
Abstract
Natural killer (NK) cells, a unique component of the innate immune system, are inherent killers of stressed and transformed cells. Based on their potent capacity to kill cancer cells and good tolerance of healthy cells, NK cells have been successfully employed in adoptive cell therapy to treat cancer patients. In recent years, the clinical success of chimeric antigen receptor (CAR)-T cells has proven the vast potential of gene-manipulated immune cells as the main force to fight cancer. Following the lessons learned from mature gene-transfer technologies and advanced strategies in CAR-T therapy, NK cells have been rapidly explored as a promising candidate for CAR-based therapy. An exponentially growing number of studies have employed multiple sources of CAR-NK cells to target a wide range of cancer-related antigens, showing remarkable outcomes and encouraging safety profiles. Clinical trials of CAR-NK cells have also shown their impressive therapeutic efficacy in the treatment of hematological tumors, but CAR-NK cell therapy for solid tumors is still in the initial stages. In this review, we present the favorable profile of NK cells as a potential platform for CAR-based engineering and then summarize the outcomes and strategies of CAR-NK therapies in up-to-date preclinical and clinical investigations. Finally, we evaluate the challenges remaining in CAR-NK therapy and describe existing strategies that can assist us in devising future prospective solutions.
Similar content being viewed by others

Introduction
Immune cells serve as the pillar of strength in antitumor and antiviral processes [1]. Through their rapid recognition and lysing of nascent transformed cells, immune cells can prevent tumorigenesis in the initial stage [2]. However, once malignant cells proliferate and metastasize uncontrollably, they change and depress the immunological responses mediated by host immune cells [3]. By infusing functionally active effector cells into immunocompromised patients, a process known as adoptive cell therapy (ACT), we can reconstruct host immunity and provide a promising strategy for disease treatment [4, 5]. Adoptive transfer of autologous immune cells that have been activated and amplified ex vivo has shown encouraging efficacy in patients with certain hematological cancers. However, the therapeutic efficiency in other tumors is far from satisfactory [6, 7]. With the advancement of gene engineering technology, cytotoxic T cells have been equipped with CARs, which endow T cells with superior and more precise killing capacity. In recent years, CAR-T cells have achieved numerous breakthroughs in cancer treatment, especially in hematologic malignancy treatment [8,9,10,11,12,13,14]. A multitude of CAR-T investigations regarding cancer treatment have progressed into the clinical trial stage, with a high rate of complete remission (CR) being exhibited, and some CAR-T cells have even developed into commercial products [15]. To date, six CAR-T products for treating hematological tumors have been approved by the US Food and Drug Administration (FDA), including Kymriah (Novartis), Yescarta (Gilead), Tecartus (Gilead), Breyanzi (Bristol Myers Squibb), Abecma (Bristol Myers Squibb and Bluebird Bio), and Carvykti (Legend and Janssen). CD19 (four products) and B-cell maturation antigen (BCMA) (two products) are the two primary antigens targeted by CAR-T cells to treat relapsed/refractory (R/R) B-cell-derived leukemia, lymphoma, and multiple myeloma [9, 13, 16,17,18,19,20,21]. Despite these promising outcomes of CAR-T cells in the treatment of hematological tumors, their limited efficacy in the treatment of solid tumors necessitates the exploration of novel strategies to help CAR-T cells break the barriers in solid neoplasm. CAR-T immunotherapy requires apheresis and time-consuming expansion of autologous immune cells from patients. For some patients with aggressively progressing cancer, costly and complicated procedures may result in delayed therapy. In addition, heavily pretreated cancer patients are unable to provide sufficient normal T cells, creating an additional barrier to CAR-T-cell development. Therefore, a surge of interest has recently focused on seeking other candidate immune cells to be engineered with CARs [22].
NK cells, a subset of innate lymphoid cells (ILCs) with diversified killing mechanisms, have recently become a focal point in the application of immunotherapy. The function of NK cells is regulated by a sophisticated array of activating and inhibitory receptors that can distinguish between healthy cells and transformed cells. The integrated signals from the engagement of these receptors and ligands can determine whether NK cells initiate killing activities against aberrant cells or maintain their tolerance of healthy cells [23, 24]. In contrast to T cells, NK cells recognize cancer cells in a human leukocyte antigen (HLA)-unrestricted manner, resulting in the lowest possibility of graft versus host disease (GVHD) development [25]. Furthermore, NK cells rarely induce severe toxicities such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) in vivo [26]. Owing to these favorable attributes, NK cells engineered with CARs can overcome many hurdles that prevent CAR-T therapy from further application. The successful adoptive transfer of allogeneic NK cells into patients further identifies NK cells as a promising platform for CAR engineering and as “off-the-shelf” products for wide application [22]. The strategies of CAR design and transduction used in CAR-T therapy are applicable in NK engineering with encouraging outcomes. To date, CAR-NK cells have shown impressive efficacy in the treatment of hematological tumors and have been widely studied in the treatment of solid tumors, with numerous breakthroughs, such as in the treatment of glioblastoma, breast cancer, and ovarian cancer [27,28,29]. In this review, we present the favorable profile of NK cells as a potential platform for CAR-based engineering and then summarize the outcomes and strategies of CAR-NK cell therapy in up-to-date preclinical and clinical investigations. Finally, we evaluate the challenges remaining in CAR-NK cell therapy and describe existing strategies that can assist us in devising future prospective solutions.
An overview of NK cell biological properties
Development and classification
NK cells are a subgroup of innate lymphoid cells (ILCs) and are identified as the first line of defense against virally infected and/or transformed cells [30]. Derived from CD34+ hematopoietic progenitor cells in bone marrow, NK cells develop in a continuous process in bone marrow as well as in some secondary lymphoid organs (SLOs), such as the spleen, tonsils, thymus, and liver [31, 32]. However, it is unclear whether NK cells differentiate in a linear or nonlinear manner [33]. The developmental stages of NK cells differ significantly among different anatomical locations. Immature NK cells are predominantly distributed in lymph nodes and intestines and have tissue-adaptation signatures, whereas terminally differentiated NK cells mainly populate the blood, bone marrow, spleen, and lungs and have improved effector function [34]. According to the expression levels of CD56 and CD16, NK cells are divided into two major subgroups: CD56brightCD16− and CD56dimCD16+ NK cells [35]. CD56bright NK cells are immature populations and are mainly distributed in SLOs. They were previously thought to be involved in immunomodulation, but recently, they have been identified with robust cytokine-releasing potential after priming with proinflammatory cytokines such as interleukin-15 (IL-15). CD56bright NK cells are more similar to helper cells, secreting abundant cytokines such as interferon-γ (IFN-γ), tumor necrosis factor-β (TNF-β) and granulocyte–macrophage colony-stimulating factor (GM-CSF) [36, 37]. CD56dim NK cells represent the final stage of NK cell maturation and constitute approximately 90% of circulating NK cells. The increased expression of CD16a (FcγRIIIa) and cytotoxic molecules in CD56dim NK cells allows them to mediate serial killing activities toward malignant cells, for example, via antibody-dependent cellular cytotoxicity (ADCC) and death receptor-mediated apoptosis [38, 39]. Recently, high-resolution sequencing technologies further revealed increased heterogeneity of NK cells in different organs, indicating that more NK subpopulations can be further defined beyond the simple delineation of CD56brightCD16− and CD56dimCD16+ NK cells [34, 40, 41].
Activation and cytotoxicity
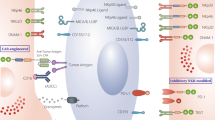
NK cells are critical for immune surveillance and antitumor responses in vivo. These biological functions are regulated by integrated signals from the stochastically expressed activating and inhibitory receptors on NK cells [42]. The activation and inhibition mechanisms of NK cells are depicted in Fig. 1. Ubiquitously expressed major histocompatibility complex class I (MHC-I) molecules (also known as HLA class I) on healthy cells can bind the inhibitory killer cell immunoglobulin-like receptors (KIRs) or NKG2A of NK cells, which can deliver predominant inhibitory signaling to maintain NK “self-tolerance” [24, 43]. However, tumor cells often downregulate their MHC-I molecule expression to evade the attack of CD8+ cytotoxic T cells, as the target recognition of CD8+ T cells relies on antigen presentation by MHC-I [44, 45]. Thus, the signaling balance of NK cells is broken, and they are inclined toward an activation state (also known as the “missing-self mechanism”). Other transformed or stressed cells expressing excessive activating ligands, such as NKG2D ligands, can directly stimulate NK cell activation through receptor‒ligand engagements called immune synapses [46]. The formulation of immune synapses can initiate firm adhesion and enable the focused delivery of lytic granules such as perforin and granzymes onto the target cells, inducing the apoptosis of target cells [45, 47]. Significantly, a single degranulation can be sufficient to lyse a target cell [48]. Following early lytic granule-mediated killing activities, delayed cell apoptosis responses can be initiated by target cells engaging with death ligands expressed on NK cells such as Fas ligand (Fasl) and TNF-related apoptosis-inducing ligand (TRAIL), conferring the serial killing ability of NK cells [49]. Additionally, CD16 is a potent activating receptor that allows NK cells to engage with antibody-opsonized target cells through ADCC. This crosslinking interaction can subsequently induce NK cells to release the cytotoxic substances mentioned above [50]. In addition to triggering their powerful killing ability, NK cells can secrete an array of cytokines and chemokines to stimulate broader cellular immune responses. For example, the IFN-γ and TNF released by activated NK cells can synergistically mediate the death of target cells [51]. IFN-γ can not only directly activate macrophages but also indirectly promote CD8+ T-cell-mediated immune responses by elevating MHC-II molecule expression on antigen-presenting cells [52]. The NK cell-dendritic cell axis also plays a critical role in tumor immunity. CCL5, XCL1, XCL2, and FLT3L secreted by NK cells are the major chemokines that recruit conventional type 1 dendritic cells (cDC1s). cDC1s can present tumor-associated antigens (TAAs) from apoptotic tumor cells to CD4+ and CD8+ T cells, thus inducing potent T-cell-mediated immune responses [52,53,54].

The mechanism of NK activation and self-tolerance. A In healthy conditions, self-HLA class I molecules of healthy cells bind the inhibitory receptors of NK cells such as KIRs and NKG2A/CD94. Dominant inhibitory signaling suppressed the cytolytic ability of NK cells to make autologous healthy cells “licensed”. B The majority of tumor cells downregulate or lost their MHC-I molecule expression to escape from the immune cells attacking. This results in decreasing tumor ligands combining with inhibitory receptors of NK cells, thus NK cells are activated to secret perforin and granzyme to lyse tumor cells. C Overexpressed activating ligands on stressed cells engage with NK cell receptors, leading to superior activating signaling surpassing inhibitory signaling. As a result, NK cells transform into activation state and initiate cell lysing. D Antibody-dependent cell-mediated cytotoxicity, ADCC. The tumor-specific Fc fragment binds CD16 (FcγRIII) of NK cells, resulting in ADCC development. In addition to ADCC, other killing mechanisms of NK cells include death-receptor-mediated and perforin/granzyme-mediated killing activities
The strength of NK cells as immunotherapy candidates
Accessibility to abundant cell sources
NK cells can be obtained from autologous and allogenic sources. Initially, autologous NK cells were the major alternative in adoptive cellular therapy owing to their safety [55, 56]. The evidence suggests that autologous NK cells are not sufficient to exert robust antitumor responses, in part due to the NK inhibitory effects mediated by self MHC-I molecules and functional impairments caused by prior heavy treatment [57, 58]. These findings encourage transitioning the focus on autologous NK cells to allogenic NK cell sources, the use of which can avoid cumbersome collection processes and satisfy clinical doses [22, 59]. These NK cell sources include peripheral blood (PB), umbilical cord blood (UCB), NK cell lines, and stem cell-derived NK cells [42]. Each source of NK cells has its own set of strengths and limitations, as summarized in Fig. 2.

The research progress, advantages, and limitations of various NK cell sources. NK cells can be obtained from 5 different sources: PB, UCB, iPSC, hESC, and NK cell lines. Most cell sources have remarkable tumor-eliminating ability and provide clinically meaningful benefit, having transitioned into in-human studies of different stages. Each source of NK cells has its own set of strengths and limitations
PB-derived NK cells, obtained through donor lymphocyte apheresis, represent a conventional option in CAR-NK investigations of cancer treatment. PB-NK cells are mainly a mature population characterized by CD56dimCD16bright cells, without obvious individual variations [60]. Additionally, PB-NK cells show relatively abundant expression of activating receptors such as NKG2D, NKp44, and NKp46, which significantly foster NK cell destruction potential against malignant cells [29]. However, the low proportion of NK cells in PB (approximately 10–15%) largely hinders the cell collection and ex vivo expansion process [34, 61]. NK cells isolated from PB are in various maturation stages and thus are characterized by heterogeneous receptor expression profiles, from maturing to fully mature phenotype variation [62, 63]. Thus, the standardization and stability of cell products are hard to guarantee.
UCB-NK cells are also a valuable and well-studied source, constituting up to 30% of UCB lymphocytes [64]. UCB-NK cells are easy to collect and can be frozen in a cell bank; thus, the incumbrances associated with the apheresis of healthy donors and time-consuming amplification can be avoided [65]. There are fewer contaminating T cells among UCB-NK cells. Furthermore, cell sorting techniques such as immunomagnetic cell separation can assist in attaining high-purity NK cells, minimizing the risk of GVHD as much as possible [66,67,68]. Additionally, UCB cells offer abundant cell sources, where hematopoietic stem cells and progenitor cells can be acquired and then differentiate into therapeutic NK cells with favorable phenotypes [69, 70]. However, compared to PB-derived NK cells, UCB-NK cells possess relatively weak cytotoxic abilities against malignant cells owing to their natural immature phenotype, represented by the CD56−CD16+ population [71, 72]. In addition, UCB-NK cells express a lower level of activating receptors and adhesion molecules (such as CD16, CD2, and CD11a) [73] and a high level of the inhibitory receptor NKG2A [74]. This feature calls for sufficient ex vivo stimulation to promote a more mature state of UCB-NK cells, thus augmenting their cytotoxicity and persistence [75].
In view of the delay in collection and complex expansion of primary cells, focus has increasingly transitioned to immortalized NK cell lines such as NK-92 [76, 77], NK-92MI [78, 79], KHYG-1 [80], and YTS [81]. NK-92 has been the most extensively studied cell line in NK-based clinical trials [82]. As they mostly lack KIR expression, NK-92 cells are more sensitive and robust in their response to tumor cells [83]. NK-92 cells are homogenous and easy to mount in desirable quantities. In addition, they can be easily genetically engineered under good manufacturing practice (GMP)-compliant methodologies, representing the industry-transformation potential of NK-92 cells. However, NK-92 cells are aneuploid and of malignant origin, thus requiring irradiation before cell infusion. Irradiation can limit the persistence of NK-92 cells and negatively impact their durable therapeutic efficacy [84]. NK-92 cells are naturally deprived of CD16, indicating a deficiency in the ADCC response [85]. Recently, high-affinity CD16 variant molecules were successfully engineered on NK-92 cells to perform a more comprehensive and robust effector function [86].
