
Abstract
N6-methyladenosine (m6A) is the most abundant mRNA modification and is catalyzed by the methyltransferase complex, in which methyltransferase-like 3 (METTL3) is the sole catalytic subunit. Accumulating evidence in recent years reveals that METTL3 plays key roles in a variety of cancer types, either dependent or independent on its m6A RNA methyltransferase activity. While the roles of m6A modifications in cancer have been extensively reviewed elsewhere, the critical functions of METTL3 in various types of cancer, as well as the potential targeting of METTL3 as cancer treatment, have not yet been highlighted. Here we summarize our current understanding both on the oncogenic and tumor-suppressive functions of METTL3, as well as the underlying molecular mechanisms. The well-documented protein structure of the METTL3/METTL14 heterodimer provides the basis for potential therapeutic targeting, which is also discussed in this review.
Similar content being viewed by others
Introduction
There are more than 170 modifications in RNA, among which N6-methyladenosine (m6A) is the most prevalent internal modification in messenger RNA (mRNA) [1,2,3,4]. Over 7000 human transcripts harbor at least one m6A site, which is found within the consensus motif RRACH (where R = A/G, H = A/C/U), and most of the m6A sites are enriched in the coding sequence (CDS) and the 3′ untranslated region (3′UTR) of mRNA, especially around the stop codons [5, 6]. Although m6A was discovered more than 40 years ago [7], it failed to spark enthusiasm in this field until the identification of FTO as an m6A demethylase in 2011 [8], which reveals that m6A can be dynamically regulated and might play vital roles in development and diseases. Since then, FTO and ALKBH5, both belonging to the AlkB family of Fe(II)/a-ketoglutarate(a-KG)-dependent dioxygenases [9], were classified as m6A “eraser” proteins that remove m6A modifications from RNA (Fig. 1a and Table 1). In contrast to being removed by “eraser” proteins, m6A can be recognized by a set of RNA-binding proteins called m6A “reader” proteins that can specifically recognize and bind to m6A-modified transcripts. The list of m6A “reader” proteins is increasing (Table 1), including the YTH domain family proteins (YTHDC1/2, YTHDF1/2/3) [28, 36, 37, 41,42,43], the insulin-like growth factor 2 mRNA-binding proteins (IGF2BP1/2/3) [38], and the heterogeneous nuclear ribonucleoproteins (hnRNPC, hnRNPG), which were shown to mediate the regulation of RNA stability, translation efficiency, RNA splicing, and RNA exporting [44,45,46,47,48,49,50].

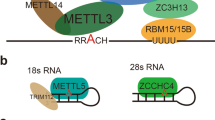
m6A RNA methylation and the structure of METTL3, the catalytic subunit of the m6A methyltransferase complex. a The writers and erasers of the dynamic m6A modification. b Schematic domain structure of METTL3. c Structure of the zinc finger domain (ZFD) of METTL3 (PDB ID: 5yz9). d Structure of the methyltransferase domain (MT-A70) of METTL3 (PDB ID: 5l6d)
The enzymes catalyzing the formation of m6A are referred to as m6A “writer” proteins, first being purified as a protein complex in 1994 by Bokar et al. [51], and were further characterized in recent years as a multicomponent m6A methyltransferase complex (MTC) comprised of a METTL3-METTL14 heterodimer core and other binding partners (Fig. 1a), such as WTAP, ZC3H13, VIRMA, and RBM15/15B [10,11,12,13,14]. In addition to the MTC, other m6A writers have also been identified in recent years, including METTL16, METTL5, and ZCCHC4 (Fig. 1a and Table 1), which are responsible for the deposition of m6A into structured RNAs, such as U6 snRNA, 28S rRNA, and 18S rRNA, and in some cases, the introns of mRNA [15,16,17,18,19,20,21,22]. The MTC core component METTL3-METTL14 heterodimer catalyzes most of m6A methylations in mRNA, with METTL3 being the only catalytic subunit that uses S-adenosylmethionine (SAM) as the methyl donor [52,53,54].
The full-length METTL3 has 580 amino acids and is comprised of a zinc finger domain (ZFD) and a methyltransferase domain (Fig. 1b), both of which are needed for the enzymatic activity. Huang et al. characterized the ZFD solution structure using nuclear magnetic resonance (NMR), showing that the domain contains two tandem CCCH-type zinc fingers (ZnF1 and ZnF2) connected by an anti-parallel β-sheet (Fig. 1c), which is responsible for target recognition, specifically for binding to single-stranded RNAs containing 5′-GGACU-3′ consensus sequence [55]. The structure of the methyltransferase domain of METTL3, named MT-A70, has been determined using X-ray crystallography in a complex with the corresponding domain of METTL14 by three independent groups [52,53,54]. It is demonstrated that METTL14 only plays a structural role for RNA-binding and stabilization of the complex, while METTL3 is the catalytically active subunit, with a co-factor binding pocket for SAM or S-adenosylhomocysteine (SAH) [52, 53]. The crystal structure reveals that the MT-A70 domain of METTL3 is formed by a Rossman fold comprising a central, curved, eight-stranded β-sheet flanked by four α- helixes, as well as an interface loop to interact with METTL14 and two gate loops that have important roles in adenosine recognition (Fig. 1d). The conserved DPPW motif (residues 395–399) of the enzyme is located in the gate loop 1, which undergoes a significant conformational change together with gate loop 2 upon SAM/SAH binding, resulting in the closure of the co-factor binding pocket [52].
