
Abstract
Background
Continual expression of PD-L1 in tumor cells is critical for tumor immune escape and host T cell exhaustion, however, knowledge on its clinical benefits through inhibition is limited in breast cancer. N6-methyladenosine (m6A) plays a crucial role in multiple biological activities. Our study aimed to investigate the regulatory role of the m6A modification in PD-L1 expression and immune surveillance in breast cancer.
Methods
MeRIP-seq and epitranscriptomic microarray identified that PD-L1 is the downstream target of METTL3. MeRIP-qPCR, absolute quantification of m6A modification assay, and RIP-qPCR were used to examine the molecular mechanism underlying METTL3/m6A/IGF2BP3 signaling axis in PD-L1 expression. B-NDG and BALB/c mice were used to construct xenograft tumor models to verify the phenotypes upon METTL3 and IGF2BP3 silencing. In addition, breast cancer tissue microarray was used to analyze the correlation between PD-L1 and METTL3 or IGF2BP3 expression.
Results
We identified that PD-L1 was a downstream target of METTL3-mediated m6A modification in breast cancer cells. METTL3 knockdown significantly abolished m6A modification and reduced stabilization of PD-L1 mRNA. Additionally, METTL3-mediated PD-L1 mRNA activation was m6A-IGF2BP3-dependent. Moreover, inhibition of METTL3 or IGF2BP3 enhanced anti-tumor immunity through PD-L1-mediated T cell activation, exhaustion, and infiltration both in vitro and in vivo. PD-L1 expression was also positively correlated with METTL3 and IGF2BP3 expression in breast cancer tissues.
Conclusion
Our study suggested that METTL3 could post-transcriptionally upregulate PD-L1 expression in an m6A-IGF2BP3-dependent manner to further promote stabilization of PD-L1 mRNA, which may have important implications for new and efficient therapeutic strategies in the tumor immunotherapy.
Similar content being viewed by others

Background
Currently, breast cancer is the leading commonly diagnosed cancer globally surpassing lung cancer [1]. Although the advances in surgeries, chemotherapy and targeted treatment approaches for breast cancer have improved overall survival rates in recent years, the therapeutic modalities are still limited for aggressive breast cancers such as the triple-negative form [2]. The rapidly growing field of cancer immunotherapy is expanding new horizons for antitumor therapy. Programmed death ligand-1 (also known as B7-H1, CD274 or PD-L1; hereafter referred to as PD-L1) is a 33 kDa type I transmembrane glycoprotein which downregulates T-cell function and cell survival by binding to programmed death-1 (PD-1) receptor; it has gained attraction in recent years [3,4,5]. PD-L1 inhibits immune-mediated rejection and assists tumor cells to evade the host immune surveillance in the tumor microenvironment [6]. Currently, atezolizumab, a monoclonal antibody that targets PD-L1, was approved in combination with nab-paclitaxel for patients with unresectable locally advanced triple-negative breast cancer (TNBC) or metastatic TNBC expressing PD-L1 [7, 8]. However, in early TNBC, complete pathological response is significantly higher among those who receive immune checkpoint inhibitor plus neoadjuvant chemotherapy, regardless of the PD-L1 levels [9]. Thus, PD-L1 expression can be dynamic [10] during the treatment course and may at least partly explain why some cancer patients with tumors lacking PD-L1 expression can respond favorably to checkpoint inhibitors therapy. Therefore, investigation of upstream regulatory mechanisms of the PD-L1 expression in breast cancer is important for an in-depth understanding of the functions of this immunosuppressive molecule. This could provide us with new strategies to reduce the cellular abundance of PD-L1.
The regulatory mechanisms controlling the PD-L1 expression are complex and multifactorial. These require intensive investigations. STAT, HIF-1, Myc, and other transcriptional factors are known to induce the expression of PD-L1 [11,12,13]. Besides, recent studies have explored the mechanisms of post-translational modifications on the regulation of PD-L1 expression. For instance, PD-L1 glycosylation triggered by B3GNT3 plays an important role in its expression; deglycosylation increases the anti-PD-L1 antibody binding affinity, leading to more accurate PD-L1 prediction and quantification in clinical outcomes [14]. In addition, poly-ubiquitination of CDK4 and SPOP promotes the degradation of PD-L1 [15], while CSN5 antagonizes its degradation [16]. Acetyltransferase DHHC3-mediated palmitoylation of the cytosolic region promotes PD-L1 expression, and palmitoylation blocks PD-L1 ubiquitination and degradation [17].
Furthermore, epigenetic regulations are also crucial in controlling gene expression. RNA methylation, a type of post-transcriptional modification, has gained widespread attention. As the common principal mRNA methylation type in mammals, N6-methyadenosine (m6A) modifications are reversible and dynamic; these are regulated by m6A “writers”, “erasers”, and “readers” (WERs). They are relevant to RNA fate as modifications manipulate the stability, translation efficiency, regulate alternative polyadenylation, and pre-mRNA splicing [18,19,20,21]. Methyltransferase-like 3 (METTL3), a section of the complex m6A methyltransferase, is essential for several biological processes, including cell differentiation, proliferation, and survival [22,23,24]. Recent studies show that m6A modification is also important in immunoregulation. METTL3 depletion promotes STAT1 and IRF1 mRNA expression in an m6A-YTHDF2-dependent manner, which in turn improves immunotherapeutic response by modulation of tumor-infiltrating cells in the intratumor microenvironment of colorectal cancer [25]. Inhibition of METTL3 weakens PD-1 blockade treatment by altering reprogramming of the bone marrow-derived macrophages [26]. Moreover, FTO-mediated m6A modification is implicated in the regulation of melanoma tumorigenesis and resistance to anti-PD-1 therapy [27]. However, the impact of m6A regulator(s) on the expression of immune-checkpoint molecules in breast cancer remains unclear and requires further investigation.
In the current study, we identified PD-L1 as a downstream target of METTL3-mediated m6A modification in breast cancer cells. Further, insulin-like growth factor 2 mRNA binding protein 3 (IGF2BP3) binding to PD-L1 was verified to recognize the m6A modification involving METTL3 stabilized PD-L1 mRNA. Moreover, METTL3 and IGF2BP3 participated in the regulation of tumor immune surveillance. Overall, we found that RNA epigenetic regulation is a novel mechanism of PD-L1 expression regulation in breast cancer. Thus, our study broadened the current molecular understanding of tumor immune surveillance.
Materials
Cell culture, generation of the stable cell line, and co-culture experiments
The human breast cancer cell lines (MDA-MB-231, HCC38, SK-BR-3), mouse breast cancer cell line 4T1 and human normal breast epithelial cell lines (MCF10A) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). MDA-MB-231 cells were cultured in L15 (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS, Hyclone, USA). HCC38 and 4T1 were preserved in 1640 medium (Gibco) enhanced with 10% FBS, and SK-BR-3 were grown in McCoys 5A medium (BI, Israel) enhanced with 10% FBS (BI, Israel). 37 °C was used to incubate the cells in a humidified 5% CO2 incubator.
