
Abstract
Antimicrobials have been widely used to prevent and treat infectious diseases and promote growth in food-production animals. However, the occurrence of antimicrobial resistance poses a huge threat to public and animal health, especially in less developed countries where food-producing animals often intermingle with humans. To limit the spread of antimicrobial resistance from food-production animals to humans and the environment, it is essential to have a comprehensive knowledge of the role of the resistome in antimicrobial resistance (AMR), The resistome refers to the collection of all antimicrobial resistance genes associated with microbiota in a given environment. The dense microbiota in the digestive tract is known to harbour one of the most diverse resistomes in nature. Studies of the resistome in the digestive tract of humans and animals are increasing exponentially as a result of advancements in next-generation sequencing and the expansion of bioinformatic resources/tools to identify and describe the resistome. In this review, we outline the various tools/bioinformatic pipelines currently available to characterize and understand the nature of the intestinal resistome of swine, poultry, and ruminants. We then propose future research directions including analysis of resistome using long-read sequencing, investigation in the role of mobile genetic elements in the expression, function and transmission of AMR. This review outlines the current knowledge and approaches to studying the resistome in food-producing animals and sheds light on future strategies to reduce antimicrobial usage and control the spread of AMR both within and from livestock production systems.
Similar content being viewed by others


Introduction
It is estimated that the world population will reach 9.1 billion in 2050, among which 70% will be urbanites [1]. Concomitantly, the consumption of meat and eggs, as well as dairy products, is predicted to increase by 73% and 58%, worldwide respectively, by 2050 [2]. In order to meet the increasing demand for animal food products, strategies should be implemented to improve growth and efficiency in ruminants, swine, and chickens. In fact, meat production in low- and middle-income countries such as Africa, Asia, and South America have increased by 68%, 64%, and 40% respectively, since 2000 [3], which is largely due to the adoption of of intensive production systems.
In such systems, antimicrobials play a vital role in increasing the health and production efficiency of livestock [4]. Since the 1950s, antimicrobials have been used in livestock and poultry production to prevent and treat diseases and to improve the feed conversion efficiency and promote growth [5]. The global total use of antimicrobials for cattle, pigs and chickens will increase from approximately 63,000 tons in 2010 to approximately 105,000 tons by 2030, an increase of up to 67% [6]. In European Union, 8,927 tons of antimicrobials were used in food-producing animals in 2004 [7]. In the US, sub-therapeutic doses of antimicrobials used in food-producing animals reached approximately 14,600 tons in 2012 [8]. In China, the world’s largest producer and consumer of antimicrobials, 29,774.09 tons of antimicrobials were used in animal husbandry in 2018, with more than half of this amount used to promote animal growth [9, 10]. Despite their benefits, there is growing evidence that large-scale use of antimicrobials in food-producing animals selects for antimicrobial resistance (AMR) bacteria in livestock [11,12,13]. The selection for AMR bacteria not only increases morbidity and mortality in food-producing animals but also increases the risk of transmission of AMR bacteria to human beings [14,15,16]. This is because the antimicrobial resistance genes (ARGs) of the bacteria in the digestive tract of food-producing animals can be transferred to bacteria that can come in contact with humans either directly or from the environment. In this regard, the World Health Organization called on its member countries to reduce the use of veterinary antimicrobial drugs in 2017 [17, 18]. Therefore, AMR is one of the most urgent challenges facing the world currently, posing a threat to health care and food safety.
Efforts have been made to limit the potential spread of AMR from food-producing animals to human beings and environment. For example, many European countries have banned the use of antimicrobials in farm animals for ‘non-therapeutic’ purposes [19]. Recently, the Chinese government launched a regulation to withdraw medicated feed additives in accordance with the National Action Plan to Combat Antimicrobial Resistance from Animal Resources (2017–2020) [20,21,22]. In addition to these policies, several strategies have been developed (e.g. the use of bacteriophages, antimicrobial peptides, or vaccines) which may be promising to replace the use of antimicrobials in food-producing animals, which have been extensively reviewed [23, 24] While these strategies are crucial to restrict the prevalence of AMR in food-producing animals, ARGs can still be detected in the animal production systems even if no antimicrobials are administered [25]. In order to further reduce the spread of ARGs from food-producing animals to humans or the environment, it is essential to clarify which ARGs are carried by the microbes (bacteria or archaea) inhabiting the digestive tract of food-producing animals. In recent years, with the development of next-generation sequencing (NGS) techniques, studies have enabled the characterization of a collection of ARGs, termed the resistome [26, 27], in a variety of environments including water, soil, as well as the digestive tract of humans and livestock [28,29,30]. This approach has greatly expanded the scope of ARG monitoring compared with traditional culture and/or polymerase chain reaction (PCR)-based techniques [31, 32]. In this review, we first introduce methods, pipelines (e.g. read- and assembly-based approaches), resources/tools, and databases for resistome identification using shotgun metagenomic sequencing techniques. We then summarize the findings on the profiles and abundance of resistome, as well as define the factors (e.g. dietary composition or use of antimicrobials) that influence the resistome in the digestive tract of swine, poultry, and ruminants (cattle and sheep). Finally, we propose future research advancements in the application of long-read sequencing in resistome analysis, role of mobile genetic elements in resistome development, and expression profiles of the resistome. We also propose host, microbial, and environmental factors that may explain the effect of antimicrobial use on resistome profiles in the digestive tract of food-producing animals, with an aim to develop strategies to control the spread of ARGs from food-producing animals to humans.
Metagenomic-based approach for resistome characterization
Traditionally, two state-of-the-art approaches have been used to detect ARGs within their bacterial host. One approach is based on culturing, antimicrobial susceptibility testing, and polymerase chain reaction. However, this approach only applies to culturable bacteria and does not enable the discovery of distantly related or unknown elements [33]. The other approach is based on whole genome sequencing, which enables the detection of complete genome in bacteria including the ARGs that it may carry [33, 34]. However, this approach can only identify ARGs of in collected isolates and may not reflect the complexity of the resistome in vivo, as the digestive tract of humans and animals harbors a complex and dynamic bacterial population. The advent and development of NGS technologies has enabled and accelerated research of environmental microbiome using shotgun metagenomics [35, 36], which also allows the analysis of the whole genome as well as identification of unknown genetic elements of both culturable and unculturable microbial species [37, 38]. More recently, shotgun metagenomics has expanded our abilities to comprehensively investigate ARGs, as a result of an increase in the availability of bacterial genome databases as well as decrease in sequencing costs.
Estimation of sample size
Prior to initiating an experiment, it is crucial for a researcher to estimate the statistical power to determine the sample size needed for a resistome studies. This is important and needed to test the research hypothesis adequately and draw meaningful conclusions [39]. To conduct power calculations in microbiome research, methods such as t-test, analysis of variance, χ2 test, and the Dirichlet-Multinomial model can be applied [40]. Some web-based pipelines are also available for sample size and power calculation in microbiome studies [41, 42]. However, the sample sizes have not been evaluated in most resistome studies in food-producing animals. One reason could be that it may be difficult to perform a formal sample size evaluation for specific food-producing animals, as the related studies are highly limited and the variation among individuals for a population is largely unknown.
Sample preparation
Another key to the success of metagenomic analysis is to obtain high-quality DNA for unbiased interpretation of microbiota in the digestive tract of food-producing animals. Therefore, proper sampling and storage processes need to be taken into account during the design of resistome studies. Under practical conditions, it is not feasible to collect and process samples on the same day, as samples are usually collected at different time points and stored at −80°C for future analysis. In this regard, appropriate methods for sample storage and DNA extraction are essential to accurately represent the gut microbiota using NGS technologies [43]. A recent study showed that sample storage reagents and DNA extraction methods interactively impact the recovery of gut microbiota from chickens and pigs [44]. Consequently, standardization of protocols for sample storage and DNA extraction are needed in the study of gut microbiota in food-producing animals.
Quality control of metagenomic reads
The most crucial step before the analysis of resistome is the quality control (QC) of metagenomic reads. Workflows to conduct QC of metagenomic analysis have been extensively reviewed by Li et al. [36], which mainly includes trimming low-quality bases and residual artificial sequences as well as depleting host DNA. Trimmomatic [45] and Cutadapt [46] are frequently used tools for adapter trimming and read filtering. Non-microbial host DNA can be a major concern for sequencing-based microbiome analysis, especially for samples that are low in microbial biomass [47, 48]. It has been shown that samples with high amounts of host DNA coupled with reduced sequencing depths lower the sensitivity of microbiome profiling within metagenomic datasets [49]. Host DNA can be depleted either experimentally or bioinformatically. For example, the use of MolYsis complete5 kit was shown to effectively remove host DNA in bovine milk samples [48]. In addition to experimental methods, several bioinformatic tools/resources such as MG-RAST [50] and TopHat2 [51] can be further used to filter bovine DNA and RNA sequences from metagenomic or metatranscriptomic datasets [52, 53]. While MG-RAST is a web-based pipeline, TopHat2 can be ran on a desk top computer, with both approaches using Bowtie aligner [54] to map contaminated reads against a host genome database. Other pipelines such as MetaWRAP [55] and Sunbeam [56] that incorporate Trimmomatic or Bowtie aligner can also be used for QC and removal of host-derived reads.
Assembly-based analysis vs. Read-based analysis
To analyze the resistome based on shotgun metagenomics, the short sequence reads generated by any sequencing platforms such as Illumina can either be directly mapped to reference databases (defined as reads-based methods), or first assembled into contigs and then annotated though comparison with reference databases (defined as assembly-based methods) [57]. For both approaches, high-quality data and reliable bioinformatics pipelines are needed. Here we provide an overview of the major steps of metagenomic-based resistome analysis (Fig. 1).