Regulation of METTL3 expression and m6A deposition
The expression of METTL3 is dysregulated in cancer via different mechanisms (Fig. 2a). It was demonstrated in pancreatic cancer that cigarette smoke condensate induces hypomethylation of METTL3 promoter and subsequently the recruitment of transcription factor NFIC to induce METTL3 overexpression [56]. An intestinal microbial metabolite, butyrate, was suggested to downregulate the expression of METTL3 and inhibit the development of colorectal cancer; however, the detailed mechanism is unclear [57]. Wang et al. showed in gastric cancer that P300 mediates histone H3 acetylation at lysine 27 (H3K27ac) and promotes METTL3 transcription [58]. It was also reported that microRNA miR-24-2 might promote METTL3 transcription; however, the detailed mechanism remains elusive [59]. Several other microRNAs, including miR-186, miR-4429, miR-600, and let-7g [60,61,62,63], were proposed to suppress METTL3 by targeting METTL3 mRNA. In addition, SUMOylation of METTL3 protein was reported to repress its methyltransferase activity without affecting the protein stability or localization, although the mechanism remains unclear [64].

Regulation on the expression of METTL3 and its function on m6A deposition. a Multi-level regulation of METTL3. b Sequester or recruitment of METTL3 by RNA binding proteins (RBP). c Histone H3K36me3 directed deposition of m6A via recruiting of METTL14 and METTL3. d Recruitment of METTL3 by transcriptional factors (TF). e METTL3 guided by long non-coding RNAs (lncRNA). M3, METTL3; M14, METTL14
The methyltransferase activity of METTL3 is guided in diverse manners (Fig. 2b–e). It is suggested that m6A deposition could be governed in cis via the sequence code and structure at the modified site [65]; however, the mechanism underlying was unknown. Recently, it was shown that histone H3 trimethylation at lysine 36 (H3K36me3) could guide m6A RNA modification co-transcriptionally through direct interaction with METTL14 and the subsequent recruitment of the MTC complex, allowing for the selective deposition of m6A in CDS and 3′UTR where H3K36me3 is often found [66]. Increasing efforts have also been paid to trans regulators; among which, zinc finger protein 217 (ZFP217) was reported to sequester METTL3 and counteracts m6A deposition on stemness associated transcripts [67], while the TGFβ signaling factor SMAD2/3 could recruit METTL3/14 complex to a subset of transcripts involved in early cell fate decisions [68]. Another transcriptional factor, CAATT-box-binding protein CEBPZ, was demonstrated to directly recruit METTL3 to chromatin [68]. A large-scale computational screening aimed at identifying RNA-binding proteins as cell-specific trans regulators of m6A, and validated experimentally that TRA2A and CAPRIN1 could interact with METTL3 [69]. Fish et al. found that the RNA-binding protein TARBP2 recruits METTL3 and deposits m6A on the introns of the target mRNA, regulating RNA splicing and stability [70]. Two long non-coding RNAs (ARHGAP5-AS1 and LINC00470) have also been revealed to guide METTL3 to specific targets [71, 72], among which, the natural anti-sense transcript ARHGAP5-AS1 mediates METTL3 to deposit m6A marks on ARHGAP5 mRNA, promoting the mRNA stabilization and inducing chemoresistance [71].
METTL3 functions as an m6A methyltransferase in cancer
Accumulating evidence in recent years has demonstrated that METTL3 plays critical roles in cancer as an m6A methyltransferase, either as an oncogene or a tumor suppressor, as summarized in Table 2.
METTL3 as an oncogene
In most cases, METTL3 was reported as an oncogene to promote the initiation and development of a variety of cancers, including hematopoietic malignancies and solid tumors, through depositing m6A modification on critical transcripts (Fig. 3).

Molecular mechanisms underlying regulation of METTL3 on target genes in cancers. METTL3 methylates target transcripts, and the resulting m6A marks recruit m6A reader proteins to determine RNA fate. The target genes of METTL3 are in italics, with those involved in the tumor-suppressive function of METTL3 being boxed. CRC, colorectal cancer; BCa, bladder cancer, NSCLC, non-small cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; HCC, hepatocellular carcinoma; GBM, glioblastoma; GC, gastric cancer; AML, acute myeloid leukemia; OM, ocular melanoma; EC, endometrial cancer; TC, thyroid carcinoma
Acute myeloid leukemia
Acute myeloid leukemia (AML) is one of the most common hematopoietic malignancies with diverse genetic and molecular abnormalities, in which the hematopoietic stem and progenitor cells (HSPCs) retain the self-renewal capacity, while the myeloid differentiation capacity is hampered [121,122,123]. Vu et al. found that METTL3 was more abundant in AML cells than in normal HSPCs, and when overexpressed, the wild-type but not the catalytically inactive form of METTL3 could inhibit the differentiation of HSPCs. In AML cells, depletion of METTL3 induced cell differentiation and apoptosis and delayed leukemia progression. The authors further demonstrated that METTL3 mediated m6A modification on MYC, BCL2, and PTEN mRNAs and promoted their translation [73].
By performing two independent CRISPR screens, Barbieri et al. showed that METTL3 was necessary for AML cell survival and further revealed that the CAATT-box-binding protein CEBPZ was required for recruitment of METTL3 to chromatin. Promoter bound METTL3 introduces m6A modification within the coding region of SP1 and SP2 transcripts and enhances their translation, thus promoting cell proliferation and AML maintenance [74].
Liver cancer
Liver cancer is the fourth most common cause of cancer-related death worldwide, among which, hepatocellular carcinoma (HCC) accounts for the majority of cases [124], and the genetic and epigenetic alterations including m6A deregulation have been widely investigated [125, 126].