Recently, there has been increased interest in stem cells such as human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) as sources of NK cells, which hold great potential to be standardized “off-the-shelf” therapies. Through multiple rounds of stromal cell coculturing or cytokine cocktail stimulation, iPSCs or hESCs can gradually develop into mature and homogeneous CD45+CD56+ NK cells [87,88,89]. Phenotype analysis has demonstrated that hESC-derived NK cells can differentiate toward nearly mature phenotypes akin to PB-NK cells [89, 90]. A study showed that hESC-NK cells have more cytolytic effects on tumor cells than UCB-NK cells [91]. iPSC-derived NK cells are weak in ADCC due to a low level of CD16 expression, although they are amenable to genetic manipulation with a high-affinity CD16 molecule to restore their ADCC mechanism [92]. Relying on their excellent properties, iPSC-derived NK cells have been investigated in a large number of preclinical and clinical studies, either as a single therapy or in combination with agents, which remedies the limitations caused by their defective phenotype and killing ability [92,93,94,95]. The unlimited reproductivity and plasticity of iPSC-NK cells offer exceptional advantages for them to be potent standardized products in a broad range of applications [96, 97]. Fate therapeutics have led the way to engineer multifunctional iPSC-NK cells, with five products being evaluated in clinical settings to treat hematological and solid malignancies [68].
Robust antitumor responses and preferable safety profile
NK cells have great potential to be broadly applied in cancer treatment. Hematological tumor cells are more accessible to NK cells and are sensitive to their responses. In the context of solid tumor cells, circulating NK cells must extravasate from the blood and traverse the tumor stroma to reach tumor beds, guided by the chemokines secreted by NK cells and other immune cells [98]. Upon entry into the tumor sites, the integrated activation signaling from NK-tumor interactions induces a series of killing activities, from cytolytic granule release and death receptor‒ligand interactions to ADCC [99]. During these processes, continuous secretion of cytokines such as IFN-γ, TNF, GM-CSF, M-CSF, IL-5, and IL-10 assists in tumor elimination by recruiting and regulating the antitumor responses of other immune cells [100]. In addition, NK cells have been identified with memory-like functions in multiple studies, which is not an attribute of innate immune cells [101, 102]. Memory-like NK cells can initiate a more rapid and robust response characterized by enhanced IFN-γ secretion. A generation scheme of memory-like NK cells that are preactivated by IL-12, IL-15, and IL-18 has been widely adopted. This special NK population has been utilized to augment and consolidate hematopoietic cell transplantation (HCT) in clinical settings, achieving promising outcomes [103,104,105,106]. A commercial memory-like NK product (WU-NK-101) developed by Wugen has also been evaluated in clinical trials (NCT05470140), showing that innovation in NK-based therapy is ongoing [107].
In addition to multiple powerful killing activities, the excellent safety performance of NK cells is another major asset making them potential immunotherapy candidates. NK cells can recognize pathologic cells in a non-HLA-restricted modality without the risk of GVHD development [108, 109] and can spare healthy cells from attacks through a “missing-self” mechanism mediated by the predominant signaling of iKIRs and NKG2A, as described above [110, 111]. The proinflammatory cytokines IL-1, IL-6, and TNF-, which are associated with cytokine storm and neurotoxicity, are also secreted at low levels by NK cells [29, 112]. A clinical study reported that ex vivo activated NK cells lasted 7 to 22 days upon infusion into patients. The short persistence of NK cells may raise doubts about durable therapeutic efficacy, but some researchers consider it an indication of controllable therapy, and the treatment efficacy may be addressed by multiple infusion doses [102]. These safety attributes of NK cells open the way for their broad application in allogeneic settings, showing the potential of these cells as off-the-shelf cellular therapy products.
Chimeric antigen receptor (CAR) design for NK cells
CAR was first introduced in T cells to endow them with target-specific recognition ability and potent killing responses [113]. CAR is a synthetic protein with three major parts: the extracellular domain, transmembrane region, and intracellular domain. The extracellular domain is composed of an antibody-derived single chain variable fragment (scFv) for antigen recognition [114, 115]. Recently, a single variable domain on a heavy chain (VHH) characterized by small size and high affinity has also been utilized for this aim [116, 117]. The hinge region connects scFv or VHH with the transmembrane region, which docks the ectodomain region of the CAR molecule to the cell membrane [118]. Intracellular signaling domains are derived from the signal transduction domains of TCRs or other activating receptors and are responsible for stimulating downstream pathways and activating CAR-carrying effector cells upon the recognition of target antigens [119, 120]. According to the number and components of the intracellular portion, CARs are traditionally classified into three generations [121] (as depicted in Fig. 3). First-generation CAR possesses only one signaling domain (usually CD3ζ) that is considered insufficient to induce a potent killing response in the absence of costimulatory domains [122, 123]. Thus, second-generation and third-generation CARs are respectively engineered with one or two costimulatory domains, such as CD28, CD137 (4-1BB), or CD134 (OX40), fused to CD3ζ, contributing to enhanced activation of effector cells [124]. However, there is no definite conclusion indicating that third-generation CAR outperforms the-second-generation CARs. Defects in CAR-effector cells, such as weak persistence and potential toxicity, promoted the development of novel-generation CARs (also known as fourth-generation CAR). Leveraging the advances in synthetic biology, innovative modules have been exploited to program CAR-engineered systems with self-supporting and safety modulation. Cytokine genes have been incorporated into CAR cassettes to support the activation and persistence of CAR-effector cells, either in autocrine or membrane-bound forms [125,126,127]. The inducible caspase 9 (iCasp9) suicide gene system serves as a “safety switch” that can induce the apoptosis of effector cells after the addition of small molecule drugs. iCasp9-incorporating CAR has been demonstrated efficient in controlling the toxicity of effector cells under unfavorable circumstances [26, 128] (as depicted in Fig. 3). Based on the lessons learned from current preclinical and clinical investigations, more sophisticated strategies have been developed to overcome the obstacles that hinder the efficacy of CAR-based therapy. For example, antigen escape is a major barrier to CAR-therapy and correlates with a poor prognosis [129]. A bispecific CAR that contains either two separate CARs targeting different antigens or a single CAR with two target-recognition domains can be a feasible modality to enhance the stringency of tumor recognition and prevent tumor evasion [130]. In addition, trogocytosis is an active process characterized by the transfer of surface molecules from target cells to effector cells. Trogocytosis can lead to the fratricide and dysfunction of CAR-T cells, with the potential for antigen-low tumor relapse [131]. Therefore, an inhibitory CAR (iCAR) directed to NK-cell-specific inhibitory receptors was introduced in NK cells to initiate a “don’t kill me” signal. The cooperation of tumor-targeting activating CAR (aCAR) and NK self-recognizing iCAR has demonstrated effective prevention of trogocytosis-mediated NK fratricide and enhanced CAR-NK cell activity [132].

The evolution of CAR design and emerging strategies on CAR-NK structure. The main distinction of the three generation CARs lies in the number and composition of intracellular domains. In addition to the T-cell based signaling domains, NK-specific receptor (such as DAP10, DAP12, 2B4) has introduced into NK cells to explore CAR-NK therapy. The novel generation CAR strategies utilize the fundamental principles of CAR signaling and innovative approaches to enhance cytotoxicity persistence, trafficking, and safety performance of CAR-NK cells, endowing them multifunctional attributes
Recently, more insights have been gained into NK cells as an alternative for CAR-targeted immunotherapy [133]. As some activating signaling moieties are shared between T cells and NK cells, such as CD3ζ, CD28, and 4-1BB, CARs conventionally designed for T cells are theoretically applicable for NK cells and have been proven effective [26, 134]. Following these encouraging outcomes, the substitution of intracellular elements of CAR with NK-biology-pertinent signaling domains has increasingly garnered interest. Some studies have adopted NK-associated DNAX-activation protein 10 (DAP10) or DAP12 as a signaling domain in place of CD3ζ [135,136,137,138]. In a comparative analysis, DAP12-based CAR resulted in an in vitro cytotoxicity of PB-derived NK cells that was superior to that of CD3ζ-based CAR [139]. Li et al. [96] assessed the functionality of nine CAR constructs (one T-like CAR and eight NK-like CARs) based on the NK-92 cell line and iPSC-derived NK cells. Several cytotoxicity assessments revealed that using the CAR containing the NKG2D transmembrane domain along with the NK-specific 2B4 costimulatory domain can endow the NK cells with the most potent killing ability and activation degree. Overall, these findings indicate that the antitumor capabilities can be further augmented based on the optimization of the CAR-NK design.
The application of CAR-NK therapy in cancer treatment
CAR-NK therapy for hematological tumors
The paradigm-shifting success of CAR-T therapy has provided valuable guidance for CAR-NK therapy. Initial investigations of CAR-NK cells primarily focused on the treatment of a variety of hematological cancers, especially B-cell derived malignancies (Table 1), which is identical to the preliminary stage of CAR-T cells [140]. CAR-NK cell therapy research continues to experience tremendous growth, with many strategies advancing from preclinical studies into the clinical stage. The clinical trials of CAR-NK therapy are summarized in Table 3.
B-cell malignancies
CD19, ubiquitously expressed in the B lymphocyte lineage but with predominant expression on malignant B cells, is the most prevalent target exploited in CAR-NK based cellular therapy. NK-92 and primary NK cells have been engineered with a variety of anti-CD19 CARs, showing enhanced targeted killing ability toward an array of B-cell leukemia, B-cell lymphoma cells, and autologous leukemia blasts from patients [169,170,171,172]. According to these studies, the different intracellular domains of CAR can confer NK cells with varying degrees of cytotoxicity. First, the first generation anti-CD19 CAR incorporating CD3ζ endows NK cells with cytotoxic ability that is superior to that of DAP10-incorporating CAR counterparts [171, 172]. Second, the addition of one costimulatory molecule, such as 4-1BB or CD28, to the CAR can further augment the anti-leukemia response of primary NK cells [171]. However, it was found in NK-92 cells that CD19.4-1BB.CD3ζ CAR-NK-92 cells are less effective in tumor killing than NK-92 cells carrying CD19.CD3ζ CAR or CD19.CD28.CD3ζ [169]. Third, anti-CD19 CARs integrated with 2B4, an NK-associated activating receptor, can markedly overcome the tumor resistance of NK cells, showing robust killing activities toward autologous leukemia cells [170]. Identical outcomes were confirmed by anti-123.2B4ζ CAR-NK cells targeting AML cells and anti-CD5.2B4ζ CAR-NK cells targeting malignant T cells, and both CAR-NK cells showed more potent cytotoxicity toward target cells than their respective counterparts incorporating the 4-1BBζ component [155, 173]. In addition, CD19 CAR-NK-92 cells are sufficient to induce potent anti-lymphoma activity toward anti-CD20 antibody-resistant BNHL cells, offering a potential treatment alternative for some drug-resistant diseases [174]. Some clinical studies have demonstrated that the expansion and persistence of adoptive effector cells are correlated with clinical response [175, 176]. Researchers have pursued methods to increase CAR-NK cell longevity for better antitumor performance. Transgenic expression of secretory interleukin (sIL)-15, a critical cytokine supporting NK cell activation and expansion, on CAR-NK cells has been tested in the treatment of hematological tumors [26, 128, 143]. For example, the poor persistence of anti-CD19 CB-CAR-NK has been addressed by transducing the autocrine IL-15 gene into CAR, showing striking efficacy in eliminating patient-derived leukemia cells. To enhance the safety performance of autocrine IL-15 CAR-NK cells, an inducible caspase-9-based suicide gene (iC9) was also introduced to the CAR design, allowing pharmacological-mediated death of iC9/CAR.19/IL15 NK cells in case of adverse responses [128]. CD20 and FLT3 are also common targets in the immunotherapy of B-cell derived malignancies [144, 148, 149]. By virtue of the inherent ADCC ability mediated by FcγRIII of NK cells, Boissel et al. compared in vitro killing performance of NK cells mediated by two modes: a CD20-redirected CAR and anti-CD20 monoclonal antibodies (mAbs). The cytolytic response of CD20-CAR-NK cells significantly outperformed that of ADCC in the presence of a panel of NK-resistant CLL cells. CD20-CAR-NK cells significantly suppressed tumor growth and prolonged the survival of CLL-bearing mice [144].
Based on the inspiring outcomes from a large number of CD19 CAR-NK preclinical studies, CD19 is the most favorable target in clinical investigation for the treatment of B-cell derived lymphoma and leukemia. The aforementioned iC9/CAR.19/IL15 CB-NK cells were administered to the patients in incremental single doses. At a median follow-up of 13.8 months, the objective response rate was 73% [8, 11], and 7 out of the patients obtained a CR. The CAR-NK infusion did not induce any symptoms of adverse events. In addition, iC9/CAR.19/IL15 CB-NK cells were found to persist in patients for as long as 1 year by quantitative polymerase chain reaction, indicating their durable efficacy and potential [26]. As CB-NK cells and PB-NK cells are heterogeneous cells from which standardized products are difficult to generate, homogenous iPSC-derived CAR-NK cells are extensively utilized in clinical settings to treat a wide range of B-cell hematological cancers. Fate Therapeutics and Allife Medical Science and Technology are the two main forces producing CAR-NK products centered around iPSC-NK cells. FT596, a multiplexed iPSC-CAR-NK cell product from Fate, is engineered with a CD19-targeting CAR, a high-affinity CD16, and IL-15/IL-15Rα. FT596 was evaluated in a phase I clinical trial for patients with relapsed/refractory (R/R) B-NHL and CLL [177]. The disclosed data exhibited equivalent outcomes of FT596 with CD19 CAR-T cells in patients with CD19-positive B-cell malignancies, achieving a 71% ORR (10/14) and 50% CRR. In combination with rituximab (anti-CD20), FT596 had a stronger ability to kill CD19- and/or CD20-positive hematological tumor cells (NCT04245722). No evidence of CRS, ICANS, or GVHD was observed in these patients, showing the potential of iPSC-CAR-NK cells for widespread clinical use [178]. Soon, Fate Therapeutics will submit an Investigational New Drug (IND) application concerning the next-generation CD19-targeting iPSC-CAR-NK cell product with five innovative synthetic controls (FT522) in mid-2023 to further upgrade the strategies implemented on iPSC-NK cells.
T-cell malignancies
The joint antigens between normal T cells and malignant T cells can induce the dysfunction and fratricide of post-infusion CAR-T cells [179]. NK cells, lacking multiple classic T-cell associated markers, can serve as an ideal alternative to treat aggressive T-lymphoid cancers [180]. CAR-NK cells have shown effectiveness in targeting several T-cell TAAs, including CD3, CD4, CD5 and CD7, among which CD5 is the investigational epicenter of targets for T-cell malignancy treatment. A third-generation CD5 CAR incorporating CD28 and 4-1BB costimulatory molecules was engineered in NK-92 cells and achieved potent anti-T-ALL responses with stable expansion ability [153]. Voynova et al. compared NK cells carrying an NK-cell-specific CAR framework (a CD8a hinge region, NKG2D transmembrane region, 2B4 costimulatory domain, and CD3ζ signaling domain) and T-cell-specific CAR framework (a hinge region, transmembrane and costimulatory domains of CD28, and a CD3ζ signaling domain) to target CD3- or CD5-positive T-ALL. The results showed that NK-specific CAR constructs can confer NK cells with more potent killing activities both in vitro and in a T-ALL xenograft mouse model. The author considered that CD5 is a better target than CD3 for CAR-based treatment of T-cell malignancies, as less antigen escape of T-ALL cells was found in the group treated with CD5 CAR-NK cells [152]. A phase I/II clinical study conducted by the M.D. Anderson Cancer Center is evaluating the safety and optimal dose of anti-CD5 CAR and IL15-transduced CB-NK cells in patients with relapsed/refractory T-cell malignances (NCT05110742). The results are currently not available. You and his colleagues engineered CD7-targeting CAR-NK-92MI cells by employing monovalent or bivalent CD7 nanobody VHH6 sequences in the CAR cassette. Both CD7-CAR-NK-92MI cells exhibited marked elevation of IFN-γ and Granzyme B in the exposure of T-ALL cell lines and primary tumor cells. In particular, bivalent CD7-CAR-NK-92MI cells had a superior inhibitory effect on T-ALL tumors in a mouse model [156]. In addition, CD7-CAR-engineered NK-92 cells have been clinically tested in patients with CD7-positive relapsed or refractory leukemia and lymphoma (NCT02742727).