For METTL3 and IGF2BP3 knockdown, lentiviral vectors harboring shRNA for knockdown and overexpression of METTL3 and IGF2BP3 and negative control underwent syncretization and the cloned into pLKO.1 vector. The plasmids were transfected using lipofectamine iMAX (Invitrogen, USA) into breast cancer cells according to the manufacturer’s protocol. shRNAs sequences are captured in Additional file 7: Table S1. Briefly, stably transfected cells were selected with 10 μg/ml puromycin (MCE, USA) for 3 weeks.
The peripheral venous blood was collected from healthy volunteers (6 ml) in heparin anticoagulant tube. Ficoll lymphocyte separation solution was added to dilute the blood. The PBMCs isolation from blood was done using gradient density centrifugation and then counted. The MDA-MB-231 cells were supplemented with fresh medium and co-cultured with activated cytokine-induced killer cells for 48 h in a ratio of 1:5 (cancer cell: cytokine-induced killer cells). After incubation, 5 mins was used to centrifuge the plates at 400 g, 5 min. The supernatant was collected and the LDH release assay (Beyotime) was performed based on the instructions from the manufacturer. Absorbance was detected at 490 nm and it was done using Biotek microplate reader. Finally, the supernatant was used for the enzyme-linked immune-sorbent assay (ELISA) for quantifying IFN-γ and IL-2 production (R&D System, Minnesota, USA) as per instructions given by the manufacturer.
MeRIP-qPCR assay and MeRIP-Seq
The m6A immunoprecipitation (MeRIP) procedure was performed according to instructions issued by the manufacturer using a Magna MeRIP™ m6A kit (#17–10,499, Merck Millipore, MA). Briefly, purified mRNA was digested by DNase I and then fragmented into ∼100 nt using RNA fragmentation reagent and incubated at 94 °C. After fragmenting, the stop buffer was added, following which standard ethanol precipitation was performed and collected. The anti-m6A antibody for 12 μg was pre-incubated with 50 μl beads in IP buffer (150 mM NaCl, 0.1% NP-40, 10 mM Tris–HCl, pH 7.4) at room temperature for 1 h. Next, 6 μg of fragment mRNAs were added to the antibody-beads mixture and incubated at 4 °C for 4 h on a rotator. After adequate washing, immunoprecipitated mixture was digested using high concentration of proteinase K, and the bound RNAs were extracted using phenol-chloroform method and ethanol precipitation and were used for qPCR analysis or library construction. qPCR analysis determined the modification of m6A in PD-L1 analysis based on precise primers (primers for MeRIP-qPCR were listed in Additional file 7: Table S1). All m6A sites of PD-L1 were predicted using SRAMP (http://www.cuilab.cn/sramp) [51]. We created primers to make sure that the target sequence included all these sites within 100 nt length. SMARTer smRNA-Seq Kit was used to perform the library constructions of IP-RNA samples with a small fraction of fragmented mRNAs used as input, and sequenced on the Illumina HiSeq X Ten platform.
Epitranscriptomic microarray analysis
Briefly, the total RNAs were immunoprecipitated using anti-N6-methyadenosine (m6A) antibody. The elute of immunoprecipitation magnetic beads was called “IP” which is m6A modified RNAs. “Sup” was the recovered supernatant which is unmodified RNAs. We labeled the “Sup” and “IP” RNA used for Cy5 and Cy3, respectively, as cRNAs. Hybridization of cRNA was done after merging to Arraystar Human m6A Epitranscriptomic Microarray (8 × 60 K, Arraystar, Rockville, MD, USA). Finally, the array was scanned using an Agilent scanner G2505C.
RNA immunoprecipitation (RIP)
RIP assay was performed using Magna RIP Kit (17–700, Millipore, MA) according to the instructions by the manufacturer. Briefly, 5 μg anti-METTL3 (Abcam, USA), anti-IGF2BP3 (Millipore, Germany) or anti-N6-methyladenosine (m6A) (Millipore, Germany) and anti-rabbit IgG (Millipore, Germany) were incubated with 50 μL magnetic beads before cell lysates were added (approximately 2 × 107 cells per sample). Then, the RNA-protein IP complexes were washed 6 times and proteinase K digestion buffer was used for incubation to remove the proteins. Finally, RNAs were extracted by phenol-chloroform RNA extraction and purified for qPCR analysis. Normalization of the relative enrichment was done to the input as: %Input =1/10 × 2Ct [IP] – Ct [input].
RNA total m6A quantification
EpiQuik™ m6A RNA Methylation Quantification Kit (Colorimetric) (Epigentek, USA) was used to colorimetrically measure the total level of m6A in breast cancer cells. Two hundred nanogram RNA was briefly combined with the capture antibody in each well which was used for subsequent detection. During multiple incubations, colorimetric measurement of m6A content was done at 450 nm wavelength and calculated based on the standard curve.
RNA stability assay
Breast cancer cells were treated as follows: 6-well plates were used to seed the cells overnight, and actinomycin D (5 μg/mL, HY-17559, MedChemExpress) was used to treat them for 0, 2, 4, 6 h. Total RNA was isolated using TRIzol and quantified by qRT-PCR. Group expression of the mRNA at the indicated times was calculated and normalized by GAPDH.
Luciferase reporter assay
cDNAs with firefly luciferase which contained full-length sequence of PD-L1 were cloned into pGL3-control vectors (Promega). For mutant 1, 2, 3, and 1–3 reporter plasmids, cytosine (C) replaced marked adenosine (A) in m6A motif. Pre-treated breast cancer cells were seeded into 6-well plates followed by co-transfection with 0.5 μg of wild-type or mutated PD-L1 reporter plasmids with 25 ng pRL-TK plasmids (renilla luciferase reporter vector) using jetPRIME Polyplus kit. After 24–36 h, cells were harvested and the luciferase activity was assessed using Dual-Glo Luciferase system (Promega). This was normalized to pRL-TK activity. Each experiment was conducted in triplicates.
Quantitative real-time PCR
TRIzol Reagent (Invitrogen, USA) was used to extract total RNA from cells as per instructions of the manufacture. The mRNA levels were examined using SYBR Premix Ex Taq (Takara, Dalian, China). GAPDH normalized the results and quantification of the mRNAs relative expression was done using the 2–∆∆Ct method. Used primers are presented in Additional file 7: Table S1.
Western blot
Extraction of total protein from breast cancer cells using pre-chilled RIPA buffer (Beyotime, Shanghai, China). After protein quantification, sample of protein of equal amounts were loaded and were 10% SDS-PAGE was applied to separate them and then moved onto 0.45 μm PVDF membranes (Millipore, USA). After blocking by 5% non-fat milk in TBST for 1.5 h, incubation of the membranes was done at 4 °C overnight using the corresponding primary antibodies. Then, secondary antibodies were applied in the incubation under room temperature for 1 h after being washed thrice with TBST. Detection of immunoblots was done using an imaging system (Bio-Rad, USA). Each antibody used in this study is captured in the Additional file 7: Table S1.
Flow Cytometry
PBS was used to wash harvested cells and they were incubated for 30 min on ice using 2% FBS and appropriate antibodies in PBS. After washing with PBS, the samples were analyzed on LSRFortessa SORP (BD Biosciences, Franklin Lakes, NJ), and data was done in FlowJo (Ashland, OR).
Immunohistochemistry
Paraffin was used to embed Xenograft tumors and then they were cut into sections of 4 μm. Section. Staining of TMA was done using eosin, hematoxylin, or incubated with primary antibodies, with the aid of ElivisionTM plus Polymer HRP immunohistochemistry kit (Maxim, Fujian, China). Capturing of images of representative fields was done on Aperio ImageScope (Leica Biosystems, Wetzlar, Germany).