It was reported that METTL3 was significantly upregulated in HCC and was associated with shorter overall survival in HCC patients, while METTL3 depletion significantly inhibited HCC tumorigenicity and metastasis. Mechanistically, METTL3 mediates m6A modification of suppressor of cytokine signaling 2 (SOCS2) and promotes its mRNA degradation via a YTHDF2-dependent pathway [76]. In accordance with this, however, via an alternative mechanism, Lin et al. showed that METTL3 regulated the epithelial-mesenchymal transition (EMT) of HCC cells by methylating the CDS of Snail and triggering polysome-mediated translation [80]. A recent study showed that m6A modification by METTL3 on RAD52 motif 1 (RDM1) mRNA represses its expression and increases HCC cell proliferation via p53 and Ras/Raf/ERK pathways [77]. METTL3 can also promote HCC by methylating non-coding RNAs. Zuo et al. revealed that METTL3-mediated m6A modification promoted the stabilization of LINC00958, which sponged miR-3619-5p to upregulate hepatoma-derived growth factor (HDGF) and thereby facilitating HCC lipogenesis and progression [78].
As the most common pediatric liver cancer, hepatoblastoma (HB) has also been reported to have aberrant m6A modification [60, 81]. Liu et al. found that METTL3 was significantly upregulated in HB, inducing enhanced m6A modification across the whole transcriptome. They further identified CTNNB1, a key component of the Wnt/β-catenin signaling pathway, as a target of METTL3 [81]. Cui et al. proposed METTL3 as an independent prognostic factor in HB patients. They showed that knockdown of METTL3 dramatically suppressed HB cell proliferation, migration, and invasion via regulating Wnt/β-catenin pathway-associated proteins [60].
Gastric cancer
Gastric cancer (GC) is the fifth most common cancer and the third most lethal malignancy worldwide [127]. The study from Wang et al. showed that the m6A level significantly increased in GC cells owing to P300-mediated METTL3 overexpression. Specifically, elevated m6A level of HDGF mRNA promoted its stability via the binding of IGF2BP3, and the upregulated HDGF activated GLUT4 and ENO2 expression to promote glycolysis and eventually tumor growth and metastasis of GC cells [58]. Consistently, another two studies also reported that the upregulation of METTL3 in GC promoted cell proliferation and migration via affecting AKT signaling pathway and MYC target genes, respectively [89, 91].
The elevated METTL3 level was correlated with tumor survival and recurrence of GC patients. Zinc finger MYM-type containing 1 (ZMYM1) was identified as a target of METTL3 and the stability of m6A-modified ZMYM1 was enhanced by HuR (also known as ELAVL1) binding, leading to E-cadherin repression and EMT progression [90]. SEC62 is another target of METTL3 that is stabilized by IGF2BP1 in an m6A-dependent manner. The upregulation of SEC62 in GC promotes cell proliferation [62].
Lung cancer
Lung cancer is the leading cause of cancer incidence and mortality worldwide [127], and non-small cell lung cancer (NSCLC) comprises the most majority (~ 85%) of all lung cancers. Jin et al. demonstrated that METTL3 increases m6A modification of both YAP and lncRNA MALAT1, which induces drug resistance and metastasis of NSCLC cells through diverse pathways [97]. During TGFβ-induced EMT of NSCLC cells, both METTL3 expression and total m6A level were upregulated [96]. METTL3 knockdown decreased the m6A modification of JUNB, one of the important EMT regulators, leading to mRNA destabilization [96]. The precursor miR-143-3p was reported as a direct target of METTL3; increased m6A level of the precursor miRNA promotes its maturation, which induces invasion of NSCLC by targeting 3′UTR of vasohibin-1 (VASH1) to inhibit its expression [95].
Colorectal cancer
Colorectal cancer (CRC) is a complex and heterogeneous carcinoma tightly related to dietary and lifestyle factors, and a number of studies have uncovered genetic alterations and epigenetic dysregulation involved in CRC [128]. It is reported in recent years that m6A RNA modification also plays key roles in CRC progression. Li et al. found that the expression of METTL3 was higher in CRC metastatic tissues and was associated with a poor prognosis. They demonstrated that SRY-box 2 (SOX2) was a target of METTL3, and the m6A in the coding region of SOX2 mRNA was recognized by IGF2BP2 to protect the mRNA from decay [98]. Another study showed that METTL3 methylated CBX8 mRNA to enhance its stability in an IGF2BP1-dependent manner and that the upregulated CBX8 could promote stemness and suppress chemosensitivity of CRC cells through regulating LGR5 expression [103]. A role of METTL3 in regulating cell metabolism in CRC was also reported, where METTL3 regulates the glycolysis pathway through depositing m6A modification on the UTR of HK2 and SLC2A1 (GLUT1) [101].
Three studies highlight the role of non-coding RNAs in mediating the function of METTL3 in CRC. Wu et al. reported that m6A methylation of lncRNA RP11 by METTL3 increased its nuclear accumulation, which triggers the migration and invasion of CRC cells via post-translational upregulation of Zeb1 [99]. Peng et al. found that overexpressed METTL3 in CRC could methylate pri-miR-1246 and promote miRNA maturation to downregulate SPRED2, leading to tumor metastasis [100]. m6A modification of the circular RNA circNSUN2 increases its export to the cytoplasm, where it stabilizes HMGA2 mRNA to promote CRC metastasis [102].
Bladder cancer
Bladder cancer (BCa) is more common in men, with a 4 times higher incidence and mortality rate than in women globally [127]. Two groups found that METTL3 was upregulated in BCa patient samples and further demonstrated that METTL3-mediated m6A modification in target transcripts, such as CDCP1 and ITGA6, promoted mRNA translation via YTHDF1/3 binding [86, 87]. Other mRNA transcripts, including AFF4, IKBKB, RELA, and MYC, were revealed to be targets of METTL3 and mediated the role of METTL3 in promoting cell proliferation, invasion, and survival [85], whereas SETD7 and KLF4 tumor suppressors were negatively regulated by METTL3 in a YTHDF2-dependent manner [88]. Recently, a study by Han et al. showed that METTL3-mediated m6A modification in pri-miR221/222 facilitates miRNA maturation, resulting in the reduction of PTEN, which ultimately leads to BCa cell proliferation [84].