Pipelines for the analysis of resistome based on metagenomic sequencing (Created with BioRender.com)
For assembly-based methods, the first step is to assemble the post-QC reads into contigs. This can be performed using De Bruijn graph-based assemblers, with MEGAHIT [58] and MetaSPAdes [59] being most frequently used [60]. A recent review described the protocols of these assemblers in detail [61]. It should be noted that multiple biological (e.g. sample origin, biomass, and representation) and technical (e.g. sequence quality, depth, and platform) factors may affect the performance of an assembler’s ability to generate high-quality contigs [62]. Thus, different assemblers should be tested on a subset of samples to identify the optimal procedure for a given dataset. For read-based approaches, ARGs can be detected either by aligning reads directly to the reference databases using aligners such as Bowtie2 [54] or Burrows-Wheeler aligner (BWA) [63], or by splitting reads into k-mers before mapping them to reference databases [57]. Specific tools such as Short Read Sequencing Typing (SRST2) [64] can be used to identify AMR in samples with insufficient reads to perform de novo assembly. More recently, tools such as Antimicrobial Resistance Identification By Assembly (ARIBA) [65] and graphing Resistance Out Of meTagenomes (GROOT) [66], have been developed that can efficiently map large-scale sequence datasets.
It should be noted that there is no consensus on which approach (read- vs. assembly-based) is more accurate and both have advantages and disadvantages [67]. For example, read-based approaches are generally faster and less computationally demanding as compared with assembly-based methods, as they bypass de novo assembly, protein-coding gene prediction and pairwise alignment to public databases. However, read based approaches lack the positional information required to analyze upstream and downstream factors of identified resistance genes. In contrast, assembly-based methods can construct whole genomes or large contigs with protein-coding genes and regulatory sequence information, enabling generation of a surrounding genomic context. This enables the analyses of co-associated genes and biological pathways that could play a role in the regulation of ARGs. Nevertheless, assembly-based methods are computationally demanding, time consuming and requires deeper genomic coverage to avoid information loss [68]. In this regard, the profiles of resistome among studies need to be interpreted and compared with caution if different analysis approach are used.
ARG reference database
In addition to data processing and analysis approaches, the prediction accuracy of ARGs also relies on the integrity of the reference databases. The ARG reference databases can be divided into two types based on their features. For example, databases such as Antibiotic Resistance Genes Database (ARDB, not curated since 2009) [69], ResFinder [70], and Comprehensive Antibiotic Resistance Database (CARD, also contains all data from ARDB) [71] have been used to detect all known sequenced ARGs, while those such as Antibiotic Resistance Genes Online (ARGO) are specifically used to detect only β-lactam and vancomycin resistance genes [72]. Secondly, the criteria for entry in the reference databases are different: while CARD requires the entries must have been published in the scientific literature [71], ResFinder lacks this requirement [70]. Thirdly, the types of entries differ between reference databases. Most reference databases include all drug resistance genes, and only a few reference databases also include drug resistance genes that arise as a result of chromosomal mutations (e.g. MUBII-TB-DB [73] and PointFinder [74]). Finally, entry format also differs among reference databases (e.g. fasta or json, etc.), download permissions, and regular maintenance frequency [75]. Therefore, it is necessary to have a full understanding of these distinguishing characteristics when selecting a suitable database for resistome analysis.
Annotation of ARGs and data process
For assembly-based methods, the metagenomic-assembled contigs are annotated for resistance determinants by predicting protein-coding regions on contigs, which are then compared against AMR reference databases using similarity-based search tools such as Basic local alignment search tool (BLAST) [76] or DIAMOND [77]. For read-based analysis, post-QC reads are directly aligned to AMR reference databases using alignment tools as mentioned above for the characterization of ARGs. The annotated ARG can be normalized to either total number of reads [78,79,80] or 16S rRNA gene copies [81,82,83,84,85]. Although the 16S rRNA gene is frequently used for normalization, it may not always yield accurate estimation of ARGs as multiple copies of this gene can be present within a genome [86]. Recently, it has been shown that using multiple single-copy marker genes [87] or comparison of each gene count with the whole content of ARGs [88] may be an alternative approach for data normalization. In summary, the methods for data normalization should be taken into consideration when comparing the resistome in the digestive tract of food-producing animals across different studies.
Identification of microbial host of ARGs using metagenomic binning
Metagenomic binning enables near-complete microbial genomes to be reconstructed from metagenomic sequencing data, which has been extensively used in the food-producing animals such as swine [89, 90], chickens [91, 92], and ruminants [93,94,95]. Technically, binning of assembled metagenomic sequences requires binners such as MaxBin [96, 97], MetaBAT [98, 99], or Concoct [100]. The obtained metagenomic-assembled genomes (MAGs) can be further refined using tools such as DASTool [101] to remove non-microbial DNA and increase predictive accuracy. The completeness and residual DNA contamination of MAGs can be calculated using CheckM v1.0.6 [102] based on lineage-specific conserved marker gene sets in each genome. Finally, the classification of MAGs can be achieved by blasting them against the reference genomic database. A recent review comprehensively evaluated the performance of 15 genome binning tools and suggested that Groopm2 [103] achieved the highest purity while MetaBat2 had higher completeness than other binners using metagenomic datasets generated from the contents of the chicken gut [104]. In recent years, metagenomic binning analysis has been applied in combination with resistome analysis to identify the microbial host of ARGs in environmental samples from public/private houses [105], lake sediments [106], and a wastewater treatment plant [107]. However, the use of binning analysis is still very limited for resistome analysis of samples collected from human, environment, and food-producing animals. One potential reason could be that binning analysis requires either genomic sequences derived from uncharacterized microorganisms (reference-dependent) or substantial computing resources (reference-independent) [108]. With the expansion in the reference database of various microorganisms as well as development of less computationally-intensive tools (e.g. MetaBMF [109]), we speculate that binning analysis will be widely used to identify microbial hosts of ARGs in the digestive tract of food-producing animals in the future.
Factors affecting the accuracy of resistome analysis
In addition to the difference in analysis approach (read- and assembly-based), reference databases (ARDB, CARD), as well as data normalization method, other factors may also affect the predictability of the resistome. First of all, there are currently at least 50 tools/pipelines that can be used to analyze the resistome [110, 111]. Since the features (e.g. read-based or assembly-based) of these analysis tools differ greatly, it is necessary to consider benchmarking of resistomes that are analyzed using different pipelines [112]. Secondly, the composition of resistome is heavily influenced by the phylogenetic profile of the bacterial population. Considering that there are still a large number of bacteria that possess unidentified ARGs, the known ARGs may only represent a small part of the true resistome. It is reasonable to assume that with future advances in sequencing as well as the expansion in ARGs and phylogenetic databases, new ARGs will be identified and characterized. In addition, sequencing depth can also affect the profile of resistomes. For example, by sampling three potential environmental ARG reservoirs (pig caeca, river sediment, effluent) and sequencing them using shotgun metagenomics, Gweon et al. [113] found that at least 80 million reads per sample were required to cover the full richness of different ARG families within these environments. In addition, Zaheer et al. [114] showed that the number of reads being assigned to ARGs increased significantly with increasing sequencing depth (from 26 to 117 million reads), with a depth of approximately 60 million being suitable to describe the resistome in feces from beef cattle. While the optimal sequencing depth needed for resistome analysis in the digestive tract of food-producing animals still deserves investigation, these studies suggest that a balance between sequencing depth and cost needs to be defined in order to obtain reasonable results.
Profiles of resistome in the digestive tract of food-producing animals
Recently, a number of studies characterizing the resistome in food-producing animals been conducted as a result of a reduction in the cost of metagenomic sequencing as well as the development of pipelines and resources for resistome analysis [75, 115]. Here we summarize the recent advances in the analysis of resistome in the digestive tract of ruminants, swine, and poultry, focusing on the fecal resistome. Feces often represent the principal conduit of ARG contact from livestock with soil, water, vegetation and humans. This is particular true for less developed countries where food-producing animals are often raised in close proximity to human habitations [116].
Resistome in the digestive tract of swine
Antimicrobials belonging to penicillin, tetracycline, and macrolide groups are commonly used in swine [117, 118]. In general, oral administration via water or feed is the most common route of antimicrobial administration in swine [119], for treatment of infectious disease, metaphylaxis, prophylaxis and growth promotion [118]. Here we summarized findings from 13 studies investigating the resistome in the digestive tract of swine (Table 1). Most of these studies investigated the effect of anti-microbial use (AMU) [120,121,122,123,124,125,126,127,128,129] on the profiles of resistome in the swine digestive tract, while only one study reported the resistome profile in the absence of AMU [130]. Two studies failed to mention if antimicrobials were used or not [30, 131].
Joyce et al. [130] investigated the resistomes of swine feces and found that the core resistome (present in all samples) contained 56 ARGs with five tetracycline resistant genes (tetW, tetQ, tet44, tet37, tet40) being the most abundant, and the accessory resistome (detected in at least one sample but not present in all samples) being comprised of 201 ARGs. This finding provides insights into the profiles of fecal resistome in swine in the absence of antimicrobial selective pressure and emphasizes that AMU is not the only factor that dictates the nature of the resistome within the digestive tract of swine.
Resistome profiles have also been shown to differ among AMU protocols. For example, a study compared the profiles of resistome in feces of pigs of differing age and found that tetracycline was the most abundant ARG for both 1- and 8-month-old pigs, followed by aminoglycoside and macrolide-lincosamide-streptogramin (MLS) [120]. These authors further found that the abundances of several classes of ARGs in feces including bleomycin, fosmidomycin, and polymyxin decreased with age (8- vs. 1-month-old), possibly as a result of the reduction in the use of these antimicrobials in older swine [120]. However, it should be noted that only 3 samples of each age were analyzed in that study, which may lead to low reproducibility of the results. In addition, Van Gompel et al. [123] found a positive association between the use of macrolides and tetracyclines and AMR in pigs raised in 9 European countries. However, they didn’t find significant associations between the high use of β-lactams and the abundance of their resistance genes in the feces of younger pigs. These authors also reported that the profiles of fecal resistome in pigs were country-specific [122], possibly a reflection of differences in AMU (frequency/dosage) among countries with it being high in Spain and Italy and low use in Denmark and the Netherlands. Administration of oxytetracycline significantly enriched the abundances of 41 ARGs in swine feces, mainly members of the tetracycline, β-lactam and multidrug (MDR) classes, with an increase in Escherichia and Prevotella within the bacterial population as compared to non-medicated pigs [124]. It was proposed that these ARG-carrying bacteria may have the potential to transfer ARGs to other susceptible bacteria in the digestive tract of swine [124]. One limitation of this study is that dietary factors, which may have a significant impact on microbiota and associated resistome, were not reported. In addition, a study investigated the effect of perinatal use of tulathromycin on the profiles of fecal resistome in pre-weaned piglets [125]. Although these authors identified a total of 127 ARGs related to 19 different classes, perinatal use of antimicrobials had no impact on the structure of fecal microbiota or the abundance of ARGs in feces from pre-weaned piglets [125]. These inconsistent findings suggest that AMU may not be the only factor that impacts the resistome profiles in the digestive tract of swine, and that other factors such as diet, growth stage, housing type and other environmental influences also play a role.
