
Abstract
A drug conjugate consists of a cytotoxic drug bound via a linker to a targeted ligand, allowing the targeted delivery of the drug to one or more tumor sites. This approach simultaneously reduces drug toxicity and increases efficacy, with a powerful combination of efficient killing and precise targeting. Antibody‒drug conjugates (ADCs) are the best-known type of drug conjugate, combining the specificity of antibodies with the cytotoxicity of chemotherapeutic drugs to reduce adverse reactions by preferentially targeting the payload to the tumor. The structure of ADCs has also provided inspiration for the development of additional drug conjugates. In recent years, drug conjugates such as ADCs, peptide‒drug conjugates (PDCs) and radionuclide drug conjugates (RDCs) have been approved by the Food and Drug Administration (FDA). The scope and application of drug conjugates have been expanding, including combination therapy and precise drug delivery, and a variety of new conjugation technology concepts have emerged. Additionally, new conjugation technology-based drugs have been developed in industry. In addition to chemotherapy, targeted therapy and immunotherapy, drug conjugate therapy has undergone continuous development and made significant progress in treating lung cancer in recent years, offering a promising strategy for the treatment of this disease. In this review, we discuss recent advances in the use of drug conjugates for lung cancer treatment, including structure-based drug design, mechanisms of action, clinical trials, and side effects. Furthermore, challenges, potential approaches and future prospects are presented.
Similar content being viewed by others
Introduction
Current status of lung cancer treatment
Cancer is a major public health problem worldwide [1,2,3]. Lung cancer, one of the most prevalent tumors with the highest mortality rate, originates in the trachea, bronchus and lungs, and its prevalence threatens human health [4, 5]. Systemic lung cancer therapy includes surgery, radiation and chemotherapy [6]. Surgery is used to remove some or all of the tumor tissue at the local site [7]. Radiation therapy has also been used to treat local tumors [8]. However, these approaches are ineffective in treating metastatic cancer, and the tumor cells cannot be completely removed. Many patients experience relapse after a short time following these treatments. Chemotherapy is the conventional treatment for lung cancer and provides certain benefits to patients [9]. However, chemotherapy lacks selectivity and can inhibit tumor cell growth and kill large numbers of normal cells, resulting in destruction of the immune system, many side effects and the rapid development of drug resistance [10, 11]. Chemotherapeutic drugs usually have a low therapeutic index and severe side effects, even after long-term development. Compared with traditional cytotoxic drugs, targeted therapy has higher efficacy and greater tolerability [12]. Lung cancer treatment has entered the era of precision targeted therapy. Typical molecular targeted therapy, represented by tyrosine kinase inhibitors (TKIs) of epithelial growth factor receptor (EGFR), has completely changed the treatment options for NSCLC [13]. Targeted therapy, represented by selective EGFR-TKIs, has great importance because it not only effectively inhibits tumor growth but also has fewer side effects than chemotherapy [14,15,16]. Although standard-of-care drugs, e.g., EGFR-TKIs, achieve a relatively high initial response in lung cancer, resistance inevitably develops after 9–12 months of treatment [17,18,19,20,21,22,23]. Immunotherapy, a novel treatment method that utilizes the human immune system to inhibit cancer cell growth, has received much attention in recent years. Immunotherapy does not directly interact with cancer cells but activates the immune system to eliminate tumors, thereby effectively treating lung cancer [24,25,26,27,28,29]. However, immunotherapy may easily cause side effects such as autoimmune disorders. Due to the instability of the tumor cell genome, the effectiveness of immunotherapy may vary from person to person. Therefore, developing drugs that combine the advantages of strong targeting and high toxin activity, which can simultaneously reduce toxic side effects and improve antitumor effects, has become a promising strategy for the treatment of lung cancer. Drug conjugates exhibit the above characteristics because they consist of cytotoxic drugs bound to targeted ligands via linkers, enabling targeted delivery of the drug to tumor sites. ADCs are the best-known drug conjugates and typically consist of a monoclonal antibody (mAb) bound to a payload via a linker. This construction combines the specificity of antibodies with the cytotoxicity of chemotherapeutic drugs, potentially reducing the severity of adverse reactions by preferentially targeting the payload to the tumor site [30]. For example, when ADCs enter the bloodstream, the antibody component can recognize the target and thus bind to lung tumor cells and enter through endocytosis. The cytotoxic drug is then released to kill tumor cells. In addition, the emergence and widespread use of drug conjugates may lead to the development of alternative approaches to overcoming TKI resistance [31,32,33].
Developmental history of ADCs and other drug conjugates
Currently, there are many cytotoxic drugs in clinical use that can effectively kill tumor cells, but they cause numerous adverse reactions due to their lack of tumor targeting, which limits their clinical application. Therefore, instead of exploring and developing additional cytotoxic drugs, repurposing the existing nonspecific cytotoxic drugs into targeted chemotherapeutic drugs is highly important for tumor treatment. One hundred years ago, Paul Ehrlich first proposed the concept of “magic bullets”: compounds that could directly bind cancer cells [34, 35], thereby curing disease. Such compounds should be effective at killing tumors but harmless to normal cells [36]. At that time, however, research progress on antibodies was subject to technological limitations [37, 38]. In 1975, Köhler and Milstein introduced hybridoma technology, enabling the production of mAbs for therapeutic purposes [39, 40]. The research and development process present many challenges throughout [38]. For example, the molecular weight of ADCs is much greater than that of other drugs, and the ability of these drugs to penetrate the cell membrane of tumor cells is limited. A recent study showed that only a small number of the ADCs that are injected into patients can ultimately reach tumor cells. Problems with delivery, antibody specificity and antibody homology have hampered the development of ADCs. As technology continues to advance, the heterogeneity and instability of ADCs remain problematic. Moreover, mAbs that originate in mice usually have immunogenicity in the human body [41,42,43]. Differences between species, including differences in target structure, function, distribution, and expression levels, as well as differences in immune system function, can cause qualitative and quantitative differences in the biological responses of antibodies in experimental animals and humans. As technology advances, the selection of genetically modified animals that can express human target proteins, the use of homologous substitute antibodies, and the use of human cells or tissues for in vitro experiments have promoted the development of drugs. At present, ADCs are no longer rare, and new technologies and drug conjugate forms have emerged [44]. mAb drugs are playing an increasingly important role in cancer treatment due to their excellent targeting specificity [45, 46]. The emergence of DNA recombination technology has enabled scientists to produce engineered antibodies, leading to the development of human–mouse chimeric antibodies, humanized antibodies and fully humanized antibodies, which have overcome the problem of immunogenicity [47]. Since then, many mAbs targeting various anticancer antigens have been developed as alternatives to traditional cancer chemotherapy [48]. Currently, ADCs play a unique role in anticancer drug treatment and cannot be overlooked [49]. Gemtuzumab ozogamicin was the first ADC approved in 2000 by the FDA for the treatment of CD33-positive acute myeloid leukemia (AML) [50]. By February 2023, 12 ADCs had been approved by the FDA, 6 for hematological malignancies [51]. In addition to approved ADCs, more than 140 ADCs are currently in clinical trials for cancer treatment, reflecting and inspiring industry-wide interest in this modality [44, 52]. The successful application of ADCs has increased the enthusiasm of scientists for developing novel ADCs. However, ADCs present problems such as high molecular weight, high immunogenicity and complex antibody production processes. With the continuous progress of chemical conjugation, protein genetic engineering and other technologies, the field of targeted delivery of drug conjugates is not limited to ADCs, and various types of drug conjugates have been generated. The “formula” for the success of antibody‒drug conjugates, that is, a carrier that targets tumors, a substance that kills tumor cells and a linker that connects the first two, has provided inspiration for additional conjugated drugs. For example, a peptide‒drug conjugate (PDC) comprises a homing peptide, cytotoxin and linker [53]. In radionuclide drug conjugates (RDCs), another innovative form of medical imaging and treatment, tumor antigen-specific targeting antibodies or small molecules are connected by linkers to radioisotopes (both imaging and radiokilling), which enables accurately guided radionuclide delivery to tumors for diagnosis or treatment. Other examples include small molecule–drug conjugates (SMDCs), virus-like drug conjugates (VDCs), antibody–oligonucleotide conjugates (AOCs), antibody–cell conjugates (ACCs), immune-stimulating antibody conjugates (ISACs), antibody fragment–drug conjugates (FDCs), antibody–degrader conjugates (ADeCs), and aptamer–drug conjugates (ApDCs). In recent years, many drug conjugates have been approved by the FDA for treatment and diagnosis (Table 1). The development of ADCs and other drug conjugates from infancy to maturity over the past 100 years is depicted in Fig. 1, including advances in tumor cell-killing substances, linkers, and tumor-targeting carriers such as peptides and radioisotopes.

The history of drug conjugates. A History of ADC development. B History of peptide development. C History of nuclear medicine development. ADC antibody‒drug conjugate, FDA Food and Drug Administration, RDC radionuclide drug conjugate
ADCs in combination with other anticancer therapies
ADCs can provide survival benefits for patients. However, most patients develop resistance to ADCs and do not achieve long-lasting cancer control. Thus, ADC treatment is insufficient for many tumor types, and many ADCs are being tested in clinical trials as part of combination therapies [54]. Combining therapeutic agents may increase the likelihood of complete remission and cure [55]. The positive therapeutic effects of combination therapy have inspired the development of the next generation of ADCs in the pharmaceutical industry worldwide, and many preclinical studies and clinical trials of ADCs in combination with other anticancer drugs have been conducted [32] (Fig. 2).

Standard-of-care treatments and combination therapy involving ADCs in lung cancer. ADC antibody‒drug conjugate, VEGFR vascular endothelial growth factor receptor
Chemotherapy and ADCs act synergistically by increasing surface-antigen expression and blocking the cell cycle. Most chemotherapeutic drugs target the S phase of the cell cycle and induce G2/M arrest [56, 57]. In past trials investigating the synergistic effects of ADCs in combination with doxorubicin or carboplatin, encouraging treatment responses were observed in both platinum-sensitive and platinum-resistant ovarian cancer patients [58, 59]. The surface antigen expression of cancer cells can be affected by chemotherapy: for example, gemcitabine can upregulate HER2 expression 14.81-fold, and G2/M phase pancreatic adenocarcinoma cells are more sensitive to gemcitabine, which corresponds to a greater likelihood of gemcitabine effectively binding with trastuzumab emtansine (T-DM1) [60]. In addition, the toxicity increased when ADCs were combined with chemotherapy due to the overlap of the off-target and off-tumor effects of the payloads. Endocrine therapy is a common therapeutic approach for hormone-sensitive cancers [61]. The possibility of combining T-DXd therapy with endocrine therapy has been proven in patients with low-HER2 breast cancer at any stage [54]. Importantly, combining ADCs with endocrine therapy does not appear to increase toxicity, and endocrine therapies seem to have favorable safety profiles [61, 62]. The mechanisms of the combined application of radiotherapy and ADC therapy include the radiation-induced generation of (neo)antigens [63,64,65], and ADCs increase the sensitivity of tumor cells to radiotherapy, among other potential mechanisms [66, 67]. Several studies have evaluated the safety of ADCs combined with radiation, and a small number of studies have reported adverse effects (AEs) associated with radiation and ADCs [68]; however, reliable data on the effectiveness and tolerability of this combination are insufficient. Combining radiation with ADCs is a promising treatment strategy; however, additional evidence on the safety of this approach is urgently needed. The effects of combination therapy with ADCs and other targeted drugs synergistically combine multiple mechanisms, such as increased cellular uptake and antitumor activity [69,70,71], the upregulation of surface antigens, synthetic lethality and combined targeting, and can overcome intratumor heterogeneity and drug resistance. The evidence suggests that the efficacy of ADCs is sensitive to the efficacy of immunotherapy [72]. Numerous preclinical studies and initial findings from early-stage clinical trials have shown improved anti-tumor effects attributable to mechanisms such as Fc-mediated effector functions [73], immunogenic cell death [74, 75], epithelial cell adhesion molecule (EpCAM) [76] and the direct activation and maturation of dendritic cells (DCs) [77, 78] for combination therapy with ADCs and immune checkpoint inhibitors (ICIs) [79,80,81]. Research has shown that both trastuzumab deruxtecan (T-DXd) and T-DM1 can maintain the inherent efficacy of trastuzumab [82, 83]. In the KATE2 trial (phase III), AEs, including one treatment-related death, were observed in metastatic breast cancer patients treated with atezolizumab in combination with T-DM1 [84]. Reaching definitive conclusions is difficult because of the lack of a randomized design for most other studies on combining ADCs and immunotherapy, and progress in ADC combination therapy will require the identification of novel tumor targets and elucidation of their pharmacological properties (Table 2).
Payload
The payload, also known as a “cytotoxic molecule” or “warhead” [85], is an important factor that affects the properties and activity of ADCs [86, 87]. Payload selection is crucial in the development of ADCs because the payload directly affects the therapeutic window and often plays a major role in clinical applications [88]. For success in designing therapeutic agents, cytotoxic drugs should have high cytotoxicity to tumor cells, but the amount of drug that can reach the tumor tissue after intravenous injection of an ADC is very limited, resulting in low intracellular concentrations [89]. The ideal payload should have a low molecular weight and a long half-life and should remain stable in the circulation and in lysosomes during endocytosis. Most approved cytotoxic payloads belong to one of the following three categories: microtubule inhibitors (such as maytansine or auristatin), DNA-damaging agents (such as doxorubicin, mitomycin, camptothecin analogs, and calicheamicin [90]), or topoisomerase inhibitors [91]. Cytotoxic payloads that can damage DNA are often very effective, while microtubule and topoisomerase inhibitors are moderately effective [92,93,94,95].
Microtubule inhibitors include maytansine and auristatin, both of which are derived from bacteria. Auristatin, similar to monomethyl auristatin E (MMAE) and monomethyl auristatin F (MMAF) [96, 97], is a synthetic compound extracted from the natural mitotic inhibitor dolastatin that can inhibit microtubule polymerization, leading to cell cycle arrest. MMAE can penetrate the cell membrane, and its cytotoxicity is 100–1000 times greater than that of standard chemotherapeutic drugs. In contrast, the more hydrophilic MMAF cannot penetrate the cell membrane; therefore, ADCs derived from MMAF are less efficient than those derived from MMAE, and their toxicity is relatively weak [98]. MMAE has been used in multiple ADCs. In 2015, the FDA approved brentuximab vedotin, an MMAE conjugate, for the treatment of Hodgkin’s lymphoma and anaplastic large cell lymphoma. Researchers prepared rituximab-Vc-MMAE, and the results showed high efficacy against CD20-positive cell lines but no effect on CD20-negative cell lines. In addition, rituximab-VC-MMAE was able to inhibit colony formation of CD20-positive cells. These data suggest that rituximab-c-MMAE may be an effective and selective drug for the treatment of B-cell lymphoma [99]. Bourillon et al. found that HER3 antibody‒drug conjugates (HER3 ADCs) based on MMAE were effectively internalized by tumor cells, increased the proportion of cells arrested in G2/M phase, which is the most radiation-sensitive phase in the cell cycle, and promoted programmed cell death in irradiated HER3-positive pancreatic cancer cells. HER3-ADCs reduced the clonogenic survival of irradiated cells by increasing the formation of DNA double-strand breaks (based on γH2AX levels) and regulating DNA damage repair. This approach may constitute a promising new strategy for the treatment of pancreatic cancer [100].
Maytansinoids are a class of ansamacrolides, and their derivatives are known as maytansinoidoids. Maytansinoids are natural products isolated from the African shrub Maytenus ovatus, and their mechanism of action is to disrupt microtubule polymerization. Maytansinoids are among the earliest cytotoxic drugs with an IC50 value in the picomolar range for tumor cells [101]. Maytansinoids and vinca alkaloids bind to the same sites on microtubules and have similar in vitro inhibition efficiencies. Due to their excellent stability and acceptable solubility in aqueous solutions, maytansinoids can be used to make ADCs [102, 103]. Maytansinoid derivatives are mainly divided into two types: DM1 and DM4 [104, 105]. DM1 maytansinoid derivatives (emtansine and mertansine) are potent drugs with broad lethal effects on xenografts of non-Hodgkin’s lymphoma in vivo [106]. DM4 drugs include soravtansine and ravtansine, which can enhance the “bystander effect” of adjacent cells in vivo, thereby eradicating tumors [107]. Effective payloads that act by damaging DNA include calicheamicin, doxorubicin, and camptothecin-like drugs. Unlike tubulin-binding agents, these effective payloads are not cell cycle specific and can exert cytotoxic effects on both proliferating and nonproliferating cells. Calicheamicin is a highly potent enediyne-class antitumor antibiotic originally isolated from Micromonospora echinospora. It can bind to the minor grooves of DNA, causing transcriptional damage, double-strand breaks, and cell apoptosis through DNA cleavage. Calicheamicin is also strongly hydrophobic, and each immunoglobulin can form drug conjugates with only a few molecules [108]. PF-06647263 is a calicheamicin-containing ADC targeting ephrin A4 and has recently entered phase I trials for triple-negative breast cancer [109]. Unlike calicheamicin, both doxorubicin and pyrrolobenzodiazepine class drugs are alkylating agents that bind to the minor groove of DNA and cause irreversible alkylation, leading to cell death [110]. A HER2-targeted ADC (trastuzumab deruxtecan, SYD985) prepared by linking trastuzumab with a new cleavable linker to the dual doxorubicin prodrug secoDUBA showed antitumor activity in preclinical breast and gastric cancer models with low HER2 expression [111, 112]. Vadastuximab talirine, a site-specific ADC currently in clinical trials, is an anti-CD33 antibody linked through engineered cysteine residues in the heavy chain that can yield a drug–to-antibody ratio (DAR) of 2. It was the first clinical ADC with a pyrrolobenzodiazepine-class drug payload and was tested in phase 1 trials in 2013 [113]. However, an efficient payload may also lead to greater safety risks. The phase III trial of vadastuximab talirine was terminated due to problems with dryness toxicity, despite achieving a complete remission rate of 70% in acute myeloid leukemia patients. The balance between efficacy and safety is a key consideration for scientists and regulatory agencies [114].
T-DXd is an ADC composed of an anti-HER2 antibody, a linker based on a cleavable tetrapeptide and a cytotoxic topoisomerase I inhibitor with cytotoxicity that has shown sustained antitumor activity in a pretreated population of HER2-positive metastatic breast cancer patients [115]. T-DXd has shown antitumor activity even in tumors with low HER2 expression. According to safety and efficacy data, the most likely recommended phase II dosage is 5.4 mg/kg or 6.4 mg/kg [116]. T-DXd also increases the antitumor immune response, as evidenced by the increased expression of DC markers, increased expression of MHC I in tumor cells and rejection of restimulated tumor cells by adaptive immune cells, indicating that T-DXDa improves the T-cell recognition of tumors. This immunostimulatory activity is distinct from the cytotoxic effect of DXd on tumor cells [117]. U3-1402 is composed of an anti-HER3 antibody (patritumab) and a DXd derivative linked together by the maleimide GGFG peptide. DX-8951 is a topoisomerase inhibitor. When U3-1402 binds to HER3 overexpressed in cancer cells, U3-1402 is cleaved by lysosomal enzymes to release DXd, which specifically inhibits topoisomerase 1 in cancer cells. Furthermore, the administration of U3-1402 significantly inhibited the growth of EGFR-TKI-resistant PC9AZDR7 xenograft tumors [118] (Table 3).
The current cytotoxic payloads of PDCs can be divided into chemical and nonchemical agents. Chemical drug therapy is one of the three major clinical treatments for malignant tumors. Tumor chemotherapeutic drugs include alkylating agents, antimetabolites, antibiotics, hormones, plant-derived drugs, platinum-based drugs, and immunomodulatory agents. Most chemotherapeutic drugs used in PDCs for tumor treatment are alkylating agents, antibiotics, and plant-derived drugs. For example, paclitaxel can be used to synthesize PDCs, as can vincristine, doxorubicin, methotrexate, and nitrogen mustard [119]. Paclitaxel is a first-line or second-line treatment for ovarian cancer, lung cancer, and other diseases, but it can cause bone marrow suppression, cardiotoxicity and drug resistance. Conjugating paclitaxel with tumor-targeting peptides can overcome these disadvantages [120]. After conjugation with peptides, paclitaxel retains significant tumor specificity. Paclitaxel-octreotide (PTX-OCT) can specifically bind to the STTR2 receptor overexpressed in tumor tissues [121]. The main pharmacophore of vincristine is the lactone ring, and the acyl group formed after lactone ring hydrolysis can interact with the nucleophilic group of topoisomerase I. However, the lactone ring of vincristine is easily hydrolyzed and becomes inactive after entering the circulatory system. Conjugating vincristine with the integrin receptor-targeting peptide ALOS4 significantly increases the stability of its lactone ring [122]. Moreover, the ALOS4–vincristine conjugate uses a peptide moiety for targeting, thus effectively reducing the systemic toxicity of vincristine by targeted delivery to tumor tissues. Doxorubicin is a commonly used chemotherapeutic drug that induces cardiotoxicity, which is closely related to drug accumulation. Resistance to doxorubicin is very common and decreases the concentration of doxorubicin in tumor cells. Studies have shown that TAT–doxorubicin has significantly greater cell toxicity to the doxorubicin-resistant cancer cell line KB-V1 than does doxorubicin alone [123]. RGD–doxorubicin can also increase the efficiency of doxorubicin delivery to tumor cells, where it exhibits significant cytotoxicity [124].
The nonchemical agents used for cytotoxic payloads include tumor necrosis factor (TNF), small interfering RNA (siRNA), and antisense oligonucleotides (AONs). TNF is a type of cytokine secreted by macrophages or lymphocytes in the body that can kill tumor cells or cause necrosis of tumor tissues, and includes TNF-α and TNF-β. Representative PDCs targeting TNF include etanercept and infliximab. Both drugs are targeted therapeutic drugs for TNF, but the underlying mechanisms are slightly different. Etanercept is an artificially synthesized fusion protein composed of the human TNF receptor and the human IgG1 Fc region [125] that can bind to TNF and prevent its binding to TNF receptors on the cell surface, thus reducing inflammation and disease. Etanercept has been approved for the treatment of inflammatory diseases such as rheumatoid arthritis, ankylosing spondylitis, and psoriasis. Infliximab is a monoclonal antibody that can specifically bind to TNF and prevent its biological activity. Therefore, infliximab is widely used to treat clonal disease, rheumatoid arthritis, inflammatory bowel disease and psoriasis. NGR–hTNF consists of TNF-α linked to peptides containing NGR, which can specifically recognize tumor neovascularization with high CD13 expression and thus deliver high concentrations of TNF-α to exert therapeutic effects on tumor tissues. A phase II clinical trial of NGR–hTNF and doxorubicin as second-line treatments for small-cell lung cancer revealed that NGR–hTNF showed effective antitumor activity in recurrent small-cell lung cancer patients [126]. siRNA is another molecule that can be used in PDCs and has great potential in gene silencing, gene expression control, and disease treatment. A representative PDC is Atu027, which is an siRNA-targeted antibody–peptide drug synthesized using Synthesis Platform technology and is used to treat patients with liver, bile, stomach, or pancreatic cancer. In addition, siRNAs can specifically knock down specific genes, thereby interfering with their expression [127]. Researchers can design corresponding siRNAs to target genes that affect the proliferation and differentiation of tumor cells, such as epidermal growth factor receptor, caspase-3, and caspase-9 [128] (Table 4).
Linkers
Linkers form the chemical connection between the antibody and the cytotoxic payload in ADCs [129]. They are a critical component of ADCs, and the linker should ideally stabilize the ADC in the bloodstream, ensuring that the ADC can reach the tumor site intact and allowing cleavage and release of the cytotoxic payload when the ADC binds to the antigen or is internalized. Although the linker itself may not be cytotoxic, its stability significantly affects the toxicity of the cytotoxic molecule. Stable linkers allow the cytotoxic agent to be precisely released upon reaching the specific target, while less stable linkers are more likely to undergo nonspecific cleavage, resulting in off-target side effects. Most dose-limiting and off-target toxicity is related to the stability of the linker molecule and the release of the payload into the systemic circulation.
Two primary linker types exist: non-cleavable and cleavable [130,131,132]. Initially, non-cleavable linkers were thought to be more useful than cleavable linkers because they can increase the stability of ADCs in plasma [133, 134], thereby decreasing the systemic toxicity risk, expanding the therapeutic window, and increasing tolerability. However, in ADCs connected by non-cleavable disulfide linkers, such as a non-cleavable succinimidyl 4-N-maleimidomethyl cyclohexane-1-carboxylate linker connecting trastuzumab to a monoclonal antibody [135], the non-cleavable linker cannot trigger bystander effector functions, and the ADC is ineffective in tumors with heterogeneous target antigen expression [136]. Cleavable linkers are sensitive to the physiological environment. The characteristics of tumor cells or their microenvironment can be used to disassociate the payload from the antibody portion. There are several mechanisms by which these chemical linkers are cleaved: enzyme-sensitive linkers [137] include valine–citrulline (Val–Cit), glutathione-cleavable triggers, and phosphatase-cleavable triggers [138]; pH-sensitive linkers [139] include hydrazone triggers [140, 141] and carbonate triggers. In addition, there are GSH-cleavable triggers [142, 143], non-cleavable linkers, Fe(II) cleavable triggers and redox-sensitive linkers. Reducing molecules such as glutathione are usually present at higher concentrations in the cytoplasm than in the extracellular space, allowing them to cleave disulfide bonds and release the payload within the cell. ADCs containing these types of linkers also typically have better solubility than those containing dipeptide linkers. Acid-cleavable linkers are hydrolyzed by the acidic environment of endosomes and lysosomes. The recognition and hydrolysis of a protease-sensitive linker is similar to the process of a peptide sequence being hydrolyzed by lysosomal proteases [144].