Absolute quantification of m6A modification
For detection of absolute m6A levels, the probes L1 (left probe) and R1 (right probe) were designed. Each probe contained a universal primer-specific sequence used for PCR amplification (red and blue) and a target-specific sequence (orange and green), which were complementary to the RNA target immediately upstream and downstream of the m6A site, respectively. Although probes L1 and R1 would hybridize adjacent to each other with the RNA around the m6A site, they could not be ligated with T3 DNA ligase which has lesser selectivity with m6A modification, reflecting the non-methylated RNA levels at this site by this way. To quantitatively determine the m6A modification fraction in RNA transcript, the non-m6A site of the same RNA transcript was selected as the reference site, as it only contains “A”. The probes L2 and R2, which were RNA complementary target immediately upstream and downstream of the non-m6A site, were respectively designed. Where non-m6A, the probes L2 and R2 would be lowered RNA transcript expression levels could be detected by PCR. Finally, the m6A modification fraction could be precisely determined by real-time fluorescence PCR signals at the m6A site and non-m6A site. The schematic diagram is captured in Additional file 5: Fig. S2 g. Primers used are listed in Additional file 7: Table S1.
Animals
B-NDG mice (4–6 weeks old, female) were supplied by Jiangsu Biocytogen Laboratory Animal Co. (BCM002F). BALB/c mice (4 weeks old, female) were supplied by the Army Medical University, Chongqing and Precision Biotech. Army Medical University and institutional review board of Daping Hospital approved all the procedures. For subcutaneous xenograft experiments in B-NDG mice, approximately 1 × 106 MDA-MB-231 and there was subcutaneous injection of the cells that resuspended in 100 μl PBS into the left flank of the mice and were divided into 11 groups randomly (each containing 5 mice). After the treatment as shown in Additional file 5: Fig. S4a, Atezolizumab (Selleck, Shanghai, China) or corresponding iso control antibody (Selleck, Shanghai, China) was injected intratumorally on day 3, 6, 9, 12, 15 post-MDA-MB-231 inoculations, and 5 × 106 cytokine-induced killer (CIK) cells were injected in the tail vein on day 7, 14, 21. Tumor sizes were measured every 2–3 days. After the feeding, tumors of the mice were removed through sacrifice. Recording of tumor weight was done as well as volume estimation according to the formula: 1/2 × (length×width2).
For peritoneal- and subcutaneous-tumor xenograft models in BALB/c mice, approximately 1 × 106 4T1 cells ([4T1, sh-control, sh-control+ISO mAb, sh-METTL3–1#, sh-METTL3–2#, sh-control+PD-L1 mAb] and 4T1 cells expressing the corresponding luciferase), per mouse suspended in 100 μl PBS were intraperitoneal injected or subcutaneously injected in the flank, respectively. The iso control and PD-L1 mAb (Bio X Cell, Beijing, China) were conducted by intraperitoneal injection (150 μg/mouse) on day 3-, 6-, 9-, 12-, 15-, 18-post-4T1 cells inoculation. FUSION FX imaging system (Vilber Lourmat, Paris, France) was used to perform bioluminescence imaging of tumor-bearing mice to evaluate the tumor growth. All the mice were monitored for survival and tumor volume. Survival was based on the number of days from the inoculation tumor cell to the day of animals were to be euthanized based on the following symptoms: hemiparesis, seizures, loss of weight (more than 20%), inability to move, and other serious neurological deficits symptoms. The results were analyzed using GraphPad Prism 7.0 software. Mice were anesthetized and sacrificed; tumors obtained were processed for IHC staining.
Statistical analysis
GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA) was applied to analyze the data. Results are presented in the form of means ± SD not less than three biological replicates. Student’s t-tests were used to compare between two groups and one-way analysis of variance (ANOVA) and for comparison of multiple groups was done using Dunnett’s test. Pearson correlation analysis was done to find the correlation between PD-L1 and METTL3 or IGF2BP3 expression levels. P-value less than 0.05 in all the tests was taken to be statistically significant.
Results
PD-L1 expression is regulated by METTL3-mediated m6A modification in breast cancer
To investigate the regulatory role of m6A modification in the post-transcriptional expression of PD-L1, we first used the m6A target database (http://m6a2target.canceromics.org/#/) [52], based on the results of diverse high-throughput sequencing and mass spectrometry, to obtain information on m6A modification in PD-L1. Predicted results showed that PD-L1 could be modified by METTL3-mediated m6A modification. Since METTL3 is an important m6A writer, we further examined whether METTL3 exerted regulatory effects on the overall m6A modification in the breast cancer cells. Indeed, the knockdown of METTL3 reduced the global m6A modification in MDA-MB-231 and HCC38 cells in the RNA methylation quantification assay (Fig. 1a). Additionally, the results from m6A epitranscriptomic microarray showed that 346 genes were hyper-methylated while 841 genes were hypo-methylated, following METTL3 inhibition as compared to the control group (Fig. 1b). The 695- and 345-m6A peaks represented the total statistical decrease and increase in the MeRIP-seq results, respectively, in METTL3 knockdown cells relative to the control (Fig. 1c). Subsequent investigation on the m6A peak distributions revealed that total m6A distribution patterns were the same in the METTL3 knockdown and control groups (Fig. 1d). The consensus motif which was highly concentrated in m6A sites was present in both the control and METTL3 knocked-down cells (Fig. 1e). At the intersection of findings from the epitranscriptomic microarray and MeRIP-seq, 30 sequences from 24 genes that harbored both hypo-methylation and down-regulated peaks were identified, including PD-L1 (CD274) (Fig. 1f). In addition, the methylation inhibitor, 3-deazaadenosine (DAA), also significantly decreased PD-L1 expression in a dose-dependent manner (Fig. 1g). Furthermore, the interaction differences between METTL3 and PD-L1 were also observed in RIP-seq data from sh-METTL3-MDA-MB-231 cells (Additional file 1: Fig. S1a). These results indicated that METTL3 is involved in the regulation of m6A modification of PD-L1 mRNA in breast cancer cells. Due to the higher expression of PD-L1 mRNA, MDA-MB-231, HCC38, SK-BR-3, and 4T1 cell lines were chosen for subsequent experiments (Additional file 1: Fig. S1b).