Pancreatic cancer
Pancreatic cancer, primarily pancreatic ductal adenocarcinoma (PDAC), is a highly lethal disease with mortality closely parallel with incidence [129]. It was suggested that a high level of METTL3 expression was associated with a high pathological stage in PDAC [104], as well as chemo- and radio-resistance in pancreatic cancer cells [130]. Zhang et al. conducted an elaborate study demonstrating that cigarette smoke condensate induced METTL3 expression and increased m6A modification on the oncogenic primary miR-25, leading to the enhanced maturation of the miRNA and the activation of AKT signaling in pancreatic duct epithelial cells to provoke malignant transformation [56].
Glioblastoma
Glioblastoma is an aggressive malignancy of astrocytes with high molecular heterogeneity and poor prognosis [131]. Visvanathan et al. reported that METTL3-mediated m6A modification on SOX2 mRNA plays crucial roles in glioma stem-like cells (GSCs) maintenance and dedifferentiation [82]. In consistence with this, Li et al. found that elevated expression of METTL3 was highly correlated with the clinical aggressiveness of malignant gliomas. They further showed that knockdown of METTL3 decreases m6A modification of the splicing factor SRSF, leading to YTHDC1-dependent nonsense-mediated mRNA decay of the SRSF transcripts and alternative splicing isoform switches in glioblastoma [83].
Prostate cancer
Prostate cancer (PCa) ranks as the second most frequent cancer worldwide in men, and it is the most lethal malignancy among men in 46 countries; however, relatively little is known regarding its etiology [127]. Cai et al. found that METTL3 is overexpressed in PCa cells and that METTL3 silencing decreases m6A modification and downregulates GLI1, an important component of the hedgehog pathway, which induces cell apoptosis [92]. Yuan et al. proposed METTL3 upregulation as a poor prognostic factor in PCa patients and revealed MYC as a METTL3 target [93]. In a recent report, Li et al. showed that METTL3 is upregulated in PCa tissues, especially those with bone metastasis. Their data suggested that METLT3-mediated m6A in Integrin β1 (ITGB1) stabilizes the mRNA via specific binding of HuR, which enhances cell motility and bone metastasis [94].
Breast cancer
A positive feedback mechanism was suggested in the progression of breast cancer, where upregulated METTL3 promoted the expression of hepatitis B X-interacting protein (HBXIP) in an m6A-dependent manner, and HBXIP inhibited miRNA let-7 g, a METTL3 negative regulator, thereby maintaining the high expression level of METTL3 and the accelerated cell proliferation in breast cancer [63]. Wang et al. also reported METTL3 overexpression in breast cancer and identified BCL2 as a target of METTL3, demonstrating that Bcl-2 translation was promoted owing to elevated m6A modification in the mRNA, which eventually promoted cancer cell proliferation [75].
Other cancers
In addition to the aforementioned cancer types, METTL3 also plays an oncogenic role in other types of cancer (Table 2). LEF1, ATAD2, c-Met, AKT, ΔNp63, and ZNF750 have been suggested as oncogenes in association with METTL3 with uncharacterized pathways [105, 107, 110, 111, 113, 114]. Several mRNAs, including DRG1, MYC, and TCF1, as well as non-coding RNA lncAROD, have been suggested as METTL3 targets that are stabilized in a m6A-dependent manner, resulting in cell proliferation and/or migration in osteosarcoma, oral squamous cell carcinoma (OSCC), thyroid carcinoma, and head and neck squamous cell carcinoma (HNSCC), respectively [106, 108, 109, 112]. It is noteworthy that in the case of OSCC, the authors concluded that the m6A modification in MYC enhanced the mRNA stability mediated by YTHDF1 [108], which was a different mechanism from that in other reports and highlighted the functional complexity of the protein.
METTL3 as a tumor suppressor
While METTL3 exhibits oncogenic functions in most cancer types, it was also reported as a tumor suppressor in some cases [132]. Li et al. detected lower expression of METTL3 in renal cell carcinoma (RCC) tissues compared with adjacent non-tumor tissues and also showed that higher expression of METTL3 might predict better survival outcome of RCC patients, possibly by promoting cell cycle arrest in G1 phase and thus suppressing tumor growth [115]. Based on a novel statistical model and the following experimental validation, Zhao et al. also identified METTL3 as a tumor suppressor gene in bladder cancer and showed that somatic mutations in METTL3 may promote cancer cell growth [120]. Cui et al. showed that the self-renewal of glioblastoma stem cell (GSC) was regulated by m6A mRNA modification and that knocking-down of METTL3 significantly promoted tumor progression and shortened the lifespan of GSC-grafted animals [116]. A similar conclusion was made by Deng et al. in CRC, where they found that METTL3 suppresses cell proliferation, migration, and invasion through p38/ERK pathways and thus is associated with longer survival time [119].
Hypomethylation of m6A resulting from decreased METTL3 expression was also observed in some cancers. A study from Jia and colleagues reported that decreased m6A level in ocular melanoma due to downregulation of METTL3 and upregulation of ALKBH5 predicted earlier recurrence and enhanced aggressiveness and also showed that METTL3-mediated m6A modification promoted the translation of HINT2, a tumor suppressor gene, in a YTHDF1-dependent mechanism [118]. Liu et al. depicted a more complicate scenario, in which they found that about 70% of endometrial tumors exhibit m6A hypomethylation, probably due to a hotspot mutation in METTL14 or reduced expression of METTL3, which resulted in downregulation of the negative AKT regulator PHLPP2 and upregulation of the positive AKT regulator mTORC2 [117].