Existing chemically cleavable linkers can be divided into pH-sensitive linkers, cathepsin-cleavable linkers, GSH-cleavable linkers, Fe(II)-cleavable triggers, photoresponsive cleavable linkers, novel enzyme-cleavable linkers and bioorthogonally cleavable linkers [129, 145, 146] (Table 5). Glutathione (GSH), which contains cysteine, is a small peptide present in the human body, and its concentration is significantly greater in tissues such as lung cancer tissues than in normal tissues. Glutathione-sensitive linking moieties are connected to drugs by disulfide bonds. When drugs containing such linking moieties reach tumor tissue, the linker is cleaved by glutathione, and the cytotoxic payload is released to exert antitumor bioactivity. Studies have shown that the low-pH insertion peptide-sulfur-sulfur-doxorubicin (pHLIP-SS-DOX) can target acidic tumor cells and reverse multidrug resistance [145]. Moreover, the study of in vitro cytotoxicity mediated by GSH demonstrated that pHLIP-SS-DOX has significant cytotoxicity at a pH of 6.0. Tumor cells proliferate and undergo metabolic reactions more rapidly than do normal cells, leading to lactate accumulation and a decrease in the pH in the tumor microenvironment to approximately 6.8, whereas the pH in the bloodstream is approximately 7.3. pH-sensitive linking moieties are designed to exploit this change in pH [147, 148]. Enzyme-sensitive linking moieties can remain stable in the circulatory system of the human body, but when they reach locations where they are surrounded by the target enzymes, they undergo specific enzyme cleavage. Research has shown that linking moieties containing the short peptide sequence GFLG can be specifically cleaved by tissue protease B, releasing doxorubicin in tumor cells [149]. MMPs are a family of proteases that can target the extracellular matrix, and various subtypes of MMPs are highly expressed in tumor tissues [150, 151]. MMP2 and MMP9 play important roles in tumor invasion and metastasis by degrading collagen fibers cleaved by collagenase. The short peptide sequence PLGLAG is an MMP2/MMP9-sensitive linking fragment that can be cleaved in tumor tissue [152]. Abnormal iron metabolism can elevate the levels of unbound ferrous iron [153, 154]. Spangler et al. reported the use of the Fe(II)-reactive 1,2,4-trioxolane scaffold (TRX) linker in ADCs [155]. The linker Val–Cit has been shown to exhibit widespread sensitivity to a variety of cathepsins, but only cathepsin B is thought to be highly expressed in cancer cells. Pyrophosphate groups can be employed as linkers to load lipotropic payloads and increase the hydrophilicity [156]. Because the pyrophosphate linker showed high stability in vivo, Kern et al. replaced the traditional Val–Cit–PAB linker with a phosphate diester structure [157]. Recently, payload release based on photoresponsive cleavable linkers has gradually emerged. Photoresponsive linkers incorporate a UV light-controlled O-nitrobenzyl group as a chemical trigger. However, ADCs that undergo cleavage by near-infrared light present challenges including self-aggregation, complex structure and photoinstability, and near-infrared light cannot penetrate skin to reach deeply into the tumor area [158]. There are also bioorthogonally cleavable linkers; for example, although SMCC is a noncleavable linker, studies have identified 2-(maleimidomethyl)-1,3-dioxane (MD) as a potent alternative to the classical SMCC linker because of its greater stability. Another report showed that the use of noncleavable linkers in MMAE-based ADCs could broaden the therapeutic window [159]. Novel chemical triggers have been developed to increase the selectivity of delivery to the tumor area. Developing linkers with simplified structures and integrated functions may be another direction for ADC research.
Drug–antibody ratio (DAR)
The DAR refers to the number of effective payload molecules carried by each antibody and is another important factor related to the activity of the ADC. Once the best linker has been selected for the ADC, it is important to determine the ideal number of conjugates to the antibody. A very low DAR will reduce efficacy, and a high DAR is associated with increased in vitro potency but may also have adverse effects on pharmacokinetic and pharmacological properties [171]. An excess of payloads on a single antibody can destabilize the structure, leading to increased hydrophobicity and toxicity. For example, the binding of doxorubicin or MMAE to an ADC at a high DAR can result in a greater degree of hydrophobicity [172], leading to increased aggregation and a higher clearance rate [173]. This effect can be offset by using hydrophilic linker molecules [174]. The synergistic “1 + 1 > 2” combination of chemotherapy and targeted drug has both increased the treatment efficacy and reduced the incidence of toxic side effects, significantly improving therapeutic outcomes.
The molecular structures of natural antibodies present two main conjugation opportunities, namely, amino conjugation to lysine (Lys) and sulfur conjugation to cysteine (Cys). Since the antibody contains at least 40 modifiable lysine residues and the drug is conjugated randomly to different lysine residues, the conjugation products may contain a complex mixture of many unique molecules. Thus, lysine conjugation results in ADCs with highly variable DARs. The conjugation of cysteine residues may overcome this problem. When all 8 sulfhydryl groups of the antibody react with small molecules, an ADC with a uniform DAR of 8 can be obtained. In theory, the more payloads the ADC carries, the stronger the antitumor effect will be during the treatment window. In reality, however, most ADCs that have been approved or are under clinical development have DARs limited to 24 [175]. In one study, the tubulin inhibitor MMAE was conjugated to the CD30 mAb via cysteine to produce ADCs with different DARs (named E2, E4, and E8, depending on the DAR). The antitumor effects of E2, E4 and E8 were tested in vitro and in vivo, and the results showed that the antitumor efficacy increased with DAR in vitro (IC50 E8 < E4 < E2), but E8 had the same antitumor efficacy as E4 in vivo. Thus, the increased DAR did not confer additional efficacy and the maximum tolerated dose (MTD) decreased with increasing DAR (MTD = 50 mg/kg, 100 mg/kg and 250 mg/kg for E8, E4, and E2, respectively) [176]. Pharmacokinetics analysis showed that the clearance rate of the ADC increased with the DAR, which explains why E8 had the same effect as E4 in vivo.
However, the DAR limit can be overcome. A high DAR can facilitate internalization of the ADC, leading to increased efficacy, but it can also increase the clearance rate, resulting in rapid drug elimination. Currently, the DARs of most clinical ADCs are between 2 and 4, which represents a balance between potency and physicochemical properties [177]. However, a recently reported approach, optimizing a cleavable linker molecule with a fleximer scaffold and combining it with an uncleavable linker molecule that is then attached to the antibody, increases the DAR while preserving the pharmacokinetic profile and drug-like properties, thus increasing treatment efficacy at lower antigen expression levels. However, this technology has been evaluated only in vitro and in preclinical models [178]. An optimized cleavable linker based on the GGFG tetrapeptide increased the DAR of T-DXd from 4.1 to 7.7. The extremely wide therapeutic window and high DAR of T-DXd enable the delivery of enough cytotoxic drugs to kill tumor cells with low HER2 expression. There also new platforms to control DARs. Such as hydrophobic (HIC) chromatcolumn- TSKgel HIC-ADC Butyl. The particle size of 5 μm and the hydrophilic nonporous polymer matrix packing is particularly suitable for DAR values of ADC drugs. Antibody deglycosylation of ADC can simplify DAR measurements with rapid DAR analysis within 15 min by deglycosylation processing and LC–MS assays, thus enabling real-time DAR monitoring to optimize the ADC synthesis process. Therefore, when the limitations of linker and conjugation technology are overcome, high DAR benefits cancer patients, and impressive efficacy against low-expression targets is expected to lead to significant changes in clinical practice in the future [179].
Resistance
In recent years, ADCs have undergone rapid development in the field of cancer treatment; however, some patients still experience disease progression after receiving ADC treatment, and the problem of drug resistance to ADCs is of increasing concern. Based on the deep understanding of drug resistance mechanisms, the development of novel ADCs and the exploration of combination treatment strategies are particularly important for further increasing the clinical efficacy of ADCs in treating cancer. The mechanisms of drug conjugate resistance are complicated, and possibilities include the following: (1) Antigen-related resistance. Downregulation of target antigen expression on the tumor cell surface prevents ADCs from exerting cytotoxic effects. For example, a decreased expression level of HER2 leads to T-DM1 drug resistance. Similarly, CD30 downregulation leads to drug resistance in anaplastic large cell lymphoma (ALCL) [180]. Thus, dual-epitope ADCs were developed to overcome such resistance (NCT03821233, NCT04695847). Paradoxically, high antigen expression may also reduce ADC effectiveness, possibly through reduced drug exposure. (2) Endocytosis and migration disorders. For optimal efficacy, ADCs must undergo endocytotic uptake by cells. Endocytosis can proceed through different pathways, including clathrin-mediated endocytosis (CME), caveolin (CAV1)-mediated endocytosis, and clathrin caveolin-independent endocytosis. T-DM1 colocalization associated with CAV1 and drug resistance was also demonstrated in an HER2 + cell line [181]. (3) Lysosomal dysfunction: The ADC enters the lysosome, where the cytotoxic drug is released by chemical or enzymatic cleavage. T-DM1 aggregation in the lysosome was observed in cells with long-term exposure to T-DM1 resistance. In such cells, the ADC reaches the lysosomal compartment but has lower proteolytic activity than in sensitive cells, which decreases the activity of lysosomal proteolytic enzymes. Therefore, all ADCs that require degradation by lysosomal acidic proteases, may be subject to this resistance mechanism [182]. (4) Drug efflux pump: A common mechanism of chemoresistance is the elimination of the drug from the cytoplasm by ATP-binding (ABC) transporters [183]. These drug efflux pumps may contribute to resistance to ADCs, as many cytotoxic drugs are substrates of ABC transporters. Multidrug Resistance Gene (MDR1) is a major driver of resistance to Val–Cit–MMAE ADCs, and significantly lower MDR1 activity is observed in AML myeloblasts with a therapeutic response to gemtuzumab ozogamicin than in nonresponders [184]. (5) Mutations in target sites: One potential mechanism of ADC resistance could be cellular target mutations of cytotoxic agents [185]. However, no ADC resistance model with mutations in tubulin, topoisomerase I, or RNA polymerase II has yet been reported. (6) Cell cycle: Cyclin B, which is involved in the G2-M transition, was also recently proposed to be involved in the T-DM1 resistance mechanism. T-DM1 induced an increase in cyclin B levels in T-DM1-sensitive HER2 + breast cancer cells but T-DM1 was not observed in cells resistant to T-DM1. Clinical trials have shown that the antitumor effect of T-DM1 is associated with cyclin B expression, so cyclin B could be used as a biomarker for T-DM1 sensitivity [185]. (7) PI3K/AKT signaling pathway: The activation of PI3K/AKT signaling is correlated with resistance to gemtuzumab ozogamicin in primary AML cells in vitro. The AKT inhibitor MK-2206 significantly increased the sensitivity of resistant cells to gemtuzumab ozogamicin [186]. A clinical trial investigating the safety of T-DM1 in combination with the PI3K inhibitor BYL719 is ongoing (NCT02038010). (8) Apoptosis dysregulation: Changes in the regulation of apoptosis may also regulate sensitivity to ADCs. Overexpression of the antiapoptotic protein BCL-2 is associated with resistance to gemtuzumab ozogamicin [187]. High expression of BCL-XL is also associated with reduced sensitivity to CD79b–Val–Cit-MMAE [188]. The administration of a BCL-2 family inhibitor increases ADC activity in vivo [189].
Based on the aforementioned resistance mechanisms, resistance to the antibody components of ADC can be conferred by downregulation or mutation of the target cell surface antigen, and resistance to payload toxicity can be conferred by increased drug efflux transporter activity. Unique resistance mechanisms specific to the mode of action of ADCs have also emerged, such as altered internalization or cell surface recycling of targeted tumor antigens, changes in the intracellular routing or processing of ADCs, and impaired release of toxic payloads into the cytoplasm. Combination therapies are more promising than single-agent therapies for overcoming drug resistance. FDA-approved ADCs provide valuable treatment options for difficult-to-treat patient populations, but drug resistance is a frequently encountered limitation, and appropriate combination therapies may increase the percentage of cancer patients who receive long-term therapeutic benefits [190].
Antibody–drug conjugates (ADCs)
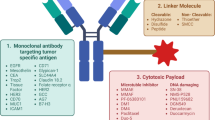
ADCs are a relatively new class of anticancer drugs [191] designed to combine the target selectivity of monoclonal antibodies with the cytotoxic properties of chemotherapeutic drugs [192]. Chemotherapy is still one of the main methods of cancer treatment, and many chemotherapeutic drugs are widely used in clinical practice; however, many adverse effects (AEs) and drug resistance problems are associated with these methods. ADCs can directly deliver cytotoxic drugs to tumor sites, transforming chemotherapy into targeted therapy. ADCs contain three essential factors: antibodies targeting specific tumor antigens, cytotoxic drugs (also known as payloads or warheads) and linkers connecting the payloads to the antibodies [32]. After an ADC enters the circulatory system, it combines with the target antigen to form a complex [193, 194]. The complex is internalized by endocytosis [195, 196], and cleavage of the linker leads to the release of the cytotoxic drug [197, 198] (Fig. 3). The antibody component can specifically recognize tumor antigens expressed at the target site, and the linker acts as a bridge to carry cytotoxic small molecules with significant lethal effects [199]. This approach combines mAb drugs and small-molecule chemical drugs, utilizing antibodies to achieve tumor targeting and efficiently eliminating tumor cells by releasing cytotoxins with strong killing effects in the target tissues. Advantages such as high activity, low toxicity and a long duration of action have allowed ADCs to greatly increase the therapeutic indices of small-molecule chemical drugs. Moreover, this approach partially solves the problems of low activity and high drug resistance associated with mAb drugs.

Mechanism of antibody–drug conjugates. ADCs bind to target antigens to form complexes, which are internalized by endocytosis. Linker cleavage leads to the release of cytotoxic drugs. ADC antibody‒drug conjugate
After the development of three generations of ADCs, they are considered a mature technology. Mylotarg, which targets CD33, is a representative first-generation ADC [200]. Mouse-derived antibodies have strong immunogenicity and are prone to inducing the production of human anti-mouse antibodies. The linker is unstable, and the toxin is quickly released into the plasma, leading to severe toxic side effects. However, the efficacy of the cytotoxic drugs is insufficient for killing tumor cells [201]. Second-generation ADCs are represented by Adcetris, which targets CD30, and Kadcyla, which targets HER2: in these ADCs, human–mouse chimeric antibodies and humanized monoclonal antibodies were used instead of mouse-derived monoclonal antibodies, along with more cytotoxic payloads and more stable linkers [202, 203]. However, the DARs are uneven [204, 205], and naked antibodies that are not bound to cytotoxic moieties enter the circulation, where they compete for conjugate antigen binding sites and reduce efficacy. In addition, the binding of excessive drug molecules to antibodies can easily cause problems such as antibody aggregation, accelerated clearance, and increased nonspecific toxicity [206, 207]. For third-generation ADCs, due to the development of fixed-point conjugation technology, DARs have been stabilized at approximately 2–4, and the stability and pharmacokinetic properties have improved [208, 209]. More hydrophilic linker modifications, such as PEGylation, are also employed in the third generation of ADCs [210, 211]. Moreover, the bystander effect [212, 213], which is achieved by the use of cleavable connectors, increases treatment efficacy and reduces systemic toxicity. A representative example is Enhertu, which targets HER2.
Target and antibody selection
The ideal target for an ADC is an antigen that is expressed only on the surface of tumor cells [214]. Targets that are preferentially expressed in tumors compared to nonmalignant tissues have a wider therapeutic window and a lower likelihood of systemic toxicity [215]. Therefore, choosing the appropriate antigen is one of the major challenges in the development of ADCs. Based on this requirement, three aspects should be considered in antigen selection: (1) high expression in tumors and low expression in healthy tissues [216]; (2) expression of the target antigen on the surface of tumor cells, making it accessible to antibodies; and (3) the existence of a pathway of intracellular transportation and a suitable internalization rate. Notably, noninternalizing ADCs can also exert therapeutic effects through an alternative “bystander effect” [217], in which a membrane-permeable drug can induce the death of neighboring cells [218].
A suitable antibody should have high target specificity, abundant target expression, and an appropriate internalization rate [219]. Due to problems such as acute hypersensitivity reactions and the side effects of neutralizing antibodies when murine antibodies are used, the antibodies currently used in ADCs are mainly humanized antibodies, which have significantly lower immunogenicity than murine and chimeric monoclonal antibodies. They also have higher solubility and a longer half-life. Nonetheless, although the use of humanized antibodies can minimize the problems encountered with mouse-derived antibodies, these problems are not completely solved [220]. Most antibodies used in clinical practice are derived from human immunoglobulin (IgG), which has a molecular weight of approximately 150 kDa and consists of two heavy chains and two light chains. Antibody derivatives can generally be divided into antigen-binding fragments (Fabs), single-chain variable fragments (scFvs) and variable domains (VHHs). Fabs and scFvs retain the size and affinity of the antigen-binding region and are smaller than conventional IgG [221], resulting in improved the pharmacokinetic properties for tumor penetration [222, 223]. Antibodies also need to bind to antigens with appropriate affinity to increase accumulation and prolong the retention time at the tumor site. However, if the retention time is too long, the paracancerous cells surrounding the solid tumor may be compromised [224].
ADCs targeting HER2/HER3
Human epidermal growth factor receptor 2 (HER2) is a receptor tyrosine kinase encoded by the ERB-B2 receptor tyrosine kinase 2 (ERBB2) gene. The HER family consists of four members: HER1 (EGFR/ErbB1), HER2, HER3 (ERBB3) and HER4 (ERBB4) [225, 226]. The HER family, especially HER2, is considered a therapeutic target in lung cancer because it is overexpressed or mutated in multiple tumors, including lung cancer, and the activation of related pathways, such as MAPK, PI3K, AKT and PKC, can lead to excessive cell proliferation [227], resulting in tumor occurrence and development. In addition to corresponding targeted therapies, ADCs can exhibit excellent antitumor activity by acting on the HER family.
T-DM1
T-DM1 consists of the HER2 monoclonal antibody trastuzumab and the microtubule inhibitor emtansine (DM1) linked via a nonreducible sulfur linker, with an average of 3.5 payload molecules per antibody [228, 229]. T-DM1 was the first ADC to be tested against advanced HER2-positive NSCLC and provides a new treatment strategy for patients with advanced HER2-positive disease [230]. A phase II clinical trial including 18 patients with advanced HER2-mutated NSCLC showed a partial response (PR) in 8 patients, with a median progression-free survival (PFS) of 5 months [231]. Another phase II study reported similar results, with an objective response rate (ORR) of 51% and a median PFS of 5 months [232] in 49 patients with HER2 mutation or overexpression. Based on these data, the National Comprehensive Cancer Network (NCCN) recommends T-DM1 as a class 2A drug for the treatment of advanced HER2-mutated NSCLC [233, 234]. However, two other phase II clinical studies showed limited efficacy of T-DM1 in HER2-positive or HER2-overexpressing NSCLC patients. In preclinical studies of HER2-immunohistochemistry score 3 + CALU-3 lung cancer cells, T-DM1 dose-dependently inhibited tumor cell growth. A phase I study investigating HER2 overexpression in 49 previously treated patients with advanced NSCLC reported ORRs of 0% and 20% for HER2 IHC2 + and 3 + , respectively, with median PFS times of 2.6 and 2.7 months [234]. In addition, T-DM1 has shown significant efficacy in the treatment of lung cancer with HER2 exon 20 insertions [235]. The main adverse effects of T-DM1 include transaminase elevation, thrombocytopenia and nausea [231].
T-DXd
T-DXd (trastuzumab deruxtecan), also known as DS-8201, is a novel HER2-targeting ADC [236] with a different mechanism of action from that of other ADCs: it binds and stabilizes topoisomerase I-DNA complexes, inducing DNA double-strand breaks and apoptosis [237]. T-DXd consists of trastuzumab, a cleavable linker, and the topoisomerase I inhibitor deruxtecan [238]. T-DXd has satisfactory membrane permeability and can not only kill HER2-positive tumor cells but also exert bystander effects to kill nearby tumor cell [239]. T-DXd has a high DAR of 8, indicating that an average of 8 effective payload molecules can be conjugated to each trastuzumab molecule [240]. T-DXd has shown good antitumor activity in patients with HER2-mutated solid tumors (except for breast and gastric cancer). The latest data showed an overall ORR of 72.7% and a median PFS of 11.3 months in 11 previously treated NSCLC patients with HER2 mutation [241]. Subsequently, an open-label, global phase II DESTINY-Lung01 clinical study was conducted for advanced NSCLC patients with HER2 overexpression or mutation. Among 91 patients with HER2-mutated NSCLC, the ORR was 55%, the disease control rate (DCR) was 92%, the median PFS was 8.2 months, and the median OS was 17.8 months [242]. In January 2021, the World Conference on Lung Cancer (WCLC) released data on HER2-overexpressing NSCLC patients treated with T-DXd. Among the 49 patients, the ORR was 24.5%, the DCR was 69%, and the median PFS was 5.4 months. Subgroup analysis revealed that the ORRs of the IHC3 + and IHC2 + groups were 20% and 25.6%, respectively, indicating that HER2 IHC expression had no significant effect on the ORR. Currently, the phase Ib DESTINY-Lung03 clinical study exploring the clinical efficacy of T-DXd combined with durvalumab and chemotherapy in newly diagnosed HER2-positive advanced NSCLC patients is ongoing. Therefore, T-DXd is more effective for treating advanced NSCLC with HER2 mutations. Regarding toxicity, the most common adverse reactions to T-DXd are gastrointestinal and hematologic toxicities, with neutropenia being the most common grade 3 adverse reaction [243]. Importantly, interstitial lung disease (ILD) was observed in 11.9% of HER2 mutation patients (all grade 2), with a median onset time of 86 days [244]. The incidence of ILD was slightly greater in the HER2-overexpressing population (16.3%), which included 3 patients with grade 5 ILD. Overall, T-DXd has good overall safety, but patients treated with T-DXd need to be closely monitored for the occurrence of ILD.
A166 and MRG003
A166 is an ADC targeting HER2 that consists of a microtubule inhibitor connected to trastuzumab via a cleavable linker. Data from a phase I clinical study evaluating A166 in 81 patients with advanced solid tumors showed an ORR greater than 60%. Regarding safety, the most common adverse reactions include keratitis, dry eye, blurred vision and decreased appetite [245].
MRG003 is a novel ADC targeting EGFR that showed remarkable preliminary efficacy in phase I clinical studies for various solid tumors. Clinical trials of MRG003 for late-stage EGFR-mutant NSCLC are still ongoing, and MRG003 is expected to become China’s first anti-EGFR ADC.
U3-1402
U3-1402 (patritumab deruxtecan) is an ADC targeting HER3, another member of the EGFR family, and consists of a humanized anti-HER3 antibody and a topoisomerase I inhibitor payload [246]. HER3 is overexpressed in 19% of NSCLCs and up to 46% of adenocarcinomas [247] and is involved in mediating resistance to EGFR tyrosine kinase inhibitors (TKIs) [248]. A phase I study enrolled 57 patients with advanced NSCLC without the T790M mutation who progressed after EGFR-TKI treatment, and almost all the patients were found to express HER3. The results showed that the ORR of U3-1402 monotherapy was 39%, with a median PFS of 8.2 months, suggesting that U3-1402 could be an important treatment option for patients with NSCLC with multidrug resistance [249]. At American Society of Clinical Oncology (ASCO) 2021, the latest data from a phase I dose escalation/expansion trial including 39 patients with locally advanced or metastatic NSCLC with EGFR mutations who had experienced disease progression after EGFR-TKI therapy were presented. The ORR was 39%, and the DCR was 72%. With a median follow-up of 10.2 months, the median DoR was 6.9 months, and the median PFS was 8.2 months [249]. Subgroup analyses also revealed the antitumor activity of U3-1402 in NSCLC patients harboring resistant EGFR mutations. A phase II study of U3-1402 is ongoing. U3-1402 lacks significant efficacy compared to that of other targeted drugs but may be a new treatment option for patients who are resistant to third-generation TKIs or who are otherwise not suitable for third-generation TKI treatment. In terms of safety, 47% of patients experienced grade 3 or higher adverse events, among which thrombocytopenia (28%) and neutropenia (19%) were the most common.
ADCs targeting Trop-2
Trophoblast cell surface antigen 2 (Trop-2) is a transmembrane protein that is closely related to cell proliferation and differentiation. Trop-2 is expressed at low or almost undetectable levels in normal tissues and overexpressed in various epithelial cancers, including NSCLC and SCLC. Trop-2 overexpression has also been shown to be associated with poor prognosis in lung adenocarcinoma [250], suggesting that Trop-2 has potential as a new target for lung cancer treatment.