PD-L1 is a downstream target of METTL3. a The global content of m6A was examined by RNA methylation quantification assay. b The starplot presented the distribution of genes with both differential (hyper or hypo) methylation level (Y axis; |fold change| ≥ 1.5) and differential (up or down) gene expression level (X axis; |fold change| ≥ 1.5) in sh-METTL3 with control groups. c Volcano plot of changed m6A peaks was identified by MeRIP-seq in control and METTL3-knockdown MDA-MB-231cells. d Distribution of total m6A peaks in sh-control and sh-METTL3 groups were shown. e Top sequence motif was identified from MeRIP-seq. f Venn diagram showed the down-modified genes following METTL3 knockdown. g The mRNA expression levels of MDA-MB-231 and HCC38 cells were tested by qRT-PCR after 3-deazaadenosine (DAA) treatment in the indicated concentration. *p < 0.05; **p < 0.01
METTL3 increases m6A modification and expression of PD-L1 mRNA
To examine the role of METTL3 in post-transcriptional modification of PD-L1, using the RIP-qPCR assay, we observed a significantly higher METTL3 enrichment with PD-L1 mRNA relative to IgG control (Additional file 2: Fig. S2a). Silencing METTL3 suppressed this enrichment in MDA-MB-231, HCC38, SK-BR-3, and 4T1 cells, while overexpression of METTL3 resulted in higher enrichment with PD-L1 mRNA (Fig. 2a, b, Additional file 2: Fig. S2b, c). Furthermore, consistent with the results of the MeRIP-seq, a significant decrease in m6A modified PD-L1 was observed by MeRIP-qPCR assay upon METTL3 disruption and METTL3 overexpression increased the levels of m6A modified PD-L1 mRNA in MDA-MB-231, HCC38 (Fig. 2c, d), SK-BR-3, and 4T1 cells (Additional file 2: Fig. S2d, e).

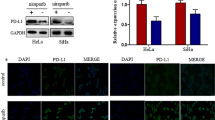
METTL3 increases m6A modification and expression of PD-L1 mRNA. a-b The interaction between METTL3 and PD-L1 mRNA was analyzed by RIP-qPCR assay in MDA-MB-231 and HCC38 cells with METTL3 knockdown or overexpression. c-d The relative levels of m6A in PD-L1 were tested by MeRIP-qPCR from MDA-MB-231 and HCC38 cells with overexpression or knockdown of METTL3. e Putative m6A modification sites in the CDS sequence of PD-L1 and synonymous mutations in the PD-L1 CDS. f Relative activity of the WT or Mut luciferase reporters in METTL3-silenced MDA-MB-231 and HCC38 cells was determined (normalized to negative control groups). g The m6A levels of three specific sites of PD-L1 (correspond to the figure e) were determined by absolute quantification of m6A modification. h PD-L1 mRNA levels were determined by qRT-PCR in MDA-MB-231 and HCC38 cells (control and METTL3 disruption) after actinomycin D treatment (normalized to 0 h). i-k PD-L1 mRNA, protein and cell surface expression levels were detected by qRT-PCR, western blot and flow cytometry in sh-Ctrl or sh-METTL3 MDA-MB-231 and HCC38 cells. Values are the mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance
To enhance the understanding of the role of m6A modification in the regulation of PD-L1 expression, the online tool SRAMP (http://www.cuilab.cn/sramp) [51] was used to predict m6A sites, and we constructed a wild-type (WT) and four mutant- (Mut1, 2, 3, 1–3) plasmids to examine the specific modifications of PD-L1. The wild-type plasmid consisted of the full-length CDS sequence with intact m6A sites, while each of the mutants had A-C mutations which eliminated the impact of m6A methylation (Fig. 2e). As shown in Fig. 2f, the relative luciferase activity of WT remarkably reduced upon METTL3 knockdown, but those for Mut groups were resistant to the effect of METTL3 silencing. To determine the m6A methylation levels at each site, we used absolute quantification in the m6A modification assay. This is a new and ultrasensitive quantitation assay for the accurate determination of m6A at single-nucleotide resolution. Its schematic diagram is shown in Additional file 2: Fig. S2 g. We found that METTL3 could significantly methylate PD-L1 mRNA especially on sites 2 and 3 (Fig. 2g). Furthermore, we found the knockdown of METTL3 led to lower mRNA stability owing to the reduced half-life of PD-L1 transcript after treatment with actinomycin D (Fig. 2h, Additional file 2: Fig. S2f). Collectively, these results suggested that METTL3 directly interacted with PD-L1 and could regulate m6A modification of PD-L1 mRNA.
Additionally, PD-L1 mRNA and protein expression levels decreased upon METTL3 knockdown in MDA-MB-231, HCC38 (Fig. 2i, j), SK-BR-3, and 4T1 cells (Additional file 2: Fig. S2h, i). We also determined the cell-surface PD-L1 expression by flow cytometry as shown in Fig. 2k and Additional file 2: Fig. S2j. Taken together, our results suggested that PD-L1 expression was regulated by METTL3-mediated m6A modification.
IGF2BP3 mediates PD-L1 mRNA expression in an m6A-dependent manner
Recently, the regulatory effects of “m6A readers” on m6A-modified transcripts were confirmed. IGF2BPs, including IGF2BP1/2/3, are a distinct family of m6A readers that can recognize and bind to thousands of mRNA transcripts targets through the m6A motif. These play a crucial role in mRNA stabilization [44]. To investigate the role of IGF2BPs in the modulation of PD-L1 mRNA, IGF2BP1–3 was knocked down in breast cancer cells, and our results showed that PD-L1 expression was significantly inhibited by IGF2BP3 knockdown (Fig. 3a, Additional file 3: Fig. S3b) instead of IGF2BP1/2 disruption (Additional file 3: Fig. S3a). Due to the lower expression of IGF2BP3 in 4T1 cells, we used MDA-MB-231, HCC38, and SK-BR-3 cells to investigate the regulatory mechanism of IGF2BP3. The total protein and membrane expressions of PD-L1 were remarkably reduced upon IGF2BP3 disruption in breast cancer cells (Fig. 3b, c, Additional file 3: Fig. S3c, d). To confirm whether IGF2BP3 was a potential reader of PD-L1 m6A methylation, direct binding interaction between IGF2BP3 and PD-L1 mRNA was evaluated by RIP-qPCR assay compared with IgG (Additional file 3: Fig. S3e). The results also showed that PD-L1 mRNA levels increased upon IGF2BP3 overexpression and decreased when IGF2BP3 was knocked down in breast cancer cell lines (Fig. 3d, e, Additional file 3: Fig. S3f, g). As shown in Additional file 3: Fig. S3h, m6A mutant sites in the PD-L1 transcript limited the binding of IGF2BP3. Moreover, METTL3 knockdown inhibited the binding interaction between IGF2BP3 and PD-L1 mRNA (Fig. 3f, Additional file 3: Fig. S3i), which suggested that IGF2BP3 could bind PD-L1 mRNA in a METTL3/m6A-dependent manner.

IGF2BP3 mediates PD-L1 mRNA expression in an m6A-dependent manner. a-c PD-L1 mRNA, protein and cell surface expression levels were tested by qRT-PCR, western blot and flow cytometry in sh-ctrl or sh-IGF2BP3 MDA-MB-231 and HCC38 cells. d-e The interaction between IGF2BP3 and PD-L1 mRNA was analyzed by RIP-qPCR assay with overexpression or knockdown of IGF2BP3. f The binding of IGF2BP3 was tested by RIP-qPCR in sh-METTL3 and control cells. g PD-L1 mRNA levels were analyzed by qRT-PCR assay in MDA-MB-231 and HCC38 cells after actinomycin D treatment. Results were presented as mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001
In addition, IGF2BP3 disruption reduced the stability of PD-L1 mRNA, and the knockdown of IGF2BP3 could reverse the increased PD-L1 stability mediated by METTL3 overexpression (Fig. 3g, Additional file 3: Fig. S3j). Moreover, western blotting results showed that overexpression of METTL3 and IGF2BP3 led to a substantial increase in PD-L1 expression, while IGF2BP3 disruption could reverse this effect. The PD-L1 expression significantly decreased due to METTL3 and IGF2BP3 deficiency; however, IGF2BP3 overexpression could not rescue the PD-L1 expression levels upon METTL3 knockdown (Additional file 3: Fig. S3k). Taken together, these results demonstrated that PD-L1 mRNA stability and expression were upregulated through the METTL3-IGF2BP3 axis in an m6A-dependent manner.