In addition to feces, the resistome in the intestinal contents of weaned piglets has been recently reported [129]. These authors reported that the resistome in the colon contained ARGs mainly associated with tetracycline, MLS, and aminoglycoside resistance. In the ileum, tetracycline was also the predominant ARG, while penem, fluoroquinolone, aminoglycoside, and cephalosporin ARGs also accounted for a large proportion of resistome. In addition, they found that administration of low-dosage antibiotics for 4 weeks had no significant impact on the structure, diversity, or diversity of resistome in either ileum or colon contents. Considering that only one sampling was available in this study, the long-term effect of exposure to antibiotics on resistome needs further investigation. Nevertheless, it should be noted that the microbiota in feces may not fully represent the microbiota within the whole gastrointestinal tract [132]). More research is needed to investigate the profiles of resistome in the various regions of the swine gastrointestinal tract in addition to that in feces so as to have a comprehensive understanding of the resistome throughout the digestive tract of swine.
Resistome in the digestive tract of chickens
In intensive poultry farming, antimicrobials such as tetracycline, bacitracin, tylosin, salinomycin, virginiamycin and bambermycin are often used [117, 133]. In US, tetracyclines represent more than two-thirds of antimicrobials administered in poultry production [134], while they represent only 37% [135] in the EU poultry industry. Amoxycillin, oxytetracycline and ceftriaxone are the most commonly used antimicrobials in poultry, followed by ofloxacin and norfloxacin in China [136]. Inclusion in feed is the most common route of administration of antimicrobials in poultry, mainly to prevent necrotic enteritis caused by Clostridium perfringens and coccidiosis [137].
Studies of the resistome in the digestive tract of chickens are relatively limited as compared to swine (Table 2). As with swine, it has been reported that tetracycline, aminoglycoside, and MLS are the most abundant ARG classes in chicken feces (20-day and 80-day old) [120, 121], despite that only 2 samples were collected from each age group. Subsequent studies confirmed that ARGs belonging to these classes were predominant, although one study reported MDR as the predominant ARG class in chicken feces [125, 137]. A total of 49 core ARGs were identified in 178 fecal samples collected from 9 European countries, with tetracycline and macrolide ARGs accounting for the majority of the resistome [121]. Although these authors reported a more diverse resistome in fecal samples from chickens than pigs [121], dietary compositions of chickens raised in different countries were unavailable, making it difficult to determine whether dietary factors may contribute to the diversity of the resistome in chickens. A recent study in China compared the fecal resistome of chickens at poultry farms to those at live poultry markets in China [139]. For both sites, ARGs conferring resistance to aminoglycoside, tetracycline, MLS, and β-lactam were more abundant than those associated with other classes. In addition, these ARG classes were more abundant in birds at the poultry market than in those on farm [139], an observation that could reflect changes in microbiome due to increased stress in birds that are marketed through live poultry trade.
A few studies also investigated the use of specific antimicrobials on the profiles of the fecal resistome in poultry. For example, administration of ampicillin led to an increase in the abundance of most β-lactam and bacitracin ARGs and a decrease in ARGs associated with tetracycline classes in chicken feces [140]. In addition, this increase in β-lactam, bacitracin-resistance, and MDR ARGs were more evident if the ampicillin was administered orally as compared to intramuscularly [140]. However, therapeutic dosages of chlortetracycline in feed increased the abundance of tetracycline resistance (tetA and tetW) and reduced multiple MDR ARGs in broiler feces [138]. This response to chlortetracycline was attributed to a decline in the population of Escherichia, a major host of MDR ARGs, and the enrichment of Bifidobacterium, which harbours more tetW [138]. These findings indicate that dosage, method of administration and type of antimicrobial all influence the profile of the resistome within the digestive tract of poultry. In addition to the fecal resistome, a study compared the profile of cecum resistome in chickens housed at low (730 m) and high (3,300 m) altitudes [141]. While tetracycline, MLS, and cephalosporin were the predominant classes of ARGs identified at both altitudes, these classes were more abundant in chickens reared at low than high altitude [141], This may reflect the lower abundance of ARGs in soil and water bacteria at high-altitude environments that could be transferred to chickens, but such a hypothesis would require further investigation.
Resistome in the digestive tract of ruminants
At least ten classes of antimicrobials are used in ruminants (e.g. tetracyclines, amphenicols, penicilins, cephalosporins), which are mainly used to prevent or treat diarrhoea, respiratory disease, navel infections, liver abscesses, foot rot and joint and uterine infections in beef and dairy cattle [142]. In dairy cows, penicillins, cephalosporins, or other beta-lactams are used to prevent and control mastitis [143]. In addition, acute puerperal metritis can be treated with penicillin/ampicillin in conjunction with oxytetracycline or ampicillin and cloxacillin [144]. The route of administration depends on the type of antimicrobial. For example, in dairy cows, penicillins, cephalosporins, or other beta-lactams are often infused into the mammary gland, whereas penicillins, macrolides, aminoglycosides, and fluoroquinolones are administered parenterally, while sulfonamides [143,144,145] are administered orally. For beef cattle, tylosin, chlortetracycline, oxytetracycline, virginiamycin, are used for liver abscess prevention, while macrolides and tetracycline’s are often administered to prevent or treat bovine respiratory disease [146]. In sheep and goats, penicillin is often used due to its low cost and the low risk associated with off-label use, as few antimicrobials are specifically registered for use in small ruminants [147,148,149].
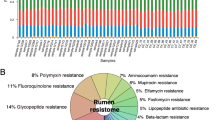
Rumen resistome
The rumen harbors a dense microbiota including bacteria, methanogens, protozoa, fungi, and phages [150]. Consequently, the resistome of this unique microbial ecosystem has been investigated in several studies [151, 152]. Recent research showed that rumen microbiota in cattle and sheep also harbor a vast reservoir of ARGs, with the abundance and gene type affected by factors such as diet and/or use of antimicrobials (Table 3). For example, Hitch et al. [153] identified 30 ARGs in the rumen of sheep that harboured a high abundance of daptomycin and colistin resistance genes, both of which represent ‘last-resort’ antimicrobials against gram-positive bacterial infections in humans [166, 167]. Although not specifically documented, there is a risk that these ARGs could be transferred to human-associated bacteria through close contact on farm, in abattoirs or consumption of contaminated meat or milk products. In addition, Auffret et al. [152] comparatively investigated the effects of diet (concentrate:forage ratio) and breed on the profiles of resistome in the rumen of beef cattle. These authors found ARGs belonging to 13 ARGs classes with macrolide, chloramphenicol, β-lactam, and aminoglycoside classes predominating. Chloramphenicol and aminoglycosides ARGs were predominant in rumen samples of beef cattle fed either high forage or high concentrate diets. However, breed had no significant effect on the profile of ARGs. This finding suggests that diet may have a more significant impact on the rumen resistome than the genetics of the host.
Two recent studies showed that the prevalence of AMR may not be necessarily associated with the use of antimicrobials in rumen of beef and dariy cattle. Thomas et al. [151] detected ARGs belonging to the macrolide (ermF and ermG) class in the rumen of beef cattle supplemented with monensin and tylosin as well as those that did not receive antimicrobials. As the period of supplementation of antimicrobials only lasted for 74 days, the resistome profile may change with longer term supplementation. More recently, Xue et al. [25] identified ARGs belonging to 26 classes of ARGs using samples collected from 49 dairy cows that did not receive antimicrobials during the experiment. They found that ARGs encoding resistance to tetracycline were the most common, followed by those encoding glycopeptide and fluoroquinolone resistance. One possible reason for this finding could be that these animals were exposed to antimicrobials shortly after birth (e.g. disease prevention or treatment), which could have had a long-term impact on the rumen resistome. As the number of related studies are very scarce, more experiments should be conducted to investigate the effect of use of antimicrobials on the profiles of the rumen resistome in ruminants such as cattle, sheep, and goats.
In addition to these metagenome-based in vivo studies, a microbial-genomics-based in vitro study analyzed 435 genomes of ruminal bacteria and archaea and identified a high abundance of genes encoding tetracycline resistance using a variety of ARGs databases (e.g. ResFinder, Resfams, ARG-ANNOT), a finding which is consistent with previous in vivo studies [168]. However, the prevalence of genes resistant to ARG classes can be influenced by the nature of the analytical approach employed. For example, the prevalence of beta-lactam and vancomycin resistant genes was only detected using Resfams, but not ResFinder or ARG-ANNOT, demonstrating that variable tools for resistome analysis may generate different results. Consequently, results that are generated with different computational pipelines need to be interpreted with caution. By aligning several genes conferring resistance to aminoglycosides, beta-lactams, macrolides, tetracyclines, as well as vancomycin to selected rumen metatranscriptomic datasets, the authors confirmed the expression of additional ARGs including tetQ, tetW, tetO, and tet37. While these findings need validation in vivo, our recent study found that tetW and tetQ were the most abundant ARG transcripts (expressed resistome) in the rumen of beef cattle (Ma et al, submitted). The expression of these tetracycline resistant genes and their functions need further investigation.