DS-1062a
DS-1062a (datopotamab deruxtecan) is an ADC in which a Trop-2-targeting antibody is connected to a topoisomerase I inhibitor payload via a tetrapeptide linker. DS-1062a may have antitumor effects on multiple types of cancer. The latest research results on the tolerability and safety of DS-1062a in treating advanced NSCLC in a clinical trial were presented at the 2021 ASCO Annual Meeting. In NSCLC patients receiving different 4.0, 6.0, and 8.0 mg/kg doses of DS-1062a, the ORRs were 31%, 20%, and 26.3%, respectively, and the DCRs were 79%, 75%, and 79%, indicating that DS-1062a has good antitumor activity in lung cancer. In another phase I study of 175 recurrent/refractory advanced NSCLC patients, the ORRs of DS-1062a at doses of 4, 6, and 8 mg/kg were 23%, 21%, and 25%, respectively, and the median PFS times were 4.3, 8.2, and 5.4 months, respectively. Treatment-related adverse reactions were dose dependent, with a grade ≥ 3 incidence of 10–34%, and included oral mucositis, nausea, fatigue, mucositis, and anemia. Among them, four patients in the 8 mg/kg group experienced grade ≥ 3 ILD. The most common grade ≥ 3 adverse reactions in patients treated with DS-1062a at different doses were oral mucositis, mucosal inflammation, nausea, fatigue, and anemia.
IMMU-132
IMMU-132 (sacituzumab govitecan) is an ADC in which the topoisomerase I inhibitor SN-38 (the active metabolite of irinotecan) is linked to a humanized anti-Trop-2 antibody via a cleavable linker with a DAR of 7.6 [251]. In a phase I clinical trial including 25 patients with standard therapy-refractory metastatic solid tumors (including NSCLC and SCLC), 2 patients achieved PR, and 16 patients had stable disease [252]. Based on these results, the trial entered a phase II exploration with a total of 495 patients enrolled. The researchers evaluated 54 patients with advanced NSCLC, with an ORR of 16.7%, a median DoR of 6.0 months, a median PFS of 4.4 months, and a median OS of 7.3 months. In another group of 62 patients with first-line chemotherapy-resistant or sensitive metastatic SCLC, the ORR was 17.7%, the median DoR was 5.7 months, the median PFS was 3.7 months, and the median OS was 7.1 months [253]. Currently, IMMU-132 is undergoing phase Ib/II clinical trials in combination with atezolizumab for NSCLC and in combination with the ATR inhibitor berzosertib for SCLC. Regarding toxicity, grade 3 adverse events included diarrhea, fatigue, anemia, nausea, and neutropenia [254]. The data above suggest that IMMU-132 may be a promising drug for treating NSCLC and SCLC.
Dato-DXd
Dato-DXd has been explored for use in lung cancer treatment and has shown broad application prospects [255]. The TROPION-Lung02 trial is an ongoing global, open cohort phase Ib study to evaluate the safety and effectiveness of Dato-DXd (4 or 6 mg/kg) + pembrolizumab (200 mg) ± platinum chemotherapy (cisplatin or carboplatin) in patients with advanced or metastatic NSCLC who are newly treated or previously treated and who have no driver gene mutations. The median follow-up times for the dual drug group and the triple drug group were 6.5 months and 4.4 months, respectively, at which times 53% and 77%, respectively, of patients in the two groups were still receiving treatment. The median treatment durations were 4.1 months and 3.0 months, respectively. In the first-line treatment groups, the ORRs in the dual drug group and the triple drug group were 62% and 50%, respectively, and the disease control rates were 100% and 90%, respectively. In the second-line treatment groups, the ORRs were 24% and 29%, respectively. This combination regimen is well tolerated and exhibits encouraging antitumor activity as a first-line treatment.
ADCs targeting c-Met
The c-Met protein is encoded by the gene mesenchymal-epithelial transition (Met) and is a tyrosine kinase receptor expressed on the surface of both epithelial and endothelial cells [256,257,258]. When activated, it promotes cell proliferation, growth, migration, and angiogenesis. The abnormal activation of the c-Met pathway in NSCLC mainly involves Met14 exon skipping mutations, Met fusion and overexpression, and MET amplification, which is also a resistance mechanism in EGFR-mutant NSCLC resistant to EGFR TKIs [259]. The incidence of MET14 exon skipping mutation is 3–4%, the incidence of primary MET amplification is approximately 3%, and the incidence of secondary amplification is 10–15%; MET amplification is associated with resistance to multiple TKIs, and the incidence of overexpression, which is a predictor of poor prognosis, is approximately 24% [244, 260,261,262].
Teliso-V
Teliso-V is an ADC composed of an anti-MET monoclonal antibody (ABT-700) linked to the cytotoxic payload MMAE, which inhibits microtubule polymerization. The key to the mechanism of action is that after antibody binding, the cytotoxic payload can be directly delivered to tumor cells, limiting potential resistance mechanisms related to intracellular signaling, such as ME3 amplification in EGFR TKI resistance. A phase I study showed that Teliso-V, either as a single agent or in combination with erlotinib, was well tolerated in patients with advanced MET-positive NSCLC and exhibited good antitumor activity both as a monotherapy and in combination with erlotinib. In a separate phase I dose escalation and expansion study, Teliso-V was shown to be effective as a single agent only in MET-positive advanced NSCLC patients [263]. However, a phase II study evaluating the efficacy of Teliso-V in patients with MET-positive advanced squamous cell NSCLC was terminated early due to severe adverse reactions and a low ORR [264]. Recently, targeted therapy has shown good antitumor activity in patients with Met14 exon skipping mutations, but there is no standard treatment that addresses Met amplification.
ABBV-399
ABBV-399 (telisotuzumab vedotin) consists of the microtubule inhibitor MMAE conjugated to a humanized anti-c-Met monoclonal antibody via a cleavable linker with a DAR of 3.1 [265]. A phase I study of 58 patients with advanced c-Met-positive NSCLC showed an ORR of 18.8%, a median DoR of 4.8 months, and a median PFS of 5.7 months [263]. Based on these encouraging results, the phase II trial SWOG S1400K was designed to evaluate the efficacy of ABBV-399 in 23 patients with c-Met-positive advanced squamous NSCLC, but the study was terminated early due to a lack of expected results [264]. Another phase II trial including 52 patients with c-Met-positive NSCLC showed that 9 patients (23%) achieved objective responses, with a median DoR of 8.7 months and a median PFS of 5.2 months [266]. ABBV-399 showed promising efficacy against nonsquamous NSCLC in a phase II study, with an ORR of 35.1% in patients with c-MET-positive, wild-type EGFR nonsquamous NSCLC, 53.8% in the high-expression group and 25% in the moderate expression group; however, ABBV-399 had only limited efficacy in the patient groups with EGFR mutation and squamous NSCLC [264]. Overall, ABBV-399 has shown encouraging efficacy in treating relapsed/refractory nonsquamous NSCLC with c-MET overexpression and wild-type EGFR, but further studies will be needed to validate its efficacy in patients with squamous NSCLC and EGFR-mutant NSCLC.
ADCs targeting DLL3
Delta-like protein 3 (DLL3) is a ligand that inhibits the Notch signaling pathway, which is involved in multiple processes associated with growth and development. DLL3 is highly expressed in 72% of primary SCLC tumor tissues and 85% of recurrent SCLC tumor tissues [267], whereas it is rarely expressed in normal tissues, making it a promising target [268, 269].
Rova-T
Rova-T (rovalpituzumab tesirine) is an ADC that targets DLL3 and consists of an anti-DLL3 monoclonal antibody, a DNA-damaging pyrrolobenzodiazepine dimer toxin, and a protease-cleavable linker [270]. In a phase I clinical trial, the ORR of 74 recurrent SCLC patients treated with Rova-T was 18%, with a median PFS of 3.1 months and a median OS of 4.6 months [271]. The TRINITY study was a phase II trial in which Rova-T was applied as a third-line treatment to 339 patients with DLL3-expressing SCLC; the ORR was 12.4%, the median PFS was 3.5 months, and the median OS was 5.6 months [272]. The TAHOE study compared the efficacy of Rova-T and topotecan as second-line treatments for SCLC [273]. The Rova-T group and the topotecan group included 296 and 148 patients, respectively. The results showed that the median PFS and OS in the Rova-T group were 3.0 months and 6.3 months, respectively, while they were 4.3 months and 8.6 months in the topotecan group. Because the PFS and OS of the Rova-T group were both worse, the study was terminated early. Another phase III MERU study was likewise terminated early due to limited efficacy [244]. Based on the results of monotherapy, a phase I/II clinical trial explored the efficacy of Rova-T in combination with nivolumab or in combination with both nivolumab and ipilimumab in 42 patients with advanced-stage SCLC; the resulting ORR was 30%, the median PFS was 4.2 months, and the median OS was 7.4 months [274]. Another phase I study evaluated the efficacy of Rova-T in combination with budesonide in 31 SCLC patients, and the ORR was 24.1% [275]. Overall, Rova-T monotherapy has limited benefits for SCLC patients, but combination therapy is expected to be effective. In terms of safety, 38 to 64% of patients experienced grade 3 or higher adverse reactions, among which the most common were platelet count reduction, pleural effusion, and elevated lipase [276].
SC-002
SC-002 is an ADC composed of a humanized anti-DLL3 monoclonal antibody linked to SC-DR002 by a cleavable linker, with a DAR of 2 [277]. Phase I studies included 35 patients with relapsed/refractory SCLC or large-cell neuroendocrine tumors, and the ORR was only 14% (5/35); for DLL3-positive patients, the ORR was only 11.8%. Overall, ADCs targeting DLL3 have proven to be unsuccessful.
ADCs targeting AXL
AXL is a receptor tyrosine kinase that promotes tumor development through multiple pathways and is associated with chemotherapy and immune therapy resistance in various types of cancer [278]. In NSCLC, AXL activation is associated with EGFR-targeted therapy resistance and lower survival rates in patients with advanced NSCLC [279]. Therefore, AXL is an attractive target for antitumor therapy. Enav (enapotamab vedotin) and BA3011 are ADCs that target this pathway. Enav consists of an anti-AXL monoclonal antibody linked to the microtubule inhibitor MMAE via a cleavable linker [280]. The most common grade 3 or higher adverse reactions observed were gastrointestinal reactions, which included constipation, colitis, diarrhea, bloating, nausea, and vomiting. However, because of the low efficacy, the clinical development was terminated.
ADCs targeting NaPi2b
NaPi2b is a sodium-dependent phosphate transporter encoded by SLC34A2 that has been shown to play a role in cell differentiation and tumorigenesis [281]. NaPi2b is highly expressed in various cancers, including lung cancer, particularly in patients who are TTF1-positive or have KRAS and EGFR mutations [282]. Because of its elevated expression in multiple cancers, NaPi2b is an attractive target for ADC development.
XMT-1536 is an ADC composed of a humanized anti-NaPi2b targeting antibody and the potent payload auristatin F-hydroxypropylamide (AF-HPA). A preclinical study showed that XMT-1536 had strong antitumor efficacy in mouse models of NSCLC and ovarian cancer [283]. Phase I/II dose escalation and expansion studies of XMT-1536 for the treatment of refractory advanced NSCLC are still ongoing.
ADCs targeting CEACAM5
CEACAM5, also known as CD66e, is a glycoprotein encoded by the carcinoembryonic antigen gene and is expressed at low levels in normal tissues but at moderate to high levels in multiple cancers, including NSCLC: 20% of nonsquamous NSCLCs exhibit high expression (> 50%), and 25% exhibit moderate expression (1–49%) [213]. SAR408701 is a novel ADC composed of a humanized anti-CEACAM5 monoclonal antibody and the microtubule inhibitor maytansinoid DM4, connected by a cleavable tetrapeptide linker, with a DAR of 3.9. In the first clinical study, which included 92 patients with advanced NSCLC for whom previous treatments had failed, SAR408701 achieved ORRs of only 7.1% in the moderate CEACAM5 expression group and 20.3% in the high CEACAM5 expression group, with a median DOR of 5.6 months. The incidence of ≥ 3 AEs was 47.8%, of which 15.2% were drug related, including keratitis (10.9%) and fatigue (4.3%). The most severe AE was dyspnea related to disease progression. A phase III clinical trial (NCT02187848) investigating combined first-line chemotherapy and immunotherapy for advanced NSCLC patients with high CEACAM5 expression is currently underway, with hopes of clinical benefit.
Antibodies play a crucial role in the internalization of ADCs into tumor cells. Therefore, identifying antibodies with high specificity and affinity for the target antigen is essential. Dual-targeting antibodies can not only increase internalization but also increase the specificity for tumor cells. Compared to single-targeting antibodies, dual-targeting antibodies have higher antitumor activity and may be a useful new research direction [284]. Additionally, converting traditional antibody frameworks into “small” peptide fragments or single-chain variable fragments can increase tissue permeability and payload transmission by reducing the molecular weight of conjugates [285]. Moreover, innovative payloads can contribute to improving the antitumor effects of ADCs. Increasing the DAR is another important method for improving the antitumor efficacy of ADCs. Preclinical studies have shown that dolaflexin technology can increase the DAR and thereby induce tumor regression [178].
Other targets in the development of ADCs for lung cancer treatment
Different ADCs targeting other transmembrane proteins or membrane receptors, including CD19, TF, PTK7, and B7-H3, are currently undergoing clinical trials for lung cancer [286] (Table 6). The composition of each ADC is shown in Table 7, and the chemical structure of each ADC is shown in Fig. 4.


Chemical structures of representative antibody‒drug conjugates (ADCs) in clinical trials for lung cancer treatment
Peptide–drug conjugates (PDCs)
Despite three generations of ADC development, many unresolved issues remain. The first-generation ADCs contained mouse-derived antibodies and uncleavable linkers. Their disadvantages included insufficient cytotoxicity and low expression of loci. The drawbacks of the second-generation ADCs included DARs that were too low or too high, narrow treatment windows and low effectiveness. The disadvantages of third-generation ADCs include the difficulty of replicating conjugation technology and the insensitivity of cancers to microtubule protein inhibitors [287]. PDCs have the advantages of easy synthesis and purification and low production costs, and they are the most promising type of drug conjugate for achieving therapeutic breakthroughs after ADC [288, 289].
Peptides, as ligand analogs, are characterized by strong targeting ability and the ability to assemble with other drugs. Assembling peptide analogs with chemotherapeutic drugs can produce PDCs with targeted delivery effects [160, 290]. Compared with ADCs, PDCs have the advantage that peptides are easier to synthesize and purify than antibodies are, leading to lower production and transportation costs. Peptide structural modification can facilitate drug design to increase bioavailability, binding affinity, and stability. Additionally, peptides have lower molecular weights than antibodies and thus can more easily penetrate the tumor matrix and enter tumor cells. The structure and composition of PDCs are simpler, and the immunogenicity is lower, which corresponds to a lower probability of an immune stress response in the body. Additionally, PDCs can be eliminated by the kidneys, which results in lower liver toxicity and higher safety. The main indications for PDCs include esophageal tumors, brain tumors, lung cancer, gastric tumors, ovarian tumors, multiple myeloma, pancreatic tumors, and advanced solid tumors [291], making PDCs a promising new generation of targeted anticancer drugs after small-molecule drugs, mAbs and ADCs.
In recent years, the U.S. FDA has approved clinical trials of several tumor-targeting peptide compounds as potential drugs [292]. The selection of tumor protein targets is a major focus of research on tumor-targeting peptides and is directly related to whether a given peptide can be used as an antitumor drug. With the rapid development of X-ray crystallography technology, computer systems, and component technology, great progress has been made in computer-aided drug design [293,294,295,296,297]. Tumor-targeting PDCs have become a new research focus for the development of antitumor drugs in recent years, as they can overcome the disadvantages of conventional chemotherapeutic drugs, particularly by providing increased selectivity between normal and tumor cells [298]. According to statistics from the U.S. clinical trial database, Aeterna Zentaris has conducted 5 phase II/III clinical trials for AEZS-108, which targets breast cancer, endometrial cancer, prostate cancer, and urothelial carcinoma; MolMed has conducted 12 phase II/III clinical trials for NGR–hTNF, which targets colon cancer, ovarian cancer, NSCLC, small-cell lung cancer, malignant thymic epithelial tumors, and metastatic adult soft tissue sarcomas [299]. However, tumor-targeting PDCs have common drawbacks, such as rapid in vivo metabolism and weak drug stability. Rational drug design using appropriate targeting peptides, linker molecules, and cytotoxic payloads can mitigate these problems to some extent [300].
Tumor-targeting peptides
Tumor-targeting peptides are predominantly synthesized via solid-phase peptide synthesis. The payload, which is the active pharmaceutical ingredient, is manufactured through processes such as synthesis, extraction, or fermentation. The linker is designed with a minimum of two functional groups to facilitate the covalent connection of the tumor-targeting peptide and the payload through chemical synthesis. Tumor-targeting peptides can specifically recognize tumor blood vessels or tumor-related receptors to achieve targeting. With advancing research techniques, many tumor-targeting peptides have been discovered [124, 126, 301,302,303,304]. The accumulation of PDCs in tumors and normal organs relies primarily on tumor-targeting peptides, which play a crucial role in molecular targeting. Compared to alternative drug delivery systems, the elimination of extraneous components from molecular drug delivery systems increases the clinical efficacy and safety in cancer patients, thereby maximizing the therapeutic outcome [305, 306].
PDCs targeting CD13
Mammalian aminopeptidase N (APN)-CD13 is an ectoenzyme found on the surface of cells and is overexpressed in lung cancer [307]. The peptide Asn-Gly-Arg (NGR) is a tumor-targeting peptide that is upregulated during angiogenesis and the formation of new blood vessels. It specifically binds to vascular cells that express APN. Currently, there are two fusion protein drugs based on the concept of PDCs that incorporate the NGR-targeting peptide: NGR–human tumor necrosis factor (hTNF) and truncated tissue factor (tTF)–NGR [308]. These drugs are currently undergoing clinical studies. In tTF–NGR, the active payload is the external domain of tTF, while Gly-Asn-Gly-Arg-Ala-His-Ala serves as the tumor-targeting peptide connected to the C-terminus of tTF. tTF–NGR has shown acceptable tolerability in low-dose clinical applications and has successfully reduced tumor perfusion. Phase I clinical trials of this treatment for solid tumors, including lung cancer, are currently underway. Although fusion protein drugs do not exactly imitate PDCs since the payload is directly linked to the tumor-targeting peptide, both NGR–hTNF and tTF–NGR exhibit targeting and therapeutic characteristics identical to those of PDCs.
Integrins
Integrins regulate various steps in tumor cell migration and invasion and affect tumor cell growth and survival during tumor cell escape and blood/lymphatic vessel infiltration [309]. Integrins consist of 24 heterodimeric cell adhesion receptors, each consisting of α and β subunits. The extracellular region of the α chain includes four extracellular domains. Arg-Gly-Asp (RGD) can bind to a total of 8 integrins [310]. Among them, ανβ3, ανβ5, α5β1 and ανβ6 are associated with cancer progression and metastasis. RGD has the highest affinity for ανβ3 and ανβ5, neither of which is expressed in normal tissues. Therefore, targeting integrins with RGD-based ligands is highly important for specifically targeting tumor cells that overexpress integrins in antiangiogenic therapy.
Several PDC candidates targeting RGD peptides, including [18F]Fluciclatide, [18F]RGD-K5 [311], and 68Ga-NOTA-bombesin (BBN)–RGD, have recently entered clinical trials as positron emission tomography (PET) tracers. Although RGD peptide sequences have many advantages, they also have several shortcomings. RGD-based anticancer drugs and imaging agents can target and bind to integrins to inhibit tumor angiogenesis, but they can also promote tumor cell adhesion, spreading and migration [312,313,314].
PDCs targeting SST
There are five subtypes of SST receptors, which are widely distributed in the brain, pancreas, and pituitary tissues. Natural SST is rapidly degraded by enzymes, and the half-life (t½) is short, less than 3 min after intravenous injection. To date, various SST analogs, such as octreotide (t½ = 2 h), have been developed as prodrugs. The affinity of octreotide for the SST2, SST5, SST3, SST1, and SST4 receptors is high (IC50 0.38–0.60 nmol/l), relatively high (IC50 6.3–7.0 nmol/l), moderate (IC50 7.1–34.5 nmol/l), low (IC50 280–1140 nmol/l), and > 1000 nmol/l, respectively. The use of octreotide and its analogs as tumor-targeting peptides and radioactive isotopes or cytotoxic molecules as effective payloads can achieve the therapeutic/diagnostic purpose of targeting SST2 receptors on tumor cell surfaces. Currently, several PDCs based on octreotide as a tumor-targeting peptide, including diagnostic agents and imaging agents such as 111In-DTPA-octreotide, 99mTc-HYNIC/EDDA-3Tyr-octreotide, 68Ga-DOTATOC, 68Ga-DOTATATE and 177Lu-DOTATATE, are on the market [315,316,317].
Other receptors
Lung cancer is closely associated with the overexpression of membrane type-1 matrix metalloproteinase (MT1-MMP). Consequently, MT1-MMP is considered a potential prognostic biomarker of lung cancer and is linked to unfavorable prognosis. Compared to ADCs, BT1718, an MT1-MMP-targeted PDC, has a low molecular weight and a favorable distribution. As a result, BT1718 can rapidly infiltrate and eliminate tumor cells, achieving a positive therapeutic effect on advanced solid tumors.
Prostate-specific membrane antigen (PSMA) is highly overexpressed in the neovasculature of prostate tumor cells and most solid tumors but not in normal blood vessels. After intravenous administration, PSMA-targeted G202, a soluble thapsigargin prodrug, is metabolized into the active cytotoxic analog of thapsigargin, known as 12-ADT-β-Asp. This mechanism obstructs the nutrient supply to tumor cells, resulting in a high concentration of 12-ADT-β-Asp at the tumor site without causing systemic toxicity.
Hepatocyte receptor A2 (EphA2) expression is generally low in healthy adult tissues but abnormally high in various solid tumors and is associated with poor prognosis. BT5528, a PDC that targets EphA2, can accumulate in tumor tissues at a minimal plasma concentration, increasing the selectivity to eradicate tumor cells while minimizing systemic toxicity.
PDCs have attracted widespread attention due to their ability to significantly improve targeting and ameliorate toxicity and resistance, but many challenges remain. First, the molecular delivery system is administered mainly by injection to prevent degradation in the gastrointestinal tract, but the inconvenience of injections results in poor patient compliance. Second, the in vivo distribution and targeting time of PDCs are limited. The short half-life of tumor-targeting peptides results in a short window of time for effective payload entry into tumor cells. The existing strategies to address this challenge include head-to-tail cyclization, disulfide bond cyclization, substitution of nonnatural amino acids, peptidomimetics, stapled peptides, and bicyclic peptides. However, these strategies must not compromise the binding of tumor-targeting peptides to receptors.
Furthermore, PDCs rely on conditions such as pH, redox status, and enzyme activity in vivo to release the payload. This dependence prevents some payloads from being released as prodrugs or, in some cases, from being released at all. Additionally, payloads modified with functional groups can exhibit significantly reduced biological activity compared to that of prodrugs. Therefore, it is necessary to demonstrate that the active targeting advantage of PDCs can counterbalance the reduced biological activity of the payload. Some candidate drugs have been terminated due to unsatisfactory clinical results, indicating the need to improve the molecular delivery systems.
From clinical diagnosis to cancer treatment, peptide-based drug delivery systems are flourishing. Formulation is still considered the key to the drug delivery process, and PDCs can currently satisfy all functional requirements for drug formulations, including absorption, distribution, metabolism and excretion. Compared to nontargeted anticancer drugs applied in clinical practice, molecular delivery systems based on PDCs exhibit significant advantages: prolonged circulation time, increased maximum tolerable doses, elevated drug accumulation in tumor cells, and increased anticancer biological activity. All of the PDCs currently in development are in specific clinical trials for lung cancer [286] (Table 8). The composition of each PDC is presented in Table 9, and the chemical structure of each PDC is shown in Fig. 5.


Chemical structures of representative peptide‒drug conjugates (PDCs) in clinical trials for lung cancer treatment
Other drug conjugates
Radionuclide drug conjugates (RDCs)
Radionuclide drug conjugates (RDCs) developed because ADC contain chemotherapy drugs, which may cause a series of toxic reactions. RDCs replace cytotoxic drugs with nucleotides, which can be conjugated with antibodies to form radionuclide–antibody conjugates (RACs) [318]. RDCs are emerging precision tumor therapy drugs that utilize tumor antigen-specific molecular carriers for delivery, accurately targeting radionuclides to tumors for brachytherapy [319]. The mechanisms of the therapeutic effect of RDCs on tumors are as follows: (1) After the radiolabeled antibody specifically targets the membrane antigen on the tumor surface, the radionuclide directly damages DNA, mitochondrial DNA, the cell membrane, etc. The surrounding cells are also exposed to radiation through cross effects. Cell damage leads to the secretion and release of cytokines, ions, ROS, RNS, or exosomes into the extracellular microenvironment. (2) The cytokines and other effector molecules released into the microenvironment bind to cell death receptors, inducing adjacent cancer cell death by a bystander effect. (3) The irradiated cells secrete DAMPs that can bind to the T-cell receptor of antigen-presenting cells, activate the immune system by binding to CD4 or CD8 T cells, and attack remote tumor cells in another type of bystander effect. Radionuclides generally include the β-emitting radionuclides 131I, 90Y, 177Lu, and 188Re and the α-emitting radionuclides 213Bi and 211At. The targets included 4 proteins related to hematological tumors, namely, CD20, CD22, CD33 and CD66, as well as 12 proteins related to solid tumors. Relevant clinical trials are currently being conducted. In recent years, the focus of RDC development has gradually shifted toward the treatment of solid tumors. However, due to issues such as difficulty in delivery caused by abnormal tumor blood vessels and nontarget organ toxicity, the development of RDCs for solid tumors is challenging. The selection of appropriate radionuclides and carriers according to the tumor type and tumor antigen is crucial for optimizing and balancing the therapeutic effect, increasing the dose absorbed by tumors and reducing the toxicity to nontarget tissues. In the past 10 years, multiple clinical trials of RDCs have been published, with 67% for the treatment of nonsolid tumors and 33% for the treatment of solid tumors. Lymphoma accounts for the vast majority (92.5%) of nonsolid tumors in clinical trials of RDCs. Among solid tumors, the types and targets covered by RDCs are more diverse. RDCs in trials for lung cancer treatment include ANG1005, ITM-41, 111In-DTPA-octreotide, 99mTc-EDDA, 68Ga-DOTATATE, 68Ga-DOTATOC, Lu177 dotatate, 68Ga-PSMA-11 and Cu-64 DOTATATE.