METTL3/IGF2BP3-downregulated antitumor immunity in breast cancer cells
Tumor-derived PD-L1 exerts significant inhibition on antitumor T-cell activation. To investigate the role of METTL3 or IGF2BP3 in PD-L1-mediated tumor immune surveillance, the cytokine-induced killer (CIK) cells were co-cultured with MDA-MB-231 cells. LDH release assay showed that METTL3-, IGF2BP3-knockdown and atezolizumab (anti-PD-L1) treatment increased the breast cancer cell sensitivity towards T-cell killing as compared to control groups, and these effects could be reversed by the overexpression of PD-L1 (Fig. 4a). In addition, T cells were activated with the increase in IL-2 and IFN-γ secretion due to METTL3 or IGF2BP3 knockdown, and by atezolizumab treatment; overexpression of PD-L1 could reverse these effects (Fig. 4b, c). Consistent with PD-L1 binding to its receptor on activated T lymphocytes resulting in T cell exhaustion, we also detected the expression of exhaustion markers, including PD-1, TIM3, and NR4A1, in the T-cells isolated from co-cultured medium. As shown in Fig. 4d-f, we found that PD-1, TIM3, and NR4A1 mRNA levels reduced significantly upon METTL3 or IGF2BP3 deficiency in MDA-MB-231 cells; overexpression of PD-L1 could reverse these effects. Our results suggested that PD-L1 could be a key mediator of METTL3/IGF2BP3-induced tumor immune surveillance in breast cancer cells.

METTL3/IGF2BP3-downregulated antitumor immunity in breast cancer cells. a The cytotoxicity was measured by lactate dehydrogenase (LDH) release assay after incubation for 48 h. b-c The IFN-γ and IL-2 protein levels in co-culture medium were measured by ELISA after 48 h co-incubation. d-f The PD-1, TIM3 and NR4A1 mRNA expression levels were tested by qRT-PCR. g Images at the end points of subcutaneous xenograft tumors formed by MDA-MB-231 cells in B-NDG mice (n = 5 for each group; scale bar, 1 cm). h Tumors weight were measured in the xenograft mice. Results were presented as mean ± SD of three independent experiments. *p < 0.05; **p < 0.01; n.s., no significance
To examine whether METTL3 or IGF2BP3 inhibition–mediated suppression of tumor progression was dependent on PD-L1 in vivo, we used MDA-MB-231 cells (stable METTL3-knockdown, IGF2BP3-knockdown, and simultaneous PD-L1 overexpression [rescue condition]) and subcutaneously injected them into female B-NDG mice for tumor xenograft. Following the treatment as described above (Additional file 4: Fig. S4a), indeed the silencing of METTL3 or IGF2BP3 could suppress tumor growth (Fig. 4g), tumor weight, and volume, similar to effects observed upon PD-L1 blockade treatment; PD-L1 overexpression was able to partially rescue tumor growth (Fig. 4g, h, Additional file 4: Fig. S4b, c). These results indicated that the knockdown of METTL3/IGF2BP3 could enhance T cell-mediated antitumor immunity to alleviate breast cancer progression by downregulating PD-L1 expression.
METTL3 knockdown enhances antitumor immunity and immune infiltration
Due to the low expression of IGF2BP3 in 4T1 cells, we constructed stable METTL3-knockdown 4T1 cell lines. To evaluate the immune regulatory effect of METTL3, a syngeneic 4T1 murine tumor model was constructed with immune-competent female BALB/c mice through peritoneal and subcutaneous tumors xenografts. First, the two METTL3-knockdown 4T1 cells were stably engineered to express luciferase for bioluminescence imaging in vivo. The bioluminescence imaging was performed at 12 and 24 days after transplantation. We found that intraperitoneally xenografted tumors in METTL3-silenced groups grew more slowly and prolonged the overall survival as compared to those in the control group, consistent with PD-L1 mAb treatment (Fig. 5a, Additional file 4: Fig. S4d, e). A concomitant decrease in PD-L1 expression was observed due to METTL3 deficiency; higher densities of CD3+, CD8+, and CD4+ T-cell infiltrations were also found in sh-METTL3–1/2# tumors as compared to control tumors which were validated by IHC staining (Fig. 5b, Additional file 4: Fig. S4f). As the therapeutic efficacy of PD-L1 mAb is dependent on the blockade of PD-1/PD-L1 interaction, we speculated that PD-L1 expression in each group was not significantly different. Additionally, in the subcutaneous xenograft models, tumoral METTL3 inhibition and anti-PD-L1 therapy could both markedly limit tumor growth of the mice as compared to the control groups (Fig. 5c, Additional file 4: Fig. S4g, h). The tumors progression were inhibited due to transplanted METTL3-knockdown 4T1 cells showed low PD-L1 levels. Consistent with the results of the intraperitoneal xenografted tumors, METTL3 disruption in transplanted tumors increased CD3+, CD4+, and CD8+ T-cell infiltrations (Fig. 5d, Additional file 4: Fig. S4i). These results suggested the METTL3 was involved in the tumor immune escape by upregulation of PD-L1 expression which inhibited intertumoral T-cell infiltration; tumoral METTL3 deficiency showed similar responses to those upon PD-L1 blockade treatment.

METTL3 knockdown enhances antitumor immunity and immune infiltration. a Bioluminescent images of intraperitoneal xenografted tumors from the indicated groups for Day 12 and Day 24. b The CD3+, CD4+ and CD8+ densities and PD-L1 expression were determined by Immunohistochemical analysis in the intraperitoneal xenografted tumors. Scale bar, 20 μm. c Images of subcutaneous tumors from the indicated groups (scale bar: 1 cm). d The CD3+, CD4+ and CD8+ densities and PD-L1 expression were determined by Immunohistochemical assay in the subcutaneous xenograft models. Scale bar, 20 μm
PD-L1 expression positively correlates with METTL3 and IGF2BP3 expression in breast cancer
To determine the clinical correlation between PD-L1 and METTL3 or IGF2BP3, IHC staining was performed to verify the protein expression of METTL3, IGF2BP3, and PD-L1 using a tissue microarray consisting of 140 breast cancer tissues. We chose four patients as representatives including two cases of positive and two cases of negative PD-L1 expression (Fig. 6a). After evaluating the respective staining intensity scores, we found that, indeed, the PD-L1 expression in breast cancer was positively associated with the expression of METTL3 or IGF2BP3 (Fig. 6b). Next, we also analyzed the expression correlation in different subtypes of breast cancer. The results indicated that the correlation of METTL3 or IGF2BP3 with PD-L1 was higher in HER2+ (HER2 positive) and TNBC as compared to other subtypes (Fig. 6c, Additional file 6: Fig. S6a). Additionally, PD-L1-positive tissues expressed higher levels of METTL3 and PD-L1-negative tissues showed a concomitant decrease in IGF2BP3 expression, which suggested the existence of a METTL3-IGF2BP3-PD-L1 regulating axis in breast cancer (Fig. 6d). Next, we found that both METTL3 and IGF2BP3 expression levels were higher in TNBC and HER2+ subtypes which represented more aggressive phenotypes (Additional file 6: Fig. S6b). Additionally, due to the lack of access to validated data for breast cancer, we used TISIDB database (http://cis.hku.hk/TISIDB/index.php) [47] to evaluate whether METTL3 and IGF2BP3 showed differential expression in clinical data between responders and non-responders undergoing anti-PD-1/PD-L1 treatment. The results showed that tumors of the responders expressed higher levels of METTL3 (three data sets) and IGF2BP3 (four data sets) (Fig. 6e), which suggested that tumors with higher METTL3 or IGF2BP3 expression were likely to be sensitive towards anti-PD-1/PD-L1 immunotherapy. Collectively, these clinical sample data also verified that the expressions of METTL3 and IGF2BP3 were positively correlated with PD-L1 and indicated that PD-L1 could be a potential downstream target of the METTL3/IGF2BP3 axis in breast cancer.

