
Abstract
Antibody-drug conjugates (ADCs) represent an important class of cancer therapies that have revolutionized the treatment paradigm of solid tumors. To date, many ongoing studies of ADC combinations with a variety of anticancer drugs, encompassing chemotherapy, molecularly targeted agents, and immunotherapy, are being rigorously conducted in both preclinical studies and clinical trial settings. Nevertheless, combination therapy does not always guarantee a synergistic or additive effect and may entail overlapping toxicity risks. Therefore, understanding the current status and underlying mechanisms of ADC combination therapy is urgently required. This comprehensive review analyzes existing evidence concerning the additive or synergistic effect of ADCs with other classes of oncology medicines. Here, we discuss the biological mechanisms of different ADC combination therapy strategies, provide prominent examples, and assess their benefits and challenges. Finally, we discuss future opportunities for ADC combination therapy in clinical practice.
Similar content being viewed by others

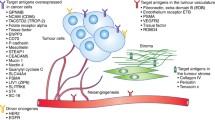

Background
In the past decade, antibody-drug conjugates (ADCs) have emerged as a transformative treatment modality for a broad spectrum of solid and hematological malignancies [1, 2]. ADCs are antibody-based macromolecular complexes comprising three main constituents: antibodies, linkers, and payloads. Their mechanism of action can be summarized as follows: when the antibody binds to the antigen on the surface of a target cell, the ADC is internalized, releasing the payload and exerting cytotoxicity [3] (Fig. 1). Following the initial approval of ADCs for solid tumors in 2013 [4], interest in this field has increased, and numerous such conjugates have been evaluated across various tumor categories.

Structure and mechanism of action of conventional ADCs. ADCs consist of three essential components: a monoclonal antibody that binds to an antigen primarily expressed on the surface of tumor cells, providing specificity in targeting tumor cells; a linker that prevents premature release of the payload in the bloodstream but instead releases it in the tumor cells; and a cytotoxic payload that triggers tumor cell death by targeting critical components such as DNA, microtubules, and topoisomerase. ADC cytotoxicity involves a series of sequential stages: ① binding of the antibody to the antigen, ② internalization of the ADC-antigen complex, ③ degradation of the ADC in the lysosomes, ④ release of the payload in the cytoplasm, ⑤ its interaction with the target; ⑥ possible discharge of a fraction of the payload into the extracellular milieu, ⑦ subsequent occurrence of the bystander effect where it is internalized by neighboring cells in the tumor microenvironment. Abbreviation: TME, tumor microenvironment
Several ADCs have shown potent anti-tumor activities against treatment-refractory cancers. To date, eight ADCs have been approved for solid tumors with different indications (Table 1). Nevertheless, even for target-positive tumor types, most patients do not achieve long-lasting disease control and develop resistance to ADCs. Thus, for many tumor types, a single treatment is insufficient and many ADCs are undergoing clinical trials with more responsive regimens.
In the realm of cancer treatment, it is widely acknowledged that the likelihood of achieving complete remission and cure is often heightened by combining therapeutic agents that operate through diverse mechanisms of action, particularly when dealing with the complexities of tumor heterogeneity [5]. The primary approach for addressing resistance and/or enhancing ADC therapies involves the integration of ADCs with different therapeutic strategies. Synergy is commonly defined as the effect of two or more agents working in combination that is greater than the expected additive effect. An additive effect is generally considered as the baseline effect for synergy detection methods. Consequently, active research is exploring the combination of ADCs with various other types of anticancer medications, such as chemotherapy, radiotherapy, endocrine therapy, targeted molecular agents, and immunotherapy, both in preclinical models and clinical trials. There is an interest in developing rational combinations that could prolong survival compared to monotherapies.
In this review, we discuss the mechanisms of different ADC combination therapies and review the ongoing clinical trials for their selection and evaluation. Finally, we outline and examine key translational, statistical, and regulatory considerations from a combination perspective, highlighting the current progress and significant challenges yet to be addressed.
ADCs combined with chemotherapy
Integrating different forms of chemotherapy with ADC has proven to be a well-accepted strategy for overcoming drug resistance and achieving favorable treatment outcomes in preclinical and clinical studies [6]. Exploring the most effective combination regimen requires a comprehensive understanding of how ADC antibodies and payloads work synergistically with chemotherapy drugs to affect the cell cycle and alter the presence of surface antigens. However, to date, many ADCs have been added to commonly used chemotherapeutic regimens merely as carriers for the delivery of toxic payloads without considering their synergistic effects, leading to mixed results in both preclinical and clinical research. This highlights the significant and unmet need for continued efforts in designing clinical trials for ADCs combined with chemotherapy. Table 2 presents a list of such trials.
Mechanism of ADCs combined with chemotherapy
According to reported findings, chemotherapy and ADCs act synergistically in ways that include targeting different phases of the cell cycle or modulating tumor cell surface antigen expression.
Cell cycle phase blockers
Many chemotherapeutic drugs are DNA-damaging agents, such as antimetabolites, platinum-based compounds, and topoisomerase inhibitors that target the S phase of the cell cycle and induce G2/M arrest, which can be effectively combined with ADC containing microtubule-disrupting payloads that target the G2/M phase of the cell cycle. This concept has been illustrated through the effective combination of carboplatin with mirvetuximab soravtansine (targeting folate receptor α with DM4), anetumab ravtansine (targeting mesothelin with DM4), or luveltamab tazevibulin (targeting folate receptor α with SC239) in ovarian cancer preclinically [7,8,9]. During early phase trials investigating the synergistic effects of ravtansine-based ADCs in combination with carboplatin or doxorubicin, positive treatment responses were observed in both platinum-sensitive and -resistant patients with ovarian cancer [10,11,12,13,14].
Improved surface-antigen expression
The choice of chemotherapeutic companion may affect the levels of surface antigens targeted by ADCs. For instance, gemcitabine can upregulate HER2 expression on pancreatic adenocarcinoma cells by 14.81 folds, predominantly within the G2/M phase. Thus, the effect of gemcitabine on DNA synthesis renders it effective against G1 and early S phase cells, whereas G2/M phase cells are more resistant. The enhanced HER2 expression in G2/M cells implies a greater likelihood of gemcitabine effectively binding with trastuzumab emtansine (T-DM1, HER2 targeted with DM1 payload), which contributes to the improved efficacy of the combination on pancreatic ductal adenocarcinoma cells [15]. Thus, gemcitabine generate synergistic effects in combination with T-DM1 through their ability to enhance antigen availability. However, it remains uncertain whether this observation holds true for other ADC-chemotherapy combinations with different targets, and whether the increased antigen expression levels are directly related to the actual available antigenic epitopes for ADC binding or even to the efficacy of ADCs.
Coordination of different drugs
The timing of administration is a significant factor to consider when designing ADC combinations, as most conjugates must be internalized by tumor cells to be effective, which involves systemic transport and cell entry processes. For example, induction of G2/M phase arrest by DNA damage requires at least 15 h for microtubule disruptors to act [16]. Wahl et al. elegantly demonstrated this concept in preclinical models of colon, lung, and breast cancers. They observed that sequential management of SGN-15 (a construct targeting the Lewis Y antigen with a doxorubicin payload) followed by paclitaxel resulted in greater DNA fragmentation than simultaneous treatment [17].This observation suggests that the sequence of drug administration may be taken into consideration when combined chemotherapy with ADCs. However, these concepts await clinical trial assessment and should be explored in light of the recognized rates of ADC internalization and cell cycle progression in individual tumor types.
Safety profile of the ADC–chemotherapy combination
Notably, the combination of ADCs and chemotherapy presents challenges related to overlapping toxicities. Substantial insights in this regard have been gained from clinical trials. For example, a study evaluating T-DM1 in combination with docetaxel (with or without pertuzumab) for HER2-positive breast cancer demonstrated dose-limiting toxicities (DLTs) and grade ≥ 3 adverse events in approximately 80% patients with metastatic breast cancer [18]. These adverse effects included neutropenia, fatigue, epistaxis, stomatitis, nausea, and diarrhea. Similarly, the combination of T-DM1 with capecitabine resulted in increased discontinuation rates without a significant improvement in response rates [19]. The combination of trastuzumab deruxtecan (T-DXd) with 5-fluorouracil (5-FU) or capecitabine resulted in notable toxicities in patients with metastatic HER2-positive gastric cancer, with dose-limiting stomatitis and a high incidence of grade ≥ 3 adverse events in DESTINYGastric03 trial [14].
Datopotamab deruxtecan (Dato-DXd) in combination with platinum-based chemotherapy and pembrolizumab resulted in substantial grade ≥ 3 toxicities in a significant proportion of patients, characterized by the common occurrence of nausea, anemia, fatigue, and stomatitis [13]. In addition, the combination of mirvetuximab soravtansine with carboplatin in a Phase Ib trial resulted in notable toxicities including nausea, vomiting, diarrhea, eye problems, fatigue, and cytopenia [10].
In summary, the results of these studies suggested a notable increase in toxicity when ADCs were combined with conventional chemotherapy. This is likely due to the overlap of toxicities resulting from the off-target and off-tumor effects of the ADC payloads.
ADCs combined with endocrine therapy
Endocrine therapy is a widely used therapeutic approach for hormone-sensitive cancers (e.g., breast and prostate cancers). It works either by blocking hormone synthesis or interfering with hormones that stimulate the growth of tumor cells. Both ADCs and endocrine therapy drugs can induce cellular effects that jointly impede tumor cell survival and proliferation. Combination therapy reduces the likelihood of tumor cells developing resistance by employing multiple drugs with distinct mechanisms of action. There have been some clinical trials related to ADC combined with endocrine therapy (Table 3), while basic research to explore the mechanism of combined action is lacking.
Safety profile of the ADC–endocrine therapy combination
The side effects of endocrine therapy are minimal [29]. This gives clinicians more confidence in adding ADC drugs to endocrine therapy, as demonstrated in the KATHERINE phase III clinical trial. In this trial, the adjuvant utilization of T-DM1 was compared with that of trastuzumab in patients with HER2-positive breast cancer with residual disease who had undergone neoadjuvant HER2-targeted therapy. Both treatment arms were permitted to include concurrent adjuvant endocrine therapy. Patients administered T-DM1 with or without endocrine therapy exhibited comparable toxicity rates of any grade. Similarly, no significant differences between the two groups were observed in terms of grade ≥ 3 adverse events (26.0% versus 24.9%), serious adverse events (12.9% versus 12.2%), and events that resulted in a T-DM1 dosage reduction (11.0% versus 15.0%) [30].
The possibility of combining endocrine therapy with T-DXd has been investigated in patients with HER2-low breast cancer at both early and advanced stages. The TALENT study is a randomized phase II trial evaluating the administration of neoadjuvant T-DXd, with or without anastrozole, in patients with early breast cancer and low HER2 expression. Interestingly, this study showed that both treatment arms exhibited similar toxicity profiles, highlighting the feasibility of this combination approach [31]. Similarly, the Phase Ib study, DESTINY-Breast08, revealed that adding anastrozole or fulvestrant to T-DXd did not result in any DLTs. This combination approach was observed to maintain a toxicity profile akin to that of the solitary administration of T-DXd in individuals diagnosed with metastatic HER2 low breast cancer.
In general, the co-administration of ADCs and endocrine therapy does not appear to result in increased toxicity. This observation is consistent with the distinct patterns of adverse effects exhibited by each agent when administered independently. This is also consistent with the favorable safety profiles of most endocrine therapies compared to other systemic cancer treatments.
ADCs combined with radiotherapy
The combination of radiotherapy and ADC includes external radiation therapy combined with ADC and radionuclide antibody conjugates (RACs). RACs, also known as radioimmunoconjugates, radioimmunotherapy, or targeted radiotherapy, are a type of medical treatment that uses specific monoclonal antibodies labelled with radioactive isotopes (radionuclides, generally beta emitters), as described in a review by Mattes [32]. Therefore, RAC is not discussed in the present review. Based on the timing of radiotherapy and ADC administration, the combination is either concomitant or sequential. Concomitant radiotherapy involves simultaneous administration of ADC and radiotherapy. However, the definition of sequential ADC administration varies across studies: the temporal span ranges from 77 to 131 days when ADC is administered before radiotherapy and 420 to 1426 days when administered after radiotherapy [33]. The fractionation regimens include conventional fractionated radiotherapy and stereotactic radiosurgery (SRS)/stereotactic body radiotherapy (SBRT). The clinical studies on ADCs combined with radiotherapy are shown in Table 4.
Mechanism of ADCs combined with radiotherapy
The synergistic mechanisms of the combined application of radiotherapy and ADC include the regulation of surface antigen expression in tumor cells by radiotherapy, an increase in radiation sensitivity of tumor cells by ADC, and other potential mechanisms, such as affecting the tumor microenvironment (TME) and vascular permeability.
Radiation induces generation of (neo)antigens
Ionizing radiation (IR) induces morphological and functional alterations in tissues [44]. Tumor cells are more prone to survival and propagation when “stress-regulated proteins” are highly upregulated by external stimuli. (Neo-) antigens expressed on the surface of cancer cells after IR exposure provide opportunities to develop cancer-targeted therapeutics. Cell adhesion molecules were the first IR-inducible proteins identified [45, 46]. However, these inducible proteins are expressed on microvascular endothelial cells rather than on tumor cells, and some are shed from the cell surface. Glucose-regulated protein 78 (GRP78) is a radiation-induced endoplasmic reticulum stress response protein that plays an important role in radioresistance, enhancement of tumor cell proliferation, protection against apoptosis, and promotion of tumor angiogenesis [47, 48]. The effectiveness of combining GRP78 antibodies with radiotherapy has been studied previously [49, 50]. The researchers discovered that antibodies targeting the functional domain of GRP78 disrupt its interactions with its binding partners. Consequently, this reduces tumor cell viability and enhances radiosensitization. Tax interacting protein 1 (TIP-1), another radiation-induced tumor-specific target, translocates to the cell plasma membrane after exposure to IR [51]. Based on this finding, Lewis et al. conjugated a high-affinity anti-TIP-1 antibody (7H5) to a payload such as monomethyl auristatin E (MMAE) with a valine-citrulline (Vc) linker to form a radiosensitizer (7H5-VcMMAE) [52]. The use of 7H5-Vc-MMAE in combination with radiotherapy resulted in a prolonged delay in tumor growth and improved the survival of A549 and H1299 non-small cell lung cancer (NSCLC) animal models. To date, the targeting of radiation-inducible antigens using ADCs combined with radiotherapy has not been explored in clinical trials. The new clinical paradigm of using IR to guide drug delivery merits further investigation in preclinical and clinical trials involving patients with radiation-resistant cancers.
