
Abstract
Background
This phase Ib study (NCT00960960) evaluated pictilisib (GDC-0941; pan-phosphatidylinositol 3-kinase inhibitor) plus paclitaxel, with and without bevacizumab or trastuzumab, or in combination with letrozole, in patients with locally recurrent or metastatic breast cancer.
Methods
This was a three-part multischedule study. Patients in parts 1 and 2, which comprised 3 + 3 dose escalation and cohort expansion stages, received pictilisib (60–330 mg) plus paclitaxel (90 mg/m2) with and without bevacizumab (10 mg/kg) or trastuzumab (2–4 mg/kg). In part 3, patients received pictilisib (260 mg) plus letrozole (2.5 mg). Primary objectives were evaluation of safety and tolerability, identification of dose-limiting toxicities (DLTs) and the maximum tolerated dose (MTD) of pictilisib, and recommendation of a phase II dosing regimen. Secondary endpoints included pharmacokinetics and preliminary antitumor activity.
Results
Sixty-nine patients were enrolled; all experienced at least one adverse event (AE). Grade ≥ 3 AEs, serious AEs, and AEs leading to death were reported in 50 (72.5%), 21 (30.4%), and 2 (2.9%) patients, respectively. Six (8.7%) patients reported a DLT, and the MTD and recommended phase II pictilisib doses were established where possible. There was no pictilisib–paclitaxel drug–drug interaction. Two (3.4%) patients experienced complete responses, and 17 (29.3%) patients had partial responses.
Conclusions
Combining pictilisib with paclitaxel, with and without bevacizumab or trastuzumab, or letrozole, had a manageable safety profile in patients with locally recurrent or metastatic breast cancer. The combination had antitumor activity, and the additive effect of pictilisib supported further investigation in a randomized study.
Trial registration
ClinicalTrials.gov, NCT00960960. Registered on August 13, 2009.
Similar content being viewed by others


Background
Despite improvements in treatment outcomes for patients with metastatic breast cancer, there is a continued unmet need to improve therapies for this patient population. Breast cancer is a heterogeneous disease, and current treatment strategies are based on disease type. Hormone receptor-positive, recurrent, or stage IV breast cancer in postmenopausal women is often managed with endocrine therapies, such as the nonsteroidal aromatase inhibitor letrozole [1], whereas primary treatment options for patients with human epidermal growth factor receptor 2 (HER2)-negative locally recurrent or metastatic breast cancer include single-agent cytotoxic chemotherapeutic agents, such as paclitaxel [1, 2]. Addition of the monoclonal antibody bevacizumab, which blocks angiogenesis by inhibiting vascular endothelial growth factor A, to paclitaxel has been shown to improve progression-free survival (PFS) and objective response rate (ORR) in patients with first-line metastatic breast cancer [1, 3]. In patients with HER2-positive locally recurrent or metastatic breast cancer, therapies include the antibody–drug conjugate ado-trastuzumab emtansine and the combination of the monoclonal antibodies trastuzumab and pertuzumab, both of which target HER2, with either docetaxel or paclitaxel [1, 2].
The phosphatidylinositol 3-kinase (PI3K) signaling pathway is deregulated in a wide variety of cancers, including breast cancer [4,5,6], and plays a key role in cell growth, survival, and migration [7]. The PI3K lipid kinases are grouped according to substrate specificity, structure, and mechanism of action into three classes (IA, IB, II, and III) [8]. Activating mutations of the catalytic subunit of PI3K (phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha [PIK3CA]), which belongs to the class IA PI3K family, are frequently observed in breast cancer [9, 10], and approximately 35–45% of cases of hormone receptor-positive breast cancer harbor mutations in this gene [11, 12]. Preclinical data suggest that activation of the PI3K pathway, via mutation of PIK3CA, loss of phosphatase and tensin homolog (PTEN) expression, or HER2 overexpression, may promote resistance to antiestrogen therapy and hormonal independence in estrogen receptor (ER)-positive models of breast cancer [13,14,15]. In addition, results from three clinical trials suggest that inhibition of both the PI3K/mammalian target of rapamycin (mTOR) and estrogen-signaling pathways may provide improved efficacy compared with single-agent endocrine therapies [16,17,18]. Thus, inhibition of the PI3K pathway has emerged as a promising strategy for treatment of breast cancer.
Pictilisib (GDC-0941) is a potent and selective oral inhibitor of class I PI3K [19] that prevents the formation of phosphatidylinositol (3,4,5)-trisphosphate, a key component of the PI3K pathway, by binding to the adenosine triphosphate-binding pocket of PI3K [19]. Pictilisib is a pan-PI3K inhibitor that inhibits all four isoforms of class I PI3Ks (p110α, p110β, p110δ, and p110γ subunits) [19] with rapid absorption following oral administration and a dose-proportional pharmacokinetic (PK) profile [20]. Pan-PI3K inhibitors may be better suited to combination therapy than inhibitors of mammalian target of rapamycin complex 1/2, and there is evidence that their activity may not be restricted to tumor types with PIK3CA mutations [21]. In contrast, isoform-specific PI3K inhibitors such as alpelisib (BYL719) and taselisib (GDC-0032), which both selectively target PI3Kα [22, 23], offer the potential specifically to block their target while limiting toxicities associated with a broader inhibition [21].
