
Abstract
The molecular mechanisms underlying cancer immune escape are a core topic in cancer immunology research. Cancer cells can escape T cell-mediated cellular cytotoxicity by exploiting the inhibitory programmed cell-death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1, CD274) immune checkpoint. Studying the PD-L1 regulatory pattern of tumor cells will help elucidate the molecular mechanisms of tumor immune evasion and improve cancer treatment. Recent studies have found that tumor cells regulate PD-L1 at the transcriptional, post-transcriptional, and post-translational levels and influence the anti-tumor immune response by regulating PD-L1. In this review, we focus on the regulation of PD-L1 in cancer cells and summarize the underlying mechanisms.
Similar content being viewed by others

Introduction
The occurrence of tumors results from gene deletion, mutation, or abnormal expression in the presence of genetic and environmental factors, which eventually lead to abnormal cell proliferation. Tumor cells can be found, recognized, and eliminated by the immune system; however, the interaction between tumor cells and immunological cells is regulated by immune activating and inhibitory molecules [1]. Tumor cells inhibit the immune response by upregulating immunosuppressive chemicals and downregulating immune-activating molecules, allowing them to avoid detection and flourish. Immune checkpoints, which consist of various receptors and their respective ligands, are essential for the initiation and termination of an effective immune response [2, 3]. Immune checkpoints (ICs), such as programmed cell death protein 1 (PD-1)/programmed cell death ligand 1 (PD-L1, CD274), lymphocyte activating 3 (LAG3), cytotoxic T-lymphocyte associated protein 4 (CTLA4), hepatitis A virus cellular receptor 2 (HAVCR2, TIM3), and T cell immunoreceptor with Ig and ITIM domains (TIGIT), serve a vital role in tumor immunoevasion [4,5,6]. For example, high expression of myeloid leukemia cell ICs in patients with acute myeloid leukemia is associated with poor prognosis [7]. Blocking the suppressive immune checkpoint proteins with specific antibodies restores the immune system’s ability to distinguish and eliminate cancer cells and achieves favorable tumor immunotherapy results. Ipilimumab, an FDA-approved ICI, which is a monoclonal antibody directed against CTLA4, was used to treat melanoma, and since then, new drugs that regulate immune checkpoint proteins have emerged. PD-1/PD-L1 immunotherapy is promising and has recently become a hot study area in tumor immunotherapy [8]. However, the mechanisms underlying immune checkpoint blockade therapy resistance and the regulation of PD-L1 are not entirely understood. To develop a more effective and durable immune checkpoint blockade therapeutic strategy, it is necessary to understand the multiple functions and intricate regulatory mechanisms of PD-L1 in cancer. In this context, we review present knowledge about the regulation of PD-L1 in terms of transcriptional, protein, and epigenetic aspects. Furthermore, we discuss the efficient investigation of the PD-L1 regulatory mechanism.
The expression and biological function of PD-L1
PD-L1, a PD-1 ligand, is a cell surface glycoprotein that belongs to the B7 co-stimulatory molecule family. Non-hematopoietic cells such as vascular endothelial cells and hepatocytes, in addition to hematopoietic cells, including T cells, B cells, and macrophages, constitutively express PD-L1 [9, 10]. PD-L1 upregulation occurs across a broad spectrum of cell types as a result of stimulation by pro-inflammatory cytokines and other factors, and PD-L1 is highly expressed in many cancer cells such as lung, ovarian, colon, and melanoma [11, 12].
Tumor cells possess abundant PD-L1, which binds to the PD-1 receptor on the surface of tumor-infiltrating lymphocytes (TILs), subsequently sending immunosuppressive signals to TILs and preventing antigen-specific CD8 + T lymphocytes from eliminating tumor cells [13,14,15,16]. Moreover, PD-L1 enables the promotion of tumor therapy through non-T cell-mediated immunity. For instance, avelumab (MSB0010718C) is a monoclonal antibody that targets PD-L1 and exhibits antibody-dependent cell-mediated cytotoxicity (ADCC), thereby significantly enhancing the killing effects of high-affinity natural killer cells in head and neck squamous cell carcinoma (HNSCC) [17, 18]. Additionally, PD-L1 is capable of promoting cancer cell proliferation. Nuclear PD-L1, coupled with transcription factor Sp1, activates the MERTK signaling pathway by regulating Gas6 mRNA synthesis and promoting Gas6 secretion, which promotes cell proliferation in non-small-cell lung cancer (NSCLC) [19]. Patients with high PD-L1 expression are more likely to have vascular invasion and tumor recurrence than those with low PD-L1 expression in rectal cancer [20]. In addition to playing a significant role in immunotherapy, PD-L1 contributes to the development of tumor drug resistance. Gemcitabine and oxaliplatin are the main chemotherapeutic agents for gallbladder cancer; nevertheless, placenta-specific protein 8 (PLAC8) can lead to the development of chemotherapy resistance through upregulation of PD-L1 expression [21]. Likewise, nuclear factor E2-related factor 2 (NRF2) promotes PD-L1 expression by inhibiting miR-1 expression, ultimately increasing the resistance of hepatocellular carcinoma (HCC) cells to sorafenib [22]. Studies have shown that radiation therapy induces systemic antitumor responses and enhances the sensitivity of refractory tumors to immunotherapy; therefore, stereotactic body radiation therapy in combination with PD-1/PD-L1 blockade may improve drug resistance in patients with advanced NSCLC [23]. The above studies suggest that PD-L1 suppresses T cell function and natural killer cells to play a role in tumor immune escape, but also promotes tumor development and tumor drug resistance (Fig. 1).
Transcriptional regulation of PD-L1
Transcription factors are proteins that bind to DNA-regulatory sequences to modulate the rate of gene transcription. PD-L1 expression is regulated by a wide range of transcription factors, containing MYC, bromodomain containing 4 (BRD4), STAT family members, nuclear factor kappa-B (NF-κB), cyclin-dependent kinase 5 (CDK5), hypoxia-inducible factor 1 subunit alpha (HIF-1α), NRF2, and cyclic AMP-dependent transcription factor 3 (AFT3) [24]. Here, we summarize the critical transcription factors involved in PD-L1 expression (Fig. 2).
MYC
MYC is a nuclear phosphoprotein involved in proliferation, apoptosis, and differentiation [25]. In a Tet-off transgenic mouse model of MYC-induced T cell acute lymphoblastic leukemia (T-ALL), PD-L1 and MYC expression were significantly correlated. Coupled with the promoter of PD-L1, MYC can directly regulate the transcription. Both inactivation and knockdown of MYC can reduce PD-L1 expression [26]. Moreover, overexpression of bridging integrator-1 (BIN1) in NSCLC could reverse PD-L1-mediated immune escape by inhibiting the expression of MYC [27]. Moreover, cell cycle protein-dependent kinase 7 (CDK7) inhibitor THZ1 could downregulate PD-L1 expression by inhibiting MYC activation, and when combined with the PD-L1 inhibitor Atezolizumab improves the outcome of NSCLC [28].
BRD4
BRD4, a member of the bromodomain and extra terminal domain (BET) family, directly binds to the PD-L1 promoter and is significantly connected with PD-L1 expression [29]. The BET inhibitor JQ1 can inhibit PD-L1 mRNA and protein expression in lymphomas and leukemia mouse models [26, 30]. Mechanistically, JQ1 can reduce the binding of BRD4 to the PD-L1 promoter. In contrast, IFN-γ can enhance BRD4 binding to the PD-L1 promoter in ovarian cancer (OC) cells [31]. Similarly, BRD4 and interferon regulatory factor 1 (IRF1) can modulate the PD-L1 transcription triggered by interferon [30, 31].
STATs
IFN-γ-induced PD-L1 expression promotes cancer immune escape, and this has been found in multiple tumor types. IFN-γ binds to the type II interferon receptor and activates janus kinase (JAK)-STAT signaling, further activating interferon response factors [32, 33]. For example, in CHL and primary mediastinal large B-cell lymphoma (PMBCL), gp24.1 amplification increases PD-L1 expression and its induction by the JAK2-STAT1-IRF1 signaling pathway [33]. Moreover, in melanoma, epigallocatechin gallate decreases PD-L1 expression by restraining STAT1 gene expression and phosphorylation followed by downregulation of IRF1 expression [34]. The JAK inhibitor JAKi was used in HNSCC cells to block STAT1 phosphorylation, thereby inhibiting the increase in PD-L1 mRNA levels induced by combined 5-FU and IFN-γ treatment [35]. The expression of inflammatory macrophages can be further increased by IFN-γ through elevating PD-L1 on these cells in a STAT1-dependent manner [36]. The STAT protein family member STAT3 binds directly to the PD-L1 promoter to activate transcription. Chimeric nucleophosmin (NPM)/anaplastic lymphoma kinase (ALK) is significantly associated with malignant cell transformation and NPM/ALK-carrying T cell lymphoma (ALK + TCL) cells abundantly express PD-L1, which is regulated by STAT3, and knocking down STAT3 can inhibit this effect. [37]. Under hypoxia, p-STAT3 interacts with PD-L1 to promote its nuclear translocation and enhances gasdermin C (GSDMC) transcription, thereby inducing pyroptosis in breast cancer (BC) cells [38]. In Epstein-Barr virus (EBV)-positive nasopharyngeal carcinoma cell lines, PD-L1 expression is greater, and latent membrane protein 1 (LMP1) activates PD-L1 by increasing STAT3 phosphorylation [39]. Conversely, water extract from sporoderm-broken spores of G. lucidum (BSGWE) can reduce PD-L1 expression in osteosarcoma by blocking STAT3 phosphorylation [40]. By downregulating CD40 and STAT3 expression, miR-502-5p can downregulate PD-L1 expression in gastric cancer (GC) cells [41].