ADCs increase the sensitivity of tumor cells to radiotherapy
The cell cycle phase plays an important role in determining the relative radiosensitivity of cells [53]. Cells are most sensitive to IR during the G2/M phase, display intermediate sensitivity in the G1 phase, and exhibit the lowest sensitivity in the later stages of the S phase. Based on these findings, many radiosensitizing drugs have been developed to increase the anti-tumor activity and optimize patient outcomes. However, in practice, the clinical utility of radiosensitizing drugs is substantially curtailed due to unintended off-target side effects. To address this critical challenge, radiation-sensitizing payloads (e.g., MMAE, MMAF, and DM1) have been conjugated to antibodies to selectively radiosensitize tumors based on antigen overexpression. These radiosensitizer-ADCs are capable of increasing radiosensitizer delivery to tumors, enhancing radiation-induced cytotoxicity, and improving tumor control [54, 55]. Furthermore, in combination with radiotherapy, radiosensitizer-ADCs have been shown to enhance the effectiveness of radiation and improve survival in preclinical tumor models of the lung, head and neck, oesophageal, breast, and pancreatic cancers [52, 54,55,56,57,58].
Other potential mechanisms
Radiotherapy not only directly acts on tumor cells but also affects the TME in a complex and dynamic manner. Several radiation-induced molecules within tumor blood vessels, including ICAM-1, E-selectin, P-selectin, and β3 integrin, reportedly have the potential to serve as therapeutic targets [59, 60]. Moreover, there is mounting evidence that the interplay between radiotherapy and the TME could be utilized to enhance the accumulation and intratumoral distribution of nanoparticles and liposome formulations mediated by changes in the vasculature and stroma, with secondary effects on hypoxia, interstitial fluid pressure, solid tissue pressure, and the recruitment and activation of bone marrow-derived myeloid cells [61,62,63]. A previous study found that vascular permeability in tumors significantly increased 24 h after irradiation at doses higher than 400 cGy, resulting in increased antibody uptake following radiation [64]. In addition to affecting the tumor blood vessels, radiotherapy also has a direct impact on blood–brain barrier permeability. Nakata et al. found that a single large dose of 20–40 Gy promoted the extravasation of serum albumin in the rat brain tissue preclinically [65]. Nevertheless, whether conventional fractionated radiotherapy, routinely used in clinics, can promote ADC penetration of the blood–brain barrier remains unknown and the underlying mechanism remains elusive. Finally, the effect of radiotherapy on the distribution of antibodies and the payload of linker-cleavable ADCs in tumors is yet to be investigated.
In summary, multiple potential synergistic benefits and underlying mechanisms of the combination of radiotherapy and ADCs deserve further exploration at both the preclinical and clinical stages.
Safety profile of the ADC–radiotherapy combination
The clinical applications of radiosensitizer-ADCs have also been evaluated. For non-CNS tumors, there is insufficient robust evidence regarding the safety profile and efficacy of ADCs in combination with different radiotherapy segmentations. Available data mainly focus on breast cancer treatments [33, 66]. The KATHERINE trial evaluated the effectiveness of adjuvant T-DM1 in patients with HER2-positive breast cancer and residual disease after neoadjuvant chemotherapy with anti-HER2 therapy [35]. The results demonstrated that adjuvant T-DM1 treatment reduces the risk of disease recurrence and death (50%). In this clinical trial, patients who underwent breast-conserving surgery and those who had locally advanced disease following mastectomy (clinical T3N+/T4Nx /TxN2-3 disease) were administered radiation within 60 days of surgery. However, a subgroup analysis specific to patients undergoing radiation was not conducted to assess the safety profile of the combined treatment. An increase in ≥ 3 grade toxicities was noted among the irradiation group in comparison to the non-irradiated group (27.4% vs. 16.2%). Comparable incidences of radiation-related cutaneous complications were observed, affecting 25.4% and 27.6% patients in the T-DM1 and trastuzumab groups, respectively. Patients who were administered T-DM1 showed a modest increase in the occurrence of radiation-induced pneumonitis and pulmonary radiation injury rates (1.5% and 0.1%, respectively), in contrast to those administered trastuzumab (0.7% and 0%) [30]. Considering the potential risk of cardiotoxicity associated with trastuzumab, a study was conducted to investigate the cardiac safety and feasibility of radiotherapy in combination with T-DM1 in a group of 116 patients (nconcurrent = 39, nsequential = 77) [34]. Roughly 95% patients receiving T-DM1 plus radiotherapy successfully adhered to ≥ 95% of the planned radiotherapy dosage with a delay of ≤ 5 days. No protocol-prespecified cardiac side effects or instances of heart failure were reported following T-DM1. However, Zolcsak et al. presented the initial safety profile associated with the concurrent use of T-DM1 and radiotherapy in a group of 14 patients diagnosed with residual invasive HER2-positive breast cancer. A dosage of 50 Gy delivered in 25 fractions was administered for adjuvant irradiation of the breast or chest wall. A reversible grade 2 decrease in left ventricular ejection fraction (LVEF) was observed in two patients [42]. Considering the mechanism of T-DM1’s action involving radiosensitization through microtubule inhibitors and in the absence of solid safety data, the delivery of concurrent radiation should be approached with caution. Recent clinical studies have revealed that T-DXd improved both progression-free survival (PFS) and overall survival (OS) compared to T-DM1, but information concerning the combined use of T-DXd and radiotherapy is scarce. T-DXd exhibited an increased incidence of drug-related interstitial lung disease (ILD) or pneumonitis (10.5% vs. 1.9%), and gastrointestinal toxicities were more frequently reported with T-DXd treatment. The combination of T-Dxd with thoracic or abdominal radiotherapy requires extreme caution. In a metastatic breast cancer setting, a case-series study assessed the toxicity of concurrent palliative radiotherapy and T-DM1 in three patients with bone metastases [39]. The radiotherapy field involved the thoracic vertebrae, sacrum, and shoulder, with a prescribed dose of 15 Gy delivered in five fractions or 8 Gy delivered in one fraction. All patients experienced substantial pain relief, and no documented adverse reactions associated with the concomitant use of radiotherapy and T-DM1 were reported. Furthermore, approximately 50% of HER2-positive metastatic breast cancer cases are associated with brain metastases [67]. In terms of CNS metastatic tumors, high-level data on the effectiveness and tolerance of concurrent administration of ADCs and brain radiation therapy are insufficient. Evidence gleaned from case reports or a small series of patients indicates that the combination of T-DM1 and concomitant whole-brain radiation therapy is manageable, without severe side effects or any increase in clinically significant toxicity [40]. However, prudence is needed when considering concurrent or sequential SRS as several cases of complications have been documented in the literature [36, 41, 43, 68, 69]. The case series presented by Carlson et al. showed that four out of seven (57.1%) patients who underwent SRS after T-DM1 administration developed radiation brain necrosis [36]. This elevated rate of clinical radiation brain necrosis is clearly unacceptable. In another study involving 45 patients diagnosed with CNS metastases from breast cancer, Stumpf et al. observed a 13.5-fold rise in the risk of radiation necrosis when T-DM1 was administered in conjunction with SRS [68]. The DEBBRAH phase II study demonstrated the feasibility of combining intracranial treatment with T-Dxd and radiation, showing manageable toxicity in patients with HER2-positive and HER2-low breast cancer who underwent whole-brain radiation therapy and/or SRS. However, the authors did not mention the timing between irradiation and the sequential administration of T-DXd [70]. Notably, significant heterogeneity was observed among these studies. Regarding primary CNS tumors, there are already some data on the effectiveness and safety outcomes of the combination of ADC and radiation treatment. An anti-EGFR antibody conjugated to MMAF, Depatuxizumab-mafodotin (Depatux-M), was administered to patients with glioblastoma (GBM) receiving standard treatment with radiotherapy plus temozolomide [37]. The safety profile of Depatux-M combined with radiotherapy and temozolomide for the treatment of newly diagnosed GBM is acceptable. However, interim analysis revealed that Depatux-M did not yield an OS benefit for the treatment of newly diagnosed EGFR-amplification GBM, notwithstanding the longer PFS. The study was terminated at the early stage. The potential reasons for these negative results are as follows: 1. Depatux-M may be ineffective in treating GBM; 2. there is a probability that Depatux-M effectively eliminated EGFR-amplification (and particularly EGFRvIII-mutant) tumor cells, improving PFS; however, resistant clones emerged and voided any OS benefit, a hypothesis supported by results from patient-derived xenografts [71]; 3. heterogeneous delivery across the blood–brain barrier limits the efficacy of Depatux-M in CNS tumors [71], and non-cleavable linkers are detrimental to drug diffusion within the tumors.
In summary, radiosensitizer-ADCs combined with radiation are a promising treatment strategy; however, there is an urgent need for high-level evidence regarding their safety.
ADCs combined with molecular targeted cancer therapy
Targeted therapies, including monoclonal antibodies, tyrosine kinase inhibitors (TKIs), and anti-angiogenic agents, have been used in clinical practice for decades to treat tumors with specific mutations, overexpression, and amplification, with clinically proven safety and efficacy. However, the efficacy of these treatments in combination with ADC remains poorly understood. In this section, we focus on the treatment strategies that combine ADCs with targeted therapeutics and discuss their potential synergistic effects and safety profiles. The corresponding clinical trials are shown in Table 5.
Mechanism of ADCs combined with targeted therapy
The combined effects of molecular-targeted drugs (including antibodies) and ADCs synergistically involve multiple mechanisms, such as improving intratumoral drug delivery by targeting tumor blood vessels, regulating tumor cell surface antigen expression, overcoming intratumoral heterogeneity and tumor drug resistance, and synthetic lethality.
Enhanced cellular uptake and anti-tumor activity
The macromolecular size of ADCs limits extravasation and leads to impaired distribution of ADCs in tumor tissues, ultimately resulting in unsatisfactory efficacy [88]. Two types of barriers affecting ADC delivery have been identified: the blood-tumor barrier, a physical barrier, and the binding-site barrier (BSB), a biological one. Within the blood-tumor barrier microenvironment, blood vessels present in solid tumors are composed of many immature and disorganized vessels, resulting in poor blood flow and hypoxia [89]. Meanwhile, high interstitial oncotic pressure collapses the tumor blood vessels, thus limiting the convective transport of ADCs from the blood into the tumor interstitial fluid [90]. The primary method for altering the tumor vasculature involves modulating angiogenesis and vessel porosity using agents such as anti-vascular endothelial growth factor antibodies such as bevacizumab. Jose F Ponte et al. showed that co-treatment with mirvetuximab soravtansine (an FRα-targeting ADC) and bevacizumab induced swift disruption of tumor microvasculature and extensive necrosis, and improved the efficacy in platinum-resistant epithelial ovarian cancer (EOC) models, emphasizing the superior bioactivity profile of the combination [9]. Another preclinical study showed that the combination of anetumab ravtansine (a mesothelin-targeting ADC) with bevacizumab improved the anti-tumor activity in ST081 and OVCAR-3 human ovarian cancer models [8]. Two studies that investigated the safety and efficacy of mirvetuximab soravtansine in combination with bevacizumab in treating advanced ovarian cancer showed that co-treatment with mirvetuximab soravtansine (6 mg/kg adjusted ideal body weight) and bevacizumab (15 mg/kg), administered intravenously once every three weeks, was well tolerated and presented encouraging efficacy in patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancers [11, 91]. However, there is not just one opinion when it comes to ADC combined with anti-angiogenesis therapy [92,93,94,95]. Arjaans et al. reported that the normalization of tumor blood vessels triggered by bevacizumab hampers antibody uptake [92]. The timeframe spanning from normalization to excessive pruning is dependent on both the dose of the anti-angiogenic agent and the duration following administration, which has proven challenging [93]. Importantly, grade 1–2 pneumonitis was detected in six patients (9%) when bevacizumab was introduced alongside mirvetuximab soravtansine, whereas no instances of pneumonitis were observed with the single use of mirvetuximab soravtansine. Therefore, there is an urgent need to rigorously design preclinical and clinical trials to explore the underlying mechanisms of this combination therapy-induced pneumonitis. Co-treatment with ADCs and bevacizumab in a non-clinical trial setting must be performed with caution because of a possible reduction in tumoral accumulation of ADCs that may be caused by bevacizumab.
The BSB is a biological barrier in tumor vasculature regions, which constrains the efficacy of high-affinity antibodies because of the successful binding of antibodies to cellular antigens at the point of extravasation, resulting in antibody sequestration and suboptimal tumor exposure [96]. Many factors, including elevated antigen expression and rapid antigen internalization combined with sluggish tumor uptake and slow interstitial diffusion of therapeutic antibodies, result in poor antibody penetration into the tumor. Transient competitive inhibition, which improves antibody distribution in solid tumors, is one strategy for overcoming BSB. The combined utilization of T-DM1 and pertuzumab showed synergistic activity in cell culture models and had an acceptable safety profile in phase Ib and II studies [97, 98]. Bordeau et al. [99] found that the co-administration of an anti-trastuzumab single domain antibody (1HE) with trastuzumab significantly increased both the penetration of trastuzumab from the vasculature and the percentage of tumor area that stained positive for trastuzumab. 1HE co-administered with a single dose of T-DM1 to NCI-N87 xenograft-bearing mice significantly enhanced T-DM1 efficacy and increased the median survival. However, results from multiple phase III clinical trials (MARIANNE, KRISTINE, and KAITLIN) have shown that the T-DM1 combined with pertuzumab (T-DM1 + P) regimen reduced grade ≥ 3 adverse events and ensured a better quality of life and the this regimen resulted in a higher chance of event-free survival and invasive disease-free survival than regimens of chemotherapy combined with trastuzumab and pertuzumab [72, 100,101,102]. However, Although monoclonal antibody combined with ADCs could overcome BSB, improve the distribution and anti-tumor efficacy of ADCs in tumor cells, and reduce toxicity, ADC is still not a replacement for standard chemotherapy.
