
Abstract
Adoptive cell therapy has received a great deal of interest in the treatment of advanced cancers that are resistant to traditional therapy. The tremendous success of chimeric antigen receptor (CAR)-engineered T (CAR-T) cells in the treatment of cancer, especially hematological cancers, has exposed CAR’s potential. However, the toxicity and significant limitations of CAR-T cell immunotherapy prompted research into other immune cells as potential candidates for CAR engineering. NK cells are a major component of the innate immune system, especially for tumor immunosurveillance. They have a higher propensity for immunotherapy in hematologic malignancies because they can detect and eliminate cancerous cells more effectively. In comparison to CAR-T cells, CAR-NK cells can be prepared from allogeneic donors and are safer with a lower chance of cytokine release syndrome and graft-versus-host disease, as well as being a more efficient antitumor activity with high efficiency for off-the-shelf production. Moreover, CAR-NK cells may be modified to target various antigens while also increasing their expansion and survival in vivo. Extensive preclinical research has shown that NK cells can be effectively engineered to express CARs with substantial cytotoxic activity against both hematological and solid tumors, establishing evidence for potential clinical trials of CAR-NK cells. In this review, we discuss recent advances in CAR-NK cell engineering in a variety of hematological malignancies, as well as the main challenges that influence the outcomes of CAR-NK cell-based tumor immunotherapies.
Similar content being viewed by others

Introduction
In recent years, adoptive cell therapy has made unprecedented advances in the treatment of many tumors to conventional therapy based on the intrinsic properties of transplanted effector cells to target specific tumor antigens and remove tumors [1, 2]. A recent tremendous advance is based on chimeric antigen receptor (CAR)-engineered immune effector cells which improved cytotoxic activity of immune cells in the defense against tumor cells [3]. CARs are synthetic proteins consisting of a single-chain variable fragment (scFv) as an extracellular antigen-binding domain, connected to a diverse range of feasible intracellular activating signaling domain(s) that are designed to present a new ability to affect cells in the recognition of specific antigens in tumor cells and the final destruction of tumor cells [3, 4]. CAR is also a strategy that enables modified T cells to recognize a variety of MHC-independent tumor antigens. Autologous human T cells were the pioneer cells used in CAR therapy and have made a significant breakthrough in the treatment of patients with hematological malignant tumors, including chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemia (ALL), Hodgkin’s lymphoma, and non-Hodgkin’s lymphoma, and other B lymphomas [5,6,7,8,9,10,11].
Although CAR-modified T cell immunotherapy has exhibited great treatment effectiveness against hematological malignancies, several obstacles restrict their further clinical application [12, 13]. It is logistically challenging to generate an autologous CAR-T cell product separately for each patient, and CAR-T cell therapy economically is unfeasible for general clinical systems. Besides, several weeks are required for the production of CAR-T cells, which results in unavoidable inefficiency in the treatment of patients with aggressive diseases. Allogeneic T cells could be able to resolve these obstacles, but their transfer even if human leukocyte antigen (HLA) is matched between donors and recipients may pose a risk of severe graft-versus-host disease (GVHD) due to minor histocompatibility of antigens with genetic polymorphism of cytokines [14, 15]. Furthermore, the expansion and long-term persistence of CAR-T cells in the human body may cause serious and/or long-term adverse effects, such as cytokine release syndrome (CRS), which may threaten patients’ life in the process of secretion of pro-inflammatory cytokines [16, 17]. Natural killer cells have been demonstrated to prevail over the above drawbacks and provide promising alternative therapeutic targets for CAR engineering in adoptive immunotherapy since they do not require prior antigen sensitization or HLA matching, so they have the potential to be used as an allogeneic off-the-shelf product in cellular therapy and have minimal or no ability to induce graft-versus-host disease (GvHD) [18,19,20].
Natural killer (NK) cells were discovered almost 50 years ago and are the first series of cytotoxic lymphocyte cells to defend against tumors in most tissues [21,22,23]. The natural function of NK cells is spontaneously triggered by specific stimulation and is ordered by the integration of signals from multiple germs line-encoded activating and inhibitory receptors which bind to homologous ligands in target tumor cells, assisting NK cells to identify malignant cells from normal cells. Then, NK cells can eliminate tumor cells directly and acutely through various pathways (Fig. 1) [22, 24, 25]. However, there are mechanisms by which the tumor escapes from the immune surveillance and inhibit the function of NK cells such as tumor microenvironment and immunosuppressive factors that prevent the expression of activating receptors and the interactions NK cells with other cells, also antigen escape ways can trigger inhibitory NK cell receptors and inhibit activating NK cell receptors [26,27,28,29,30]. To overcome these mechanisms, scientists have applied strategies directed at boosting the antitumor properties of NK cells and preventing immune escape through genetic engineering and the development of CAR-engineered NK cells [18, 31, 32]. Herein, we will discuss the advantages of CAR-NK cell immunotherapy and the challenges faced by CAR-NK cells, which must be removed before CAR-NK cell therapy replaces previous strategies, and then focuses on opportunities that highlight the efficacy of CAR-modified NK cells from preclinical studies in hematological malignancies.

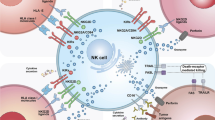
The NK cell-mediated cytotoxicity against tumor cells by affecting both the innate and adaptive immunity. As the innate killer cells, NK cells can identify the stress-induced ligands and reduced MHC I expression rates by own activating and inhibitory receptors, respectively (A). Upon activation, NK cells can stimulate the elimination of the tumor cells by the release of granules with granzyme B and perforin, the death receptor/ligand interaction and also ADCC (B). Further, activated NK cells can produce a spectrum of cytokine and chemokine that trigger the DC maturation and recruitment leading to the modulation of T cell response, more importantly the cytotoxic CD8+ T cell and Th cell induction (C). Natural killer (NK) cell, antibody-dependent cell cytotoxicity (ADCC), TNF-related apoptosis-inducing ligand (TRAIL), T helper (Th) cell, dendritic cell (DC)
NK cell biology and adoptive tumor immunotherapy with NK cells
NK cells are critical cells of the innate immune system with the ability to perform cytotoxicity functions against tumor and virally infected cells quickly, spontaneously, and efficaciously without any previous sensitization, making them a desirable candidate for cancer immunotherapy [33,34,35]. The level of expression of the CD56 antigen surface, tissue location, and immune functions distinguish two main subgroups of NK cells: CD56bright cells and CD56dim cells [36, 37]. The CD56dim cells are fully mature and are the most common NK subgroup found in the peripheral blood and spleen. These cells predominantly have an immediate natural cytotoxic immune response to infected and also tumor cells. Furthermore, CD56dim cells express the CD16 (FcRIII) receptor, which is responsible for antibody-dependent cell-mediated cytotoxicity against antibody-opsonized tumor or virus-infected cells (ADCC) [38]. In contrast to CD56dim cells, CD56bright cells are more immature, have the maximum reproductive potential, are found in fewer subgroups in the peripheral blood, and are predominantly located in secondary lymphoid organs. These cells primarily regulate the immune system by secreting cytokines such as interferon, tumor necrosis factor (TNF), and granulocyte macrophage colony-stimulating factor, among others [39]. Exocytosis of lytic granules, the release of pro-inflammatory cytokines, and cytotoxicity are all mediated by the interaction of signals obtained from two receptors with opposing effects, “germline-encoded inhibitory and activating receptors.” These receptors bind to corresponding homologous ligands on target cells, such as classical or non-classical major histocompatibility complex (MHC) class I, allowing NK cells to distinguish between transformed and normal cells [36, 40, 41]. The activating NK cell receptors that stimulate the destroying effects of these cells include the natural cytotoxicity receptor (NCR) family (NKp30, NKp44, and NKp46), the C-type lectin-like activating receptors (NKG2D, CD94/NKG2F, CD94/NKG2E, CD94/NKG2C, and CD161), activating killer immunoglobulin receptors (KIR2DS1, KIR2DS4 and KIR2DL4), FcγRc IIIA (CD16), and costimulatory receptor DNAM-1 (CD226) [42]. Killer cell immunoglobulin-like receptors (KIRs) and the heterodimeric C-type lectin receptor (NKG2A) are germline-encoded inhibitory receptors that engage with MHC molecules on normal cells and transmit inhibitory signals to NK cells, resulting in the development of NK cell tolerance to normal cells under healthy conditions and preventing NK cell-mediated lysis of normal cells [43, 44].
When healthy cells become infected with a virus or undergo transformation and malignancy, the expression of cell-surface molecules changes, preventing inhibitory reactions and activating NK cells, which then eliminate the transformed cells. Two theories explain how NK cells contribute to tumor immunosurveillance: missing self and induced self [45]. When NK cells recognize malignantly transformed cells with reduced expression of MHC class I molecules, they do not receive inhibitory signals from their inhibitory receptors, allowing these abnormal cells to be quickly lysed, according to the missing-self hypothesis [35, 46]. Induced self-mechanism displays increased expression of stress ligands on transformed cells due to elevated cellular stress factors and DNA damage, which can bind to NK cell-stimulating receptors such as NKG2D, resulting in NK cell activation and tumor cell destruction. As a result, the destruction of tumor cells by NK cells is mediated by a mixture of two pathways [47, 48]. Activated NK cells can mediate cytotoxicity against cancerous cells in a variety of ways. Via intrinsic cytotoxic action, formation of an immunologic synapse with transformed cells, and then exocytosis of cytotoxic granules containing perforin and granzymes, as well as the secretion of immune pro-inflammatory cytokines, activated NK cells may lyse tumor cells directly and acutely in a non-specific manner [49, 50]. Another pathway involves Fas ligand or TNF-related apoptosis-inducing ligand (TRAIL) “members of the tumor necrosis factor (TNF) family” interacting with tumor cells and inducing apoptosis [51, 52]. Target cells can also be eliminated by other pathways, such as antibody-dependent cell cytotoxicity (ADCC), which causes IgG-opsonized cells to be eliminated after binding to the CD16 receptor on NK cells, resulting in a robust immune response to tumors [50, 53]. NK cells also secrete cytokines including INF-gamma, which have pleiotropic effects and regulate antitumor responses in the tumor microenvironment by influencing the activity of activated T cells, macrophages, and dendritic cells [54].