NF-κB
NF-κB is a homologous or heterologous dimer formed by members of the Rel family, and it plays a key role in cell growth, apoptosis, and immune responses through transcriptional regulation. LMP1 may induce PD-L1 expression in natural killer/T-cell lymphoma cells through regulation of the MAPK/NF-KB signaling pathway and is associated with poor prognosis [42]. IFN-γ stimulates NF-κB in melanoma cells to induce the expression of PD-L1, and inhibiting NF-κB expression lowers IFN-γ-induced PD-L1 but not constitutive PD-L1 expression [43]. In prostate cancer, RelB, a major member of the NF-κB family, upregulates PD-L1 mainly by binding to the NF-κB element located in the promoter of PD-L1 [44]. Furthermore, knockdown of histone deacetylase 5 (HDAC5) results in notable activation of NF-κB signaling, thus significantly increasing PD-L1 expression in pancreatic cancer (PC) [45].
CDK5
CDK5 is a cyclin-dependent kinase that is widely expressed in many malignancies, allowing cancer cells to escape immune detection. The CDK5 inhibitor roscovitine prevents IFN-induced PD-L1 expression in medulloblastoma. Inhibition of CDK5 causes the PD-L1 transcriptional repressors IRF2 and IRF2BP2 to compete with IRF1 for binding to regions in the promoter of PD-L1 [46].
HIF-1α
HIF-1α is a master regulator of the response to hypoxia via its activation of the transcription of many genes. Under hypoxic conditions, there was a positive correlation between the HIF-1α and PD-L1 expression levels together with the inactivation of T cells. In addition to proliferation and inhibition of apoptosis, HIF-1α has an immunosuppressive function [47, 48]. HIF-1α and PD-L1 expression levels were positively correlated with T cell inactivation under hypoxic circumstances, and inhibiting PD-L1 expression improved bone marrow-derived suppressor cells (MDSCs)-mediated T cell activation [49]. The combination of the HIF-1α inhibitor PX-478 and an anti-PD-L1 antibody increased dendritic cell (DC) and CD8 + T cell activation and dramatically suppressed tumor growth in a glioma mouse model. Mechanistically, HIF-1α activates PD-L1 transcription by binding to the hypoxia response element HRE-4 in the PD-L1 proximal promoter [50, 51]. In both acute myeloid leukemia (AML) mouse model and AML cell lines including MOLM-13 and THP1, PD-L1 boosts glycolytic metabolism, which enhances cell proliferation and tumor formation via the Akt/mTOR/HIF-1α signaling pathway [52].
NRF2
NRF2 is a crucial transcription factor for antioxidant and detoxification enzymes that protect cells from damage caused by oxidative stress [53]. This protein decreases the expression of miR-1, leading to the elevation of PD-L1 in HCC patients treated with sorafenib [22]. The management of colon cancer has been transformed by the development of ICIs. Nonetheless, aberrant upregulation of PD-L1 promotes chemoresistance and results in poor prognosis, while NRF2 inhibition remarkably lowers PD-L1 mRNA expression in sensitive and resistant cells [54].
ATF3
ATF3 is a common stress-inducible transcription factor that is instrumental in modulating oncogenesis, immunity, and metabolism. In melanoma and NSCLC cells, adenosine A1 receptor (ADORA1) inhibition promotes PD-L1 gene expression through ATF3 binding to the PD-L1 promoter directly. Similarly, Sophora alopecuroides Linn impedes PD-L1 expression by enhancing ADORA1 activation in NSCLC [55, 56].
In addition to the above-mentioned, directly regulating transcription factors, some genes such as phosphatase and tensin homolog (PTEN), mTOR, and p53, which are closely related to tumor development, can indirectly regulate PD-L1 transcription and thus promote tumor immune escape [52, 57, 58]. In conclusion, the above transcription factors regulate PD-L1 expression, influence tumor immune escape, and facilitate tumor progression and the development of drug resistance.
Epigenetic regulation of PD-L1
DNA methylation, histone modification, non-coding RNA–mediated regulation, and N6-methyladenosine are the most common epigenetic regulation mechanisms. Epigenetic regulation is important for modulating several cellular functions, including PD-L1 expression (Fig 2).
DNA methylation
DNA methylation occurs when a methyl group is added to the C5 position of a DNA cytosine loop to produce 5-methylcytosine (5mC), and it regulates gene expression by controlling chromatin structure and DNA stability and conformation [59]. Recent research has linked DNA methylation of the PD-L1 promoter to PD-L1 mRNA expression in various malignancies [60,61,62,63]. PD-L1 expression can be controlled by DNA methylation, and when its promoter is hypermethylated, it improves the overall survival of melanoma patients [62, 64]. Furthermore, the demethylating drug decitabine dose-dependently increases PD-L1 mRNA levels in leukemia cells [39]. Thus, inhibitors of epigenetic regulation may improve the effectiveness of anti-PD-1/PD-L1 antibodies.
Histone modification
Recently, histone modification, particularly histone acetylation, has also been implicated in the transcriptional regulation of PD-L1. TET2, a ten-eleven translocation (TET) family member, recruits histone deacetylases (HDAC1/2) to deacetylate the histone modification H3K27ac on the PD-L1 promoter and consequently restraining PD-L1 transcription in BC [53]. HDAC3 overexpression suppressed PD-L1 and inhibited CD3+ T cell proliferation [65]. Suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor, reduced PD-L1 expression in lung cancer cells in a dose-dependent manner [66]. These investigations suggest combining HDAC inhibitors with PD-1/PD-L1 inhibitors may improve therapeutic efficacy.
Apart from histone acetylation, histone methylation in the PD-L1 promoter can influence PD-L1 transcription. The histone methyltransferase EZH2 represses PD-L1 transcription by histone H3 lysine 27 trimethylation (H3K27me3) of the PD-L1 promoter [67]. In contrast, the histone methyltransferase lysine methyltransferase 2 A (MLL1) promotes PD-L1 transcription through histone H3 lysine 4 trimethylation (H3K4me3) of the PD-L1 promoter [68].
MicroRNAs
MicroRNAs (miRNAs) are a type of non-coding, single-stranded RNA molecules. Some miRNAs can bind to the 3’UTR of PD-L1 mRNA (shown in Table 1) e.g., miR-34 and miR-513, to degrade PD-L1 mRNA or inhibit PD-L1 translation [69,70,71]. In NSCLC, p53 increases miR-34 expression to reduce PD-L1 expression. MiR-34 can bind directly to the 3’UTR of PD-L1 mRNA to suppress PD-L1 production and antagonize T cell exhaustion [58]. Unlike miR-34, miR-197 indirectly represses PD-L1 expression via CDC28 protein kinase regulatory subunit 1B (CKS1B)/STAT3 in chemotherapy-resistant NSCLC [72].
LncRNA and circRNA
Long non-coding RNA (lncRNA) and circular RNA (circRNA) are crucial types of ncRNAs that are sensitive to the tumor immune response. LncRNAs are involved in a variety of cellular processes and molecular signaling cascades via their modulation of PD-L1 expression at the epigenetic, transcriptional, and post-transcriptional levels [73, 74]. Some circRNAs regulate the function of miRNAs as microRNA sponges and are essential to transcriptional regulation [75].
The lncRNA HOTTIP stimulates neutrophil IL-6 secretion to allow STAT3 phosphorylation, which increases PD-L1 expression in OC, limiting T cell activity and eventually accelerating tumor immune escape [76]. By interrupting miR-21/IL-6 crosstalk, the lncRNA small nucleolar RNA host gene 12 (SNHG12) promotes the PD-L1 expression regulated by IL-6R [77]. Compared with SNHG12, inhibition of the lncRNA SNHG20 downregulates PD-L1 expression through the ATM/JAK/PD-L1 pathway, affecting the epithelial-mesenchymal transition (EMT) and metastasis in esophageal cancer [78]. In DLBCL, the lncRNA SNHG14 increases the expression of zinc finger E-box binding homeobox 1 (ZEB1) by sponging miR-5590-3p. Subsequently, SNHG14 and PD-L1 are transcriptionally activated by ZEB1, thereby promoting tumor immune evasion [79]. Through EIF4A3-mediated E2F transcription factor 1 (E2F1) upregulation, the lncRNA cancer susceptibility candidate 11 (CASC11) inhibited NF-κB and PI3K/AKT/mTOR pathway activation to mediate PD-L1 expression and encourage tumor progression in a mouse model of HCC with lung metastasis [80]. In OC, the EMX2OS/miR-654/AKT3/PD-L1 axis is associated with aggressiveness, and PD-L1 upregulation reverses the anti-cancer functions of miR-654. miR-654 is sponged and downregulated by lncRNA EMX2OS, while, in contrast, AKT3 and PD-L1 are upregulated. [81]. The lncRNA NUTM2A-AS1 directly targets miR-376a, which increases TET1 and HIF-1α expression followed by an increase in PD-L1 expression, thereby promoting gastric carcinogenesis and drug resistance [82]. Similarly, Hoxa-AS2 binds to miR-519 and results in significant upregulation of HIF-1α and PD-L1, which prominently promotes the progression of nasopharyngeal carcinoma including its proliferation, migration, and invasive ability [83]. In oral squamous carcinoma (OSCC), the lncRNA IFITM4P enhances the binding of histone 3 lysine 4 demethylase KDM5A to the PTEN promoter to diminish PTEN transcription, boosting PD-L1 expression. It also recruits SAM and SH3 domain containing protein 1 (SASH1) in the cytoplasm to bind and phosphorylate TAK1 (Thr187), increasing NF-κB phosphorylation to stimulate PD-L1 transcription [84]. Conversely, the expression of PD-L1 is extremely hindered by LncMX1-215 through the blocking of GCN5-mediated acetylation of H3K27 [85].
Hsa_circ_0000190 enhances PD-L1 mRNA-mediated expression of soluble PD-L1 and promotes non-small cell lung carcinogenesis and immune evasion [86]. In addition, HasCircRNA-002178 is capable of enhancing PD-L1 expression by inducing T cell exhaustion via sponging miR-34 in LUAD cells [87]. CDR1-AS remarkably increases the expression of CMTM4 and CMTM6, the pivotal regulators of PD-L1 protein, and concurrently upregulates PD-L1 expression [88] (Table 2).