In summary, the dose and time window of ADCs combined with anti-angiogenesis therapy should be further explored in future studies. The establishment of a reasonable model is crucial. Current data show that naked antibodies combined with ADC can overcome BSB to increase tumor penetration and anti-tumor tumor effects; however, they are still unable to replace traditional chemotherapy in clinical settings.
Upregulation of surface antigens and overcoming intratumor heterogeneity and drug resistance
Intratumor heterogeneity is a key factor contributing to therapeutic failure. Furthermore, the appearance of compensatory pathways in tumor therapy is one of the mechanisms of drug resistance, which is frequently accompanied by the downregulation of surface antigens [103]. Tumor heterogeneity and drug resistance are major challenges in cancer treatment and research. The different sites of action of monoclonal antibodies and TKI make combination therapy a potential strategy for overcoming these difficulties. The addition of a TKI to a combinational target blockade may provide greater selectivity, with a potentially improved therapeutic index.
Data on TKI that can overcome ADC resistance are scarce. Recently, a preclinical study on T-DM1 resistance was reported. PLK1, a key cell cycle regulator, was upregulated in both acquired and primary T-DM1 resistance models. And inhibition of PLK1 using volasertib led to T-DM1 re-sensitization both in vitro and in vivo [104]. ADCs may also be effective companions for modulating resistance mechanisms of targeted drugs [105,106,107]. Patients with NSCLC frequently develop acquired drug resistance to EGFR TKIs [108]. HER3 is a unique pseudokinase member of the ERBB family. It dimerizes with other ERBB family members (EGFR and HER2) and is frequently overexpressed in EGFR-mutant NSCLC [109]. Haikala et al. reported that EGFR inhibition by osimertinib leads to increased HER3 membrane expression and promotes HER3-DXd ADC internalization and efficacy, supporting the clinical development of an EGFR inhibitor/HER3-DXd combination in EGFR-mutant lung cancer preclinically [106]. Another example is the co-administration of the EGFR-TKI osimertinib and T-DM1, which contributed to an synergistic anti-tumor effect, where T-DM1 was able to delay or overcome osimertinib resistance in EGFR-mutant NSCLC models [105]. In melanomas, AXL-high cells are resistant to MAPK pathway inhibitors, whereas AXL-low cells are sensitive to them. Heterogeneous tumors show partial therapeutic responses, allowing for the emergence of drug-resistant clones that often express high levels of the receptor tyrosine kinase AXL [110]. Boshuizen et al. [111] found that AXL-107-MMAE and MAPK pathway inhibitors cooperatively inhibited tumor growth by eliminating distinct populations in heterogeneous melanoma cell pools in a preclinical study. Furthermore, the BRAF/MEK inhibitors potentiated the efficacy of AXL-107-MMAE by inducing AXL transcription [111]. In acute myeloid leukemia, a preclinical study found a promising and potent anti-leukemic strategy involving the co-administration of midostaurin (a TKI that inhibits the FLT3 pathway) and a novel FLT3-targeting ADC [112]. The mechanisms behind TKI inducing drug resistance and regulating surface antibody expression are complicated and vary with different drugs. HER2-targeting TKIs (lapatinib, neratinib, tucatinib, and poziotinib) have been shown to increase the efficacy of T-DM1. However, while lapatinib enhances HER2 abundance via robust transcriptional upregulation and reduced ubiquitination, neratinib downregulates surface HER2 abundance by stimulating internalization and endocytosis. The effectiveness of tucatinib on cell surface HER2 is still intricate, and poziotinib upregulates the exon 20 mutant, but not wild-type HER2, suggesting synergistic mechanisms independent of HER2 surface density [21, 113,114,115,116,117,118,119].
Several clinical trials have explored the use of TKI in combination with ADCs. The TEAL study showed that employing a combination of T-DM1, lapatinib, and nab-paclitaxel for the neoadjuvant treatment of patients with HER2-positive breast cancer yielded improved responses compared to the standard paclitaxel, trastuzumab, and pertuzumab combination, which was accentuated in the traditionally challenging hormone receptor-positive subset [21]. The combination regimen of T-DM1/T-DXd and tucatinib for advanced breast cancer progression with prior taxane and trastuzumab showed acceptable toxicity and preliminary anti-tumor activity in patients with ERBB2/HER2-positive metastatic breast cancer with and without brain metastases [74, 79, 120]. Moreover, phase III trials testing T-DM1 or T-DXd with tucatinib (HER2CLIMB-02 and HER2CLIMB-04) are ongoing [79, 120], and we look forward to their deterministic results.
Synthetic lethality and combined targeting
Synthetic lethality is a promising and clinically effective therapeutic strategy for tumors with defects in DNA homologous recombination repair pathways. Given the recent focus on DNA damage response (DDR) pathways in cancer therapy, several DDR proteins, including ATR, ATM, DNA-PK, CHK1, CHK2, Wee1, and PARP, have been extensively explored as promising synthetic lethality targets for anticancer drug development [121,122,123]. While PARP inhibitors first achieved clinical approval in 2014, inhibitors targeting other DDR proteins are currently under intense clinical investigation.
A subset of ADCs incorporates topoisomerase I (TOPO 1) inhibitors as payloads. For instance, TOPO 1, an enzyme, initiates the cleavage of one strand of double-stranded DNA, resulting in partial unwinding and subsequent reannealing of the strand to relieve tension. Camptothecin and its derivatives bind to the TOPO 1/DNA complex and prevent proper reannealing. This disruption can lead to cell death owing to the accumulation of partially cleaved DNA. SN38, a semi-synthetic derivative of camptothecin, is the active component of irinotecan and has been used in sacituzumab govitecan, a clinically approved TROP2-targeting ADC [124]. Another camptothecin derivative, DXd, is a derivative of exatecan that is approximately 10 times more potent than SN38, with an IC50 value of 310 nM. This potent compound is used as a payload in HER2-targeting (DS-8201a) and TROP2-targeting (DS1062) ADCs.
PARP-1, the most abundant member of the PARP protein family, has been observed to co-localizes with Topo I throughout the cell cycle. However, when DNA damage occurs, PARP-1 dissociates from Topo I, leading to decreased enzymatic activity [125]. Combining a TOPO I inhibitor with a PARP inhibitor (PARPi) results in the accumulation of double-stranded DNA breaks (DSBs) by retarding the homologous recombination repair pathways that effectively and precisely repair DNA damage. This DSB accumulation ultimately triggers apoptosis and cell death. In addition, in cells lacking functional BRCA1/2 genes or those deficient in the homologous recombination repair mechanism, an alternative but less precise DNA damage repair pathway known as non-homologous end-joining emerges. This more error-prone pathway further compromises cells toward irreparable DNA damage and apoptosis [126]. Recent research has also shown that combining CPT-11 (the prodrug of SN-38) and PARPi achieved synergistic inhibition in both BRCA1 wild-type and BRCA1 mutant triple-negative breast cancer (TNBC) cell lines in vitro [127]. Concurrently, sacituzumab govitecan combined with olaparib, rucaparib, or talazoparib also synergistically inhibited tumor cell growth and increased DSBs in HCC1806 TNBC tumors harboring mutations in the BRCA1/2 genes, as well as in those with wild-type counterparts [128].
Safety profile of ADCs combined with targeted therapies
Among the 15 types of ADC currently approved, the combination of T-DM1 and targeted agents has the greatest evidence of efficacy and safety. A phase Ib trial evaluated the combination of T-DM1 and the HER2 TKI, tucatinib. Although this combination is well-tolerated, it is associated with frequent gastrointestinal and hepatic toxicities. In particular, 37% of the patients experienced at least one clinically significant adverse event, and 56% required discontinuation of tucatinib [74]. More information is expected from the ongoing phase III HER2CLIMB-02 trial evaluating T-DM1 with tucatinib. Compared with T-DM1 alone, these results may provide more insight into the additional toxicity associated with this combination. Another Ib study evaluated the combination of intermittent inhibition of T-DM1 and CDK4/6 using riboflavin; however, no DLTs were observed. However, ribociclib dose reductions were required in 58% of the patients due to thrombocytopenia or neutropenia, and one patient experienced grade 2 QTcF prolongation [129]. In a phase Ib study involving patients with metastatic TNBC, sacituzumab govitecan was administered in combination with talazoparib, a PARP inhibitor. This study demonstrated several DLTs primarily caused by severe myelosuppression, as described in the initial study results. In particular, the majority of enrolled patients experienced febrile neutropenia [83]. Finally, a phase Ib study evaluated the effects of adding bevacizumab to mirvetuximab soravtansine. This study included patients with platinum-resistant ovarian cancer [11]. The combination resulted in a toxicity profile similar to that of ADC alone [130]. However, it is worth noting that grade 1–2 pneumonitis was observed in six patients (9%) with the addition of bevacizumab. This was in contrast to the absence of pneumonitis when mirvetuximab soravtansine was administered alone.
In summary, although we observed promising results from combining TKIs and ADCs in preclinical models and clinical trials, the underlying molecular interplay is still far from being completely understood. A better mechanistic understanding is helpful for the selection of drug combinations and the management of potential side effects.
ADCs combined with immunotherapy
Accumulating evidence suggests that ADCs are sensitive to the effectiveness of immunotherapeutic agents [131]. Combining immunotherapy with ADCs is a current trend in clinical practice, with a number of preclinical studies and initial findings from early-stage clinical trials showing improved anti-tumor effects [131]. The clinical trials are listed in Table 6. We still await the outcomes of large-cohort randomized phase III clinical trials to demonstrate the determinant evidence of this combination’s efficacy compared with that of conventional treatments.
Mechanism of ADCs combined with immunotherapy
The underlying mechanisms are diverse and encompass Fc-mediated effector functions, initiation of immunogenic cell death (ICD), maturation of dendritic cells (DCs), enhancement of T cell infiltration, reinforcement of immunological memory, and expression of immunomodulatory proteins such as programmed death ligand 1 (PD-L1) and major histocompatibility complex (MHC) [132,133,134,135,136]. Multiple studies have revealed that ADCs exert a stronger effect in immunocompetent animal models than in immunodeficient models, indicating their significant immunomodulatory capabilities [137, 138]. These findings provide a basis for devising clinical trials that incorporate low doses of ADCs as immunostimulants to improve the efficacy of immunotherapy without causing adverse effects.
Fc-mediated effector functions
In the design of an ADC, the antibody component plays a multifaceted role, instead of solely delivering cytotoxic agents to cancer cells. Its unique Y-shaped structure has many additional functions, including regulation of innate immune responses. While the antigen-binding fragments of an antibody are responsible for recognizing the target antigen and determining its specificity, the crystallizable fragment (Fc) interacts with immune cells and regulates the duration of the antibody’s circulation in the bloodstream. The Fc region, in fact, plays key roles in several vital functions, including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cell-mediated phagocytosis, and complement-dependent cytotoxicity. The effectiveness of the initial two actions relies on Fcγ receptors (FcγRs), which are present on natural killer cells, macrophages, and various other immune cells. Conversely, complement-dependent cytotoxicity is triggered by C1q protein.
The IgG antibody family consists of IgG1, IgG2, IgG3, and IgG4 subclasses, each of which exert different effects on factors such as antibody solubility, half-life, interaction with the C1q protein, and binding strength to FcγRs. The IgG1 subclass of antibodies is most commonly used in the 15 clinically approved types of ADCs [166]. The reason for selecting IgG1 as the backbone of ADC is because it has a long half-life of approximately 21 days, similar to IgG2 and IgG4, but is characterized by its enhanced ability to activate the complement system and bind to FcγRs. In contrast, IgG2 and IgG4 have limited efficacy in triggering effector functions, and are used strategically in antibody design when eliciting an immune response is not the primary goal [167]. In contrast, IgG3 is the most immunogenic subclass capable of eliciting an immune response. However, these antibodies are typically bypassed in the design of ADCs because of their short half-lives (approximately seven days).
In preclinical models, both T-DXd and T-DM1 exhibit the ability to maintain functions inherent to unconjugated trastuzumab, including triggering ADCC associated with the IgG1 isotype [137, 168]. In addition to these therapeutic benefits, Fc-mediated effector functions give rise to undesirable side effects. For instance, T-DM1 is internalized by megakaryocytes through its interaction with FcγRIIA, which could potentially lead to the development of thrombocytopenia, a known side effect of this agent [169].
Modulating the ability of an ADC to engage the immune system may involve engineering the Fc region. One approach is to produce afucosylated IgGs, which enhance ADCC by increasing the binding affinity for FcγRIIIa [170, 171]. Conversely, the Fc region can be modified by introducing mutations that impact effector functions, yielding what is known as "Fc silent antibodies" [138]. MEDI4276, for instance, employs this strategy with three mutations in its Fc domain to curtail FcγR binding, aiming to minimize thrombocytopenia as observed with T-DM1 [172].
Another approach that involves the interaction between Fc and FcγRs takes place in the TME, in which the tumor-associated macrophages (TAMs) constitute a significant proportion. In preclinical models, non-targeted ADCs have been shown to be effectively engulfed by TAMs. Through the engagement of FcγRs, TAMs internalize and process these ADCs, resulting in the release of the cytotoxic payloads within the TME. This results in the killing of neighboring tumor cells which is called “bystander effect”. This mechanism may enhance the efficacy of ADCs against tumors that exhibit heterogeneity or low levels of target antigens [173].
However, it is important to recognize that this mechanism of antigen-independent ADC uptake into non-malignant cells within the TME could exacerbate the toxicity of ADCs. For instance, it could potentially lead to more rapid clearance of ADCs and reduced overall efficacy. Another theoretical concern related to the release of cytotoxic payload within the TME and the subsequent bystander effect is the potential destruction of local T cells, which can negatively affect the efficacy of immune checkpoint inhibitors (ICIs).
In summary, while the interaction of Fc and FcγRs between ADCs and the TME represents a potential avenue for enhancing ADC activity against tumors with challenging characteristics, careful consideration must be given to balance efficacy with potential drawbacks such as increased toxicity and interference with immune checkpoint inhibition.