Fecal resistome
Tetracycline is the most abundant ARG class in feces of ruminants, and MLS along with aminoglycoside ARGs have been also reported to account for a large proportion of the fecal resistome of ruminants (Table 3). In general, the diversity of the fecal resistome decreases with increasing age in cattle [154]. While trimethoprim and aminoglycoside classes were only identified in feces from calves, tetracycline superfamily alignments were only identified in mature cattle, with macrolide efflux pumps and lincosamide nucleotidyltransferases also more common in adult cattle than calves [155].
Numerous studies have evaluated the effect of the use of antimicrobials on the profiles of the fecal resistome in cattle. In general, more abundant tetracycline, macrolide, and aminoglycoside ARGs were detected in feedlot cattle administered antimicrobials in feed as compared to those that were not [156]. Specifically, the use of ceftiofur, a 3rd-generation cephalosporin for the treatment of respiratory disease and foot rot [169], enriched for β-lactam ARGs [157, 158]. Similarly, cattle fed chlortetracycline showed a significant increase in the relative abundance of tetracycline ARGs in feces [158]. Nevertheless, some studies found no difference in the profiles or abundance of fecal resistome in feedlot cattle under conventional vs. raised without antimicrobials production conditions [159]. For example, supplementation of tylosin, a macrolide that inhibits protein synthesis in bacteria [170], did not affect the abundance of these ARGs within resistome of feces from beef cattle [160]. Moreover, no difference was observed in the profiles of fecal resistome of cattle injected with or without tulathromycin, a macrolide used to prevent and treat bovine respiratory disease [171, 172]. This was in spite of the composition of both the microbiome and resistome changing over 11 days post-injection [161]. The difference in these findings may be attributed to the dosage of antimicrobial used, as administration of low-dosage of oxytetracycline (100–200 μg per day) in veal calves did not result in increased tetM resistance levels as was observed in high-dosage (1 g per day) group after 42 days of administration [162]. Based on these findings, administration of antimicrobials may not necessarily impact the profiles of resistome in fecal microbiota of ruminants. However, the transmission of ARGs from fecal to environmental microbiota (e.g. soil and water) in ruminants administered antimicrobials needs further investigation.
Future focus on resisome analysis in the digestive tract of food-producing animals
Although some progress has been made, our current knowledge of resistome in the digestive tract of food-producing animals is still limited and more efforts are warranted. Here we propose several potential approaches that could be used to advance our understanding of the resistome within food-producing animals (Fig. 2).

Future directions of resistome analysis in the digestive tract of food-producing animals (Created with BioRender.com)
Application of long-read sequencing techniques
While short-read sequencers such as Illumina’s HiSeq and MiSeq [173] produce reads of up to 600 bases, long-read sequencing technologies, featured by Pacific Biosciences single-molecule real-time sequencing and Oxford Nanopore Technologies’ nanopore sequencing, routinely generate reads more than 10 kb [174]. Compared with short-read sequencing, long-read sequencing improves de novo assembly, mapping certainty, transcript isoform identification, and detection of structural variants [175]. Moreover, it is difficult to determine the exact genomic context of ARGs using short-read sequencing, while long read sequencing offers a solution to this problem as repeat regions can be spanned and defined [176]. Attempts have been made to use long read sequencing to characterize resistome associated with foods [177], stormwater [178], feces and preterm babies [179]. Along with resistome mapping tools specifically developed for long-read sequencing such as poreFUME [180] and ARGpore [181], the results of the above studies showed that long-read sequencing can be used to establish the context of ARGs within the resistome. Additionally, the combination of long-read (Oxford Nanopore) and short-read (Illumina) metagenomic sequencing has been proven to be a promising approach to comprehensively investigate the resistome in environmental samples such as sewage sludge [182] or in bacterial isolates [176]. Long-read sequencing looks particularly promising for further characterizing factors that regulate the expression of ARGs and the role mobile genetic elements play in their dispersion among bacterial members within microbiomes [183]. With improvements in error correction and continued reductions in sequencing cost, we foresee long-read sequencing as a promising approach to further investigating the resistome profiles in the digestive tract of food-producing animals.
Application of single-cell sequencing techniques
Single-cell sequencing refers to the sequencing the genome or transcriptome of a single cell to characterize the genomic, transcriptomic and metabolomic function of single cells [184]. This is accomplished by the physical separation of single cells from environmental samples, followed by sequencing and assembly of their individual genomes which are then subject to a series of downstream analysis. Single-cell sequencing has proven to be a powerful tool for studying unculturable organisms and delineating complex populations since its first inception in 2005 [185]. However, for a long time, the widespread use of single-cell sequencing was limited due to its high cost. With the continuous efforts made in lowering detection costs, single-cell sequencing is increasingly used in various fields such as cancer research, immunology, and microbiology [186]. The bioinformatics pipelines of single-cell RNA sequencing technologies have been recently reviewed [187]. Lan et al. performed high-throughput single-cell genome sequencing (SiC-seq) on synthetic communities to analyze the distribution of ARGs, virulence factors and phage sequences in environmental microbial communities [188]. The work showed that single-cell sequencing technologies can play an important role in the identification of antimicrobial resistant microorganisms.
Application of culturomic techniques
Culturomics refer to a culture-dependent approach to study complex microbial ecosystems such as the human and animal intestinal tract. This approach can be used to complement metagenomics by overcoming the depth bias inherent in metagenomic sequencing [189]. Culturomics use high-throughput tools such as matrix-assisted laser desorption ionization time-of-Flight (MALDI-TOF) mass spectrometry to comprehensively identify bacteria from environmental samples to the strain level [190]. McLain et al. summarized the use of culturomics for identification of AMR in agroecosystems and suggested that culture as well as isolation of individual microbes carrying ARGs are essential for determining multi-antimicrobial resistance phenotypes [191]. A recent review by Bilen et al. [192] updated the inventory of prokaryotes (from 2,172 to 2,776 species) from different human body sites that was originally published by Hugon et al. [193]. In this update, culturomics contributed up to 66.2% of the updates to this repertoire. This review concluded that culturomics proved useful in identifying new ARG-carrying bacteria. Despite these efforts, the use of culturomics to detect AMR in the digestive tract of food-producing animals has not been reported. We therefore speculate that culturomics could be an ideal complementary approach to metagenomics sequencing to study the resistome in the digestive tract of food-producing animals.
Identification of resistome on mobile genetic element
An important route for bacteria to acquire antimicrobial resistance is through horizontal gene transfer (HGT) [194,195,196], mediated by mobile genetic elements (MGE) such as plasmids [197]. It has been shown that plasmids are one of the MGEs that are abundantly present in bovine rumen bacterial populations [198]. In addition, plasmid-mediated resistance genes such as qnrA, blaCTX-M and mcr-1 have been identified within the resistome of swine [199]. Although metagenomic approaches have been used to characterize plasmid associated ARGs in activated sludge [200, 201] and the human gut [202], the profiles of plasmid-associated ARGs have not been extensively examined in food-producing animals. Our recent study showed that a total of 90 ARGs belonging to 15 classes were plasmid-associated in the rumen of beef cattle, with tetracycline, aminoglycoside, and MLS being the ARGs most often associated with MGE’s (Ma et al., submitted). With the continuous curation of reference databases such as a CLAssification of Mobile genetic Elements (ACLAME) [203]), PlasmidFinder [204], and MOB-suite [205], there is a need to characterize the role of plasmid-associated ARGs within the reistome within the digestive tract of food-producing animals.
Investigation into the expressions of resistome
Current studies of the resistome in food-producing animals are mainly based on technologies such as metagenomic sequencing. However, expression of ARGs within the resistome and the factors that influence it are still largely unclear in food-producing animals. Recent studies have used metagenomics and metatranscriptomics to assess the resistome of wastewater treatment plants and revealed that the location of the plant not only affects the types of ARGs present, but also their expression [206]. Expression of ARGs in the fecal resistome of chickens and pigs has been examined, but only in a limited number of samples (n = 6) for each species [30]. Our recent study showed that the expressed ARGs in the rumen of beef cattle only represented less than 1% of the resistome, with tetW and mefA exhibiting the highest level of expression (Ma et al., submitted). Moreover, we found significant differences in the abundance of microbial ARGs (metagenomic profiling) but not expressed ARGs (metatranscriptomic profiling) in the rumen in different beef breeds. In this regard, more studies based on metatranscriptomics are needed to understand the expression of the resistome in the digestive tract of food-producing animals. Although numerous tools have been developed for resistome analysis, they have been designed for metagenomic datasets and their suitability for metatranscriptomic datasets needs further validation.
Functions of the resistome in addition to AMR
Other than transmission of AMR, studies have shown that the ARGs may also have other functions that impact both host and the microbiome within the digestive tract of food-producing animals. For example, some AMR elements such as efflux pumps may regulate amino acid, fatty acid or nucleotide metabolism of microbiota while also conferring intrinsic antimicrobial resistance [207,208,209]. A study based on metagenomic sequencing also revealed that dairy cattle managed in the same manner and fed the same diet, but with high and low milk protein yields exhibited different rumen resistome profiles (resistotypes) [25]. Specifically, these authors reported that the abundance of 128 ARGs differed in dairy cows with high and low milk protein yield. In particular, cows with low milk protein yield had a higher abundance of mfd (encoding a transcription-repair coupling factor involved in strand-specific DNA repair [210]) and sav1866 (encoding a multidrug export ATP-binding/permease protein [211]). While this study revealed a potential relationship between ARGs and host production, a recent study by our group found a positive correlation between the abundance of multiple subtypes of expressed ARGs (e.g. tetW and tetQ) and various metabolism pathways within the active rumen microbiome (Ma et al., submitted). In addition, we found that higher expression of tetW may be associated with increased stability of the active rumen microbiota, which may be more resistant to external perturbations such as the administration of antimicrobials in feed. However, as the above studies are based on correlation, they may not reflect causation, and the exact roles ARGs in maintaining or regulating host/microbial functions needs further elucidation. A future goal could be to differentiate those ARGs that may play an ancillary role in microbiome functions that influence host productivity from those that specifically result in the transmission of AMR.