Small-molecule–drug conjugates (SMDCs)
SMDCs are usually composed of targeted molecules, linkers and effector molecules [320]. In fact, due to the excessive segmentation in the field of drug conjugates, there is also crossover between different drug concepts, and PDCs are examples of SMDCs. The largest difference between SMDCs and ADCs lies in the targeting ligand. The ligand of an ADC is a macromolecular antibody that binds to antigens, while the ligand of an SMDC is a relatively low-molecular-weight organic functional group that binds to transporters with high selectivity. Small-molecule ligands also determine the pharmacokinetic characteristics of SMDCs, as they can easily penetrate and spread evenly into tumor tissue without aggregating in either tumors or normal cells. The small amount of off-target drugs will be quickly expelled from the body, which also decreases toxicity to normal cells. The mechanisms of action of SMDCs and ADCs also highly similar. For example, SMDCs targeting folate receptors cannot enter cells through the reduced folate carrier channels by which normal cells absorb folate. Instead, similar to ADCs, they bind to high-affinity folate receptors and enter cells by endocytosis; they are then cleaved and release cytotoxic molecules, exerting a killing effect, while the folate receptors cycle back to the cell surface [321].
PEN-866 is an SMDC developed by Saisheng Pharmaceutical that is currently undergoing phase II clinical trials for the treatment of solid tumors in the United States. PEN-866 carries SN-38, an active metabolite of the topoisomerase inhibitor irinotecan, into the tumor and accumulates in the tumor by the selective binding of the small molecule with the intracellular target heat shock protein 90 (HSP90). SN-38 is cleaved and released over time, preventing adverse reactions caused by systemic irinotecan exposure. Clinical data show that at the recommended dose (175 mg/m2), no DLT was observed in patients who received PEN-866, and only one patient experienced uncomplicated G3-grade transient neutropenia. Comparing these results to the occurrence of ≥ grade 3 neutropenia events in 53.8% of patients treated with irinotecan shows that the safety advantage of PEN-866 was significant. Currently, there are several SMDCs in clinical trials for lung cancer treatment, including PEN-866, EC-1456, MBC-11, CBP-1008, vintafolide, BMS-753493, and EC0489EC0225.
Virus-like drug conjugates (VDCs)
In VDCs, viral capsids designed to form noninfectious protein nanoparticles (VLPs) act as efficient delivery carriers [322]. In some studies, VLPs from human papillomavirus or HPV were selectively attached to the surface of modified heparan sulfate proteoglycans (HSPGs) to target solid tumor cells or metastatic foci instead of normal tissues. AU-001 is a VDC produced by this mechanism, in which virus-like components selectively bind to HSPG. Conjugated infrared light-activated cytotoxic drugs selectively destroy tumor cells under irradiation, leading to acute necrosis of tumor cells and activation of the immune system to produce antitumor responses.
Antibody–oligonucleotide conjugates (AOCs)
In AOCs, antibodies are used to deliver therapeutic oligonucleotides (siRNAs, PMOs, etc.) to specific cells or tissues, thereby reducing the amount of the drugs needed to treat diseases and addressing the challenges of targeting and oligonucleotide delivery [323]. The conjugation of oligonucleotides with targeted ligands can also improve the pharmacokinetic properties of oligonucleotides and expand their application scope. Technically speaking, AOCs use antibodies as the delivery medium for small molecules, proteins and other functional molecules. Based on this concept, the AOC product AOC1001 was developed for the treatment of ankylosing myotonic dystrophy type 1 (DM1). AOC1001 consists of three parts: a full-length monoclonal antibody targeting transferrin receptor 1 (TfR1), a linker, and siRNA targeting DMPK mRNA. The indication for AOC1001 is DM1. TfR1 is widely expressed on the cell surface and can transport iron into the cell. Muscle cells require a large amount of iron, which makes TfR1 particularly useful for delivering drugs to muscle cells. The design principle of AOC1001 is to treat DM1 by knocking down the expression of mutated DMPK to release Muscleblind-like (MBNL) and enable it to function normally. MBNL proteins are RNA-binding proteins that were first discovered in Drosophila and play important roles in the development of muscles and eyes, as well as in the pathogenesis of human myotonic dystrophy.
Antibody‒cell conjugates (ACCs)
ACC technology uses a 5' NHS ester ssDNA linker to conjugate the amino groups of antibodies to cell surface proteins, and two linkers bind to form double-stranded DNA, thus completing the conjugation of immune cells and antibodies [324]. ACCs are similar to CAR-T cells in that they provide targets for cell therapy. The difference is that ACCs require only a chemical reaction for conjugation and do not require genetic modification. This powerful cell therapy approach has the potential to significantly increase the efficacy of NK cells by unlocking multiple receptor signaling pathways, such as γδ T cells, which have the ability to recognize T cells and participate in tumor killing [325]. This approach may enable ACC NK therapy to overcome the challenges of effectively targeting solid tumors with cell therapy. The two ACCs currently under development are ACE1702 and ACE1655.
Immune-stimulating antibody conjugates (ISACs)
The technical requirements of ISACs are similar to those of ADCs, except that the ISAC payload is a congenital immune agonist or regulator with the ability to transform immunologically cold tumors into immunologically hot tumors [326]. ISACs can activate immune killing and therapeutic sensitivity by modulating immune stimulation and the microenvironment [327]. The drugs used in this approach mainly include the Toll-like receptor agonist (TLR) class ISACs SBT6050, SBT6290 and BDC-1001 [328]; the STING agonist ISAC XMT-2056 [329]; and the Treg cell regulatory ISAC ADCT-301 [330, 331]. The current core candidate drug BDC-1001 is a Boltbody-based drug™. The immune-stimulating antibody of this platform is conjugated with Bolt’s proprietary TLR 7/8 double agonist through a noncleavable linker, which is biologically similar to the anti-HER-2 drug trastuzumab and is used to treat HER2-positive solid tumors. Another SMDC in clinical trials for lung cancer treatment is BDC-2034.
Antibody fragment–drug conjugates (FDCs)
As the name suggests, FDCs use smaller antibody fragments instead of larger antibody molecules [332]. It is generally believed that antibody fragments are relatively easy to detect and that a higher DAR can be achieved using biotechnology [333]. Compared with ADCs, FDCs have the following advantages: the ability to maximize drug efficacy by promoting higher DARs for the delivery of many active drug molecules; small size, which can enable rapid and uniform tumor penetration and thus faster therapeutic effects; rapid clearance from normal tissues and the circulation because of the small size and lack of Fc [334]; a lack of unnecessary molecular interactions that inhibit drug activity; suitability for most antibody fragment forms, which results in high versatility; the ability to reverse engineer the entire mAb and ADC to facilitate drug conjugate manufacturing, increase solubility, and improve the formulation for the next-generation FDC while retaining the stability and binding function of the scFv; and improved pharmacokinetics/kinetics of the entire mAb. As a next-generation cancer treatment method, FDCs can overcome many limitations of existing treatment options and have great market potential.
Antibody–degrader conjugates (ADeCs)
ADeCs are currently in the early development stage. The technical principle is the use of protein degradation agents as payloads, combining the tumor specificity of ADCs and the applicability of PROTAC molecular catalysts for the treatment of solid tumors with low target protein expression. A representative ADeC is ORM-5029, developed by Orum Therapeutics. This drug, like ADCs, shares the ability to specifically target tumor cells and can accurately deliver its payload of a new protein-degrading agent to the cell interior to degrade intracellular target proteins. In addition, drugs such as AnDC-0003, AnDC-multiple, TD-0001, and IO-0001 are still under development for the treatment of solid tumors, including lung cancer.
Aptamer–drug conjugates (ApDCs)
In ApDCs, the antibody in an ADC is replaced with an aptamer. The linker connects the aptamer with the drug molecule, which exerts a therapeutic effect. The aptamer serves as a recognition ligand, guiding the therapeutic drug to a disease site or regulating the biological function of targeted biomarkers [335]. Nucleic acid aptamers are oligonucleotide sequences identified using live cell-based index enriched ligand system evolution technology (Cell SELEX) that can bind to various targets with high affinity and specificity [336]. Compared with antibodies, aptamers have many advantages: (1) the high efficiency of aptamer screening, which takes only a few days to several months; (2) the ability to bind toxins or antigens with low immunogenicity for which corresponding antibodies cannot be found; (3) relatively mature solid-phase synthesis technology, with low cost and small batch differences; (4) ease of modification; (5) better thermal and chemical stability; (6) a smaller molecular weight and thus better tissue permeability; and (7) almost no immunogenicity, with no immune side effects. Aptamers are often used in combination with various therapies, such as chemotherapy, phototherapy, toxins, gene therapy, and vaccines. To date, researchers have designed and developed various nucleic acid ApDCs and nucleic acid aptamer-functionalized nanomedicines and have confirmed their potential to significantly promote drug enrichment in tumor lesions. Despite these unique advantages, the sensitivity of nucleic acid ApDCs to nucleases results in short half-lives in vivo, and nonspecific protein adsorption causes nucleic acid aptamer-functionalized nanodrugs to have poor pharmacokinetic behavior. These problems limit the implementation of antitumor drugs based on nucleic acid aptamers in vivo [337]. One study revealed that tumor-targeted chemotherapy achieved by ApDC nanomicelles can increase the antitumor immune response. Therefore, a multivalent ApDC (ApMDC), an amphiphilic terminal dendritic macromolecule composed of hydrophilic aptamers and hydrophobic single dendrites anchored to four anticancer drugs through acid-sensitive junctions, was designed and synthesized. By co-self-assembly with ApMDC analogs, in which the aptamers are replaced by polyethylene glycol, the surface aptamer density of these nanomicelles can be adjusted to optimize the balance between blood circulation time and tumor-targeting ability. The optimized nanomicelles can promote the immunogenic cell death of tumor cells, thereby significantly increasing the tumor-specific immune response to checkpoint blockade in immune-active tumor-bearing mice. Other drugs are still being developed.
Various other drug conjugates currently in clinical trials for lung cancer treatment are presented in Fig. 6. The compositions of these drugs are shown in Table 10, and the chemical structures are shown in Fig. 7.

Drug conjugates in trials for lung cancer treatment. ADCs antibody‒drug conjugates, PDCs peptide‒drug conjugates, RDCs radionuclide drug conjugates, SMDCs small molecule–drug conjugates, ACCs antibody–cell conjugates, ISACs immune-stimulating antibody conjugates, VDCs virus–like drug conjugates, ADeCs antibody–degrader conjugates

Chemical structures of other representative drug conjugates in clinical trials for lung cancer treatment
Toxicities and side effects
The use of drug conjugates in the treatment of lung cancer has been limited by their potential for toxicity [338, 339]. This review summarizes the toxicities of drug conjugates that underwent lung cancer clinical trials in which the primary endpoint events were AEs or serious AEs (SAEs) in Table 11. Although drug conjugates have shown great promise, some studies have resulted in SAEs, and in the NCT03245736 study, 100% of patients experienced other (not including serious) AEs. Nervous system disorders were the most common side effects, and other AEs included blood and lymphatic system disorders; metabolic and nutritional disorders; and respiratory, thoracic and mediastinal disorders. However, the all-cause mortality in this trial was 0. In addition, all of the patients in the NCT02001623 trial experienced AEs, and the most common side effect was nausea. Overall, the all-cause mortality was within acceptable limits. Several lung cancer clinical trials have had primary endpoint events of AEs/SAEs (NCT00346385, NCT02673060, NCT01002924, NCT02277717, NCT02874664, NCT02552121, and NCT03913741); however, the results from these trials have not been disclosed. It is necessary to fully understand the adverse reactions caused by drug conjugates and to establish corresponding safety management strategies and evaluation methods. In addition to tumor therapy, drug conjugates are still in clinical trials and preclinical testing for nontumor indications such as immunity and infection [220].
Because many of the toxicities associated with drug conjugates are dose related, researchers have expended substantial effort to optimize doses and administration patterns to improve the therapeutic indices of drug conjugates. At present, typical dose optimization strategies include adjustment of the upper limit of dose, adjustment of the upper limit of treatment duration, graded dose administration, patient treatment response-guided dose adjustment, and random-effect dose research [340].
The optimization of drug conjugate structures is important for maximizing the efficacy and safety of formulations and can affect tolerance [341]. In addition to common drug conjugates, probe–drug conjugates can reduce the incidence of targeted and nontumor toxicity [342]. Because the vast majority of payloads are released in the circulatory system, the toxicity of drug conjugates is currently similar to that of chemical drugs. The most important aspects of optimization are the tumor-targeted delivery of drug conjugates and the use of cleaved linkers to increase the bystander effect [343]. Reducing the risk of AEs after drug conjugate treatment is an important step in clinical management. The genomic parameters of drugs may also affect their reactivity. The inclusion of drug genome maps in early drug design trials may be a reasonable approach to ensure that safety is not excessively affected by genetic variations among populations or individuals.
Outlook
Challenges in drug conjugate development
The currently approved drug conjugates are much more potent than conventional chemotherapeutic agents. The development and application of drug conjugates could have a unique impact on lung cancer treatment. A legitimate question, therefore, is to what extent drug conjugates can optimize conventional cytotoxic chemotherapy, at least for some indications [344]. There are still some challenges, and the current limitations of drug conjugates include cost, drug resistance, and instability.
Drug resistance
The mechanisms of drug conjugate resistance are complicated and can involve [340] antigen-related resistance, endocytosis and migration disorders, lysosomal dysfunction, drug efflux pump activity, mutations in target sites, the cell cycle, the PI3K/AKT signaling pathway and apoptosis dysregulation [345]. However, there are currently no effective treatments to counteract drug conjugate resistance [346, 347]. Combination therapy with other drugs, switching to drug conjugates with different targets, or developing new payload drugs are potential approaches to reversing resistance [182, 348, 349].
High heterogeneity
Due to the high heterogeneity and dynamic changes in target antigens expressed by tumor cells, it is necessary to select appropriate drug conjugates targeting tumor tissue-specific antigens. Among the existing ADCs, only a few have shown promising efficacy in target-enriched patients, such as an ORR of 55% for T-DM1 and T-DXd in treating advanced HER2-mutated NSCLC. However, these agents have limited efficacy against NSCLC with other mutation types, such as HER2-overexpressing NSCLC. Currently, the majority of ADCs lack effective biomarkers for predicting efficacy. In the future, relevant studies need to be conducted to explore the selection of patients who will benefit from ADCs. In addition, multiple patient-related factors, including baseline organ function, the presence of comorbidities, and polymorphisms of enzymes involved in ADC metabolism, can influence the pharmacokinetics and pharmacodynamics of these drugs. The analysis of patient heterogeneity may facilitate the development of personalized treatment plans and improve outcomes [350].
Instability
In general, the presence of lysine and cysteine residues on antibodies provide reactive sites for conjugation [351, 352]. Early ADCs were typically randomly conjugated via lysine or cysteine residues [353], but this approach can lead to many problems [354]. The stability of these drug conjugates is sometimes insufficient, which can cause premature payload release and off-target toxicity [355,356,357,358].
DAR limitations
The DAR determines the amount of the payload that can be delivered to the tumor, directly affecting the safety and effectiveness of drug conjugates. Simply put, the effectiveness of drug conjugates is directly linked to the DAR. The DAR can be understood as the amount of ammunition carried by the magic bullet ADC, where the higher the value of the DAR is, the stronger the antitumor efficacy. Although a high DAR represents a large drug-loading capacity, a drug conjugate with a high DAR is also more likely to be recognized by the human immune system as a foreign object and cleared by the body, thus reducing the effectiveness. A high DAR can also easily lead to drug release in the circulatory system, resulting in high toxicity. Therefore, the DAR of most drug conjugates is limited to 2–4.
Rapid intracellular disintegration
Rapid disintegration has a critical impact on drug conjugates. The cytotoxic drugs released by the cleavage of a cleavable linker can penetrate the cell membrane and kill the surrounding tumor cells via a process called the bystander effect. In contrast, for a noncleaved linker, even if the antibody is degraded by proteases, amino acid residues remain connected to the linker and the cytotoxic drug. The resulting charged metabolites cannot effectively pass through the cell membrane and therefore usually do not exert a bystander effect.
How to address these challenges
In the long term, the development of ADCs is mainly aimed at updating linker payloads, but in the short term, antibody forms, including monoclonal antibodies, double antibodies, multiantibodies and existing linker payloads, remain dominant. To address the above challenges, there are currently numerous systematic treatment strategies to ensuring safety while improving treatment efficiency.
Overcoming drug resistance
Dual-payload ADCs may be a promising class of drugs to address the clinical challenges of tumor heterogeneity and drug resistance [359]. By accurately controlling the ratio of the two drugs and simultaneously delivering two synergistic toxins to cancer cells, the overall therapeutic effect can be increased, resulting in a higher response rate. Simultaneously, due to the different mechanisms of action of the toxins, the incidence of drug resistance is significantly reduced [360]. For example, in preclinical studies, a single anti-HER2 ADC that includes both MMAE and MMAF [273, 361] exhibited significantly higher antitumor activity than the simultaneous administration of corresponding single-toxin ADCs and even achieved complete remission [362]. However, the design of dual-toxin drug conjugates has not yet been validated in clinical trials. Due to the potential for synergistic (1 + 1 > 2) toxicity, the safety phase is an important focus.
The administration method can also affect the occurrence of drug resistance. In early clinical trials, ADCs were mainly administered as single drugs. Currently, treatment options that combine conventional chemotherapy and other targeted drugs are being explored in clinical practice. In addition, the order of treatment may be an important factor. One study revealed that patients who had previously received trastuzumab/pertuzumab had a worse response to T-DM1 than did those who had not received these two antibodies [363]. In a preclinical T-DM1-resistant model, however, the combination of trastuzumab and pertuzumab was found to be effective [364].
Combinatorial strategies are under investigation to assess the efficacy of drug conjugates delivered in association with partner drugs [365]. Combinations of ADCs with TKIs directed against the same target to increase internalization or overcome TKI resistance or with immunotherapeutic agents for potential synergistic effects in lung cancer can also be evaluated as strategies [366, 367].
Solving the heterogeneity issue by target selection and the use of bispecific antibodies (BsAbs)
Strategies for using drug conjugates in lung cancer treatment involve either biomarker-driven or biomarker-agnostic approaches. Different expression levels and cutoff values might affect the efficacy of treatment across different trials. Conversely, HER2 mutations, not HER2 overexpression, have been associated with the response of lung cancer to anti-HER2 ADCs [368], and stronger established drivers might represent better therapeutic targets [369].
According to the latest research reports, combining BsAb technology with ADC technology is currently a new direction in the field of ADCs [370, 371]. At present, there are two main BsAb design strategies: (1) Many targets, such as HER2, have promising tumor expression profiles, but poor internalization and poor lysosome transport limit their full potential as effective ADC targets. An ADC with dual specificity for the two nonoverlapping epitopes of the HER2 protein has been designed and is called a biparental ADC. This structure can increase the cross-linking of cell surface receptors and the aggregation of receptors, thus promoting the internalization and lysosome delivery of ADCs and thereby increasing their efficacy. However, it should be noted that not all nonoverlapping antibodies are equally effective at promoting internalization and lysosome transport, and specific epitopes and spatial directions can even reduce lysosomal transport. In addition, there are some designs that achieve the same function by combining weak internalization targets with strong internalization targets to form bispecific ADCs. (2) To increase the selectivity for tumors over normal tissues, an appropriate combination of two specific ADCs with optimized affinity can be selected, which expanding the therapeutic indices and increases the safety and effectiveness of ADCs. In addition, antigen selection is critical to drug efficacy, and several factors need to be considered in the selection of target antigens: the degree of antigen expression in tumors and healthy tissues; the physiological function of antigens in normal cells and tumor cells; the endocytosis of the antigen and the mechanism involved; if, where and how the antigen is released; the potential impact of antigen shedding on the effectiveness of the ADC; and the antigenic cycle and its influence on the mechanism of ADC action.
Increasing stability by adjusting the structure of drug conjugates
Ideally, drug conjugates can maintain their integrity and stability in the blood circulation before entering the target cell, and many methods, including conjugation site selection and linker modification, have been developed to increase drug conjugate stability. In fact, it has been reported that less than 1% of administered ADCs reach human tumors, and the rest may cause unnecessary toxicity. Usually, each component can be modified to increase stability [372, 373], but research has shown that adjusting the conjugation sites and the length and steric hindrance of the linker are more effective general methods [374, 375]. By selecting conjugation or attachment sites with high steric hindrance, spatial shielding by the antibodies can be established [376].
Linkers influence the stability and pharmacokinetics (PK) of a given drug conjugate [377], and linkers can be selected for tumor-specific release, allowing drug release in both the tumor microenvironment and tumor cells without affecting the half-life of the drug conjugate in circulation. Due to the presence of tissue proteases in the tumor microenvironment, peptide linkers that are sensitive to tissue proteases have this characteristic. Improvements in linker development could include the use of (1) payload-masking linkers [378], (2) hydrophilic linkers [379], (3) branched linkers to increase the DAR [380], (4) tandem cleavage linkers, and (5) dual-sensitivity linkers [381]. Payload modifications that might increase the therapeutic benefit of next-generation drug conjugates include the creation of (1) prodrug-based payloads to mitigate off-tumor toxicity [382, 383], (2) hydrophilic cytotoxic payloads [384, 385], and (3) bifunctional payloads to increase antitumor efficacy [386, 387].
Raising the DAR by modifying the drug conjugation mode and linker
Concerning DARs and pharmacokinetic characteristics [388, 389], most current drug conjugates use highly potent cytotoxic warheads, which generally produce the expected effect when the DAR is 2–4 [390]. Through modification of the conjugation mode and linker, T-DXd and sacituzumab govite can achieve a DAR of approximately 8, indicating that additional cytotoxic molecules can bind to an antibody without affecting its solubility, aggregation tendency, or pharmacokinetic characteristics [391, 392]. This ability has important implications: compounds with lower potency but different mechanisms of action may serve as effective payloads for drug conjugates. Alternatively, using drug conjugates with the same payload but lower DARs might increase efficacy due to the ability to increase the dosage.
Guaranteeing rapid intracellular disintegration by targeting mutant proteins
Recent research has indicated that rapid intracellular disintegration critically affects the cytotoxicity of drug conjugates. Compared to wild-type proteins, mutated proteins typically have greater stability against rapid intracellular disintegration but are more prone to internalization and degradation. Thus, drug conjugates targeting mutated proteins may have significant clinical effects [393]. In addition, designing cleavable linkers to increase intracellular disintegration presents a challenge.
Prospects
There are multiple comprehensive treatment methods for lung cancer, and the extent to which drug conjugates offer advantages over traditional treatments is unclear [394]. Although measuring the impact of a novel anticancer agent in the clinic can be difficult, drug conjugates have had a pronounced impact on lung cancer treatment. The approval of 27 drug conjugates by the FDA and the encouraging clinical performance of other drug conjugate candidates have attracted increasing attention in the field, and many studies on lung cancer have shown promising results for drug conjugates. Even if drug conjugate development is more complex than unconjugated drug development [395], the challenges encountered in the development of drug conjugates are being overcome. We expect the number of approved drug conjugates to increase substantially in the coming years, and we anticipate much better treatment effects for lung cancer (Fig. 8).

Measures to address the difficulties associated with drug conjugate development and application. DAR drug-to-antibody ratio
Availability of data and materials
Not applicable.
Abbreviations
- ACC:
-
Antibody cell conjugate
- ADC:
-
Antibody drug conjugate
- ADeC:
-
Antibody degrader conjugate
- AOC:
-
Antibody oligonucleotide conjugate
- AON:
-
Antisense oligonucleotide
- ApDC:
-
Aptamer drug conjugate
- DAR:
-
Drug-to-antibody ratio
- DCR:
-
Disease control rate
- DLL3:
-
Delta-like protein 3
- EGFR:
-
Epithelial growth factor receptor
- FDA:
-
Food and Drug Administration
- FDC:
-
Antibody fragment drug conjugate
- GSH:
-
Glutathione
- hTNF:
-
Human tumor necrosis factor
- ILD:
-
Interstitial lung disease
- ISAC:
-
Immune-stimulating antibody conjugate
- mAb:
-
Monoclonal antibody
- MMAE:
-
Monomethyl auristatin E
- MMAF:
-
Monomethyl auristatin F
- MTX:
-
Methotrexate
- NA:
-
Not available
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- PBD:
-
Pyrrolobenzodiazepines
- PDC:
-
Peptide drug conjugate
- PET:
-
Positron emission tomography
- PFS:
-
Progression free survival.
- PSMA:
-
Prostate-specific membrane antigen
- RDC:
-
Radionuclide drug conjugate
- siRNA:
-
Small interfering RNA
- SMDC:
-
Small-molecule drug conjugate
- TKI:
-
Tyrosine kinase inhibitor
- TTP:
-
Tumor-targeting peptide
- VDC:
-
Virus-like drug conjugate
- Val–Cit:
-
Valine–citrulline
- VEGFR:
-
Vascular endothelial growth factor receptor
- WCLC:
-
World Conference on Lung Cancer
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q, et al. Recent advances in targeted strategies for triple-negative breast cancer. J Hematol Oncol. 2023;16(1):100.