Various NK cell-based adoptive immunotherapies have emerged as a result of the significant antitumor function of NK cells. Increase the activity of endogenous NK cells with cytokines, especially IL-2, is one of the primary strategies in tumor therapy. In vitro and In vivo, cytotoxicity and proliferation ability of NK cells enhance after treatment with exogenous cytokines such as Interleukins-2, -12, -15, -18, and -21. In patients with lung cancer, systemic administration of IL-2 was found to improve NK cell cytotoxicity. Unfortunately, some limitations, such as extreme cytokine toxicity and IL-2-based T lymphocyte regulator stimulation, restrict the strategy’s effectiveness and clinical use [55, 56]. Another strategy for enhancing NK cell activation and cytolytic properties has been the design of therapeutic monoclonal antibodies that bind to and suppress inhibitory receptors on NK cells [57, 58]. Adoptive cell therapy (ACT) has been developed in recent years to overcome the limitations of previous therapies. This approach involves isolating NK cells from autologous or allogeneic sources, expanding them in vitro, enhancing their activation and cytotoxicity by cytokine priming or co-culture of isolated NK cells with feeder cell lines, and then returning these modified NK cells to the patient’s body [59, 60]. Since self-HLA molecules on tumor cells suppress the cytotoxicity of autologous NK cells, NK cells from allogeneic origins seem to be a potential alternative therapeutic target for adoptive immunotherapy [61]. In recent years, emerging research has shown that allogeneic NK cells can be used to treat a wide variety of hematological malignancies and solid tumors [60]. Furthermore, it has previously been explored whether genetically modified NK cells, such as those engineered to express tumor-targeting CARs, could improve their effectiveness in destroying tumor cells [2].
CAR-NK cell generation
Source of NK cells for the manufacture of CAR-NK cells
Functional NK cells for adoptive cell-based immunotherapy can be obtained from a variety of cell sources, and then be modified to establish CAR-redirected NK cells (Fig. 2) [62]. Allogeneic NK cells can be generated from a variety of sources, including peripheral blood (PB) [63], easily accessible NK cell lines [62, 64], and, more recently, stem cells such as umbilical cord blood (UCB) [65], human embryonic stem cells (hESCs) [66], and induced pluripotent stem cells (iPSCs) [67, 68]. Each source has its own set of advantages and disadvantages. Peripheral blood NK cells collected from a donor by lymphocyte apheresis have favorable features of relief and safety, as well as expressing a huge number of activating receptors such as CD16, NKG2D, and the NCRs (NKp44 and NKp46) equipped with KIRs after activation, which plays an important role in NK cell licensing and has a powerful destructive ability against abnormal cells [69, 70]. However, collecting NK cells from peripheral blood (PB) is time-consuming and expensive; on the other hand, peripheral blood mononuclear cells (PBMCs) contain a low percentage of NK cells under normal conditions [71]. As a result, techniques for extending and activating NK cells have appeared. Among these, stimulatory cytokine exposure or co-culture of NK cells with feeder cells are existing methods for obtaining a large number of NK cells and activation [37, 72]. Clonal NK cell lines, such as NK-92, NK-YS, NKL, NKG, KHYG-1, and others, are also essential cellular origins for allogeneic CAR-NK cell therapy [73]. NK-92 cells are well-defined and have effective anticancer properties [74]. NK-92 cells secrete more perforin, granzyme, and several cytotoxic cytokines than PB NK cells. Furthermore, unlike PB NK cells, these cells can be quickly multiplied without the use of feeder cells or under difficult and complicated conditions, and they generate a more homogeneous cell population [75, 76]. However, NK-92 cells lack the expression of several common activating receptors, such as NKp44 and NKp46, and the lack of inhibitory KIRs may limit their potent cytotoxic effects [77]. Consequently, they lack CD16 (FCRIII) expression and hence are unable to mediate antibody-dependent cell cytotoxicity (ADCC) [78]. Moreover, to mitigate the likelihood of perpetual allogeneic tumor engraftment and tumorigenicity, the NK-92 cell line must be irradiated before infusion, which inhibits their durability and expansion in the host [78]. Another potential source of allogeneic NK cells and “off-the-shelf” products for immunotherapy treatment is umbilical cord blood (UCB). UCB NK cells are naturally immature, with lower levels of activating receptors including NKp46, NKG2C, IL-2R, DNAM-1, CD57, adhesion molecules as CD2, CD11a, CD18, CD62L, and CD16, and a higher level of the inhibitory receptor NKG2A [79, 80]. So, following ex vivo amplification and activation of these cells, which require exposure to combinations of stimulatory cytokines or donors, they mature and express a broad range of activating and inhibitory receptors [81]. The significant advantages of UCB NK cells include their relative speed and convenience in the collection, significant proliferative ability, and reduced probability of GvHD [82, 83]. CD34-positive cells, such as iPSCs, have recently been identified as an additional source of NK cell production. Under standardized culture conditions, iPSC-NK cells have a significant amplification potential to generate a large amount of homogeneous NK cells with potent cytotoxic activity [84,85,86]. These cells have a normal rate of killer activating receptors such as NKG2D, NKp46, Fas, and TRAIL expression, but a lower rate of KIR expression than peripheral blood NK cells [87]. Moreover, iPSC-NK cells are surprisingly simple to genetically modify with altered characteristics, making them a promising technique in immunotherapy [78].

The CAR-NK cell production process. Procured or established NK cells derived from multiple sources, such as PBMC, UCB, HSCs, ESCs, and iPSC can be modified with CAR-expressing vectors, and then be cultured in NK cell-specific expansion media with particular cytokines to finally generate CAR-redirected NK cells. Umbilical cord blood (UCB), peripheral blood mononuclear cell (PBMC), induced pluripotent stem cell (iPSCs), embryonic stem cell (ESC), hematopoietic stem cell (HSC), chimeric antigen receptor-natural killer (CAR-NK) cells
Structure of CAR
The design of an appropriate CAR construct for the genetic modification of NK cells, similar to CAR-T cells, helps to increase the cytotoxic potential of these cells for specific antigens in targeted cells. CAR-NK is composed of a main CAR-T construct, an extracellular antigen identification domain (typically a single-chain variable antibody fragment (scFv)), a transmembrane domain, and an intracellular signaling domain [88]. The CAR’s evolution is currently summarized in four generations. In the first generation of CARs, both CAR-T and CAR-NK typically use CD3ζ as a single activation intracellular signaling region [89]. Following that, as second- and third-generation CARs, additional cost-stimulatory signaling regions, such as CD28 or CD137 (4-1BB), were introduced to improve the efficiency of NK cells (Fig. 3) [90]. While 4-1BB-composing CARs are used in both T and NK cells, the role of CD28-composing CARs in NK cells has received less attention [91]. As a result, subsequent studies have designed CAR structures based on NK cell characteristics and the use of more specific costimulatory regions such as DAP10, DAP12, or 2B4 (Fig. 3) [92,93,94]. The main intracellular domains for signal transmission of NK stimulatory receptors are DNAX-activation protein 12 (DAP12) and DAP10 (Fig. 3). DAP12 is required for the activating receptors NKG2C, NKp44, and KIR, while DAP10 is necessary for NKG2D costimulatory signaling [95]. Anti-CD19 CARs containing both signaling domains DAP10 and CD3ζ showed a more efficient cytotoxicity response than either domain alone in the CAR-NK structure [96]. Moreover, DAP12-incorporated CAR constructs show more apparent promise in a range of primary NK cells or NK92 cell lines than NK cells with CD3ζ-containing CAR constructs [97]. Besides that, using the 2B4, a prominent NK-specific costimulatory domain, to generate anti-CD5 CAR-NK cells resulted in rapid proliferation, specific cytotoxicity, and stronger anti-malignant efficacy against T cell malignancies in vitro and T-ALL xenograft mice [98]. The function of CAR-NK cells in tumor inhibition has been stated not only by the ability of the CAR construct to detect tumor-specific antigens but also by their natural receptors, which are not antigen-specific, for the transmission of stimulatory signals within NK cells and the activation of various killing mechanisms have a decisive role to play in explaining the behavior of NK cells in the tumor microenvironment [99]. Recently, the fourth generation of CARs carrying a transgenic “payload”' such as IL-2 or IL-15 has been engineered to improve CAR-NK cell proliferation, longevity, and cytotoxicity against antigen-negative tumor cells. To date, the data published affirm the specific features of these latest CAR constructs as a possible immunotherapeutic approach to modulating the tumor microenvironment [100,101,102].