N6-methyladenosine
N6-methyladenosine (m6A) is a common mRNA modification that regulates mRNA stability, localization, transport, shearing, and translation. METTL3 mediates the RNA methylation modification process and functionally catalyzes m6A mRNA methylation [89]. By increasing the m6A content in protein arginine methyltransferase 5 (PRMT5) and PD-L1, METTL3 promotes the metastasis and proliferation of OSCC [90]. Through a m6A-dependent mechanism, METTL3 positively regulates IGF2BP3 to increase the stability and expression of PD-L1 mRNA in BC [91]. YTH domain family proteins (YTHDF) recognize and bind m6A in mRNA, YTHDF1 mediates translation to promote translation efficiency, and YTHDF2 mediates degradation to control the half-life of target transcripts, ensuring efficient protein production from m6A-tagged dynamic transcripts [92]. Recent evidence suggests that m6A demethylases fat mass and obesity-associated protein (FTO) and ALKBH5 induce m6A mRNA demethylation [93]. ALKBH5 deficiency enhances m6A modification in the PD-L1 3’UTR region, thereby promoting PD-L1 mRNA degradation in a YTHDF2-dependent manner [94]. Conversely, FTO promotes PD-L1 expression by inducing m6A demethylation of mRNA in colon cancer cells; however, whether m6A demethylase acts directly on PD-L1 mRNA remains uncertain [95] (Table 3).
PD-L1 regulation at the protein level
The ultimate mechanism regulating PD-L1 expression is post-translational regulation, which is influenced by ubiquitination, phosphorylation, glycosylation, acetylation, palmitoylation, autophagy, and other factors (Fig 2).
Ubiquitination
Ubiquitination is a common post-translational modification that controls the stability of proteins [96]. TNFα secreted by macrophages can positively regulate PD-L1 protein at the post-translational level without affecting its transcription. TNFα activates NF-κB through the nuclear translocation and downstream transactivation of p65. Subsequently, p65 activates COP9 signalosome 5 (CSN5) transcription and promotes CSN5 expression. CSN5 binds PD-L1 and deubiquitinates PD-L1, enhancing PD-L1 stability, and evading T cell immune surveillance [97]. Additionally, berberine binds to CSN5 and diminishes its deubiquitination activity, leading to PD-L1 ubiquitination and degradation and promoting anti-tumor immunity [98]. LPS or high-cholesterol diet (HCD)-induced macrophage infiltration significantly activates the C-C motif chemokine ligand 5 (CCL5)-P65/STAT3-CSN5-PD-L1 signaling pathway in azoxymethane-induced CC mouse models and is correlated with poor prognosis [99]. In contrast to CSN5, the deubiquitinase USP8 removes TNF receptor associated factor 6 (TRAF6)-mediated K63-linked ubiquitination, thereby promoting PD-L1 degradation in mouse models of lung and colon cancer [100]. Ubiquilin 4 (UBQLN4) suppresses PD-L1 ubiquitination and promotes protein stability in melanoma. Albendazole stimulates tumor immune function by reducing UBQLN4 expression and, as a result, facilitating PD-L1 protein degradation [101]. Overexpression of the E3 ligase ITCH in melanoma cells ubiquitinates and suppresses MAPK-induced PD-L1 expression, boosts CD8 + cell production, and promotes antitumor effects [102]. The CMTM family has a significant impact on the immune system and is involved in the occurrence and development of tumors. A quintessential example is that EMT transcription factor SNAI1 promotes PD-L1 expression in BC by positively regulating CMTM6 [103]. Two groups used whole-genome CRISPR–Cas9 screening and haploid gene screening based on the fluorescence-activated cell sorting technology to jointly discover that CMTM6 is a key protein that modulates PD-L1 stability. CMTM6 interacts with PD-L1 and co-localizes at endosomes and the plasma membrane. CMTM6 protects PD-L1 from lysosomal-mediated degradation, increases its stability, and enhances the ability of tumor cells to suppress immune responses. Inhibiting CMTM6 expression can diminish PD-L1 expression and greatly limit the tumor cell’s ability to block T cell activity, but it has little effect on the MHC class I molecules [104]. Mechanistically, CMTM6 inhibits the ubiquitination of PD-L1 and prolongs its half-life. In tumor cells, CMTM6 is involved in BCLAF1-dependent PD-L1 upregulation through inhibition of ionizing radiation-induced PD-L1 ubiquitination [105]. Similar to CMTM6, CMTM4 also functions to regulate PD-L1 and is an alternative regulator of this protein [106] (Table 4).
Glycosylation
Glycosylation of membrane receptor proteins influences not only the interaction of ligands and receptors but also protein activity [107]. In human tumor tissues and cancer cell lines, PD-L1 is glycosylated. Glycogen synthase kinase 3β (GSK3β) is associated with non-glycosylated PD-L1 and triggers PD-L1 degradation by β-TrCP. PD-L1 N192, N200, and N219 glycosylation can antagonize GSK3β binding, thereby increasing the stability of PD-L1 [108]. Similarly, the N-glycosyltransferase subunit STT3 can increase the glycosylation and stability of PD-L1, resulting in cancer cells evading the immune system [109, 110]. Moreover, a spliced isoform, FKBP51s, and sigma-1 receptor, SIGMAR1, increased PD-L1 expression by promoting PD-L1 glycosylation and stability [111, 112].
Phosphorylation
Non-glycosylated PD-L1 is an unstable protein whose T180 and S184 residues are easily phosphorylated by GSK3β and then bound by the E3 ubiquitin ligase β-TrCP, resulting in PD-L1 degradation in the cytoplasm [113]. For instance, in basal-like breast cancer, suppression of GSK3β activity by epidermal growth factor stabilizes PD-L1 [114]. IL-6-activated JAK1 phosphorylates PD-L1 Tyr112 and recruits the N-glycosyltransferase STT3A, which catalyzes PD-L1 glycosylation and stabilizes PD-L1 [115]. Another important player regulating PD-L1 phosphorylation is AMP-activated protein kinase (AMPK). By directly phosphorylating S195 of PD-L1 in response to metformin activation of AMPK, aberrant PD-L1 glycosylation is induced, which causes endoplasmic reticulum accumulation and ER-related destruction [116].
Acetylation and palmitoylation
Acetylation is a common post-translational modification of proteins. PD-L1 is acetylated by p300 enzyme at Lys263 affecting nuclear translocation. HDAC2 enzyme catalyzes PD-L1 deacetylation and binds to huntingtin interacting protein 1 related (HIP1R), thereby translocating to the nucleus. Accumulation of nuclear PD-L1 may promote tumor cells to evade immune surveillance during metastasis [117]. Palmitoyltransferase ZDHHC3 (DHHC3) catalyzes PD-L1 palmitoylation in colon and breast malignancies and stabilizes PD-L1 by suppressing ubiquitination, which prevents lysosomal degradation of PD-L1. Using 2-bromopalmitate, the suppression of PD-L1 palmitoylation or the silencing of DHHC3 triggers anticancer immunity [118, 119].
Autophagy
Autophagy is a membrane transport process involved in the metabolism of intracellular component degradation that is activated under nutrient or energy deprivation conditions. Under adverse conditions, such as hypoxia, a shortage of growth hormones, or reactive oxygen species (ROS), autophagy also allows cells to survive [120, 121]. With an in-depth study of autophagy in the tumor immune response, the relationship between PD-L1 and autophagy in tumors has been explored. PD-L1 upregulates beclin 1 (BECN1), a crucial molecule in autophagy regulation, and enhances the autophagy of OC cells [122]. Interestingly, IFN-γ promotes PD-L1 expression by inhibiting autophagy via p62/ sequestosome 1 (SQSTM1) accumulation and NF-κB activation [123] (Table 2).
Targeting PD-L1 and the PD-L1 regulatory pathway for cancer immunotherapy
Cancer immunotherapy involving PD-1/PD-L1 blocking antibodies has brought considerable therapeutic advantages to patients with advanced-stage cancer [124]. Research on anti-PD-L1 antibodies has been conducted in a variety of tumors, including NSCLC, SCLC, melanoma, HCC, BC, head and neck squamous cell carcinoma, gastric and gastroesophageal junction cancer, OSCC, urothelial carcinoma (UC), renal cell carcinoma (RCC) [125]. FDA-approved anti-PD-1/PD-L1 applications are quickly expanding across various tumor types. The PD-L1 level in tumors is an essential factor influencing the therapeutic efficacy of anti-PD-1/PD-L1 therapy [126, 127]. The combination of PD-L1 and PD-1 results in the loss of the killing ability of tumor-infiltrating lymphocytes, which leads to uninhibited tumor growth. Therefore, the destruction of the interaction between PD-L1 and PD-1 manifests tremendous potential in releasing the lethality of the immune system to cancer cells [128, 129]. Here, we summarize the research progress of small-molecule agents that target the PD-L1/PD-1 axis and PD-L1 inhibitors (Table 3).
Numerous small molecule agents targeting epigenetic regulation directly or indirectly downregulate PD-L1 and increase immunotherapy effects. Notably, The STAT family is critical in the control of PD-L1. For instance, Stri-201, a small molecule inhibitor targeting STAT3, can effectively inhibit the expression of PD-L1 in the human tongue squamous cell carcinoma cell line CAL27 cells [130]. Nexturastat and tubastatin A inhibited HDAC6-mediated STAT3 activity in melanoma, leading to PD-L1 expression reduction [131]. The EIF4F inhibitor silvestrol enhances IFN-γ-induced PD-L1 transcription and elevates anti-tumor immunomodulatory effects in melanoma [132]. Contrary to silvestrol, verteporfin blocks the STAT1-IRF1- tripartite motif containing 28 (TRIM28) signaling cascade and induces autophagy-mediated degradation of the Golgi apparatus, thereby efficiently downregulating PD-L1 expression [133]. By impairing MLL1, the epipolythiodioxopiperazine metabolite verticillin A lowers the amount of H3K4me3 in the PD-L1 promoter, transcriptionally hindering PD-L1 expression and enhancing the effectiveness of PD-L1/PD-1 immunotherapy in PC patients [134]. By targeting c-MYC, cisplatin inhibits miR-145 expression, which results in upregulation of PD-L1 in OC [135]. Transforming growth factor-beta (TGF-β) affects PD-L1 inhibitors to some extent and induces drug resistance. YM101 can suppress TGF-β and PD-L1, which dramatically enhances the anti-tumor effect [136]. Furthermore, Mn2+ acts synergistically with YM101 to facilitate the conversion of non-inflammatory to immune inflammatory tumors and greatly antagonizes PD-1/PD-L1 drug resistance [137].