Thai AA, Solomon BJ, Sequist LV, Gainor JF, Heist RS. Lung cancer. Lancet. 2021;398(10299):535–54.
Liu SM, Zheng MM, Pan Y, Liu SY, Li Y, Wu YL. Emerging evidence and treatment paradigm of non-small cell lung cancer. J Hematol Oncol. 2023;16(1):40.
Cortes J, Perez-Garcia JM, Llombart-Cussac A, Curigliano G, El Saghir NS, Cardoso F, et al. Enhancing global access to cancer medicines. CA Cancer J Clin. 2020;70(2):105–24.
Gadaleta E, Thorn GJ, Ross-Adams H, Jones LJ, Chelala C. Field cancerization in breast cancer. J Pathol. 2022;257(4):561–74.
Tfayli AH, Sfeir PM, Youssef BY, Khuri FR. Locally advanced lung cancer. CA Cancer J Clin. 2021;71(6):461–5.
Mei T, Wang T, Deng Q, Gong Y. The safety of combining immune checkpoint inhibitors and platinum-based chemotherapy for the treatment of solid tumors: a systematic review and network meta-analysis. Front Immunol. 2023;14:1062679.
Shi N, Wong AKC, Wong FKY, Sha L. Mobile health application-based interventions to improve self-management of chemotherapy-related symptoms among people with breast cancer who are undergoing chemotherapy: a systematic review. Oncologist. 2023;28(4):e175–82.
Filetti M, Lombardi P, Giusti R, Falcone R, Scotte F, Giannarelli D, et al. Efficacy and safety of antiemetic regimens for highly emetogenic chemotherapy-induced nausea and vomiting: a systematic review and network meta-analysis. Cancer Treat Rev. 2023;115: 102512.
Kroeze SGC, Pavic M, Stellamans K, Lievens Y, Becherini C, Scorsetti M, et al. Metastases-directed stereotactic body radiotherapy in combination with targeted therapy or immunotherapy: systematic review and consensus recommendations by the EORTC-ESTRO OligoCare consortium. Lancet Oncol. 2023;24(3):e121–32.
Shi Y, Hu X, Zhang S, Lv D, Wu L, Yu Q, et al. Efficacy, safety, and genetic analysis of furmonertinib (AST2818) in patients with EGFR T790M mutated non-small-cell lung cancer: a phase 2b, multicentre, single-arm, open-label study. Lancet Respir Med. 2021;9(8):829–39.
Zhuang H. Research progress on the impact of radiation on TKI resistance mechanisms in NSCLC. J Cancer. 2018;9(20):3797–801.
Fu Y, Zhang Y, Lei Z, Liu T, Cai T, Wang A, et al. Abnormally activated OPN/integrin alphaVbeta3/FAK signalling is responsible for EGFR-TKI resistance in EGFR mutant non-small-cell lung cancer. J Hematol Oncol. 2020;13(1):169.
Shi K, Wang G, Pei J, Zhang J, Wang J, Ouyang L, et al. Emerging strategies to overcome resistance to third-generation EGFR inhibitors. J Hematol Oncol. 2022;15(1):94.
Li J, Li P, Shao J, Liang S, Wan Y, Zhang Q, et al. Emerging role of noncoding RNAs in EGFR TKI-resistant lung cancer. Cancers (Basel). 2022;14(18):4423.
Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–25.
Rahman SU, Huang Y, Zhu L, Chu X, Junejo SA, Zhang Y, et al. Tea polyphenols attenuate liver inflammation by modulating obesity-related genes and down-regulating COX-2 and iNOS expression in high fat-fed dogs. BMC Vet Res. 2020;16(1):234.
Wu YL, Zhang L, Fan Y, Zhou J, Zhang L, Zhou Q, et al. Randomized clinical trial of pembrolizumab vs chemotherapy for previously untreated Chinese patients with PD-L1-positive locally advanced or metastatic non-small-cell lung cancer: KEYNOTE-042 China Study. Int J Cancer. 2021;148(9):2313–20.
Garon EB, Kim JS, Govindan R. Pemetrexed maintenance with or without pembrolizumab in non-squamous non-small cell lung cancer: a cross-trial comparison of KEYNOTE-189 versus PARAMOUNT, PRONOUNCE, and JVBL. Lung Cancer. 2021;151:25–9.
Gadgeel S, Rodriguez-Abreu D, Speranza G, Esteban E, Felip E, Domine M, et al. Updated analysis from KEYNOTE-189: pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol. 2020;38(14):1505–17.
Paz-Ares LG, Ciuleanu TE, Pluzanski A, Lee JS, Gainor JF, Otterson GA, et al. Safety of first-line nivolumab plus ipilimumab in patients with metastatic NSCLC: a pooled analysis of CheckMate 227, CheckMate 568, and CheckMate 817. J Thorac Oncol. 2023;18(1):79–92.
Kennedy LB, Salama AKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. 2020;70(2):86–104.
Shimabukuro-Vornhagen A, Boll B, Schellongowski P, Valade S, Metaxa V, Azoulay E, et al. Critical care management of chimeric antigen receptor T-cell therapy recipients. CA Cancer J Clin. 2022;72(1):78–93.
Kandra P, Nandigama R, Eul B, Huber M, Kobold S, Seeger W, et al. Utility and drawbacks of chimeric antigen receptor T cell (CAR-T) therapy in lung cancer. Front Immunol. 2022;13: 903562.
Gainor JF. Adjuvant PD-L1 blockade in non-small-cell lung cancer. Lancet. 2021;398(10308):1281–3.
Sun JM, Shen L, Shah MA, Enzinger P, Adenis A, Doi T, et al. Pembrolizumab plus chemotherapy versus chemotherapy alone for first-line treatment of advanced oesophageal cancer (KEYNOTE-590): a randomised, placebo-controlled, phase 3 study. Lancet. 2021;398(10302):759–71.
Wakelee H, Liberman M, Kato T, Tsuboi M, Lee SH, Gao S, et al. Perioperative pembrolizumab for early-stage non-small-cell lung cancer. N Engl J Med. 2023;389(6):491–503.
Mao S, Yang S, Liu X, Li X, Wang Q, Zhang Y, et al. Molecular correlation of response to pyrotinib in advanced NSCLC with HER2 mutation: biomarker analysis from two phase II trials. Exp Hematol Oncol. 2023;12(1):53.
Tolcher AW. Antibody drug conjugates: lessons from 20 years of clinical experience. Ann Oncol. 2016;27(12):2168–72.
Tarantino P, Carmagnani Pestana R, Corti C, Modi S, Bardia A, Tolaney SM, et al. Antibody–drug conjugates: Smart chemotherapy delivery across tumor histologies. CA Cancer J Clin. 2022;72(2):165–82.
De Cecco M, Galbraith DN, McDermott LL. What makes a good antibody–drug conjugate? Expert Opin Biol Ther. 2021;21(7):841–7.
Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8(6):473–80.
Schwartz RS. Paul Ehrlich’s magic bullets. N Engl J Med. 2004;350(11):1079–80.
Yamada K, Ito Y. Recent chemical approaches for site-specific conjugation of native antibodies: technologies toward next-generation antibody–drug conjugates. ChemBioChem. 2019;20(21):2729–37.
Jain N, Smith SW, Ghone S, Tomczuk B. Current ADC linker chemistry. Pharm Res. 2015;32(11):3526–40.
Tsuchikama K, An Z. Antibody–drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018;9(1):33–46.
Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–7.
Kaplon H, Crescioli S, Chenoweth A, Visweswaraiah J, Reichert JM. Antibodies to watch in 2023. MAbs. 2023;15(1):2153410.
Aires da Silva F, Corte-Real S, Goncalves J. Recombinant antibodies as therapeutic agents: pathways for modeling new biodrugs. BioDrugs. 2008;22(5):301–14.
Matsumura Y. Cancer stromal targeting therapy to overcome the pitfall of EPR effect. Adv Drug Deliv Rev. 2020;154–155:142–50.
Hock MB, Thudium KE, Carrasco-Triguero M, Schwabe NF. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015;17(1):35–43.
Abdollahpour-Alitappeh M, Lotfinia M, Gharibi T, Mardaneh J, Farhadihosseinabadi B, Larki P, et al. Antibody–drug conjugates (ADCs) for cancer therapy: strategies, challenges, and successes. J Cell Physiol. 2019;234(5):5628–42.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 2023;41(8):1501–10.
Acchione M, Kwon H, Jochheim CM, Atkins WM. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody–drug conjugates. MAbs. 2012;4(3):362–72.
Almagro JC, Fransson J. Humanization of antibodies. Front Biosci. 2008;13:1619–33.
Redman JM, Hill EM, AlDeghaither D, Weiner LM. Mechanisms of action of therapeutic antibodies for cancer. Mol Immunol. 2015;67(2 Pt A):28–45.
Rassy E, Delaloge S. A second-generation antibody–drug conjugate to treat HER2-positive breast cancer. Lancet. 2023;401(10371):80–1.
Zaytsev D, Girshova L, Ivanov V, Budaeva I, Motorin D, Badaev R, et al. Rapid efficacy of gemtuzumab ozogamicin in refractory AML patients with pulmonary and kidney failure. Biology (Basel). 2020;9(2):28.
Najminejad Z, Dehghani F, Mirzaei Y, Mer AH, Saghi SA, Abdolvahab MH, et al. Clinical perspective: antibody–drug conjugates for the treatment of HER2-positive breast cancer. Mol Ther. 2023;31(7):1874–903.
Panowski S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6(1):34–45.
Wu M, Huang W, Yang N, Liu Y. Learn from antibody–drug conjugates: consideration in the future construction of peptide‒drug conjugates for cancer therapy. Exp Hematol Oncol. 2022;11(1):93.
Wei Q, Li P, Yang T, Zhu J, Sun L, Zhang Z, et al. The promise and challenges of combination therapies with antibody–drug conjugates in solid tumors. J Hematol Oncol. 2024;17(1):1.
Pomeroy AE, Schmidt EV, Sorger PK, Palmer AC. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer. 2022;8(11):915–29.
Quanz M, Hagemann UB, Zitzmann-Kolbe S, Stelte-Ludwig B, Golfier S, Elbi C, et al. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget. 2018;9(75):34103–21.
Ponte JF, Ab O, Lanieri L, Lee J, Coccia J, Bartle LM, et al. Mirvetuximab Soravtansine (IMGN853), a folate receptor alpha-targeting antibody–drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia. 2016;18(12):775–84.
Moore KN, O’Malley DM, Vergote I, Martin LP, Gonzalez-Martin A, Malek K, et al. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRalpha)-targeting antibody–drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecol Oncol. 2018;151(1):46–52.
O’Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRalpha)-targeting antibody–drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2020;157(2):379–85.
Kan S, Koido S, Okamoto M, Hayashi K, Ito M, Kamata Y, et al. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer. 2015;15:726.
Mamounas EP, Untch M, Mano MS, Huang CS, Geyer CE Jr, von Minckwitz G, et al. Adjuvant T-DM1 versus trastuzumab in patients with residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer: subgroup analyses from KATHERINE. Ann Oncol. 2021;32(8):1005–14.
Risbridger GP, Davis ID, Birrell SN, Tilley WD. Breast and prostate cancer: more similar than different. Nat Rev Cancer. 2010;10(3):205–12.
Bennardo L, Passante M, Cameli N, Cristaudo A, Patruno C, Nistico SP, et al. Skin manifestations after ionizing radiation exposure: a systematic review. Bioengineering (Basel). 2021;8(11):153.
Dadey DYA, Kapoor V, Hoye K, Khudanyan A, Collins A, Thotala D, et al. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin Cancer Res. 2017;23(10):2556–64.
Lewis CD, Singh AK, Hsu FF, Thotala D, Hallahan DE, Kapoor V. Targeting a radiosensitizing antibody–drug conjugate to a radiation-inducible antigen. Clin Cancer Res. 2021;27(11):3224–33.
Hingorani DV, Doan MK, Camargo MF, Aguilera J, Song SM, Pizzo D, et al. Precision chemoradiotherapy for HER2 tumors using antibody conjugates of an auristatin derivative with reduced cell permeability. Mol Cancer Ther. 2020;19(1):157–67.
Adams SR, Yang HC, Savariar EN, Aguilera J, Crisp JL, Jones KA, et al. Anti-tubulin drugs conjugated to anti-ErbB antibodies selectively radiosensitize. Nat Commun. 2016;7:13019.
Salvestrini V, Kim K, Caini S, Alkner S, Ekholm M, Skytta T, et al. Safety profile of trastuzumab-emtansine (T-DM1) with concurrent radiation therapy: a systematic review and meta-analysis. Radiother Oncol. 2023;186: 109805.
Cilliers C, Guo H, Liao J, Christodolu N, Thurber GM. Multiscale modeling of antibody–drug conjugates: connecting tissue and cellular distribution to whole animal pharmacokinetics and potential implications for efficacy. AAPS J. 2016;18(5):1117–30.
Gilbert L, Oaknin A, Matulonis UA, Mantia-Smaldone GM, Lim PC, Castro CM, et al. Safety and efficacy of mirvetuximab soravtansine, a folate receptor alpha (FRalpha)-targeting antibody–drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2023;170:241–7.
Oosting SF, Brouwers AH, van Es SC, Nagengast WB, Oude Munnink TH, Lub-de Hooge MN, et al. 89Zr-bevacizumab PET visualizes heterogeneous tracer accumulation in tumor lesions of renal cell carcinoma patients and differential effects of antiangiogenic treatment. J Nucl Med. 2015;56(1):63–9.
Cao W, Xing H, Li Y, Tian W, Song Y, Jiang Z, et al. Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark Res. 2022;10(1):38.
Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody–drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123(20):3128–38.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25(6):920–8.
Bauzon M, Drake PM, Barfield RM, Cornali BM, Rupniewski I, Rabuka D. Maytansine-bearing antibody–drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. Oncoimmunology. 2019;8(4): e1565859.
Liu Y, Wang Y, Sun S, Chen Z, Xiang S, Ding Z, et al. Understanding the versatile roles and applications of EpCAM in cancers: from bench to bedside. Exp Hematol Oncol. 2022;11(1):97.
Martin K, Muller P, Schreiner J, Prince SS, Lardinois D, Heinzelmann-Schwarz VA, et al. The microtubule-depolymerizing agent ansamitocin P3 programs dendritic cells toward enhanced anti-tumor immunity. Cancer Immunol Immunother. 2014;63(9):925–38.
Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med. 2015;7(315): 315ra188.
Huang L, Wang R, Xie K, Zhang J, Tao F, Pi C, et al. A HER2 target antibody drug conjugate combined with anti-PD-(L)1 treatment eliminates hHER2+ tumors in hPD-1 transgenic mouse model and contributes immune memory formation. Breast Cancer Res Treat. 2022;191(1):51–61.
Xia L, Wen L, Qin Y, Dobson HE, Zhang T, Comer FI, et al. HER2-targeted antibody–drug conjugate induces host immunity against cancer stem cells. Cell Chem Biol. 2021;28(5):610–24.
Ma W, Xue R, Zhu Z, Farrukh H, Song W, Li T, et al. Increasing cure rates of solid tumors by immune checkpoint inhibitors. Exp Hematol Oncol. 2023;12(1):10.
Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128(2):347–56.
Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen BQ, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21(1):123–33.
Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Kim SB, et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020;21(10):1283–95.
Khera E, Cilliers C, Smith MD, Ganno ML, Lai KC, Keating TA, et al. Quantifying ADC bystander payload penetration with cellular resolution using pharmacodynamic mapping. Neoplasia. 2021;23(2):210–21.
Jin Y, Schladetsch MA, Huang X, Balunas MJ, Wiemer AJ. Stepping forward in antibody–drug conjugate development. Pharmacol Ther. 2022;229: 107917.
Zhao P, Zhang Y, Li W, Jeanty C, Xiang G, Dong Y. Recent advances of antibody drug conjugates for clinical applications. Acta Pharm Sin B. 2020;10(9):1589–600.
Conilh L, Sadilkova L, Viricel W, Dumontet C. Payload diversification: a key step in the development of antibody–drug conjugates. J Hematol Oncol. 2023;16(1):3.
Chau CH, Steeg PS, Figg WD. Antibody–drug conjugates for cancer. Lancet. 2019;394(10200):793–804.
Kaytor MD, Wilkinson KD, Warren ST. Modulating huntingtin half-life alters polyglutamine-dependent aggregate formation and cell toxicity. J Neurochem. 2004;89(4):962–73.
Ferraro E, Drago JZ, Modi S. Implementing antibody–drug conjugates (ADCs) in HER2-positive breast cancer: state of the art and future directions. Breast Cancer Res. 2021;23(1):84.
Cheung-Ong K, Giaever G, Nislow C. DNA-damaging agents in cancer chemotherapy: serendipity and chemical biology. Chem Biol. 2013;20(5):648–59.
Elmroth K, Nygren J, Martensson S, Ismail IH, Hammarsten O. Cleavage of cellular DNA by calicheamicin gamma1. DNA Repair (Amst). 2003;2(4):363–74.
Pommier Y. DNA topoisomerase I inhibitors: chemistry, biology, and interfacial inhibition. Chem Rev. 2009;109(7):2894–902.
Gregson SJ, Howard PW, Hartley JA, Brooks NA, Adams LJ, Jenkins TC, et al. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J Med Chem. 2001;44(5):737–48.
Koga Y, Manabe S, Aihara Y, Sato R, Tsumura R, Iwafuji H, et al. Antitumor effect of antitissue factor antibody-MMAE conjugate in human pancreatic tumor xenografts. Int J Cancer. 2015;137(6):1457–66.
Yao X, Jiang J, Wang X, Huang C, Li D, Xie K, et al. A novel humanized anti-HER2 antibody conjugated with MMAE exerts potent anti-tumor activity. Breast Cancer Res Treat. 2015;153(1):123–33.
Dan N, Setua S, Kashyap VK, Khan S, Jaggi M, Yallapu MM, et al. Antibody–drug conjugates for cancer therapy: chemistry to clinical implications. Pharmaceuticals (Basel). 2018;11(2):327–44.
Abdollahpour-Alitappeh M, Hashemi Karouei SM, Lotfinia M, Amanzadeh A, Habibi-Anbouhi M. A developed antibody–drug conjugate rituximab-vcMMAE shows a potent cytotoxic activity against CD20-positive cell line. Artif Cells Nanomed Biotechnol. 2018;46(sup2):1–8.
Bourillon L, Bourgier C, Gaborit N, Garambois V, Lles E, Zampieri A, et al. An auristatin-based antibody–drug conjugate targeting HER3 enhances the radiation response in pancreatic cancer. Int J Cancer. 2019;145(7):1838–51.
Chen H, Lin Z, Arnst KE, Miller DD, Li W. Tubulin inhibitor-based antibody–drug conjugates for cancer therapy. Molecules. 2017;22(8):1281.
Nittoli T, Delfino F, Kelly M, Carosso S, Markotan T, Kunz A, et al. Antibody drug conjugates of cleavable amino-benzoyl-maytansinoids. Bioorg Med Chem. 2020;28(23): 115785.
Yang H, Ganguly A, Cabral F. Inhibition of cell migration and cell division correlates with distinct effects of microtubule inhibiting drugs. J Biol Chem. 2010;285(42):32242–50.
Campos MP, Konecny GE. The target invites a foe: antibody–drug conjugates in gynecologic oncology. Curr Opin Obstet Gynecol. 2018;30(1):44–50.
Lopus M. Antibody-DM1 conjugates as cancer therapeutics. Cancer Lett. 2011;307(2):113–8.
Shen Y, Yang T, Cao X, Zhang Y, Zhao L, Li H, et al. Conjugation of DM1 to anti-CD30 antibody has potential antitumor activity in CD30-positive hematological malignancies with lower systemic toxicity. MAbs. 2019;11(6):1149–61.
Wu Y, Li W, Chen X, Wang H, Su S, Xu Y, et al. DOG1 as a novel antibody–drug conjugate target for the treatment of multiple gastrointestinal tumors and liver metastasis. Front Immunol. 2023;14:1051506.
Mills A, Gago F. Structural and mechanistic insight into DNA bending by antitumour calicheamicins. Org Biomol Chem. 2021;19(30):6707–17.
Damelin M, Bankovich A, Park A, Aguilar J, Anderson W, Santaguida M, et al. Anti-EFNA4 calicheamicin conjugates effectively target triple-negative breast and ovarian tumor-initiating cells to result in sustained tumor regressions. Clin Cancer Res. 2015;21(18):4165–73.
Yao HP, Zhao H, Hudson R, Tong XM, Wang MH. Duocarmycin-based antibody–drug conjugates as an emerging biotherapeutic entity for targeted cancer therapy: pharmaceutical strategy and clinical progress. Drug Discov Today. 2021;26(8):1857–74.
van der Lee MM, Groothuis PG, Ubink R, van der Vleuten MA, van Achterberg TA, Loosveld EM, et al. The preclinical profile of the duocarmycin-based HER2-targeting ADC SYD985 predicts for clinical benefit in low HER2-expressing breast cancers. Mol Cancer Ther. 2015;14(3):692–703.
Abuhelwa Z, Alloghbi A, Alqahtani A, Nagasaka M. Trastuzumab deruxtecan-induced interstitial lung disease/pneumonitis in ERBB2-positive advanced solid malignancies: a systematic review. Drugs. 2022;82(9):979–87.
Kung Sutherland MS, Walter RB, Jeffrey SC, Burke PJ, Yu C, Kostner H, et al. SGN-CD33A: a novel CD33-targeting antibody–drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122(8):1455–63.
Drake PM, Rabuka D. Recent developments in ADC technology: preclinical studies signal future clinical trends. BioDrugs. 2017;31(6):521–31.
Modi S, Saura C, Yamashita T, Park YH, Kim SB, Tamura K, et al. Trastuzumab deruxtecan in previously treated HER2-positive breast cancer. N Engl J Med. 2020;382(7):610–21.
Doi T, Shitara K, Naito Y, Shimomura A, Fujiwara Y, Yonemori K, et al. Safety, pharmacokinetics, and antitumour activity of trastuzumab deruxtecan (DS-8201), a HER2-targeting antibody–drug conjugate, in patients with advanced breast and gastric or gastro-oesophageal tumours: a phase 1 dose-escalation study. Lancet Oncol. 2017;18(11):1512–22.
Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody–drug conjugate, trastuzumab Deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17(7):1494–503.
Yonesaka K, Takegawa N, Watanabe S, Haratani K, Kawakami H, Sakai K, et al. An HER3-targeting antibody–drug conjugate incorporating a DNA topoisomerase I inhibitor U3–1402 conquers EGFR tyrosine kinase inhibitor-resistant NSCLC. Oncogene. 2019;38(9):1398–409.
Mousavizadeh A, Jabbari A, Akrami M, Bardania H. Cell targeting peptides as smart ligands for targeting of therapeutic or diagnostic agents: a systematic review. Colloids Surf B Biointerfaces. 2017;158:507–17.
Deng X, Mai R, Zhang C, Yu D, Ren Y, Li G, et al. Discovery of novel cell-penetrating and tumor-targeting peptide‒drug conjugate (PDC) for programmable delivery of paclitaxel and cancer treatment. Eur J Med Chem. 2021;213: 113050.
Chen X, Zhang XY, Shen Y, Fan LL, Ren ML, Wu YP. Synthetic paclitaxel-octreotide conjugate reversing the resistance of A2780/Taxol to paclitaxel in xenografted tumor in nude mice. Oncotarget. 2016;7(50):83451–61.
Redko B, Tuchinsky H, Segal T, Tobi D, Luboshits G, Ashur-Fabian O, et al. Toward the development of a novel non-RGD cyclic peptide drug conjugate for treatment of human metastatic melanoma. Oncotarget. 2017;8(1):757–68.
Zhang P, Cheetham AG, Lock LL, Cui H. Cellular uptake and cytotoxicity of drug-peptide conjugates regulated by conjugation site. Bioconjug Chem. 2013;24(4):604–13.
Gilad Y, Noy E, Senderowitz H, Albeck A, Firer MA, Gellerman G. Dual-drug RGD conjugates provide enhanced cytotoxicity to melanoma and non-small lung cancer cells. Biopolymers. 2016;106(2):160–71.
Hagihara Y, Saerens D. Engineering disulfide bonds within an antibody. Biochim Biophys Acta. 2014;1844(11):2016–23.
Gregorc V, Cavina R, Novello S, Grossi F, Lazzari C, Capelletto E, et al. NGR-hTNF and Doxorubicin as second-line treatment of patients with small cell lung cancer. Oncologist. 2018;23(10):1133.
He S, Cen B, Liao L, Wang Z, Qin Y, Wu Z, et al. A tumor-targeting cRGD-EGFR siRNA conjugate and its anti-tumor effect on glioblastoma in vitro and in vivo. Drug Deliv. 2017;24(1):471–81.
Hammond SM, Hazell G, Shabanpoor F, Saleh AF, Bowerman M, Sleigh JN, et al. Systemic peptide-mediated oligonucleotide therapy improves long-term survival in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2016;113(39):10962–7.
Su Z, Xiao D, Xie F, Liu L, Wang Y, Fan S, et al. Antibody–drug conjugates: recent advances in linker chemistry. Acta Pharm Sin B. 2021;11(12):3889–907.
Birrer MJ, Moore KN, Betella I, Bates RC. Antibody–drug conjugate-based therapeutics: state of the science. J Natl Cancer Inst. 2019;111(6):538–49.