Myeloid malignancies
CAR-T-cell therapy has been extensively investigated for acute myeloid leukemia (AML) and multiple myeloma (MM), achieving impressive efficacy by targeting antigens such as BCMA, CD33, CD38, IL-3 receptor alpha chain (CD123), and CD138 [181]. Benefiting from sufficient research on CAR-T-cell therapy, recent paradigm-changing CAR-NK therapy has brought new hope in the treatment of myeloid malignancies.
CD123, the most-studied target in AML, has been targeted by several NK cell types engineered with CARs, including NK92, PB-derived, and CB-derived NK cells. Depending on the third- or fourth-generation anti-CD123 CARs, NK cell products can efficiently lyse AML cells in vitro and suppress tumors in AML xenograft mice, providing a firm foundation for further clinical transformation [165, 167, 182, 183]. CD38 is a typical molecule expressed on AML cells, NK cells, and some myeloid cells [184, 185]. The researchers used a natural CD38-low-expression NK cell line KHYG-1 to bear CD38-CAR, whose efficacy to lyse AML cells was greatly enhanced. To further minimize CAR-NK self-fratricide, the CD38 knockout technique was conducted on primary NK cells before anti-CD38 CAR transduction. Cytokine-induced memory-like (CIML) NK cells, characterized by enhanced and prolonged responses to tumor restimulation, have shown promising therapeutic effects on treating relapsed/refractory AML, according to recent clinical reports [104, 105, 186, 187]. Investigators have armed CIML NK cells with a CAR targeting a neoepitope (NPM1c), an AML-specific nucleophosmin-1 (NPM1) gene mutant, [146], resulting in more precise and robust killing as well as minimal on-target off-tumor effects [168]. The most commonly used virus-mediated CAR transfer modes require laborious virus production, and the virus quality is variable from lot to lot. An investigation applied a nonviral-form piggyBac transposon technology in the engineering of CAR NK cells to boost transduction efficiency and manufacturing stability. Utilizing this system, the team has successfully engineered PB-derived NK cells with an NKG2D-CAR cassette including the IL-15 gene, achieving synergistically enhanced anti-AML activity and improved in vivo persistence [162]. It is important to note that AML stem cells have also evolved mechanisms to evade NK cell recognition, such as the downregulation of NKG2D ligands, which may, in turn, limit the efficacy of CAR NK cell therapy [189].
CD138, a primary membrane protein on MM cells, is targeted by the first-generation CD138 CAR-NK-92MI cells. The results demonstrated enhanced and selective cytotoxicity toward CD138-positive MM cells in vitro and rapid regression of tumors in the subcutaneous injection mouse model [158]. CS1, which colocalizes with CD138 and ubiquitously exists on MM cells, is also a potential target for MM treatment. CS1 CAR-NK was analyzed in CS1-expressing MM cells and MM-bearing mice. Elevated secretion of cytokines and obvious tumor eradication were observed in a CS1-expression-dependent manner [160]. NKG2D, possessing a broad spectrum of ligand types on multiple cancer cells, was designed as a scFv component of CAR. The results showed that NKG2D CAR-NK cells could mediate strong antitumor responses in MM xenograft mice [161].
Clinical trials for T-cell-derived hematologic cancer are relatively limited. In 2018, a first-in-man phase I trial of CD33-CAR-NK-92 cells was initiated by Tang et al. to treat a small cohort of R/R AML patients (n = 3) (NCT02944162) [82]. Third-generation CARs, including CD28 and 4-1BB costimulatory domains, were transferred to NK-92 cells with a high transduction efficiency of up to 90%. Three patients received three incremental doses of CD33-CAR-NK-92 cells every other day, peaking at 5 × 109 cells. Only moderate to high fevers or low-grade CRS were observed in the three patients, and these adverse effects were quickly relieved within two days. Then, sponsored by Xinqiao Hospital of Chongqing (China), a phase I clinical trial of CD33-CAR-NK-92 cell therapy was initiated in 2021 to evaluate the safety and efficacy of CAR-NK-92 cells in combination with the cancer medication cytoxan and the antineoplastic drug fludarabine in AML patients. The results are still pending. FT576, another pipeline product of Fate Therapeutics, aims to treat patients with relapsed/refractory multiple myeloma. Similar to FT596, FT576 encompasses a BCMA-targeting CAR, high-affinity CD16 and IL-15/IL-15Rα. In addition, CD38 is knocked out to mitigate the negative response induced by NK cell fratricide. According to its interim phase I clinical data, FT576 as a monotherapy or a combination therapy with daratumumab (anti-CD38 antibody) shows effective anti-myeloma activity [190].
CAR-NK cell therapy for solid tumors
The application of cellular adoptive immunotherapy for hematological tumors has shown encouraging outcomes, but obstacles remain in the treatment of solid tumors. NK cells possess favorable attributes, such as the ability to infiltrate tumors and intrinsic recognition and killing of tumor cells, attracting investigators’ enthusiasm for exploiting CAR-NK cells in the treatment of solid tumors. A large number of CAR-NK preclinical studies have been conducted to test CAR-NK cell therapy efficacy for a series of solid tumors, mainly breast cancer (BC), ovarian cancer (OC), glioblastoma (GBM), and some types of gastrointestinal malignancies (Table 2). A few CAR-NK cell studies have moved into the clinical stage, and these studies are summarized in Table 3.
Human epidermal growth factor receptor 2 (HER2) is highly expressed in breast cancer [191], GBM [192], and renal cell carcinoma (RCC) [193]; thus, it serves as an attractive target for CAR-engineered NK cells. Portillo et al. used healthy donor and patient-derived NK cells to carry Her2.CD28.CD3ζ CAR. Under the condition of immunosuppressive factors such as TGF-β and PGE2, Her2 CAR-NK cells had robust cytotoxic effects on a series of HER2-positive breast cancer cells, and minimal to no toxic effects on healthy tissue cells were found [194]. Oxidative stress is also an immunosuppressive factor in the TME that can induce the dysfunction and death of NK cells [195, 196]. Peroxiredoxin-1 (PRDX1), a critical element of antioxidative defense, is absent in NK cells [197]. The investigators constructed NK-92 cells with stable PRDX1 expression and then engineered anti-PD-L1 CARs, which can support NK cell function in the presence of oxidative stress and induce potent killing of PD-L1-positive breast cancer cells [198]. In addition, NK cells that co-expressed anti-HER2-CAR and soluble PD-1 (designated sPD-1-CAR-NK cells) could suppress the interactions of PD-1/PD-L1 and significantly augment immunological anticancer efficacy in PD-L1+Her2+ breast cancer cells [199]. The first clinical trial of CAR-NK cells in Germany deployed Her2 CAR-NK-92 cells harboring CD28 and CD3ζ signaling domains (NK-92/5.28.z cells) to target recurrent HER2-positive glioblastoma [193, 200] (NCT03383978). The main objective was to evaluate the safety and tolerability of NK-92/5.28.z cells. NK-92/5.28.z cells were injected into the resection cavity during surgery in the dose-escalation scheme. To date, no toxicity has been observed at any dose.
EGFR, which is closely associated with tumor progression and migration, is expressed in 50% of GBM patients [201]. Intracranial injection of anti-EGFR.CD28. CD3ζ CAR-NK cells in the GBM mouse model showed obvious tumor repression and extended the overall survival rate. However, GBM tumors with EGFR amplification are often accompanied by a self-active EGFR mutant form, EGFRvIII, which contributes to heterogeneity and treatment resistance [202]. In 2018, Murakami et al. introduced an EGFRvIII-targeted CAR into the novel NK cell line KHYG-1, named EvCAR-KHYG-1 cells. Appreciable antitumor ability was observed in an EGFRvIII-dependent manner when cocultured with the GBM cell line U87MG [203]. To facilitate the homing of CAR-NK cells to the EGFRvIII-expressing tumor site, EGFRvIII-specific CARs with concomitant expression of the chemokine receptor CXCR4 on NK cells displayed redirected and evident migration to GBMs and significantly enhanced the mouse survival time in comparison with EGFR-CAR-NK alone [204]. In addition to CAR-NK cell monotherapy, combined therapies can also achieve outstanding effects. For example, taking advantage of the effective tumor-lysing ability of oncolytic viruses (OVs), an IL-15/IL-15Rα complex fusion protein was engineered on an OV (OV-IL15C) to synergistically favor the cytotoxicity, persistence, and infiltration of EGFR-CAR-NK cells. Better tumor regression-inducing capability was achieved than with either monotherapy [205].
CAR-NK cells are also employed in targeting HER2 [206], mesothelin (MSLN) [207], folate receptor-α (FRα) [208], and CD133 [209] to treat ovarian cancer. Mesothelin (MSLN)-targeted CAR-NK92 cells were identified as having potent cytotoxicity against MSLN-positive ovarian cell lines such as OVCAR-3 and SKOV3. The conspicuous tumor elimination ability in both subcutaneous and intraperitoneal tumor models further indicated that MSLN is a reliable target for future ovarian cancer treatments. Moreover, a novel dual-CAR was engineered into NK92 cells for the simultaneous targeting of MSLN and CD24; these cells could promote the apoptosis of both ovarian cell lines and primary ovarian tumor cells, and off-target effects were greatly minimized [210]. In clinical settings, anti-MSLN CAR-NK cells were developed by Allife Medical Science and Technology Company for patients with epithelial ovarian cancer (NCT03692637). This study is not yet recruiting, and no additional information is available. CAR-NK therapy targeting claudin-6 for ovarian cancer treatment is in the phase I/II stage of clinical trials, and the results are still pending [211].
Gastrointestinal cancers such as pancreatic cancer (PC), hepatocellular carcinoma (HCC) and colorectal cancer (CRC) have also been largely evaluated in recent years. Pancreatic ductal adenocarcinoma (PDAC) accounts for 90% of all kinds of PC, and its immunosuppressive stroma greatly hinders the infiltration of immune cells. Through bioinformatic integration of patient-derived samples, folate receptor α (FRα) and death receptor 4 (DR4) have been identified as optimal targets in the treatment of PDAC. A study confirmed that FRα-redirected CD27.CD3ζ CAR-NK92 cells with surface-displayed TRAIL vastly enhanced tumor-selective apoptosis both in vitro and in xenograft mice [212].
GPC3 is overexpressed in hepatocellular carcinoma (HCC) cells but is undetectable in normal tissues [213, 214]; therefore, it represents an ideal immunotherapeutic target. Hu9F2, a humanized anti-GPC3 scFv, was incorporated into CAR to redirect NK92 cells, killing HCC cell lines with gradually decreasing GPC3 expression. Selective lysis of GPC3-positive HCC cells was observed in vitro and in multiple HCC xenograft mouse models [215]. CD147 is expressed in several cell types and is particularly upregulated in pathological cells, including HCC cells. To minimize the on-target/off-tumor toxicity of CAR-NK cells, Tseng et al. used a logic-gated (log) synthetic notch to regulate the killing of dual-targeting (GPC3 and CD147) CAR-NK cells. Specifically, synthetic notch-mediated CAR-NK cells could recognize and eliminate double-positive (GPC3+CD147+) HCC cells but remained inactivated against single-positive (GPC3−CD147+ or GPC3+CD147−) HCC cells. In a human CD147-transgenic mouse model, the median survival time of mice treated with dual-targeted CAR NK cells was significantly extended, and there was no additional on-target/off-tumor effect during the observation period [216].
NKG2D CAR-NK cells incorporating the cytoplasmic domain of DAP12 (NKG2Dp CAR) have been employed to treat colorectal cancer (CRC), resulting in effective elimination of CRC cell lines and prolonging the survival time of CRC-bearing mice model [139]. Furthermore, the researchers successfully tested the feasibility of NKG2D CAR-NK cells in three patients with chemotherapy-refractory metastatic colorectal cancer (NCT03415100). Multiple doses of NKG2Dp CAR-NK cells were intraperitoneally infused into two patients with severe CRC burdens; the NKG2Dp CAR-NK cells contributed to a significant reduction in the volume of ascites and tumor regression. In the third patient with metastatic colon cancer in the liver, intraperitoneal infusion together with ultrasound-guided percutaneous injections of NKG2Dp CAR-NK cells induced obvious tumor regression in the liver. Additionally, another clinical trial of NKG2D CAR-NK cell therapy was initiated in 2022 to assess its safety and efficacy in patients with refractory metastatic colorectal cancer (NCT05213195). The detailed data have not been disclosed. Carcinoembryonic antigen (CEA) is another CRC target. CAR-NK-92MI cells have potent cytotoxicity against CEA-positive CRC cells. The administration of the histone deacetylase inhibitor sodium butyrate (NaB) or the methylation inhibitor 5-azacytidine (5-AZA) can induce CEA expression on CRC cells, which further enhances anti-CEA CAR-NK-92MI cell-induced cytotoxicity. This combination therapy showed potential to be clinically applied in terminal-stage colorectal cancer treatment [217].
The remaining CAR-NK cells in clinical trials target a diverse set of antigens, such as PSMA for prostate cancer, DLL3 for small-cell lung cancer, and roundabout homolog 1 (ROBO1) and MUC1 for advanced solid cancers. In summary, the number of clinical studies on solid tumors is still limited. The results of these clinical studies are still awaited to determine the persistence and durable response of CAR-NK cells. Additionally, it is noteworthy to observe whether the risk of tumor escape in CAR-NK cell therapy would be lower than that in CAR-T-cell therapy, as CAR-NK cells possess CAR-independent innate killing ability.
Strategies to address the limitations of CAR-NK cell therapy
Despite the remarkable outcomes of CAR-NK cells in treating a series of tumors, there are still obstacles in the field. Here, we focus on the major challenges that significantly restrict the production and therapeutic efficacy of CAR-NK cells (Fig. 4), sketching a developmental path for broader application.

The challenges of CAR-NK existing in the process from lab production to tumor infiltration. The unsatisfactory CAR transduction efficiency and limited proliferation ability add barriers to CAR-NK production. Multiple approaches including virus-mediated and non-viral-mediated transduction have been utilized to boost CAR expression and stability. The ex-vivo expansion are mainly stimulated by cytokines or feeder cell system with limited potential. Upon infusion into the body, the trafficking and infiltration abilities are impeded by the disruptive chemokines/chemokine receptors axis in the dysregulated tumor vasculature. In tumor bed, suppressive cells (Treg cells, Breg cells and MDSCs) and soluble inhibitory cytokines (TGF-β, IL-10 and IL-6) can disrupt NK cell effector functions. The harsh TME owing to the nutrient deficiency, hypoxia, and acidic conditions can further suppress and dampen NK activities
CAR-NK cell transduction efficiency
The most commonly used transduction systems in CAR-NK cell therapy are also lentiviruses (LVs) and retroviruses (RVs) [242]. However, LV and RV-mediated CAR transduction into NK cells has not shown the same satisfactory efficiency as that into T cells [140]. Other viruses being investigated for transduction, such as vaccinia and adenoviruses, are not suitable for NK cells, as they can alter the cytotoxic phenotype of NK cells and possess weaker transduction capability [243].