The common CAR constructions utilized in CAR-NK cell generation. CAR molecules on NK cells include three chief fragments, comprising an antigen detection domain (ScFv or NKG2D), and transmembrane domain concomitant with the signaling domain. First-generation CARs only include CD3ζ or DAP12 as the signaling domain, and CD3ζ seems to be a more efficient signaling domain than DAP10, while DAP12 can stimulate NK cell functions more powerful than CD3ζ. Besides, second-generation CARs express a second signaling domain, including CD28 or 4-1BB in association with CD3ζ. Finally, third-generation CARs include two costimulatory signaling domains. Importantly, respecting the mechanism by which NKG2D induces NK cells, an exclusive CAR construct including NKG2D as the ectodomain that connects DAP10 and CD3ζ as chief signaling domains has been progressed. Chimeric antigen receptor-natural killer (CAR-NK) cells, natural killer group 2D (NKG2D)
CAR transduction into NK cells
To progress in the field of CAR-NK cell immunotherapy, an appropriate method for transferring genetic material into NK cells must be used. So far, two kinds of vectors based on viral or non-viral systems have been extensively used in CAR gene transduction. Vectors based on retroviruses or lentiviruses are more commonly used in genetic modification strategies. During retroviral RNA genome integration, viral RNAs are reverse-transcribed into double-stranded cDNA, which is then semi-randomly inserted into the host genome with the assistance of viral integrase [78]. Vectors developed by these viral pathogens have shown considerable advantages, which are determined by their features and making them comparatively convenient to inject into host cells. Typically, an adequate amount of retroviral generate is used for efficacious gene transfer in the manufacturing process, and vectors are stably integrated into the host DNA. Retroviral vectors can hold transgenes of acceptable sizes (7–8 kb) [103], and their transduction efficiency for primary NK cells from peripheral blood is high (43–93%) [96]. However, this approach has several potential drawbacks, including vector instability and the failure to infect non-dividing cells. Besides that, retroviral vectors integrate into the host genome at random, raising the possibility of insertional mutagenesis and other harmful implications [4, 104]. In comparison to retroviral vectors, lentivirus-based vectors infect both dividing and non-dividing cells and allow for the introduction of larger transgenes up to 10 kb in size, as well as a lower likelihood of insertion mutagenesis, but have a lower capacity to inject CARs into primary NK cells, which needs an overhaul [54, 103]. The transduction efficiency of lentivirus-based vectors, on the other hand, is reasonable for NK cells derived from cord blood [105]. Resistance of NK cells to viral-based gene delivery and lower transfecting efficiency of NK cells compared to T cells stem from NK cells’ natural capacity, as an essential cytotoxic member of the innate immune system, to defend against viral infection guided by pattern recognition receptor signaling [106, 107]. As a result, suppressing innate immune responses to viral infection can improve lentivirus transfection of NK cells and establish viral vectors as an effective and safe strategy for NK cell gene modification. According to research, the use of BX-795, an inhibitor of 3-phosphoinositide-dependent kinase 1 (PDK1), negatively controls signaling pathways of RIG-I, MDA-5, and TLR3 that are involved in the antiviral response, and thus enhances the transduction efficiency of lentiviral vectors by 3.8-fold on average [108].
When compared to viral approaches to gene alteration, non-viral-based gene modifications are more stable, easier to synthesize, can transduce larger genetic payloads (> 100 kb), and are cost-effective for therapeutic use. As a result, non-viral vectors have emerged as a more effective alternative to viral vector-based CAR integration techniques [109,110,111,112]. Transposon-based mechanisms, such as the Sleeping Beauty (SB) transposon vector, have been used successfully to deliver transgene into the host genome as an alternative to viral vectors. Transposon vectors are made up of a transposon with the CAR gene sequence flanked by inverted terminal repeats (ITRs) and a transposase (e.g., SB100X) that binds to ITRs and mediates random transposon entry into the host genome [4, 113, 114]. SB transposon systems have been developed to produce safe and effective CAR-T cells for preclinical and clinical research [115, 116]. However, the feasibility of using an SB transposon system to deliver DNA plasmids as CAR sequence carriers into NK cells is debatable and unknown [4]. Moreover, genetically modifying NK cells through electroporation of DNA or mRNA plasmids is a promising non-integrating strategy. While electroporation of DNA plasmids has low efficiency, electroporation of mRNA is an accurate and cost-effective tactic in clinical applications [105, 117]. Xiao et al. observed that > 95% of primary NK cells expressed NKG2D RNA CAR and that NK cell viability after mRNA electroporation was greater than 90% before infusion [118]. Notably, mRNA electroporation efficiencies of 80–90% were observed in both ex vivo-expanded NK cells and primary resting (non-cytokine stimulated) human NK cells [119]. Nonetheless, owing to constant permeability in the membrane and the failure of cell homeostasis, electrical pulses in the electroporation tactic will often result in the loss of a variety of cells [120, 121]. Besides that, a major drawback of most non-viral gene vectors, such as electroporation, is the transgene’s unstable and transient expression, which could necessitate additional doses of adoptively transferred immune cells to achieve efficient therapeutic function; nevertheless, additional doses may be problematic and have their conflicting consequences [122,123,124].
To conclude, since transferring genetic material into NK cells and scaling up these transformed cell products is challenging, selecting a more appropriate and efficient technique that adheres to existing good manufacturing practices (cGMP) standards is an essential step toward successful clinical trials.
Advantages of CAR-NK cell immunotherapy
Despite the recent success of CAR-T cell immunotherapy in the treatment of hematological tumors [125, 126], FDA approval is a major step in the evolution of genetically engineered cell-based tumor therapies. However, there are also flaws in the extensive therapeutic application of CAR-T cell therapy. On the other hand, the distinct biological properties of NK cells, as well as some benefits of NK cell therapy over CAR-T cell therapy, have sparked significant interest in the application of CAR-NK cells and their development for cancer immunotherapy [99, 127, 128].
First, preclinical and some phase I/II clinical trials have shown that allogeneic NK cell infusions decrease the risk of GVHD and serious harmful impacts. As a result, NK cells are acceptable CAR drivers that are not restricted to autologous cells [60, 129,130,131,132,133]. Second, mature NK cells have a comparatively shorter lifespan in the bloodstream, reducing the possibility of profound and long-term cell deficiency due to cellular memory responses and on-target/off-tumor effects such as B cell deficiency (in the case of CD19-targeting CAR-T cells) [133, 134]. Third, the cytokines secreted by activated NK cells were assumed to be safer and typically consisted of IFN- and granulocyte macrophage colony-stimulating factor (GMCSF), which eliminated the risk of cytokine storm and extreme neurotoxicity caused by pro-inflammatory cytokines such as TNF-, IL-1, and IL-6 in CAR-T cell therapy [127, 135]. Fourth, CAR-NK cells maintain their natural intrinsic ability to recognize stress-evoked ligands presented on tumor cells through their native activating receptors, such as natural cytotoxicity receptors (NKp46, NKp44, and NKp30), NKG2D, and DNAM-1, unlike CAR-T cells, which eliminate cancer target cells only by identifying the tumor-associated antigen through scFv in CAR-related mechanisms. These stress-induced ligands are found on tumor cells during an initial interaction with immune cells or during long-term treatment. As a result, CAR-expressing NK cells can effectively eradicate heterogeneous malignancy in which some malignant cells lack CAR-targeted specific antigen via both CAR-dependent and NK cell receptor-dependent pathways [9, 136, 137]. Fifth, the minimal risk for alloreactivity and GVHD potentially permits allogenic CAR-NK cells to be procured from various sources, including PB, UCB, hESCs, iPCSs, and even NK-92 cell lines. For instance, homogeneous NK92 cells provide an “off-the-shelf” CAR-engineered manufacture for wider clinical applications, while the CAR-NK cells generated from genetically modified human iPSCs can be produced a homogeneous population and have presented strong anticancer capacity and proliferative capability in preclinical studies [4, 138].
The challenges faced by CAR-NK cells
Despite the potential advantages of NK cells, there are several challenges to using CAR-NK cells in clinical trials. The first reluctance stems from the structure of the current CARs used in NK cells. The position of the epitopes binding the current CAR, as well as their distance from the CAR-NK cell’s surface reduces these cells’ ability to bind antigens and stimulate CAR-NK cells. To eliminate melanoma, Li et al. generated an optimized construct of CARs for NK cell activation and cytotoxicity by concentrating on intracellular excitation adapter molecules [76, 139].
The second issue comes from the lack of an efficient gene transfer approach in NK cells. Although viral transfection is a promising method for T cells, it results in low levels of transgene expression in NK cells and adversely impacts their survival, as stated in previous sections. Non-viral vectors minimize these disadvantages and are regarded as desirable and safe alternatives. However, it is unclear whether all of these non-viral vectors are appropriate for CAR-NK constructs [38, 62, 140].
The third concern with using NK cells for immunotherapy is that they are highly vulnerable to freezing and thawing, and their activation and cytotoxicity are greatly reduced after thawing. Many studies have shown that incubating frozen NK cells with cytokines such as IL-2 may restore their function. As a result, strategies for desirable cryopreservation and optimum restoration of activity of frozen CAR-NK cells for adoptive therapy must be investigated [141, 142].