PD-L1 expression positively correlates with METTL3 and IGF2BP3 expression in breast cancer. a The expressions of PD-L1, METTL3 and IGF2BP3 were analyzed by IHC in a tissue microarray containing of 140 breast cancer tissues. Four Cases as representative IHC staining with positive- and negative-PD-L1 were shown. Scale bars, 100 μm. b The correlation of PD-L1 with METTL3 and IGF2BP3 in all breast cancer tissues (n = 140) were analyzed by IHC scores. Proportion scores were recorded as 0, 1, 2, 3, 4 corresponding to < 5%, 5–25%, 25–50%, 50–75%, and ≥ 75%. Intensity scores were recorded as 0, 1, 2, 3 corresponding to negative, weak, moderate, and strong staining. Finally, IHC scores was calculated as “proportion score × intensity score”. c The correlation between PD-L1 and METTL3 or IGF2BP3 were analyzed in HER2+ (n = 26) and TNBC (n = 27) subtypes. Spearman’s rank correlation test was used to analyze the P value. d Number of cases of METTL3 and IGF2BP3 were presented in two categories (PD-L1 positive and PD-L1 negative) in 140 tissues. e The differential expression of METTL3 or IGF2BP3 between responders and non-responders in cilnial data sets. The Y-axis represents the log2 Fold change values (responders vs. non-responders). f A schematic model illustrating the mechanism of METTL3/IGF2BP3-mediated N6-methyladenosine modification of PD-L1 mRNA in breast cancer
Discussion
PD-L1 is a major co-inhibitory immune-checkpoint protein and the PD1/ PD-L1 axis can inhibit the killing effect of cytotoxic T cells in the tumor microenvironment, further resulting in tumor immune escape [5, 6, 28]. Given the knowledge of these mechanisms, the microenvironment and immune-mediated factors in certain breast cancers have become significant for the development of treatment strategies [29]. Initially, breast cancer was not considered as an immunogenic tumor, however, recent studies show that aggressive triple-negative breast cancer, resistant to chemotherapy with poor prognosis, are immunogenic [29]; they are responsive to immunotherapy [30, 31]. Atezolizumab in combination with nab-paclitaxel is effective in unresectable, metastatic, or locally advanced TNBC, where the tumor is PD-L1-positive. These patients may thus benefit from immunotherapy [32]. However, objective responses to PD-L1 blockade therapy in breast cancer trials are not very encouraging [33]. Continual PD-L1 expression may affect its efficacy in clinical application. Therefore, further studies on the regulatory mechanisms of PD-L1 expression in breast cancer are needed. These could advance the current molecular understanding of immunoregulation and provide better immunotherapy strategies.
In this study, we found a critical role for m6A RNA modifications in the regulation of PD-L1 expression, stability, and T-cell-mediated killing in breast cancer. Thus, post-transcriptional modifications may be a promising therapeutic strategy for immunoregulation. N6-methyladenosine (m6A) modifications are prominent internal chemical modifications of RNA that are involved in multiple cellular activities, including RNA stability, protein translation, and molecular structure switching, which profoundly regulate several physiological processes and disease pathogenesis [18,19,20,21, 34,35,36]. Our results showed that m6A modification plays an important role in tumor immune evasion by upregulating PD-L1 expression and stability in breast cancer. First, using multidimensional sequencing technology, we identified PD-L1 as a potential direct downstream target of METTL3-mediated m6A alteration in breast cancer cells. The inhibition of METTL3 indeed led to PD-L1 downregulation, decreased binding and m6A levels, which was confirmed by qRT-PCR, MeRIP-qPCR, RIP-qPCR, and luciferase assays. We also evaluated the exact methylation level of each m6A site in the CDS sequence of PD-L1 mRNA at a single-nucleotide resolution using a new method for absolute quantification of m6A levels. Additionally, METTL3 knockdown could functionally improve T-cell killing and inhibit T-cell exhaustion to enhance immune evasion by downregulating PD-L1 expression. In addition to investigating the mechanism of PD-L1 m6A modification and in vitro cellular functions, we also analyzed the potential therapeutical effects of knocking down METTL3 in immunodeficient and immunocompetent mice models as compared to anti-PD-L1 treatment. The results suggested that silencing METTL3 eliminated the progression of xenograft tumors and increased the abundance of infiltrating immune cell types in the tumor microenvironment. Our results were similar to that of a previous study which shows that FTO depletion inhibits the expression of PD-L1 in colon cancer cells through m6A modification [43]. However, our study aimed at investigating the detailed specific molecular mechanisms underlying PD-L1 m6A modification and the impact of METTL3 on immunoregulation in breast cancer.
METTL3, through its methyltransferase activity, influences several biological processes and plays multiple roles in cancers. Previous studies suggest that diverse signals and pathways in cancers are regulated by METTL3, including cell proliferation, invasion, metastasis, and drug resistance [22,23,24,25, 37,38,39,40,41,42]. In breast cancer, METTL3 enhances the expression of HBXIP and induces positive feedback of HBXIP/let-7 g/METTL3/HBXIP signaling axis on cell proliferation [39]. In addition, METTL3 deficiency also influences macrophage reprogramming and enhances tumor progression in mouse models, thereby, eliminating the efficacy of PD-1 blockade treatment [26]; mRNA methylation mediated by METTL3/14 sensitizes pMMR-MSI-L colorectal cancer immunity to anti-PD-1 treatment by increasing STAT1 and IRF1 expression in an m6A-dependent manner [25]. Our study laid a strong case for a novel mechanism of METTL3 function as an immunomodulator in the tumor microenvironment of breast cancer, in addition to regulating tumor progression.