To enhance viral-mediated transduction efficiency, some small molecular compounds are used to reduce the repulsion of NK cells to foreign viral particles [244]. Negative charges on the surface of both the virus and the target cell are detrimental for transduction [245]. Thus, some cationic polymers, such as protamine sulfate, polybrene, and dextran, can promote transduction efficiency by positively charging cells [244, 246]. RetroNectin, a recombinant human fibronectin fragment, can vastly enhance the transduction efficiency of NK cells by colocalizing viruses and cells in close proximity [141, 247]. Similarly, Vectofusin-1 can promote CAR transfer by augmenting the adhesion of the virus to the cellular plasma membrane. There is no consensus on which is a superior transduction enhancer [248]. BX795 is an inhibitor of TBK1/IKKɛ, which are critical kinases involved in antiviral response signaling pathways [249, 250]. This functional inhibitor has been reported to greatly enhance gene transfer efficiency in primary immune cells [251, 252]. Other compounds have been utilized in various immune cells to improve transduction, such as phytohemagglutinin (PHA) [253], prostaglandin E2 (PGE2), and phorbol 12-myristate 13-acetate (PMA), but further mechanistic description is lacking.
Vesicular stomatitis virus (VSV) G-protein has been widely used for pseudotyping lentiviruses, as VSV-G has broad cell tropism [254]. Low-density lipid receptor (LDL-R), the main receptor of VSV-G, has a low level of expression on NK cells [255, 256]. This may be an explanation for the low transduction efficiency of VSV-G pseudotyped lentiviruses (VSV-G-LVs) into NK cells. Statins are widely prescribed medications for CLL patients, but they were recently identified with the function of upregulating LDL-R expression on immune cells, including NK cell lines and primary NK cells [256, 257]. Theoretically, transduction efficiency can be enhanced through statin administration. However, not all statins are suitable for boosting VSV-G-LV transduction efficiency, as most statins can negatively affect cell viability. In a comparative study, rosuvastatin was found to be the most potent substance to augment transduction, and its suppressive effect can be reversed in the presence of GGPP [256]. In addition to regulating LDL-R expression on target cells, employing other glycoproteins to pseudotype the viral vectors is also a feasible strategy to improve virus-mediated transduction efficiency [118]. The virtually unanimous conclusion is that the expression level of lentivirus receptors on target cells has a positive correlation with the transduction efficiency of the virus. BaEV, MV-, and RD114-pseudotyped viruses have been tested in NK cell transduction, and BaEV showed the best performance in large part due to the high expression of its receptors on NK cells [171, 255, 258].
Due to the risk of viral insertional mutagenesis and quality variability in large-scale viral production [259, 260], nonviral transduction methods have gained more attention in recent years.
Electroporation is the most commonly used nonviral transfection method. It is considered a safer transduction approach because it induces short-term gene expression [261, 262]. Electroporating DNA into cells showed a limited transduction rate, but superior performance was presented in electroporating mRNA, which can reach up to a 95% transduction rate with minimal damage to cell viability [149, 263, 264]. The transfection efficiency is significantly enhanced in electroporating NK-92 cells but with limited improvement in transducing CB-NK and PB-NK cells [263, 265,266,267]. Several preclinical results have demonstrated the efficacy of electroporation-based CAR-NK cells in the treatment of both solid and hematologic tumors [76, 139, 268]. Nucleofection is a transduction method based on electroporation, depending on the specific electric pulse used to directly deliver DNA into the cell nucleus, regardless of the cell division phase [269]. Nucleofection has been employed to induce CAR-NK cells to express a range of CARs targeting ROR in solid tumors [270] or CD20 in hematologic tumors [148, 149]. However, the transience of CAR expression induced by electroporation-based methods necessitates the transfusion of cell products into patients within seven days. The cell membrane damage and cell death caused by electric pulses are also major concerns for the continuous expansion of electroporation in clinical settings [271].
Two nonviral transposon systems, namely, Sleeping Beauty (SB) and PiggyBac (PB), can provide long-term transgene expression by inserting foreign genes by “a cut-and-paste” mechanism [272,273,274,275]. SB and PB systems have several advantages over virus-mediated transduction approaches: (1) more random gene integration; (2) a large capacity for foreign genes; and (3) cost-effective production of the basic components [276, 277]. These attributes make the SB and PB systems attractive tools for CAR-based therapy. In recent years, transposon systems have been mainly applied to generate CAR-T cells in preclinical and clinical settings [278,279,280,281,282] but are in the minority of systems used in the CAR-NK engineering field. NK-92 cell lines were easier to engineer with CARs by transposon-based methods, and the resulting products showed effective antitumor responses [138, 283]. Recently, investigators have successfully engineered PB-NK cells with NKG2D CAR and the IL-15 gene using the PB system [162]. There are also some drawbacks to transposon-mediated methods, such as uncontrolled transposition events and transgene remobilization in target cells [284, 285]. Additionally, the transfer of transposon components (such as transposage and gene vectors) needs to be promoted by a virus or electroporation, which can lead to the negative effects mentioned above [272, 286].
CRISPR/Cas9 is a potent genetic modification technique that has been widely applied in cellular immunotherapy [287]. The CRISPR/Cas9 system generally consists of two components: a single guide RNA (sgRNA) and Cas nuclease protein [288]. The precise and highly efficient gene-editing process is initiated through the recognition of specific gene loci by the sgRNA, followed by interaction with Cas9. CRISPR/Cas9 has been utilized in the CAR-T therapy field to address multifaceted issues such as generating allogeneic CAR-T cells and overcoming CAR-T cell exhaustion and the negative factors of the TME [289,290,291]. Similarly, CRISPR/Cas9 was initially adopted to disrupt or insert functionally relevant genes to improve CAR-NK cell performance. Researchers have successfully knocked out CD38 to prevent NK cell fratricide [164] and conducted triple editing (disruption of ADAM17 and PDCD1, knock-in of CD16), achieving high manipulation efficiency and enhanced function [292]. More recently, CRISPR/Cas9 has also been utilized to realize highly efficient and locus-specific CAR transduction into immune cells. Directing CD19 CAR to the T-cell receptor α constant (TRAC) locus by CRISPR/Cas9 resulted in consistent CAR expression in human peripheral blood T cells as well as improved effector responses [293]. One group combined CRISPR/Cas9 with an adeno-associated virus (AVV)-mediated gene-delivery approach to insert anti-CD33 CAR to a safe-harbor locus of primary NK cells, acquiring a mean expression of 68% CAR-positive NK cells and enhanced anti-AML activity [294].
Other emerging transduction strategies also include lipid nanoparticle (LNP)- and charge-altering releasable transporter (CART)-based transduction. LNPs and CARTs serve as protective carriers of nucleic acids, infusing into the cells without degradation by the nucleases. Once entering the cell cytosol, these substances can transform into a positively charged state to allow the release of internal mRNA and then proceed with protein expression. These strategies have been demonstrated to be effective in anti-CD19 CAR transduction into NK cells [295,296,297]. Altogether, these strategies have vast potential as genetic engineering tools in cellular immunotherapy but are still in their infancy of development. More investigations are required to test the safety performance and persistence of these cell products.
CAR-NK cell expansion and persistence
Large amounts of NK cells are required for clinical therapy to achieve sufficient responses. However, the weak in vitro expansion of NK cells significantly hinders CAR-NK cell production and broad application. Autologous NK cells from patients account for a smaller proportion of cells in PB, causing additional difficulty for NK cell expansion [298]. A common expansion method relies on a series of cytokines for stimulation, such as IL-2, IL-12, IL-15, IL-18, and IL-21 [299]. A specific cytokine cocktail can tune NK cells to a particular phenotype. For example, the combination of IL-12, IL-15, and IL-18 can facilitate the generation of memory-like NK cells, which exhibit optimal in vivo persistence and antitumor activity [103, 104, 187, 300]. Nevertheless, the proliferation of cytokine-induced NK cells is associated with limited fold changes. Furthermore, solely depending on cytokines, NK cells easily become cytokine-susceptible and cytokine-addicted, which may raise a major concern for in vivo persistence and vitality in the absence of abundant cytokines [301].
Feeder cells serve as large-scale culture systems that combine cytokine stimulation and receptor-mediated activation [299]. K562 cells are representative feeder cells. Other cells, such as the Epstein‒Barr virus-transformed lymphoblastoid cell line (EBV‑LCL), 721.221, and PBMCs, are also exploited as feeder cells [205, 302, 303]. They are generally engineered to express membrane-bound cytokines (IL-2, IL-15, and IL-21) and/or ligands of NK activating receptors (4-1BBL, OX40L, and HLA-E), which can synergistically promote persistent expansion and antitumor activity [303,304,305,306,307]. K562 cells expressing IL-21 and 4-1BBL have been tested clinically and are considered safe in patients [303]. Compared to the sole cytokine culture system, feeder cells can significantly extend the number of fold changes and alleviate the dysfunction and apoptosis of NK cells induced by cytokine deficiency post-infusion. However, the majority of feeder cells are derived from cancer cell lines and thus must be lethally irradiated prior to infusion. Many concerns are arising about whether surviving feeder cells and other unknown factors could pose potential risks in the context of a complex body environment.
To circumvent the administration of feeder cells to support activation and proliferation, several groups are endeavoring to manipulate CAR plasmids incorporating cytokine transgenes to facilitate expansion and persistence [248,249,250]. The expression form of cytokine gene cassettes can be either membrane-bound or constitutively autocrine. A team from MD Anderson Cancer Center managed to engineer CAR-CB-NK cells to express IL-15 in a constitutively autocrine manner, demonstrating enhanced proliferation and in vivo persistence. There were no signs indicating elevation of systematic IL-15 or other toxicities [26, 128]. In another study, ectopic expression of IL-15 significantly prolonged the persistence of NKG2D CAR- NK cells both in vitro and in vivo. Additionally, the effector function of CAR-PB-NK cells was also significantly facilitated in AML mouse models [162].
The trafficking and infiltration capabilities of CAR-NK cells
It is easier for infused CAR-NK cells to come into contact with hematological cancer cells in circulating peripheral blood; however, in their trafficking to solid tumor sites, multifaceted obstacles are encountered. The trafficking ability and infiltration amounts of NK cells have prognostic value for improved clinical outcomes [309,310,311].
To surmount the anatomical barriers in the treatment of solid tumors, orthotopic injections such as intraperitoneal injections, anterior prostatic lobe injections, and other ultrasound-guided injections have demonstrated effective tumor elimination in CAR-NK cell therapy without tissue damage [139, 192, 312]. In a phase I clinical trial in 9 patients with recurrent HER2-positive GB, NK-92/5.28.z cells targeting Her2 were injected into the wall of the resection cavity during relapse surgery. The disease progression of 5 patients was suppressed, lasting for 7 to 37 weeks, and no signs of dose-limiting toxicities were observed, demonstrating the feasibility and safety of intracranial injection of HER2-targeted CAR-NK cells [313].
The commonly used injection method is intravenous (i.v.) injection. CAR-NK cells need to extravasate from the blood and migrate to the solid tumor bed. This homing process is regulated by the dynamic chemokine receptor-and-ligand interactions between NK cells and tumor cells [314, 315]. Thus, increasing the expression of chemokine receptors on NK cells is a major strategy initially applied in NK cell-based therapies. A growing body of studies has equipped NK cells with chemokine receptors (CCRs), such as chemokine receptor chemokine (C-X-C motif) receptor 2 (CXCR2) [316, 317], CXCR4 [318] and CXCR7 [319], to match their cognate ligands expressed on tumor cells, and improved chemoattraction in the antitumor response of NK cells was shown. Concomitant expression of the CXCR1 or CXCR4 transgene on CAR-NK cells has also demonstrated enhanced migration to CD19+ hematological tumors [320], glioblastoma [321] and ovarian tumors [322]. The release level of chemokines in the TME varies greatly following different kinetics of the tumors, indicating the need for artificial infusion of sufficient chemokines to attract NK cells [318]. The feasible approaches include direct local administration of stimulation factors [318] or delivery of fusion proteins loaded with chemokine ligands [323]. The latter can release chemokine ligands upon engagement with tumor cells, thus facilitating the connection of chemokine receptor/ligand axes. However, these chemokine interactions are versatile and vary in different milieus, possibly causing a totally reversed effect (either promoting or diminishing) on the trafficking ability of NK cells [324, 325]. Thus, more efforts should be made to investigate the comprehensive mechanisms and responses of the chemokine interplay in the intricate context of the human body.
As hinted above, the complex TME is another major barrier that hinders the homing and function of CAR-NK cells. The obstructive factors existing in the TME network can be generally divided into three parts: counterproductive cells, immunosuppressive soluble substances, and a harsh metabolic milieu [180]. In the normal environment of tissues, immune cells such as myeloid cells, regulatory T (Treg) cells, or regulatory B (Breg) cells can provide positive feedback to the proinflammatory cytokines secreted from NK cells. They similarly produce cytokines such as IL-12, IL-15, and IL-18 to promote NK cell growth, maturation, and functionality [53, 326]. However, the formulation and exacerbation of the TME in solid tumors render some immune cells, including Treg cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), to have suppressive roles. Under TME pressure, NK cells can also be forced to transform to have suppressive phenotypes, which discount their activity and infiltration ability [325]. Traitorous immune cells release inhibitory cytokines such as transforming growth factor beta 1 (TGF-β), prostaglandin E2 (PGE2), and IL-10 or “absorb” cytokines that are favorable for NK cells (IL-2), both of which directly or indirectly impede NK cell response and survival [327,328,329,330]. Thus, circumvention of these negative modulators is critical for CAR-NK cell therapy efficacy in vivo [331]. Pharmacological interventions such as chemotherapies have been adopted to eliminate MDSCs and Treg cells [233, 332, 333]. Additionally, NK cells were manipulated with the NKG2D.ζ CAR construct to target NKG2DL-expressing MDSCs, and cytotoxicity toward MDSCs in the xenograft TME model was observed. NKG2D.ζ CAR-NK cells have been tested in clinical studies to show that they can effectively eliminate intertumoral MDSCs in neuroblastoma patients and facilitate the infiltration and efficacy of infused CAR-T cells [334].
Excessive TGF-β is produced by immunosuppressive cells and tumor cells themselves in the TME. Pathologic levels of TGF-β are often correlated with serious disease progression and an impaired immune system [335,336,337,338]. Multiple strategies have been employed to disrupt TGF-β signaling to preserve NK cell therapy efficacy. Genetically engineering negative TGF-β receptors into NK cells [339,340,341] or knocking out TGF-β signaling-related downstream mediators on NK cells [342] successfully diminished or eliminated the blocking effects of TGF-β on NK cells. In vivo studies have identified enhanced NK cytolytic efficacy in GBM-engrafted mice after silencing TGF-β signaling. Additionally, molecular kinase inhibitors [341, 343, 344] and monoclonal antibodies [345] targeting TGF-β have exhibited equivalent antagonistic effects and improved the performance of infiltrating NK cells. Other TME-abundant suppressive soluble factors, including adenosine, PGE2, IL-10, and IL-37, are likewise potential targets to reverse the unfavorable TME [346, 347]. Adenosine is a metabolic byproduct in response to hypoxia in the TME, and its accumulation can paralyze NK cell functional activity [348]. The mainstream strategies to circumvent the negative effects of adenosine are targeting the hypoxic ectoenzyme CD73 [219, 349,350,351,352] and/or knocking out A2A receptors expressed on immune cells [352,353,354,355]. The results showed a plunging concentration of adenosine in the TME as well as the revival of NK cells. A triple-functional NK was manipulated with a locally released CD73 antibody fragment concomitant with dual-targeting (NKG2D and GD2) CAR expression to target GBM [219]. Enhanced cytolytic ability and persistence of intratumoral NK cells have been observed. Regional regulation of adenosine did not cause metabolic disorders in the whole body. Lever aging eminent preclinical performances, successional clinical trials using CD73 blocking antibodies have been carried out to overcome the dilemma of treating solid tumors (NCT04148937, NCT03454451, and NCT03616886).