Post-translational modifications are critical to PD-L1 stability; therefore, targeting this process may lead to irreversible PD-L1 degradation. Proteolysis targeting chimeras (PROTACs) induce PD-L1 ubiquitination and subsequent lysosomal degradation by recruiting E3 ligases. PROTACs can induce PD-L1 protein degradation in various malignant cells in vivo in a proteasome-dependent manner [138, 139]. Similarly, the peptide PD-LYSO, which contains the lysosome-sorting signal as well as the PD-L1-binding sequence of HIP1R, causes PD-L1 expression to be reduced in tumor cells [140]. CSN5 is required for PD-L1 stabilization owing to its inhibition of the ubiquitination of PD-L1. Thus, the CSN5 inhibitor curcumin increases tumor cell susceptibility to CTLA4 therapy by lowering PD-L1 expression [97]. Protein kinase AMPK agonists or ketogenic diets promote PD-L1 phosphorylation and disrupt its interaction with CMTM4, which contributed to inducing PD-L1 degradation and enhancing anti-CTLA4 immunotherapy in mouse tumor models [141].
Some chemotherapy drugs are closely correlated with PD-L1 regulation. For instance, the DNA demethylation drug decitabine induced DNA hypomethylation in CC, directly upregulated PD-L1 expression, stimulated more antitumor effects, and significantly enhanced the immunotherapeutic effects of PD-L1 [142]. This phenomenon was similarly observed in mouse models of leukemia, HNSCC, and NSCLC [60, 143, 144]. Temozolomide can trigger STAT3 activation, subsequently increasing PD-L1 expression [145]. In melanoma, regorafenib inhibits JAK1/2-STAT1 and MAPK signaling by targeting the RET-Src axis, consequently attenuating IFN-γ-induced PD-L1 expression [146]. A previous study has reported that the glucocorticoid receptor inhibitor mifepristone and dual ICB act synergistically in PC to inhibit PD-L1 expression while promoting cytotoxic T cell infiltration and activity, enhancing antitumor immunity [147]. Metformin activates AMPK, which phosphorylates S195 of PD-L1, resulting in abnormal PD-L1 glycosylation [109]. In contrast, metformin reduces the abundance of PD-L1 by disrupting electrostatic interactions, thereby promoting the dissociation of the cytoplasmic structural domain membrane of PD-L1 [148]. Aberrant activation and expression of receptor tyrosine kinases (RTKs) are relevant to numerous human cancers. MET was confirmed as a specific RTK in PC, and it is abundant in PC tissues and positively correlates with PD-L1 levels. Notably, the MET inhibitor capmatinib has a pivotal function in restraining PD-L1 expression and stopping tumor progression [149]. By inhibiting UBQLN4 and promoting ubiquitination in melanoma cells, Albendazole has an anti-tumor immunological impact, culminating in PD-L1 protein degradation [101].
The majority of the small molecule drugs mentioned above target not just PD-L1, but also additional proteins that influence tumor survival, proliferation, and metabolism. This gives them advantages over PD-L1 antibodies. Small molecule medications, however, may also activate alternate pathways or feedback mechanisms that control PD-L1 expression, producing ineffective anti-tumor effects. Consequently, concurrently in-depth comprehension of PD-L1 antibodies is still required. There are currently four types of PD-L1 inhibitors approved by the FDA, including Avelumab, Atezolizumab, Durvalumab, and Cemiplimab (Table 4). Avelumab (MSB0010718C) is a fully human IgG1 monoclonal antibody that can mediate ADCC by targeting PD-L1 [18]. It is effective in clinical trials for treating metastatic merkel cell carcinoma (MCC), metastatic UC, RCC and is well tolerated by patients [150,151,152,153]. Remarkably, Avelumab was the first drug licensed by the FDA for the treatment of MCC. Currently, atezolizumab (MPDL3208A), a humanized human and murine cross-reactive therapeutic PD-L1 antibody, is in clinical trials for patients with NSCLC and locally advanced and metastatic urothelial carcinoma [154, 155]. Durvalumab is a PD-L1-targeting immunosuppressant that the European Medicines Agency approved for the consolidation of locally advanced PD-L1-positive NSCLC following chemoradiotherapy [156]. While maintenance chemotherapy is more successful than duvacizumab in patients with hormone receptor-positive and HER2-negative breast cancer, duvacizumab increases overall survival (OS) in PD-L1-positive TNBC[157]. Cemiplimab is presently approved by the FDA for the treatment of patients with metastatic cutaneous squamous cell carcinoma (CSCC) or locally advanced unresectable CSCC and NSCLC [158, 159].
Conclusions
Cancer immunotherapy with PD-1/PD-L1 blocking antibodies has ushered in a new era of cancer treatment, but only a fraction of patients have shown objective clinical responses. Currently, FDA-approved PD-L1 inhibitors are indicated for locally advanced or metastatic UC, NSCLC, MCC, and CSSS. PD-L1 expression in tumors, microsatellite instability, tumor mutational burden, and tumor-infiltrating lymphocytes are biomarkers that indicate how well PD-1/PD-L1 inhibitors work [160]. Treatment with small-molecule drugs that modulate PD-L1 expression is a favorable strategy for cancer therapy; nevertheless, it can be a double-edged sword. On the one hand, induction of PD-L1 expression in tumor cells may increase the sensitivity of cancer cells to PD-1/PD-L1 immune checkpoint blockade, such as Poly-(ADP-ribose) polymerase (PARP) inhibitors in breast cancer and MET proto-oncogene, receptor tyrosine kinase (MET) inhibitors in HCC. Nevertheless, inducing PD-L1 expression artificially may also promote immunosuppression. On the other hand, the combination of PD-L1-targeting small-molecule drugs with anti-PD-1/PD-L1 antibodies may produce synergistic anticancer activity, but it may also render the antibodies ineffective due to loss of immune checkpoint expression. As noted in this review, the expression of PD-L1 is governed by a number of regulatory mechanisms. Transcription factors, oncogenes, tumor suppressor genes, and microRNAs all regulate PD-L1, affecting anti-tumor immune responses. Selecting one or the other to alter immune checkpoint expression may be inefficient, as other regulatory elements may compensate and become overactivated. Therefore, further preclinical and clinical studies will be needed to advance the understanding of tumor immune evasion mechanisms and the search for optimal combination therapies.
In conclusion, understanding the underlying regulatory pathways will improve current immunotherapies by manipulating PD-L1 expression.

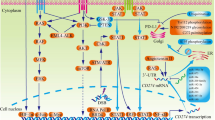
The expression and biological function of PD-L1. PD-L1 is expressed in hematopoietic cells, including T cells, B cells, DCs, macrophages, mast cells, and many non-hematopoietic cell types. PD-1 binds to PD-L1 to induce cancer cell immune escape, proliferation, drug resistance, and autophagy, and PD-1/PD-L1 blockade can inhibit these functions

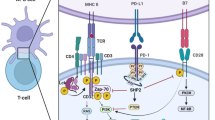
Overview of the regulatory mechanisms involved in PD-L1 expression. By attaching to the PD-L1 promoter, numerous transcription factors contribute to the increase of PD-L1 expression. N6-methyladenosine increases PD-L1 expression while DNA methylation, histone modification, and autophagy suppress it. MicroRNAs, including miR-138, miR-138-5p, miR-152, and others shown in Table 1, suppress PD-L1 by directly binding to the 3’UTR of PD-L1 mRNA. LncRNAs and circRNAs are also relevant to PD-L1 expression and tumor immune escape. PD-L1 is upregulated by glycosylation and palmitoylation, which stabilize PD-L1 protein, while ubiquitination, phosphorylation, and acetylation exert the opposite effect
Availability of data and materials
Not applicable.