In preclinical studies, pictilisib had antitumor activity in breast cancer models harboring PIK3CA mutations and/or amplification of HER2, although several models without these mutations were also sensitive to pictilisib treatment [24]. Pictilisib was found to increase the antitumor activity of taxanes, with an associated increase in apoptotic cell death, in multiple breast cancer xenografts [25] and, in combination with trastuzumab, synergistically inhibited cell proliferation and the PI3K signaling pathway in HER2-amplified breast cancer cell lines [26]. Pictilisib was also reported to inhibit the growth of activated human endothelial cells, suggesting the potential for antiangiogenic activity [27].
Single-agent pictilisib was well tolerated and showed evidence of antitumor activity in a phase I study of 60 patients with solid tumors at doses ≥ 100 mg [20]. In addition, the PK profile of single-agent pictilisib was dose-proportional, with a maximum tolerated dose (MTD) of 330 mg administered orally daily [20]. Several studies have investigated the effect of PI3K inhibition in patients with breast cancer and alterations of the PI3K pathway. The phase III BOLERO-2 trial demonstrated that the mTOR inhibitor everolimus, when combined with an aromatase inhibitor, improved PFS in hormone receptor-positive advanced breast cancer previously treated with nonsteroidal aromatase inhibitors [17], although there was no statistically significant improvement in overall survival [28].
This open-label, multischedule phase Ib study aimed to evaluate the safety and PK of pictilisib in combination with paclitaxel, with and without bevacizumab or trastuzumab, or letrozole, in patients with locally recurrent or metastatic breast cancer. In addition, we sought to establish a recommended phase II dose for each treatment combination regimen.
Methods
Patients
Eligible patients were ≥ 18 years with histologically or cytologically confirmed locally recurrent or metastatic adenocarcinoma of the breast. Inclusion criteria specified that patients had HER2-negative tumors, unless in the cohort that received trastuzumab, where all patients were required to have HER2-positive tumors and an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1. Patients who received letrozole were postmenopausal and required to have hormone receptor-positive disease. Adequate hematologic and end-organ function was required, in addition to disease measurable by Response Evaluation Criteria In Solid Tumors (RECIST) v1.0.
Patients who had received more than two prior chemotherapy regimens for locally recurrent or metastatic breast cancer were not eligible for inclusion in the arms that received pictilisib + paclitaxel ± bevacizumab or trastuzumab treatment (parts 1 and 2). Patients were eligible for enrollment in the pictilisib + letrozole arm (part 3) if they were currently receiving letrozole for the treatment of advanced or metastatic breast cancer, but they were excluded if they had received more than one prior chemotherapy regimen or more than two prior endocrine therapy regimens for locally recurrent or metastatic breast cancer. Patients with known hypersensitivity to paclitaxel were excluded.
Patients were not eligible for bevacizumab treatment if they had inadequately controlled hypertension, significant vascular disease within 6 months prior to the first dose of study treatment, history of hemoptysis within 1 month prior to the first dose of study treatment, or evidence of bleeding diathesis or significant coagulopathy. Patients were not eligible for trastuzumab treatment if they had a history of grade ≥ 3 hypersensitivity to the antibody, or grade ≥ 1 with the most recent trastuzumab infusion before study entry, or continued requirement for prolonged trastuzumab infusions (> 30 minutes) to prevent infusion-related reactions. Patients with a history of exposure to anthracyclines (cumulative doses > 500 mg doxorubicin, > 900 mg liposomal doxorubicin, > 900 mg epirubicin, > 120 mg mitoxantrone, and > 90 mg idarubicin; if another anthracycline or more than one anthracycline was used, the cumulative dose could not exceed the equivalent of 500 mg doxorubicin) and cardiopulmonary dysfunction were also excluded.
Study design and treatment
This was an open-label, multicenter, phase Ib dose escalation study (ClinicalTrials.gov registration number NCT00960960) performed in three parts. Parts 1 and 2 comprised two stages (a 3 + 3 dose escalation stage and a cohort expansion stage), with the dose escalation stage designed to evaluate the safety, tolerability, and PK of pictilisib in combination with paclitaxel, or with paclitaxel plus bevacizumab or trastuzumab. Part 3 had a 3 + 3 dose escalation enrollment design and assessed the combination of pictilisib and letrozole. Patients were assigned in the order in which they were enrolled.
In part 1, patients received oral pictilisib (at an initial dose of 60 mg) administered daily on days 1–21 of each 28-day cycle (“21 + 7” schedule) and 90 mg/m2 intravenous paclitaxel (cohort 1) or 90 mg/m2 intravenous paclitaxel plus 10 mg/kg intravenous bevacizumab (all subsequent cohorts). On study treatment days, pictilisib was administered prior to paclitaxel or bevacizumab. Paclitaxel was administered on days 1, 8, and 15 of each 28-day cycle, and bevacizumab was administered on days 1 and 15 of each 28-day cycle.
In part 2, patients received oral pictilisib (daily for 5 of 7 consecutive days [“5 + 2” schedule]) in combination with 90 mg/m2 intravenous paclitaxel. Once the MTD had been established, two additional arms were opened to determine the MTD for pictilisib in combination with paclitaxel plus 10 mg/kg intravenous bevacizumab or 2–4 mg/kg intravenous trastuzumab. The starting dose for pictilisib in combination with paclitaxel plus bevacizumab was at or below the MTD for pictilisib plus paclitaxel, whereas the starting dose for pictilisib in combination with paclitaxel plus trastuzumab was at least one dose level below the MTD for pictilisib plus paclitaxel alone. Paclitaxel was administered on days 1, 8, and 15 of each 28-day cycle; bevacizumab was administered on days 1 and 15 of each 28-day cycle; and trastuzumab was administered on days 1, 8, 15, and 22 of each 28-day cycle.