Hypoxia, a typical hallmark of the TME, develops as a result of the malignant outgrowth of barely vascularized solid tumor tissues [356]. Restricted oxygen concentrations and deficient nutrients in solid tumor regions induce the downregulation of NK cell functional molecule expression (such as activation/inhibitory receptors, cytokines and death receptors) and substantially suppress their killing performance and migration ability [357, 358]. Nevertheless, relying on the hypoxic TME, tumor cells can escape immune cell monitoring and attack [359, 360]. Thus, mitigating intratumor hypoxia is a potential strategy to improve the treatment of solid tumors. Multiple pharmacological and physical strategies have been widely investigated in a series of preclinical and clinical studies to directly adjust the hypoxic state [361,362,363,364,365,366,367]. For example, hypoxia-activated prodrugs (HAPs) [368] or inhibitors targeting the hypoxia-inducible factor (HIF) protein family may be applied, or patients may even be physically exposed to a hyper oxygenated environment [369]. These strategies may provide directions to ameliorate and transform infiltrating-tumoral CAR-NK cell functional and metabolic exhaustion. We can also draw inspiration from a recent novel hypoxia-sensing CAR-T cell structure, which made full use of deleterious hypoxia as a switch to favor itself, launching a CAR-mediated killing procedure [370, 371]. This approach may establish a good pattern for the future design of CAR-NK cells to overcome hypoxia. Another unfavorable condition is the low pH in the TME. As a result of hypoxia, more anaerobic glycolysis reacts to support the activity of tumor cells; thus, increasingly accumulated lactic acid can damage the functionality of NK cells and promote suppressive immune cells [372]. The depletion of excessive metabolites is a direct strategy. However, it is noteworthy that preventing excessive immune metabolite modulation and maintaining a physiologic balance of the inner environment would be critically important for NK cell function.
Conclusion and future perspectives
Paradigm shifting CAR-T-cell therapy has pioneered the development of the CAR technique and yielded promising outcomes in treating hematological tumors. Benefiting from the technologies and valuable lessons learned from CAR-T cell therapy, CAR-NK cell therapy has advanced rapidly with continuous innovations. Preclinical and early clinical outcomes have demonstrated the vast potential of CAR-NK cells as “off-the-shelf” products for cancer treatment. To date, numerous strategies have been applied in CAR-NK cell therapies to address the challenges discussed above, and satisfactory outcomes have been observed in preclinical studies. However, some of these strategies are difficult to translate into clinically approved procedures, such as the systematic infusion of NK-cell-stimulating cytokines. In contrast, multiplexed CAR-NK cell design systems or combinatorial approaches based on radiotherapy and other FDA-approved drugs may hold great potential to overcome the barriers in the CAR-NK cell therapy field and provide clinical benefit. With the development of cutting-edge technologies such as single-cell RNA sequence analysis (scRNA-seq), we have access to elucidating key parameters associated with CAR-NK cell biological function and therapeutic efficacy, which may provide investigators and clinicians with critical insights into how to optimize the promise of NK cell-based cancer therapy. In the coming years, clinical translation-oriented research and in-depth clinical testing of CAR-NK cell therapies are urgently needed to determine their potential market authorization.
Availability of data and materials
Not applicable.
Abbreviations
- ACT:
-
Adoptive cell therapy
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- BCMA:
-
B cell maturation antigen
- CAR:
-
Chimeric antigen receptor
- Cas9:
-
CRISPR-associated system 9
- CCR:
-
Chemokine receptors
- CEA:
-
Carcinoembryonic antigen
- CLL:
-
Chronic lymphocytic leukemia
- CIML:
-
Cytokine-induced memory-like NK cells
- c-Met:
-
Cellular-mesenchymal epithelial transition factor
- CR:
-
Complete remission
- CRS:
-
Cytokine release syndrome
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- CSC:
-
Cancer stem cell
- CSPG4:
-
Chondroitin sulfate proteoglycan 4
- DLBCL:
-
Diffuse large B-cell lymphoma
- DLL3:
-
Delta-like ligand 3
- EBV-LCL:
-
Epstein–Barr virus-transformed lymphoblastoid cell line
- EGFR:
-
Epidermal growth factor receptor
- EGFRvIII:
-
EGFR variant III
- EPCAM:
-
Epithelial cell adhesion molecule
- EPHA2:
-
EPH Receptor A2
- ER:
-
Estrogen receptor
- FasL:
-
Fas ligand
- FcγRIII:
-
Fragment crystallizable receptor III
- FDA:
-
US Food and Drug Administration
- FLT3:
-
FMS-like tyrosine kinase 3
- FRα:
-
Folate receptor alpha
- Fzd:
-
Frizzled
- GM-CSF:
-
Granulocyte-macrophage colony-stimulating factor
- GVHD:
-
Graft-versus-host disease
- GPC3:
-
Glypican-3
- Her2:
-
Human epidermal growth factor receptor 2
- hESC:
-
Human embryonic stem cells
- HLA:
-
Human leukocyte antigen
- IFN-γ:
-
Interferon-γ
- iPSC:
-
Induced pluripotent stem cell
- IS:
-
Immunologic synapse
- iKIRs:
-
Inhibitory killer cell immunoglobulin-like receptors
- mAb:
-
Monoclonal antibody
- M-CSF:
-
Macrophage colony-stimulating factor
- MHC:
-
Major histocompatibility complex
- MSLN:
-
Mesothelin
- MM:
-
Multiple myeloma
- NCR:
-
Natural cytotoxicity receptor
- NK:
-
Natural killer
- NSG:
-
NOD scid gamma
- PB:
-
Peripheral blood
- ORR:
-
Objective response rate
- PR:
-
Progesterone receptor
- PSMA:
-
Prostate-specific membrane antigen
- PSCA:
-
Prostate stem cell antigen
- PSMA:
-
Prostate specific membrane antigen
- RCC:
-
Renal cell carcinoma
- SCLC:
-
Small cell lung cancer
- scFv:
-
Single-chain antibody variable fragment
- scRNA-seq:
-
Single-cell RNA sequence analysis
- SLO:
-
Secondary lymphoid organs
- TAA:
-
Tumor-associated antigen
- TF:
-
Tissue factor
- TGF-β:
-
Transforming growth factor-beta
- TME:
-
Tumor microenvironment
- TNBC:
-
Triple‑negative breast cancer
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- UCB:
-
Umbilical cord blood
References
June CH, Sadelain M. Chimeric antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20(11):651–68.
Liu Q, Liao Q, Zhao Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017;17:68.
Papaioannou NE, Beniata OV, Vitsos P, Tsitsilonis O, Samara P. Harnessing the immune system to improve cancer therapy. Ann Transl Med. 2016;4(14):261.
Wang Y, Wang M, Wu HX, Xu RH. Advancing to the era of cancer immunotherapy. Cancer Commun (Lond). 2021;41(9):803–29.
Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–7.
Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-lasting complete responses in patients with metastatic melanoma after adoptive cell therapy with tumor-infiltrating lymphocytes and an attenuated IL2 regimen. Clin Cancer Res. 2016;22(15):3734–45.
June CH, Riddell SR, Schumacher TN. Adoptive cellular therapy: a race to the finish line. Sci Transl Med. 2015;7(280):280ps7.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322–31.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25.
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang ML, Arnason JE, et al. Pivotal safety and efficacy results from transcend NHL 001, a multicenter phase 1 study of lisocabtagene maraleucel (liso-cel) in relapsed/refractory (R/R) large B cell lymphomas. Blood. 2019;134(Supplement_1):241.
Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. 2023;22(1):20.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. 2022;28(2):325–32.
Locke FL, Ghobadi A, Jacobson CA, Miklos DB, Lekakis LJ, Oluwole OO, et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(1):31–42.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–52.
Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–24.
Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16.
Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. 2022;22(10):557–75.
Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. “Off-the-shelf” allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19(3):185–99.
Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9(5):495–502.
Malmberg KJ, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren HG. Natural killer cell-mediated immunosurveillance of human cancer. Semin Immunol. 2017;31:20–9.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382(6):545–53.
Wrona E, Borowiec M, Potemski P. CAR-NK cells in the treatment of solid tumors. Int J Mol Sci. 2021;22(11):5899.
Zhang L, Meng Y, Feng X, Han Z. CAR-NK cells for cancer immunotherapy: from bench to bedside. Biomark Res. 2022;10:12.
Marofi F, Saleh MM, Rahman HS, Suksatan W, Al-Gazally ME, Abdelbasset WK, et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res Ther. 2021;12:374.
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, et al. Innate lymphoid cells: 10 years on. Cell. 2018;174(5):1054–66.
Freud AG, Yu J, Caligiuri MA. Human natural killer cell development in secondary lymphoid tissues. Semin Immunol. 2014;26(2):132–7.
Scoville SD, Freud AG, Caligiuri MA. Modeling human natural killer cell development in the era of innate lymphoid cells. Front Immunol. 2017;8:360.
Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. 2019;10:2078.
Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, et al. Tissue determinants of human NK cell development, function, and residence. Cell. 2020;180(4):749-763.e13.
Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. 2017;47(5):820–33.
Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. 2017;127(11):4042–58.
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97(10):3146–51.
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22(11):633–40.
Berrien-Elliott MM, Jacobs MT, Fehniger TA. Allogeneic natural killer cell therapy. Blood. 2023;141(8):856–68.
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, et al. High-dimensional single-cell analysis identifies organ-specific signatures and conserved NK cell subsets in humans and mice. Immunity. 2018;49(5):971-986.e5.
Brownlie D, Scharenberg M, Mold JE, Hård J, Kekäläinen E, Buggert M, et al. Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood. Proc Natl Acad Sci USA. 2021;118(11): e2016580118.
Myers JA, Miller JS. Exploring the NK cell platform for cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(2):85–100.
Yokoyama WM, Kim S. How do natural killer cells find self to achieve tolerance? Immunity. 2006;24(3):249–57.
Dhatchinamoorthy K, Colbert JD, Rock KL. Cancer immune evasion through loss of MHC class I antigen presentation. Front Immunol. 2021;12: 636568.
Maskalenko NA, Zhigarev D, Campbell KS. Harnessing natural killer cells for cancer immunotherapy: dispatching the first responders. Nat Rev Drug Discov. 2022;21(8):559–77.
Joncker NT, Shifrin N, Delebecque F, Raulet DH. Mature natural killer cells reset their responsiveness when exposed to an altered MHC environment. J Exp Med. 2010;207(10):2065–72.
Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8(9):713–25.
Gwalani LA, Orange JS. Single Degranulations in NK cells can mediate target cell killing. J Immunol. 2018;200(9):3231–43.
Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216(9):2113–27.
Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015;6:368.
Wang R, Jaw JJ, Stutzman NC, Zou Z, Sun PD. Natural killer cell-produced IFN-γ and TNF-α induce target cell cytolysis through up-regulation of ICAM-1. J Leukoc Biol. 2012;91(2):299–309.
Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18(11):671–88.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell. 2018;172(5):1022-1037.e14.
Kyrysyuk O, Wucherpfennig KW. Designing cancer immunotherapies that engage T cells and NK cells. Annu Rev Immunol. 2023;41:17–38.
Lizana-Vasquez GD, Torres-Lugo M, Dixon RB, Powderly JD, Warin RF. The application of autologous cancer immunotherapies in the age of memory-NK cells. Front Immunol. 2023;14:1167666.
Nahi H, Chrobok M, Meinke S, Gran C, Marquardt N, Afram G, et al. Autologous NK cells as consolidation therapy following stem cell transplantation in multiple myeloma. Cell Rep Med. 2022;3(2): 100508.
Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The rise of allogeneic natural killer cells as a platform for cancer immunotherapy: recent innovations and future developments. Front Immunol. 2017;8:631.
Parkhurst MR, Riley JP, Dudley ME, Rosenberg SA. Adoptive transfer of autologous natural killer cells leads to high levels of circulating natural killer cells but does not mediate tumor regression. Clin Cancer Res. 2011;17(19):6287–97.
Daher M, Melo Garcia L, Li Y, Rezvani K. CAR-NK cells: the next wave of cellular therapy for cancer. Clin Transl Immunol. 2021;10(4): e1274.
Strauss-Albee DM, Fukuyama J, Liang EC, Yao Y, Jarrell JA, Drake AL, et al. Human NK cell repertoire diversity reflects immune experience and correlates with viral susceptibility. Sci Transl Med. 2015;7(297):297ra115.
Martín-Antonio B, Suñe G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural killer cells: angels and devils for immunotherapy. Int J Mol Sci. 2017;18(9):1868.
Lundqvist A, Kremer V. Markers and function of human NK cells in normal and pathological conditions. Cytom B Clin Cytom. 2017;92(2):98–9.
Béziat V, Descours B, Parizot C, Debré P, Vieillard V. NK cell terminal differentiation: correlated stepwise decrease of NKG2A and acquisition of KIRs. PLoS ONE. 2010;5(8): e11966.
Kotylo PK, Baenzinger JC, Yoder MC, Engle WA, Bolinger CD. Rapid analysis of lymphocyte subsets in cord blood. Am J Clin Pathol. 1990;93(2):263–6.
Domogala A, Madrigal JA, Saudemont A. Cryopreservation has no effect on function of natural killer cells differentiated in vitro from umbilical cord blood CD34(+) cells. Cytotherapy. 2016;18(6):754–9.
Eapen M, Rubinstein P, Zhang MJ, Stevens C, Kurtzberg J, Scaradavou A, et al. Outcomes of transplantation of unrelated donor umbilical cord blood and bone marrow in children with acute leukaemia: a comparison study. Lancet. 2007;369(9577):1947–54.
Atsuta Y, Suzuki R, Nagamura-Inoue T, Taniguchi S, Takahashi S, Kai S, et al. Disease-specific analyses of unrelated cord blood transplantation compared with unrelated bone marrow transplantation in adult patients with acute leukemia. Blood. 2009;113(8):1631–8.
Lamerskok N, Panella D, Georgoudaki AM, Liu H, Özkazanc D, Kučerová L, et al. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J Hematol Oncol. 2022;15(1):164.
Goldenson BH, Zhu H, Wang YM, Heragu N, Bernareggi D, Ruiz-Cisneros A, et al. Umbilical cord blood and iPSC-derived natural killer cells demonstrate key differences in cytotoxic activity and KIR profiles. Front Immunol. 2020;11: 561553.
Fang F, Xie S, Chen M, Li Y, Yue J, Ma J, et al. Advances in NK cell production. Cell Mol Immunol. 2022;19(4):460–81.
Harris DT, Schumacher MJ, Locascio J, Besencon FJ, Olson GB, DeLuca D, et al. Phenotypic and functional immaturity of human umbilical cord blood T lymphocytes. Proc Natl Acad Sci USA. 1992;89(21):10006–10.
Sarvaria A, Jawdat D, Madrigal JA, Saudemont A. Umbilical cord blood natural killer cells, their characteristics, and potential clinical applications. Front Immunol. 2017;8:329.
Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. 2012;73(3):248–57.
Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. 2010;116(19):3853–64.
Damele L, Spaggiari GM, Parodi M, Mingari MC, Vitale M, Vitale C. Cord blood-derived natural killer cell exploitation in immunotherapy protocols: more than a promise? Cancers (Basel). 2022;14(18):4439.