Abbreviations
- PD-1:
-
Programmed cell-death protein 1
- PD-L1, CD274:
-
Programmed cell death 1 ligand 1
- ICs:
-
Immune-checkpoint
- CTLA4:
-
Cytotoxic T-lymphocyte associated protein 4
- LAG3:
-
Lymphocyte activating 3
- HAVCR2, TIM3:
-
Hepatitis A virus cellular receptor 2
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- TILs:
-
Tumor-infiltrating lymphocytes
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- HNSCC:
-
Head and neck squamous cell carcinoma
- NSCLC:
-
Non-small-cell lung cancer
- PLAC8:
-
Placenta-specific protein 8
- NRF2:
-
Nuclear factor E2-related factor 2
- HCC:
-
Hepatocellular carcinoma
- BRD4:
-
Bromodomain containing 4
- NF-κB:
-
Nuclear factor kappa-B
- CDK5:
-
Cyclin-dependent kinase 5
- HIF-1α:
-
Hypoxia inducible factor 1 subunit alpha
- AFT3:
-
Cyclic AMP-dependent transcription factor 3
- T-ALL:
-
T cell acute lymphoblastic leukemia
- BIN1:
-
Bridging integrator-1
- CDK7:
-
Cell cycle protein-dependent kinase 7
- BET:
-
Bromodomain and extra terminal domain
- OC:
-
Ovarian cancer
- IRF1:
-
Interferon regulatory factor 1
- CHL:
-
Classical Hodgkin lymphoma
- PMBCL:
-
Primary mediastinal large B-cell lymphoma
- JAK:
-
Janus kinase
- EGCG:
-
Epigallocatechin gallate
- NPM:
-
Nucleophosmin
- ALK:
-
Anaplastic lymphoma kinase
- ALK + TCL:
-
NPM/ALK-carrying T cell lymphoma
- GSDMC:
-
Gasdermin C
- BC:
-
Breast cancer
- EBV:
-
Epstein-Barr virus
- LMP1:
-
Latent membrane protein 1
- BSGWE:
-
Water extract of sporoderm-broken spores of G. lucidum
- GC:
-
Gastric cancer
- MAPK:
-
Mitogen activated kinase-like protein
- PI3K:
-
Phosphatidylinositol 3-kinase
- HDAC5:
-
Histone deacetylase 5
- PC:
-
Pancreatic cancer
- MDSCs:
-
Myeloid-derived suppressor cells
- AML:
-
Acute myeloid leukemia
- mTOR:
-
Mechanistic target of rapamycin kinase
- ADORA1:
-
Adenosine A1 receptor
- PTEN:
-
Phosphatase and tensin homolog
- 5mC:
-
5-methylcytosine
- HDAC:
-
Histone deacetylase
- TET:
-
Ten-eleven translocation
- SAHA:
-
Suberoylanilide hydroxamic acid
- H3K27me3:
-
Histone H3 lysine 27 trimethylation
- MLL1:
-
Lysine methyltransferase 2 A
- H3K4me3:
-
Histone H3 lysine 4 trimethylation
- miRNAs:
-
microRNAs
- CKS1B:
-
CDC28 protein kinase regulatory subunit 1B
- HOXC4:
-
Homeobox C4
- MAGI2:
-
Membrane-associated guanylate kinase inverted 2
- LATS2:
-
Large tumor suppressor kinase 2
- YAP1:
-
Yes1 associated transcriptional regulator
- ARID3A:
-
AT-rich interaction domain 3 A
- KDM1A:
-
Lysine-specific demethylase 1 A
- CC:
-
Colorectal cancer
- LUAD:
-
Lung adenocarcinoma
- MM:
-
Malignant mesothelioma
- LC:
-
Lung carcinogenesis
- DLBCL:
-
Diffuse large B cell lymphoma
- lncRNA:
-
Long non-coding RNA
- circRNA:
-
Circular RNA
- SNHG12:
-
Small nucleolar RNA host gene 12
- EMT:
-
Epithelial-Mesenchymal Transition
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- E2F1:
-
E2F transcription factor 1
- CASC11:
-
Cancer susceptibility candidate 11
- OSCC:
-
Oral squamous carcinoma
- m6A:
-
N6-methyladenosine
- PRMT5:
-
Protein arginine methyltransferase 5
- YTHDF:
-
YTH domain family proteins
- FTO:
-
Fat mass and obesity-associated protein
- CSN5:
-
COP9 signalosome 5
- HCD:
-
High-cholesterol diet
- CCL5:
-
C-C motif chemokine ligand 5
- TRAF6:
-
TNF receptor associated factor 6
- UBQLN4:
-
Ubiquilin 4
- GSK3β:
-
Glycogen synthase kinase 3β
- AMPK:
-
AMP-activated catalytic subunit alpha 1
- HIP1R:
-
Huntingtin interacting protein 1 related
- ROS:
-
Reactive oxygen species
- BECN1:
-
Beclin 1
- SQSTM1:
-
Sequestosome 1
- UC:
-
Urothelial carcinoma
- TNBC:
-
Triple-negative breast cancer
- RCC:
-
Renal cell carcinoma
- TRIM28:
-
Tripartite motif containing 28
- TGF-β:
-
Transforming growth factor-beta
- PROTACs:
-
Proteolysis targeting chimeras
- RTKs:
-
Receptor tyrosine kinases
- MCC:
-
Merkel cell carcinoma
- OS:
-
Overall survival
- CSCC:
-
Cutaneous squamous cell carcinoma
- PARP:
-
Poly-(ADP-ribose) polymerase
- MET:
-
MET proto-oncogene, receptor tyrosine kinase
- ORR:
-
Objective response rate
- mPFS:
-
Median progression-free survival
- mOS:
-
Median overall survival
References
Topalian SL, Drake CG and Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015. 27(4): p. 450–61.
Mohammadi P, Hesari M, Chalabi M, Salari F and Khademi F. An overview of immune checkpoint therapy in autoimmune diseases. Int Immunopharmacol. 2022. 107: p. 108647.
Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol. 2020;11:167.
Yu X, Gao R, Li Y and Zeng C. Regulation of PD-1 in T cells for cancer immunotherapy. Eur J Pharmacol. 2020. 881: p. 173240.
Rezaei M, Tan J, Zeng C, Li Y, Ganjalikhani-Hakemi M. TIM-3 in leukemia; immune response and beyond. Front Oncol. 2021;11:753677.
Marin-Acevedo JA, Kimbrough EO and Lou Y. Next generation of immune checkpoint inhibitors and beyond. J Hematol Oncol. 2021. 14(1): p. 45.
Chen C, Liang C, Wang S, Chio CL, Zhang Y, Zeng C, et al. Expression patterns of immune checkpoints in acute myeloid leukemia. J Hematol Oncol. 2020. 13(1): p. 28.
Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8(328):328rv4.
Iwai Y, Hamanishi J, Chamoto K and Honjo T. Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci. 2017. 24(1): p. 26.
Sharpe AH, Wherry EJ, Ahmed R and Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007. 8(3): p. 239–45.
Yamaguchi H, Hsu JM, Yang WH and Hung MC. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat Rev Clin Oncol. 2022. 19(5): p. 287–305.
Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med. 2016;375(18):1767–78.
Nguyen LT and Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015. 15(1): p. 45–56.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002. 8(8): p. 793–800.
Martin-Orozco N, Wang YH, Yagita H, Dong C. Cutting edge: programmed death (PD) ligand-1/PD-1 interaction is required for CD8 + T cell tolerance to tissue antigens. J Immunol. 2006;177(12):8291–5.
Chen B, Hu J, Hu X, Chen H, Bao R, Zhou Y, et al. DENR controls JAK2 translation to induce PD-L1 expression for tumor immune evasion. Nat Commun. 2022;13(1):2059.
Friedman J, Padget M, Lee J, Schlom J, Hodge J, and Allen C. Direct and antibody-dependent cell-mediated cytotoxicity of head and neck squamous cell carcinoma cells by high-affinity natural killer cells. Oral Oncol. 2019. 90: p. 38–44.
Boyerinas B, Jochems C, Fantini M, Heery CR, Gulley JL, Tsang KY, et al. Antibody-dependent cellular cytotoxicity activity of a novel anti-PD-L1 antibody avelumab (MSB0010718C) on human tumor cells. Cancer Immunol Res. 2015;3(10):1148–57.
Du W, Zhu J, Zeng Y, Liu T, Zhang Y, Cai T, et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Differ. 2021. 28(4): p. 1284–1300.
Saigusa S, Toiyama Y, Tanaka K, Inoue Y, Mori K, Ide S, et al. Implication of programmed cell death ligand 1 expression in tumor recurrence and prognosis in rectal cancer with neoadjuvant chemoradiotherapy. Int J Clin Oncol. 2016. 21(5): p. 946–952.
Gong K, Gong ZJ, Lu PX, Ni XL, Shen S, Liu H, et al. PLAC8 overexpression correlates with PD-L1 upregulation and acquired resistance to chemotherapies in gallbladder carcinoma. Biochem Biophys Res Commun. 2019. 516(3): p. 983–990.
Li D, Sun FF, Wang D, Wang T, Peng JJ, Feng JQ, et al. Programmed death ligand-1 (PD-L1) regulated by NRF-2/MicroRNA-1 regulatory axis enhances drug resistance and promotes tumorigenic properties in sorafenib-resistant hepatoma cells. Oncol Res. 2020;28(5):467–81.
Chen Y, Gao M, Huang Z, Yu J and Meng X. SBRT combined with PD-1/PD-L1 inhibitors in NSCLC treatment: a focus on the mechanisms, advances, and future challenges. J Hematol Oncol. 2020. 13(1): p. 105.
Yi M, Niu M, Xu L, Luo S and Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021. 14(1): p. 10.
Zeng C, Liu S, Lu S, Yu X, Lai J, Wu Y, et al. The c-Myc-regulated lncRNA NEAT1 and paraspeckles modulate imatinib-induced apoptosis in CML cells. Mol Cancer. 2018. 17(1): p. 130.
Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016. 352(6282): p. 227–31.
Wang J, Jia Y, Zhao S, Zhang X, Wang X, Han X, et al. BIN1 reverses PD-L1-mediated immune escape by inactivating the c-MYC and EGFR/MAPK signaling pathways in non-small cell lung cancer. Oncogene. 2017. 36(45): p. 6235–6243.
Wang J, Zhang R, Lin Z, Zhang S, Chen Y, Tang J, et al. CDK7 inhibitor THZ1 enhances antiPD-1 therapy efficacy via the p38alpha/MYC/PD-L1 signaling in non-small cell lung cancer. J Hematol Oncol. 2020. 13(1): p. 99.
Chen C, Xu L, Gao R, Wang S, Zhang Y, Wang C, et al. Transcriptome-Based Co-Expression of BRD4 and PD-1/PD-L1 Predicts Poor Overall Survival in Patients With Acute Myeloid Leukemia. Front Pharmacol. 2020. 11: p. 582955.
Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18(9):2162–74.
Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–37.
Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, et al. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19(6):1189–201.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010. 116(17): p. 3268–77.
Ravindran Menon D, Li Y, Yamauchi T, Osborne DG, Vaddi PK, Wempe MF, et al. EGCG inhibits tumor growth in melanoma by targeting JAK-STAT signaling and its downstream PD-L1/PD-L2-PD1 axis in tumors and enhancing cytotoxic T-cell responses. Pharmaceuticals (Basel). 2021;14(11):1081.
Lailler C, Lamuraglia M, Racine F, Louandre C, Godin C, Chauffert B, et al. DNA damage response- and JAK-dependent regulation of PD-L1 expression in head and neck squamous cell carcinoma (HNSCC) cells exposed to 5-fluorouracil (5-FU). Transl Oncol. 2021. 14(8): p. 101110.