In part 3, patients were treated with 260 mg pictilisib plus 2.5 mg letrozole by continuous daily dosing in 28-day cycles.
Either the MTD or a lower dose was selected as the recommended phase II dose. This was dependent on both the MTD-defining dose-limiting toxicities (DLTs) and the adverse events (AEs) reported during the DLT observation period and beyond in all patients treated at a given dose. Study treatment was discontinued in patients who experienced disease progression or unacceptable toxicity or who were not compliant with the study protocol.
Tumor assessments were performed according to RECIST v1.0. In parts 1 and 2, assessments were performed at screening and at the end of cycles 2, 5, 8, and 11 and every three cycles thereafter for patients who were on the study for > 1 year. Objective responses were confirmed ≥ 4 weeks after the initial documentation. In part 3, assessments were performed at screening and on day 1 of cycle 4 (± 7 days) and on day 1 (± 7 days) every three cycles thereafter.
Safety assessment
AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.0 [29]. AEs were recorded until 30 days after the last dose of study treatment or until initiation of another anticancer therapy, whichever occurred first.
DLT was defined as one of the following AEs, if occurring during the DLT assessment window (following the first dose of pictilisib and including evaluations prior to dosing on day 1 of cycle 2) and considered by the investigator to be related to study treatment: grade ≥ 3 nonhematologic, nonhepatic major organ AE; grade ≥ 4 thrombocytopenia lasting > 48 hours or associated with clinically significant bleeding; grade ≥ 3 fasting hyperglycemia; grade ≥ 4 neutropenia lasting ≥ 7 days; grade ≥ 3 febrile neutropenia; grade ≥ 3 total bilirubin, alkaline phosphatase (ALP), or hepatic transaminases (alanine aminotransferase or aspartate aminotransferase); grade ≥ 2 lung diffusing capacity concomitant with a decrease of ≥ 20% from baseline.
AEs that were not considered DLTs included grade 3 nausea, vomiting, or diarrhea that resolved to grade ≤ 1 with optimal medical management within 3 days, grade 3 hypertension for patients receiving bevacizumab, grade 3 fasting hyperglycemia that resolved to grade ≤ 1 within 7 days (with or without antihyperglycemic therapy), and grade 3 fasting hyperglycemia within 3 days of glucocorticoid use. For patients with grade 1 hepatic transaminase levels at baseline, a hepatic transaminase elevation > 7.5 times the upper limit of normal (ULN) was considered a DLT. For patients with grade 1 ALP levels at baseline, an elevation > 7.5 times the ULN was considered a DLT.
The DLT assessment window followed the first dose of pictilisib and included evaluations prior to dosing on day 1 of cycle 2. The MTD was exceeded if a DLT was observed in at least one-third of patients or if greater than one-third of patients in a cohort missed ≥ 5 days of pictilisib for drug-related AEs.
PK analysis
Blood samples were collected after single and multiple doses of pictilisib, paclitaxel, and letrozole for PK evaluations. Plasma concentrations of pictilisib, paclitaxel, 6α-hydroxy paclitaxel (6α-OH-paclitaxel; cytochrome P450 2C8 [CYP2C8]-formed metabolite of paclitaxel), and letrozole were determined using validated liquid chromatography-tandem mass spectrometry (LC-MS/MS) methods, and PK parameters were estimated using noncompartmental analysis (WinNonlin 6.4; Pharsight, Mountain View, CA USA).
Outcomes
The primary endpoints for the treatment combinations of pictilisib with letrozole alone, paclitaxel alone, and paclitaxel in combination with bevacizumab or trastuzumab, were safety and tolerability, DLTs, MTD, and identification of a recommended phase II dosing regimen. The secondary endpoints were PK of pictilisib and preliminary antitumor activity (ORR, duration of response [DoR], and PFS). Exploratory objectives included exploration of the potential relationship between PI3K pathway alterations and antitumor activity, as well as identification of the potential role of polymorphisms in drug metabolism enzyme and transporter genes in the PK disposition, and/or response to pictilisib, standard-of-care chemotherapy regimens, or antiestrogen agents.
Biomarker assessments
Mutational analysis of PIK3CA was performed using RT-PCR assays as previously described [30]. Nucleotide substitutions in the amino acids E542 (K), E545 (A, G, D, or K), Q546 (E, K, L, or R), and H1047 (L, R, or Y) or the wild-type alleles were detected. PTEN expression was examined with immunohistochemistry as previously described (clone 138G6; Cell Signaling Technology, Danvers, MA, USA), and an H-score was assigned to each sample on the basis of the percentage of cells staining at four different levels of intensity (0, 1+, 2+, or 3+) [31].
Statistical methods
Final analysis was performed on cumulative clinical data collected until the last patient’s last visit. The efficacy-evaluable population, which was the basis for ORR analysis, was defined as treated patients with baseline measurable disease and at least one postbaseline tumor assessment, or discontinuation of the study due to disease progression or death within 30 days of treatment initiation. All analyses were based on the safety-evaluable population, which was defined as all enrolled patients who received any dose of pictilisib. This study was designed not with regard to explicit power and type I error considerations, but to obtain preliminary safety and PK information in this patient population. The data cutoff for all analyses was December 1, 2015.