Immunogenic cell death
Based on the initial stimulus, cancer cell death can either activate the immune system (immunogenic) or go unnoticed by it (non-immunogenic). ICD is a regulatory process characterized by the induction of stress within the endoplasmic reticulum and cellular structures. This process is accompanied by changes in the cell surface composition and the subsequent release of soluble mediators, which follow a precise spatiotemporal sequence and ultimately lead to cell death [174, 175].
The ability of a drug to induce ICD and establish an immunological memory is often predicted by its ability to induce damage-associated molecular patterns (DAMPs) in vitro [176, 177]. A wide array of anticancer therapeutics, including traditional chemotherapy, radiotherapy, and targeted anticancer agents, has demonstrated the potential to induce DAMPs [178,179,180]. Only a small fraction (< 10%) of all chemotherapeutic agents, such as anthracyclines [181, 182] and oxaliplatin [183], are classified as ICD-inducing drugs. The majority of cytotoxic payloads employed in ADCs exhibit the ability to activate immune cells both in the laboratory and in living organisms, which not only improves their anti-tumor tumor efficacy, but also synergistically enhances the effect of ICIs in preclinical models [184].
In mouse models, ADCs with payloads such as maytansine, pyrrolobenzodiazepine, and tubulysin have shown the ability to induce ICD [135] trigger immune modulation, and establish immune memory. These ADCs not only exhibit potent cytotoxicity but also synergize with various ICIs. Notably, these ADCs exhibited significantly greater anti-tumor activity in immunocompetent mice than in immunocompromised mice, highlighting the role of the immune system in their efficacy. Similarly, a newly developed anti-HER2 ADC containing a potent anthracycline derivative payload (T-PNU) increased DAMP expression and enhanced efficacy when combined with an anti-PD-1 drug in a breast cancer model that developed resistance to other HER2-targeted therapies. Notably, the efficacy of T-PNU was significantly reduced when CD8 + T cells were depleted, confirming the critical role of the adaptive immune system in regulating the anticancer activity of T-PNU. In addition, T-PNU appeared to promote the formation of an immunological memory in tumor-bearing animals, resulting in protection against tumor rechallenge [132]. Such an ICD-induced intrinsic inflammatory response has also been observed with brentuximab vedotin [185], ladiratuzumab vedotin [186], and enapotamab vedotin [136], which are three ADCs that share the same MMAE payload. This unique property further enhances the efficacy of ICIs.
Direct activation and maturation of dendritic cells
Mature DCs play a pivotal role in tumor immunity by acting as antigen-presenting cells capable of activating anti-tumor T cell responses through the MHC class II complex [187]. However, cancer cells often develop immunosuppression by inhibiting DC maturation or causing them to become dysfunctional, ultimately resulting in immune evasion [188]. Overcoming these barriers is essential for improving immunotherapy outcomes in clinical practice.
Previous research has shown that compounds that destabilize microtubules are capable of inducing the phenotypic and functional maturation of DCs, which was not observed with microtubule-stabilizing compounds such as taxanes [189]. This phenomenon appears to be a common feature of this class of compounds, indicating their potential use as "immunostimulatory" agents. This immunostimulatory effect was first reported in vinblastine [190] and subsequently in many other microtubule-disrupting agents, such as maytansinoids (e.g., ansamitocin P3 and its synthetic derivative DM1) and dolastatins (from which auristatins are derived), which are frequently used as payloads in ADCs [133, 191]. In preclinical models, these payloads have been demonstrated to directly trigger DC activation and maturation. Importantly, these potent immunoregulatory effects were observed even without cancer cell death, indicating an independent binary mode of action. The complete therapeutic efficacy of these ADCs includes payload cytotoxicity and immunoregulatory functions; the latter strongly depends on an intact host immune system, which is significantly diminished in immunocompromised mouse models.
Preclinical studies combining tubule inhibitor-based ADCs with ICIs have confirmed that these two types of treatment modalities can work synergistically to increase therapeutic efficacy rather than have additive effects [134, 191]. In cases where tumors responded completely to the combination treatment, mice showed protection upon rechallenge with the same tumor, indicating the successful establishment of immunological memory. Notably, an analysis of paired samples from 28 patients with breast cancer who underwent short-term preoperative treatment with T-DM1 as part of the WSG-ADAPT protocol sub-trial revealed significant increases in the number and density of tumor-infiltrating T cells [168]. There is evidence that topoisomerase I inhibitors can also act as immunomodulators by activating DCs [192], as exemplified by T-DXd, an HER2-targeted ADC that carries the exatecan derivative DXd, a topoisomerase I inhibitor, as payload. T-DXd has been found to significantly increase the presence of tumor-infiltrating DCs and the expression of markers indicative of maturation and activation, leading to an increase in tumor-infiltrating CD8 + T cells, along with increased expression of PD-L1 and MHC class I on tumor cells. Notably, in a CT26-HER2 tumor model, the combination of T-DXd with an anti-PD-1 agent proved to be more effective than either treatment alone, possibly because of the immunomodulatory changes induced by T-DXd [134]. It is worth noting that the T-DXd payload possesses a tenfold more potent topoisomerase I inhibitor activity compared to SN-38, which may contribute to a more robust immunologic effect compared to other agents in the same class [168].
Combining ADCs and ICIs
Currently, HER2-targeted ADCs are under intense clinical investigation for their synergistic effects in combination with ICIs; however, many trials are still ongoing, and determinant evidence is very limited [137, 193, 194]. The only published randomized trial evaluating the combination of an ADC and ICI is the KATE2 trial. This study evaluated the efficacy of T-DM1 plus atezolizumab and compared it with T-DM1 plus placebo. The study was conducted in patients who had previously been treated for HER2-positive breast cancer and, disappointingly, the combination therapy did not result in a statistically significant improvement in PFS in the overall patient population, with a median PFS of 8.2 months in the combination arm and 6.2 months in the control arm (P = 0.33). However, a trend suggesting potential benefit in a specific subset of patients with positive PD-L1 expression was observed, where median PFS was 8.5 months for the combination arm and 4.1 months for the control arm (P = 0.099). This indicated that adding an ICI to HER2-targeted treatment for HER2 + breast cancer may be particularly beneficial in the PD-L1-positive subset [77, 195,196,197,198,199,200,201,202,203,204]. Most cancer patients enrolled in published studies had not previously received treatment with an ICI. Thus, we are uncertain about the synergistic benefits that ADC may provide when combined with ICIs in tumor types that are known to respond well to immunotherapy. However, currently available clinical data suggest significantly improved response rates, as shown by a comparison with the historical efficacy results achieved with standalone immunotherapy in these specific tumor types [132, 133, 135, 136, 184,185,186,187,188,189,190].
After witnessing remarkable improvements in efficacy in specific cancer contexts, certain combinations will likely be adopted as new standards of care, potentially replacing traditional cytotoxic treatment approaches. To illustrate this theory, the combination of enfortumab vedotin, an ADC targeting nectin 4, and pembrolizumab has been evaluated as a first-line treatment option for individuals diagnosed with locally advanced or metastatic urothelial cancer (as demonstrated in the EV-103/KEYNOTE-869 study; ClinicalTrials.gov identifier: NCT03288545) [184]. In this patient population, the combination achieved an impressive objective response rate of 73% and extended the PFS to 12.3 months. This result led to a breakthrough designation granted by the US FDA, specifically for patients ineligible for cisplatin-based therapy. Moreover, other cancers, including cervical cancer, triple-negative breast cancer, Hodgkin's lymphoma, and primary mediastinal large B cell lymphoma, have shown encouraging results when treated with a combination of anti-PD-1 antibodies and ADCs targeting specific markers, such as tissue factor, TROP2, and CD30 [205]. This novel strategy of combination therapy holds great promise, particularly for older and frail patients who are at an increased risk of experiencing severe side effects from traditional chemotherapy regimens [187, 188, 190, 206,207,208]. In the coming months and years, we expect to see more trial results that employ various immunotherapeutic agents to enhance ADC activity.
Safety profile of ADCs combined with immunotherapy
In the phase III KATE2 trial, a significant increase in adverse events, including one treatment-related death, was observed in the combination arm that combined atezolizumab and T-DM1 in patients with previously treated HER2-positive metastatic breast cancer. The frequencies of clinically significant adverse events (33% vs. 19%) and most adverse reactions, particularly fever (35% vs. 16%, including several hospitalizations), increased after the introduction of atezolizumab [140]. Similarly, randomized data are now available for enfortumab vedotin, with and without pembrolizumab, in 149 patients with advanced-stage urothelial carcinoma. The introduction of pembrolizumab resulted in a higher occurrence of clinically significant (23.7% vs. 15.1%) and fatal (3.9% vs. 2.7%) adverse events, and an overall increase in the incidence of all adverse events. Notably, an increase in serious skin reactions was observed [209]. Nevertheless, the combination received accelerated approval from the FDA in April 2023 for patients with locally advanced or metastatic urothelial carcinoma, particularly those who could not receive cisplatin-based chemotherapy. This approval was based on the efficacy of the combination. However, the lack of a randomized design in most other studies on ADC and ICI combination therapies makes it difficult to reach definitive conclusions regarding the many unanswered questions. To date, there have been no alarming signs of increased toxicity induced by ICIs in combination with T-DXd [77, 210, 211], Dato-DXd [204], or sacituzumab-govitecan [212], all of which have additive toxicity profiles. These safety profiles include similar rates and intensities of ILD, which frequently occurs with DXd-based ADC treatments. Interestingly, the addition of ICIs does not appear to increase the incidence of ILD associated with ADCs [141, 175, 177, 213]. Ongoing randomized phase III trials (NCT05629585, NCT05382286, and NCT05633654) are expected to offer additional insights into this domain. These trials may further clarify the toxicity patterns of treatment approaches that combine ICIs with ADCs beyond T-DM1.
Conclusions
Monotherapy with ADCs has exhibited transformative anti-tumor efficacy across a broad spectrum of solid and hematological malignancies. The present landscape is characterized by substantial efforts from both the academic and industrial sectors focused on advancing the understanding of ADC combination therapy, which entails the progress of next-generation ADCs by identifying novel tumor targets and clarifying their pharmacological properties.
Notably, the combination of ADCs with chemotherapy or chemoimmunotherapy regimens, excluding ICIs, has yielded demonstrable survival advantages over the established standard regimens in randomized investigations of hematological malignancies. Although the combination of ADCs with ICIs has exhibited encouraging outcomes, as exemplified by the FDA breakthrough designations of enfortumab vedotin and pembrolizumab for cisplatin-ineligible urothelial cancer, the envisioned survival improvements and underlying biological mechanisms for solid tumors remain elusive within the context of randomized controlled trials.
Furthermore, ADC combinations with targeted agents, particularly inhibitors targeting the HER2 and DDR pathways, hold substantial promise, although their potential is contingent upon validation through more mature datasets. The constrained success witnessed thus far in combination therapy with first-generation ADCs (e.g. T-DM1) can be attributed to many factors that encompass the indiscriminate expression of target molecules, resulting in off-tumor side effects on normal tissues, overlapping toxicities, limited efficacy, and unclear procedures conferring resistance. The landscape of ADC-based combination therapies remains dynamic, with current challenges underscoring the complexities of target expression, tumor heterogeneity, and the intricate interplay of therapeutic modalities.
In addition to understanding the pharmacological properties (e.g. DAR and bystander effect) to improve ADC efficacy, there is also a strong need to stratify patients with high response rates and detect relevant predictive biomarker profiles. Exploring preclinical experiments in carefully characterized patient-derived xenograft models and conducting clinical trials in window-of-opportunity contexts could facilitate the identification of promising ADC-based combinations in clinical practice. More strategic methodologies are required to effectively identify suitable ADC-based combination approaches for selected patient cohorts and tumor types. This will not only capitalize on the refinement of ADC design and properties, but also leverage well-informed patient selection strategies to optimize therapeutic outcomes.
Availability of data and materials
Not applicable.
References
DeVita VT, Rosenberg SA. Two hundred years of cancer research. N Engl J Med. 2012;366:2207–14.
Criscitiello C, Morganti S, Curigliano G. Antibody-drug conjugates in solid tumors: a look into novel targets. J Hematol Oncol. 2021;14.
Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18:327–44.
Verma S, Miles D, Gianni L, Krop IE, Welslau M, Baselga J, et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N Engl J Med. 2012;367:1783–91.
Pomeroy AE, Schmidt EV, Sorger PK, Palmer AC. Drug independence and the curability of cancer by combination chemotherapy. Trends Cancer. 2022;8:915–29.
Yardley DA. Drug resistance and the role of combination chemotherapy in improving patient outcomes. Int J Breast Cancer. 2013;2013:1–15.
Abrahams C, Krimm S, Li X, Zhou S, Hanson J, Masikat MR, et al. Abstract NT-090: Preclinical activity and safety of STRO-002, a novel ADC targeting folate receptor alpha for ovarian and endometrial cancer. Clinical Cancer Res. 2019;25:NT-090. https://doi.org/10.1158/1557-3265.OVCASYMP18-NT-090
Quanz M, Hagemann UB, Zitzmann-Kolbe S, Stelte-Ludwig B, Golfier S, Elbi C, et al. Anetumab ravtansine inhibits tumor growth and shows additive effect in combination with targeted agents and chemotherapy in mesothelin-expressing human ovarian cancer models. Oncotarget. 2018;9:34103–21.
Ponte JF, Ab O, Lanieri L, Lee J, Coccia J, Bartle LM, et al. Mirvetuximab Soravtansine (IMGN853), a folate receptor alpha-targeting antibody-drug conjugate, potentiates the activity of standard of care therapeutics in ovarian cancer models. Neoplasia. 2016;18:775–84.
Moore KN, O’Malley DM, Vergote I, Martin LP, Gonzalez-Martin A, Malek K, et al. Safety and activity findings from a phase 1b escalation study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin in patients with platinum-sensitive ovarian cancer. Gynecol Oncol. 2018;151:46–52.
O’Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2020;157:379–85.
Bulat I, Moore KN, Haceatrean A, Chung JW, Rajagopalan P, Xia C, et al. Abstract 5571: Phase Ib study of anti-mesothelin antibody drug conjugate anetumab ravtansine in combination with pegylated liposomal doxorubicin in platinum-resistant ovarian, fallopian tube, or primary peritoneal cancer. 2018;36:5571–5571.