Loke P and Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A. 2003. 100(9): p. 5336–41.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc Natl Acad Sci U S A. 2008. 105(52): p. 20852–7.
Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020. 22(10): p. 1264–1275.
Fang W, Zhang J, Hong S, Zhan J, Chen N, Qin T, et al. EBV-driven LMP1 and IFN-gamma up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget. 2014. 5(23): p. 12189–202.
He J, Zhang W, Di T, Meng J, Qi Y, Li G, et al. Water extract of sporoderm-broken spores of Ganoderma lucidum enhanced pd-l1 antibody efficiency through downregulation and relieved complications of pd-l1 monoclonal antibody. Biomed Pharmacother. 2020. 131: p. 110541.
You W, Liu X, Yu Y, Chen C, Xiong Y, Liu Y, et al. miR-502-5p affects gastric cancer progression by targeting PD-L1. Cancer Cell Int. 2020. 20(1): p. 395.
Bi XW, Wang H, Zhang WW, Wang JH, Liu WJ, Xia ZJ, et al. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol. 2016. 9(1): p. 109.
Gowrishankar K, Gunatilake D, Gallagher SJ, Tiffen J, Rizos H, and Hersey P. Inducible but not constitutive expression of PD-L1 in human melanoma cells is dependent on activation of NF-kappaB. PLoS One. 2015. 10(4): p. e0123410.
Zhang Y, Zhu S, Du Y, Xu F, Sun W, Xu Z, et al. RelB upregulates PD-L1 and exacerbates prostate cancer immune evasion. J Exp Clin Cancer Res. 2022. 41(1): p. 66.
Zhou Y, Jin X, Yu H, Qin G, Pan P, Zhao J, et al. HDAC5 modulates PD-L1 expression and cancer immunity via p65 deacetylation in pancreatic cancer. Theranostics. 2022. 12(5): p. 2080–2094.
Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, et al. Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science. 2016. 353(6297): p. 399–403.
Chen J, Jiang CC, Jin L and Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016. 27(3): p. 409–16.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014. 211(5): p. 781–90.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014. 211(5): p. 781–90.
Ding XC, Wang LL, Zhang XD, Xu JL, Li PF, Liang H, et al. The relationship between expression of PD-L1 and HIF-1α in glioma cells under hypoxia. J Hematol Oncol. 2021. 14(1): p. 92.
Noman MZ and Chouaib S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology. 2015. 3(12): p. e954463-e954463.
Ma P, Xing M, Han L, Gan S, Ma J, Wu F, et al. High PDL1 expression drives glycolysis via an Akt/mTOR/HIF1alpha axis in acute myeloid leukemia. Oncol Rep. 2020. 43(3): p. 999–1009.
Shen Y, Liu L, Wang M, Xu B, Lyu R, Shi YG, et al. TET2 inhibits PD-L1 gene expression in breast cancer cells through histone deacetylation. Cancers (Basel). 2021;13(9):2207.
Payandeh Z, Pirpour Tazehkand A, Mansoori B, Khaze V, Asadi M, Baradaran B, et al. The impact of Nrf2 silencing on Nrf2-PD-L1 axis to overcome oxaliplatin resistance and migration in colon cancer cells. Avicenna J Med Biotechnol. 2021;13(3):116–22.
Liu H, Kuang X, Zhang Y, Ye Y, Li J, Liang L, et al. ADORA1 inhibition promotes tumor immune evasion by regulating the ATF3-PD-L1 axis. Cancer Cell. 2020;37(3):324-339 e8.
Chen S, Ma S, Wang H, Shao X, Ding B, Guo Z, et al. Unraveling the mechanism of alkaloids from Sophora alopecuroides Linn combined with immune checkpoint blockade in the treatment of non-small cell lung cancer based on systems pharmacology. Bioorg Med Chem. 2022. 64: p. 116724.
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007. 13(1): p. 84–8.
Cortez MA, Ivan C, Valdecanas D, Wang X, Peltier HJ, Ye Y, et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst. 2016. 108(1):djv303.
Angeloni A and Bogdanovic O. Enhancer DNA methylation: implications for gene regulation. Essays Biochem. 2019. 63(6): p. 707–715.
Franzen A, Vogt TJ, Muller T, Dietrich J, Schrock A, Golletz C, et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget. 2018. 9(1): p. 641–650.
Goltz D, Gevensleben H, Dietrich J and Dietrich D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology. 2017. 6(1): p. e1257454.
Micevic G, Thakral D, McGeary M and Bosenberg MW. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019. 32(3): p. 435–440.
Amini M, Hejazi M, Ghorban K, Mokhtarzadeh A and Baradaran B. Identification of functional methylated CpG loci in PD-L1 promoter as the novel epigenetic biomarkers for primary gastric cancer. Gene. 2021. 772: p. 145376.
Emran AA, Chatterjee A, Rodger EJ, Tiffen JC, Gallagher SJ, Eccles MR, et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol. 2019;40(4):328–44.
Wang H, Fu C, Du J, Wang H, He R, Yin X, et al. Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J Exp Clin Cancer Res. 2020. 39(1): p. 29.
Rathinavelu A. Regulation of PD-L1 expression by histone deacetylase inhibitor SAHA in lung cancer cells. Biomed J Sci Tech Res. 2019;19(4):14519–22.
Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM, Zhou ZG, et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J Immunother Cancer. 2019. 7(1): p. 300.
Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J Natl Cancer Inst. 2017;109(6):djw283.
Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015. 27(3): p. 443–52.
Gong AY, Zhou R, Hu G, Li X, Splinter PL, O’Hara SP, et al. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J Immunol. 2009. 182(3): p. 1325–33.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014. 5: p. 5241.
Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. 2015. 23(4): p. 717–27.
Jiang W, Pan S, Chen X, Wang ZW and Zhu X. The role of lncRNAs and circRNAs in the PD-1/PD-L1 pathway in cancer immunotherapy. Mol Cancer. 2021. 20(1): p. 116.
Guo Y, Xie Y, Luo Y. The role of long non-coding RNAs in the tumor immune microenvironment. Front Immunol. 2022;13:851004.
Rong D, Sun H, Li Z, Liu S, Dong C, Fu K, et al. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget. 2017. 8(42): p. 73271–73281.
Shang A, Wang W, Gu C, Chen C, Zeng B, Yang Y, et al. Long non-coding RNA HOTTIP enhances IL-6 expression to potentiate immune escape of ovarian cancer cells by upregulating the expression of PD-L1 in neutrophils. J Exp Clin Cancer Res. 2019. 38(1): p. 411.
Qian M, Ling W and Ruan Z. Long non-coding RNA SNHG12 promotes immune escape of ovarian cancer cells through their crosstalk with M2 macrophages. Aging (Albany NY). 2020. 12(17): p. 17122–17136.
Zhang C, Jiang F, Su C, Xie P and Xu L. Upregulation of long noncoding RNA SNHG20 promotes cell growth and metastasis in esophageal squamous cell carcinoma via modulating ATM-JAK-PD-L1 pathway. J Cell Biochem. 2019. 120(7): p. 11642–11650.
Zhao L, Liu Y, Zhang J, Liu Y and Qi Q. LncRNA SNHG14/miR-5590-3p/ZEB1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating PD-1/PD-L1 checkpoint. Cell Death Dis. 2019. 10(10): p. 731.
Song H, Liu Y, Li X, Chen S, Xie R, Chen D, et al. Long noncoding RNA CASC11 promotes hepatocarcinogenesis and HCC progression through EIF4A3-mediated E2F1 activation. Clin Transl Med. 2020. 10(7): p. e220.
Duan M, Fang M, Wang C, Wang H and Li M. LncRNA EMX2OS Induces Proliferation, Invasion and Sphere Formation of Ovarian Cancer Cells via Regulating the miR-654-3p/AKT3/PD-L1 Axis. Cancer Manag Res. 2020. 12: p. 2141–2154.
Wang J, Yu Z, Wang J, Shen Y, Qiu J, and Zhuang Z. LncRNA NUTM2A-AS1 positively modulates TET1 and HIF-1A to enhance gastric cancer tumorigenesis and drug resistance by sponging miR-376a. Cancer Med. 2020. 9(24): p. 9499–9510.
Wang S, You H and Yu S. Long non-coding RNA HOXA-AS2 promotes the expression levels of hypoxia-inducible factor-1alpha and programmed death-ligand 1, and regulates nasopharyngeal carcinoma progression via miR-519. Oncol Lett. 2020. 20(5): p. 245.
Shi L, Yang Y, Li M, Li C, Zhou Z, Tang G, et al. LncRNA IFITM4P promotes immune escape by up-regulating PD-L1 via dual mechanism in oral carcinogenesis. Mol Ther. 2022. 30(4): p. 1564–1577.
Ma H, Chang H, Yang W, Lu Y, Hu J, and Jin S. A novel IFNalpha-induced long noncoding RNA negatively regulates immunosuppression by interrupting H3K27 acetylation in head and neck squamous cell carcinoma. Mol Cancer. 2020. 19(1): p. 4.
Luo YH, Yang YP, Chien CS, Yarmishyn AA, Adekunle Ishola A, Chien Y, et al. Circular RNA hsa_circ_0000190 facilitates the tumorigenesis and immune evasion by upregulating the expression of soluble PD-L1 in non-small-cell lung cancer. Int J Mol Sci. 2021;23(1):64.
Wang J, Zhao X, Wang Y, Ren F, Sun D, Yan Y, et al. circRNA-002178 act as a ceRNA to promote PDL1/PD1 expression in lung adenocarcinoma. Cell Death Dis. 2020;11(1):32.
Tanaka E, Miyakawa Y, Kishikawa T, Seimiya T, Iwata T, Funato K, et al. Expression of circular RNA CDR1AS in colon cancer cells increases cell surface PDL1 protein levels. Oncol Rep. 2019. 42(4): p. 1459–1466.
Zeng C, Huang W, Li Y and Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020. 13(1): p. 117.