Fourth, the reality that infused cells do not persist in the absence of cytokine assistance is perhaps the most significant challenge of appropriate CAR therapies. While these features of NK cells can be beneficial, they may also restrict the effectiveness of NK cell immunotherapy. For infused NK cells to survive and proliferate in vivo, exogenous cytokines must be administered sequentially [143]. Exogenous cytokines, on the other hand, have undesirable side effects and can promote other immune subsets such as regulatory T cells, which may be immunosuppressive to NK cells [144, 145]. A unique approach is to incorporate cytokine transgenes into CAR-engineered NK cells, which continuously supply cytokine support [146, 147]. CAR.19/IL-15-transduced CB NK cells generated intense interleukin (IL)-15, which assisted their extended survival and enhanced proliferation in vivo. Furthermore, CAR.19/IL15+ CB NK cells outperformed CAR.19-transduced NK cells missing IL-15 in terms of antitumor function and survival in the elimination of B cell malignancies [148].
Preclinical studies of CAR-NK cell in hematological malignancies
Even though CAR-T cell immunotherapy has reported promising results in the treatment of patients with hematological malignancies, treatment-related toxicity and side effects remain serious barriers. Natural killer (NK) cells, as an effective effector cell in innate immunity, have a great deal of antitumor capacity and have more promising prospects in the immunotherapy of hematologic malignancies. The purpose of creating CAR-NK cells is to use a novel strategy for activating NK cells and improving their antitumor properties by genetic modification to eventually produce “off-the-shelf” antitumor immunotherapeutic products [149]. Lymphomas and leukemias are a large group of hematological malignancies characterized by clonal growth and dysfunction of lymphoid and myeloid cells at different stages of maturation and commitment, with varying clinical outcomes and prognoses [150, 151]. A large number of preclinical trials have revealed that CAR-NK can effectively treat lymphomas and leukemias by targeting antigens such as CD19, CD20, CD7, and CD5 (Tables 1 and 2) [148, 158, 163, 174].
CAR-NK cells in leukemia
Extensive studies on the application of CAR-NK cells in the treatment of various forms of Leukemia have just been initiated. By lentiviral gene transfer to CD19-specific CARs that attacked CD19-positive cells and carried a CD3 signaling endodomain either alone or with a costimulatory domain, Oelser et al. transduced a heterogeneous effector cell population defined as cytokine-induced killer (CIK) cells (CD28 or CD137). In vitro, these targeted CIK cells exhibited selective cytotoxicity against cancer cell lines and Primary Pre-B- acute lymphoblastic leukemia (ALL) blasts [155].. Other research groups later confirmed that CAR-engineered NK92 cells had an antileukemic function and could overcome B cell acute and chronic leukemia resistance to parental NK cells. The NK-92 cell lines can be transfected with high transduction efficiency and minimal effect on cell viability using electroporation of CD19-CAR mRNA [156]. In vitro, Boissel et al. reported that NK-92 cells expressing anti-CD19 CAR had effective cytolytic activity against previously resistant CD19-positive B-ALL cell lines and primary chronic lymphocytic leukemia (CLL) [158]. NK-92 cells were also transduced with a lentiviral gene that encoded an FMS-like tyrosine kinase 3 (FLT3)-specific CAR with a composite CD28-CD3 signaling domain and an inducible caspase-9 (iCasp9) suicide gene to improve therapeutic utility and safety. FLT3-specific CAR-NK-92 cells with FLT3 expression and CAR activation were used to target B-ALL cell lines and primary blasts that had altered or lost CD19 expression and were resistant to parental NK-92 in this study. In addition, FLT3-specific CAR-NK-92 cells expressing the iCasp9 suicide gene were readily inactivated in the event of serious side effects [162]. A group of researchers looked into the ability of CAR-engineered NK-92 cells and parental NK-92 cells expressing FcRIII plus anti-CD20 monoclonal antibodies (MAbs) like rituximab to kill NK cell-resistant B-lymphoid leukemia. In vitro observations indicate that CD20-specific CAR-NK-92 cells more effectively eliminated NK cell-resistant primary CLL cells than NK-92 and ADCC arising from it with the participation of anti-CD20 MAbs. They employed two ALL cell lines, BCR-ABL1+ SUP-B15 (CD19 + CD20−) and TMD-5 (CD19 + CD20+), to create two xenotransplant mouse models of residual leukemia for in vivo studies. SUP-B15 cells were killed by intravenous injection of CD19-specific CAR-NK-92, but TMD-5 cells resisted the immune attack. Interfemoral injection of CD19-specific CAR-NK-92, on the other hand, was a method of removing TMD-5 cells from the bone marrow environment. As a consequence, it seems that the process of administration affects the treatment outcome, owing to permeability and local concentration [158]. UCB is a noteworthy and allogeneic source of NK cells in addition to NK-92. CAR-NK was transduced from CB with a structure that included coding segments for CAR for CD19, interleukin (IL)-15 (to increase cell proliferation and persistence), and an inducible caspase-9 (iC9)-based suicide gene, according to Liu and his colleagues. The CAR-modified NK’s antitumor activity and survival were significantly improved by the synthesis of IL-15, compared to normal CAR-NK, and the expression of iC9 improved the cells’ safety [101].
CAR-NK has also shown promise as a successful curative therapy for aggressive T cell malignancies. CARs targeting CD5 or CD3 that are delivered into the NK-92 cell line have been found to have efficient cytotoxic activity against primary peripheral T cell lymphoma cells and T cell leukemia cell lines [163, 175]. Among these, CD5 is one of the significant characteristic markers and potent target for T cell malignancy, and NK cells, which are CD5-negative, may be efficacious for the immunotherapy of T cell malignancy. Chen et al. used lentiviral gene transfer to transform human NK cell line NK-92 into anti-CD5 CAR (CD5CAR), which could target several T cell leukemia and lymphoma cell lines as well as primary tumor cells and contained the intracellular signaling domain CD3 zeta either with two costimulatory domains (CD28 and 4-1BB). They demonstrated that CD5CAR-NK-92 cells specifically target CD5+ tumor cell lines and CD5+ primary tumor cells in vitro and exert consistent, potentially cytotoxic effects. In addition, an in vivo study of T-ALL xenograft mouse models revealed effective tumor progression restriction and control, supporting the efficacy of CAR-NK in T cell malignancies [163]. Besides, in another research, two anti-CD5 CAR plasmids with distinct costimulatory domains were designed; one utilized T cell-related activating receptor-4-1BB (BB.z) and the other utilized NK-cell-related activating receptor-2B4 (2B4.z). By the upregulation of activation markers and cytotoxic granule release, 2B4.z-NK cells demonstrated rapid proliferation and superior antitumor potency in both malignant CD5+ cell lines and primary CD5+ malignant cells in vitro. Moreover, employing mouse xenograft models to investigate the cytotoxic effects, it was found that CAR-2B4-NK cells were more effective than CAR-4-1BB-NK cells [98].
Acute myeloid leukemia (AML) is a heterogeneous aggressive disorder characterized by high proliferation, clonal expansion, and faulty and irregular cell differentiation of hematopoietic progenitors/precursors in the bone marrow and peripheral blood. The lack of satisfactory findings in AML immunotherapy is due in part to the shared expression of phenotypic markers with normal HSCs and in part to AML gene expression heterogeneity [176]. CAR-NK cells may have the ability to overcome treatment challenges and improve the results of AML immunotherapy because AML blasts contain ligands that can be detected by activating receptors in NK cells [177]. According to research, CD123 CAR-NK cells could be a promising immunotherapy component for treating all acute leukemias with high CD123 expression, including AML and B-ALL. In vitro, the NK cell line NK92 and PB NK cells engineered with self-inactivating (SIN) alpha-retroviral vectors to express the third-generation anti-CD123 CAR constructs showed strong antitumor activity against the CD123+ AML cell line KG-1a and primary human leukemia cells, according to the findings. While CD123-CAR-NK-92 cells and CD123-CAR-NK cells both improved antileukemic function as compared to unmodified or EGFP-modified NK cells, CD123-CAR.NK-92 cells showed significantly greater activity against AML cells [170]. In another analysis, peripheral blood NK cells were expanded with irradiated (25 Gy) autologous feeder cells in the presence of IL-21, and then engineered with alpha-retroviral self-inactivating (SIN) vectors to express anti-CD123 CAR with CD28 and 4-1BB (CD137) costimulatory signaling endodomains, as well as the CD3f signaling domain. In vitro, Klob et al. found that these modified cells had improved antitumor function, with enhanced degranulation and higher secretion of tumor necrosis factor-alpha, interferon-gamma, and granzyme A and B [169]. CB NK cells were reprogrammed by retroviral transduction to express anti-CD123 CARs comprising coding parts for interleukin (IL)-15 (to support NK cellular survival and proliferation) and inducible caspase-9-based suicide gene (iC9), in other preclinical models. When compared to non-transduced NK cells, these modified NK cells displayed more antileukemic activity and higher secretion of IFN-gamma and TNF-α in vitro against CD123 + ve AML cell lines (THP-1 and MOLM-14) [171]. CD4 is another target expressed in certain AML subsets. The CD4CAR lentiviral vector was used to design third-generation CAR-NK cells (CD28.BB), which successfully eradicated CD4-expressing AML cells in vitro and demonstrated robust antileukemic activity in a CD4+ AML xenograft mouse model [173]. Besides, the first-in-human clinical trial used CD33 as a therapeutic target, which is expressed on both healthy and malignant myeloid cells, to develop a third-generation CAR lentiviral designed to reprogramme NK92 cells. In a GMP facility, CD33 CAR-NK cells were expanded. Infusion of CD33 CAR-NK cells was shown to be safe in this phase I/II study, but no reaction to treatment was seen in patients with acute myeloid leukemia (AML) (NCT02892695 and NCT02944162).