Roex G, Campillo-Davo D, Flumens D, Shaw PAG, Krekelbergh L, De Reu H, et al. Two for one: targeting BCMA and CD19 in B-cell malignancies with off-the-shelf dual-CAR NK-92 cells. J Transl Med. 2022;20(1):124.
Cao B, Liu M, Huang J, Zhou J, Li J, Lian H, et al. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int J Biol Sci. 2021;17(14):3850–61.
Hong S, Yu C, Wang P, Shi Y, Cao W, Cheng B, et al. Glycoengineering of NK cells with glycan ligands of CD22 and selectins for B-cell lymphoma therapy. Angew Chem Int Ed Engl. 2021;60(7):3603–10.
Yang S, Cao B, Zhou G, Zhu L, Wang L, Zhang L, et al. Targeting B7-H3 immune checkpoint with chimeric antigen receptor-engineered natural killer cells exhibits potent cytotoxicity against non-small cell lung cancer. Front Pharmacol. 2020;11:1089.
Yagita M, Huang CL, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14(5):922–30.
Han J, Chu J, Keung Chan W, Zhang J, Wang Y, Cohen JB, et al. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci Rep. 2015;5:11483.
Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res. 2018;8(6):1083–9.
Gong JH, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8(4):652–8.
Suck G, Odendahl M, Nowakowska P, Seidl C, Wels WS, Klingemann HG, et al. NK-92: an “off-the-shelf therapeutic” for adoptive natural killer cell-based cancer immunotherapy. Cancer Immunol Immunother. 2016;65(4):485–92.
Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. 2018;2018:4054815.
Snyder KM, Hullsiek R, Mishra HK, Mendez DC, Li Y, Rogich A, et al. Expression of a recombinant high affinity IgG Fc receptor by engineered NK cells as a docking platform for therapeutic mAbs to target cancer cells. Front Immunol. 2018;9:2873.
Bernarreggi D, Pouyanfard S, Kaufman DS. Development of innate immune cells from human pluripotent stem cells. Exp Hematol. 2019;71:13–23.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJN, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2(4):274–83.
Woll PS, Martin CH, Miller JS, Kaufman DS. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J Immunol. 2005;175(8):5095–103.
Lehmann D, Spanholtz J, Osl M, Tordoir M, Lipnik K, Bilban M, et al. Ex vivo generated natural killer cells acquire typical natural killer receptors and display a cytotoxic gene expression profile similar to peripheral blood natural killer cells. Stem Cells Dev. 2012;21(16):2926–38.
Woll PS, Grzywacz B, Tian X, Marcus RK, Knorr DA, Verneris MR, et al. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent in vivo antitumor activity. Blood. 2009;113(24):6094–101.
Zhu H, Blum RH, Bjordahl R, Gaidarova S, Rogers P, Lee TT, et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood. 2020;135(6):399–410.
Bachanova V, Ghobadi A, Patel K, Park JH, Flinn IW, Shah P, et al. Safety and efficacy of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood. 2021;138(Supplement 1):823.
Bjordahl R, Gaidarova S, Goodridge JP, Mahmood S, Bonello G, Robinson M, et al. FT576: a novel multiplexed engineered off-the-shelf natural killer cell immunotherapy for the dual-targeting of CD38 and Bcma for the treatment of multiple myeloma. Blood. 2019;134(Supplement_1):3214.
Patel K, Bachanova V, Goodman AM, Pagel JM, Griffis K, Anderson M, et al. Phase I study of FT516, an off-the-shelf iPSC-derived NK cell therapy, in combination with rituximab in patients with relapsed/refractory B-cell lymphoma. Blood. 2021;138(Supplement 1):3873.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23(2):181-192.e5.
Karagiannis P, Kim SI. iPSC-derived natural killer cells for cancer immunotherapy. Mol Cells. 2021;44(8):541–8.
Ran GH, Lin YQ, Tian L, Zhang T, Yan DM, Yu JH, et al. Natural killer cell homing and trafficking in tissues and tumors: from biology to application. Signal Transduct Target Ther. 2022;7(1):205.
Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer. 2020;20(8):437–54.
Moreira A, Alari-Pahissa E, Munteis E, Vera A, Zabalza A, Llop M, et al. Adaptive features of natural killer cells in multiple sclerosis. Front Immunol. 2019;10:2403.
O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH. T cell- and B cell-independent adaptive immunity mediated by natural killer cells. Nat Immunol. 2006;7(5):507–16.
Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457(7229):557–61.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, et al. Cytokine activation induces human memory-like NK cells. Blood. 2012;120(24):4751–60.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8(357):357ra123.
Berrien-Elliott MM, Cashen AF, Cubitt CC, Neal CC, Wong P, Wagner JA, et al. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 2020;10(12):1854–71.
Shapiro RM, Birch GC, Hu G, Vergara Cadavid J, Nikiforow S, Baginska J, et al. Expansion, persistence, and efficacy of donor memory-like NK cells infused for posttransplant relapse. J Clin Invest. 2022;132(11): e154334.
Sullivan R, Mathyer M, Govero J, Dean J, Martens A, Zhou Y, et al. 188 development of WU-NK-101, a feeder cell-free expanded allogeneic memory NK cell product with potent anti-tumor activity. J Immunother Cancer. 2021;9(Suppl 2):A200.
Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115(21):4293–301.
Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130(16):1857–68.
Moretta A, Bottino C, Vitale M, Pende D, Biassoni R, Mingari MC, et al. Receptors for Hla class-I molecules in human natural killer cells. Annu Rev Immunol. 1996;14(1):619–48.
Mahaweni NM, Ehlers FAI, Bos GMJ, Wieten L. Tuning natural killer cell anti-multiple myeloma reactivity by targeting inhibitory signaling via KIR and NKG2A. Front Immunol. 2018;9:2848.
Hunter BD, Jacobson CA. CAR T-cell associated neurotoxicity: mechanisms, clinicopathologic correlates, and future directions. J Natl Cancer Inst. 2019;111(7):646–54.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Rc L, Mv M. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61.
Milone MC, Xu J, Chen SJ, Collins MA, Zhou J, Powell DJ, et al. Engineering enhanced CAR T-cells for improved cancer therapy. Nat Cancer. 2021;2(8):780–93.
Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, et al. Nanobody-based CAR-T cells for cancer immunotherapy. Biomark Res. 2022;10:24.
Gong L, Li Y, Cui K, Chen Y, Hong H, Li J, et al. Nanobody-engineered natural killer cell conjugates for solid tumor adoptive immunotherapy. Small. 2021;17(45): e2103463.
Gong Y, Klein Wolterink RGJ, Wang J, Bos GMJ, Germeraad WTV. Chimeric antigen receptor natural killer (CAR-NK) cell design and engineering for cancer therapy. J Hematol Oncol. 2021;14:73.
Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195.
Yilmaz A, Cui H, Caligiuri MA, Yu J. Chimeric antigen receptor-engineered natural killer cells for cancer immunotherapy. J Hematol Oncol. 2020;13(1):168.
Abreu TR, Fonseca NA, Gonçalves N, Moreira JN. Current challenges and emerging opportunities of CAR-T cell therapies. J Control Release. 2020;319:246–61.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–98.
Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-cells forward. Nat Rev Clin Oncol. 2016;13(6):370–83.
Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood Rev. 2016;30(3):157–67.
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–54.
Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71(17):5697–706.
Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med. 2018;24(3):352–9.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32(2):520–31.
Majzner RG, Mackall CL. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 2018;8(10):1219–26.
Cronk RJ, Zurko J, Shah NN. Bispecific chimeric antigen receptor T cell therapy for B cell malignancies and multiple myeloma. Cancers (Basel). 2020;12(9):2523.
Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–6.
Li Y, Basar R, Wang G, Liu E, Moyes JS, Li L, et al. KIR-based inhibitory CARs overcome CAR-NK cell trogocytosis-mediated fratricide and tumor escape. Nat Med. 2022;28(10):2133–44.
Ruppel KE, Fricke S, Köhl U, Schmiedel D. Taking lessons from CAR-T cells and going beyond: tailoring design and signaling for CAR-NK cells in cancer therapy. Front Immunol. 2022;13: 822298.
Tran AC, Zhang D, Byrn R, Roberts MR. Chimeric zeta-receptors direct human natural killer (NK) effector function to permit killing of NK-resistant tumor cells and HIV-infected T lymphocytes. J Immunol. 1995;155(2):1000–9.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194(7):3201–12.
Chang YH, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73(6):1777–86.
Peng Y, Zhang W, Chen Y, Zhang L, Shen H, Wang Z, et al. Engineering c-Met-CAR NK-92 cells as a promising therapeutic candidate for lung adenocarcinoma. Pharmacol Res. 2023;188: 106656.
Wang J, Lupo KB, Chambers AM, Matosevic S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J Immunother Cancer. 2018;6(1):136.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27(6):1114–25.
Baghery Saghchy Khorasani A, Yousefi AM, Bashash D. CAR NK cell therapy in hematologic malignancies and solid tumors; obstacles and strategies to overcome the challenges. Int Immunopharmacol. 2022;110: 109041.
Müller S, Bexte T, Gebel V, Kalensee F, Stolzenberg E, Hartmann J, et al. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front Immunol. 2020;10:3123.
Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2020;34(4):1102–15.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137(5):624–36.
Boissel L, Betancur-Boissel M, Lu W, Krause DS, Van Etten RA, Wels WS, et al. Retargeting NK-92 cells by means of CD19- and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2(10): e26527.
Ureña-Bailén G, Dobrowolski JM, Hou Y, Dirlam A, Roig-Merino A, Schleicher S, et al. Preclinical evaluation of CRISPR-edited CAR-NK-92 cells for off-the-shelf treatment of AML and B-ALL. Int J Mol Sci. 2022;23(21):12828.
Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145(7):1935–45.
Müller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother. 2008;57(3):411–23.
Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell non-Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3(4):333–44.
Chu Y, Yahr A, Huang B, Ayello J, Barth M, Cairo MS. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology. 2017;6(9): e1341031.
Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. 2016;7(35):56219–32.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8(68):112783–96.
Voynova E, Hawk N, Flomerfelt FA, Telford WG, Gress RE, Kanakry JA, et al. Increased activity of a NK-specific CAR-NK framework targeting CD3 and CD5 for T-cell leukemias. Cancers (Basel). 2022;14(3):524.
Chen KH, Wada M, Pinz KG, Liu H, Lin KW, Jares A, et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31(10):2151–60.
Raikar SS, Fleischer LC, Moot R, Fedanov A, Paik NY, Knight KA, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology. 2017;7(3): e1407898.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12(1):49.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9(1):64–78.
Tassev DV, Cheng M, Cheung NK. Retargeting NK92 cells using an HLA-A2-restricted, EBNA3C-specific chimeric antigen receptor. Cancer Gene Ther. 2012;19(2):84–100.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8(2):297–310.
Luanpitpong S, Poohadsuan J, Klaihmon P, Issaragrisil S. Selective cytotoxicity of single and dual anti-CD19 and anti-CD138 chimeric antigen receptor-natural killer cells against hematologic malignancies. J Immunol Res. 2021;2021:5562630.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28(4):917–27.
Leivas A, Valeri A, Córdoba L, García-Ortiz A, Ortiz A, Sánchez-Vega L, et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021;11(8):146.
Du Z, Ng YY, Zha S, Wang S. piggyBac system to co-express NKG2D CAR and IL-15 to augment the in vivo persistence and anti-AML activity of human peripheral blood NK cells. Mol Ther Methods Clin Dev. 2021;23:582–96.
Albinger N, Pfeifer R, Nitsche M, Mertlitz S, Campe J, Stein K, et al. Primary CD33-targeting CAR-NK cells for the treatment of acute myeloid leukemia. Blood Cancer J. 2022;12(4):61.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2022;107(2):437.
Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28(10):897–913.
Caruso S, De Angelis B, Del Bufalo F, Ciccone R, Donsante S, Volpe G, et al. Safe and effective off-the-shelf immunotherapy based on CAR.CD123-NK cells for the treatment of acute myeloid leukaemia. J Hematol Oncol. 2022;15(1):163.
Morgan MA, Kloos A, Lenz D, Kattre N, Nowak J, Bentele M, et al. Improved activity against acute myeloid leukemia with chimeric antigen receptor (CAR)-NK-92 cells designed to target CD123. Viruses. 2021;13(7):1365.
Dong H, Ham JD, Hu G, Xie G, Vergara J, Liang Y, et al. Memory-like NK cells armed with a neoepitope-specific CAR exhibit potent activity against NPM1 mutated acute myeloid leukemia. Proc Natl Acad Sci USA. 2022;119(25): e2122379119.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19(2):235–49.
Altvater B, Landmeier S, Pscherer S, Temme J, Schweer K, Kailayangiri S, et al. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin Cancer Res. 2009;15(15):4857–66.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106(1):376–83.
Soldierer M, Bister A, Haist C, Thivakaran A, Cengiz SC, Sendker S, et al. Genetic engineering and enrichment of human NK cells for CAR-enhanced immunotherapy of hematological malignancies. Front Immunol. 2022;13: 847008.
Christodoulou I, Ho WJ, Marple A, Ravich JW, Tam A, Rahnama R, et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J Immunother Cancer. 2021;9(12): e003894.
Ravi D, Sarkar S, Purvey S, Passero F, Beheshti A, Chen Y, et al. Interaction kinetics with transcriptomic and secretory responses of CD19 CAR natural killer cell therapy in CD20 resistant non-Hodgkin lymphoma. Leukemia. 2020;34(5):1291–304.
Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using IL-2 diphtheria toxin fusion protein. Blood. 2014;123(25):3855–63.
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139.
Goodridge JP, Mahmood S, Zhu H, Gaidarova S, Blum R, Bjordahl R, et al. FT596: translation of first-of-kind multi-antigen targeted off-the-shelf CAR-NK cell with engineered persistence for the treatment of B cell malignancies. Blood. 2019;134(Supplement_1):301–301.
Fate therapeutics showcases positive interim phase 1 data from FT596 off-the-shelf, iPSC-derived CAR NK cell program for relapsed/refractory B-cell lymphoma at 2021 ASH annual meeting | Fate Therapeutics, Inc. https://ir.fatetherapeutics.com/news-releases/news-release-details/fate-therapeutics-showcases-positive-interim-phase-1-data-ft596. Accessed 17 June 2023.
Alcantara M, Tesio M, June CH, Houot R. CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia. 2018;32(11):2307–15.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11(1):45–58.
Qu C, Zhang H, Cao H, Tang L, Mo H, Liu F, et al. Tumor buster—where will the CAR-T cell therapy ‘missile’ go? Mol Cancer. 2022;21(1):201.
Kloess S, Oberschmidt O, Dahlke J, Vu XK, Neudoerfl C, Kloos A, et al. Preclinical assessment of suitable natural killer cell sources for chimeric antigen receptor natural killer-based “off-the-shelf” acute myeloid leukemia immunotherapies. Hum Gene Ther. 2019;30(4):381–401.
Development and evaluation of NK-CD123 CAR against high risk acute myeloid leukemia | Semantic Scholar. https://www.semanticscholar.org/paper/Development-and-Evaluation-of-NK-CD123-CAR-Against-Sinha-Seth/c393be76c4cf6a6b291ae71f6748a958aa4bb23e. Accessed 15 June 2022.
Drent E, Groen RWJ, Noort WA, Themeli M, Lammerts van Bueren JJ, Parren PWHI, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. 2016;101(5):616–25.