Limiting the use of antimicrobials to control host, microbial, and environmental transmission of ARGs
Efforts have been made to minimize or limit the spread of AMR by reducing AMU in food producing animals. Indeed, several of the above-mentioned studies showed a reduction in the diversity or abundance of ARGs after eliminating AMU in food-producing animals. However, other studies also found no difference between the resistome profiles of food-processing animals that received or did not receive antimicrobials. We propose several factors that may impact resistome as a result of the use of antimicrobials in food-producing animals (Fig. 3). First, several recent studies showed that the gut microbiome is heavily influenced by the host in cattle [212,213,214], swine [215] and poultry [216, 217]. This raises the possibility that the host genetics may also affect the profiles of microbial-associated resistome in the digestive tract of livestock and poultry. Although Auffret et al. [152] did not find significant impact of cattle breed on the fecal resistome, our recent research found that crossbred cattle had a less diverse resistome as compared to purebred cattle (Ma et al., submitted). In addition to breed, age may also influence the impact of antimicrobials on the resistome, as the gut microbiota may be less resilient to external perturbations such as the use of antimicrobials in early life, with potential life-long impacts on the gut microbiome [218, 219]. We thus speculate that the profiles of microbial-associated resistome may also be more likely to change in response to the use of antimicrobials at an early age in food-producing animals.

Host (breed and age), microbial (sample origin and type), and environmental (water, soil, diet, and companion animal) factors that may impact the effect of administration of antimicrobials on the diversity, composition, and abundance of resistome in the digestive tract of food-producing animal (Created with BioRender.com)
Companion animals as a source of ARGs in food animal production systems
In addition, it is noticeable that ARGs can also be transmitted between companion animals and food production animals and humans. Companion animals such as dogs and cats are present on most farms [220], making the transmission of ARGs possible among them and food-producing animals as well as human beings. According to a recent review, β-lactams such as amoxicillin are the most commonly used antimicrobials for dogs and cats in European countries including UK, Italy, and four Nordic countries [221]. A recent study investigated the profiles of resistome in the digestive tract of dogs and cats and found that tetracycline and aminoglycoside resistance genes were the most abundant among the 23 classes of ARGs, indicating that the gut microbiota of dogs and cats is also a reservoir of ARGs [222]. We thus speculate that there could be many shared ARGs between these companion and food-producing animals living together in farms considering the similarity of the profiles of predominant ARGs (e.g. tetW and tetQ) in their digestive tract. In horses, antimicrobials (e.g. penicillins) are used mainly to treat equine skin infections [223] and colitis/diarrhea [224]. However, studies of the resistome in the digestive-tract of horses are still limited. A study showed that prophylactic administration of a macrolide antimicrobial with rifampin promoted MDR genes in Rhodococcus equi and commensals, which could potentially infect or colonize other animals [225]. We therefore suggest that more studies are warranted to simultaneously investigate the profiles and associations of resistome in the digestive tract of both food-producing and companion animals living in the same farm, in order to assess the potential risks of transmissions of ARGs among them and human beings.
Conclusions
Based on metagenomic sequencing, it has been shown that there are abundant ARGs in the digestive tract of food-producing animals. The existence of these ARGs may not always be directly related to AMU, but is undoubtedly influenced by the use of injectable antimicrobials or their administration through feed or water. In most studies, feces were used to investigate the impact of antimicrobials on the diversity, profile and abundance of ARGs within the resistome. While fecal samples are easy to collect and are often used as a proxy of the microbial population within the digestive tract, composition of the microbiota differ across segments within the digestive tract [132, 163, 226, 227]. Additionally, compared to fecal microbiota, which originates from digesta, mucosa-associated microbiota can directly interact with the host where mucosa-associated microbiota are more likely to contact antimicrobials within the blood stream. It has also been shown that mucosa-associated and digesta-associated bacterial populations in the ileum of swine respond differently to antimicrobials [227]. It can therefore be speculated that, the response to the administration of antimicrobials may differ between mucosa- and digesta-associated microbiomes resulting in differences in associated resistome of these populations. In this regard, more investigations into the change in the resistome in mucosa-associated microbiota induced by antimicrobials in food-producing animals are warranted. In the future, it is necessary to further utilize emerging techniques for the analysis of resistome such as long-read sequencing, with special focuses on the expression of ARGs and the role of MGE in the dissemination of AMR in the gut of different food producing animals. Such information will be pivotal in defining the risk of spread of ARGs from food-producing animals to humans, and to develop effective strategies to reduce the threat of ARGs to human health and the environment.
Availability of data and materials
Not applicable
Abbreviations
- ACLAME:
-
A CLAssification of Mobile genetic Elements
- AMR:
-
Antimicrobial resistance
- AMU:
-
Anti-microbial use
- ARDB:
-
Antibiotic Resistance Genes Database
- ARGs:
-
Antimicrobial resistance genes
- ARGO:
-
Antibiotic Resistance Genes Online
- ARIBA:
-
Antimicrobial Resistance Identification By Assembly
- BLAST:
-
Basic local alignment search tool
- BWA:
-
Burrows-Wheeler aligner
- CARD:
-
Comprehensive Antibiotic Resistance Database
- GROOT:
-
Graphing Resistance Out Of meTagenomes
- HGT:
-
Horizontal gene transfer
- MAGs:
-
Metagenomic-assembled genomes
- MALDI-TOF:
-
Matrix-assisted laser desorption ionization time-of-flight
- MDR:
-
Multidrug resistance
- MGE:
-
Mobille genomic element
- MLS:
-
Macrolide-lincosamide-streptogramin
- NGS:
-
Next-generation sequencing
- PCR:
-
Polymerase chain reaction
- QC:
-
Quality control
- SiC-seq:
-
Single-cell genome sequencing
- SRST2:
-
Short read sequencing typing
References
Northoff E. 2050 A third more mouths to feed. Food and Agriculture Organization of the United Nations; 2016.
McLeod A. World livestock 2011–livestock in food security. Food and Agriculture Organization of the United Nations; 2011.
Van Boeckel TP, Pires J, Silvester R, Zhao C, Song J, Criscuolo NG, et al. Global trends in antimicrobial resistance in animals in low- and middle-income countries. Science. 2019;365:eaaw1944.
Marshall BM, Levy SB. Food animals and antimicrobials: impacts on human health. Clin Microbiol Rev. 2011;24:718–33.
Cogliani C, Goossens H, Greko C. Restricting antimicrobial use in food animals: lessons from Europe: banning nonessential antibiotic uses in food animals is intended to reduce pools of resistance genes. Microbe. 2011;6:274–9.
Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, et al. Global trends in antimicrobial use in food animals. Proc Natl Acad Sci U S A. 2015;112:5649–54.
European Centre for Disease Prevention and Control (ECDC), European Food Safety Authority (EFSA), European Medicines Agency (EMA). ECDC/EFSA/EMA second joint report on the integrated analysis of the consumption of antimicrobial agents and occurrence of antimicrobial resistance in bacteria from humans and food-producing animals. ESFA J. 2017;15(7):e04872.
US Food Drug Administration. FDA Annual Summary Report on Antimicrobials Sold or Distributed in 2012 for Use in Food-Producing Animals. https://www.aasv.org/news/story.php?id=7555. 2014.
Hu Y, Cheng H. Research opportunities for antimicrobial resistance control in China’s factory farming. Environ Sci Technol. 2014;48:5364–5.
Ministry of Agriculture and Rural Affairs of the People’s Republic of China. Report on the use of veterinary antibiotics of China in 2018. Official Vet Bull. 2019;21:57–9.
Campagnolo ER, Johnson KR, Karpati A, Rubin CS, Kolpin DW, Meyer MT, et al. Antimicrobial residues in animal waste and water resources proximal to large-scale swine and poultry feeding operations. Sci Total Environ. 2002;299:89–95.
Heuer H, Solehati Q, Zimmerling U, Kleineidam K, Schloter M, Müller T, et al. Accumulation of sulfonamide resistance genes in arable soils due to repeated application of manure containing sulfadiazine. Appl Environ Microbiol. 2011;77:2527–30.
Hao H, Cheng G, Iqbal Z, Ai X, Hussain HI, Huang L, et al. Benefits and risks of antimicrobial use in food-producing animals. Front Microbiol. 2014;5:288.
Acar JF. Consequences of bacterial resistance to antibiotics in medical practice. Clin Infect Dis. 1997;24:S17–8.
Cosgrove SE, Sakoulas G, Perencevich EN, Schwaber MJ, Karchmer AW, Carmeli Y. Comparison of mortality associated with methicillin-resistant and methicillin-susceptible staphylococcus aureus bacteremia: a meta-analysis. Clin Infect Dis. 2003;36:53–9.
Hawkey PM. The growing burden of antimicrobial resistance. J Antimicrob Chemother. 2008;62:1–9.
Tang KL, Caffrey NP, Nóbrega DB, Cork SC, Ronksley PE, Barkema HW, et al. Restricting the use of antibiotics in food-producing animals and its associations with antibiotic resistance in food-producing animals and human beings: A systematic review and meta-analysis. Lancet Planet Health. 2017;1:e316–7.
World Health Organization. WHO Guidelines on Use of Medically Important Antimicrobials in Food-Producing Animals. 2017.
Van Boeckel TP, Glennon EE, Chen D, Gilbert M, Robinson TP, Grenfell BT, et al. Reducing antimicrobial use in food animals. Science. 2017;357(6358):1350–2.
Announcement of the Ministry of Agriculture and Rural People’s Republic of China No. 194. Beijing: China Ministry of Agriculture and Rural Affairs. 2019. http://www.xmsyj.moa.gov.cn/zcjd/201907/t20190710_6320678.htm.
National action plan to combat animal resources antimicrobial resistance (2017-2020). Beijing: China Ministry of Agriculture and Rural Affairs. 2017. http://www.moa.gov.cn/nybgb/2017/dqq/201801/t20180103_6133925.htm.
Xiao Y, Li L. China’s national plan to combat antimicrobial resistance. Lancet Infect Dis. 2016;16:1216–8.
Marquardt RR, Li S. Antimicrobial resistance in livestock: advances and alternatives to antibiotics. Anim Front. 2018;8(2):30–7.
Nowakiewicz A, Zięba P, Gnat S, Matuszewski Ł. Last call for replacement of antimicrobials in animal production: modern challenges, opportunities, and potential solutions. Antibiotics. 2020;9(12):883.