CAR-NK cells in lymphoma
Non-Hodgkin’s lymphomas (NHLs) are a heterogeneous group of lymphoproliferative malignancies that originate in B-, T-, or natural killer (NK) lymphocytes. One of the appealing molecules to target B cell non-Hodgkin lymphoma (B-NHL) is CD20. Rituximab, a chimeric anti-CD20 antibody, is effective in treating mature B-NHL. However, certain B cell cancers are vulnerable to it or resistant to it. Chu et al. successfully modified and expanded peripheral blood NK cells ex vivo following anti-CD20 CAR mRNA nucleofection and demonstrated that anti-CD20 CAR-NK cells improved dramatically cytotoxicity against CD20+ B-NHL cells in vitro, including both rituximab-sensitive and rituximab-resistant B-NHL cells, and prolonged survival and reduced cancer size in xenotransplant (NSG) mice [160]. Peripheral T cell lymphomas (PTCLs) are a type of non-lymphoma Hodgkin’s (NHL) that comes up from mature T cells that express CD4 antigen widely and uniformly. As a result, CD4 may be an ideal candidate for CAR immunotherapy [178]. Pinz et al. developed the CD4CAR, a third-generation CD4-specific CAR with a single variable Fc chain in the extracellular domain for CD4 antigen recognition and signaling domains of CD28, 4-1BB, and CD3. CD4CAR-NK-92 cells were found to be directly and ruthlessly destroyed. CD4+ human T cell leukemia and lymphoma cell lines from adults and children, as well as patient samples, were studied ex vivo. The use of a third-generation CAR structure prolonged the lifespan of CAR-NK cells in vivo substantially suppressed lymphoma cell proliferation and greatly improved the survival of a xenogeneic mouse model [165]. Furthermore, it was discovered that anti-CD20-CAR mRNA electroporated PB NK cells and romidepsin had synergistic cytotoxic activities in both in vitro and in vivo one of mature B cell (CD20+) non-Hodgkin lymphoma (B-NHL) type, including Burkitt lymphoma (BL). Since romidepsin can significantly increase anti-CD20 CAR-expanded peripheral blood NK cells (exPBNK) in vitro cytotoxicity through NKG2D by inducing the expression of NKG2D ligands MICA/B in both rituximab-sensitive and rituximab-resistant BL cells [161]. Besides that, in another study, CD20-specific CAR-NK-92 injected directly into subcutaneous Daudi (Burkitt’s lymphoma) lymphoma xenografts suppressed local tumor development more effectively than CD19-specific CAR-NK-92 [158]., and in a subcutaneous Raji Burkitt’s lymphoma xenograft in a mouse model, CAR-modified CIK cells revealed potent antitumor activity and remarkably controlled disease development [155].
Role of CAR-NK cells against multiple myeloma
Multiple myeloma (MM) is a malignant neoplasm of plasma cells that is characterized by clonal expansion of plasma cells in the bone marrow and the presence of monoclonal antibodies in the serum or urine. Over the last decade, treatment and prognosis methods for multiple myeloma have advanced dramatically. Since MM cells are extremely heterogeneous and the surface antigen for MM stem cells is unclear, finding an appropriate therapeutic target is a critical component of using CAR-NK cells to treat MM [179, 180]. The cell-surface glycoprotein CS1 (also defined as CD319, CRACC, and SLAMF7) is expressed in leukocytes, NK cells, and normal plasma cells. However, its expression was upregulated in MM, suggesting that c-Maf-mediated interaction with bone marrow stromal cells is responsible for MM cell adhesion, unrestricted proliferation, and tumorigenicity [181, 182]. New evidence shows that CS1 may be a potential target for CAR design. In this regard, a lentiviral construct of a CS1-specific CAR with a CD28-CD3z costimulatory signaling domain was designed and transduced into the NK-92 and NKL NK cell lines and their anti-MM functions were evaluated in vitro and an in vivo xenograft murine model of MM. The findings showed that CS1-CAR-NK cells could effectively eliminate CS1+ MM cells in vitro by enhancing MM cell removal and IFN secretion [166]. Furthermore, adoptive transfer of NK-92 cells expressing CS1-CAR directly reduced the growth of IM9 MM cells and resulted in long-term survival in aggressive MM xenograft murine models [166].
CD138 (syndecan-1) is an integral membrane protein and an early diagnostic antigen of MM cells that mediates MM development and proliferation, in addition to CS1. CD138’s upregulated expression on MM cells suggests that it may be a good choice for CAR immunotherapy. Jiang et al. used a lentiviral vector containing a CD138-specific CAR fused to the CD3z chain as a signaling domain to modify NK-92MI cells. When compared to the empty vector-transduced NK-92MI cells, these reprogrammed NK-92MI cells showed significantly increased CD107a expression and production of granzyme B, IFN- against CD138-positive human MM cell lines (RPMI8226, U266, and NCI-H929). In the xenograft NOD-SCID mouse model, irradiation of NK-92 cells was shown to block further cell proliferation while also exerting specific antitumor activity [167]. Since the current CAR manufacturing strategies include the formation of a specialized antibody domain to recognize cancer cells by CAR-engineered cells. It is likely, though, that using this technique would result in the loss of tumor antigen. In this case, primary NK cells from MM patients were expanded and transduced with a novel type of CAR include natural killer group 2 member D (NKG2D) CARs carrying 4-1BB and CD3z signaling domains showing considerably cytotoxic activity against the majority of MM cell lines. These findings pave the way for further research and development of this novel CAR build as a treatment option for aggressive MM [168].
CRISPR-Cas9 application in CAR-NK cell-based therapies
Clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) has recently developed as an encouraging technology for genome edition [183]. This technique depends on the insertion of Cas9 protein in association with guide RNA into the NK cells [184]. It can be utilized to exactly delete, repair, or insert genes in a particular locus and thereby hold promise to establish more effective antitumor NK cells. For the first time, this strategy was exploited in primary NK cells to impair the CD38 gene for avoiding the fratricide of NK cells when these cells were utilized concomitant with daratumumab (anti-CD38), as CD38 is typically expressed not only on NK cells but also on MM and AML cells [185]. It has been suggested that this technique could result in manufacturing more potent and persistent CAR-NK cells. For example, the use of homology-directed repair (HDR) templates improved the knock-in efficiencies about 75% using K562-IL-21-expanded NK cells [186]. As well, CAR expression induced by an endogenous promotor following CRISPR/Cas9-specific locus knock-in methods leads to the ameliorated and prolonged CAR expression in redirected effector cells in vivo, with a memory-like phenotype and less expression of exhaustion markers [187]. Further, Daher et al. found that CRISPR/Cas9-mediated knockout of cytokine-inducible SH2-containing protein (CIS) as a negative regulator of IL-15 signaling improved CAR-NK cell activity in vitro and in animal models partially by augmented aerobic glycolysis [188].
Conclusions
Extensive research over the last few decades has shown the safety and credibility of using NK cells to treat cancer. However, NK cell therapy is still vulnerable to immunosuppressive mechanisms. To resolve immunosuppression, improve cancer cell targeting, and finally enhance the antitumor effects of NK cells in cancer immunotherapy, acceptable and effective gene manipulation systems are required. Although the CAR-NK cells have shown great promise as a new strategy in adoptive immunotherapy with effective anticancer function against refractory malignancies in various clinical trials (Table 3), but the architecture of CAR-NK cells yet requires further investigations. It is important to choose the opposite target and consider which NK cell source and which kind of CAR construct can improve the efficiency of CAR-NK cells. Besides, the components and regimen of the culture medium that are suitable for NK cells from various sources, improvement of NK cell expansion, and then manufacturing CAR-NK cells with memory properties in vivo for tumor surveillance remain to be determined. Moreover, the long-term anticancer efficiency following administration to patients is still unclear. CRISPR/Cas9-based genetic modifications are noteworthy innovations that might increase the cytotoxicity efficiency and safety of CAR-NK cells by specifically editing primary NK cell genes and constructing stably transduced NK cells. Nonetheless, considering advanced efforts to resolve remaining obstacles, as well as the rapid developments achieved by robust preclinical investigation and clinical research, CAR-NK cell products are expected to make a strong and important contribution in cancer therapy.
Availability of data and materials
Not applicable
Abbreviations
- CAR:
-
Chimeric antigen receptor
- GvHD:
-
Graft-versus-host disease
- CAR-NK:
-
CAR-transduced NK cells
- NK cells:
-
Natural killer cells
- ICIs:
-
Immune checkpoint inhibitors
- ACTs:
-
Adaptive cell therapies
- NHL:
-
Non-Hodgkin lymphomas
- DLBCL:
-
Diffused large B cell lymphoma
- FL:
-
Follicular lymphoma
- ALL:
-
Lymphoblastic leukemia
- MM:
-
Multiple myeloma
- AML:
-
Acute myeloid leukemia
- CRS:
-
Cytokine release syndrome
- CTLs:
-
Cytotoxic T lymphocytes
- NCRs:
-
Cytotoxicity receptors
- SHP-1:
-
SH-2 containing protein tyrosine phosphatase
- ITIMs:
-
Immunoreceptor tyrosine-based inhibitory motifs
- hESCs:
-
Human embryonic stem cells
- SCGM:
-
Stem cell growth medium
- b-NHL:
-
B cell non-Hodgkin’s lymphoma
- RCC:
-
Renal cell carcinoma
- CAIX:
-
Carbonic anhydrase IX
- EpCAM:
-
Epithelial cell adhesion molecule
- iCAS-9:
-
Inducible caspase 9
References
Lin C-Y, Gobius I, Souza-Fonseca-Guimaraes F. Natural killer cell engineering–a new hope for cancer immunotherapy. In: Seminars in Hematology. Amsterdam: Elsevier; 2020.