METTL3 promotes cancer independent of catalytic activity
Elevated expression of METTL3 was detected in lung cancer cells, which was associated with cancer cell growth, survival, and invasion [133]. In-depth investigation found that METTL3 promoted the translation of certain oncogenic transcripts such as EGFR and TAZ, independent of its catalytic activity, but through recruiting eukaryotic translation initiation factor 3 (eIF3) to the translation initiation complex [133]. In a following report from the same group, the authors further revealed that the N-terminal fragment of METTL3 (1–200 amino acids) interacted directly with the eIF3 subunit h (eIF3h) and that the mutated METTL3 (A155P) abrogated the interaction and the ability to promote mRNA translation was severely compromised [134]. The authors thus proposed a novel “mRNA looping” mechanism, in which METTL3 binds to the m6A-modified 3′UTR of target mRNA and then recruits eIF3h as well as translation initiation factors such as CBP800 and eIF4e, thus facilitating the ribosome recycling and promoting translation efficiency [133, 134].
A recent work by Hua et al. also presented a similar mechanism, in which METTL3 promoted translation of the receptor tyrosine kinase AXL, independent of its methyltransferase activity. The authors also showed that upregulated METTL3 in ovarian carcinoma was significantly associated with tumor grade and overall survival rate [135]. It is noteworthy that METTL3 only binds to approximately 22% of the m6A sites [14], suggesting a mechanism of selectivity of METTL3 towards its targets for translational regulation, which needs to be further elucidated in the future.
Targeting of METTL3 for potential clinical application
Based on the emerging data on the roles and the molecular mechanisms in cancer, m6A regulators have attracted growing investigation as therapeutic targets [136]. As discussed above, METTL3 plays both oncogenic and tumor-suppressive roles in human cancers. Although still limited, activators and inhibitors targeting METTL3 have been reported recently. A structure-based virtual screening of compound databases carried out by Selberg et al. discovered four small molecules displaying the potency of enhancing the activity of the METTL3-METTL14 complex, among which the most effective one increased the relative m6A level by 21.4 ± 12.9% in the following cellular assays [137]. Simultaneous docking analysis showed that the compound interacted with SAM in close proximity in the active center of METTL3, which might increase the binding affinity of SAM and also lower the energy barrier of the substrate RNA methylation reaction [137].
Another study performed by Bedi et al. used co-factor mimicking approach to screen a library of 4000 adenosine analogs and derivatives and identified seven compounds showing diverse activities in two in vitro assays; among them, one compound (illustrated in Fig. 4a) has the most favorable inhibitory potency (IC50 = 8.7 μM) [138]. This is the first study reporting METTL3 inhibitors; however, several issues remain to be addressed. First, there is a lack of cellular activity data; thus, the actual effect of the compounds on m6A level is unknown. Second, the potential use of the compounds needs to be carefully evaluated because of the poor cell permeability and pharmacokinetics of adenosine analogs. Third, the co-factor binding pockets are common to the wide range of Rossmann fold enzymes; therefore, the selectivity of the inhibitors also requires further investigation.

Potential strategies for targeting of METTL3 in cancer. a A reported substrate competitive inhibitor of METTL3. b Protein-protein interaction (PPI)-based drug design. c Proteolysis-targeting chimera (PROTAC) for the degradation of METTL3 protein
As discussed above, the oncogenic function of METTL3 relies on the heterodimer structure formed with METTL14 in most cases; therefore, it will be a reasonable option to design inhibitors based on protein-protein interaction (PPI) strategy (Fig. 4b), as having been successfully applied in other drug targets [139]. Proteolysis targeting chimera (PROTAC) is another emerging biotechnology to degrade a wide range of proteins specifically [140], which is a bifunctional molecule consisting of a ligand of the target protein and a covalently linked ligand of an E3 ubiquitin ligase (Fig. 4c). Whether this strategy could be applied for degrading METTL3 in cancers warrants further investigation. In addition, structural analysis suggested that the substrate-binding site and SAM-binding site of METTL3 are merged into a large pocket [141]; therefore, the development of bisubstrate inhibitors that simultaneously occupy both of the binding sites might be an alternative strategy for future METTL3 targeting.
Overall, targeting of METTL3 for clinical application is still in its infancy. With the increasing knowledge regarding the regulation, functions, and mechanisms of METTL3 in cancer, it is promising to develop METTL3 targeted agents in the near future.
Conclusions and perspectives
It is widely accepted that several biological capabilities are regarded as hallmarks of cancer, including uncontrolled proliferation, cell death resistance, angiogenesis, invasion and metastasis, metabolism dysregulation, and immune escape [142]. The accumulating studies in recent years have revealed diverse pathways in cancers that are affected by METTL3, mostly focused on cell proliferation, cell death resistance, invasion, and metastasis (Table 2). However, emerging evidence also suggests that METTL3 plays vital roles in other biological processes, such as angiogenesis [58] and metabolism dysregulation [58, 101]. A recent publication showed that m6A modification of viral RNA enables the virus to avoid detection by innate immunity [143]; thus, there is a possibility that METTL3 might also mediate immune escape in cancers. Further studies are warranted to fully uncover the roles and mechanisms of METTL3 in affecting every hallmark of cancer.
It should be emphasized that although this review focuses mostly on METTL3, other components of the MTC, especially METTL14, should be taken into account when studying the m6A-dependent function of METTL3 in cancer. It is reasonably thought that proteins in the MTC should exhibit similar roles in the same pathological conditions; however, this is not always the case as reported. For example, METTL14 plays similar roles as METTL3 in some types of cancer, such as AML [122, 144, 145], breast cancer [146], and endometrial cancer [117], but exhibits opposite functions in other cancers, such as liver cancer [147, 148]. How could components in the same protein complex play opposite roles in the same cancer type needs to be carefully investigated and addressed. It is also noticeable that in glioblastoma, bladder cancer, and colorectal cancer, METTL3 was reported to play either oncogenic or/and tumor-suppressive functions by different groups, which may be explained by tumor heterogeneity and/or different model systems used for the study, and more further comprehensive and detailed studies are warranted to gain a better view.