m6A reader proteins, such as YTHDF1/2/3, YTHDC1/2, and IGF2BP1/2/3, can bind to modified motifs of each target to exert diverse biological effects and influence the genetic information flow [44, 45]. The IGF2BP (IGF2BP1/2/3) family is especially pivotal for recognizing m6A modifications and regulates mRNA stabilization and translation. We identified that only IGF2BP3 could significantly decrease the expression and stability of PD-L1 in breast cancer cells. We investigated the role of IGF2BP3 as a reader of PD-L1 m6A methylation and through IGF2BP3-RIP analysis we found an enrichment of PD-L1 mRNA; this interaction was interrupted due to METTL3 deficiency. Furthermore, IGF2BP3 preferentially recognizes the m6A modification and influences METTL3-mediated regulation to prevent PD-L1 degradation. Correspondingly, we also found that IGF2BP3 knockdown was necessary to promote T cell-induced immune attack in breast cancer cells. Since the expression of IGF2BP3 could not be detected in the immunocompetent BALB/c mice, we used the TIMER [46] and TISIDB databases [47] for the analyses. The results indicated that IGF2BP3 expression was positively correlated with infiltration of CD8+, CD4+ T cells, and B cells in each of the breast cancer subtypes (Additional file 5: Fig. S5a, b), which was similar to the effect of PD-L1 (CD274). It has been reported that IGF2BP3, a well-known oncoprotein, is post-transcriptionally active and is involved in tumor growth, metastasis, survival, and chemo-resistance in the gastric, liver, and breast cancers and self-renewal and tumor initiation in cancer stem cells [48,49,50]. Our findings thus enriched the understanding of the function of m6A reader protein-mediated immunoregulation in breast cancer.
Additionally, we used the tissue microarray of breast cancer and found that METTL3, IGF2BP3, and PD-L1 were positively correlated by IHC staining. More importantly, the higher expression of METTL3 or IGF2BP3 was also found in patients who received PD-1/PD-L1 blockade therapy and seemed to show better responses. However, because of the limited clinical data, the efficacy of clinical trials in breast cancer could not be adequately captured in this study. Thus, further larger sample sizes are needed for clinical investigation and examination of in-detailed mechanisms underlying METTL3/IGF2BP3-induced antitumor immunity.
Taken together, the present study on METTL3/IGF2BP3-mediated N6-methyladenosine modification of PD-L1 mRNA and antitumor immunity provided a novel mechanism for m6A regulator-induced immunosuppression in breast cancer, which may have potential application as a novel therapeutic target.
Conclusion
In conclusion, our study has illustrated the critical role of METTL3-mediated m6A modification in PD-L1 mRNA stabilization in an IGF2BP3-associated manner in breast cancer cells. Our findings broaden knowledge of the epi-transcriptional regulation mechanisms of PD-L1 expression and the functional value of m6A methyltransferase in tumor immune surveillance which may have important implications for new and efficient therapeutic strategies in the tumor immunotherapy.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Abbreviations
- m6A:
-
N6-methyladenosine
- METTL3:
-
Methyltransferase-like 3
- IGF2BP3:
-
Insulin-like growth factor-2 mRNA-binding protein 3
- MeRIP:
-
Methylated RNA immunoprecipitation
- RIP:
-
RNA immunoprecipitation
- DAA:
-
3-Deazaadenosine
- IgG:
-
Immunoglobulin G
- CDS:
-
Protein-coding transcripts region
- ISO:
-
Iso control antibody
- TNBC:
-
Triple-negative breast cancer
- HER2 + :
-
HER2-positive breast cancer
- IHC:
-
Immunohistochemistry
- IL-2:
-
Interleukin-2
- PD-1:
-
Programmed death receptor-1
- PD-L1:
-
Programmed death-ligand 1
- IFN-γ:
-
Interferonγ
- TIM3:
-
Hepatitis A virus cellular receptor 2
- NR4A1:
-
Nuclear receptor subfamily 4, group A, member 1
References
Sung H, Ferlay J, Siegel R, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. 2021.
Li ZL, Zhang HL, Huang Y, Huang JH, Sun P, Zhou NN, et al. Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat Commun. 2020;1:3806.
Esteva F, Hubbard-Lucey V, Tang J, Pusztai L. Immunotherapy and targeted therapy combinations in metastatic breast cancer. Lancet Oncol. 2019;20:e175–86.
Postow M, Callahan M, Wolchok JD. Immune checkpoint blockade in Cancer therapy. J Clin Oncol. 2015;33:1974–82.
Topalian S, Drake C, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–12.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–21.
Schmid P, Rugo HS, Adams S, et al. Atezolizumab plus nab-paclitaxel as frst-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efcacy results from a randomised, doubleblind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020;21:44–59.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N Engl J Med. 2020;382(9):810–21.
Burr ML, Sparbier CE, Chan Y-C, Williamson JC, Woods K, Beavis PA, et al. CMTM6 maintains the expression of PD-L1 and regulatesanti-tumour immunity. Nature. 2017;549:101–5.
Bellucci R, Martin A, Bommarito D, Wang K, Hansen S, Freeman G, et al. Interferon-γ-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology. 2015;4:e1008824.
Noman M, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:781–90.
Casey S, Tong L, Li Y, Do R, Walz S, Fitzgerald K, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–31.
Li C, Lim S, Chung E, Kim Y, Park A, Yao J, et al. Eradication of triple-negative breast Cancer cells by targeting glycosylated PD-L1. Cancer Cell. 2018;33:187–201.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira N, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–5.
Lim S, Li C, Xia W, Cha J, Chan L, Wu Y, et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–39.
Yao H, Lan J, Li C, Shi H, Brosseau J, Wang H, et al. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306–17.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169:1187–200.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–9.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Liu J, Harada BT, He C. Regulation of gene expression by N6-methyladenosine in cancer. Trends Cell Biol. 2019;29:487–99.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N6-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28(5):507–17.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–5.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m6A methyltransferase METTL3 promotes translation in human Cancer cells. Mol Cell. 2016;62:335–45.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m6A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39:e104514.
Yin H, Zhang X, Yang P, Zhang X, Peng Y, Li D, et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat Commun. 2021;12:1394.
Yang S, Wei J, Cui Y, Park G, Shah P, Deng Y, et al. m6A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Tsutsumi S, Saeki H, Nakashima Y, Ito S, Oki E, Morita M, et al. Programmed death-ligand 1 expression at tumor invasive front is associated with epithelial-mesenchymal transition and poor prognosis in esophagealsquamous cell carcinoma. Cancer Sci. 2017;108:1119–27.
Adams S, Gatti-Mays ME, Kalinsky K, Korde LA, Sharon E, Amiri-Kordestani L, et al. Current landscape of immunotherapy in breast cancer: a review. JAMA Oncol. 2019;5:1205–14.
Polk A, Svane IM, Andersson M, Nielsen D. Checkpoint inhibitors in breast cancer - current status. Cancer Treat Rev. 2018;63:122–34.
Villadolid J, Amin A. Immune checkpoint inhibitors in clinical practice: update on management of immune-related toxicities. Transl Lung Cancer Res. 2015;4:560–75.
Emens L, Adams S, Barrios C, Diéras V, Iwata H, Loi S, et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann Oncol. 2021;32(8):983–93.
Kyte JA, Andresen NK, Russnes HG, Fretland SØ, Falk RS, Lingjærde OC, et al. ICON: a randomized phase IIb study evaluating immunogenic chemotherapy combined with ipilimumab and nivolumab in patients with metastatic hormone receptor positive breast cancer. J Transl Med. 2020;18:269.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m6A mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Patil DP, et al. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–76.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–70.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193–205.
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. m6A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12:135.
Tsuruta N, Tsuchihashi K, Ohmura H, Yamaguchi K, Ito M, Ariyama H, et al. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem Biophys Res Commun. 2020;10:530.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Li T, Fan J, Wang B, Traugh N, Chen Qianming, Liu JS, et al. TIMER: A web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer Res. 2017;77:e108–10.