Rezvani K, Rouce R, Liu E, Shpall E. Engineering natural killer cells for cancer immunotherapy. Mol Ther. 2017;25:1769–81.
Glienke W, Esser R, Priesner C. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21.
Hu Y, Tian Z-G, Zhang C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin. 2018;39:167–76.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–8.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor–modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33.
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 2012;119:2709–20.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18.
Guedan S, Ruella M, June CH. Emerging cellular therapies for cancer. Annu Rev Immunol. 2019;37:145–71.
Chow VA, Gopal AK, Maloney DG, Turtle CJ, Smith SD, Ujjani CS, et al. Outcomes of patients with large B-cell lymphomas and progressive disease following CD19-specific CAR T-cell therapy. Am J Hematol. 2019;94:E209–13.
Mehta RS, Rezvani K. Chimeric antigen receptor expressing natural killer cells for the immunotherapy of cancer. Front Immunol. 2018;9:283.
Kalaitsidou M, Kueberuwa G, Schütt A, Gilham DE. CAR T-cell therapy: toxicity and the relevance of preclinical models. Immunotherapy. 2015;7:487–97.
Cheng Q, Ma S, Lin D, Mei Y, Gong H, Lei L, et al. The S1P 1 receptor-selective agonist CYM-5442 reduces the severity of acute GVHD by inhibiting macrophage recruitment. Cell Mol Immunol. 2015;12:681–91.
Xu X-J, Tang Y-M. Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett. 2014;343:172–8.
Yakoub-Agha I, Moreau A-S, Ahmad I, Borel C, Hadhoum N, Masouridi-Levrat S, et al. Management of cytokine release syndrome in adult and pediatric patients undergoing CAR-T cell therapy for hematological malignancies: recommendation of the French Society of Bone Marrow and Cellular Therapy (SFGM-TC). Bull Cancer. 2019;106:S102–9.
Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146–53.
Caligiuri MA. Human natural killer cells. Blood. 2008;112:461–9.
Morandi F, Yazdanifar M, Cocco C, Bertaina A, Airoldi I. Engineering the Bridge between innate and adaptive immunity for cancer immunotherapy: focus on γδ T and NK cells. Cells. 2020;9:1757.
Locatelli F, Moretta F, Brescia L, Merli P. Natural killer cells in the treatment of high-risk acute leukaemia. In: Seminars in immunology. Amsterdam: Elsevier; 2014. p. 173–9.
Farag SS, Caligiuri MA. Human natural killer cell development and biology. Blood Rev. 2006;20:123–37.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10.
Jelenčić V, Šestan M, Kavazović I, Lenartić M, Marinović S, Holmes TD, et al. NK cell receptor NKG2D sets activation threshold for the NCR1 receptor early in NK cell development. Nat Immunol. 2018;19:1083–92.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–36.
Kochan G, Escors D, Breckpot K, Guerrero-Setas D. Role of non-classical MHC class I molecules in cancer immunosuppression. Oncoimmunology. 2013;2:e26491.
Guerra N, Tan YX, Joncker NT, Choy A, Gallardo F, Xiong N, et al. NKG2D-deficient mice are defective in tumor surveillance in models of spontaneous malignancy. Immunity. 2008;28:571–80.
Vitale M, Cantoni C, Pietra G, Mingari MC, Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur J Immunol. 2014;44:1582–92.
Davis ZB, Felices M, Verneris MR, Miller JS. Natural killer cell adoptive transfer therapy: exploiting the first line of defense against cancer. Cancer J (Sudbury, Mass). 2015;21:486.
Bruno A, Ferlazzo G, Albini A, Noonan DM. A think tank of TINK/TANKs: tumor-infiltrating/tumor-associated natural killer cells in tumor progression and angiogenesis. J Natl Cancer Inst. 2014;106:1–13.
Siegler EL, Zhu Y, Wang P, Yang L. Off-the-shelf CAR-NK cells for cancer immunotherapy. Cell Stem Cell. 2018;23:160–1.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–7.
Lanier LL. Up on the tightrope: natural killer cell activation and inhibition. Nat Immunol. 2008;9:495–502.
Malmberg K-J, Carlsten M, Björklund A, Sohlberg E, Bryceson YT, Ljunggren H-G. Natural killer cell-mediated immunosurveillance of human cancer. In: Seminars in immunology. New York City: Elsevier; 2017. p. 20–9.
Campbell KS, Hasegawa J. Natural killer cell biology: an update and future directions. J Allergy Clin Immunol. 2013;132:536–44.
Cheng M, Chen Y, Xiao W, Sun R, Tian Z. NK cell-based immunotherapy for malignant diseases. Cell Mol Immunol. 2013;10:230–52.
Koepsell SA, Miller JS, McKenna DH Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53:404–10.
Rezvani K, Rouce RH. The application of natural killer cell immunotherapy for the treatment of cancer. Front Immunol. 2015;6:578.
Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126:458–65.
Shifrin N, Raulet DH, Ardolino M. NK cell self tolerance, responsiveness and missing self recognition. In: Seminars in immunology. New York City: Elsevier; 2014. p. 138–44.
Sivakumar P, Gunturi A, Salcedo M, Schatzle J, Lai W, Kurepa Z, et al. Cutting edge: expression of functional CD94/NKG2A inhibitory receptors on fetal NK1. 1+ Ly-49− cells: a possible mechanism of tolerance during NK cell development. J Immunol. 1999;162:6976–80.
Konjević G, Vuletić A, Martinović KM. Natural killer cell receptors: alterations and therapeutic targeting in malignancies. Immunol Res. 2016;64:25–35.
Benson DM, Caligiuri MA. Killer immunoglobulin-like receptors and tumor immunity. Cancer Immunol Res. 2014;2:99–104.
Kumar S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology. 2018;154:383–93.
Ljunggren H-G, Kärre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–44.
Kärre K. NK cells, MHC class I molecules and the missing self. Scand J Immunol. 2002;55:221–8.
Chouaib S, Pittari G, Nanbakhsh A, El Ayoubi H, Amsellem S, Bourhis J-H, et al. Improving the outcome of leukemia by natural killer cell-based immunotherapeutic strategies. Front Immunol. 2014;5:95.
Diefenbach A, Raulet DH. Innate immune recognition by stimulatory immunoreceptors. Curr Opin Immunol. 2003;15:37–44.
Screpanti V, Wallin RP, Ljunggren H-G, Grandien A. A central role for death receptor-mediated apoptosis in the rejection of tumors by NK cells. J Immunol. 2001;167:2068–73.
Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18:671–88.
Wajant H. The Fas signaling pathway: more than a paradigm. Science. 2002;296:1635–6.
Waring P, Müllbacher A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol Cell Biol. 1999;77:312–7.
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56bright subset. Blood. 2001;97:3146–51.
Shimasaki N, Coustan-Smith E, Kamiya T, Campana D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy. 2016;18:1422–34.
Hu W, Wang G, Huang D, Sui M, Xu Y. Cancer immunotherapy based on natural killer cells: current progress and new opportunities. Front Immunol. 2019;10:1205.
Li Y, Yin J, Li T, Huang S, Yan H, Leavenworth J, et al. NK cell-based cancer immunotherapy: from basic biology to clinical application. Sci China Life Sci. 2015;58:1233–45.
Romain G, Senyukov V, Rey-Villamizar N, Merouane A, Kelton W, Liadi I, et al. Antibody Fc engineering improves frequency and promotes kinetic boosting of serial killing mediated by NK cells. Blood. 2014;124:3241–9.
Benson DM Jr, Bakan CE, Zhang S, Collins SM, Liang J, Srivastava S, et al. IPH2101, a novel anti-inhibitory KIR antibody, and lenalidomide combine to enhance the natural killer cell versus multiple myeloma effect. Blood. 2011;118:6387–91.
Lin C, Zhang J. Reformation in chimeric antigen receptor based cancer immunotherapy: Redirecting natural killer cell. Biochim Biophys Acta (BBA) Rev Cancer. 2018;1869:200–15.
Lupo KB, Matosevic S. Natural killer cells as allogeneic effectors in adoptive cancer immunotherapy. Cancers. 2019;11:769.
Shevtsov M, Multhoff G. Immunological and translational aspects of NK cell-based antitumor immunotherapies. Front Immunol. 2016;7:492.
Glienke W, Esser R, Priesner C, Suerth JD, Schambach A, Wels WS, et al. Advantages and applications of CAR-expressing natural killer cells. Front Pharmacol. 2015;6:21.
Verneris MR, Miller JS. The phenotypic and functional characteristics of umbilical cord blood and peripheral blood natural killer cells. Br J Haematol. 2009;147:185–91.
Oelsner S, Friede ME, Zhang C, Wagner J, Badura S, Bader P, et al. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy. 2017;19:235–49.