Levy B, Paz-Ares L, Rixe O, Su W-C, Yang T-Y, Tolcher A, et al. MA13.07 TROPION-Lung02: initial results for datopotamab deruxtecan plus pembrolizumab and platinum chemotherapy in advanced NSCLC. J Thoracic Oncol 2022;17:S91.
Janjigian YY, Oh D-Y, Rha SY, Lee KW, Steeghs N, Chao Y, et al. Abstract 295: Dose-escalation and dose-expansion study of trastuzumab deruxtecan (T-DXd) monotherapy and combinations in patients (pts) with advanced/metastatic HER2+ gastric cancer (GC)/gastroesophageal junction adenocarcinoma (GEJA): DESTINY-Gastric03. 2022;40:295–295.
Kan S, Koido S, Okamoto M, Hayashi K, Ito M, Kamata Y, et al. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer. 2015;15.
Tseng CJ, Wang YJ, Liang YC, Jeng JH, Lee W Sen, Lin JK, et al. Microtubule damaging agents induce apoptosis in HL 60 cells and G2/M cell cycle arrest in HT 29 cells. Toxicology. 2002;175:123–42.
Wahl AF, Donaldson KL, Mixan BJ, Trail PA, Siegall CB. Selective tumor sensitization to taxanes with the mAb-drug conjugate cBR96-doxorubicin. Int J Cancer. 2001;93:590–600.
Martin M, Fumoleau P, Dewar JA, Albanell J, Limentani SA, Campone M, et al. Trastuzumab emtansine (T-DM1) plus docetaxel with or without pertuzumab in patients with HER2-positive locally advanced or metastatic breast cancer: results from a phase Ib/IIa study. Ann Oncol. 2016;27:1249–56.
Cortés J, Diéras V, Lorenzen S, Montemurro F, Riera-Knorrenschild J, Thuss-Patience P, et al. Efficacy and safety of trastuzumab emtansine plus capecitabine vs trastuzumab emtansine alone in patients with previously treated ERBB2 (HER2)-positive metastatic breast cancer: a phase 1 and randomized phase 2 trial. JAMA Oncol. 2020;6:1203–9.
Patel TA, Ensor J, Rodriguez AA, Belcheva A, Darcourt JG, Niravath PA, et al. Phase ib study of trastuzumab emtansine (TDM1) in combination with lapatinib and nab-paclitaxel in metastatic HER2-neu overexpressed breast cancer patients: Stela results. J Clin Oncol. 2018;36:1035–1035.
Patel TA, Ensor JE, Creamer SL, Boone T, Rodriguez AA, Niravath PA, et al. A randomized, controlled phase II trial of neoadjuvant ado-trastuzumab emtansine, lapatinib, and nab-paclitaxel versus trastuzumab, pertuzumab, and paclitaxel in HER2-positive breast cancer (TEAL study). Breast Cancer Res. 2019;21.
López-Miranda E, Pérez-García JM, Di Cosimo S, Brain E, Ravnik M, Escrivá-De-romaní S, et al. Trastuzumab emtansine plus non-pegylated liposomal doxorubicin in HER2-positive metastatic breast cancer (thelma): a single-arm, multicenter, phase Ib trial. Cancers (Basel). 2020;12:1–14.
Zimmer AS, Steinberg SM, Smart DD, Gilbert MR, Armstrong TS, Burton E, et al. Temozolomide in secondary prevention of HER2-positive breast cancer brain metastases. Future Oncol. 2020;16:899–909.
Planchard D, Brahmer JR, Yang JC, Chen Y-M, Lee K-Y, Suksombooncharoen T, et al. Abstract CT572: Phase 1b dose-escalation and dose-expansion study evaluating trastuzumab deruxtecan (T-DXd) in combination with durvalumab and cisplatin, carboplatin, or pemetrexed in advanced or metastatic, HER2-overexpressing, nonsquamous non-small cell lung cancer (NSCLC): DESTINY-Lung03. Cancer Res. 2022;82:CT572–CT572. https://doi.org/10.1158/1538-7445.AM2022-CT572
O’Donnell PH, Milowsky MI, Petrylak DP, Hoimes CJ, Flaig TW, Mar N, et al. Enfortumab vedotin with or without pembrolizumab in cisplatin-ineligible patients with previously untreated locally advanced or metastatic urothelial cancer. J Clin Oncol. 2023.
Le X, Carneiro BA, Hong DS, Birnbaum AE, Taylor MH, Patel SA, et al. innovaTV 207: New combination dosing cohorts in the open label phase 2 study of tisotumab vedotin in solid tumors. 2022;40:TPS6100–TPS6100.
Lassman AB, Van Den Bent MJ, Gan HK, Reardon DA, Kumthekar P, Butowski N, et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: results from an international phase I multicenter trial. Neuro Oncol. 2019;21:106–14.
O’Malley DM, Oaknin A, Matulonis UA, Mantia-Smaldone G, Lim PC, Castro CM, et al. Abstract 5504: Mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients (pts) with platinum-agnostic ovarian cancer: final analysis. 2021;19:8–10.
Risbridger GP, Davis ID, Birrell SN, Tilley WD. Breast and prostate cancer: more similar than different. Nat Rev Cancer. 2010;10:205–12.
Mamounas EP, Untch M, Mano MS, Huang CS, Geyer CE, von Minckwitz G, et al. Adjuvant T-DM1 versus trastuzumab in patients with residual invasive disease after neoadjuvant therapy for HER2-positive breast cancer: subgroup analyses from KATHERINE. Ann Oncol. 2021;32:1005–14.
Hurvitz SA, Wang LS, Chan D, Phan V, Lomis T, McAndrew NP, et al. TRIO-US B-12 TALENT: Phase II neoadjuvant trial evaluating trastuzumab deruxtecan with or without anastrozole for HER2-low, HR+ early-stage breast cancer. 2022;40:TPS623–TPS623.
Mattes MJ. Radionuclide-antibody conjugates for single-cell cytotoxicity. Cancer. John Wiley and Sons Inc.; 2002. p. 1215–23.
Salvestrini V, Kim K, Caini S, Alkner S, Ekholm M, Skyttä T, et al. Safety profile of trastuzumab-emtansine (T-DM1) with concurrent radiation therapy: a systematic review and meta-analysis. Radiother Oncol. 2023;186: 109805.
Krop IE, Suter TM, Dang CT, Dirix L, Romieu G, Zamagni C, et al. Feasibility and cardiac safety of trastuzumab emtansine after anthracycline-based chemotherapy as (neo)adjuvant therapy for human epidermal growth factor receptor 2-positive early-stage breast cancer. J Clin Oncol. 2015;33:1136–42.
von Minckwitz G, Huang C-S, Mano MS, Loibl S, Mamounas EP, Untch M, et al. Trastuzumab emtansine for residual invasive HER2-positive breast cancer. N Engl J Med. 2019;380:617–28. https://doi.org/10.1056/NEJMoa1814017.
Carlson JA, Nooruddin Z, Rusthoven C, Elias A, Borges VF, Diamond JR, et al. Trastuzumab emtansine and stereotactic radiosurgery: an unexpected increase in clinically significant brain edema. Neuro Oncol. 2014;16:1006–9. https://doi.org/10.1093/neuonc/not329.
Lassman AB, Pugh SL, Wang TJC, Aldape K, Gan HK, Preusser M, et al. Depatuxizumab-mafodotin in EGFR-amplified newly diagnosed glioblastoma: a phase III randomized clinical trial. Neuro Oncol. 2022;
Narita Y, Muragaki Y, Kagawa N, Asai K, Nagane M, Matsuda M, et al. Safety and efficacy of depatuxizumab mafodotin in Japanese patients with malignant glioma: a nonrandomized, phase 1/2 trial. Cancer Sci. 2021;112:5020–33.
Géraud A, Xu HP, Beuzeboc P, Kirova YM. Preliminary results of the concurrent use of radiotherapy for bone metastases and trastuzumab emtansine in patients with HER2-positive metastatic breast cancer. Cancer/Radiothérapie. 2016;20:312–3.
Jacot W, Pons E, Frenel JS, Guiu S, Levy C, Heudel PE, et al. Efficacy and safety of trastuzumab emtansine (T-DM1) in patients with HER2-positive breast cancer with brain metastases. Breast Cancer Res Treat. 2016;157:307–18. https://doi.org/10.1007/s10549-016-3828-6.
Geraud A, Xu HP, Beuzeboc P, Kirova YM. Preliminary experience of the concurrent use of radiosurgery and T-DM1 for brain metastases in HER2-positive metastatic breast cancer. J Neurooncol. 2017;131:69–72. https://doi.org/10.1007/s11060-016-2265-z.
Zolcsák Z, Loirat D, Fourquet A, Kirova YM. Adjuvant trastuzumab emtansine (T-DM1) and concurrent radiotherapy for residual invasive HER2-positive breast cancer: single-center preliminary results. Am J Clin Oncol Cancer Clin Trials. 2020;43:895–901.
Mills MN, Walker C, Thawani C, Naz A, Figura NB, Kushchayev S, et al. Trastuzumab emtansine (T-DM1) and stereotactic radiation in the management of HER2+ breast cancer brain metastases. BMC Cancer. 2021;21:1–8. https://doi.org/10.1186/s12885-021-07971-w.
Bennardo L, Passante M, Cameli N, Cristaudo A, Patruno C, Nisticò SP, et al. Skin manifestations after ionizing radiation exposure: a systematic review. Bioengineering 2021;8:153.
Hariri G, Zhang Y, Fu A, Han Z, Brechbiel M, Tantawy MN, et al. Radiation-guided P-selectin antibody targeted to lung cancer. Ann Biomed Eng. 2008;36:821–30. https://doi.org/10.1007/s10439-008-9444-9.
Shamay Y, Elkabets M, Li H, Shah J, Brook S, Wang F, et al. P-selectin is a nanotherapeutic delivery target in the tumor microenvironment. Sci Transl Med. 2016. https://doi.org/10.1126/scitranslmed.aaf7374.
Dong D, Ni M, Li J, Xiong S, Ye W, Virrey JJ, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. https://doi.org/10.1158/0008-5472.CAN-07-2950.
Lin TY, Chang JTC, Wang HM, Chan SH, Chiu CC, Lin CY, et al. Proteomics of the radioresistant phenotype in head-and-neck cancer: GP96 as a novel prediction marker and sensitizing target for radiotherapy. Int J Radiat Oncol Biol Phys. 2010;78:246–56.
Dadey DYA, Kapoor V, Hoye K, Khudanyan A, Collins A, Thotala D, et al. Antibody targeting GRP78 enhances the efficacy of radiation therapy in human glioblastoma and non-small cell lung cancer cell lines and tumor models. Clin Cancer Res. 2017;23:2556–64.
Leo R, Emmanuelle M, Valentina D, Eline M, Karin V, Constantin L, et al. A GRP78-Directed monoclonal antibody recaptures response in refractory multiple myeloma with extramedullary involvement. Clin Cancer Res. 2016;22:4341–9. https://doi.org/10.1158/1078-0432.CCR-15-3111.
Wang H, Yan H, Fu A, Han M, Hallahan D, Han Z. TIP-1 Translocation onto the cell plasma membrane is a molecular biomarker of tumor response to ionizing radiation. PLoS ONE. 2010;5:e12051. https://doi.org/10.1371/journal.pone.0012051.
Lewis CD, Singh AK, Hsu FF, Thotala D, Hallahan DE, Kapoor V. Targeting a radiosensitizing antibody-drug conjugate to a radiation-inducible antigen. Clin Cancer Res. 2021;27:3224–33.
Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59:928–42.
Buckel L, Savariar EN, Crisp JL, Jones KA, Hicks AM, Scanderbeg DJ, et al. Tumor radiosensitization by monomethyl auristatin E: mechanism of action and targeted delivery. Cancer Res. 2015;75:1376–87.
Hingorani DV, Doan MK, Camargo MF, Aguilera J, Song SM, Pizzo D, et al. Precision chemoradiotherapy for HER2 tumors using antibody conjugates of an auristatin derivative with reduced cell permeability. Mol Cancer Ther. 2020;19:157–67.
Norbury CJ, Hickson ID. Cellular responses to DNA damage. Annu Rev Pharmacol Toxicol. 2001;41:367–401. https://doi.org/10.1146/annurev.pharmtox.41.1.367.
Bourillon L, Bourgier C, Gaborit N, Garambois V, Llès E, Zampieri A, et al. An auristatin-based antibody-drug conjugate targeting HER3 enhances the radiation response in pancreatic cancer. Int J Cancer. 2019;145:1838–51. https://doi.org/10.1002/ijc.32273.
Adams SR, Yang HC, Savariar EN, Aguilera J, Crisp JL, Jones KA, et al. Anti-tubulin drugs conjugated to anti-ErbB antibodies selectively radiosensitize. Nature Communications 2016 7:1. 2016;7:1–11.
Stacy DR, Lu B, Hallahan DE. Radiation-guided drug delivery systems. Expert Rev Anticancer Ther. 2004;4:283–8.
Hallahan DE, Geng L, Cmelak AJ, Chakravarthy AB, Martin W, Scarfone C, et al. Targeting drug delivery to radiation-induced neoantigens in tumor microvasculature. J Control Release. 2001;74:183–91.
Stapleton S, Jaffray D, Milosevic M. Radiation effects on the tumor microenvironment: implications for nanomedicine delivery. Adv Drug Deliv Rev. 2017;109:119–30.
Stolarz AJ, Chhetri BP, Borrelli MJ, Jenkins S V., Jamshidi-Parsian A, Phillips JH, et al. Liposome formulation for tumor-targeted drug delivery using radiation therapy. Int J Mol Sci. 2022;23.
Geng L, Osusky K, Konjeti S, Fu A, Hallahan D. Radiation-guided drug delivery to tumor blood vessels results in improved tumor growth delay. J Control Release. 2004;99:369–81.
Kalofonos H, Rowlinson G, Epenetos2 AA. Enhancement of monoclonal antibody uptake in human colon tumor xenografts following irradiation. Cancer Res. 1990;50:159–63.