Ru B, Wong C, Tong Y, Zhong J, Zhong S, Wu W, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019;35:4200–2.
Zhou Y, Huang T, Siu H, Wong C, Dong Y, Wu F, et al. IGF2BP3 functions as a potential oncogene and is a crucial target of miR-34a in gastric carcinogenesis. Mol Cancer. 2017;16:77.
Samanta S, Sun H, Goel H, Pursell B, Chang C, Khan A, et al. IMP3 promotes stem-like properties in triple-negative breast cancer by regulating SLUG. Oncogene. 2016;35:1111–21.
Wang S, Chim B, Su Y, Khil P, Wong M, Wang X, et al. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes Dev. 2019;33:1048–68.
Zhou Y, Zeng P, Li YH, Zhang Z, Cui Q. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sitesbased on sequence-derived features. Nucleic Acids Res. 2016;44:e91.
Deng S, Zhang H, Zhu K, Li X, Ye Y, Li R, et al. M6A2Target: a comprehensive database for targets of m6Awriters, erasers and readers. Brief Bioinform. 2021;22.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Key R&D Program (2018YFC1313400), Science and Technology Innovation Enhancement Project of Army Medical University (2019CXJSB017 and 2019XYY21) and Chonqing Science and Technology Bureau (No. 2021MSXM338).
Author information
Authors and Affiliations
Contributions
Conceptualization and design: Xiang Xu, Donglin Luo, Weijun Wan, and Xiang Ao; Methodology development: Quan Chen, Yang Yu, Wei Xing, Wei Guo and Luoquan Ao; Data acquisition (provided animals, provided access to facilities, etc.): Xiaofeng Wu, Chengxiu Pu, Xueting Hu, Zhuo Chen Zhan Li and Mengwei Yao; Data analysis and interpretation (e.g., statistical analysis, bioinformatics, and other computational analyses): Weijun Wan, and Xiang Ao; Writing and/or revision of the manuscript: Xiang Xu, Weijun Wan, and Xiang Ao; Administrative, technical, or material support (e.g., reporting or organizing data, constructing databases): Wei Xing, and Luoquan Ao; Study supervision: Xiang Xu, Donglin Luo; Other (e.g. supervision of in vivo animal work): Xiaofeng Wu, Chengxiu Pu, and Xueting Hu; All authors read and approved this manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of the Daping Hospital of Army Medical University. All animal experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animal by International Committees. Every effort was made to minimize the numbers and suffering of the included animals.
Consent for publication
The authors confirm that they have obtained written consent from each patient to publish the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
PD-L1 is a downstream target of METTL3. a RIP-seq demonstrated the METTL3 binding profile in PD-L1 gene. b PD-L1 mRNA expression was tested by qRT-PCR in several breast cancer cells.
Additional file 2: Fig. S2.
METTL3 increases N6-methyladenosine modification and expression of PD-L1 mRNA. a Enrichment of METTL3 on PD-L1 mRNA was analyzed by RIP-qPCR in breast cancer cells compared to IgG. b-c The interaction between METTL3 and PD-L1 mRNA was analyzed by RIP-qPCR assay in MDA-MB-231 and HCC38 cells with overexpression or knockdown of METTL3. d-e The relative levels of m6A in PD-L1 were tested by MeRIP-qPCR in SK-BR-3 and HCC38 cells with knockdown or overexpression of METTL3. f The mRNA lifetime of PD-L1 transcripts in breast cancer cells with (shMETTL3) or without (sh-control) METTL3 silencing. g Schematic representation of experiment for absolute quantification of m6A modification. h-j The expression levels of PD-L1 were analyzed by qRT-PCR, western blot and flow cytometry in SK-BR-3 and HCC38 cells transfected with or wihthout sh-METTL3. *p < 0.05; **p < 0.01; ***p < 0.001.
Additional file 3: Fig. S3.
IGF2BP3 mediates the mRNA expression of PD-L1 in m6A-dependent manner. a The mRNA level of PD-L1 was tested by qRT-PCR in MDA-MB-231 cell with knockdown of IGF2BP1 or IGF2BP2. b-d The expression levels of PD-L1 mRNA, protein and membrane were investigated by qRT-PCR, flow cytometry and western blot in SK-BR-3 cells. e Enrichment of IGF2BP3 on PD-L1 mRNA was analized by RIP-qPCR in breast cancer cells compared to IgG. f-g The interaction between IGF2BP3 and PD-L1 mRNA was analyzed by RIP-qPCR assay in SK-BR-3 cells. h The interaction between IGF2BP3 and PD-L1 mRNA with m6A mutation was detected by RIP-qPCR in breast cancer cells. i Enrichment of IGF2BP3 on PD-L1 mRNA was detected by RIP-qPCR assay in control and METTL3-knockdown cells. j PD-L1 mRNA levels were analyzed by qRT-PCR assay in SK-BR-3 cells after actinomycin D treatment. k The expression of PD-L1 protein was determined by western blot with transfection of indicated genes.*p < 0.05; **p < 0.01; ***p < 0.001; n.s., no significance.
Additional file 4: Fig. S4.
METTL3/IGF2BP3-downregulated antitumor immunity in breast cancer cells. a The schematic illustrates the protocol for administration in B-NDG mice. b-c The volume of tumors were measured in the MDA-MB-231 cells-constructed xenograft models with indicated treatment. d The effect of METTL3 disruption on tumour growth was verified by luciferase activities assay in BALB/c peritonealtumor xenograft models. e The survival times were recorded and visualized using Kaplan-Meier survival curve. f CD3/ 4/ 8-positive cell number per high-power field (HPF) using immunohistochemistry (n = 3). g-h Volume and weight of tumors were determined in the subcutaneous-tumor with knockdown of METTL3. i CD3/ 4/ 8-positive cell number per high-power field (HPF) using immunohistochemistry (n = 3).
Additional file 5: Fig. S5.
IGF2BP3 expression positively correlates with infiltration levels of immune cells in breast cancer. a Scatterplots of correlation between IGF2BP3 or PD-L1 expression and abundance of immune infiltration from TIMER database in breast cancer subtypes were shown. b IGF2BP3 expression had significant positive correlations with act-B Tcells/ CD8 T cells/ CD4 T cells, as with PD-L1.
Additional file 6: Fig. S6.
The posotive correlation of PD-L1 with METTL3 and IGF2BP3. a The correlation between PD-L1 and METTL3 or IGF2BP3 in luminal A (n = 41) and luminal B (n = 38) subtypes were analyzed by Spearman’s rank correlation test. b The expression of METTL3 and IGF2BP3 in luminal A, luminal B, HER2+ and TNBC subtypes.
Additional file 7: Table S1.
Sequences of primers and antibodies used in this study. Table S2. The list of down-modified genes in the intersection of epitranscriptomic microarray and MeRIP-seq.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wan, W., Ao, X., Chen, Q. et al. METTL3/IGF2BP3 axis inhibits tumor immune surveillance by upregulating N6-methyladenosine modification of PD-L1 mRNA in breast cancer. Mol Cancer 21, 60 (2022). https://doi.org/10.1186/s12943-021-01447-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-021-01447-y