Xue M-Y, Xie Y-Y, Zhong Y-F, Liu J-X, Guan LL, Sun H-Z. Ruminal resistome of dairy cattle is individualized and the resistotypes are associated with milking traits. Anim Microbiome. 2021;3:18.
Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5:175–86.
Perry JA, Wright GD. Forces shaping the antibiotic resistome. BioEssays. 2014;36:1179–84.
Dias MF, Fernandes GR, da Paiva MC, Salim ACM, Santos AB, Nascimento AMA. Exploring the resistome, virulome and microbiome of drinking water in environmental and clinical settings. Water Res. 2020;174(1):115630.
Nesme J, Simonet P. The soil resistome: a critical review on antibiotic resistance origins, ecology and dissemination potential in telluric bacteria. Environ Microbiol. 2015;17(4):913–30.
Wang Y, Hu Y, Liu F, Gao J, Lv N, Zhu B, et al. Integrated metagenomic and metatranscriptomic profiling reveals differentially expressed resistomes in human, chicken, and pig gut microbiomes. Environ Int. 2020;138:105649.
Baquero F. Metagenomic epidemiology: a public health need for the control of antimicrobial resistance. Clin Microbiol Infec. 2012;18:67–73.
Miller RR, Montoya V, Gardy JL, Patrick DM, Tang P. Metagenomics for pathogen detection in public health. Genome Med. 2013;5:81.
Founou LL, Founou RC, Essack SY. Antibiotic resistance in the food chain: a developing country-perspective. Front Microbiol. 2016;7:1881.
Ho J, Yeoh YK, Barua N, Chen Z, Lui G, Wong SH, et al. Systematic review of human gut resistome studies revealed variable definitions and approaches. Gut Microbes. 2020;12:1.
Aguiar-Pulido V, Huang W, Suarez-Ulloa V, Cickovski T, Mathee K, Narasimhan G. Metagenomics, metatranscriptomics, and metabolomics approaches for microbiome analysis. Evol Bioinform. 2016;12:5–16.
Li F, Neves ALA, Ghoshal B, Guan LL. Symposium review: Mining metagenomic and metatranscriptomic data for clues about microbial metabolic functions in ruminants. J Dairy Sci. 2018;101:5605–18.
Allen HK. Antibiotic resistance gene discovery in food-producing animals. Curr Opin Microbiol. 2014;19:25–9.
Thanner S, Drissner D, Walsh F. Antimicrobial resistance in agriculture. mBio. 2016;7:e02227–15.
Casals-Pascual C, González A, Vázquez-Baeza Y, Song SJ, Jiang L, Knight R. Microbial diversity in clinical microbiome studies: sample size and statistical power considerations. Gastroenterology. 2020;158:1524–8.
Xia Y, Sun J, Chen D. Power and sample size calculations for microbiome data. Statistical analysis of microbiome data with R Singapore. Singapore: Springer; 2018. p. 129–66. https://doi.org/10.1007/978-981-13-1534-3.
Kelly BJ, Gross R, Bittinger K, Sherrill-Mix S, Lewis JD, Collman RG, et al. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics. 2015;31(15):2461–8.
Mattiello F, Verbist B, Faust K, Raes J, Shannon WD, Bijnens L, et al. A web application for sample size and power calculation in case-control microbiome studies. Bioinformatics. 2016;32(13):2038–40.
Zhou X, Nanayakkara S, Gao J-L, Nguyen K-A, Adler CJ. Storage media and not extraction method has the biggest impact on recovery of bacteria from the oral microbiome. Sci Rep. 2019;9:14968.
Wegl G, Grabner N, Köstelbauer A, Klose V, Ghanbari M. Toward best practice in livestock microbiota research: a comprehensive comparison of sample storage and DNA extraction strategies. Front Microbiol. 2021;12:627539.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
Fricker AM, Podlesny D, Fricke WF. What is new and relevant for sequencing-based microbiome research? A mini-review. J Adv Res. 2019;19:105–12.
Yap M, Feehily C, Walsh CJ, Fenelon M, Murphy EF, McAuliffe FM, et al. Evaluation of methods for the reduction of contaminating host reads when performing shotgun metagenomic sequencing of the milk microbiome. Sci Rep. 2020;10:21665.
Pereira-Marques J, Hout A, Ferreira RM, Weber M, Pinto-Ribeiro I, van Doorn LJ, et al. IMPACT of host DNA and sequencing depth on the taxonomic resolution of whole metagenome sequencing for microbiome analysis. Front Microbiol. 2019;10:1277.
Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server-a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9:386.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36.
Shabat SK, Sasson G, Doron-Faigenboim A, Durman T, Yaacoby S, Miller MEB, et al. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016;10:2958–72.
Li F, Guan LL. Metatranscriptomic profiling reveals link- ages between the active rumen microbiome and feed efficiency in beef cattle. Appl Environ Microbiol. 2017;83:e00061–17.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Uritskiy GV, DiRuggiero J, Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6(1):158.
Clarke EL, Taylor LJ, Zhao C, Connell A, Lee J-J, Fett B, et al. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome. 2019;7:46.
Boolchandani M, D’Souza AW, Dantas G. Sequencing-based methods and resources to study antimicrobial resistance. Nat Rev Genet. 2019;20:356–70.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31:1674–6.
Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–34.
Ayling M, Clark MD, Leggett RM. New approaches for metagenome assembly with short reads. Brief Bioinformatics. 2020;21:584–94.
Lapidus AL, Korobeynikov AI. Metagenomic data assembly–the way of decoding unknown microorganisms. Front Microbiol. 2021;12:613791.
Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, et al. Critical Assessment of Metagenome Interpretation—a benchmark of metagenomics software. Nat Methods. 2017;14:1063–71.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Inouye M, Dashnow H, Raven L-A, Schultz MB, Pope BJ, Tomita T, et al. SRST2: rapid genomic surveillance for public health and hospital microbiology labs. Genome Med. 2014;6:90.
Hunt M, Mather AE, Sánchez-Busó L, Page AJ, Parkhill J, Keane JA, et al. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genom. 2017;3:e000131.
Rowe WPM, Winn MD. Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics. 2018;34:3601–8.
Carr R, Borenstein E. Comparative analysis of functional metagenomic annotation and the mappability of short reads. PLoS ONE. 2014;9:e105776.
Henson J, Tischler G, Ning Z. Next-generation sequencing and large genome assemblies. Pharmacogenomics. 2012;13:901–15.
Liu B, Pop M. ARDB-antibiotic resistance genes database. Nucleic Acids Res. 2009;37:D443–7.
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–4.
Jia B, Raphenya AR, Alock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–73.
Scaria J, Chandramouli U, Verma SK. Antibiotic Resistance Genes Online (ARGO): a Database on vancomycin and beta-lactam resistance genes. Bioinformation. 2005;1:5–7.
Flandrois JP, Lina G, Dumitrescu O. MUBII-TB-DB: a database of mutations associated with antibiotic resistance in Mycobacterium tuberculosis. BMC Bioinformatics. 2014;15:107.
Zankari E, Allesoe R, Joensen KG, Cavaco LM, Lund O, Aarestrup FM. PointFinder: a novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J Antimicrob Chemother. 2017;72:2764–8.
Hendriksen RS, Bortolaia V, Tate H, Tyson GH, Aarestrup FM, McDermott PF. Using genomics to track global antimicrobial resistance. Front Microbiol. 2019;7:242.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Nesme J, Cécillon S, Delmont TO, Monier JM, Vogel TM, Simonet P. Large-scale metagenomic-based study of antibiotic resistance in the environment. Curr Biol. 2014;24:1096–100.
Port JA, Cullen AC, Wallace JC, Smith MN, Faustman EM. Metagenomic frameworks for monitoring antibiotic resistance in aquatic environments. Environ Health Perspect. 2014;122:222–8.
Tang J, Bu Y, Zhang XX, Huang K, He X, Ye L, et al. Metagenomic analysis of bacterial community composition and antibiotic resistance genes in a wastewater treatment plant and its receiving surface water. Ecotoxicol Environ Saf. 2016;132:260–9.
Petersen TN, Rasmussen S, Hasman H, Carøe C, Bælum J, Schultz CA, et al. Metagenomic analysis of toilet waste from long distance flights; A step towards global surveillance of infectious diseases and antimicrobial resistance. Sci Rep. 2015;5:1–9.
Bengtsson-Palme J, Hammarén R, Pal C, Östman M, Björlenius B, Flach CF, et al. Elucidating selection processes for antibiotic resistance in sewage treatment plants using metagenomics. Sci Total Environ. 2016;572:697–712.
Ju F, Li B, Ma L, Wang Y, Huang D, Zhang T. Antibiotic resistance genes and human bacterial pathogens: Co-occurrence, removal, and enrichment in municipal sewage sludge digesters. Water Res. 2016;91:1–10.
Liu X, Geng S, Chan EWC, Chen S. Increased prevalence of Escherichia coli strains from food carrying blaNDM and mcr-1-bearing plasmids that structurally resemble those of clinical strains, China, 2015 to 2017. Euro Surveill. 2019;24:1800113.
Liu J, Taft DH, Maldonado-Gomez MX, Johnson D, Treiber ML, Lemay DG, et al. The fecal resistome of dairy cattle is associated with diet during nursing. Nat Commun. 2019;10:1–15.
Větrovský T, Baldrian P. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS ONE. 2013;8(2):e57923.
Chu BT, Petrovich ML, Chaudhary A, Wright D, Murphy B, Wells G, et al. Metagenomics reveals the impact of wastewater treatment plants on the dispersal of microorganisms and genes in aquatic sediments. Appl Environ Microbiol. 2018;84:e02168–17.
Calle ML. Statistical analysis of metagenomics data. Genomics Inform. 2019;17(1):e6.
Wang W, Hu H, Zijlstra RT, Zheng J, Gänzle MG. Metagenomic reconstructions of gut microbial metabolism in weanling pigs. Microbiome. 2019;7:48.