Results
Patient characteristics
Overall, 69 patients were enrolled in the study (August 2009 to December 2015), with 20 patients in part 1 (pictilisib + paclitaxel ± bevacizumab), 18 patients in part 2A (pictilisib + paclitaxel), 15 patients in part 2B (pictilisib + paclitaxel + bevacizumab), 9 patients in part 2C (pictilisib + paclitaxel + trastuzumab), and 7 patients in part 3 (pictilisib + letrozole) (Fig. 1). At final analysis, all patients had discontinued study treatment because of an AE (21.7%), progressive disease (58.0%), physician decision (13.0%), patient decision (5.8%), or sponsor termination of the study (1.4%). Baseline characteristics were well balanced among treatment groups (Table 1). The median age of all patients was 54.0 years (range, 30–76 years), and the majority of patients (71.0%) had hormone receptor-positive disease.

Participant flow diagram. AE Adverse event, PD Progressive disease
Safety
The safety profile of all dosing regimens examined in this dose-finding trial was assessed. All patients experienced at least one AE (Table 2), and the most common AEs (reported in ≥ 30% of patients) were diarrhea (78.3%), nausea (62.3%), fatigue (59.4%), alopecia (52.2%), rash (50.7%), neutropenia (44.9%), stomatitis (37.7%), vomiting (33.3%), decreased appetite (33.3%), and cough (30.4%). The most common AEs related to any study drug (≥ 15% of patients) were diarrhea (75.4%), nausea (58.0%), fatigue (56.5%), alopecia (52.2%), rash (46.4%), neutropenia (44.9%), and stomatitis (36.2%) (Additional file 1: Table S1). The majority of patients experienced a grade ≥ 3 AE (n = 50; 72.5%) (Table 2), and the most common (in at least two patients) were neutropenia (n = 19), rash (n = 7), peripheral neuropathy (n = 4), hypophosphatemia (n = 3), decreased lung diffusing capacity (n = 3), dyspnea (n = 2), hypertension (n = 2), diarrhea (n = 2), nausea (n = 2), pneumonia (n = 2), increased blood glucose (n = 2), decreased appetite (n = 2), pulmonary embolism (n = 2), nail disorder (n = 2), and deep vein thrombosis (n = 2). In addition, most patients (n = 43; 62.3%) experienced at least one grade ≥ 3 AE related to any study drug (Additional file 1: Table S1). Overall, serious AEs were reported in 30.4% of patients (n = 21) (Table 2 and Additional file 1: Table S2) and those reported in at least two patients included pneumonia (n = 2), nausea (n = 2), decreased lung diffusing capacity (n = 2), and pulmonary embolism (n = 2).
Fifteen patients (21.7%) had an AE that led to discontinuation of any study drug (Table 2), and the most common were peripheral neuropathy (n = 5), decreased lung diffusing capacity (n = 4), rash (n = 3), neutropenia (n = 2), paresthesia (n = 2), pulmonary embolism (n = 2), deep vein thrombosis (n = 2), and hypertension (n = 2). In the case of pictilisib, 18 patients (26.1%) experienced AEs leading to withdrawal (Table 2) and those in at least two patients were decreased lung diffusing capacity (n = 4), rash (n = 3), and deep vein thrombosis (n = 2). AEs that led to pictilisib dose reduction included grade 2 neutropenia (n = 3) and grades 1 and 3 rash (n = 1 and n = 2, respectively). Thirty-nine (56.5%) patients had their pictilisib dose interrupted owing to an AE, whereas six patients (8.7%) had their dose reduced (Table 2). Withdrawal of paclitaxel, bevacizumab, and trastuzumab occurred in 21 patients (33.9%), 14 patients (40.0%), and two patients (22.2%), respectively (Table 2).
AEs of special interest included pneumonitis (3 [4.3%] patients), hyperglycemia or increased blood glucose (15 [21.7%] patients), left ventricular dysfunction (1 [1.4%] patient), and decreased carbon monoxide-diffusing capacity (5 [7.2%] patients) (Additional file 1: Table S3). Grade ≥ 3 AEs of special interest in these patients were reported for hyperglycemia or increased blood glucose (4 [5.8%] patients), left ventricular dysfunction (1 [1.4%] patient), and decreased carbon monoxide-diffusing capacity (3 [4.3%] patients) (Additional file 1: Table S3).
Two patients (2.9%) experienced AEs that led to a fatal outcome (Table 2). One patient had grade 5 left ventricular dysfunction, considered by the investigator to be related to pictilisib, bevacizumab, and paclitaxel. The other patient experienced grade 5 worsened ECOG PS, which was considered by the investigator to be related to pictilisib and unrelated to letrozole.
Overall, six patients (8.7%) reported DLTs (Table 2 and Additional file 1: Table S4). In part 1, one DLT was reported with 60 mg pictilisib, whereas none were observed with the 100-mg dose (pictilisib administered on the “21 + 7” dosing schedule and in combination with paclitaxel and bevacizumab). In part 2A, one DLT was observed in a patient treated with 250 mg pictilisib (“5 + 2” dosing schedule and in combination with paclitaxel), whereas there were two DLTs at the next dose level (330 mg pictilisib); thus, the MTD was exceeded. In part 2B, there was one reported DLT in a patient treated with 250 mg pictilisib (“5 + 2” dosing schedule and administered in combination with paclitaxel and bevacizumab), whereas in part 2C a DLT was observed in one patient treated with 260 mg pictilisib (“5 + 2” dosing schedule and administered in combination with paclitaxel plus trastuzumab). There were no DLTs reported in part 3 (260 mg pictilisib administered continuously with letrozole).