In conclusion, METTL3 affects a broad range of biological processes and plays diverse roles in cancers, either dependent or independent of its methyltransferase activity, while the therapeutic targeting of METTL3 is just in the early stage. Continuing efforts are still needed to design and optimize strategies for targeting of METTL3 for cancer treatment.
Availability of data and materials
Not applicable.
Abbreviations
- m6A:
-
N6-methyladenosine
- METTL3:
-
Methyltransferase-like 3
- METTL14:
-
Methyltransferase-like 14
- ALKBH5:
-
AlkB Homolog 5
- FTO:
-
Fat mass and obesity associated
- YTHDC:
-
YT521-B homology domain containing
- YTHDF:
-
YT521-B homology domain family
- IGF2BP:
-
Insulin-like growth factor 2 mRNA-binding protein
- HNRNPC:
-
Heterogeneous nuclear ribonucleoproteins C
- HNRNPG:
-
Heterogeneous nuclear ribonucleoproteins G
- MTC:
-
m6A methyltransferase complex
- NMR:
-
Nuclear magnetic resonance
- AML:
-
Acute myeloid leukemia
- HSPCs:
-
Hematopoietic stem and progenitor cells
- HCC:
-
Hepatocellular carcinoma
- CRC:
-
Colorectal cancer
- GC:
-
Gastric cancer
- NSCLC:
-
Non-small cell lung cancer
- BCa:
-
Bladder cancer
- PDAC:
-
Pancreatic ductal adenocarcinoma
- GSCs:
-
Glioma stem-like cells
- PCa:
-
Prostate cancer
- OSCC:
-
Oral squamous cell carcinoma
- HNSCC:
-
Head and neck squamous cell carcinoma
- RCC:
-
Renal cell carcinoma
- eIF3:
-
Eukaryotic translation initiation factor 3
- PROTAC:
-
Proteolysis targeting chimera
References
Huang H, Weng H, Deng X, Chen J. RNA modifications in cancer: functions, mechanisms, and therapeutic implications. Annual Review of Cancer Biology. 2020;4:221–40.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–17.
Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat Cell Biol. 2019;21(5):552–9.
Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m(6)a RNA methylation in cancer. J Hematol Oncol. 2018;11(1):48.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1(1):37–42.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m(6)a mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)a methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028–38.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. M(6)a RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, et al. Methylation of structured RNA by the m(6)a writer METTL16 is essential for mouse embryonic development. Mol Cell. 2018;71(6):986–1000.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C, et al. Human METTL16 is a N(6)-methyladenosine (m(6)a) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18(11):2004–14.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)a methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169(5):824–35.
Ignatova VV, Stolz P, Kaiser S, Gustafsson TH, Lastres PR, Sanz-Moreno A, et al. The rRNA m(6)a methyltransferase METTL5 is involved in pluripotency and developmental programs. Genes Dev. 2020;34(9-10):715–29.
van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47(15):7719–33.
Pinto R, Vagbo CB, Jakobsson ME, Kim Y, Baltissen MP, O’Donohue MF, et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020;48(2):830–46.
Ren W, Lu J, Huang M, Gao L, Li D, Wang GG, et al. Structure and regulation of ZCCHC4 in m(6)A-methylation of 28S rRNA. Nat Commun. 2019;10(1):5042.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15(1):88–94.
Mauer J, Sindelar M, Despic V, Guez T, Hawley BR, Vasseur JJ, et al. FTO controls reversible m(6)am RNA methylation during snRNA biogenesis. Nat Chem Biol. 2019;15(4):340–7.
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)a, m(6)am, and m(1)a demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973–85.
Bartosovic M, Molares HC, Gregorova P, Hrossova D, Kudla G, Vanacova S. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3'-end processing. Nucleic Acids Res. 2017;45(19):11356–70.
Xu C, Liu K, Tempel W, Demetriades M, Aik W, Schofield CJ, et al. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289(25):17299–311.
Feng C, Liu Y, Wang G, Deng Z, Zhang Q, Wu W, et al. Crystal structures of the human RNA demethylase Alkbh5 reveal basis for substrate recognition. J Biol Chem. 2014;289(17):11571–83.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. Elife. 2017;6:e31311.
Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, et al. Nuclear m(6)a reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–19.
Mao Y, Dong L, Liu XM, Guo J, Ma H, Shen B, et al. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat Commun. 2019; 10(1): 5332.
Kretschmer J, Rao H, Hackert P, Sloan KE, Hobartner C, Bohnsack MT. The m(6)a reader protein YTHDC2 interacts with the small ribosomal subunit and the 5'-3' exoribonuclease XRN1. RNA. 2018;24(10):1339–50.
Liu T, Wei Q, Jin J, Luo Q, Liu Y, Yang Y, et al. The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 2020.
Han B, Yan S, Wei S, Xiang J, Liu K, Chen Z, et al. YTHDF1-mediated translation amplifies Wnt-driven intestinal stemness. EMBO Rep. 2020;21(4):e49229.
Li M, Zhao X, Wang W, Shi H, Pan Q, Lu Z, et al. Ythdf2-mediated m(6)a mRNA clearance modulates neural development in mice. Genome Biol. 2018;19(1):69.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, et al. YTHDF2 destabilizes m(6)A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, et al. Cytoplasmic m(6)a reader YTHDF3 promotes mRNA translation. Cell Res. 2017;27(3):444–7.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–99.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10(11):927–9.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Zhao Y, Shi Y, Shen H, Xie W. M(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13(1):35.
Livneh I, Moshitch-Moshkovitz S, Amariglio N, Rechavi G, Dominissini D. The m(6)a epitranscriptome: transcriptome plasticity in brain development and function. Nat Rev Neurosci. 2020;21(1):36–51.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24.
Peer E, Moshitch-Moshkovitz S, Rechavi G, Dominissini D. The epitranscriptome in translation regulation. Cold Spring Harb Perspect Biol. 2019;11(8):a032623.