Mohit E, Rafati S. Chemokine-based immunotherapy: delivery systems and combination therapies. Immunotherapy. 2012;4(8):807–40.
Rao C, Rangan VS, Deshpande S. Challenges in antibody–drug conjugate discovery: a bioconjugation and analytical perspective. Bioanalysis. 2015;7(13):1561–4.
Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, et al. Antibody–drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66(6):3214–21.
Oflazoglu E, Stone IJ, Gordon K, Wood CG, Repasky EA, Grewal IS, et al. Potent anticarcinoma activity of the humanized anti-CD70 antibody h1F6 conjugated to the tubulin inhibitor auristatin via an uncleavable linker. Clin Cancer Res. 2008;14(19):6171–80.
Gordon MR, Canakci M, Li L, Zhuang J, Osborne B, Thayumanavan S. Field guide to challenges and opportunities in antibody–drug conjugates for chemists. Bioconjug Chem. 2015;26(11):2198–215.
Chari RV, Miller ML, Widdison WC. Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed Engl. 2014;53(15):3796–827.
van Berkel SS, van Delft FL. Enzymatic strategies for (near) clinical development of antibody–drug conjugates. Drug Discov Today Technol. 2018;30:3–10.
Pillow TH, Sadowsky JD, Zhang D, Yu SF, Del Rosario G, Xu K, et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem Sci. 2017;8(1):366–70.
Wang Y, Fan S, Xiao D, Xie F, Li W, Zhong W, et al. Novel silyl ether-based acid-cleavable antibody-MMAE conjugates with appropriate stability and efficacy. Cancers (Basel). 2019;11(7):957.
Corti C, Giugliano F, Nicolo E, Ascione L, Curigliano G. Antibody–drug conjugates for the treatment of breast cancer. Cancers (Basel). 2021;13(12):2898.
Parigger J, Zwaan CM, Reinhardt D, Kaspers GJ. Dose-related efficacy and toxicity of gemtuzumab ozogamicin in pediatric acute myeloid leukemia. Expert Rev Anticancer Therapy. 2016;16(2):137–46.
Zhang D, Fourie-O’Donohue A, Dragovich PS, Pillow TH, Sadowsky JD, Kozak KR, et al. Catalytic cleavage of disulfide bonds in small molecules and linkers of antibody–drug conjugates. Drug Metab Dispos. 2019;47(10):1156–63.
Pallardo FV, Markovic J, Garcia JL, Vina J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol Aspects Med. 2009;30(1–2):77–85.
Doronina SO, Bovee TD, Meyer DW, Miyamoto JB, Anderson ME, Morris-Tilden CA, et al. Novel peptide linkers for highly potent antibody–auristatin conjugate. Bioconjug Chem. 2008;19(10):1960–3.
Song Q, Chuan X, Chen B, He B, Zhang H, Dai W, et al. A smart tumor targeting peptide‒drug conjugate, pHLIP-SS-DOX: synthesis and cellular uptake on MCF-7 and MCF-7/Adr cells. Drug Deliv. 2016;23(5):1734–46.
Jeffrey SC, Nguyen MT, Moser RF, Meyer DL, Miyamoto JB, Senter PD. Minor groove binder antibody conjugates employing a water soluble beta-glucuronide linker. Bioorg Med Chem Lett. 2007;17(8):2278–80.
Chang M, Zhang F, Wei T, Zuo T, Guan Y, Lin G, et al. Smart linkers in polymer-drug conjugates for tumor-targeted delivery. J Drug Target. 2016;24(6):475–91.
Burns KE, Hensley H, Robinson MK, Thevenin D. Therapeutic efficacy of a family of pHLIP-MMAF conjugates in cancer cells and mouse models. Mol Pharm. 2017;14(2):415–22.
Chen Z, Zhang P, Cheetham AG, Moon JH, Moxley JW Jr, Lin YA, et al. Controlled release of free doxorubicin from peptide‒drug conjugates by drug loading. J Control Release. 2014;191:123–30.
Min J, Feng Q, Liao W, Liang Y, Gong C, Li E, et al. IFITM3 promotes hepatocellular carcinoma invasion and metastasis by regulating MMP9 through p38/MAPK signaling. FEBS Open Bio. 2018;8(8):1299–311.
Huang H, Jin H, Zhao H, Wang J, Li X, Yan H, et al. RhoGDIbeta promotes Sp1/MMP-2 expression and bladder cancer invasion through perturbing miR-200c-targeted JNK2 protein translation. Mol Oncol. 2017;11(11):1579–94.
Qin SY, Feng J, Rong L, Jia HZ, Chen S, Liu XJ, et al. Theranostic GO-based nanohybrid for tumor induced imaging and potential combinational tumor therapy. Small. 2014;10(3):599–608.
Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–55.
Spangler B, Fontaine SD, Shi Y, Sambucetti L, Mattis AN, Hann B, et al. A novel tumor-activated prodrug strategy targeting ferrous iron is effective in multiple preclinical cancer models. J Med Chem. 2016;59(24):11161–70.
Spangler B, Kline T, Hanson J, Li X, Zhou S, Wells JA, et al. Toward a ferrous iron-cleavable linker for antibody–drug conjugates. Mol Pharm. 2018;15(5):2054–9.
Kern JC, Dooney D, Zhang R, Liang L, Brandish PE, Cheng M, et al. Novel phosphate modified cathepsin B linkers: improving aqueous solubility and enhancing payload scope of ADCs. Bioconjug Chem. 2016;27(9):2081–8.
Kern JC, Cancilla M, Dooney D, Kwasnjuk K, Zhang R, Beaumont M, et al. Discovery of pyrophosphate diesters as tunable, soluble, and bioorthogonal linkers for site-specific antibody–drug conjugates. J Am Chem Soc. 2016;138(4):1430–45.
Li J, Xiao D, Xie F, Li W, Zhao L, Sun W, et al. Novel antibody–drug conjugate with UV-controlled cleavage mechanism for cytotoxin release. Bioorg Chem. 2021;111: 104475.
Wang Y, Liu L, Fan S, Xiao D, Xie F, Li W, et al. Antibody–drug conjugate using ionized Cys-Linker-MMAE as the potent payload shows optimal therapeutic safety. Cancers (Basel). 2020;12(3):744.
Vrettos EI, Mezo G, Tzakos AG. On the design principles of peptide‒drug conjugates for targeted drug delivery to the malignant tumor site. Beilstein J Org Chem. 2018;14:930–54.
Gregson SJ, Masterson LA, Wei B, Pillow TH, Spencer SD, Kang GD, et al. Pyrrolobenzodiazepine dimer antibody–drug conjugates: synthesis and evaluation of noncleavable drug-linkers. J Med Chem. 2017;60(23):9490–507.
Alas M, Saghaeidehkordi A, Kaur K. Peptide–drug conjugates with different linkers for cancer therapy. J Med Chem. 2021;64(1):216–32.
Caculitan NG, Dela Cruz Chuh J, Ma Y, Zhang D, Kozak KR, Liu Y, et al. Cathepsin B is dispensable for cellular processing of cathepsin B-cleavable antibody–drug conjugates. Cancer Res. 2017;77(24):7027–37.
Wei B, Gunzner-Toste J, Yao H, Wang T, Wang J, Xu Z, et al. Discovery of peptidomimetic antibody–drug conjugate linkers with enhanced protease specificity. J Med Chem. 2018;61(3):989–1000.
Bargh JD, Walsh SJ, Isidro-Llobet A, Omarjee S, Carroll JS, Spring DR. Sulfatase-cleavable linkers for antibody–drug conjugates. Chem Sci. 2020;11(9):2375–80.
Xiao D, Zhao L, Xie F, Fan S, Liu L, Li W, et al. A bifunctional molecule-based strategy for the development of theranostic antibody–drug conjugate. Theranostics. 2021;11(6):2550–63.
Kolodych S, Michel C, Delacroix S, Koniev O, Ehkirch A, Eberova J, et al. Development and evaluation of beta-galactosidase-sensitive antibody–drug conjugates. Eur J Med Chem. 2017;142:376–82.
Nani RR, Gorka AP, Nagaya T, Kobayashi H, Schnermann MJ. Near-IR light-mediated cleavage of antibody–drug conjugates using cyanine photocages. Angew Chem Int Ed Engl. 2015;54(46):13635–8.
Zang C, Wang H, Li T, Zhang Y, Li J, Shang M, et al. A light-responsive, self-immolative linker for controlled drug delivery via peptide- and protein-drug conjugates. Chem Sci. 2019;10(39):8973–80.
Wang X, Liu Y, Fan X, Wang J, Ngai WSC, Zhang H, et al. Copper-Triggered Bioorthogonal Cleavage Reactions for Reversible Protein and Cell Surface Modifications. J Am Chem Soc. 2019;141(43):17133–41.
Lu Z, Ren Y, Yang L, Jia A, Hu Y, Zhao Y, et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharm Sin B. 2021;11(5):1246–60.
Burke PJ, Hamilton JZ, Jeffrey SC, Hunter JH, Doronina SO, Okeley NM, et al. Optimization of a PEGylated glucuronide-monomethylauristatin E linker for antibody–drug conjugates. Mol Cancer Ther. 2017;16(1):116–23.
Sun X, Ponte JF, Yoder NC, Laleau R, Coccia J, Lanieri L, et al. Effects of drug-antibody ratio on pharmacokinetics, biodistribution, efficacy, and tolerability of antibody-maytansinoid conjugates. Bioconjug Chem. 2017;28(5):1371–81.
Lyon RP, Bovee TD, Doronina SO, Burke PJ, Hunter JH, Neff-LaFord HD, et al. Reducing hydrophobicity of homogeneous antibody–drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol. 2015;33(7):733–5.
Nakada T, Sugihara K, Jikoh T, Abe Y, Agatsuma T. The latest research and development into the antibody–drug conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 cancer therapy. Chem Pharm Bull (Tokyo). 2019;67(3):173–85.
Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–70.
Sau S, Alsaab HO, Kashaw SK, Tatiparti K, Iyer AK. Advances in antibody–drug conjugates: a new era of targeted cancer therapy. Drug Discov Today. 2017;22(10):1547–56.
Yurkovetskiy AV, Bodyak ND, Yin M, Thomas JD, Clardy SM, Conlon PR, et al. Dolaflexin: a novel antibody–drug conjugate platform featuring high drug loading and a controlled bystander effect. Mol Cancer Ther. 2021;20(5):885–95.
Modi S, Park H, Murthy RK, Iwata H, Tamura K, Tsurutani J, et al. Antitumor activity and safety of trastuzumab Deruxtecan in patients with HER2-low-expressing advanced breast cancer: results from a phase Ib study. J Clin Oncol. 2020;38(17):1887–96.
Al-Rohil RN, Torres-Cabala CA, Patel A, Tetzlaff MT, Ivan D, Nagarajan P, et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: a novel finding. J Cutan Pathol. 2016;43(12):1161–6.
Sung M, Tan X, Lu B, Golas J, Hosselet C, Wang F, et al. Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T-DM1). Mol Cancer Ther. 2018;17(1):243–53.
Rios-Luci C, Garcia-Alonso S, Diaz-Rodriguez E, Nadal-Serrano M, Arribas J, Ocana A, et al. Resistance to the antibody–drug conjugate T-DM1 is based in a reduction in lysosomal proteolytic activity. Cancer Res. 2017;77(17):4639–51.
Corbett S, Huang S, Zammarchi F, Howard PW, van Berkel PH, Hartley JA. The role of specific ATP-binding cassette transporters in the acquired resistance to pyrrolobenzodiazepine dimer-containing antibody–drug conjugates. Mol Cancer Ther. 2020;19(9):1856–65.
Cianfriglia M. The biology of MDR1-P-glycoprotein (MDR1-Pgp) in designing functional antibody drug conjugates (ADCs): the experience of gemtuzumab ozogamicin. Ann Ist Super Sanita. 2013;49(2):150–68.
Hua G, Zhang X, Zhang M, Wang Q, Chen X, Yu R, et al. Real-world circulating tumor DNA analysis depicts resistance mechanism and clonal evolution in ALK inhibitor-treated lung adenocarcinoma patients. ESMO Open. 2022;7(1): 100337.
Rosen DB, Harrington KH, Cordeiro JA, Leung LY, Putta S, Lacayo N, et al. AKT signaling as a novel factor associated with in vitro resistance of human AML to gemtuzumab ozogamicin. PLoS ONE. 2013;8(1): e53518.
Moore J, Seiter K, Kolitz J, Stock W, Giles F, Kalaycio M, et al. A Phase II study of Bcl-2 antisense (oblimersen sodium) combined with gemtuzumab ozogamicin in older patients with acute myeloid leukemia in first relapse. Leuk Res. 2006;30(7):777–83.
Dornan D, Bennett F, Chen Y, Dennis M, Eaton D, Elkins K, et al. Therapeutic potential of an anti-CD79b antibody–drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood. 2009;114(13):2721–9.
Zoeller JJ, Vagodny A, Daniels VW, Taneja K, Tan BY, DeRose YS, et al. Navitoclax enhances the effectiveness of EGFR-targeted antibody–drug conjugates in PDX models of EGFR-expressing triple-negative breast cancer. Breast Cancer Res. 2020;22(1):132.
Collins DM, Bossenmaier B, Kollmorgen G, Niederfellner G. Acquired resistance to antibody–drug conjugates. Cancers (Basel). 2019;11(3):394.
Wagland R, Richardson A, Ewings S, Armes J, Lennan E, Hankins M, et al. Prevalence of cancer chemotherapy-related problems, their relation to health-related quality of life and associated supportive care: a cross-sectional survey. Support Care Cancer. 2016;24(12):4901–11.
Criscitiello C, Morganti S, Curigliano G. Antibody–drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol. 2021;14(1):20.
Damelin M, Zhong W, Myers J, Sapra P. Evolving strategies for target selection for antibody–drug conjugates. Pharm Res. 2015;32(11):3494–507.
Diamantis N, Banerji U. Antibody–drug conjugates—an emerging class of cancer treatment. Br J Cancer. 2016;114(4):362–7.
Kovtun YV, Goldmacher VS. Cell killing by antibody–drug conjugates. Cancer Lett. 2007;255(2):232–40.
Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs. 2013;5(1):13–21.
Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular released payload influences potency and bystander-killing effects of antibody–drug conjugates in preclinical models. Cancer Res. 2016;76(9):2710–9.
Coats S, Williams M, Kebble B, Dixit R, Tseng L, Yao NS, et al. Antibody–drug conjugates: future directions in clinical and translational strategies to improve the therapeutic index. Clin Cancer Res. 2019;25(18):5441–8.
Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody–drug conjugates: a comprehensive review. Mol Cancer Res. 2020;18(1):3–19.
Siegel MM, Tabei K, Kunz A, Hollander IJ, Hamann RR, Bell DH, et al. Calicheamicin derivatives conjugated to monoclonal antibodies: determination of loading values and distributions by infrared and UV matrix-assisted laser desorption/ionization mass spectrometry and electrospray ionization mass spectrometry. Anal Chem. 1997;69(14):2716–26.
Hoffmann RM, Coumbe BGT, Josephs DH, Mele S, Ilieva KM, Cheung A, et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology. 2018;7(3): e1395127.
Prince HM, Kim YH, Horwitz SM, Dummer R, Scarisbrick J, Quaglino P, et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): an international, open-label, randomised, phase 3, multicentre trial. Lancet. 2017;390(10094):555–66.
Erickson HK, Widdison WC, Mayo MF, Whiteman K, Audette C, Wilhelm SD, et al. Tumor delivery and in vivo processing of disulfide-linked and thioether-linked antibody-maytansinoid conjugates. Bioconjug Chem. 2010;21(1):84–92.
Lucas AT, Price LSL, Schorzman AN, Storrie M, Piscitelli JA, Razo J, et al. Factors affecting the pharmacology of antibody–drug conjugates. Antibodies (Basel). 2018;7(1):10.
Strop P, Delaria K, Foletti D, Witt JM, Hasa-Moreno A, Poulsen K, et al. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat Biotechnol. 2015;33(7):694–6.
Costa MJ, Kudaravalli J, Ma JT, Ho WH, Delaria K, Holz C, et al. Optimal design, anti-tumour efficacy and tolerability of anti-CXCR4 antibody drug conjugates. Sci Rep. 2019;9(1):2443.
Wakileh GA, Bierholz P, Kotthoff M, Skowron MA, Bremmer F, Stephan A, et al. Molecular characterization of the CXCR4 / CXCR7 axis in germ cell tumors and its targetability using nanobody-drug-conjugates. Exp Hematol Oncol. 2023;12(1):96.
Yurkovetskiy AV, Yin M, Bodyak N, Stevenson CA, Thomas JD, Hammond CE, et al. A polymer-based antibody-vinca drug conjugate platform: characterization and preclinical efficacy. Cancer Res. 2015;75(16):3365–72.
Simmons JK, Burke PJ, Cochran JH, Pittman PG, Lyon RP. Reducing the antigen-independent toxicity of antibody–drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicol Appl Pharmacol. 2020;392: 114932.
Shao T, Chen T, Chen Y, Liu X, Chen YL, Wang Q, et al. Construction of paclitaxel-based antibody–drug conjugates with a PEGylated linker to achieve superior therapeutic index. Signal Transduct Target Ther. 2020;5(1):132.
Fu Z, Li S, Han S, Shi C, Zhang Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct Target Ther. 2022;7(1):93.
Shim H. Bispecific antibodies and antibody–drug conjugates for cancer therapy: technological considerations. Biomolecules. 2020;10(3):360.
Decary S, Berne PF, Nicolazzi C, Lefebvre AM, Dabdoubi T, Cameron B, et al. Preclinical activity of SAR408701: a novel anti-CEACAM5-maytansinoid antibody–drug conjugate for the treatment of CEACAM5-positive epithelial tumors. Clin Cancer Res. 2020;26(24):6589–99.
Xu S. Internalization, trafficking, intracellular processing and actions of antibody–drug conjugates. Pharm Res. 2015;32(11):3577–83.
Li X, Zhang Y, Li B, Li J, Qiu Y, Zhu Z, et al. An immunomodulatory antibody–drug conjugate (ADC) targeting BDCA2 strongly suppresses pDC function and glucocorticoid responsive genes. Rheumatology (Oxford). 2024;63(1):242–50.
DaSilva JO, Yang K, Surriga O, Nittoli T, Kunz A, Franklin MC, et al. A biparatopic antibody–drug conjugate to treat MET-expressing cancers, including those that are unresponsive to MET pathway blockade. Mol Cancer Ther. 2021;20(10):1966–76.
Singh AP, Seigel GM, Guo L, Verma A, Wong GG, Cheng HP, et al. Evolution of the systems pharmacokinetics-pharmacodynamics model for antibody–drug conjugates to characterize tumor heterogeneity and in vivo bystander effect. J Pharmacol Exp Ther. 2020;374(1):184–99.
Singh D, Dheer D, Samykutty A, Shankar R. Antibody drug conjugates in gastrointestinal cancer: from lab to clinical development. J Control Release. 2021;340:1–34.
Trenevska I, Li D, Banham AH. Therapeutic antibodies against intracellular tumor antigens. Front Immunol. 2017;8:1001.
Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327–44.
Lamb YN. Inotuzumab Ozogamicin: first global approval. Drugs. 2017;77(14):1603–10.
Schumacher D, Helma J, Schneider AFL, Leonhardt H, Hackenberger CPR. Nanobodies: chemical functionalization strategies and intracellular applications. Angew Chem Int Ed Engl. 2018;57(9):2314–33.
Hayat SMG, Sahebkar A. Antibody–drug conjugates: smart weapons against cancer. Arch Med Sci. 2020;16(5):1257–62.
Chu Y, Zhou X, Wang X. Antibody–drug conjugates for the treatment of lymphoma: clinical advances and latest progress. J Hematol Oncol. 2021;14(1):88.
Del Re M, Cucchiara F, Petrini I, Fogli S, Passaro A, Crucitta S, et al. erbB in NSCLC as a molecular target: current evidences and future directions. ESMO Open. 2020;5(4): e000724.
Wang Z. ErbB receptors and cancer. Methods Mol Biol. 2017;1652:3–35.
Jebbink M, de Langen AJ, Boelens MC, Monkhorst K, Smit EF. The force of HER2—a druggable target in NSCLC? Cancer Treat Rev. 2020;86: 101996.
Garcia-Alonso S, Ocana A, Pandiella A. Trastuzumab emtansine: mechanisms of action and resistance, clinical progress, and beyond. Trends Cancer. 2020;6(2):130–46.
Kmietowicz Z. NICE approves trastuzumab emtansine after deal with drug company. BMJ. 2017;357: j2930.
Sandmann A, Sasse F, Muller R. Identification and analysis of the core biosynthetic machinery of tubulysin, a potent cytotoxin with potential anticancer activity. Chem Biol. 2004;11(8):1071–9.
Li BT, Shen R, Buonocore D, Olah ZT, Ni A, Ginsberg MS, et al. Ado-trastuzumab emtansine for patients with HER2-mutant lung cancers: results from a phase II basket trial. J Clin Oncol. 2018;36(24):2532–7.
Li BT, Michelini F, Misale S, Cocco E, Baldino L, Cai Y, et al. HER2-mediated internalization of cytotoxic agents in ERBB2 amplified or mutant lung cancers. Cancer Discov. 2020;10(5):674–87.
Hotta K, Aoe K, Kozuki T, Ohashi K, Ninomiya K, Ichihara E, et al. A phase II study of trastuzumab emtansine in HER2-positive non-small cell lung cancer. J Thorac Oncol. 2018;13(2):273–9.
Peters S, Stahel R, Bubendorf L, Bonomi P, Villegas A, Kowalski DM, et al. Trastuzumab emtansine (T-DM1) in patients with previously treated HER2-overexpressing metastatic non-small cell lung cancer: efficacy, safety, and biomarkers. Clin Cancer Res. 2019;25(1):64–72.
Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, et al. HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci U S A. 2009;106(2):474–9.
Cortes J, Kim SB, Chung WP, Im SA, Park YH, Hegg R, et al. Trastuzumab Deruxtecan versus trastuzumab emtansine for breast cancer. N Engl J Med. 2022;386(12):1143–54.
Oostra DR, Macrae ER. Role of trastuzumab emtansine in the treatment of HER2-positive breast cancer. Breast Cancer (Dove Med Press). 2014;6:103–13.
Wolska-Washer A, Robak T. Safety and tolerability of antibody–drug conjugates in cancer. Drug Saf. 2019;42(2):295–314.
Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107(7):1039–46.
Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. 2016;22(20):5097–108.
Tsurutani J, Iwata H, Krop I, Janne PA, Doi T, Takahashi S, et al. Targeting HER2 with Trastuzumab Deruxtecan: a dose-expansion, phase I study in multiple advanced solid tumors. Cancer Discov. 2020;10(5):688–701.
Li BT, Smit EF, Goto Y, Nakagawa K, Udagawa H, Mazieres J, et al. Trastuzumab Deruxtecan in HER2-mutant non-small-cell lung cancer. N Engl J Med. 2022;386(3):241–51.
Riudavets M, Sullivan I, Abdayem P, Planchard D. Targeting HER2 in non-small-cell lung cancer (NSCLC): a glimpse of hope? An updated review on therapeutic strategies in NSCLC harbouring HER2 alterations. ESMO Open. 2021;6(5): 100260.
Reuss JE, Gosa L, Liu SV. Antibody drug conjugates in lung cancer: state of the current therapeutic landscape and future developments. Clin Lung Cancer. 2021;22(6):483–99.
Zhang J, Liu R, Gao S, Li W, Chen Y, Meng Y, et al. Phase I study of A166, an antibody–drug conjugate in advanced HER2-expressing solid tumours. NPJ Breast Cancer. 2023;9(1):28.
Koganemaru S, Kuboki Y, Koga Y, Kojima T, Yamauchi M, Maeda N, et al. U3–1402, a novel HER3-targeting antibody–drug conjugate, for the treatment of colorectal cancer. Mol Cancer Ther. 2019;18(11):2043–50.
Campbell MR, Amin D, Moasser MM. HER3 comes of age: new insights into its functions and role in signaling, tumor biology, and cancer therapy. Clin Cancer Res. 2010;16(5):1373–83.
Mishra R, Patel H, Alanazi S, Yuan L, Garrett JT. HER3 signaling and targeted therapy in cancer. Oncol Rev. 2018;12(1):355.
Janne PA, Baik C, Su WC, Johnson ML, Hayashi H, Nishio M, et al. Efficacy and safety of Patritumab Deruxtecan (HER3-DXd) in EGFR inhibitor-resistant, EGFR-mutated non-small cell lung cancer. Cancer Discov. 2022;12(1):74–89.
Chen R, Manochakian R, James L, Azzouqa AG, Shi H, Zhang Y, et al. Emerging therapeutic agents for advanced non-small cell lung cancer. J Hematol Oncol. 2020;13(1):58.
Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Arrojo R, Liu D, et al. Sacituzumab Govitecan (IMMU-132), an anti-Trop-2/SN-38 antibody–drug conjugate: characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug Chem. 2015;26(5):919–31.
Starodub AN, Ocean AJ, Shah MA, Guarino MJ, Picozzi VJ Jr, Vahdat LT, et al. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab Govitecan, for the treatment of diverse metastatic solid tumors. Clin Cancer Res. 2015;21(17):3870–8.
Bardia A, Messersmith WA, Kio EA, Berlin JD, Vahdat L, Masters GA, et al. Sacituzumab govitecan, a Trop-2-directed antibody–drug conjugate, for patients with epithelial cancer: final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann Oncol. 2021;32(6):746–56.