Nakata H, Yoshimine T, Murasawa A, Kumura E, Harada K, Ushio Y, et al. Early blood-brain barrier disruption after high-dose single-fraction irradiation in rats. Acta Neurochir (Wien). 1995;136:82–7.
Debbi K, Grellier N, Loganadane G, Boukhobza C, Mahé M, Cherif MA, et al. Interaction between radiation therapy and targeted therapies in HER2-positive breast cancer: literature review, levels of evidence for safety and recommendations for optimal treatment sequence. Cancers 2023;15:2278.
Brufsky AM, Mayer M, Rugo HS, Kaufman PA, Tan-Chiu E, Tripathy D, et al. Central nervous system metastases in patients with HER2-positive metastatic breast cancer: incidence, treatment, and survival in patients from registHER. Clin Cancer Res. 2011;17:4834–43. https://doi.org/10.1158/1078-0432.CCR-10-2962.
Stumpf PK, Cittelly DiM, Robin TP, Carlson JA, Stuhr KA, Contreras-Zarate MJ, et al. Combination of trastuzumab emtansine and stereotactic radiosurgery results in high rates of clinically significant radionecrosis and dysregulation of aquaporin-4. Clinical Cancer Res. 2019;25:3946–53. https://doi.org/10.1158/1078-0432.CCR-18-2851
Id Said B, Chen H, Jerzak KJ, Warner E, Myrehaug S, Tseng CL, et al. Trastuzumab emtansine increases the risk of stereotactic radiosurgery-induced radionecrosis in HER2 + breast cancer. J Neurooncol. 2022;159:177–83. https://doi.org/10.1007/s11060-022-04055-y.
Pérez-García JM, Vaz Batista M, Cortez P, Ruiz-Borrego M, Cejalvo JM, de la Haba-Rodriguez J, et al. Trastuzumab deruxtecan in patients with central nervous system involvement from HER2-positive breast cancer: the DEBBRAH trial. Neuro Oncol. 2023;25:157–66. https://doi.org/10.1093/neuonc/noac144.
Marin B, Porath KA, Jain S, Kim M, Conage-pough JE, Oh J, et al. Heterogeneous delivery across the blood-brain barrier limits the efficacy of an EGFR-targeting antibody drug conjugate in glioblastoma. Neuro Oncol. 2021;23:2042–53.
Perez EA, Barrios C, Eiermann W, Toi M, Im YH, Conte P, et al. Trastuzumab emtansine with or without pertuzumab versus trastuzumab plus taxane for human epidermal growth factor receptor 2-positive, advanced breast cancer: primary results from the phase III MARIANNE study. J Clin Oncol. 2017;35:141–8.
Sartore-Bianchi A, Lonardi S, Martino C, Fenocchio E, Tosi F, Ghezzi S, et al. Pertuzumab and trastuzumab emtansine in patients with HER2-amplified metastatic colorectal cancer: the phase II HERACLES-B trial. ESMO Open. 2020;5.
Borges VF, Ferrario C, Aucoin N, Falkson C, Khan Q, Krop I, et al. Tucatinib combined with ado-trastuzumab emtansine in advanced ERBB2/HER2-positive metastatic breast cancer: a Phase 1b clinical trial. JAMA Oncol. 2018;4:1214–20.
Jain S, Shah AN, Santa-Maria CA, Siziopikou K, Rademaker A, Helenowski I, et al. Phase I study of alpelisib (BYL-719) and trastuzumab emtansine (T-DM1) in HER2-positive metastatic breast cancer (MBC) after trastuzumab and taxane therapy. Breast Cancer Res Treat. 2018;171:371–81.
Goel S, Pernas S, Tan-Wasielewski Z, Barry WT, Bardia A, Rees R, et al. Ribociclib plus trastuzumab in advanced HER2-positive breast cancer: results of a phase 1b/2 trial. Clin Breast Cancer. 2019;19:399–404.
Galsky MD, Conte G Del, Foti S, Yu EY, Machiels J-PH, Doger B, et al. Primary analysis from DS8201-A-U105: A phase 1b, two-part, open-label study of trastuzumab deruxtecan (T-DXd) with nivolumab (nivo) in patients (pts) with HER2-expressing urothelial carcinoma (UC). 2022;40:438–438.
Borghaei H, Besse B, Bardia A, Mazieres J, Popat S, Augustine B, et al. Abstract TPS1100: Trastuzumab deruxtecan (T-DXd; DS-8201) in combination with pembrolizumab in patients with advanced/metastatic breast or non-small cell lung cancer (NSCLC): A phase Ib, multicenter, study. 2020;38:TPS1100–TPS1100.
Hurvitz S, Vahdat L, Harbeck N, Wolff AC, Tolaney SM, Loi S, et al. Abstract OT-28–01: HER2CLIMB-02: A randomized, double-blind, phase 3 study of tucatinib or placebo with T-DM1 for unresectable locally-advanced or metastatic HER2+ breast cancer. Cancer Res. 2021;81:OT-28–01. https://doi.org/10.1158/1538-7445.SABCS20-OT-28-01
Zhou L, Xu H, Li S, Yan X, Li J, Wu X, et al. Study RC48-C014: Preliminary results of RC48-ADC combined with toripalimab in patients with locally advanced or metastatic urothelial carcinoma. J Clin Oncol. 2022;40:515–515.
Andre F, Hamilton EP, Loi S, Schmid P, Anders CK, Yu T, et al. Abstract TPS1096: Trastuzumab deruxtecan (T-DXd) combinations in patients with HER2-positive advanced or metastatic breast cancer: a phase 1b/2, open-label, multicenter, dose-finding and dose-expansion study (DESTINY-Breast07). 2021;39:TPS1096–TPS1096.
Carey LA, Krop I, Ramos J, Feng W, Hamilton E. 331TiP HER2CLIMB-04: phase II trial of tucatinib + trastuzumab deruxtecan in patients with HER2+ locally advanced or metastatic breast cancer with and without brain metastases. Ann Oncol. 2021;32:S510–1.
Bardia A, Coates JT, Spring L, Sun S, Juric D, Thimmiah N, et al. Abstract 2638: Sacituzumab Govitecan, combination with PARP inhibitor, Talazoparib, in metastatic triple-negative breast cancer (TNBC): Translational investigation. Cancer Res. 2022;82:2638–2638. https://doi.org/10.1158/1538-7445.AM2022-2638.
McGregor BA, Kwak L, Mantia C, Ravi A, Berchuck JE, Ravi P, et al. Sacituzumab govitecan (SG) plus enfortumab vedotin (EV) for metastatic urothelial carcinoma (UC) progressing on platinum-based chemotherapy and PD1/L1 inhibitors (ICB): Double antibody drug conjugate (DAD) phase I trial. J Clin Oncol 2022;40:TPS588–TPS588.
Naumann RW, Martin LP, Oaknin A, Spira AI, Hamilton EP, Schilder RJ, et al. STRO-002-GM2: A phase 1, open-label, safety, pharmacokinetic, and preliminary efficacy study of STRO-002, an anti-folate receptor alpha (FolRα) antibody-drug conjugate (ADC), in combination with bevacizumab in patients with advanced epithelial ovarian cancer (EOC, including fallopian tube or primary peritoneal cancers). 2022;40:TPS5622–TPS5622.
Camidge DR, Barlesi F, Goldman JW, Morgensztern D, Heist R, Vokes E, et al. A Phase 1b study of telisotuzumab vedotin in combination with nivolumab in patients with NSCLC. JTO Clin Res Rep. 2021;3.
Beckwith HC, Medgyesy DC, Abraham J, Nanda R, Tkaczuk KHR, Krop IE, et al. SGNLVA-001: A phase I open-label dose escalation and expansion study of SGN-LIV1A administered weekly in breast cancer. J Clin Oncol 2020;38:TPS1104–TPS1104.
Cilliers C, Guo H, Liao J, Christodolu N, Thurber GM. Multiscale modeling of antibody-drug conjugates: connecting tissue and cellular distribution to whole animal pharmacokinetics and potential implications for efficacy. AAPS J. 2016;18:1117–30.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
Heldin CH, Rubin K, Pietras K, Östman A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–13.
Gilbert L, Oaknin A, Matulonis UA, Mantia-Smaldone GM, Lim PC, Castro CM, et al. Safety and efficacy of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2023;170:241–7.
Arjaans M, Munnink THO, Oosting SF, Van Scheltinga AGTT, Gietema JA, Garbacik ET, et al. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 2013;73:3347–55.
Huang Y, Stylianopoulos T, Duda DG, Fukumura D, Jain RK. Benefits of vascular normalization are dose- and time-dependent. Cancer Res. 2013;73:7144–6.
Arjaans M, Oosting SF, Schröder CP, De Vries EGE. Bevacizumab-induced vessel normalization hampers tumor uptake of antibodies–response. Cancer Res. 2013;73:7147–8.
Oosting SF, Brouwers AH, Van Es SC, Nagengast WB, Munnink THO, Lub-De Hooge MN, et al. 89Zr-bevacizumab PET visualizes heterogeneous tracer accumulation in tumor lesions of renal cell carcinoma patients and differential effects of antiangiogenic treatment. J Nucl Med. 2015;56:63–9.
Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–8.
Phillips GDL, Fields CT, Li G, Dowbenko D, Schaefer G, Miller K, et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: critical role for neuregulin blockade in antitumor response to combination therapy. Clin Cancer Res. 2014;20:456–68.
Miller KD, Diéras V, Harbeck N, Andre F, Mahtani RL, Gianni L, et al. Phase IIa trial of trastuzumab emtansine with pertuzumab for patients with human epidermal growth factor receptor 2-positive, locally advanced, or metastatic breast cancer. J Clin Oncol. 2014;32:1437–44.
Bordeau BM, Yang Y, Balthasar JP. Transient competitive inhibition bypasses the binding site barrier to improve tumor penetration of trastuzumab and enhance T-DM1 efficacy. Cancer Res. 2021;81:4145–54.
Krop IE, Im SA, Barrios C, Bonnefoi H, Gralow J, Toi M, et al. Trastuzumab emtansine plus pertuzumab versus taxane plus trastuzumab plus pertuzumab after anthracycline for high-risk human epidermal growth factor receptor 2-positive early breast cancer: the phase III KAITLIN study. J Clin Oncol. 2022;40:438–48.
Hurvitz SA, Martin M, Symmans WF, Jung KH, Huang CS, Thompson AM, et al. Neoadjuvant trastuzumab, pertuzumab, and chemotherapy versus trastuzumab emtansine plus pertuzumab in patients with HER2-positive breast cancer (KRISTINE): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2018;19:115–26.
Hurvitz SA, Martin M, Jung KH, Huang CS, Harbeck N, Valero V, et al. Neoadjuvant trastuzumab emtansine and pertuzumab in human epidermal growth factor receptor 2-positive breast cancer: three-year outcomes from the phase III KRISTINE study. J Clin Oncol. 2019;37:2206–16.
Hunter FW, Barker HR, Lipert B, Rothé F, Gebhart G, Piccart-Gebhart MJ, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2020;122:603–12.
Saatci Ö, Borgoni S, Akbulut Ö, Durmuş S, Raza U, Eyüpoǧlu E, et al. Targeting PLK1 overcomes T-DM1 resistance via CDK1-dependent phosphorylation and inactivation of Bcl-2/xL in HER2-positive breast cancer. Oncogene. 2018;37:2251–69.
La Monica S, Cretella D, Bonelli M, Fumarola C, Cavazzoni A, Digiacomo G, et al. Trastuzumab emtansine delays and overcomes resistance to the third-generation EGFR-TKI osimertinib in NSCLC EGFR mutated cell lines. J Exp Clin Cancer Res. 2017;36.
Haikala HM, Lopez T, Köhler J, Eser PO, Xu M, Zeng Q, et al. EGFR inhibition enhances the cellular uptake and antitumor-activity of the HER3 antibody-drug conjugate HER3-DXd. Cancer Res. 2022;82:130–41.
Kayatani H, Ohashi K, Ninomiya K, Makimoto G, Nishii K, Higo H, et al. Beneficial effect of erlotinib and trastuzumab emtansine combination in lung tumors harboring EGFR mutations. Biochem Biophys Res Commun. 2020;532:341–6.
Wu SG, Shih JY. Management of acquired resistance to EGFR TKI-targeted therapy in advanced non-small cell lung cancer. Mol Cancer. 2018;17.
Johnson M, Garassino MC, Mok T, Mitsudomi T. Treatment strategies and outcomes for patients with EGFR-mutant non-small cell lung cancer resistant to EGFR tyrosine kinase inhibitors: focus on novel therapies. Lung Cancer. 2022;170:41–51.
Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014;4:816–27.
Boshuizen J, Koopman LA, Krijgsman O, Shahrabi A, Van Den Heuvel EG, Ligtenberg MA, et al. Cooperative targeting of melanoma heterogeneity with an AXL antibody-drug conjugate and BRAF/MEK inhibitors. Nat Med. 2018;24:203–12.
Roas M, Vick B, Kasper MA, Able M, Polzer H, Gerlach M, et al. Targeting FLT3 with a new-generation antibody-drug conjugate in combination with kinase inhibitors for treatment of AML. Blood. 2023;141:1023–35.
Scaltriti M, Verma C, Guzman M, Jimenez J, Parra JL, Pedersen K, et al. Lapatinib, a HER2 tyrosine kinase inhibitor, induces stabilization and accumulation of HER2 and potentiates trastuzumab-dependent cell cytotoxicity. Oncogene. 2009;28:803–14.
Collins DM, Gately K, Hughes C, Edwards C, Davies A, Madden SF, et al. Tyrosine kinase inhibitors as modulators of trastuzumab-mediated antibody-dependent cell-mediated cytotoxicity in breast cancer cell lines. Cell Immunol. 2017;319:35–42.
O’Brien NA, Huang HKT, McDermott MSJ, Madrid AM, Luo T, Ayala R, et al. Tucatinib has selective activity in HER2-positive cancers and significant combined activity with approved and novel breast cancer-targeted therapies. Mol Cancer Ther. 2022;21:751–61.