Yang Y, Hsu PJ, Chen YS, Yang YG. Dynamic transcriptomic m(6)a decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616–24.
Patil DP, Pickering BF, Jaffrey SR. Reading m(6)a in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28(2):113–27.
Liao S, Sun H, Xu C. YTH domain: a family of N(6)-methyladenosine (m(6)a) readers. Genomics Proteomics Bioinformatics. 2018;16(2):99–107.
Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269(26):17697–704.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–8.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17.
Sledz P, Jinek M. Structural insights into the molecular mechanism of the m(6)a writer complex. Elife. 2016;5:e18434.
Huang J, Dong X, Gong Z, Qin LY, Yang S, Zhu YL, et al. Solution structure of the RNA recognition domain of METTL3-METTL14 N(6)-methyladenosine methyltransferase. Protein Cell. 2018;10(4):272–84.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10(1):1858.
Zhu W, Si Y, Xu J, Lin Y, Wang JZ, Cao M, et al. Methyltransferase like 3 promotes colorectal cancer proliferation by stabilizing CCNE1 mRNA in an m6A-dependent manner. J Cell Mol Med. 2020;24(6):3521–33.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m(6)a modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–205.
Yang Y, Song S, Meng Q, Wang L, Li X, Xie S, et al. miR24-2 accelerates progression of liver cancer cells by activating Pim1 through tri-methylation of histone H3 on the ninth lysine. J Cell Mol Med. 2020;24(5):2772–90.
Cui X, Wang Z, Li J, Zhu J, Ren Z, Zhang D, et al. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/beta-catenin signalling pathway. Cell Prolif. 2020;53(3):e12768.
Wei W, Huo B, Shi X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag Res. 2019;11:1177–87.
He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m(6)A-caused stabilization of SEC62. Biochem Biophys Res Commun. 2019;517(4):581–7.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7 g. Cancer Lett. 2018;415:11–9.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46(10):5195–208.
Garcia-Campos MA, Edelheit S, Toth U, Safra M, Shachar R, Viukov S, et al. Deciphering the “m(6)a code” via antibody-independent quantitative profiling. Cell. 2019;178(3):731–47.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, et al. Histone H3 trimethylation at lysine 36 guides m6A RNA modification co-transcriptionally. Nature. 2019;567(7748):414–9.
Aguilo F, Zhang F, Sancho A, Fidalgo M, Di Cecilia S, Vashisht A, et al. Coordination of m(6)a mRNA methylation and gene transcription by ZFP217 regulates pluripotency and reprogramming. Cell Stem Cell. 2015;17(6):689–704.
Bertero A, Brown S, Madrigal P, Osnato A, Ortmann D, Yiangou L, et al. The SMAD2/3 interactome reveals that TGFbeta controls m(6)a mRNA methylation in pluripotency. Nature. 2018;555(7695):256–9.
An S, Huang W, Huang X, Cun Y, Cheng W, Sun X, et al. Integrative network analysis identifies cell-specific trans regulators of m6A. Nucleic Acids Res. 2020;48(4):1715–29.
Fish L, Navickas A, Culbertson B, Xu Y, Nguyen HCB, Zhang S, et al. Nuclear TARBP2 drives oncogenic dysregulation of RNA splicing and decay. Mol Cell. 2019;75(5):967–81.
Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019;10(6):383.
Yan J, Huang X, Zhang X, Chen Z, Ye C, Xiang W, et al. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem Biophys Res Commun. 2020;521(4):887–93.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N(6)-methyladenosine (m(6)a)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature. 2017;552(7683):126–31.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Chen SL, Liu LL, Wang CH, Lu SX, Yang X, He YF, et al. Loss of RDM1 enhances hepatocellular carcinoma progression via p53 and Ras/Raf/ERK pathways. Mol Oncol. 2020;14(2):373–86.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Xu HF, Wang H, Zhao W, Fu SR, Li Y, Ni WJ, et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10(13):5671–86.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m(6)a methylation regulates the epithelial mesenchymal transition of cancer cells and translation of snail. Nat Commun. 2019;10(1):2065.
Liu L, Wang J, Sun G, Wu Q, Ma J, Zhang X, et al. M(6)a mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol Cancer. 2019;18(1):188.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m(6)a modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33.
Li F, Yi Y, Miao Y, Long W, Long T, Chen S, et al. N(6)-methyladenosine modulates nonsense-mediated mRNA decay in human glioblastoma. Cancer Res. 2019;79(22):5785–98.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18(1):110.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The m(6)a methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38(19):3667–80.
Jin H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N(6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207.
Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, et al. Dynamic m(6)a mRNA methylation reveals the role of METTL3-m(6)A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38(24):4755–72.
Xie H, Li J, Ying Y, Yan H, Jin K, Ma X, et al. METTL3/YTHDF2 m(6) a axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. J Cell Mol Med. 2020;24(7):4092–104.
Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 promotes the proliferation and mobility of gastric cancer cells. Open Med (Wars). 2019;14:25–31.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18(1):142.
Yang DD, Chen ZH, Yu K, Lu JH, Wu QN, Wang Y, et al. METTL3 promotes the progression of gastric cancer via targeting the MYC pathway. Front Oncol. 2020;10:115.
Cai J, Yang F, Zhan H, Situ J, Li W, Mao Y, et al. RNA m(6)a methyltransferase METTL3 promotes the growth of prostate cancer by regulating hedgehog pathway. Onco Targets Ther. 2019;12:9143–52.
Yuan Y, Du Y, Wang L, Liu X. The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J Cancer. 2020;11(12):3588–95.
Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin β1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am J Cancer Res. 2020;10(3):1012–25.
Wang, Deng Q, Lv Z, Ling Y, Hou X, Chen Z, et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer. 2019;18(1):181.