Ai Y, Liu S, Luo H, Wu S, Wei H, Tang Z, et al. METTL3 intensifies the progress of oral squamous cell carcinoma via modulating the m6A amount of PRMT5 and PD-L1. J Immunol Res. 2021;2021:6149558.
Wan W, Ao X, Chen Q, Yu Y, Ao L, Xing W, et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N(6)-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol Cancer. 2022. 21(1): p. 60.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014. 505(7481): p. 117–20.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)A demethylase ALKBH5 regulates PD-L1 expression and tumor immunoenvironment in intrahepatic cholangiocarcinoma. Cancer Res. 2021;81(18):4778–93.
Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020. 530(1): p. 235–239.
Faktor J, Pjechová M, Hernychová L, Vojtěšek B. Protein ubiquitination research in oncology. Klin Onkol. 2019;32(Supplementum 3):56–64.
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30(6):925–39.
Liu Y, Liu X, Zhang N, Yin M, Dong J, Zeng Q, et al. Berberine diminishes cancer cell PD-L1 expression and facilitates antitumor immunity via inhibiting the deubiquitination activity of CSN5. Acta Pharm Sin B. 2020. 10(12): p. 2299–2312.
Liu C, Yao Z, Wang J, Zhang W, Yang Y, Zhang Y, et al. Correction: macrophage-derived CCL5 facilitates immune escape of colorectal cancer cells via the p65/STAT3-CSN5-PD-L1 pathway. Cell Death Differ. 2020;27(7):2293.
Xiong W, Gao X, Zhang T, Jiang B, Hu MM, Bu X, et al. USP8 inhibition reshapes an inflamed tumor microenvironment that potentiates the immunotherapy. Nat Commun. 2022. 13(1): p. 1700.
Zhu L, Kuang X, Zhang G, Liang L, Liu D, Hu B, et al. Albendazole induces immunotherapy response by facilitating ubiquitin-mediated PD-L1 degradation. J Immunother Cancer. 2022. 10(5):e003819.
Yang Z, Wang Y, Liu S, Deng W, Lomeli SH, Moriceau G, et al. Enhancing PD-L1 Degradation by ITCH during MAPK Inhibitor Therapy Suppresses Acquired Resistance. Cancer Discov. 2022.
Xiao M, Hasmim M, Lequeux A, Moer KV, Tan TZ, Gilles C, et al. Epithelial to mesenchymal transition regulates surface PD-L1 via CMTM6 and CMTM7 induction in breast cancer. Cancers (Basel). 2021;13(5):1165.
Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017. 549(7670): p. 101–105.
Ma Z, Wang H, Meng F, Han Y, Chen Y, Xiao M, et al. Role of BCLAF-1 in PD-L1 stabilization in response to ionizing irradiation. Cancer Sci. 2021. 112(10): p. 4064–4074.
Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017. 549(7670): p. 106–110.
Cheung JC and Reithmeier RA. Scanning N-glycosylation mutagenesis of membrane proteins. Methods. 2007. 41(4): p. 451–9.
Daver N, Basu S, Garcia-Manero G, Cortes JE, Ravandi F, Ning J, et al. Defining the immune checkpoint landscape in patients (pts) with acute myeloid leukemia (AML). Blood. 2016;128(22):2900.
Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9(1):1908.
Ruan Z, Liang M, Lai M, Shang L, Deng X, and Su X. KYA1797K down-regulates PD-L1 in colon cancer stem cells to block immune evasion by suppressing the beta-catenin/STT3 signaling pathway. Int Immunopharmacol. 2020. 78: p. 106003.
Maher CM, Thomas JD, Haas DA, Longen CG, Oyer HM, Tong JY, et al. Small-molecule sigma1 modulator induces autophagic degradation of PD-L1. Mol Cancer Res. 2018;16(2):243–55.
D’Arrigo P, Russo M, Rea A, Tufano M, Guadagno E, Del Basso De Caro ML, et al. A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget. 2017. 8(40): p. 68291–68304.
Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res. 2018;78(22):6349–53.
Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016. 7: p. 12632.
Chan LC, Li CW, Xia W, Hsu JM, Lee HH, Cha JH, et al. IL-6/JAK1 pathway drives PD-L1 Y112 phosphorylation to promote cancer immune evasion. J Clin Invest. 2019. 129(8): p. 3324–3338.
Cha JH, Yang WH, Xia W, Wei Y, Chan LC, Lim SO, et al. Metformin promotes antitumor immunity via endoplasmic-reticulum-associated degradation of PD-L1. Mol Cell. 2018;71(4):606-620 e7.
Gao Y, Nihira NT, Bu X, Chu C, Zhang J, Kolodziejczyk A, et al. Acetylation-dependent regulation of PD-L1 nuclear translocation dictates the efficacy of anti-PD-1 immunotherapy. Nat Cell Biol. 2020. 22(9): p. 1064–1075.
Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019. 3(4): p. 306–317.
Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, et al. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019. 29(1): p. 83–86.
Antonioli M, Di Rienzo M, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42(1):28–41.
Xia H, Green DR and Zou W. Autophagy in tumour immunity and therapy. Nat Rev Cancer. 2021. 21(5): p. 281–297.
Gao H, Zhang J and Ren X. PD-L1 regulates tumorigenesis and autophagy of ovarian cancer by activating mTORC signaling. Biosci Rep. 2019. 39(12):BSR20191041.
Wang X, Wu WKK, Gao J, Li Z, Dong B, Lin X, et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J Exp Clin Cancer Res. 2019. 38(1): p. 140.
Wu X, Gu Z, Chen Y, Chen B, Chen W, Weng L, et al. Application of PD-1 blockade in cancer immunotherapy. Comput Struct Biotechnol J. 2019;17:661–74.
Bagchi S, Yuan R, Engleman EG. Immune checkpoint inhibitors for the treatment of cancer: clinical impact and mechanisms of response and resistance. Annu Rev Pathol. 2021;16:223–49.
Frydenlund N and Mahalingam M. PD-L1 and immune escape: insights from melanoma and other lineage-unrelated malignancies. Hum Pathol. 2017. 66: p. 13–33.
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, et al. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48(3):434–52.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022. 15(1): p. 24.
Bu LL, Yu GT, Wu L, Mao L, Deng WW, Liu JF, et al. STAT3 induces immunosuppression by upregulating PD-1/PD-L1 in HNSCC. J Dent Res. 2017;96(9):1027–34.
M L, P PV, T K, M P, E S, J P, et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol Oncol. 2016. 10(5): p. 735–750.
Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat Med. 2018. 24(12): p. 1877–1886.
Liang J, Wang L, Wang C, Shen J, Su B, Marisetty AL, et al. Verteporfin inhibits PD-L1 through autophagy and the STAT1-IRF1-TRIM28 signaling axis, exerting antitumor efficacy. Cancer Immunol Res. 2020;8(7):952–65.
Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, et al. The MLL1 H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J Natl Cancer Inst. 2017. 109(6):djw283.
Sheng Q, Zhang Y, Wang Z, Ding J, Song Y, and Zhao W. Cisplatin-mediated down-regulation of miR-145 contributes to up-regulation of PD-L1 via the c-Myc transcription factor in cisplatin-resistant ovarian carcinoma cells. Clin Exp Immunol. 2020. 200(1): p. 45–52.
Yi M, Zhang J, Li A, Niu M, Yan Y, Jiao Y, et al. The construction, expression, and enhanced anti-tumor activity of YM101: a bispecific antibody simultaneously targeting TGF-beta and PD-L1. J Hematol Oncol. 2021. 14(1): p. 27.
Yi M, Niu M, Zhang J, Li S, Zhu S, Yan Y, et al. Combine and conquer: manganese synergizing anti-TGF-beta/PD-L1 bispecific antibody YM101 to overcome immunotherapy resistance in non-inflamed cancers. J Hematol Oncol. 2021. 14(1): p. 146.
Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc. 2021;143(2):593–8.
Wang Y, Zhou Y, Cao S, Sun Y, Dong Z, Li C, et al. In vitro and in vivo degradation of programmed cell death ligand 1 (PD-L1) by a proteolysis targeting chimera (PROTAC). Bioorg Chem. 2021. 111: p. 104833.
Wang H, Yao H, Li C, Shi H, Lan J, Li Z, et al. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nat Chem Biol. 2019. 15(1): p. 42–50.
Dai X, Bu X, Gao Y, Guo J, Hu J, Jiang C, et al. Energy status dictates PD-L1 protein abundance and anti-tumor immunity to enable checkpoint blockade. Mol Cell. 2021. 81(11): p. 2317–2331 e6.
Huang KC, Chiang SF, Chen WT, Chen TW, Hu CH, Yang PC, et al. Decitabine augments chemotherapy-induced PD-L1 upregulation for PD-L1 blockade in colorectal cancer. Cancers (Basel). 2020;12(2):462.
Yang H, Bueso-Ramos C, DiNardo C, Estecio MR, Davanlou M, Geng QR, et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia. 2014. 28(6): p. 1280–8.
Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, and Zhang Y. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget. 2017. 8(60): p. 101535–101544.
Wang S, Yao F, Lu X, Li Q, Su Z, Lee JH, et al. Temozolomide promotes immune escape of GBM cells via upregulating PD-L1. Am J Cancer Res. 2019. 9(6): p. 1161–1171.
Wu RY, Kong PF, Xia LP, Huang Y, Li ZL, Tang YY, et al. Regorafenib promotes antitumor immunity via inhibiting PD-L1 and IDO1 expression in melanoma. Clin Cancer Res. 2019;25(14):4530–41.
Deng Y, Xia X, Zhao Y, Zhao Z, Martinez C, Yin W, et al. Glucocorticoid receptor regulates PD-L1 and MHC-I in pancreatic cancer cells to promote immune evasion and immunotherapy resistance. Nat Commun. 2021. 12(1): p. 7041.
Wen M, Cao Y, Wu B, Xiao T, Cao R, Wang Q, et al. PD-L1 degradation is regulated by electrostatic membrane association of its cytoplasmic domain. Nat Commun. 2021. 12(1): p. 5106.