Abraham J, Montero AJ, Jankowitz RC, Salkeni MA, Beumer JH, Kiesel BF, et al. Safety and efficacy of T-DM1 plus neratinib in patients with metastatic HER2-positive breast cancer: NSABP foundation trial FB-10. J Clin Oncol. 2019;37:2601–9.
Murthy RK, Loi S, Okines A, Paplomata E, Hamilton E, Hurvitz SA, et al. Tucatinib, trastuzumab, and capecitabine for HER2-positive metastatic breast cancer. N Engl J Med. 2020;382:597–609.
Robichaux JP, Elamin YY, Vijayan RSK, Nilsson MB, Hu L, He J, et al. Pan-cancer landscape and analysis of ERBB2 mutations identifies poziotinib as a clinically active inhibitor and enhancer of T-DM1 activity. Cancer Cell. 2019;36:444-457.e7.
Kulukian A, Taylor J, Jain N, Olson D, Zaval M, Thurman R, et al. Abstract PS10–08: Tucatinib potentiates the activity of the antibody-drug conjugate T-DM1 in preclinical models of HER2-positive breast cancer. Cancer Res. 2021;81:PS10–08. https://doi.org/10.1158/1538-7445.SABCS20-PS10-08
Krop IE, Ramos J, Zhang C, Hamilton EP. HER2CLIMB-04: Phase 2 open label trial of tucatinib plus trastuzumab deruxtecan in patients with HER2+ unresectable locally advanced or metastatic breast cancer with and without brain metastases (trial in progress). 2021;39:TPS1097–TPS1097.
Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8.
Carrassa L, Damia G. DNA damage response inhibitors: Mechanisms and potential applications in cancer therapy. Cancer Treat Rev. 2017;60:139–51.
Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7:20–37.
Zhu S, Wu Y, Song B, Yi M, Yan Y, Mei Q, et al. Recent advances in targeted strategies for triple-negative breast cancer. J Hematol Oncol. 2023;16.
Yung TMC, Sato S, Satoh MS. Poly(ADP-ribosyl)ation as a DNA damage-induced post-translational modification regulating poly(ADP-ribose) polymerase-1-topoisomerase I interaction. J Biol Chem. 2004;279:39686–96.
Plummer R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin Cancer Res. 2010;16:4527–31.
Boerner JL, Nechiporchik N, Mueller KL, Polin L, Heilbrun L, Boerner SA, et al. Protein expression of DNA damage repair proteins dictates response to topoisomerase and PARP inhibitors in triple-negative breast cancer. PLoS ONE. 2015;10.
Cardillo TM, Sharkey RM, Rossi DL, Arrojo R, Mostafa AA, Goldenberg DM. Synthetic lethality exploitation by an anti-trop-2-SN-38 antibody-drug conjugate, IMMU-132, plus PARP inhibitors in BRCA1/2-wild-type triple-negative breast cancer. Clin Cancer Res. 2017;23:3405–15.
Spring LM, Clark SL, Li T, Goel S, Tayob N, Viscosi E, et al. Phase 1b clinical trial of ado-trastuzumab emtansine and ribociclib for HER2-positive metastatic breast cancer. NPJ Breast Cancer. 2021;7.
Matulonis UA, Lorusso D, Oaknin A, Pignata S, Dean A, Denys H, et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J Clin Oncol. 2023;41:2436–45.
Nicolò E, Giugliano F, Ascione L, Tarantino P, Corti C, Tolaney SM, et al. Combining antibody-drug conjugates with immunotherapy in solid tumors: current landscape and future perspectives. Cancer Treat Rev. 2022;106.
D’Amico L, Menzel U, Prummer M, Müller P, Buchi M, Kashyap A, et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J Immunother Cancer. 2019;7.
Müller P, Martin K, Theurich S, Schreiner J, Savic S, Terszowski G, et al. Microtubule-depolymerizing agents used in antibody-drug conjugates induce antitumor immunity by stimulation of dendritic cells. Cancer Immunol Res. 2014;2:741–55.
Iwata TN, Ishii C, Ishida S, Ogitani Y, Wada T, Agatsuma T. A HER2-targeting antibody-drug conjugate, trastuzumab deruxtecan (DS-8201a), enhances antitumor immunity in a mouse model. Mol Cancer Ther. 2018;17:1494–503.
Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero A, et al. Antibody-drug conjugates bearing pyrrolobenzodiazepine or tubulysin payloads are immunomodulatory and synergize with multiple immunotherapies. Cancer Res. 2017;77:2686–98.
Boshuizen J, Pencheva N, Krijgsman O, DanielaD’Empaire Altimari, Castro PG, De Bruijn B, et al. Cooperative targeting of immunotherapy-resistant melanoma and lung cancer by an AXL-targeting antibody-drug conjugate and immune checkpoint blockade. Cancer Res. 2021;81:1775–87.
Junttila TT, Li G, Parsons K, Phillips GL, Sliwkowski MX. Trastuzumab-DM1 (T-DM1) retains all the mechanisms of action of trastuzumab and efficiently inhibits growth of lapatinib insensitive breast cancer. Breast Cancer Res Treat. 2011;128:347–56.
Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114–26.
Hamilton EP, Kaklamani V, Falkson C, Vidal GA, Ward PJ, Patre M, et al. Abstract PD1–05: Atezolizumab in combination with trastuzumab emtansine or with trastuzumab and pertuzumab in patients with HER2-positive breast cancer and atezolizumab with doxorubicin and cyclophosphamide in HER2-negative breast cancer: safety and biomarker outcomes from a multi-cohort Phase Ib study. Cancer Res. 2020;80:PD1–05. https://doi.org/10.1158/1538-7445.SABCS19-PD1-05
Emens LA, Esteva FJ, Beresford M, Saura C, De Laurentiis M, Kim SB, et al. Trastuzumab emtansine plus atezolizumab versus trastuzumab emtansine plus placebo in previously treated, HER2-positive advanced breast cancer (KATE2): a phase 2, multicentre, randomised, double-blind trial. Lancet Oncol. 2020;21:1283–95.
Waks AG, Keenan TE, Li T, Tayob N, Wulf GM, Richardson ET, et al. Phase Ib study of pembrolizumab in combination with trastuzumab emtansine for metastatic HER2-positive breast cancer. J Immunother Cancer. 2022;10.
Janjigian YY, Viglianti N, Liu F, Mendoza-Naranjo A, Croydon L. A phase Ib/II, multicenter, open-label, dose-escalation, and dose-expansion study evaluating trastuzumab deruxtecan (T-DXd, DS-8201) monotherapy and combinations in patients with HER2-overexpressing gastric cancer (DESTINY-Gastric03). J Clin Oncol. 2021;39:TPS261–TPS261.
Peters S, Dafni U, Perol M, Felip E, Polito L, Pal N, et al. VP2-2021: effectiveness of PD-(L)1 inhibitors alone or in combination with platinum-doublet chemotherapy in first-line (1L) non-squamous non-small cell lung cancer (Nsq-NSCLC) with high PD-L1 expression using real-world data. Ann Oncol. 2021;32:687–8.
Powles T, Yu EY, Iyer G, Campbell MT, Loriot Y, Santis M De, et al. Phase 2 clinical study evaluating the efficacy and safety of disitamab vedotin with or without pembrolizumab in patients with HER2-expressing urothelial carcinoma (RC48G001). J Clin Oncol. 2023;41:TPS594–TPS594.
Rogerson S, Turner H, Bilkhu A, Monturus E, Knott A, Restuccia E, et al. Diversity, inclusion, and patient (pt)-centricity in the randomized, double-blind, phase III ASTEFANIA study of ado-trastuzumab emtansine (T-DM1) ± atezolizumab in pts with HER2-positive early breast cancer (EBC) with residual invasive disease after preoperative chemotherapy and anti-HER2 therapy. J Clin Oncol. 2022;40:e12504–e12504.
Loi S, Schneeweiss A, Song E, Harries M, De Laurentiis M, Li Y, et al. 329TiP KATE3: A phase III study of trastuzumab emtansine (T-DM1) in combination with atezolizumab or placebo in patients with previously treated HER2-positive and PD-L1–positive locally advanced or metastatic breast cancer. Ann Oncol. 2021;32:S509.
Schmid P, Im S-A, Armstrong A, Park YH, Chung W-P, Nowecki Z, et al. BEGONIA: Phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from arm 1, d+paclitaxel (P), and arm 6, d+trastuzumab deruxtecan (T-DXd). J Clin Oncol. 2021;39:1023–1023.
Johnson ML, Solomon BJ, Awad MM, Cho BC, Gainor JF, Goldberg SB, et al. MORPHEUS: A phase Ib/II multi-trial platform evaluating the safety and efficacy of cancer immunotherapy (CIT)-based combinations in patients (pts) with non-small cell lung cancer (NSCLC). J Clin Oncol. 2018;36:TPS9105–TPS9105.
Drakaki A, Kalebasty AR, Lee J-L, Martin-Liberal J, Kim M, Shin SJ, et al. Phase Ib/II umbrella trial to evaluate the safety and efficacy of multiple 2L cancer immunotherapy (CIT) combinations in advanced/metastatic urothelial carcinoma (mUC): MORPHEUS-mUC. J Clin Oncol. 2020;38:TPS591–TPS591.
Mittendorf E, Tolaney S, Wileyto P, DeMeo M, Rugo H, Nanda R, et al. Abstract OT-33–01: Combination ipatasertib and atezolizumab to prevent recurrence in triple negative breast cancer(TNBC): A phase II single arm trial. Cancer Res. 2021;81:OT-33–01. https://doi.org/10.1158/1538-7445.SABCS20-OT-33-01
Garrido-Castro AC, Keenan TE, Li T, Lange P, Callahan C, Guerriero J, et al. Saci-IO TNBC: Randomized phase II trial of sacituzumab govitecan (SG) +/− pembrolizumab in PD-L1—metastatic triple-negative breast cancer (mTNBC). J Clin Oncol. 2021;39:TPS1106–TPS1106.
Garrido-Castro AC, Keenan TE, Li T, Lange P, Callahan C, Guerriero J, et al. Saci-IO HR+: Randomized phase II trial of sacituzumab govitecan (SG) +/− pembrolizumab in PD-L1+ hormone receptor-positive (HR+) / HER2- metastatic breast cancer (MBC). J Clin Oncol. 2021;39:TPS1102–TPS1102.
Subudhi SK, Bendell JC, Carducci MA, Kopp LM, Scott J, Grady MM, et al. ARC-6: A phase 1b/2, open-label, randomized platform study to evaluate efficacy and safety of etrumadenant (AB928)-based treatment combinations in patients with metastatic castrate-resistant prostate cancer (mCRPC). J Clin Oncol. 2021;39:5039–5039.
Jain RK, Yang Y, Chadha J, Chatwal MS, Kish JA, Raymond S, et al. Phase I/II study of ipilimumab plus nivolumab combined with sacituzumab govitecan in patients with metastatic cisplatin-ineligible urothelial carcinoma. J Clin Oncol. 2023;41:521–521.
Garon EB, Liu S V., Owen SP, Reck M, Neal JW, Vicente D, et al. EVOKE-02: A phase 2 study of sacituzumab govitecan (SG) plus pembrolizumab (pembro) with or without platinum chemotherapy in first-line metastatic non–small cell lung cancer (NSCLC). J Clin Oncol. 2022;40:TPS9146–TPS9146.
Hoffman-Censits J, Grivas P, Powles T, Martincic D, Hawley J, Tyroller K, et al. 665 JAVELIN Bladder Medley: a phase 2 trial of avelumab in combination with other antitumor drugs as first-line maintenance therapy for advanced urothelial carcinoma. J Immunother Cancer. 2022;10:A694–A694.
Meric-Bernstam F, Janjigian Y, Lang J, Ciombor KK, Ray-Coquard I, Oza A, et al. Abstract CT058: TROPION-PanTumor03: Phase 2, multicenter study of datopotamab deruxtecan (Dato-DXd) as monotherapy and in combination with anticancer agents in patients (pts) with advanced/metastatic solid tumors. Cancer Res. 2023;83:CT058–CT058. https://doi.org/10.1158/1538-7445.AM2023-CT058
Goto Y, Su W-C, Levy BP, Rixe O, Yang T-Y, Tolcher AW, et al. TROPION-Lung02: Datopotamab deruxtecan (Dato-DXd) plus pembrolizumab (pembro) with or without platinum chemotherapy (Pt-CT) in advanced non-small cell lung cancer (aNSCLC). J Clin Oncol. 2023;41:9004–9004.
Borghaei H, Waqar SN, Bruno DS, Kitazono S, Wakuda K, Spira AI, et al. TROPION-Lung04: Phase 1b, multicenter study of datopotamab deruxtecan (Dato-DXd) in combination with immunotherapy ± carboplatin in advanced/metastatic non-small cell lung cancer (mNSCLC). J Clin Oncol. 2023;41:TPS3158–TPS3158.
Necchi A, Bedke J, Galsky MD, Shore ND, Plimack ER, Xylinas E, et al. Phase 3 KEYNOTE-905/EV-303: Perioperative pembrolizumab (pembro) or pembro + enfortumab vedotin (EV) for muscle-invasive bladder cancer (MIBC). J Clin Oncol. 2023;41:TPS585–TPS585.
Hoimes CJ, Flaig TW, Milowsky MI, Friedlander TW, Bilen MA, Gupta S, et al. Enfortumab vedotin plus pembrolizumab in previously untreated advanced urothelial cancer. J Clin Oncol. 2023;41:22–31.
Powles T, Drakaki A, Teoh JY-C, Grande E, Fontes-Sousa M, Porta C, et al. A phase 3, randomized, open-label, multicenter, global study of the efficacy and safety of durvalumab (D) + tremelimumab (T) + enfortumab vedotin (EV) or D + EV for neoadjuvant treatment in cisplatin-ineligible muscle-invasive bladder cancer (MIBC) (VOLGA). J Clin Oncol. 2022;40:TPS579–TPS579.
Hoimes CJ, Bedke J, Loriot Y, Nishiyama H, Fang X, Kataria RS, et al. KEYNOTE-B15/EV-304: Randomized phase 3 study of perioperative enfortumab vedotin plus pembrolizumab versus chemotherapy in cisplatin-eligible patients with muscle-invasive bladder cancer (MIBC). J Clin Oncol. 2021;39:TPS4587–TPS4587.