Wanna-Udom S, Terashima M, Lyu H, Ishimura A, Takino T, Sakari M, et al. The m6A methyltransferase METTL3 contributes to transforming growth factor-beta-induced epithelial-mesenchymal transition of lung cancer cells through the regulation of JUNB. Biochem Biophys Res Commun. 2020;524(1):150–5.
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. M(6)a mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12(1):135.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112.
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, et al. M(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18(1):87.
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, et al. Upregulated METTL3 promotes metastasis of colorectal cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38(1):393.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. M(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19(1):72.
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, et al. N(6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10(1):4695.
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, et al. M(6)a modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18(1):185.
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang J, et al. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathol Res Pract. 2019;215(11):152666.
Miao W, Chen J, Jia L, Ma J, Song D. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem Biophys Res Commun. 2019;516(3):719–25.
Ling Z, Chen L, Zhao J. m6A-dependent upregulation of DRG1 by METTL3 and ELAVL1 promotes growth, migration, and colony formation in osteosarcoma. Biosci Rep. 2020;40(4):BSR20200282.
Zhou L, Yang C, Zhang N, Zhang X, Zhao T, Yu J. Silencing METTL3 inhibits the proliferation and invasion of osteosarcoma by regulating ATAD2. Biomed Pharmacother. 2020;125:109964.
Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, et al. METTL3 facilitates oral squamous cell carcinoma tumorigenesis by enhancing c-Myc stability via YTHDF1-mediated m(6)a modification. Mol Ther Nucleic Acids. 2020;20:1–12.
Wang K, Jiang L, Zhang Y, Chen C. Progression of thyroid carcinoma is promoted by the m6A methyltransferase METTL3 through regulating m(6)a methylation on TCF1. Onco Targets Ther. 2020;13:1605–12.
Luo G, Xu W, Zhao Y, Jin S, Wang S, Liu Q, et al. RNA m(6) a methylation regulates uveal melanoma cell proliferation, migration, and invasion by targeting c-met. J Cell Physiol. 2020.
Liang S, Guan H, Lin X, Li N, Geng F, Li J. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol Lett. 2020;19(4):3197–204.
Ban Y, Tan P, Cai J, Li J, Hu M, Zhou Y, et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(6):1282–96.
Zhou R, Gao Y, Lv D, Wang C, Wang D, Li Q. METTL3 mediated m(6)a modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating DeltaNp63. Biochem Biophys Res Commun. 2019;515(2):310–7.
Zhang P, He Q, Lei Y, Li Y, Wen X, Hong M, et al. M(6)A-mediated ZNF750 repression facilitates nasopharyngeal carcinoma progression. Cell Death Dis. 2018;9(12):1169.
Li X, Tang J, Huang W, Wang F, Li P, Qin C, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8(56):96103–16.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. M(6)a RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. M(6)a mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Jia R, Chai P, Wang S, Sun B, Xu Y, Yang Y, et al. M(6)a modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol Cancer. 2019;18(1):161.
Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, et al. M(6)a methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco Targets Ther. 2019;12:4391–402.
Zhao S, Liu J, Nanga P, Liu Y, Cicek AE, Knoblauch N, et al. Detailed modeling of positive selection improves detection of cancer driver genes. Nat Commun. 2019;10(1):3399.
Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–52.
Weng H, Huang H, Chen J. RNA N (6)-methyladenosine modification in normal and malignant hematopoiesis. Adv Exp Med Biol. 2019;1143:75–93.
Liu Y, Cheng Z, Pang Y, Cui L, Qian T, Quan L, et al. Role of microRNAs, circRNAs and long noncoding RNAs in acute myeloid leukemia. J Hematol Oncol. 2019;12(1):51.
Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380(15):1450–62.
Ogunwobi OO, Harricharran T, Huaman J, Galuza A, Odumuwagun O, Tan Y, et al. Mechanisms of hepatocellular carcinoma progression. World J Gastroenterol. 2019;25(19):2279–93.
Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X, et al. Epitranscriptomics in liver disease: basic concepts and therapeutic potential. J Hepatol. 2020;73:664.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507.
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388(10039):73–85.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9.
Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–401.
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)a methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–45.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–65.
Boriack-Sjodin PA, Ribich S, Copeland RA. RNA-modifying proteins as anticancer drug targets. Nat Rev Drug Discov. 2018;17(6):435–53.
Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, et al. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. 2019;26(13):3762–71.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Caflisch A, Sledz P. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020;15(9):744–8.
Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov. 2016;15(8):533–50.
Schapira M, Calabrese MF, Bullock AN, Crews CM. Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov. 2019;18(12):949–63.
Schapira M. Structural chemistry of human RNA methyltransferases. ACS Chem Biol. 2016;11(3):575–82.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, et al. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol. 2020;5(4):584–98.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)a modification. Cell Stem Cell. 2018;22(2):191–205.
Martin GH, Park CY. Meddling with METTLs in normal and leukemia stem cells. Cell Stem Cell. 2018;22(2):139–41.
Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, et al. Cross-talk among writers, readers, and erasers of m(6)a regulates cancer growth and progression. Sci Adv. 2018;4(10):eaar8263.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N(6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65(2):529–43.
Huang H, Weng H, Chen J. M(6)a modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer Cell. 2020;37(3):270–88.
Acknowledgements
We apologize to the colleagues whose work could not be cited due to space constraints.
Funding
This work was supported in part by the GRMH-GDL Start-up Research Grant (1101201204 to H.W.), the National Natural Science Foundation of China (81770158 to C.Z.), and the Pearl River S&T Nova Program of Guangzhou, China (201906010002 to C.Z.).
Author information
Authors and Affiliations
Contributions
W.H. collected related literature. C.Z. and W.H. wrote the manuscript. Y.L. and H.W. provided guidance and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zeng, C., Huang, W., Li, Y. et al. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol 13, 117 (2020). https://doi.org/10.1186/s13045-020-00951-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00951-w