Mihara K, Yanagihara K, Takigahira M, Imai C, Kitanaka A, Takihara Y, et al. Activated T-cell-mediated immunotherapy with a chimeric receptor against CD38 in B-cell non-Hodgkin lymphoma. J Immunother. 2009;32(7):737–43.
Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16(2):112–23.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209(13):2351–65.
Falini B, Tiacci E, Martelli MP, Ascani S, Pileri SA. New classification of acute myeloid leukemia and precursor-related neoplasms: changes and unsolved issues. Discov Med. 2010;10(53):281–92.
Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572(7768):254–9.
2004-FT57676.pdf. https://fatetherapeutics.com/wp-content/uploads/2022/12/2004-FT57676.pdf. Accessed 12 Mar 2023.
Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–50.
Zhang C, Burger MC, Jennewein L, Genßler S, Schönfeld K, Zeiner P, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;108(5):djv375. https://doi.org/10.1093/jnci/djv375.
Schönfeld K, Sahm C, Zhang C, Naundorf S, Brendel C, Odendahl M, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8.
Portillo AL, Hogg R, Poznanski SM, Rojas EA, Cashell NJ, Hammill JA, et al. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. iScience. 2021;24(6): 102619.
Izawa S, Kono K, Mimura K, Kawaguchi Y, Watanabe M, Maruyama T, et al. H2O2 production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: possible mechanisms of NK cell dysfunction. Cancer Immunol Immunother. 2011;60(12):1801–10.
Harlin H, Hanson M, Johansson CC, Sakurai D, Poschke I, Norell H, et al. The CD16− CD56(bright) NK cell subset is resistant to reactive oxygen species produced by activated granulocytes and has higher antioxidative capacity than the CD16+ CD56(dim) subset. J Immunol. 2007;179(7):4513–9.
Xu H, Zhao H, Ding C, Jiang D, Zhao Z, Li Y, et al. Celastrol suppresses colorectal cancer via covalent targeting peroxiredoxin 1. Signal Transduct Target Ther. 2023;8:51.
Klopotowska M, Bajor M, Graczyk-Jarzynka A, Kraft A, Pilch Z, Zhylko A, et al. PRDX-1 supports the survival and antitumor activity of primary and CAR-modified NK cells under oxidative stress. Cancer Immunol Res. 2022;10(2):228–44.
Xia W, Chen J, Hou W, Chen J, Xiong Y, Li H, et al. Engineering a HER2-CAR-NK cell secreting soluble programmed cell death protein with superior antitumor efficacy. Int J Mol Sci. 2023;24(7):6843.
Nowakowska P, Romanski A, Miller N, Odendahl M, Bonig H, Zhang C, et al. Clinical grade manufacturing of genetically modified, CAR-expressing NK-92 cells for the treatment of ErbB2-positive malignancies. Cancer Immunol Immunother. 2018;67(1):25–38.
Oprita A, Baloi SC, Staicu GA, Alexandru O, Tache DE, Danoiu S, et al. Updated insights on EGFR signaling pathways in glioma. Int J Mol Sci. 2021;22(2):E587.
An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene. 2018;37(12):1561–75.
Murakami T, Nakazawa T, Natsume A, Nishimura F, Nakamura M, Matsuda R, et al. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018;38(9):5049–56.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, Schmitz M, Schackert G, Pastan I, Temme A. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38(5):197.
Ma R, Lu T, Li Z, Teng KY, Mansour AG, Yu M, et al. An oncolytic virus expressing IL15/IL15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021;81(13):3635–48.
Uherek C, Tonn T, Uherek B, Becker S, Schnierle B, Klingemann HG, et al. Retargeting of natural killer-cell cytolytic activity to ErbB2-expressing cancer cells results in efficient and selective tumor cell destruction. Blood. 2002;100(4):1265–73.
Cao B, Liu M, Wang L, Liang B, Feng Y, Chen X, et al. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem Biophys Res Commun. 2020;524(1):96–102.
Ao X, Yang Y, Li W, Tan Y, Guo W, Ao L, et al. Anti-αFR CAR-engineered NK-92 cells display potent cytotoxicity against αFR-positive ovarian cancer. J Immunother. 2019;42(8):284–96.
Klapdor R, Wang S, Hacker U, Büning H, Morgan M, Dörk T, et al. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28(10):886–96.
Klapdor R, Wang S, Morgan M, Dörk T, Hacker U, Hillemanns P, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. 2019;20(3):E660.
Du H, Yang X, Fan J, Du X. Claudin 6: therapeutic prospects for tumours, and mechanisms of expression and regulation (Review). Mol Med Rep. 2021;24(3):677.
Lee YE, Ju A, Choi HW, Kim JC, Kim EE, Kim TS, et al. Rationally designed redirection of natural killer cells anchoring a cytotoxic ligand for pancreatic cancer treatment. J Control Release. 2020;326:310–23.
Gao H, Li K, Tu H, Pan X, Jiang H, Shi B, et al. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin Cancer Res. 2014;20(24):6418–28.
Fu SJ, Qi CY, Xiao WK, Li SQ, Peng BG, Liang LJ. Glypican-3 is a potential prognostic biomarker for hepatocellular carcinoma after curative resection. Surgery. 2013;154(3):536–44.
Yu M, Luo H, Fan M, Wu X, Shi B, Di S, et al. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol Ther. 2018;26(2):366–78.
Tseng HC, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11(1):4810.
Shiozawa M, Chang CH, Huang YC, Chen YC, Chi MS, Hao HC, et al. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018;19:27.
Genßler S, Burger MC, Zhang C, Oelsner S, Mildenberger I, Wagner M, et al. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology. 2015;5(4): e1119354.
Wang J, Toregrosa-Allen S, Elzey BD, Utturkar S, Lanman NA, Bernal-Crespo V, et al. Multispecific targeting of glioblastoma with tumor microenvironment-responsive multifunctional engineered NK cells. Proc Natl Acad Sci USA. 2021;118(45): e2107507118.
Chaudhry K, Geiger A, Dowlati E, Lang H, Sohai DK, Hwang EI, et al. Co-transducing B7H3 CAR-NK cells with the DNR preserves their cytolytic function against GBM in the presence of exogenous TGF-β. Mol Ther Methods Clin Dev. 2022;27:415–30.
Lin YZ, Lee CC, Cho DY, Wang YL, Chen CY, Weng CY, et al. Suppression of breast cancer cells resistant to a pure anti-estrogen with CAR-transduced natural killer cells. Am J Cancer Res. 2021;11(9):4455–69.
Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61.
Liu H, Yang B, Sun T, Lin L, Hu Y, Deng M, et al. Specific growth inhibition of ErbB2-expressing human breast cancer cells by genetically modified NK-92 cells. Oncol Rep. 2015;33(1):95–102.
Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, Chen L, Wang Y, Wang H, Yi L, Elder JB. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7(19):27764.
Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci. 2020;111(5):1478–90.
Klapdor R, Wang S, Morgan MA, Zimmermann K, Hachenberg J, Büning H, et al. NK cell-mediated eradication of ovarian cancer cells with a novel chimeric antigen receptor directed against CD44. Biomedicines. 2021;9(10):1339.
Wu L, Liu F, Yin L, Wang F, Shi H, Zhao Q, et al. The establishment of polypeptide PSMA-targeted chimeric antigen receptor-engineered natural killer cells for castration-resistant prostate cancer and the induction of ferroptosis-related cell death. Cancer Commun (Lond). 2022;42(8):768–83.
Wang F, Wu L, Yin L, Shi H, Gu Y, Xing N. Combined treatment with anti-PSMA CAR NK-92 cell and anti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin Transl Med. 2022;12(6): e901.
Montagner IM, Penna A, Fracasso G, Carpanese D, Dalla Pietà A, Barbieri V, et al. Anti-PSMA CAR-engineered NK-92 cells: an off-the-shelf cell therapy for prostate cancer. Cells. 2020;9(6):E1382.
Dai Y, Liu Y, Hu Y, Liu W, Ma J, Lu N, et al. STING agonist cGAMP enhances anti-tumor activity of CAR-NK cells against pancreatic cancer. Oncoimmunology. 2022;11(1):2054105.
Teng KY, Mansour AG, Zhu Z, Li Z, Tian L, Ma S, et al. Off-the-shelf prostate stem cell antigen-directed chimeric antigen receptor natural killer cell therapy to treat pancreatic cancer. Gastroenterology. 2022;162(4):1319–33.
Schnalzger TE, de Groot MH, Zhang C, Mosa MH, Michels BE, Röder J, et al. 3D model for CAR-mediated cytotoxicity using patient-derived colorectal cancer organoids. EMBO J. 2019;38(12): e100928.
Zhang Q, Zhang H, Ding J, Liu H, Li H, Li H, et al. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:4263520.
Liu M, Huang W, Guo Y, Zhou Y, Zhi C, Chen J, et al. CAR NK-92 cells targeting DLL3 kill effectively small cell lung cancer cells in vitro and in vivo. J Leukoc Biol. 2022;112(4):901–11.
Nilsson MB, Yang Y, Heeke S, Patel SA, Poteete A, Udagawa H, et al. CD70 is a therapeutic target upregulated in EMT-associated EGFR tyrosine kinase inhibitor resistance. Cancer Cell. 2023;41(2):340-355.e6.
Grote S, Chan KCH, Baden C, Bösmüller H, Sulyok M, Frauenfeld L, et al. CD276 as a novel CAR NK-92 therapeutic target for neuroblastoma. Adv Cell Gene Therapy. 2021;4(1): e105.
Zhang C, Röder J, Scherer A, Bodden M, Pfeifer Serrahima J, Bhatti A, et al. Bispecific antibody-mediated redirection of NKG2D-CAR natural killer cells facilitates dual targeting and enhances antitumor activity. J Immunother Cancer. 2021;9(10): e002980.
Jan CI, Huang SW, Canoll P, Bruce JN, Lin YC, Pan CM, et al. Targeting human leukocyte antigen G with chimeric antigen receptors of natural killer cells convert immunosuppression to ablate solid tumors. J Immunother Cancer. 2021;9(10): e003050.
Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med. 2012;16(3):569–81.
Kailayangiri S, Altvater B, Spurny C, Jamitzky S, Schelhaas S, Jacobs AH, et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology. 2017;6(1): e1250050.
Huang Y, Zeng J, Liu T, Xu Q, Song X, Zeng J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag Res. 2020;12:3247–55.
Hu Y, Tian ZG, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39(2):167–76.
Savan R, Chan T, Young HA. Lentiviral gene transduction in human and mouse NK cell lines. Methods Mol Biol. 2010;612:209–21.
Davis HE, Morgan JR, Yarmush ML. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys Chem. 2002;97(2–3):159–72.
Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J. 2004;86(2):1234–42.
Nanbakhsh A, Best B, Riese M, Rao S, Wang L, Medin J, et al. Dextran enhances the lentiviral transduction efficiency of murine and human primary NK cells. J Vis Exp. 2018;15(131):55063.
Hanenberg H, Xiao XL, Dilloo D, Hashino K, Kato I, Williams DA. Colocalization of retrovirus and target cells on specific fibronectin fragments increases genetic transduction of mammalian cells. Nat Med. 1996;2(8):876–82.
Fenard D, Ingrao D, Seye A, Buisset J, Genries S, Martin S, et al. Vectofusin-1, a new viral entry enhancer, strongly promotes lentiviral transduction of human hematopoietic stem cells. Mol Ther Nucleic Acids. 2013;2: e90.
Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G. Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med. 2004;199(12):1651–8.
Hemmi H, Takeuchi O, Sato S, Yamamoto M, Kaisho T, Sanjo H, et al. The roles of two IkappaB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med. 2004;199(12):1641–50.
Li L, Gao Y, Srivastava R, Wang W, Xiong Q, Fang Z, et al. Lentiviral delivery of combinatorial CAR/CRISPRi circuit into human primary T cells is enhanced by TBK1/IKKɛ complex inhibitor BX795. J Transl Med. 2020;18(1):363.
Sutlu T, Nyström S, Gilljam M, Stellan B, Applequist SE, Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum Gene Ther. 2012;23(10):1090–100.
Micucci F, Zingoni A, Piccoli M, Frati L, Santoni A, Galandrini R. High-efficient lentiviral vector-mediated gene transfer into primary human NK cells. Exp Hematol. 2006;34(10):1344–52.
Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013;110(18):7306–11.
Bari R, Granzin M, Tsang KS, Roy A, Krueger W, Orentas R, et al. A Distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front Immunol. 2019;10:2001.
Gong Y, Klein Wolterink RGJ, Janssen I, Groot AJ, Bos GMJ, Germeraad WTV. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol Ther Methods Clin Dev. 2020;17:634–46.
Amirache F, Lévy C, Costa C, Mangeot PE, Torbett BE, Wang CX, et al. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood. 2014;123(9):1422–4.
Colamartino ABL, Lemieux W, Bifsha P, Nicoletti S, Chakravarti N, Sanz J, et al. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front Immunol. 2019;10:2873.
Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–42.
Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–50.
Imamura M, Shook D, Kamiya T, Shimasaki N, Chai SMH, Coustan-Smith E, et al. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124(7):1081–8.
Streltsova MA, Barsov E, Erokhina SA, Kovalenko EI. Retroviral gene transfer into primary human NK cells activated by IL-2 and K562 feeder cells expressing membrane-bound IL-21. J Immunol Methods. 2017;450:90–4.
Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17(3):147–54.
Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266.
Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019;9:18729.
Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53(5):958–65.
Ingegnere T, Mariotti FR, Pelosi A, Quintarelli C, De Angelis B, Tumino N, et al. Human CAR NK cells: a new non-viral method allowing high efficient transfection and strong tumor cell killing. Front Immunol. 2019;10:957.
Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy. 2012;14(7):830–40.
Ren R, Guo J, Liu G, Kang H, Machens HG, Schilling AF, et al. Nucleic acid direct delivery to fibroblasts: a review of nucleofection and applications. J Biol Eng. 2022;16:30.
Elmacken M, Awasthi A, Ayello J, van de Ven C, Luo W, Liao Y, et al. Neuroblastoma and Ewing’s sarcoma associated with ROR1 expression can be effectively targeted with NK cells modified to express an anti ROR1 chimeric antigen receptor. Biol Blood Marrow Transplant. 2015;21(2):S95–7.
Batista Napotnik T, Polajžer T, Miklavčič D. Cell death due to electroporation—a review. Bioelectrochemistry. 2021;141: 107871.
Kebriaei P, Izsvák Z, Narayanavari SA, Singh H, Ivics Z. Gene therapy with the sleeping beauty transposon system. Trends Genet. 2017;33(11):852–70.
Yusa K. piggyBac Transposon. Microbiol Spectr. 2015;3(2):MDNA3-0028-2014.
Woodard LE, Wilson MH. piggyBac-ing models and new therapeutic strategies. Trends Biotechnol. 2015;33(9):525–33.
Mátés L, Chuah MKL, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, et al. Molecular evolution of a novel hyperactive sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;41(6):753–61.
Kumar D, Anand T, Talluri TR, Kues WA. Potential of transposon-mediated cellular reprogramming towards cell-based therapies. World J Stem Cells. 2020;12(7):527–44.
Skipper KA, Andersen PR, Sharma N, Mikkelsen JG. DNA transposon-based gene vehicles—scenes from an evolutionary drive. J Biomed Sci. 2013;20(1):92.
Nakazawa Y, Huye LE, Salsman VS, Leen AM, Ahmed N, Rollins L, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol Ther. 2011;19(12):2133–43.
Kawakami K, Largaespada DA, Ivics Z. Transposons as tools for functional genomics in vertebrate models. Trends Genet. 2017;33(11):784–801.