Heist RS, Guarino MJ, Masters G, Purcell WT, Starodub AN, Horn L, et al. Therapy of advanced non-small-cell lung cancer with an SN-38-Anti-Trop-2 drug conjugate, Sacituzumab Govitecan. J Clin Oncol. 2017;35(24):2790–7.
Levy BP, Felip E, Reck M, Yang JC, Cappuzzo F, Yoneshima Y, et al. TROPION-Lung08: phase III study of datopotamab deruxtecan plus pembrolizumab as first-line therapy for advanced NSCLC. Future Oncol. 2023;19(21):1461–72.
Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29(10):2085–91.
Lee JK, Madison R, Classon A, Gjoerup O, Rosenzweig M, Frampton GM, et al. Characterization of non-small-cell lung cancers with MET Exon 14 skipping alterations detected in tissue or liquid: clinicogenomics and real-world treatment patterns. JCO Precis Oncol. 2021;5:122.
Kron A, Scheffler M, Heydt C, Ruge L, Schaepers C, Eisert AK, et al. Genetic heterogeneity of MET-aberrant NSCLC and its impact on the outcome of immunotherapy. J Thorac Oncol. 2021;16(4):572–82.
Schmid S, Fruh M, Peters S. Targeting MET in EGFR resistance in non-small-cell lung cancer-ready for daily practice? Lancet Oncol. 2020;21(3):320–2.
Wolf J, Seto T, Han JY, Reguart N, Garon EB, Groen HJM, et al. Capmatinib in MET Exon 14-mutated or MET-amplified non-small-cell lung cancer. N Engl J Med. 2020;383(10):944–57.
Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5(8):850–9.
Bubendorf L, Dafni U, Schobel M, Finn SP, Tischler V, Sejda A, et al. Prevalence and clinical association of MET gene overexpression and amplification in patients with NSCLC: results from the European Thoracic Oncology Platform (ETOP) Lungscape project. Lung Cancer. 2017;111:143–9.
Strickler JH, Weekes CD, Nemunaitis J, Ramanathan RK, Heist RS, Morgensztern D, et al. First-in-human phase I, dose-escalation and -expansion study of telisotuzumab vedotin, an antibody–drug conjugate targeting c-Met, in patients with advanced solid tumors. J Clin Oncol. 2018;36(33):3298–306.
Waqar SN, Redman MW, Arnold SM, Hirsch FR, Mack PC, Schwartz LH, et al. A Phase II study of telisotuzumab vedotin in patients with c-MET-positive Stage IV or recurrent squamous cell lung cancer (LUNG-MAP Sub-study S1400K, NCT03574753). Clin Lung Cancer. 2021;22(3):170–7.
Wang J, Anderson MG, Oleksijew A, Vaidya KS, Boghaert ER, Tucker L, et al. ABBV-399, a c-Met antibody–drug conjugate that targets both MET-amplified and c-Met-overexpressing tumors, irrespective of MET pathway dependence. Clin Cancer Res. 2017;23(4):992–1000.
Camidge DR, Morgensztern D, Heist RS, Barve M, Vokes E, Goldman JW, et al. Phase I Study of 2- or 3-week dosing of Telisotuzumab Vedotin, an antibody–drug conjugate targeting c-Met, monotherapy in patients with advanced non-small cell lung carcinoma. Clin Cancer Res. 2021;27(21):5781–92.
Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, et al. A DLL3-targeted antibody–drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7(302):302.
Owen DH, Giffin MJ, Bailis JM, Smit MD, Carbone DP, He K. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol. 2019;12(1):61.
Rudin CM, Reck M, Johnson ML, Blackhall F, Hann CL, Yang JC, et al. Emerging therapies targeting the delta-like ligand 3 (DLL3) in small cell lung cancer. J Hematol Oncol. 2023;16(1):66.
Rosenberg JE, O’Donnell PH, Balar AV, McGregor BA, Heath EI, Yu EY, et al. Pivotal trial of enfortumab vedotin in urothelial carcinoma after platinum and anti-programmed death 1/programmed death ligand 1 therapy. J Clin Oncol. 2019;37(29):2592–600.
Rudin CM, Pietanza MC, Bauer TM, Ready N, Morgensztern D, Glisson BS, et al. Rovalpituzumab tesirine, a DLL3-targeted antibody–drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017;18(1):42–51.
Morgensztern D, Besse B, Greillier L, Santana-Davila R, Ready N, Hann CL, et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin Cancer Res. 2019;25(23):6958–66.
Blackhall F, Jao K, Greillier L, Cho BC, Penkov K, Reguart N, et al. Efficacy and safety of rovalpituzumab tesirine compared with topotecan as second-line therapy in DLL3-high SCLC: results from the phase 3 TAHOE study. J Thorac Oncol. 2021;16(9):1547–58.
Malhotra J, Nikolinakos P, Leal T, Lehman J, Morgensztern D, Patel JD, et al. A phase 1–2 study of rovalpituzumab tesirine in combination with nivolumab plus or minus ipilimumab in patients with previously treated extensive-stage SCLC. J Thorac Oncol. 2021;16(9):1559–69.
Calvo E, Spira A, Miguel M, Kondo S, Gazzah A, Millward M, et al. Safety, pharmacokinetics, and efficacy of budigalimab with rovalpituzumab tesirine in patients with small cell lung cancer. Cancer Treat Res Commun. 2021;28: 100405.
Serzan MT, Farid S, Liu SV. Drugs in development for small cell lung cancer. J Thorac Dis. 2020;12(10):6298–307.
Morgensztern D, Johnson M, Rudin CM, Rossi M, Lazarov M, Brickman D, et al. SC-002 in patients with relapsed or refractory small cell lung cancer and large cell neuroendocrine carcinoma: phase 1 study. Lung Cancer. 2020;145:126–31.
Pei JP, Wang Y, Ma LP, Wang X, Liu L, Zhang Y, et al. AXL antibody and AXL-ADC mediate antitumor efficacy via targeting AXL in tumor-intrinsic epithelial-mesenchymal transition and tumor-associated M2-like macrophage. Acta Pharmacol Sin. 2023;44(6):1290–303.
Yoshimura A, Yamada T, Serizawa M, Uehara H, Tanimura K, Okuma Y, et al. High levels of AXL expression in untreated EGFR-mutated non-small cell lung cancer negatively impacts the use of osimertinib. Cancer Sci. 2023;114(2):606–18.
Boshuizen J, Koopman LA, Krijgsman O, Shahrabi A, van den Heuvel EG, Ligtenberg MA, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody–drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24(2):203–12.
Banerjee S, Drapkin R, Richardson DL, Birrer M. Targeting NaPi2b in ovarian cancer. Cancer Treat Rev. 2023;112: 102489.
Heynemann S, Yu H, Churilov L, Rivalland G, Asadi K, Mosher R, et al. NaPi2b expression in a large surgical Non-Small Cell Lung Cancer (NSCLC) cohort. Clin Lung Cancer. 2022;23(2):e90–8.
Bodyak ND, Mosher R, Yurkovetskiy AV, Yin M, Bu C, Conlon PR, et al. The Dolaflexin-based antibody–drug conjugate XMT-1536 targets the solid tumor lineage antigen SLC34A2/NaPi2b. Mol Cancer Ther. 2021;20(5):896–905.
Andreev J, Thambi N, Perez Bay AE, Delfino F, Martin J, Kelly MP, et al. Bispecific antibodies and antibody–drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol Cancer Ther. 2017;16(4):681–93.
Zhuang C, Guan X, Ma H, Cong H, Zhang W, Miao Z. Small molecule-drug conjugates: a novel strategy for cancer-targeted treatment. Eur J Med Chem. 2019;163:883–95.
Parra ER, Villalobos P, Zhang J, Behrens C, Mino B, Swisher S, et al. Immunohistochemical and image analysis-based study shows that several immune checkpoints are co-expressed in non-small cell lung carcinoma tumors. J Thorac Oncol. 2018;13(6):779–91.
Adams E, Wildiers H, Neven P, Punie K. Sacituzumab govitecan and trastuzumab deruxtecan: two new antibody–drug conjugates in the breast cancer treatment landscape. ESMO Open. 2021;6(4): 100204.
Cho YS, Kim GC, Lee HM, Kim B, Kim HR, Chung SW, et al. Albumin metabolism targeted peptide‒drug conjugate strategy for targeting pan-KRAS mutant cancer. J Control Release. 2022;344:26–38.
Heh E, Allen J, Ramirez F, Lovasz D, Fernandez L, Hogg T, et al. Peptide drug conjugates and their role in cancer therapy. Int J Mol Sci. 2023;24(1):829.
Hoppenz P, Els-Heindl S, Beck-Sickinger AG. Peptide-drug conjugates and their targets in advanced cancer therapies. Front Chem. 2020;8:571.
Kalimuthu K, Lubin BC, Bazylevich A, Gellerman G, Shpilberg O, Luboshits G, et al. Gold nanoparticles stabilize peptide-drug-conjugates for sustained targeted drug delivery to cancer cells. J Nanobiotechnol. 2018;16(1):34.
Ulapane KR, Kopec BM, Moral MEG, Siahaan TJ. Peptides and drug delivery. Adv Exp Med Biol. 2017;1030:167–84.
Lin SM, Lin SC, Hsu JN, Chang CK, Chien CM, Wang YS, et al. Structure-based stabilization of non-native protein-protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J Med Chem. 2020;63(6):3131–41.
Min W, Hou Z, Zhang F, Xie S, Yuan K, Dong H, et al. Computational discovery and biological evaluation of novel inhibitors targeting histone-lysine N-methyltransferase SET7. Bioorg Med Chem. 2020;28(7): 115372.
Luan X, Yuan H, Song Y, Hu H, Wen B, He M, et al. Reappraisal of anticancer nanomedicine design criteria in three types of preclinical cancer models for better clinical translation. Biomaterials. 2021;275: 120910.
Li K, Zhu J, Xu L, Jin J. Rational design of novel phosphoinositide 3-kinase gamma (PI3Kgamma) selective inhibitors: a computational investigation integrating 3D-QSAR, molecular docking and molecular dynamics simulation. Chem Biodivers. 2019;16(7): e1900105.
El Kerdawy AM, Osman AA, Zaater MA. Receptor-based pharmacophore modeling, virtual screening, and molecular docking studies for the discovery of novel GSK-3beta inhibitors. J Mol Model. 2019;25(6):171.
Bohme D, Krieghoff J, Beck-Sickinger AG. Double methotrexate-modified neuropeptide Y analogues express increased toxicity and overcome drug resistance in breast cancer cells. J Med Chem. 2016;59(7):3409–17.
Gregorc V, Gaafar RM, Favaretto A, Grossi F, Jassem J, Polychronis A, et al. NGR-hTNF in combination with best investigator choice in previously treated malignant pleural mesothelioma (NGR015): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2018;19(6):799–811.
Cox N, Kintzing JR, Smith M, Grant GA, Cochran JR. Integrin-targeting knottin peptide-drug conjugates are potent inhibitors of tumor cell proliferation. Angew Chem Int Ed Engl. 2016;55(34):9894–7.
Zhang M, Xu H. Peptide-assembled nanoparticles targeting tumor cells and tumor microenvironment for cancer therapy. Front Chem. 2023;11:1115495.
Salem AF, Wang S, Billet S, Chen JF, Udompholkul P, Gambini L, et al. Reduction of circulating cancer cells and metastases in breast-cancer models by a potent EphA2-agonistic peptide-drug conjugate. J Med Chem. 2018;61(5):2052–61.
Toplak A, Teixeirade Oliveira EF, Schmidt M, Rozeboom HJ, Wijma HJ, Meekels LKM, et al. From thiol-subtilisin to omniligase: design and structure of a broadly applicable peptide ligase. Comput Struct Biotechnol J. 2021;19:1277–87.
Yan W, Li SX, Wei M, Gao H. Identification of MMP9 as a novel key gene in mantle cell lymphoma based on bioinformatic analysis and design of cyclic peptides as MMP9 inhibitors based on molecular docking. Oncol Rep. 2018;40(5):2515–24.
Ge J, Zhang Q, Zeng J, Gu Z, Gao M. Radiolabeling nanomaterials for multimodality imaging: new insights into nuclear medicine and cancer diagnosis. Biomaterials. 2020;228: 119553.
Ma L, Wang C, He Z, Cheng B, Zheng L, Huang K. Peptide-drug conjugate: a novel drug design approach. Curr Med Chem. 2017;24(31):3373–96.
Chang YW, Chen SC, Cheng EC, Ko YP, Lin YC, Kao YR, et al. CD13 (aminopeptidase N) can associate with tumor-associated antigen L6 and enhance the motility of human lung cancer cells. Int J Cancer. 2005;116(2):243–52.
Valentinis B, Porcellini S, Asperti C, Cota M, Zhou D, Di Matteo P, et al. Mechanism of action of the tumor vessel targeting agent NGR-hTNF: role of both NGR peptide and hTNF in cell binding and signaling. Int J Mol Sci. 2019;20(18):4511.
Arosio D, Manzoni L, Corno C, Perego P. Integrin-targeted peptide- and peptidomimetic-drug conjugates for the treatment of tumors. Recent Pat Anticancer Drug Discov. 2017;12(2):148–68.
Nieberler M, Reuning U, Reichart F, Notni J, Wester HJ, Schwaiger M, et al. Exploring the role of RGD-recognizing integrins in cancer. Cancers (Basel). 2017;9(9):116.
Provost C, Prignon A, Rozenblum-Beddok L, Bruyer Q, Dumont S, Merabtene F, et al. Comparison and evaluation of two RGD peptides labelled with (68)Ga or (18)F for PET imaging of angiogenesis in animal models of human glioblastoma or lung carcinoma. Oncotarget. 2018;9(27):19307–16.
Lv X, Liu Z, Xu L, Song E, Song Y. Tetrachlorobenzoquinone exhibits immunotoxicity by inducing neutrophil extracellular traps through a mechanism involving ROS-JNK-NOX2 positive feedback loop. Environ Pollut. 2021;268(Pt B): 115921.
Robitaille MC, Christodoulides JA, Liu J, Kang W, Byers JM, Raphael MP. Problem of diminished cRGD surface activity and what can be done about it. ACS Appl Mater Interfaces. 2020;12(17):19337–44.
Shen Z, Liu T, Yang Z, Zhou Z, Tang W, Fan W, et al. Small-sized gadolinium oxide based nanoparticles for high-efficiency theranostics of orthotopic glioblastoma. Biomaterials. 2020;235: 119783.
Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of (177)Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med. 2017;376(2):125–35.
Liu Q, Zang J, Yang Y, Ling Q, Wu H, Wang P, et al. Head-to-head comparison of (68)Ga-DOTATATE PET/CT and (18)F-FDG PET/CT in localizing tumors with ectopic adrenocorticotropic hormone secretion: a prospective study. Eur J Nucl Med Mol Imaging. 2021;48(13):4386–95.
Zhu W, Cheng Y, Wang X, Yao S, Bai C, Zhao H, et al. Head-to-head comparison of (68)Ga-DOTA-JR11 and (68)Ga-DOTATATE PET/CT in patients with metastatic, well-differentiated neuroendocrine tumors: a prospective study. J Nucl Med. 2020;61(6):897–903.
Kitson SL, Cuccurullo V, Ciarmiello A, Mansi L. Targeted therapy towards cancer-a perspective. Anticancer Agents Med Chem. 2017;17(3):311–7.
Martiniova L, Zielinski RJ, Lin M, DePalatis L, Ravizzini GC. The role of radiolabeled monoclonal antibodies in cancer imaging and ADC treatment. Cancer J. 2022;28(6):446–53.
Zana A, Puig-Moreno C, Bocci M, Gilardoni E, Di Nitto C, Principi L, et al. A comparative analysis of fibroblast activation protein-targeted small molecule-drug, antibody-drug, and peptide-drug conjugates. Bioconjug Chem. 2023;34(7):1205–11.
Leamon CP, Vlahov IR, Reddy JA, Vetzel M, Santhapuram HK, You F, et al. Folate-vinca alkaloid conjugates for cancer therapy: a structure-activity relationship. Bioconjug Chem. 2014;25(3):560–8.
Dutta K, Das R, Medeiros J, Thayumanavan S. Disulfide bridging strategies in viral and nonviral platforms for nucleic acid delivery. Biochemistry. 2021;60(13):966–90.
Dugal-Tessier J, Thirumalairajan S, Jain N. Antibody-oligonucleotide conjugates: a twist to antibody–drug conjugates. J Clin Med. 2021;10(4):838.
Frank MJ, Olsson N, Huang A, Tang SW, Negrin RS, Elias JE, et al. A novel antibody-cell conjugation method to enhance and characterize cytokine-induced killer cells. Cytotherapy. 2020;22(3):135–43.
Li HK, Hsiao CW, Yang SH, Yang HP, Wu TS, Lee CY, et al. A novel off-the-shelf trastuzumab-armed NK cell therapy (ACE1702) using antibody-cell-conjugation technology. Cancers (Basel). 2021;13(11):2724.
Fang S, Brems BM, Olawode EO, Miller JT, Brooks TA, Tumey LN. Design and characterization of immune-stimulating imidazo[4,5-c]quinoline antibody–drug conjugates. Mol Pharm. 2022;19(9):3228–41.
Ackerman SE, Pearson CI, Gregorio JD, Gonzalez JC, Kenkel JA, Hartmann FJ, et al. Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nat Cancer. 2021;2(1):18–33.
Uematsu S, Akira S. Toll-like receptors and innate immunity. J Mol Med (Berl). 2006;84(9):712–25.
Amouzegar A, Chelvanambi M, Filderman JN, Storkus WJ, Luke JJ. STING agonists as cancer therapeutics. Cancers (Basel). 2021;13(11):2695.
He L, Wang L, Wang Z, Li T, Chen H, Zhang Y, et al. Immune modulating antibody–drug conjugate (IM-ADC) for cancer immunotherapy. J Med Chem. 2021;64(21):15716–26.
Shi F, Su J, Wang J, Liu Z, Wang T. Activation of STING inhibits cervical cancer tumor growth through enhancing the anti-tumor immune response. Mol Cell Biochem. 2021;476(2):1015–24.
Jolivet L, Ait Mohamed Amar I, Horiot C, Boursin F, Colas C, Letast S, et al. Intra-domain cysteines (IDC), a new strategy for the development of original antibody fragment-drug conjugates (FDCs). Pharmaceutics. 2022;14(8):1524.
Deonarain MP, Xue Q. Tackling solid tumour therapy with small-format drug conjugates. Antib Ther. 2020;3(4):237–45.
Subedi GP, Barb AW. The structural role of antibody N-glycosylation in receptor interactions. Structure. 2015;23(9):1573–83.
Sun Y, Geng X, Ma Y, Qin Y, Hu S, Xie Y, et al. Artificial base-directed in vivo formulation of aptamer-drug conjugates with albumin for long circulation and targeted delivery. Pharmaceutics. 2022;14(12):2781.
Li Y, Zhao J, Xue Z, Tsang C, Qiao X, Dong L, et al. Aptamer nucleotide analog drug conjugates in the targeting therapy of cancers. Front Cell Dev Biol. 2022;10:1053984.
Zhu G, Niu G, Chen X. Aptamer-drug conjugates. Bioconjug Chem. 2015;26(11):2186–97.
Donaghy H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody–drug conjugates. MAbs. 2016;8(4):659–71.
Masters JC, Nickens DJ, Xuan D, Shazer RL, Amantea M. Clinical toxicity of antibody drug conjugates: a meta-analysis of payloads. Invest New Drugs. 2018;36(1):121–35.
Mahalingaiah PK, Ciurlionis R, Durbin KR, Yeager RL, Philip BK, Bawa B, et al. Potential mechanisms of target-independent uptake and toxicity of antibody–drug conjugates. Pharmacol Ther. 2019;200:110–25.
Matikonda SS, McLaughlin R, Shrestha P, Lipshultz C, Schnermann MJ. Structure-activity relationships of antibody–drug conjugates: a systematic review of chemistry on the trastuzumab scaffold. Bioconjug Chem. 2022;33(7):1241–53.
Wu P, Prachyathipsakul T, Huynh U, Qiu J, Jerry DJ, Thayumanavan S. Optimizing conjugation chemistry, antibody conjugation site, and surface density in antibody-nanogel conjugates (ANCs) for cell-specific drug delivery. Bioconjug Chem. 2023;27:10.
Colombo R, Rich JR. The therapeutic window of antibody drug conjugates: a dogma in need of revision. Cancer Cell. 2022;40(11):1255–63.
Shefet-Carasso L, Benhar I. Antibody-targeted drugs and drug resistance–challenges and solutions. Drug Resist Updat. 2015;18:36–46.
Hafeez U, Parakh S, Gan HK, Scott AM. Antibody–drug conjugates for cancer therapy. Molecules. 2020;25(20):4764.
Singh AP, Sharma S, Shah DK. Quantitative characterization of in vitro bystander effect of antibody–drug conjugates. J Pharmacokinet Pharmacodyn. 2016;43(6):567–82.
Abelman RO, Wu B, Spring LM, Ellisen LW, Bardia A. Mechanisms of resistance to antibody–drug conjugates. Cancers (Basel). 2023;15(4):1278.
Loganzo F, Tan X, Sung M, Jin G, Myers JS, Melamud E, et al. Tumor cells chronically treated with a trastuzumab-maytansinoid antibody–drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther. 2015;14(4):952–63.
Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature. 2019;575(7782):299–309.
Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC toxicity and strategies to increase ADC tolerability. Cancers (Basel). 2023;15(3):713.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody–drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37.
Brun MP, Gauzy-Lazo L. Protocols for lysine conjugation. Methods Mol Biol. 2013;1045:173–87.
Matsuda Y, Mendelsohn BA. An overview of process development for antibody–drug conjugates produced by chemical conjugation technology. Expert Opin Biol Ther. 2021;21(7):963–75.
Fukunaga A, Maeta S, Reema B, Nakakido M, Tsumoto K. Improvement of antibody affinity by introduction of basic amino acid residues into the framework region. Biochem Biophys Rep. 2018;15:81–5.
Beck A, D’Atri V, Ehkirch A, Fekete S, Hernandez-Alba O, Gahoual R, et al. Cutting-edge multi-level analytical and structural characterization of antibody–drug conjugates: present and future. Expert Rev Proteomics. 2019;16(4):337–62.
Chen R, Hou J, Newman E, Kim Y, Donohue C, Liu X, et al. CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol Cancer Ther. 2015;14(6):1376–84.
Strop P, Liu SH, Dorywalska M, Delaria K, Dushin RG, Tran TT, et al. Location matters: site of conjugation modulates stability and pharmacokinetics of antibody drug conjugates. Chem Biol. 2013;20(2):161–7.
Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody–drug conjugates. Nat Biotechnol. 2012;30(2):184–9.
Yamazaki CM, Yamaguchi A, Anami Y, Xiong W, Otani Y, Lee J, et al. Antibody–drug conjugates with dual payloads for combating breast tumor heterogeneity and drug resistance. Nat Commun. 2021;12(1):3528.
Guo J, Kumar S, Chipley M, Marcq O, Gupta D, Jin Z, et al. Characterization and higher-order structure assessment of an interchain cysteine-based ADC: impact of drug loading and distribution on the mechanism of aggregation. Bioconjug Chem. 2016;27(3):604–15.
Best RL, LaPointe NE, Azarenko O, Miller H, Genualdi C, Chih S, et al. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol Appl Pharmacol. 2021;421: 115534.
Lopez de Sa A, Diaz-Tejeiro C, Poyatos-Racionero E, Nieto-Jimenez C, Paniagua-Herranz L, Sanvicente A, et al. Considerations for the design of antibody drug conjugates (ADCs) for clinical development: lessons learned. J Hematol Oncol. 2023;16(1):118.
Bon G, Pizzuti L, Laquintana V, Loria R, Porru M, Marchio C, et al. Loss of HER2 and decreased T-DM1 efficacy in HER2 positive advanced breast cancer treated with dual HER2 blockade: the SePHER Study. J Exp Clin Cancer Res. 2020;39(1):279.
Damaschin C, Goergen H, Kreissl S, Plutschow A, Breywisch F, Mathas S, et al. Brentuximab vedotin-containing escalated BEACOPP variants for newly diagnosed advanced-stage classical Hodgkin lymphoma: follow-up analysis of a randomized phase II study from the German Hodgkin Study Group. Leukemia. 2022;36(2):580–2.
Yu J, Song Y, Tian W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J Hematol Oncol. 2020;13(1):45.
Nicolo E, Giugliano F, Ascione L, Tarantino P, Corti C, Tolaney SM, et al. Combining antibody–drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat Rev. 2022;106: 102395.
Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. Synthetic lethality exploitation by an anti-Trop-2-SN-38 antibody–drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res. 2017;23(13):3405–15.
Tarantino P, Curigliano G, Tolaney SM. Navigating the HER2-low paradigm in breast oncology: new standards. Future Horizons Cancer Discov. 2022;12(9):2026–30.
Passaro A, Peters S. Targeting HER2-mutant NSCLC—the light is on. N Engl J Med. 2022;386(3):286–9.
de Goeij BE, Vink T, Ten Napel H, Breij EC, Satijn D, Wubbolts R, et al. Efficient payload delivery by a bispecific antibody–drug conjugate targeting HER2 and CD63. Mol Cancer Ther. 2016;15(11):2688–97.