The MTD was defined in part 2A as 250 mg pictilisib (“5 + 2” dosing schedule) in combination with paclitaxel and was not established in all other arms. The MTD or maximum administered dose and recommended phase II doses of pictilisib were 100 mg (when administered with paclitaxel and bevacizumab [“21 + 7” dosing schedule]), 250 mg (when administered with paclitaxel or paclitaxel plus bevacizumab [“5 + 2” dosing schedule]), or 260 mg (when administered with paclitaxel and trastuzumab [“5 + 2” dosing schedule] or letrozole [continuous dosing schedule]).
PK analysis
In vitro data suggest that pictilisib has a moderate potential to inhibit the CYP2C8-mediated metabolism of paclitaxel to 6α-OH-paclitaxel. In this study, a consistent 6α-OH-paclitaxel:paclitaxel AUC ratio was observed across all pictilisib dose levels (Fig. 2), suggesting there was no drug–drug interaction between pictilisib and paclitaxel. In addition, no differences in the PK of pictilisib or letrozole were observed in any of the treatment combination regimens compared with historical single-agent data (data not shown).

Plasma 6α-OH-paclitaxel/paclitaxel AUC ratio as a function of pictilisib dose. Patients with evaluable 6α-OH-paclitaxel and paclitaxel PK after multiple doses of paclitaxel and pictilisib were pooled across all paclitaxel treatment arms (parts 1 and 2, n = 49). Black lines represent the median ratio for each dose level, and dots represent individual subject ratios. PK Pharmacokinetics
Clinical activity
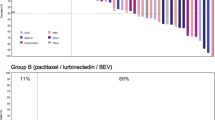
Fifty-eight (84.1%) patients were included in the efficacy-evaluable population for ORR analysis. Complete responses were observed in two patients (3.4%) overall, in parts 1 (5.3%; paclitaxel + bevacizumab) and 2A (5.9%; pictilisib + paclitaxel) (Table 3). Partial responses were observed in patients treated with pictilisib + paclitaxel ± bevacizumab (part 1; 21.1%), pictilisib + paclitaxel (part 2A; 17.6%), pictilisib + paclitaxel + bevacizumab (part 2B; 53.8%), pictilisib + paclitaxel + trastuzumab (part 2C; 33.3%), and pictilisib + letrozole (part 3; 33.3%) (Table 3). The majority of patients showed signs of tumor shrinkage (Fig. 3a–d).

Waterfall plot of maximum percentage changes from baseline in SLD for target lesions. Maximum percentage changes are shown in (a) part 1 (pictilisib + paclitaxel ± bevacizumab), (b) parts 2A and B (2A: pictilisib + paclitaxel; 2B: pictilisib + paclitaxel + bevacizumab), (c) part 2C (pictilisib + paclitaxel + trastuzumab), and (d) part 3 (pictilisib + letrozole). PTEN categories were defined as PTEN loss (PTEN H-score ≥ 0 but ≤ 100) or nonloss (PTEN H-score > 100). CR Complete response, MND Mutation not detectable, PD Progressive disease, PIK3CA Phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit-alpha, PR Partial response, PTEN Phosphatase and tensin homolog, SD Stable disease, SLD Sum of the longest diameters, UE Unevaluable
The median DoR was 8.9 months (95% CI, 6.47–11.10) among five responders treated with pictilisib + paclitaxel ± bevacizumab (part 1) and 8.8 months (95% CI, 4.40–15.34) among seven responders treated with pictilisib + paclitaxel + bevacizumab (part 2B) (Table 3).
In all treated patients (N = 69), median PFS ranged from 5.0 months (95% CI, 3.71–NE) in patients treated with pictilisib + paclitaxel (part 2A; n = 18) to 14.8 months (95% CI, 3.52–16.62) in patients treated with pictilisib + paclitaxel + trastuzumab (part 2C; n = 9) (Table 3).
PIK3CA mutation status and PTEN expression were evaluated in 24 formalin-fixed, paraffin-embedded tumor samples from parts 2A, 2B and 2C. Four of the 24 samples were evaluable for PTEN only (n = 2) or PIK3CA only (n = 2). PTEN expression was reduced or absent in five of the 22 samples examined, whereas 10 of 22 samples harbored a PIK3CA mutation. Among the patients in part 2A and 2B with evaluable tissue and either a PIK3CA mutation or PTEN loss, five of 11 (45.5%) had a complete or partial response compared with two of seven (28.6%) patients without these alterations (Fig. 3b).
Discussion
This phase Ib study evaluated the safety and PK of the pan-PI3K inhibitor pictilisib in combination with paclitaxel, with and without bevacizumab or trastuzumab, or letrozole, in patients with locally recurrent or metastatic breast cancer. At a dose of 260 mg, pictilisib had a manageable safety profile, when combined with paclitaxel, with and without bevacizumab or trastuzumab (“21 + 7” or “5 + 2” dosing schedules) or in combination with 2.5 mg letrozole. The MTD of pictilisib was exceeded in patients treated with 330 mg pictilisib plus paclitaxel. Antitumor activity was observed across all treatment arms. The PK analysis suggested no evidence of a clinical drug–drug interaction between agents in each of the evaluated treatment combination regimens. Taking the potential benefit–risk balance into consideration, 260 mg pictilisib was selected as the recommended phase II dose.