Hodge R, Narayanavari SA, Izsvák Z, Ivics Z. Wide awake and ready to move: 20 years of non-viral therapeutic genome engineering with the sleeping beauty transposon system. Hum Gene Ther. 2017;28(10):842–55.
Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Invest. 2020;130(11):6021–33.
Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with sleeping beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther. 2022;30(10):3155–75.
Batchu RB, Gruzdyn OV, Tavva PS, Kolli BK, Dachepalli R, Weaver DW, et al. Engraftment of mesothelin chimeric antigen receptor using a hybrid sleeping beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery. 2019;166(4):503–8.
Huang X, Haley K, Wong M, Guo H, Lu C, Wilber A, et al. Unexpectedly high copy number of random integration but low frequency of persistent expression of the sleeping beauty transposase after trans delivery in primary human T cells. Hum Gene Ther. 2010;21(11):1577–90.
Liang Q, Kong J, Stalker J, Bradley A. Chromosomal mobilization and reintegration of sleeping beauty and PiggyBac transposons. Genesis. 2009;47(6):404–8.
Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-cell engineering using non-viral sleeping beauty transposition from minicircle vectors. Leukemia. 2017;31(1):186–94.
Elmas E, Saljoughian N, de Souza Fernandes Pereira M, Tullius BP, Sorathia K, Nakkula RJ, et al. CRISPR gene editing of human primary NK and T cells for cancer immunotherapy. Front Oncol. 2022;12: 834002.
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–21.
Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long terminal repeat CRISPR-CAR-coupled “universal” T cells mediate potent anti-leukemic effects. Mol Ther. 2018;26(5):1215–27.
Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559(7714):405–9.
Müller TR, Jarosch S, Hammel M, Leube J, Grassmann S, Bernard B, et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep Med. 2021;2(8): 100374.
Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A genetically engineered primary human natural killer cell platform for cancer immunotherapy. Mol Ther. 2020;28(1):52–63.
Eyquem J, Mansilla-Soto J, Giavridis T, Van Der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7.
Naeimi Kararoudi M, Likhite S, Elmas E, Schwartz M, Sorathia K, Yamamoto K, et al. CD33 targeting primary CAR-NK cells generated by CRISPR mediated gene insertion show enhanced anti-AML activity. Blood. 2020;136:3.
McKinlay CJ, Benner NL, Haabeth OA, Waymouth RM, Wender PA. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc Natl Acad Sci USA. 2018;115(26):E5859–66.
Wilk AJ, Weidenbacher NLB, Vergara R, Haabeth OAW, Levy R, Waymouth RM, et al. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Adv. 2020;4(17):4244–55.
Billingsley MM, Singh N, Ravikumar P, Zhang R, June CH, Mitchell MJ. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020;20(3):1578–89.
Becker PSA, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, et al. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65(4):477–84.
Granzin M, Wagner J, Köhl U, Cerwenka A, Huppert V, Ullrich E. Shaping of natural killer cell antitumor activity by ex vivo cultivation. Front Immunol. 2017;8:458.
Boieri M, Ulvmoen A, Sudworth A, Lendrem C, Collin M, Dickinson AM, et al. IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. Oncoimmunology. 2017;6(3): e1274478.
Jacobs B, Pfefferle A, Clement D, Berg-Larsen A, Saetersmoen ML, Lorenz S, et al. Induction of the BIM short splice variant sensitizes proliferating NK cells to IL-15 withdrawal. J Immunol. 2019;202(3):736–46.
Kiani Z, Dupuy FP, Bruneau J, Lebouché B, Zhang CX, Jackson E, et al. HLA-F on HLA-Null 721.221 cells activates primary NK cells expressing the activating killer Ig-like receptor KIR3DS1. J Immunol. 2018;201(1):113–23.
Ojo EO, Sharma AA, Liu R, Moreton S, Checkley-Luttge MA, Gupta K, et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci Rep. 2019;9(1):14916.
Liu LL, Béziat V, Oei VYS, Pfefferle A, Schaffer M, Lehmann S, et al. Ex vivo expanded adaptive NK cells effectively kill primary acute lymphoblastic leukemia cells. Cancer Immunol Res. 2017;5(8):654–65.
Gurney M, Kundu S, Pandey S, O’Dwyer M. Feeder cells at the interface of natural killer cell activation, expansion and gene editing. Front Immunol. 2022;13: 802906.
Abdolahi S, Ghazvinian Z, Muhammadnejad S, Ahmadvand M, Aghdaei HA, Ebrahimi-Barough S, et al. Adaptive NK cell therapy modulated by anti-PD-1 antibody in gastric cancer model. Front Pharmacol. 2021;12: 733075.
Kweon S, Phan MTT, Chun S, Yu H, Kim J, Kim S, et al. Expansion of human NK cells using K562 cells expressing OX40 ligand and short exposure to IL-21. Front Immunol. 2019;10:879.
Lu T, Ma R, Dong W, Teng KY, Kollath DS, Li Z, et al. Off-the-shelf CAR natural killer cells secreting IL-15 target spike in treating COVID-19. Nat Commun. 2022;13(1):2576.
Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, et al. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79(12):2320–8.
Ishigami S, Natsugoe S, Tokuda K, Nakajo A, Che X, Iwashige H, et al. Prognostic value of intratumoral natural killer cells in gastric carcinoma. Cancer. 2000;88(3):577–83.
Donskov F, von der Maase H. Impact of immune parameters on long-term survival in metastatic renal cell carcinoma. J Clin Oncol. 2006;24(13):1997–2005.
Alkins R, Burgess A, Kerbel R, Wels WS, Hynynen K. Early treatment of HER2-amplified brain tumors with targeted NK-92 cells and focused ultrasound improves survival. Neuro Oncol. 2016;18(7):974–81.
Burger MC, Forster MT, Romanski A, Straßheimer F, Macas J, Zeiner PS, et al. Intracranial injection of NK cells engineered with a HER2-targeted chimeric antigen receptor in patients with recurrent glioblastoma. Neuro Oncol. 2023. https://doi.org/10.1093/neuonc/noad087.
Mlecnik B, Tosolini M, Charoentong P, Kirilovsky A, Bindea G, Berger A, et al. Biomolecular network reconstruction identifies T-cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138(4):1429–40.
Castriconi R, Carrega P, Dondero A, Bellora F, Casu B, Regis S, et al. Molecular mechanisms directing migration and retention of natural killer cells in human tissues. Front Immunol. 2018;9:2324.
Gao L, Yang L, Zhang S, Ge Z, Su M, Shi Y, et al. Engineering NK-92 cell by upregulating CXCR2 and IL-2 Via CRISPR-Cas9 improves its antitumor effects as cellular immunotherapy for human colon cancer. J Interferon Cytokine Res. 2021;41(12):450–60.
Kremer V, Ligtenberg MA, Zendehdel R, Seitz C, Duivenvoorden A, Wennerberg E, et al. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer. 2017;5(1):73.
Wennerberg E, Kremer V, Childs R, Lundqvist A. CXCL10-induced migration of adoptively transferred human natural killer cells toward solid tumors causes regression of tumor growth in vivo. Cancer Immunol Immunother. 2015;64(2):225–35.
Somanchi SS, Somanchi A, Cooper LJN, Lee DA. Engineering lymph node homing of ex vivo-expanded human natural killer cells via trogocytosis of the chemokine receptor CCR7. Blood. 2012;119(22):5164–72.
Jamali A, Hadjati J, Madjd Z, Mirzaei HR, Thalheimer FB, Agarwal S, et al. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front Immunol. 2020;11:2028.
Müller N, Michen S, Tietze S, Töpfer K, Schulte A, Lamszus K, et al. Engineering NK cells modified with an EGFRvIII-specific chimeric antigen receptor to overexpress CXCR4 improves immunotherapy of CXCL12/SDF-1α-secreting glioblastoma. J Immunother. 2015;38(5):197–210.
Ng YY, Tay JCK, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85.
Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res. 2019;7(2):219–29.
Bonanni V, Antonangeli F, Santoni A, Bernardini G. Targeting of CXCR3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J Immunother Cancer. 2019;7:290.
Ponzetta A, Benigni G, Antonangeli F, Sciumè G, Sanseviero E, Zingoni A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res. 2015;75(22):4766–77.
Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force.” Blood. 2005;106(7):2252–8.
Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015;367(2):103–7.
Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31(6):220–7.
Mao Y, Sarhan D, Steven A, Seliger B, Kiessling R, Lundqvist A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin Cancer Res. 2014;20(15):4096–106.
Gasteiger G, Hemmers S, Firth MA, Le Floc’h A, Huse M, Sun JC, et al. IL-2-dependent tuning of NK cell sensitivity for target cells is controlled by regulatory T cells. J Exp Med. 2013;210(6):1167–78.
Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, et al. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53(6):1215-1229.e8.
Murray S, Lundqvist A. Targeting the tumor microenvironment to improve natural killer cell-based immunotherapies: on being in the right place at the right time, with resilience. Hum Vaccines Immunother. 2015;12(3):607–11.
Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of MDSC trafficking with SX-682, a CXCR1/2 inhibitor, enhances NK-cell immunotherapy in head and neck cancer models. Clin Cancer Res. 2020;26(6):1420–31.
Parihar R, Rivas C, Huynh M, Omer B, Lapteva N, Metelitsa LS, et al. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR-T cell activity against solid tumors. Cancer Immunol Res. 2019;7(3):363–75.
Li MO, Wan YY, Sanjabi S, Robertson AKL, Flavell RA. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146.
Dong M, Blobe GC. Role of transforming growth factor-beta in hematologic malignancies. Blood. 2006;107(12):4589–96.
Timmins MA, Ringshausen I. Transforming growth factor-beta orchestrates tumour and bystander cells in B-cell non-hodgkin lymphoma. Cancers (Basel). 2022;14(7):1772.
Dasgupta S, Bhattacharya-Chatterjee M, O’Malley BW, Chatterjee SK. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. 2005;175(8):5541–50.
Rouce RH, Shaim H, Sekine T, Weber G, Ballard B, Ku S, et al. The TGF-β/SMAD pathway is an important mechanism for NK cell immune evasion in childhood B-acute lymphoblastic leukemia. Leukemia. 2016;30(4):800–11.
Yvon ES, Burga R, Powell A, Cruz CR, Fernandes R, Barese C, et al. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: implications for adoptive immunotherapy for glioblastoma. Cytotherapy. 2017;19(3):408–18.
Shaim H, Shanley M, Basar R, Daher M, Gumin J, Zamler DB, et al. Targeting the αv integrin/TGF-β axis improves natural killer cell function against glioblastoma stem cells. J Clin Invest. 2021;131(14): 142116.
Wang QM, Tang PMK, Lian GY, Li C, Li J, Huang XR, et al. Enhanced cancer immunotherapy with Smad3-silenced NK-92 cells. Cancer Immunol Res. 2018;6(8):965–77.
Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS ONE. 2018;13(1): e0191358.
Kim TD, Lee SU, Yun S, Sun HN, Lee SH, Kim JW, et al. Human microRNA-27a* targets Prf1 and GzmB expression to regulate NK-cell cytotoxicity. Blood. 2011;118(20):5476–86.
Cabo M, Santana-Hernández S, Costa-Garcia M, Rea A, Lozano-Rodríguez R, Ataya M, et al. CD137 costimulation counteracts TGFβ Inhibition of NK-cell antitumor function. Cancer Immunol Res. 2021;9(12):1476–90.
Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. 2014;44(6):1582–92.
Sarhan D, Hippen KL, Lemire A, Hying S, Luo X, Lenvik T, et al. Adaptive NK cells resist regulatory T-cell suppression driven by IL37. Cancer Immunol Res. 2018;6(7):766–75.
Jin D, Fan J, Wang L, Thompson LF, Liu A, Daniel BJ, et al. CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 2010;70(6):2245–55.
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci USA. 2010;107(4):1547–52.
Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol Rev. 2017;276(1):121–44.
Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27(8):2411-2425.e9.
Yang R, Elsaadi S, Misund K, Abdollahi P, Vandsemb EN, Moen SH, et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J Immunother Cancer. 2020;8(1): e000610.
Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res. 2018;78(4):1003–16.
Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell. 2016;30(3):391–403.
Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. 2021;12(1):3236.
Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, et al. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer. 2018;17(1):120.
Terrén I, Orrantia A, Vitallé J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol. 2019;10:2278.
Parodi M, Raggi F, Cangelosi D, Manzini C, Balsamo M, Blengio F, et al. Hypoxia modifies the transcriptome of human NK cells, modulates their immunoregulatory profile, and influences NK cell subset migration. Front Immunol. 2018;9:2358.
Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, et al. Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cells. 2019;8(9):1083.
Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las RJ, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updat. 2021;59: 100787.
Iliopoulos O, Jonasch E, Donskov F, Narayan V, Maughan BL, Oudard S, et al. Phase II study of the oral hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 for Von Hippel-Lindau (VHL) disease-associated clear cell renal cell carcinoma (ccRCC). JCO. 2021;39(6_suppl):333.
Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med. 2021;27(5):802–5.
Choueiri TK, Powles T, Burotto M, Escudier B, Bourlon MT, Zurawski B, et al. Nivolumab plus cabozantinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2021;384(9):829–41.
Terranova-Barberio M, Pawlowska N, Dhawan M, Moasser M, Chien AJ, Melisko ME, et al. Exhausted T cell signature predicts immunotherapy response in ER-positive breast cancer. Nat Commun. 2020;11(1):3584.
Rodriguez CP, Wu QV, Voutsinas J, Fromm JR, Jiang X, Pillarisetty VG, et al. A phase II trial of pembrolizumab and vorinostat in recurrent metastatic head and neck squamous cell carcinomas and salivary gland cancer. Clin Cancer Res. 2020;26(4):837–45.
Graham K, Unger E. Overcoming tumor hypoxia as a barrier to radiotherapy, chemotherapy and immunotherapy in cancer treatment. Int J Nanomed. 2018;13:6049–58.
Choueiri TK, Kaelin WG. Targeting the HIF2-VEGF axis in renal cell carcinoma. Nat Med. 2020;26(10):1519–30.
Hegde A, Jayaprakash P, Couillault CA, Piha-Paul S, Karp D, Rodon J, et al. A phase I dose-escalation study to evaluate the safety and tolerability of evofosfamide in combination with ipilimumab in advanced solid malignancies. Clin Cancer Res. 2021;27(11):3050–60.
Stępień K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol. 2016;33(9):101.
Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med. 2021;2(4): 100227.
Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833.
Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor-derived lactate modifies antitumor immune response: effect on myeloid-derived suppressor cells and NK cells. J Immunol. 2013;191(3):1486–95.
Acknowledgements
The authors regret that it was not possible to include many interesting studies in the field due to limited space.
Funding
This work was supported by the National Natural Science Foundation of China (81972878 and 82172733), the National Key Research and Development Program of China (2016YFC1303403 and 2020YFC0860200), and the Key Research and Development Program of Sichuan Province (2020YFS0008).
Author information
Authors and Affiliations
Contributions
WW conceived and presented the article idea and supervised the whole work. YZ collected the information, wrote and harmonized the manuscript. WZ was a major contributor in designing the figures and editing the manuscript. JY and JY participated in collecting data and reviewing the manuscript. WW provided important suggestions for manuscript writing. All authors participated in the work and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Zhou, W., Yang, J. et al. Chimeric antigen receptor engineered natural killer cells for cancer therapy. Exp Hematol Oncol 12, 70 (2023). https://doi.org/10.1186/s40164-023-00431-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-023-00431-0