Dovgan I, Koniev O, Kolodych S, Wagner A. Antibody-oligonucleotide conjugates as therapeutic, imaging, and detection agents. Bioconjug Chem. 2019;30(10):2483–501.
Schumacher FF, Nunes JP, Maruani A, Chudasama V, Smith ME, Chester KA, et al. Next generation maleimides enable the controlled assembly of antibody–drug conjugates via native disulfide bond bridging. Org Biomol Chem. 2014;12(37):7261–9.
Morais M, Forte N, Chudasama V, Baker JR. Application of next-generation maleimides (NGMs) to site-selective antibody conjugation. Methods Mol Biol. 2019;2033:15–24.
Bargh JD, Isidro-Llobet A, Parker JS, Spring DR. Cleavable linkers in antibody–drug conjugates. Chem Soc Rev. 2019;48(16):4361–74.
Gikanga B, Adeniji NS, Patapoff TW, Chih HW, Yi L. Cathepsin B cleavage of vcMMAE-based antibody–drug conjugate is not drug location or monoclonal antibody carrier specific. Bioconjug Chem. 2016;27(4):1040–9.
Su D, Zhang D. Linker design impacts antibody–drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front Pharmacol. 2021;12: 687926.
Chan SY, Gordon AN, Coleman RE, Hall JB, Berger MS, Sherman ML, et al. A phase 2 study of the cytotoxic immunoconjugate CMB-401 (hCTM01-calicheamicin) in patients with platinum-sensitive recurrent epithelial ovarian carcinoma. Cancer Immunol Immunother. 2003;52(4):243–8.
Viricel W, Fournet G, Beaumel S, Perrial E, Papot S, Dumontet C, et al. Monodisperse polysarcosine-based highly-loaded antibody–drug conjugates. Chem Sci. 2019;10(14):4048–53.
Evans N, Grygorash R, Williams P, Kyle A, Kantner T, Pathak R, et al. Incorporation of hydrophilic macrocycles into drug-linker reagents produces antibody–drug conjugates with enhanced in vivo performance. Front Pharmacol. 2022;13: 764540.
Gandhi AV, Randolph TW, Carpenter JF. Conjugation of emtansine onto trastuzumab promotes aggregation of the antibody–drug conjugate by reducing repulsive electrostatic interactions and increasing hydrophobic interactions. J Pharm Sci. 2019;108(6):1973–83.
Chuprakov S, Ogunkoya AO, Barfield RM, Bauzon M, Hickle C, Kim YC, et al. Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody–drug conjugates. Bioconjug Chem. 2021;32(4):746–54.
Gregson SJ, Barrett AM, Patel NV, Kang GD, Schiavone D, Sult E, et al. Synthesis and evaluation of pyrrolobenzodiazepine dimer antibody–drug conjugates with dual beta-glucuronide and dipeptide triggers. Eur J Med Chem. 2019;179:591–607.
Weidle UH, Tiefenthaler G, Georges G. Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genomics Proteomics. 2014;11(2):67–79.
Satomaa T, Pynnonen H, Vilkman A, Kotiranta T, Pitkanen V, Heiskanen A, et al. Hydrophilic auristatin glycoside payload enables improved antibody–drug conjugate efficacy and biocompatibility. Antibodies (Basel). 2018;7(2):15.
Tsui CK, Barfield RM, Fischer CR, Morgens DW, Li A, Smith BAH, et al. CRISPR-Cas9 screens identify regulators of antibody–drug conjugate toxicity. Nat Chem Biol. 2019;15(10):949–58.
Kumar A, Kinneer K, Masterson L, Ezeadi E, Howard P, Wu H, et al. Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody–drug conjugates with two distinct warheads. Bioorg Med Chem Lett. 2018;28(23–24):3617–21.
Nilchan N, Li X, Pedzisa L, Nanna AR, Roush WR, Rader C. Dual-mechanistic antibody–drug conjugate via site-specific selenocysteine/cysteine conjugation. Antib Ther. 2019;2(4):71–8.
Matsuda Y, Mendelsohn BA. Recent advances in drug-antibody ratio determination of antibody–drug conjugates. Chem Pharm Bull (Tokyo). 2021;69(10):976–83.
White JB, Fleming R, Masterson L, Ruddle BT, Zhong H, Fazenbaker C, et al. Design and characterization of homogenous antibody–drug conjugates with a drug-to-antibody ratio of one prepared using an engineered antibody and a dual-maleimide pyrrolobenzodiazepine dimer. MAbs. 2019;11(3):500–15.
Habara H, Okamoto H, Nagai Y, Oitate M, Takakusa H, Watanabe N. Transition of average drug-to-antibody ratio of trastuzumab deruxtecan in systemic circulation in monkeys using a hybrid affinity capture liquid chromatography-tandem mass spectrometry. Biopharm Drug Dispos. 2023;44(5):380–4.
Nagai Y, Oitate M, Shiozawa H, Ando O. Comprehensive preclinical pharmacokinetic evaluations of trastuzumab deruxtecan (DS-8201a), a HER2-targeting antibody–drug conjugate, in cynomolgus monkeys. Xenobiotica. 2019;49(9):1086–96.
Okamoto H, Oitate M, Hagihara K, Shiozawa H, Furuta Y, Ogitani Y, et al. Pharmacokinetics of trastuzumab deruxtecan (T-DXd), a novel anti-HER2 antibody–drug conjugate, in HER2-positive tumour-bearing mice. Xenobiotica. 2020;50(10):1242–50.
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. 2018;834:188–96.
Perego G, Ghidini A, Luciani A, Petrelli F. Antibody–drug conjugates in treating older patients suffering from cancer: what is the real value? Hum Vaccin Immunother. 2021;17(12):5575–8.
Dumontet C, Reichert JM, Senter PD, Lambert JM, Beck A. Antibody–drug conjugates come of age in oncology. Nat Rev Drug Discov. 2023;22(8):641–61.
Sekimizu M, Iguchi A, Mori T, Koga Y, Kada A, Saito AM, et al. Phase I clinical study of brentuximab vedotin (SGN-35) involving children with recurrent or refractory CD30-positive Hodgkin’s lymphoma or systemic anaplastic large cell lymphoma: rationale, design and methods of BV-HLALCL study: study protocol. BMC Cancer. 2018;18(1):122.
Esser L, Weiher H, Schmidt-Wolf I. Increased efficacy of brentuximab vedotin (SGN-35) in combination with cytokine-induced killer cells in lymphoma. Int J Mol Sci. 2016;17(7):1056.
Kumar A, Casulo C, Advani RH, Budde E, Barr PM, Batlevi CL, et al. Brentuximab vedotin combined with chemotherapy in patients with newly diagnosed early-stage unfavorable-risk Hodgkin lymphoma. J Clin Oncol. 2021;39(20):2257–65.
Connors JM, Jurczak W, Straus DJ, Ansell SM, Kim WS, Gallamini A, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378(4):331–44.
Gong IY, Yan AT, Earle CC, Trudeau ME, Eisen A, Chan KKW. Comparison of outcomes in a population-based cohort of metastatic breast cancer patients receiving anti-HER2 therapy with clinical trial outcomes. Breast Cancer Res Treat. 2020;181(1):155–65.
Ocana A, Amir E, Pandiella A. Dual targeting of HER2-positive breast cancer with trastuzumab emtansine and pertuzumab: understanding clinical trial results. Oncotarget. 2018;9(61):31915–9.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, et al. Inotuzumab Ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740–53.
Tvito A, Rowe JM. Inotuzumab ozogamicin for the treatment of acute lymphoblastic leukemia. Expert Opin Biol Ther. 2017;17(12):1557–64.
Hibma JE, Kantarjian HM, DeAngelo DJ, Boni JP. Effect of inotuzumab ozogamicin on the QT interval in patients with haematologic malignancies using QTc-concentration modelling. Br J Clin Pharmacol. 2019;85(3):590–600.
Ohana Z, Serraes S, Elder C, Katusa N. Cytogenetic guided therapy using blinatumomab and inotuzumab ozogamicin in a patient with relapse/refractory acute lymphoblastic leukemia. J Oncol Pharm Pract. 2022;28(5):1269–75.
Nobre CF, Newman MJ, DeLisa A, Newman P. Moxetumomab pasudotox-tdfk for relapsed/refractory hairy cell leukemia: a review of clinical considerations. Cancer Chemother Pharmacol. 2019;84(2):255–63.
Feurtado J, Kreitman RJ. Moxetumomab Pasudotox: clinical experience in relapsed/refractory hairy cell leukemia. Clin J Oncol Nurs. 2019;23(3):E52–9.
Abou Dalle I, Ravandi F. Moxetumomab pasudotox for the treatment of relapsed and/or refractory hairy cell leukemia. Expert Rev Hematol. 2019;12(9):707–14.
Kreitman RJ, Dearden C, Zinzani PL, Delgado J, Robak T, le Coutre PD, et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): long-term follow-up from the pivotal trial. J Hematol Oncol. 2021;14(1):35.
Burke JM, Morschhauser F, Andorsky D, Lee C, Sharman JP. Antibody–drug conjugates for previously treated aggressive lymphomas: focus on polatuzumab vedotin. Expert Rev Clin Pharmacol. 2020;13(10):1073–83.
Dere RC, Beardsley RL, Lu D, Lu T, Ku GH, Man G, et al. Integrated summary of immunogenicity of polatuzumab vedotin in patients with relapsed or refractory B-cell non-Hodgkin’s lymphoma. Front Immunol. 2023;14:1119510.
Malecek MK, Watkins MP, Bartlett NL. Polatuzumab vedotin for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma. Expert Opin Biol Ther. 2021;21(7):831–9.
Azizi A, Houshyar R, Mar N. Use of enfortumab vedotin in an HIV-positive patient with urothelial carcinoma. J Oncol Pharm Pract. 2022;28(5):1226–9.
Kita Y, Ito K, Sano T, Hashimoto K, Mochizuki T, Shiraishi Y, et al. Clinical practice pattern in patients with advanced urothelial cancer who had progressed on pembrolizumab in the pre-enfortumab vedotin era. Int J Urol. 2022;29(7):647–55.
Hanna KS. Enfortumab vedotin to treat urothelial carcinoma. Drugs Today (Barc). 2020;56(5):329–35.
Mantia CM, Sonpavde G. Enfortumab vedotin-ejfv for the treatment of advanced urothelial carcinoma. Expert Rev Anticancer Therapy. 2022;22(5):449–55.
Lacouture ME, Patel AB, Rosenberg JE, O’Donnell PH. Management of dermatologic events associated with the nectin-4-directed antibody–drug conjugate enfortumab vedotin. Oncologist. 2022;27(3):e223–32.
Powles T, Rosenberg JE, Sonpavde GP, Loriot Y, Duran I, Lee JL, et al. Enfortumab vedotin in previously treated advanced urothelial carcinoma. N Engl J Med. 2021;384(12):1125–35.
Ocana A, Amir E, Pandiella A. HER2 heterogeneity and resistance to anti-HER2 antibody–drug conjugates. Breast Cancer Res. 2020;22(1):15.
Li Z, Guo S, Xue H, Li L, Guo Y, Duan S, et al. Efficacy and safety of trastuzumab deruxtecan in the treatment of HER2-low/positive advanced breast cancer: a single-arm meta-analysis. Front Pharmacol. 2023;14:1183514.
Kunte S, Abraham J, Montero AJ. Novel HER2-targeted therapies for HER2-positive metastatic breast cancer. Cancer. 2020;126(19):4278–88.
Zhang C, Xiang Y, Wang J, Yan D. Comparison of the efficacy and safety of third-line treatments for advanced gastric cancer: a systematic review and network meta-analysis. Front Oncol. 2023;13:1118820.
Seligson JM, Patron AM, Berger MJ, Harvey RD, Seligson ND. Sacituzumab Govitecan-hziy: an antibody–drug conjugate for the treatment of refractory, metastatic, triple-negative breast cancer. Ann Pharmacother. 2021;55(7):921–31.
Xie J, Li S, Li Y, Li J. Cost-effectiveness of sacituzumab govitecan versus chemotherapy in patients with relapsed or refractory metastatic triple-negative breast cancer. BMC Health Serv Res. 2023;23(1):706.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N Engl J Med. 2021;384(16):1529–41.
Syed YY. Sacituzumab govitecan: first approval. Drugs. 2020;80(10):1019–25.
Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med. 2019;380(8):741–51.
Tzogani K, Penttila K, Lahteenvuo J, Lapvetelainen T, Lopez Anglada L, Prieto C, et al. EMA review of belantamab mafodotin (Blenrep) for the treatment of adult patients with relapsed/refractory multiple myeloma. Oncologist. 2021;26(1):70–6.
Markham A. Belantamab mafodotin: first approval. Drugs. 2020;80(15):1607–13.
Baines AC, Ershler R, Kanapuru B, Xu Q, Shen G, Li L, et al. FDA approval summary: belantamab mafodotin for patients with relapsed or refractory multiple myeloma. Clin Cancer Res. 2022;28(21):4629–33.
Ferron-Brady G, Rathi C, Collins J, Struemper H, Opalinska J, Visser S, et al. Exposure-response analyses for therapeutic dose selection of belantamab mafodotin in patients with relapsed/refractory multiple myeloma. Clin Pharmacol Ther. 2021;110(5):1282–92.
Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21(2):207–21.
Gomes-da-Silva LC, Kepp O, Kroemer G. Regulatory approval of photoimmunotherapy: photodynamic therapy that induces immunogenic cell death. Oncoimmunology. 2020;9(1):1841393.
Gottardi M, Simonetti G, Sperotto A, Nappi D, Ghelli Luserna di Rora A, Padella A, et al. Therapeutic targeting of acute myeloid leukemia by gemtuzumab ozogamicin. Cancers (Basel). 2021;13(18):4566.
Goldenson BH, Goodman AM, Ball ED. Gemtuzumab ozogamicin for the treatment of acute myeloid leukemia in adults. Expert Opin Biol Ther. 2021;21(7):849–62.
Dohner H, Weber D, Krzykalla J, Fiedler W, Kuhn MWM, Schroeder T, et al. Intensive chemotherapy with or without gemtuzumab ozogamicin in patients with NPM1-mutated acute myeloid leukaemia (AMLSG 09–09): a randomised, open-label, multicentre, phase 3 trial. Lancet Haematol. 2023;10(7):e495–509.
Dhunputh C, Strullu M, Petit A, Merched M, Pasquet M, Azarnoush S, et al. Single-dose (4.5 mg/m(2) ) gemtuzumab ozogamicin in combination with fludarabine, cytarabine and anthracycline as reinduction therapy in relapsed or refractory paediatric acute myeloid leukaemia. Br J Haematol. 2022;198(2):373–81.
Amadori S, Suciu S, Selleslag D, Aversa F, Gaidano G, Musso M, et al. Gemtuzumab ozogamicin versus best supportive care in older patients with newly diagnosed acute myeloid leukemia unsuitable for intensive chemotherapy: results of the randomized Phase III EORTC-GIMEMA AML-19 trial. J Clin Oncol. 2016;34(9):972–9.
Lee A. Loncastuximab tesirine: first approval. Drugs. 2021;81(10):1229–33.
Goparaju K, Caimi PF. Loncastuximab tesirine for treatment of relapsed or refractory diffuse large B cell lymphoma. Expert Opin Biol Ther. 2021;21(11):1373–81.
Kahl BS, Hamadani M, Radford J, Carlo-Stella C, Caimi P, Reid E, et al. A phase I study of ADCT-402 (Loncastuximab Tesirine), a novel pyrrolobenzodiazepine-based antibody–drug conjugate, in relapsed/refractory B-cell non-hodgkin lymphoma. Clin Cancer Res. 2019;25(23):6986–94.
Chen M, Yao K, Cao M, Liu H, Xue C, Qin T, et al. HER2-targeting antibody–drug conjugate RC48 alone or in combination with immunotherapy for locally advanced or metastatic urothelial carcinoma: a multicenter, real-world study. Cancer Immunol Immunother. 2023;72(7):2309–18.
Peng Z, Liu T, Wei J, Wang A, He Y, Yang L, et al. Efficacy and safety of a novel anti-HER2 therapeutic antibody RC48 in patients with HER2-overexpressing, locally advanced or metastatic gastric or gastroesophageal junction cancer: a single-arm phase II study. Cancer Commun (Lond). 2021;41(11):1173–82.
Xu Y, Wang Y, Gong J, Zhang X, Peng Z, Sheng X, et al. Phase I study of the recombinant humanized anti-HER2 monoclonal antibody-MMAE conjugate RC48-ADC in patients with HER2-positive advanced solid tumors. Gastric Cancer. 2021;24(4):913–25.
Heitz N, Greer SC, Halford Z. A review of tisotumab vedotin-tftv in recurrent or metastatic cervical cancer. Ann Pharmacother. 2023;57(5):585–96.
Tisotumab Vedotin-tftv. Am J Health Syst Pharm. 2022; 79(3):120–2.
Aschenbrenner DS. New drug treats cervical cancer. Am J Nurs. 2022;122(1):21.
Matulonis UA, Birrer MJ, O’Malley DM, Moore KN, Konner J, Gilbert L, et al. Evaluation of prophylactic corticosteroid eye drop use in the management of corneal abnormalities induced by the antibody–drug conjugate mirvetuximab soravtansine. Clin Cancer Res. 2019;25(6):1727–36.
Heo YA. Mirvetuximab soravtansine: first approval. Drugs. 2023;83(3):265–73.
Kaur R, Kaur G, Gill RK, Soni R, Bariwal J. Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem. 2014;87:89–124.
Walczak CE. Microtubule dynamics and tubulin interacting proteins. Curr Opin Cell Biol. 2000;12(1):52–6.
Beukhof CM, Brabander T, van Nederveen FH, van Velthuysen MF, de Rijke YB, Hofland LJ, et al. Peptide receptor radionuclide therapy in patients with medullary thyroid carcinoma: predictors and pitfalls. BMC Cancer. 2019;19(1):325.
Oshima N, Akizawa H, Kawashima H, Zhao S, Zhao Y, Nishijima KI, et al. Redesign of negatively charged (111)In-DTPA-octreotide derivative to reduce renal radioactivity. Nucl Med Biol. 2017;48:16–25.
Hubalewska-Dydejczyk A, Fross-Baron K, Mikolajczak R, Maecke HR, Huszno B, Pach D, et al. 99mTc-EDDA/HYNIC-octreotate scintigraphy, an efficient method for the detection and staging of carcinoid tumours: results of 3 years’ experience. Eur J Nucl Med Mol Imaging. 2006;33(10):1123–33.
Ferro-Flores G, Luna-Gutierrez M, Ocampo-Garcia B, Santos-Cuevas C, Azorin-Vega E, Jimenez-Mancilla N, et al. Clinical translation of a PSMA inhibitor for (99m)Tc-based SPECT. Nucl Med Biol. 2017;48:36–44.
Poletto G, Cecchin D, Sperti S, Filippi L, Realdon N, Evangelista L. Head-to-head comparison between peptide-based radiopharmaceutical for PET and SPECT in the evaluation of neuroendocrine tumors: a systematic review. Curr Issues Mol Biol. 2022;44(11):5516–30.
Simsek DH, Sanli Y, Kuyumcu S, Basaran B, Mudun A. (68)Ga-DOTATATE PET-CT imaging in carotid body paragangliomas. Ann Nucl Med. 2018;32(4):297–301.
Tremblay S, Beaudoin JF, Belissant Benesty O, Ait-Mohand S, Dumulon-Perreault V, Rousseau E, et al. (68)Ga-DOTATATE prepared from cyclotron-produced (68)Ga: an integrated solution from cyclotron vault to safety assessment and diagnostic efficacy in neuroendocrine cancer patients. J Nucl Med. 2023;64(2):232–8.
Aalbersberg EA, de Wit-van der Veen BJ, Versleijen MWJ, Saveur LJ, Valk GD, Tesselaar MET, et al. Influence of lanreotide on uptake of (68)Ga-DOTATATE in patients with neuroendocrine tumours: a prospective intra-patient evaluation. Eur J Nucl Med Mol Imaging. 2019;46(3):696–703.
Tarkin JM, Joshi FR, Evans NR, Chowdhury MM, Figg NL, Shah AV, et al. Detection of atherosclerotic inflammation by (68)Ga-DOTATATE PET compared to [(18)F]FDG PET imaging. J Am Coll Cardiol. 2017;69(14):1774–91.
Wang Y, Cheetham AG, Angacian G, Su H, Xie L, Cui H. Peptide-drug conjugates as effective prodrug strategies for targeted delivery. Adv Drug Deliv Rev. 2017;110–111:112–26.
Basu S, Parghane RV, Banerjee S. Availability of both [(177)Lu]Lu-DOTA-TATE and [(90)Y]Y-DOTATATE as PRRT agents for neuroendocrine tumors: can we evolve a rational sequential duo-PRRT protocol for large volume resistant tumors? Eur J Nucl Med Mol Imaging. 2020;47(4):756–8.
Kato A, Nakamoto Y, Ishimori T, Hayakawa N, Ueda M, Temma T, et al. Diagnostic performance of (68)Ga-DOTATOC PET/CT in tumor-induced osteomalacia. Ann Nucl Med. 2021;35(3):397–405.
Chen SH, Chang YC, Hwang TL, Chen JS, Chou WC, Hsieh CH, et al. 68Ga-DOTATOC and 18F-FDG PET/CT for identifying the primary lesions of suspected and metastatic neuroendocrine tumors: a prospective study in Taiwan. J Formos Med Assoc. 2018;117(6):480–7.
Pizzuto DA, Muller J, Muhlematter U, Rupp NJ, Topfer A, Mortezavi A, et al. The central zone has increased (68)Ga-PSMA-11 uptake: “Mickey Mouse ears” can be hot on (68)Ga-PSMA-11 PET. Eur J Nucl Med Mol Imaging. 2018;45(8):1335–43.
Huo H, Shen S, He D, Liu B, Yang F. Head-to-head comparison of (68)Ga-PSMA-11 PET/CT and (68)Ga-PSMA-11 PET/MRI in the detection of biochemical recurrence of prostate cancer: summary of head-to-head comparison studies. Prostate Cancer Prostat Dis. 2023;26(1):16–24.
Ferda J, Hes O, Hora M, Ferdova E, Pernicky J, Rudnev V, et al. Assessment of prostate carcinoma aggressiveness: relation to (68)Ga-PSMA-11-PET/MRI and Gleason Score. Anticancer Res. 2023;43(1):449–53.
Zhao Y, Xia Y, Liu H, Wang Z, Chen Y, Zhang W. Potential applications of (68)Ga-PSMA-11 PET/CT in the evaluation of salivary gland uptake function: preliminary observations and comparison with (99m)TcO(4) (-) salivary gland scintigraphy. Contrast Media Mol Imaging. 2020;2020:1097516.
Carlsen EA, Johnbeck CB, Binderup T, Loft M, Pfeifer A, Mortensen J, et al. (64)Cu-DOTATATE PET/CT and prediction of overall and progression-free survival in patients with neuroendocrine neoplasms. J Nucl Med. 2020;61(10):1491–7.
Loft M, Carlsen EA, Johnbeck CB, Johannesen HH, Binderup T, Pfeifer A, et al. (64)Cu-DOTATATE PET in patients with neuroendocrine neoplasms: prospective, head-to-head comparison of imaging at 1 hour and 3 hours after injection. J Nucl Med. 2021;62(1):73–80.
Carlsen EA, Johnbeck CB, Loft M, Pfeifer A, Oturai P, Langer SW, et al. Semiautomatic tumor delineation for evaluation of (64)Cu-DOTATATE PET/CT in patients with neuroendocrine neoplasms: prognostication based on lowest lesion uptake and total tumor volume. J Nucl Med. 2021;62(11):1564–70.
Mateos MV, Blade J, Bringhen S, Ocio EM, Efebera Y, Pour L, et al. Melflufen: a peptide-drug conjugate for the treatment of multiple myeloma. J Clin Med. 2020;9(10):3120.
Lindberg J, Nilvebrant J, Nygren PA, Lehmann F. Progress and future directions with peptide-drug conjugates for targeted cancer therapy. Molecules. 2021;26(19):6042.
FDA approves pluvicto/locametz for metastatic castration-resistant prostate cancer. J Nucl Med. 2022; 63(5):13N.
Acknowledgements
We are all thankful to all the researchers who are dedicated to cancer therapy research. Figdraw was used to construct the figures.
Funding
Financial support was supported by the National Natural Science Foundation of China (Nos. 81772477, 81201848, 82173684, 82204181 and 82270104); Fundamental Research Program of Shanxi Province (No. 202303021221192); high level key discipline construction project of the National Administration of Traditional Chinese Medicine-Resource Chemistry of Chinese Medicinal Materials (No.zyyzdxk-2023083); the Key R&D Program of Jiangsu Province (No. BE2023840); Nanjing University of Chinese Medicine National Natural Science Foundation of China Counterpart Funding (No. XPT82204181); Jiangsu Provincial Health Commission (No. Z2021057) and Young Doctors' Innovation and Development Program (No. HXQNJJ-2023-010).
Author information
Authors and Affiliations
Contributions
L.Z. wrote the manuscript, Y.L.L. and W.L. assisted in the definition of intellectual content. S.L.W. constructed all the figures, and L.L.W., P.D.Z. and G.S.Z. assisted with the tables. H.G.L., W.K.L. and S.W. critically commented on the manuscript. All the authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, L., Lu, Y., Liu, W. et al. Drug conjugates for the treatment of lung cancer: from drug discovery to clinical practice. Exp Hematol Oncol 13, 26 (2024). https://doi.org/10.1186/s40164-024-00493-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-024-00493-8