Previous clinical trials of pictilisib have reported inconsistent results. The first-in-human phase I trial of single-agent pictilisib in patients with advanced solid tumors demonstrated evidence of antitumor activity in patients with gastrointestinal stromal tumors, cervical cancer, melanoma, colorectal cancer, cholangiocarcinoma, breast cancer, and ovarian cancer, and it showed that pictilisib was well tolerated [20]. Addition of pictilisib to anastrozole in patients with ER-positive, HER2-negative early breast cancer in the OPPORTUNE study significantly decreased tumor cell proliferation [32]. The randomized phase II PEGGY trial (NCT01740336) did not show any benefit from the addition of pictilisib to paclitaxel in patients with hormone receptor-positive, HER2-negative locally recurrent or metastatic breast cancer [33]. Moreover, a randomized phase II trial (FERGI; NCT01437566) in patients with ER-positive, HER2-negative, endocrine-resistant breast cancer found that the addition of pictilisib to fulvestrant did not significantly improve PFS [34]. Although the phase III BELLE-2 study (NCT01610284) reported modest improvements in median PFS (1.9 months) in patients with hormone receptor-positive metastatic breast cancer treated with the pan-PI3K inhibitor buparlisib (BKM120) in combination with fulvestrant (versus placebo plus fulvestrant) [35], combining a PI3K inhibitor with different therapies is challenging. In both the PEGGY and FERGI studies, efficacy was likely limited by the higher incidence of AEs and dose reductions/discontinuations owing to AEs with pictilisib treatment [33, 34]. As a result, further development of pictilisib by the sponsor is not planned.
Exploratory biomarker analyses in this study showed a numerical difference in ORR between tumors that harbored a PIK3CA mutation or had PTEN loss (45.5%) and those without these alterations (28.6%); however, this study was not powered to distinguish antitumor activity between these patient groups, and results were interpreted with caution owing to small patient numbers. Previous studies found little evidence for a link between PIK3CA mutations and antitumor activity with pictilisib [32,33,34].
Although there was evidence of antitumor activity with pictilisib in this patient population, the sample size was limited. Thus, because efficacy was not the primary endpoint and the study was not powered to detect meaningful differences between study arms, the conclusions of the observed antitumor activity are limited.
Targeting the PI3K pathway with isoform-specific inhibitors may decrease dose modifications caused by toxicity. Alpelisib showed some single-agent activity, with a favorable safety profile, in patients with PIK3CA-mutant advanced breast cancer [36]. In addition, alpelisib in combination with fulvestrant demonstrated clinical activity in patients with ER-positive, PIK3CA-mutated, locally advanced or metastatic breast cancer [37], whereas alpelisib in combination with letrozole was well tolerated, with evidence of clinical activity in patients with ER-positive metastatic breast cancer that was refractory to endocrine therapy [38]. Taselisib, a potent and selective PI3K inhibitor that has greater selectivity for mutant PI3Kα isoforms than wild-type PI3Kα [23, 39, 40], has single-agent activity in tumors with PIK3CA mutations [41]. Thus, taselisib is currently being evaluated in combination with fulvestrant in postmenopausal women with ER-positive, HER2-negative, PIK3CA-mutant, locally advanced or metastatic breast cancer (ClinicalTrials.gov identifier NCT02340221) [42].
Conclusions
The combination of pictilisib with paclitaxel, with and without bevacizumab or trastuzumab, or letrozole, in this phase Ib study had an acceptable safety profile with manageable toxicities, with evidence of antitumor activity in patients with locally recurrent or metastatic breast cancer. The effect of pictilisib in combination with paclitaxel supported further investigation in a randomized clinical study.
Abbreviations
- 6α-OH-paclitaxel:
-
6α-Hydroxy paclitaxel
- AE:
-
Adverse event
- ALP:
-
Alkaline phosphatase
- CR:
-
Complete response
- DLT:
-
Dose-limiting toxicities
- DoR:
-
Duration of response
- ECOG PS:
-
Eastern Cooperative Oncology Group Performance Status
- ER:
-
Estrogen receptor
- HER2:
-
Human epidermal growth factor receptor 2
- MND:
-
Mutation not detectable
- MTD:
-
Maximum tolerated dose
- mTOR:
-
Mammalian target of rapamycin
- NE:
-
Nonevaluable
- ORR:
-
Objective response rate
- PD:
-
Progressive disease
- PFS:
-
Progression-free survival
- PI3K:
-
Phosphatidylinositol 3-kinase
- PIK3CA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha
- PK:
-
Pharmacokinetics
- PR:
-
Partial response
- PTEN:
-
Phosphatase and tensin homolog
- RECIST:
-
Response Evaluation Criteria In Solid Tumors
- SAE:
-
Serious adverse event
- SD:
-
Stable disease
- SLD:
-
Sum of the longest diameters
- UE:
-
Unevaluable
- ULN:
-
Upper limit of normal
References
National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®): Breast Cancer. Version 2. 2016. https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed 14 Oct 2016.
Cardoso F, Costa A, Norton L, Senkus E, Aapro M, André F, et al. ESO-ESMO 2nd International Consensus Guidelines for Advanced Breast Cancer (ABC2). Breast. 2014;23(5):489–502.
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–76.
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–510.
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–62.
Ma CX, Ellis MJ. The Cancer Genome Atlas: clinical applications for breast cancer. Oncology (Williston Park). 2013;27(12):1263–9. 1274–1279
Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–7.
Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22(7)):267–72.
Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3(8):772–5.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304(5670):554.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68(15):6084–91.
Arthur LM, Turnbull AK, Renshaw L, Keys J, Thomas JS, Wilson TR, et al. Changes in PIK3CA mutation status are not associated with recurrence, metastatic disease or progression in endocrine-treated breast cancer. Breast Cancer Res Treat. 2014;147(1):211–9.
Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35.
Miller TW, Perez-Torres M, Narasanna A, Guix M, Stal O, Perez-Tenorio G, et al. Loss of phosphatase and tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009;69(10):4192–201.
Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–61.
Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27(16):2630–7.
Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366(6):520–9.
Bachelot T, McCool R, Duffy S, Glanville J, Varley D, Fleetwood K, et al. Comparative efficacy of everolimus plus exemestane versus fulvestrant for hormone-receptor-positive advanced breast cancer following progression/recurrence after endocrine therapy: a network meta-analysis. Breast Cancer Res Treat. 2014;143(1):125–33.
Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51(18)):5522–32.
Sarker D, Ang JE, Baird R, Kristeleit R, Shah K, Moreno V, et al. First-in-human phase I study of pictilisib (GDC-0941), a potent pan-class I phosphatidylinositol-3-kinase (PI3K) inhibitor, in patients with advanced solid tumors. Clin Cancer Res. 2015;21(1):77–86.
Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13(5):1021–31.
Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014;13(5):1117–29.
Olivero AG, Heffron TP, Baumgardner M, Belvin M, Ross LB, Blaquiere N, et al. Discovery of GDC-0032: a beta-sparing PI3K inhibitor active against PIK3CA mutant tumors [abstract]. Cancer Res. 2013;73(8 Suppl)):Abstract DDT02–1.
O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16(14)):3670–83. A published erratum appears in Clin Cancer Res. 2011;17(7):2066–7
Wallin JJ, Guan J, Prior WW, Lee LB, Berry L, Belmont LD, et al. GDC-0941, a novel class I selective PI3K inhibitor, enhances the efficacy of docetaxel in human breast cancer models by increasing cell death in vitro and in vivo. Clin Cancer Res. 2012;18(14):3901–11.
Junttila TT, Akita RW, Parsons K, Fields C, Phillips GDL, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–40.
Raynaud FI, Eccles SA, Patel S, Alix S, Box G, Chuckowree I, et al. Biological properties of potent inhibitors of class I phosphatidylinositide 3-kinases: from PI-103 through PI-540, PI-620 to the oral agent GDC-0941. Mol Cancer Ther. 2009;8(7):1725–38.
Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014;25(12):2357–62.
U.S. Department of Health and Human Services, National Institutes of Health (NIH), National Cancer Institute (NCI). Common Terminology Criteria for Adverse Events (CTCAE). Version 3.0. 9 Aug 2006. https://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 14 Oct 2016.
Patel R, Tsan A, Tam R, Desai R, Spoerke J, Schoenbrunner N, et al. Mutation scanning using MUT-MAP, a high-throughput, microfluidic chip-based, multi-analyte panel. PLoS One. 2012;7:e51153.
Spoerke JM, O’Brien C, Huw L, Koeppen H, Fridlyand J, Brachmann RK, et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res. 2012;18(24):6771–83.
Schmid P, Pinder SE, Wheatley D, Macaskill J, Zammit C, Hu J, et al. Preoperative window of opportunity study of the PI3K inhibitor pictilisib (GDC-0941) plus anastrozole vs anastrozole alone in patients with ER+, HER2-negative operable breast cancer (OPPORTUNE study) [abstract]. Cancer Res. 2015;75(9 Suppl):Abstract S2–03.
Vuylsteke P, Huizing M, Petrakova K, Roylance R, Laing R, Chan S, et al. Pictilisib PI3Kinase inhibitor (a phosphatidylinositol 3-kinase [PI3K] inhibitor) plus paclitaxel for the treatment of hormone receptor-positive, HER2-negative, locally recurrent, or metastatic breast cancer: interim analysis of the multicentre, placebo-controlled, phase II randomised PEGGY study. Ann Oncol. 2016;27:2059–66.
Krop IE, Mayer IA, Ganju V, Dickler M, Johnston S, Morales S, et al. Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016;17:811–21.
Baselga J, Im S, Baselga J, Im SA, Iwata H, Cortés J, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:904–16.
Janku F, Juric D, Cortes J, Rugo H, Burris HA, Schuler M, et al. Phase I study of the PI3Kα inhibitor BYL719 plus fulvestrant in patients with PIK3CA-altered and wild type ER+/HER2-locally advanced or metastatic breast cancer [abstract]. Cancer Res. 2015;75(9 Suppl)):Abstract PD5.
Juric D, Gonzalez-Angulo AM, Burris HA, Schuler M, Schellens J, Berlin J, et al. Preliminary safety, pharmacokinetics and anti-tumor activity of BYL719, an alpha-specific PI3K inhibitor in combination with fulvestrant: results from a phase I study. Cancer Res. 2013;73(24 Suppl):Abstract P2–16-14.
Mayer IA, Abramson VG, Formisano L, Balko JM, Estrada MV, Sanders ME, et al. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER /HER2-negative metastatic breast cancer. Clin Cancer Res. 2017;23(1):26–34.
Wallin JJ, Edgar KA, Guan J, Sampath D, Nannini M, Belvin M, et al. The PI3K inhibitor GDC-0032 is selectively potent against PIK3CA mutant breast cancer cell lines and tumors [abstract]. Cancer Res. 2013;73(24 Suppl)):Abstract P2–17-01.
Edgar KA, Song K, Schmidt S, Kirkpatrick DS, Phu L, Nannini M, et al. The PI3K inhibitor, taselisib (GDC-0032), has enhanced potency in PIK3CA mutant models through a unique mechanism of action [abstract]. Cancer Res. 2016;76(14 Suppl):Abstract 370.