Mehta RS, Shpall EJ, Rezvani K. Cord blood as a source of natural killer cells. Front Med. 2016;2:93.
Wilber A, Linehan JL, Tian X, Woll PS, Morris JK, Belur LR, et al. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells. 2007;25:2919–27.
Ni Z, Knorr DA, Bendzick L, Allred J, Kaufman DS. Expression of chimeric receptor CD4ζ by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells. 2014;32:1021–31.
Knorr DA, Ni Z, Hermanson D, Hexum MK, Bendzick L, Cooper LJ, et al. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl Med. 2013;2:274–83.
Shoae-Hassani A, Behfar M, Mortazavi-Tabatabaei SA, Ai J, Mohseni R, Hamidieh AA. Natural killer cells from the subcutaneous adipose tissue underexpress the NKp30 and NKp44 in obese persons and are less active against major histocompatibility complex class I non-expressing neoplastic cells. Front Immunol. 2017;8:1486.
Kim S, Poursine-Laurent J, Truscott SM, Lybarger L, Song Y-J, Yang L, et al. Licensing of natural killer cells by host major histocompatibility complex class I molecules. Nature. 2005;436:709–13.
Martín-Antonio B, Suñe G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural killer cells: angels and devils for immunotherapy. Int J Mol Sci. 2017;18:1868.
Bae DS, Lee JK. Development of NK cell expansion methods using feeder cells from human myelogenous leukemia cell line. Blood Res. 2014;49:154.
Yagita M, Huang C, Umehara H, Matsuo Y, Tabata R, Miyake M, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000;14:922–30.
Gong J-H, Maki G, Klingemann HG. Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells. Leukemia. 1994;8:652–8.
Klingemann H, Boissel L, Toneguzzo F. Natural killer cells for immunotherapy–advantages of the NK-92 cell line over blood NK cells. Front Immunol. 2016;7:91.
Zhang C, Oberoi P, Oelsner S, Waldmann A, Lindner A, Tonn T, et al. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front Immunol. 2017;8:533.
Hermanson DL, Kaufman DS. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front Immunol. 2015;6:195.
Matosevic S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J Immunol Res. 2018;2018:4054815.
Luevano M, Daryouzeh M, Alnabhan R, Querol S, Khakoo S, Madrigal A, et al. The unique profile of cord blood natural killer cells balances incomplete maturation and effective killing function upon activation. Hum Immunol. 2012;73:248–57.
Björkström NK, Riese P, Heuts F, Andersson S, Fauriat C, Ivarsson MA, et al. Expression patterns of NKG2A, KIR, and CD57 define a process of CD56dim NK-cell differentiation uncoupled from NK-cell education. Blood. 2010;116:3853–64.
Hu Y, Tian Z, Zhang C. Natural killer cell-based immunotherapy for cancer: Advances and prospects. Engineering. 2019;5:106–14.
Oran B, Shpall E. Umbilical cord blood transplantation: a maturing technology. Hematol Am Soc Hematol Educ Program Book. 2012;2012:215–22.
Nomura A, Takada H, Jin C-H, Tanaka T, Ohga S, Hara T. Functional analyses of cord blood natural killer cells and T cells: a distinctive interleukin-18 response. Exp Hematol. 2001;29:1169–76.
Zhu H, Kaufman DS. An improved method to produce clinical-scale natural killer cells from human pluripotent stem cells. In: In Vitro Differentiation of T-Cells. Berlin: Springer; 2019. p. 107–19.
Bernareggi D, Pouyanfard S, Kaufman DS. Development of innate immune cells from human pluripotent stem cells. Exp Hematol. 2019;71:13–23.
Bock AM, Knorr D, Kaufman DS. Development, expansion, and in vivo monitoring of human NK cells from human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs). JoVE. 2013;4:e50337.
Saetersmoen ML, Hammer Q, Valamehr B, Kaufman DS, Malmberg K-J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. In: Seminars in immunopathology. New York City: Springer; 2019. p. 59–68.
Fan M, Li M, Gao L, Geng S, Wang J, Wang Y, et al. Chimeric antigen receptors for adoptive T cell therapy in acute myeloid leukemia. J Hematol Oncol. 2017;10:1–14.
Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunol Rev. 2014;257:127–44.
Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25.
Lang S, Vujanovic NL, Wollenberg B, Whiteside TL. Absence of B7. 1-CD28/CTLA-4-mediated co-stimulation in human NK cells. Eur J Immunol. 1998;28:780–6.
Billadeau DD, Upshaw JL, Schoon RA, Dick CJ, Leibson PJ. NKG2D-DAP10 triggers human NK cell–mediated killing via a Syk-independent regulatory pathway. Nat Immunol. 2003;4:557–64.
Lanier LL, Corliss BC, Wu J, Leong C, Phillips JH. Immunoreceptor DAP12 bearing a tyrosine-based activation motif is involved in activating NK cells. Nature. 1998;391:703–7.
Nakajima H, Colonna M. 2B4: an NK cell activating receptor with unique specificity and signal transduction mechanism. Hum Immunol. 2000;61:39–43.
Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11:45–58.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83.
Töpfer K, Cartellieri M, Michen S, Wiedemuth R, Müller N, Lindemann D, et al. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J Immunol. 2015;194:3201–12.
Xu Y, Liu Q, Zhong M, Wang Z, Chen Z, Zhang Y, et al. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J Hematol Oncol. 2019;12:1–13.
Davies JO, Stringaris K, Barrett AJ, Rezvani K. Opportunities and limitations of natural killer cells as adoptive therapy for malignant disease. Cytotherapy. 2014;16:1453–66.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020;382:545–53.
Qian H, Chen Y, Huang T, Liu T, Li X, Jiang G, et al. Combined application of Embelin and tumor necrosis factor-related apoptosis-inducing ligand inhibits proliferation and invasion in osteosarcoma cells via caspase-induced apoptosis. Oncol Lett. 2018;15:6931–40.
Carlsten M, Childs RW. Genetic manipulation of NK cells for cancer immunotherapy: techniques and clinical implications. Front Immunol. 2015;6:266.
Walther W, Stein U. Viral vectors for gene transfer. Drugs. 2000;60:249–71.
Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–42.
Boissel L, Betancur M, Lu W, Wels WS, Marino T, Van Etten RA, et al. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk Lymphoma. 2012;53:958–65.
Littwitz E, Francois S, Dittmer U, Gibbert K. Distinct roles of NK cells in viral immunity during different phases of acute Friend retrovirus infection. Retrovirology. 2013;10:1–7.
Schmidt P, Raftery MJ, Pecher G. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Front Immunol. 2021;11:3404.
Clark K, Plater L, Peggie M, Cohen P. Use of the Pharmacological Inhibitor BX795 to Study the Regulation and Physiological Roles of TBK1 and IκB Kinase ϵ: a distinct upstream kinase mediates Ser-172 phosphorylation and activation. J Biol Chem. 2009;284:14136–46.
Magnani CF, Mezzanotte C, Cappuzzello C, Bardini M, Tettamanti S, Fazio G, et al. Preclinical efficacy and safety of CD19CAR cytokine-induced killer cells transfected with sleeping beauty transposon for the treatment of acute lymphoblastic leukemia. Hum Gene Ther. 2018;29:602–13.
Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov. 2005;4:581–93.
Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev. 2009;109:259–302.
Schleef M. Minicircle and miniplasmid dna vectors: the future of non-viral and viral gene transfer. Toronto: Wiley; 2013.
Zayed H, Izsvák Z, Walisko O, Ivics Z. Development of hyperactive sleeping beauty transposon vectors by mutational analysis. Mol Ther. 2004;9:292–304.
Rostovskaya M, Fu J, Obst M, Baer I, Weidlich S, Wang H, et al. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res. 2012;40:e150.
Monjezi R, Miskey C, Gogishvili T, Schleef M, Schmeer M, Einsele H, et al. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. 2017;31:186–94.
Thokala R, Olivares S, Mi T, Maiti S, Deniger D, Huls H, et al. Redirecting specificity of T cells using the sleeping beauty system to express chimeric antigen receptors by mix-and-matching of VL and VH domains targeting CD123+ tumors. PLoS One. 2016;11:e0159477.
Heintz N, Gong S. Two-step bacterial artificial chromosome (BAC) engineering: Electroporation of competent BAC host cells with the recombinant shuttle vector. Cold Spring Harb Protoc. 2020;2020:pdb. prot098079.
Xiao L, Cen D, Gan H, Sun Y, Huang N, Xiong H, et al. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol Ther. 2019;27:1114–25.
Li L, Liu LN, Feller S, Allen C, Shivakumar R, Fratantoni J, et al. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010;17:147–54.
Grund EM, Muise-Helmericks RC. Cost efficient and effective gene transfer into the human natural killer cell line, NK92. J Immunol Methods. 2005;296:31–6.
Rubinsky B. Irreversible electroporation in medicine. Technol Cancer Res Treat. 2007;6:255–9.
Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers. 2020;12:706.
Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–61.
Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther. 2013;24:717–27.
Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12:81.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med. 2019;380:1726–37.
Klingemann H. Are natural killer cells superior CAR drivers? Oncoimmunology. 2014;3:e28147.
Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–52.
Passweg J, Tichelli A, Meyer-Monard S, Heim D, Stern M, Kühne T, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia. 2004;18:1835–8.
Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood. 2010;115:4293–301.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–100.
Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol. 2010;28:955.
Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–68.
Kalos M, Nazimuddin F, Finklestein JM, Gupta M, Kulikovskaya I, Ambrose DE, et al. Long-term functional persistence, B cell aplasia and anti-leukemia efficacy in refractory B cell malignancies following T cell immunotherapy using CAR-redirected T cells targeting CD19. Washington, DC: American Society of Hematology; 2013.
Hunter BD, Jacobson CA. CAR T-cell associated neurotoxicity: mechanisms, clinicopathologic correlates, and future directions. J Natl Cancer Inst. 2019;111:646–54.
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5:1282–95.
Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. 2015;12:292–302.
Minetto P, Guolo F, Pesce S, Greppi M, Obino V, Ferretti E, et al. Harnessing NK cells for cancer treatment. Front Immunol. 2019;10:2836.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192. e185.
Tettamanti S, Marin V, Pizzitola I, Magnani CF, Giordano Attianese GM, Cribioli E, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD 123-specific chimeric antigen receptor. Br J Haematol. 2013;161:389–401.
Van Ostaijen-Ten Dam MM, Prins H-J, Boerman GH, Vervat C, Pende D, Putter H, et al. Preparation of cytokine-activated NK cells for use in adoptive cell therapy in cancer patients: protocol optimization and therapeutic potential. J Immunother. 2016;39:90–100.
Domogala A, Madrigal JA, Saudemont A. Cryopreservation has no effect on function of natural killer cells differentiated in vitro from umbilical cord blood CD34+ cells. Cytotherapy. 2016;18:754–9.
Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res. 2009;69:4010–7.
Pedroza-Pacheco I, Madrigal A, Saudemont A. Interaction between natural killer cells and regulatory T cells: perspectives for immunotherapy. Cell Mol Immunol. 2013;10:222–9.
Waldmann TA, Lugli E, Roederer M, Perera LP, Smedley JV, Macallister RP, et al. Safety (toxicity), pharmacokinetics, immunogenicity, and impact on elements of the normal immune system of recombinant human IL-15 in rhesus macaques. Blood. 2011;117:4787–95.
Konstantinidis KV, Alici E, Aints A, Christensson B, Ljunggren H-G, Dilber MS. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp Hematol. 2005;33:159–64.
Liu E, Tong Y, Dotti G, Savoldo B, Muftuoglu M, Kondo K, et al. Cord blood derived natural killer cells engineered with a chimeric antigen receptor targeting CD19 and expressing IL-15 have long term persistence and exert potent anti-leukemia activity. Washington, DC: American Society of Hematology; 2015.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–31.
Xu K-l, Cheng H. CAR-NK cell therapeutics for hematologic malignancies: hope is on the horizon. Blood Sci. 2019;1:156–60.
Curran EK, Godfrey J, Kline J. Mechanisms of immune tolerance in leukemia and lymphoma. Trends Immunol. 2017;38:513–25.
Ghione P, Moskowitz AJ, De Paola NE, Horwitz SM, Ruella M. Novel immunotherapies for T cell lymphoma and leukemia. Curr Hematol Malig Rep. 2018;13:494–506.
Chang Y-H, Connolly J, Shimasaki N, Mimura K, Kono K, Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–86.
Herrera L, Santos S, Vesga M, Anguita J, Martin-Ruiz I, Carrascosa T, et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019;9:1–10.
Quintarelli C, Sivori S, Caruso S, Carlomagno S, Falco M, Boffa I, et al. Efficacy of third-party chimeric antigen receptor modified peripheral blood natural killer cells for adoptive cell therapy of B-cell precursor acute lymphoblastic leukemia. Leukemia. 2020;34:1102–15.
Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genßler S, Rettinger E, et al. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int J Cancer. 2016;139:1799–809.
Boissel L, Betancur M, Wels WS, Tuncer H, Klingemann H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk Res. 2009;33:1255–9.
Gang M, Marin ND, Wong P, Neal CC, Marsala L, Foster M, et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood. 2020;136:2308–18.
Boissel L, Betancur M, Lu W, Krause D, Van Etten R, Wels W, et al. Retargeting NK-92 cells by means of CD19-and CD20-specific chimeric antigen receptors compares favorably with antibody-dependent cellular cytotoxicity. Oncoimmunology. 2013;2:e26527.
Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified NK-92MI cells expressing a chimeric CD16-BB-ζ or CD64-BB-ζ receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget. 2017;8:37128.
Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ aggressive B-cell non–Hodgkin lymphoma by anti-CD20 CAR mRNA-modified expanded natural killer cells in vitro and in NSG mice. Cancer Immunol Res. 2015;3:333–44.
Chu Y, Yahr A, Huang B, Ayello J, Barth M, Cairo MS. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology. 2017;6:e1341031.
Oelsner S, Waldmann A, Billmeier A, Röder J, Lindner A, Ullrich E, et al. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int J Cancer. 2019;145:1935–45.
Chen K, Wada M, Pinz K, Liu H, Lin K, Jares A, et al. Preclinical targeting of aggressive T-cell malignancies using anti-CD5 chimeric antigen receptor. Leukemia. 2017;31:2151–60.
Raikar SS, Fleischer LC, Moot R, Fedanov A, Paik NY, Knight KA, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology. 2018;7:e1407898.
Pinz KG, Yakaboski E, Jares A, Liu H, Firor AE, Chen KH, et al. Targeting T-cell malignancies using anti-CD4 CAR NK-92 cells. Oncotarget. 2017;8:112783.
Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–27.
Jiang H, Zhang W, Shang P, Zhang H, Fu W, Ye F, et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol Oncol. 2014;8:297–310.
Leivas A, Valeri A, Rio P, Fernandez L, Pañero I, Lee DA, et al. Activated and expanded natural killer cells expressing an NKG2D-CAR efficiently target multiple myeloma cells. Blood. 2017;130:4466.
Klöß S, Oberschmidt O, Morgan M, Dahlke J, Arseniev L, Huppert V, et al. Optimization of human NK cell manufacturing: fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum Gene Ther. 2017;28:897–913.
Kloess S, Oberschmidt O, Dahlke J, Vu X-K, Neudoerfl C, Kloos A, et al. Preclinical Assessment of Suitable Natural Killer Cell Sources for Chimeric Antigen Receptor Natural Killer–Based “Off-the-Shelf” Acute Myeloid Leukemia Immunotherapies. Hum Gene Ther. 2019;30:381–401.
Sinha C, Seth A, Kahali B, Cunningham L. Development and evaluation of NK-CD123 CAR against high risk acute myeloid leukemia. Biol Blood Marrow Transplant. 2017;23:S253.
Kerbauy LN, Ang S, Liu E, Banerjee PP, Wu Y, Shaim H, et al. Cord blood NK cells engineered to express a humanized CD123-targeted chimeric antigen receptor (CAR) and IL-15 as off-the-shelf therapy for acute myeloid leukemia. Blood. 2017;130:4453.
Salman H, Pinz KG, Wada M, Shuai X, Yan LE, Petrov JC, et al. Preclinical targeting of human acute myeloid leukemia using CD4-specific chimeric antigen receptor (CAR) T cells and NK cells. J Cancer. 2019;10:4408.
You F, Wang Y, Jiang L, Zhu X, Chen D, Yuan L, et al. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am J Cancer Res. 2019;9:64.
Chen KH, Wada M, Firor AE, Pinz KG, Jares A, Liu H, et al. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget. 2016;7:56219.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52.
Baragano Raneros A, López-Larrea C, Suárez-Álvarez B. Acute myeloid leukemia and NK cells: two warriors confront each other. Oncoimmunology. 2019;8:e1539617.
Dearden C. How I treat prolymphocytic leukemia. Blood. 2012;120:538–51.
Campbell KS, Cohen AD, Pazina T. Mechanisms of NK cell activation and clinical activity of the therapeutic SLAMF7 antibody, elotuzumab in multiple myeloma. Front Immunol. 2018;9:2551.
Pittari G, Vago L, Festuccia M, Bonini C, Mudawi D, Giaccone L, et al. Restoring natural killer cell immunity against multiple myeloma in the era of new drugs. Front Immunol. 2017;8:1444.
Hsi ED, Steinle R, Balasa B, Szmania S, Draksharapu A, Shum BP, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14:2775–84.
Malaer JD, Mathew PA. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am J Cancer Res. 2017;7:1637.
Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res. 2014;42:W401–7.
Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genomics. 2020;19:175–82.
Gurney M, Stikvoort A, Nolan E, Kirkham-McCarthy L, Khoruzhenko S, Shivakumar R, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020; Online ahead of print.
Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther. 2020;28:52–63.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Cortes AKN, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. 2021;137:624–36.
Acknowledgements
Not applicable
Funding
No Funders.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception and the main idea of the work. F.M, M.M.S, H.S.R, W.S, M.E.A.G, W.K.A, L.T, A.V.Y, A.H, M.Y, R.M, and A.N drafted the main text, figures, and tables. F.M.KH supervised the work and provided the comments and additional scientific information. Y.P., B.B, and M.N also reviewed and revised the text. All authors read and approved the final version of the work to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
There is no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Marofi, F., Saleh, M.M., Rahman, H.S. et al. CAR-engineered NK cells; a promising therapeutic option for treatment of hematological malignancies. Stem Cell Res Ther 12, 374 (2021). https://doi.org/10.1186/s13287-021-02462-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-021-02462-y