Chen C, Zhou Y, Fu H, Xiong X, Fang S, Jiang H, et al. Expanded catalog of microbial genes and metagenome-assembled genomes from the pig gut microbiome. Nat Commun. 2021;12:1106.
Glendinning L, Stewart RD, Pallen MJ, Watson KA, Watson M. Assembly of hundreds of novel bacterial genomes from the chicken caecum. Genome Biol. 2020;21:34.
Gilroy R, Ravi A, Getino M, Pursley I, Horton DL, Alikhan NF, et al. Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. PeerJ. 2021;9:e10941.
Stewart RD, Auffret MD, Warr A, Wiser AH, Press MO, Langford KW, et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat Commun. 2018;9:870.
Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M. Compendium of 4,941 rumen metagenome- assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol. 2019;37:953–61.
Xie F, Jin W, Si H, Yuan Y, Tao Y, Liu J, et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome. 2021;9:137.
Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2:26.
Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32(4):605–7.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359.
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6.
Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3:836–43.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–55.
Imelfort M, Parks D, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW. GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ. 2014;2:e603.
Yue Y, Huang H, Qi Z, Dou H-M, Liu X-Y, Han T-F, et al. Evaluating metagenomics tools for genome binning with real metagenomic datasets and CAMI datasets. BMC Bioinformatics. 2020;21:334.
Mahnert A, Moissl-Eichinger C, Zojer M, Bogumill D, Mizrahi I, Rattei T, et al. Man-made microbial resistances in built environments. Nat Comm. 2019;10:968.
Chen H, Liu C, Teng Y, Zhang Z, Chen Y, Yang Y. Environmental risk characterization and ecological process determination of bacterial antibiotic resistome in lake sediments. Environ Int. 2021;147:106345.
Liu Z, Klümper U, Liu Y, Yang Y, Wei Q, Lin J-G, et al. Metagenomic and metatranscriptomic analyses reveal activity and hosts of T antibiotic resistance genes in activated sludge. Environ Int. 2019;129:208–20.
Laczny CC, Kiefer C, Galata V, Fehlmann T, Backes C, Keller A. BusyBee Web: metagenomic data analysis by bootstrapped supervised binning and annotation. Nucleic Acids Res. 2017;45:W171–9.
Ma T, Xiao D, Xing X. MetaBMF: a scalable binning algorithm for large-scale reference-free metagenomic studies. Bioinformatics. 2020;36(2):356–63.
Zankari E, Hasman H, Kaas RS, Seyfarth AM, Agersø Y, Lund O, et al. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother. 2013;68:771–7.
Clausen PT, Zankari E, Aarestrup FM, Lund O. Benchmarking of methods for identification of antimicrobial resistance genes in bacterial whole genome data. J Antimicrob Chemother. 2016;71:2484–8.
Xavier BB, Das AJ, Cochrane G, Ganck SD, Kumar-Singh S, Aarestrup FM, et al. Consolidating and exploring antibiotic resistance gene data resources. J Clin Microbiol. 2016;54:851–9.
Gweon HS, Shaw LP, Swann J, De Maio N, AbuOun M, Niehus R, et al. The impact of sequencing depth on the inferred taxonomic composition and AMR gene content of metagenomic samples. Environ Microbiome. 2019;14:7. https://doi.org/10.1186/s40793-019-0347-1.
Zaheer R, Noyes N, Polo RO, Cook SR, Marinier E, Domselaar GV, et al. Impact of sequencing depth on the characterization of the microbiome and resistome. Sci Rep. 2018;8:5890.
Gupta CL, Tiwari RK, Cytryn E. Platforms for elucidating antibiotic resistance in single genomes and T complex metagenomes. Environ Int. 2020;138:105667.
Graham DW, Bergeron G, Bourassa MW, Dickson J, Gomes F, Howe A, et al. Complexities in understanding antimicrobial resistance across domesticated animal, human, and environmental systems. Ann N Y Acad Sci. 2019;1441:17–30.
Cuong NV, Padungtod P, Thwaites G, Carrique-Mas JJ. Antimicrobial usage in animal production: a review of the literature with a focus on low- and middle-income countries. Antibiotics. 2018;7:75.
Lekagul A, Tangcharoensathien V, Yeung S. Patterns of antibiotic use in global pig production: A systematic review. Vet Anim Sci. 2019;7:100058.
Callens B, Persoons D, Maes D, Laanen M, Postma M, Boyen F, et al. Prophylactic and metaphylactic antimicrobial use in Belgian fattening pig herds. Prev Vet Med. 2012;106:53–62.
Li B, Yang Y, Ma L, Ju F, Guo F, Tiedje JM, et al. Metagenomic and network analysis reveal wide distribution and co-occurrence of environmental antibiotic resistance genes. ISME J. 2015;9:2450–502.
Ma L, Xia Y, Li B, Yang Y, Li L-G, Tiedje JM, et al. Metagenomic assembly reveals hosts of antibiotic resistance genes and the shared resistome in pig, chicken and human feces. Environ Sci Technol. 2016;50(1):420–7.
Munk P, Knudsen BE, Lukjancenko O, Duarte ASR, Van Gompel L, Luiken REC, et al. Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat Microbiol. 2018;3:898–908. https://doi.org/10.1038/s41564-018-0192-9.
Van Gompel L, Luiken REC, Sarrazin S, Munk P, Knudsen B, Hansen RB, et al. The antimicrobial resistome in relation to antimicrobial use and biosecurity in pig farming, a metagenome-wide association study in nine European countries. J Antimicrob Chemother. 2019;74:865–76.
Ghanbari M, Klose V, Crispie F, Cotter PD. The dynamics of the antibiotic resistome in the feces of freshly weaned pigs following therapeutic administration of oxytetracycline. Sci Rep. 2019;9:4062.
Zeineldin MM, Megahed A, Blair B, Burton B, Aldridge B, Lowe J. Negligible impact of perinatal tulathromycin metaphylaxis on the developmental dynamics of fecal microbiota and their accompanying antimicrobial resistome in piglets. Front Microbiol. 2019;10:726.
Zeng J, Pan Y, Yang J, Hou M, Zeng Z, Xiong W. Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ Int. 2019;126:346–54.
Wang C, Li P, Yan Q, Chen L, Li T, Zhang W, et al. Characterization of the pig gut microbiome and antibiotic resistome in industrialized feedlots in China. mSystems. 2019;4:e00206–19.
Mencía-Ares O, Cabrera-Rubio R, Cobo-Díaz JF, Álvarez-Ordóñez A, Gómez-García M, Puente H, et al. Antimicrobial use and production system shape the fecal, environmental, and slurry resistomes of pig farms. Microbiome. 2020;8:164.
Hu Q, Liu C, Zhang D, Wang R, Qin L, Xu Q. Effects of low-dose antibiotics on gut immunity and antibiotic resistomes in weaned piglets. Front Immunol. 2020;11:903.
Joyce A, McCarthy CGP, Murphy S, Walsh F. Antibiotic resistomes of healthy pig faecal metagenomes. Microb Genom. 2019;5:e000272.
Lim SK, Kim D, Moon DC, Cho Y, Rho M. Antibiotic resistomes discovered in the gut microbiomes of Korean swine and cattle. Gigascience. 2020;9:1–11.
Panasevich MR, Wankhade UD, Chintapalli SV, Shankar K, Rector RS. Cecal versus fecal microbiota in Ossabaw swine and implications for obesity. Physiol Genomics. 2018;50:355–68.
Diarra MS, Malouin F. Antibiotics in canadian poultry productions and anticipated alternatives. Front Microbiol. 2014;5:282.
Gonzalez Ronquillo M, Angeles Hernandez JC. Antibiotic and synthetic growth promoters in animal diets: review of impact and analytical methods. Food Contr. 2017;72:255e67.
Carvalho IT, Santos L. Antibiotics in the aquatic environments: a review of the European scenario. Environ Int. 2016;94:736e57.
Xu J, Sangthong R, McNeil E, Tang R, Chongsuvivatwong V. Antibiotic use in chicken farms in northwestern China. Antimicrob Resist Infect Control. 2020;9:10.
CSCRA. Système canadien de surveillance de la résistance aux antimicrobiens – Rapport de 2016. Ottawa: Gouvernement du Canada; 2016.
Xiong W, Wang Y, Sun Y, Ma L, Zeng Q, Jiang X, et al. Antibiotic-mediated changes in the fecal microbiome of broiler chickens define the incidence of antibiotic resistance genes. Microbiome. 2018;6:34.
Wang Y, Lyu N, Liu F, Liu WJ, Bi Y, Zhang Z, et al. More diversified antibiotic resistance genes in chickens and workers of the live poultry markets. Environ Int. 2021;153:106534.
Zhou Y, Li Y, Zhang L, Wu Z, Huang Y, Yan H, et al. Antibiotic administration routes and oral exposure to antibiotic resistant bacteria as key drivers for gut microbiota disruption and resistome in poultry. Front Microbiol. 2020;11:1319.
Kumar H, Park W, Lim D, Srikanth K, Kim J-M, Jia X-Z, et al. Whole metagenome sequencing of cecum microbiomes in Ethiopian indigenous chickens from two different altitudes reveals antibiotic resistance genes. Genomics. 2020;112:1988–99.
Earley B, Arguello A, O’Riordan E, Crosson P, Cappelleri A, McGee M. Antimicrobial drug usage from birth to 180 days of age in Irish dairy calves and in suckler beef calves. J Appl Anim Res. 2019;47:474–85.
Department of Agriculture (US) Fort Collins (CO): USDA, Animal and Plant Health Inspection Service, Veterinary Services, National Animal Health Monitoring System; 2008. Sep, [cited 2010 Nov 11]. Dairy 2007 part III: reference of dairy cattle health and management practices in the United States, 2007.
Nak Y, Dagalp SB, Cetin C, Nak D, Alkan F, Borum E, et al. Course and severity of postpartum metritis cases following antibiotic and PGF2α administration in postpartum metritis cows infected with BoHV-4. Transbound Emerg. Dis. 2011;58:31–6.