Juric D, Krop I, Ramanathan RK, Xiao J, Sanabria S, Wilson TR, et al. GDC-0032, a beta isoform-sparing PI3K inhibitor: results of a first-in-human phase Ia dose escalation study [abstract]. Cancer Res. 2013;73(8 Suppl):Abstract LB-64.
Baselga J, Cortes J, De Laurentiis M, Diéras V, Harbeck N, Hsu JY, et al. SANDPIPER: phase III study of the PI3-kinase (PI3K) inhibitor taselisib (GDC-0032) plus fulvestrant in patients (pts) with oestrogen receptor (ER)-positive, HER2-negative locally advanced or metastatic breast cancer (BC) enriched for pts with PIK3CA mutant tumors [abstract]. Ann Oncol. 2016;27(Suppl 6):Abstract 313TiP.
Schöffski P, Cresta S, Mayer IA, Wildiers H, Rooney I, Apt D, Gendreau S, Morrissey K, Lackner M, Spoerke J, Winer E. Tolerability and anti-tumor activity of the oral PI3K inhibitor GDC-0941 in combination with paclitaxel, with and without bevacizumab or trastuzumab in patients with locally recurrent or metastatic breast cancer [abstract]. Cancer Res. 2015;75(9 Suppl):P5-19-10.
Acknowledgements
We thank the patients, their families, the nurses, and the investigators who participated in this study. Support for third-party writing assistance, furnished by Islay Steele, PhD, of Health Interactions, was provided by F. Hoffmann-La Roche Ltd.
Funding
This study was funded by Genentech Inc./F. Hoffmann-La Roche Ltd.
Availability of data and materials
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://www.clinicalstudydatarequest.com). Further details on Roche's criteria for eligible studies are available here (https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Disclosure of prior publication
These analyses were presented previously in part in abstract form [43].
Author information
Authors and Affiliations
Contributions
PS, SG, and KMM were involved in the conception and design of the study. SG, JMS, and SMS were involved in development of the methodology used. PS, SC, IAM, HW, SD, and EW were involved in acquisition of data (provided animals, acquired and managed patients, provided facilities). PS, HW, SG, KMM, VWN, and SMS were involved in analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis). SG, IR, JMS, VWN, and SMS were involved in administrative, technical, or material support (i.e., reporting or organizing data, constructing databases). SMS and EW were involved in study supervision. All authors were involved in the writing, review, and/or revision of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with good clinical practice guidelines and the Declaration of Helsinki. Approval of the protocol and any accompanying material provided to the patients was obtained from independent ethics committees at participating institutions (Table 4). All patients provided written informed consent.
Consent for publication
Not applicable.
Competing interests
PS has received travel support from Roche for presentation of parts of this work at the 37th San Antonio Breast Cancer Symposium in 2014, institutional research support from Genentech for exploring pictilisib in patient-derived xenograft models of sarcoma, and institutional technical support from Roche (donation of a LightCycler). In addition, and outside the submitted work, PS has received honoraria (institutional support) from Daiichi Sankyo, Eisai, Eli Lilly, Medpace, Novartis, and Swedish Orphan Biovitrium; consulting or advisory role institutional support from Sixth Element Capital, Adaptimmune, Amcure, AstraZeneca, Bayer, Blueprint Medicines, BMS, Boehringer Ingelheim, Cristal Therapeutics, Daiichi Sankyo, Eisai, Eli Lilly, Epizyme, Genzyme, Ipsen, Loxo Oncology, Medpace, Nektar, Novartis, Philogen, PIQUR Therapeutics, and Plexxikon; speaker’s bureau institutional support from Bayer, Eisai, Eli Lilly, GSK, Novartis, PharmaMar, and Swedish Orphan Biovitrium; research funding (institutional support) from Bayer, Blueprint Medicines, CoBioRes NV, Exelixis, GSK, Novartis, and Plexxikon; and institutional support for travel, accommodation, and expenses from Sixth Element Capital, Adaptimmune, Amcure, AstraZeneca, Bayer, Blueprint Medicines, BMS, Boehringer Ingelheim, Cristal Therapeutics, Daiichi Sankyo, Eisai, Eli Lilly, Epizyme, Genzyme, GSK, Ipsen, Loxo Oncology, Medpace, Nektar, Novartis, PharmaMar, Philogen, PIQUR Therapeutics, Plexxikon, and Swedish Orphan Biovitrium. IAM has received research support from Novartis and Pfizer and advisory board honoraria from Novartis. HW has received advisory board honoraria, speaker‘s fees, and travel support from Roche. SG, IR, KMM, JMS, VWN, and SMS are employees of Genentech/Roche. EW has received research support from Genentech, an advisory board honorarium from Genentech, and scientific advisory board support from Leap Pharmaceuticals. SC and SD declare that they have no potential conflicts of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. All-grade AEs related to any study drug occurring in ≥ 15% of all patients and corresponding grade ≥ 3 AEs. Table S2. SAEs related to any study drug (regardless of causality). Table S3. AEs and grade ≥ 3 AEs of special interest (regardless of causality). Table S4. Summary of DLTs observed during the study. (DOCX 66 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Schöffski, P., Cresta, S., Mayer, I.A. et al. A phase Ib study of pictilisib (GDC-0941) in combination with paclitaxel, with and without bevacizumab or trastuzumab, and with letrozole in advanced breast cancer. Breast Cancer Res 20, 109 (2018). https://doi.org/10.1186/s13058-018-1015-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13058-018-1015-x