Porter R, Tayob N, Polak M, Sawyer H, Gardner J, Campos S, et al. TP015/#1566 A phase 2, two-stage, study of mirvetuximab soravtansine (IMGN853) in combination with pembrolizumab in patients with microsatellite stable (MSS) recurrent or persistent endometrial cancer (EC). Int J Gynecol Cancer. 2022;32:A229.3-A230.
Ahnert JR, Taylor MH, O’Reilly EM, Zhang J, Doebele RC, Ben Y, et al. A phase 1/2 dose-escalation and expansion study of a conditionally active anti-AXL humanized monoclonal antibody (BA3011) in patients with advanced solid tumors. J Clin Oncol. 2018;36:TPS12126–TPS12126.
Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014;5.
Hoffmann RM, Coumbe BGT, Josephs DH, Mele S, Ilieva KM, Cheung A, et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology. 2017;7.
Ogitani Y, Aida T, Hagihara K, Yamaguchi J, Ishii C, Harada N, et al. DS-8201a, A novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin Cancer Res. 2016;22:5097–108.
Uppal H, Doudement E, Mahapatra K, Darbonne WC, Bumbaca D, Shen BQ, et al. Potential mechanisms for thrombocytopenia development with trastuzumab emtansine (T-DM1). Clin Cancer Res. 2015;21:123–33.
Tai YT, Mayes PA, Acharya C, Zhong MY, Cea M, Cagnetta A, et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128–38.
Beck A, Reichert JM. Marketing approval of mogamulizumab: a triumph for glyco-engineering. MAbs. 2012;4:419–25.
Pegram MD, Hamilton EP, Tan AR, Storniolo AM, Balic K, Rosenbaum AI, et al. First-in-human, phase 1 dose-escalation study of biparatopic anti-HER2 antibody-drug conjugate MEDI4276 in patients with HER2-positive advanced breast or gastric cancer. Mol Cancer Ther. 2021;20:1442–54.
Li F, Ulrich M, Jonas M, Stone IJ, Linares G, Zhang X, et al. Tumor-associated macrophages can contribute to antitumor activity through FcγR-mediated processing of antibody-drug conjugates. Mol Cancer Ther. 2017;16:1347–54.
Garg AD, Galluzzi L, Apetoh L, Baert T, Birge RB, Bravo-San Pedro JM, et al. Molecular and translational classifications of DAMPs in immunogenic cell death. Front Immunol. 2015;6.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Kepp O, Tartour E, Vitale I, Vacchelli E, Adjemian S, Agostinis P, et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014;3.
Sukkurwala AQ, Adjemian S, Senovilla L, Michaud M, Spaggiari S, Vacchelli E, et al. Screening of novel immunogenic cell death inducers within the NCI Mechanistic Diversity Set. Oncoimmunology. 2014;3.
Rios-Doria J, Durham N, Wetzel L, Rothstein R, Chesebrough J, Holoweckyj N, et al. Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia. 2015;17:661–70.
Galluzzi L, Senovilla L, Zitvogel L, Kroemer G. The secret ally: immunostimulation by anticancer drugs. Nat Rev Drug Discov. 2012;11:215–33.
Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, et al. The BRAF and MEK inhibitors dabrafenib and trametinib: effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res. 2015;21:1639–51.
Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61.
Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: the TONIC trial. Nat Med. 2019;25:920–8.
Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;29:482–91.
Bauzon M, Drake PM, Barfield RM, Cornali BM, Rupniewski I, Rabuka D. Maytansine-bearing antibody-drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. Oncoimmunology. 2019;8.
Cao AT, Law C-L, Gardai SJ, Heiser RA. Abstract 5588: Brentuximab vedotin-driven immunogenic cell death enhances antitumor immune responses, and is potentiated by PD1 inhibition in vivo. Cancer Res. 2017;77:5588–5588. https://doi.org/10.1158/1538-7445.AM2017-5588.
Cao AT, Higgins S, Stevens N, Gardai SJ, Sussman D. Abstract 2742: additional mechanisms of action of ladiratuzumab vedotin contribute to increased immune cell activation within the tumor. Cancer Res. 2018;78:2742–2742. https://doi.org/10.1158/1538-7445.AM2018-2742.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Hargadon KM. Tumor-altered dendritic cell function: implications for anti-tumor immunity. Front Immunol. 2013;4.
Martin K, Müller P, Schreiner J, Prince SS, Lardinois D, Heinzelmann-Schwarz VA, et al. The microtubule-depolymerizing agent ansamitocin P3 programs dendritic cells toward enhanced anti-tumor immunity. Cancer Immunol Immunother. 2014;63:925–38.
Tanaka H, Matsushima H, Nishibu A, Clausen BE, Takashima A. Dual therapeutic efficacy of vinblastine as a unique chemotherapeutic agent capable of inducing dendritic cell maturation. Cancer Res. 2009;69:6987–94.
Müller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med. 2015;7.
Tanaka H, Matsushima H, Mizumoto N, Takashima A. Classification of chemotherapeutic agents based on their differential in vitro effects on dendritic cells. Cancer Res. 2009;69:6978–86.
Huang L, Wang R, Xie K, Zhang J, Tao F, Pi C, et al. A HER2 target antibody drug conjugate combined with anti-PD-(L)1 treatment eliminates hHER2+ tumors in hPD-1 transgenic mouse model and contributes immune memory formation. Breast Cancer Res Treat. 2022;191:51–61.
Xia L, Wen L, Qin Y, Dobson HE, Zhang T, Comer FI, et al. HER2-targeted antibody-drug conjugate induces host immunity against cancer stem cells. Cell Chem Biol. 2021;28:610-624.e5.
Friedlander TW, Milowsky MI, Bilen MA, Srinivas S, McKay RR, Flaig TW, et al. Study EV-103: update on durability results and long term outcome of enfortumab vedotin + pembrolizumab in first line locally advanced or metastatic urothelial carcinoma (la/mUC). 2021;39:4528–4528.
Sheng X, Zhou L, He Z, Guo H, Yan X, Li S, et al. Abstract 4581: Preliminary results of a phase Ib/II combination study of RC48-ADC, a novel humanized anti-HER2 antibody-drug conjugate (ADC) with toripalimab, a humanized IgG4 mAb against programmed death-1 (PD-1) in patients with locally advanced or metastatic urothelial carcinoma. 2022;40:4518–4518.
Calvo E, Spira A, Miguel M de, Kondo S, Gazzah A, Millward M, et al. Safety, pharmacokinetics, and efficacy of budigalimab with rovalpituzumab tesirine in patients with small cell lung cancer. Cancer Treat Res Commun. 2021;28.
Malhotra J, Nikolinakos P, Leal T, Lehman J, Morgensztern D, Patel JD, et al. A phase 1–2 study of rovalpituzumab tesirine in combination with nivolumab plus or minus ipilimumab in patients with previously treated extensive-stage SCLC. J Thorac Oncol. 2021;16:1559–69.
O’Malley DM, Oaknin A, Matulonis UA, Mantia-Smaldone G, Lim PC, Castro CM, et al. Abstract5504: Mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients (pts) with platinum-agnostic ovarian cancer: final analysis. 2021;19:8–10.
Lorusso D, Vergote I, O’Cearbhaill RE, Westermann AM, Banerjee SN, Nieuwenhuysen E Van, et al. Abstract 5507: Tisotumab vedotin (TV) + pembrolizumab (pembro) in first-line (1L) recurrent or metastatic cervical cancer (r/mCC): Interim results of ENGOT Cx8/GOG 3024/innovaTV 205. 2022;40:5507–5507.
Schmid P, Nunes AT, Dry H, Dougherty R, Karwe V, Scheuring UJ. BEGONIA: Phase 1b/2, open-label, platform study of the safety and efficacy of durvalumab (D) ± paclitaxel (P) with novel oncology therapies for first-line metastatic triple-negative breast cancer (mTNBC): Addition of arm 7, D + datopotamab deruxtecan (Dato-DXd; DS-1062). 2021;39:TPS1105–TPS1105.
Boni V, Alemany C, Meisel JL, Sinha R, Sterrenberg D, Tkaczuk KHR, et al. SGNLVA-002: Single arm, open label, phase Ib/II study of ladiratuzumab vedotin (LV) in combination with pembrolizumab for first-line treatment of patients with unresectable locally-advanced or metastatic triple-negative breast cancer. Annals of Oncology. 2019;30:iii63.
Cheson BD, Bartlett NL, LaPlant B, Lee HJ, Advani RJ, Christian B, et al. Brentuximab vedotin plus nivolumab as first-line therapy in older or chemotherapy-ineligible patients with Hodgkin lymphoma (ACCRU): a multicentre, single-arm, phase 2 trial. Lancet Haematol. 2020;7:e808–15.
Schmid P, Jung KH, Wysocki PJ, Jassem J, Ma CX, Fernandes R, et al. 166MO Datopotamab deruxtecan (Dato-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic triple-negative breast cancer (a/mTNBC): Initial results from BEGONIA, a phase Ib/II study. Ann Oncol. 2022;33:S199.
Fuentes-Antrás J, Genta S, Vijenthira A, Siu LL. Antibody-drug conjugates: in search of partners of choice. Trends Cancer. 2023;9:339–54.
Vergote IB, Monk BJ, O’Cearbhaill RE, Westermann AM, Banerjee S, Collins DC, et al. 723MO Tisotumab vedotin (TV) + carboplatin (Carbo) in first-line (1L) or + pembrolizumab (Pembro) in previously treated (2L/3L) recurrent or metastatic cervical cancer (r/mCC): Interim results of ENGOT-Cx8/GOG-3024/innovaTV 205 study. Ann Oncol. 2021;32:S726–7.
Zinzani PL, Santoro A, Gritti G, Brice P, Barr PM, Kuruvilla J, et al. Nivolumab combined with brentuximab vedotin for relapsed/refractory primary mediastinal large B-cell lymphoma: efficacy and safety from the phase II checkmate 436 study. J Clin Oncol. 2019;37:3081–9.
Advani RH, Moskowitz AJ, Bartlett NL, Vose JM, Ramchandren R, Feldman TA, et al. Brentuximab vedotin in combination with nivolumab in relapsed or refractory Hodgkin lymphoma: 3-year study results. Blood. 2021;138:427–38.
Rosenberg JE, Milowsky M, Ramamurthy C, Mar N, McKay RR, Friedlander T, et al. LBA73 Study EV-103 Cohort K: Antitumor activity of enfortumab vedotin (EV) monotherapy or in combination with pembrolizumab (P) in previously untreated cisplatin-ineligible patients (pts) with locally advanced or metastatic urothelial cancer (la/mUC). Ann Oncol. 2022;33:S1441.
Schmid P, Im S-A, Armstrong A, Park YH, Chung W-P, Nowecki Z, et al. BEGONIA: Phase 1b/2 study of durvalumab (D) combinations in locally advanced/metastatic triple-negative breast cancer (TNBC)—Initial results from arm 1, d+paclitaxel (P), and arm 6, d+trastuzumab deruxtecan (T-DXd). 2021;39:1023–1023.
Hamilton E, Shapiro CL, Petrylak D, Boni V, Martin M, Conte G Del, et al. Abstract PD3–07: Trastuzumab deruxtecan (T-DXd; DS-8201) with nivolumab in patients with HER2-expressing, advanced breast cancer: A 2-part, phase 1b, multicenter, open-label study. Cancer Res. 2021;81:PD3–07. https://doi.org/10.1158/1538-7445.SABCS20-PD3-07
Grivas P, Pouessel D, Park CH, Barthélémy P, Bupathi M, Petrylak DP, et al. TROPHY-U-01 Cohort 3: Sacituzumab govitecan (SG) in combination with pembrolizumab (Pembro) in patients (pts) with metastatic urothelial cancer (mUC) who progressed after platinum (PLT)-based regimens. 2022;40:434–434.
Schmid P, Wysocki P, Park YH, Jassem J, Jung KH, Lord S, et al. Abstract PD11–08: PD11–08 Trastuzumab deruxtecan (T-DXd) + durvalumab (D) as first-line (1L) treatment for unresectable locally advanced/metastatic hormone receptor-negative (HR−), HER2-low breast cancer: updated results from BEGONIA, a phase 1b/2 study. Cancer Res. 2023;83:PD11–08. https://doi.org/10.1158/1538-7445.SABCS22-PD11-08
Acknowledgements
Not applicable.
Funding
National Natural Science Foundation of China (NSFC; 82172598, 82074245, 82373782, 82303963). The National Key R&D Program of China (2021YFA0910100). The Natural Science Foundation of Zhejiang Province, China (LZ22H310001, LQ21H160005). The 551 Health Talent Training Project of the Health Commission of Zhejiang Province. The Agricultural and Social Development Research Project of Hangzhou Municipal Science and Technology Bureau (2022ZDSJ0474). The Zhejiang Leading Innovation and Entrepreneurship Team (2022R01006). The Key Laboratory of Prevention, Diagnosis and Therapy of Upper Gastrointestinal Cancer of Zhejiang Province (2022E10021). Medical Science and Technology Project of Zhejiang Province (WKJ-ZJ-2104). Zhejiang Provincial Research Center for Upper Gastrointestinal Track Center (JBZX-202006). Program of Zhejiang Provincial TCM Sci-Tech Plan (GZY-ZJ-KJ-230003).
Author information
Authors and Affiliations
Contributions
P.G., X.C. and J.Y. conceived and designed the review. T.Y. and J.Z. performed analyses of ADC combined with endocrine therapy and chemotherapy. L.S., Z.Z., L.W. and X.T. performed analyses of ADC combined with targeted therapy. J.C., C.H, J.X. and L.M. performed analyses of ADC combined with immunotherapy. T.S. and J.F. interpreted and discussed the results. Q.W. and P.L. wrote the manuscript. All authors approved this manuscript for publication.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
QW, TY, ZWZ, XDC and PG are co-inventors of a patent application based on this study. The other authors have declared no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wei, Q., Li, P., Yang, T. et al. The promise and challenges of combination therapies with antibody-drug conjugates in solid tumors. J Hematol Oncol 17, 1 (2024). https://doi.org/10.1186/s13045-023-01509-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01509-2