-E60.
Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. 2022;34(4):595-614.e14.
Wang D, Prager BC, Gimple RC, Aguilar B, Alizadeh D, Tang H, et al. CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discov. 2021;11(5):1192–211.
Gurusamy D, Henning AN, Yamamoto TN, Yu Z, Zacharakis N, Krishna S, et al. Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell. 2020;37(6):818-833.e9.
Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell. 2019;178(5):1189-1204.e23.
Legut M, Gajic Z, Guarino M, Daniloski Z, Rahman JA, Xue X, et al. A genome-scale screen for synthetic drivers of T cell proliferation. Nature. 2022;603(7902):728–35.
Carnevale J, Shifrut E, Kale N, Nyberg WA, Blaeschke F, Chen YY, et al. RASA2 ablation in T cells boosts antigen sensitivity and long-term function. Nature. 2022;609(7925):174–82.
Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell. 2017;8(9):634–43.
Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26(2):111–7.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377(26):2531–44.
Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23(1):91–103.
Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT, et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. 2022;40(1):53-69.e9.
An F, Wang H, Liu Z, Wu F, Zhang J, Tao Q, et al. Influence of patient characteristics on chimeric antigen receptor T cell therapy in B-cell acute lymphoblastic leukemia. Nat Commun. 2020;11(1):5928.
Ghassemi S, Durgin JS, Nunez-Cruz S, Patel J, Leferovich J, Pinzone M, et al. Rapid manufacturing of non-activated potent CAR T cells. Nat Biomed Eng. 2022;6(2):118–28.
Agarwalla P, Ogunnaike EA, Ahn S, Froehlich KA, Jansson A, Ligler FS, et al. Bioinstructive implantable scaffolds for rapid in vivo manufacture and release of CAR-T cells. Nat Biotechnol. 2022;40(8):1250–8.
Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol. 2018;36(2):160–9.
Zhang C, He J, Liu L, Wang J, Wang S, Liu L, et al. Novel CD19 chimeric antigen receptor T cells manufactured next-day for acute lymphoblastic leukemia. Blood Cancer J. 2022;12(6):96.
Rossig C, Pule M, Altvater B, Saiagh S, Wright G, Ghorashian S, et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia. 2017;31(5):1087–95.
Park AK, Fong Y, Kim S-I, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med. 2020;12(559):eaaz1863.
Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367(6476):446–53.
Acknowledgements
Figdraw was used for image production.
Funding
This work was supported by National Natural Science Foundation of China (No. 82020108004 and 81873424), Natural Science Foundation of Chongqing, China (No. CSTB2022NSCQ-MSX1287), Special Funding for the Frontiers of Military Medical Basics (No. 2018YQYLY002), and Key Technical Innovation Projects in Clinical Fields of Xinqiao Hospital (No. 2018JSLC0020).
Author information
Authors and Affiliations
Contributions
YL and LA collected the related literature, prepared the figures and tables, and drafted the manuscript. RH provided critical revision of the manuscript and figures. HY and JX participated in the design of the review. XW and XZ designed the review, critically revised and supervised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
CRISPR system-based screenings for CAR-T cell optimization.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., An, L., Huang, R. et al. Strategies to enhance CAR-T persistence. Biomark Res 10, 86 (2022). https://doi.org/10.1186/s40364-022-00434-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-022-00434-9