Li E, Huang X, Zhang G and Liang T. Combinational blockade of MET and PD-L1 improves pancreatic cancer immunotherapeutic efficacy. J Exp Clin Cancer Res. 2021. 40(1): p. 279.
Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol. 2016. 17(10): p. 1374–1385.
Apolo AB, Infante JR, Balmanoukian A, Patel MR, Wang D, Kelly K, et al. Avelumab, an anti-programmed death-ligand 1 antibody, in patients with refractory metastatic urothelial carcinoma: results from a multicenter, phase Ib study. J Clin Oncol. 2017;35(19):2117–24.
Motzer RJ, Penkov K, Haanen J, Rini B, Albiges L, Campbell MT, et al. Avelumab plus Axitinib versus Sunitinib for advanced renal-cell carcinoma. N Engl J Med. 2019;380(12):1103–15.
D'Angelo SP, Bhatia S, Brohl AS, Hamid O, Mehnert JM, Terheyden P, et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J Immunother Cancer. 2020. 8(1):e000674.
Jotte R, Cappuzzo F, Vynnychenko I, Stroyakovskiy D, Rodriguez-Abreu D, Hussein M, et al. Atezolizumab in combination with carboplatin and nab-paclitaxel in advanced squamous NSCLC (IMpower131): results from a randomized phase III trial. J Thorac Oncol. 2020;15(8):1351–60.
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A, et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016. 387(10031): p. 1909–1920.
Faivre-Finn C, Spigel DR, Senan S, Langer C, Perez BA, Özgüroğlu M, et al. Impact of prior chemoradiotherapy-related variables on outcomes with durvalumab in unresectable Stage III NSCLC (PACIFIC). Lung Cancer. 2021. 151: p. 30–38.
Bachelot T, Filleron T, Bieche I, Arnedos M, Campone M, Dalenc F, et al. Durvalumab compared to maintenance chemotherapy in metastatic breast cancer: the randomized phase II SAFIR02-BREAST IMMUNO trial. Nat Med. 2021. 27(2): p. 250–255.
Migden MR, Khushalani NI, Chang ALS, Lewis KD, Schmults CD, Hernandez-Aya L, et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: results from an open-label, phase 2, single-arm trial. Lancet Oncol. 2020. 21(2): p. 294–305.
Moreno V, Garrido P, Papadopoulos KP, De Miguel Luken MJ, Gil-Martin M, Aljumaily R, et al. Tolerability and antitumor activity of cemiplimab, a human monoclonal anti-PD-1, as monotherapy in patients with pretreated non-small cell lung cancer (NSCLC): Data from the Phase 1 NSCLC expansion cohort. Lung Cancer. 2021. 155: p. 151–155.
Grossman JE, Vasudevan D, Joyce CE and Hildago M. Is PD-L1 a consistent biomarker for anti-PD-1 therapy? The model of balstilimab in a virally-driven tumor. Oncogene. 2021. 40(8): p. 1393–1395.
Zhao L, Yu H, Yi S, Peng X, Su P, Xiao Z, et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget. 2016. 7(29): p. 45370–45384.
Ashizawa M, Okayama H, Ishigame T, Thar Min AK, Saito K, Ujiie D, et al. miRNA-148a-3p Regulates Immunosuppression in DNA Mismatch Repair-Deficient Colorectal Cancer by Targeting PD-L1. Mol Cancer Res. 2019. 17(6): p. 1403–1413.
Liu L, Yu T, Jin Y, Mai W, Zhou J, Zhao C. MicroRNA-15a carried by mesenchymal stem cell-derived extracellular vesicles inhibits the immune evasion of colorectal cancer cells by regulating the KDM4B/HOXC4/PD-L1 axis. Front Cell Dev Biol. 2021;9:629893.
Zhu J, Chen L, Zou L, Yang P, Wu R, Mao Y, et al. MiR-20b, -21, and – 130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum Immunol. 2014. 75(4): p. 348–53.
Wang Y, Wang D, Xie G, Yin Y, Zhao E, Tao K, et al. MicroRNA-152 regulates immune response via targeting B7-H1 in gastric carcinoma. Oncotarget. 2017. 8(17): p. 28125–28134.
Xie G, Li W, Li R, Wu K, Zhao E, Zhang Y, et al. Helicobacter pylori promote B7-H1 expression by suppressing miR-152 and miR-200b in gastric cancer cells. PLoS One. 2017;12(1):e0168822.
Li Z, Suo B, Long G, Gao Y, Song J, Zhang M, et al. Exosomal miRNA-16-5p derived from M1 macrophages enhances T cell-dependent immune response by regulating PD-L1 in gastric cancer. Front Cell Dev Biol. 2020;8:572689.
Xie WB, Liang LH, Wu KG, Wang LX, He X, Song C, et al. MiR-140 expression regulates cell proliferation and targets PD-L1 in NSCLC. Cell Physiol Biochem. 2018;46(2):654–63.
Li L, Zhang Q and Lian K. Circular RNA circ_0000284 plays an oncogenic role in the progression of non-small cell lung cancer through the miR-377-3p-mediated PD-L1 promotion. Cancer Cell Int. 2020. 20(1): p. 247.
Tang D, Zhao D, Wu Y, Yao R, Zhou L, Lu L, et al. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J Cell Mol Med. 2018. 22(8): p. 3847–3856.
Fujita Y, Yagishita S, Hagiwara K, Yoshioka Y, Kosaka N, Takeshita F, et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol Ther. 2015;23(4):717–27.
Huang J, Weng Q, Shi Y, Mao W, Zhao Z, Wu R, et al. MicroRNA-155-5p suppresses PD-L1 expression in lung adenocarcinoma. FEBS Open Bio. 2020. 10(6): p. 1065–1071.
Costa C, Indovina P, Mattioli E, Forte IM, Iannuzzi CA, Luzzi L, et al. P53-regulated miR-320a targets PDL1 and is downregulated in malignant mesothelioma. Cell Death Dis. 2020. 11(9): p. 748.
Zhang Q, Pan J, Xiong D, Wang Y, Miller MS, Sei S, et al. Pulmonary aerosol delivery of let-7b microRNA confers a striking inhibitory effect on lung carcinogenesis through targeting the tumor immune microenvironment. Adv Sci (Weinh). 2021;8(17):e2100629.
Lin YZ, Liu SH, Wu WR, Shen YC, Wang YL, Liao CC, et al. miR-4759 suppresses breast cancer through immune checkpoint blockade. Comput Struct Biotechnol J. 2022. 20: p. 241–251.
Yao X, Tu Y, Xu Y, Guo Y, Yao F, and Zhang X. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J Cell Mol Med. 2020. 24(17): p. 9560–9573.
Dou D, Ren X, Han M, Xu X, Ge X, Gu Y, et al. Cancer-associated fibroblasts-derived exosomes suppress immune cell function in breast cancer via the miR-92/PD-L1 pathway. Front Immunol. 2020;11:2026.
Xu S, Tao Z, Hai B, Liang H, Shi Y, Wang T, et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun. 2016. 7: p. 11406.
Jia L, Xi Q, Wang H, Zhang Z, Liu H, Cheng Y, et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun. 2017. 488(2): p. 425–431.
Zheng W, Lai G, Lin Q, Issah MA, Fu H, Shen JA. miR-129-5P/ARID3A negative feedback loop modulates diffuse large B cell lymphoma progression and immune evasion through regulating the PD-1/PD-L1 checkpoint. Front Cell Dev Biol. 2021;9:735855.
Wang Y, Cao K. KDM1A promotes immunosuppression in hepatocellular carcinoma by regulating PD-L1 through demethylating MEF2D. J Immunol Res. 2021;2021:9965099.
Yee D, Shah KM, Coles MC, Sharp TV and Lagos D. MicroRNA-155 induction via TNF-alpha and IFN-gamma suppresses expression of programmed death ligand-1 (PD-L1) in human primary cells. J Biol Chem. 2017. 292(50): p. 20683–20693.
Chen Y, Xie C, Zheng X, Nie X, Wang Z, Liu H, et al. LIN28/let-7/PD-L1 pathway as a target for cancer immunotherapy. Cancer Immunol Res. 2019;7(3):487–97.
Noman MZ and Chouaib S. Targeting hypoxia at the forefront of anticancer immune responses. Oncoimmunology. 2014. 3(12): p. e954463.
Deng S, Hu Q, Zhang H, Yang F, Peng C, Huang C. HDAC3 inhibition upregulates PD-L1 expression in B-cell lymphomas and augments the efficacy of anti-PD-L1 therapy. Mol Cancer Ther. 2019;18(5):900–8.
Luo YH, Yang YP, Chien CS, Yarmishyn AA, Ishola AA, Chien Y, et al. Circular RNA hsa_circ_0000190 facilitates the tumorigenesis and immune evasion by upregulating the expression of soluble PD-L1 in non-small-cell lung cancer. Int J Mol Sci. 2022;23(1):64.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the National Natural Science Foundation of China (No. 81770158, 82070152), the Pearl River S&T Nova Program of Guangzhou, China (No. 201906010002), Guangdong Province Science and Technology Project (No. 2020A0505100042) (No. 2020A1515110310), Guangdong Basic and Applied Basic Research Foundation (No. 2021A1515110140), Special Funds for the Cultivation of Guangdong College Students’ Scientific and Technological Innovation (“Climbing Program” Special Funds.) (No. pdjh2022a0055), and National Innovation and Entrepreneurship Training Program for Undergraduate (No. 202110559101) (No. 202210559061).
Author information
Authors and Affiliations
Contributions
ZDL and XBY collected the related literature, prepared the figures and drafted the manuscript. LX, YQL, and CWZ participated in the design of the review. ZDL, XBY, and CWZ critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Z., Yu, X., Xu, L. et al. Current insight into the regulation of PD-L1 in cancer. Exp Hematol Oncol 11, 44 (2022). https://doi.org/10.1186/s40164-022-00297-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00297-8