Ferroni L, Lovito C, Scoccia E, Dalmonte G, Sargenti M, Pezzotti G, et al. Antibiotic consumption on dairy and beef cattle farms of central italy based on paper registers. Antibiotics. 2020;9(5):273.
Cameron A, McAllister TA. Antimicrobial usage and resistance in beef production. J Anim Sci Biotechno. 2016;7:68.
Santman-Berends I, Luttikholt S, Van den Brom R, Van Schaik G, Gonggrijp M, Hage H, et al. Estimation of the Use of Antibiotics in the Small Ruminant Industry in the Netherlands in 2011 and 2012. PLoS One. 2014;9:8.
Avery BP, Rajić A, McFall M, Reid-Smith RJ, Deckert AJ, Irwin RJ, et al. Antimicrobial use in the Alberta sheep industry. Can J Vet Res. 2008;72:137–42.
Moon CS, Berke O, Avery BP, McEwen SA, Reid-Smith RJ, Scott L, et al. Characteristics of drug use on sheep farms in Ontario. Canada. Can Vet J. 2010;51:1373–8.
Morgavi DP, Kelly WJ, Janssen PH, Attwood GT. Rumen microbial (meta) genomics and its application to ruminant production. Animal. 2013;7(Suppl 1):184–201.
Thomas M, Webb M, Ghimire S, Blair A, Olson K, Fenske GJ, et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Rep. 2017;7:12257.
Auffret MD, Dewhurst RJ, Duthie CA, Rooke JA, Wallace RJ, Freeman TC, et al. The rumen microbiome as a reservoir of antimicrobial resistance and pathogenicity genes is directly affected by diet in beef cattle. Microbiome. 2017;5:159.
Hitch TCA, Thomas BJ, Friedersdorff JCA, Ougham H, Creevey CJ. Deep sequence analysis reveals the ovine rumen as a reservoir of antibiotic resistance genes. Environ Pollut. 2018;235:571–5.
Noyes NR, Yang X, Linke LM, Magnuson RJ, Cook SR, Zaheer R, et al. Characterization of the resistome in manure, soil and wastewater from dairy and beef production systems. Sci Rep. 2016;8:24645.
Noyes NR, Yang X, Linke LM, Magnuson RJ, Dettenwanger A, Cook SR, et al. Resistome diversity in cattle and the environment decreases during beef production. eLife. 2016;5:e13195.
Rovira P, McAllister T, Lakin SSM, Cook SR, Doster E, Noyes NR, et al. Characterization of the microbial resistome in conventional and “raised without antibiotics” beef and dairy production systems. Front Microbiol. 2019;10:1980.
Chambers L, Yang Y, Littier H, Ray P, Zhang T, Pruden A, et al. Metagenomic analysis of antibiotic resistance genes in dairy cow feces following therapeutic administration of third generation cephalosporin. PLoS One. 2015;10:e0133764.
Weinroth MD, Scott HM, Norby B, Loneragan GH, Noyes NR, Rovira P, et al. Effects of ceftiofur and chlortetracycline on the resistomes of feedlot cattle. Appl Environ Microbiol. 2018;84:e00610–8.
Vikram A, Rovira P, Agga GE, Arthur TM, Bosilevac JM, Wheeler TL, et al. Impact of “raised without antibiotics” beef cattle production practices on occurrences of antimicrobial resistance. Appl Environ Microbiol. 2017;83:e01682–17.
Weinroth MD, Martin JN, Doster E, Geornaras I, Parker JK, Carlson CR, et al. Investigation of tylosin in feed of feedlot cattle and effects on liver abscess prevalence, and fecal and soil microbiomes and resistomes. J Anim Sci. 2019;97:4567–78.
Doster E, Rovira P, Noyes NR, Burgess BA, Yang X, Weinroth MD, et al. Investigating effects of tulathromycin metaphylaxis on the fecal resistome and microbiome of commercial feedlot cattle early in the feeding period. Front Microbiol. 2018;9:1715.
Keijser BJF, Agamennone V, van den Broek TJ, Caspers M, van de Braak AA, Bomers R, et al. Dose-dependent impact of oxytetracycline on the veal calf microbiome and resistome. BMC Genomics. 2019;20:65.
Salaheen S, Kim SW, Hovingh E, Kessel JASV, Haley BJ. Metagenomic analysis of the microbial communities and resistomes of veal calf feces. Front Microbiol. 2021;11:609950.
Huebner KL, Martin JN, Weissend CJ, Holzer KL, Parker JK, Lakinn SM, et al. Effects of a Saccharomyces cerevisiae fermentation product on liver abscesses, fecal microbiome, and resistome in feedlot cattle raised without antibiotics. Sci Rep. 2019;9:2559.
Zaheer R, Lakin SM, Polo RO, Cook SR, Larney FJ, Morley PS, et al. Comparative diversity of microbiomes and resistomes in beef feedlots, downstream environments and urban sewage influent. BMC Microbiol. 2019;19:197.
Bialvaei AZ, Samadi KH. Colistin, mechanisms and prevalence of resistance. Curr Med Res Opin. 2015;31(4):707–21.
Gotsbacher MP, Cho S, Kwon HJ, Karuso P. Daptomycin, a last-resort antibiotic, binds ribosomal protein S19 in humans. Proteome Sci. 2017;15:16.
Sabino YNV, Santana MF, Oyama LB, Santos FG, Moreira AJS, Huws SA, et al. Characterization of antibiotic resistance genes in the species of the rumen microbiota. Nat Commun. 2019;10:5252.
Hornish RE, Katarski S. Cephalosporins in veterinary medicine-ceftiofur use in food animals. Curr Top Med Chem. 2002;2:717–31.
Nakajima Y. Mechanisms of bacterial resistance to macrolide antibiotics. J Infect Chemother. 1999;5:61–74.
O’Connor AM, Yuan C, Cullen JN, Coetzee JF, da Silva N, Wang C. A mixed treatment meta-analysis of antibiotic treatment options for bovine respiratory disease-An update. Prev Vet Med. 2016;132:130–9.
Abell KM, Theurer ME, Larson RL, White BJ, Apley M. A mixed treatment comparison meta-analysis of metaphylaxis treatments for bovine respiratory disease in beef cattle. J Anim Sci. 2017;95:626–35.
Goodwin S, McPherson JD, McCombie WR. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17(6):333–51.
Pollard MO, Gurdasani D, Mentzer AJ, Porter T, Sandhu MS. Long reads: their purpose and place. Hum Mol Genet. 2018;27(R2):234–41.
Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020;21:30.
Berbers B, Saltykova A, Garcia-Graells C, Philipp P, Arella F, Marchal K, et al. Combining short and long read sequencing to characterize antimicrobial resistance genes on plasmids applied to an unauthorized genetically modified Bacillus. Sci Rep. 2020;10:4310.
Solcova M, Demnerova K, Purkrtova S. Application of Nanopore sequencing (Minion) for the analysis of bacteriome and resistome of bean sprouts. Microorganisms. 2021;9:937.
Białasek M, Miłobedzka A. Revealing antimicrobial resistance in stormwater with MinION. Chemosphere. 2020;258:127392.
Leggett RM, Alcon-Giner C, Heavens D, Caim S, Brook TC, Kujawska M, et al. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat Microbiol. 2020;5:430–42.
van der Helm E, Imamovic L, Ellabaan MMH, van Schaik W, Koza A, Sommer MOA. Rapid resistome mapping using nanopore sequencing. Nucleic Acids Res. 2017;45:e61.
Xia Y, Li AD, Deng Y, Jiang XT, Li LG, Zhang T. MinION Nanopore sequencing enables correlation between resistome phenotype and genotype of coliform bacteria in municipal sewage. Front Microbiol. 2017;8:2105.
Che Y, Xia Y, Liu L, Li A-D, Yang Y, Zhang T. Mobile antibiotic resistome in wastewater treatment plants revealed by Nanopore metagenomic sequencing. Microbiome. 2019;7:44.
Kwon HJ, Chen Z, Evans P, Meng J, Chen Y. Characterization of mobile genetic elements using long-read sequencing for tracking listeria monocytogenes from food processing environments. Pathogens. 2020;9(10):822.
Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med. 2020;52:1419–27.
Raghunnathan A, Ferguson HR Jr, Bornarth CJ, Song W, Driscoll M and Lasken RS. Genomic DNA amplification from a single bacterium. Appl Environ Microbiol. 2005;71(6):3342–7. https://doi.org/10.1128/AEM.71.6.3342-3347.2005.
Tang X, Huang Y, Lei J, Luo H, Zhu X. The single-cell sequencing: new developments and medical applications. Cell Biosci. 2019;9:53.
Hwang B, Lee JH, Bang D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med. 2018;50:96.
Lan F, Demaree B, Ahmed N, Abate AR. Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat Biotechnol. 2017;35:640–6.
Lagier J-C, Armougom F, Million M, Hugon P, Pagnier I, Robert C, et al. Microbial culturomics: paradigm shift in the human gut microbiome study. Clin Microbiol Infect. 2012;18:1185–93.
Nowrotek M, Jalowiecki Ł, Harnisz M, Płaza GA. Culturomics and metagenomics: In understanding of environmental resistome. Front Environ Sci Eng. 2019;13(3):40.
McLain JE, Cytryn E, Durso LM, Young S. Culture-based methods for detection of antibiotic resistance in agroecosystems: Advantages, challenges, and gaps in knowledge. J Environ Qual. 2016;45(2):432–40.
Bilen M, Dufour JC, Lagier JC, Cadoret F, Daoud Z, Dubourg G, et al. The contribution of culturomics to the repertoire of isolated human bacterial and archaeal species. Microbiome. 2018;6(1):94.
Hugon P, Dufour JC, Colson P, Fournier PE, Sallah K, Raoult D. A comprehensive repertoire of prokaryotic species identified in human beings. Lancet Infect Dis. 2015;15(10):1211–9.
Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer accelerates genome innovation and evolution. Mol Biol Evol. 2003;20:1598–602.
McCarthy AJ, Loeffler A, Witney AA, Gould KA, Lloyd D, Lindsay JA. Extensive horizontal gene transfer during Staphylococcus aureus co-colonization in vivo. Genome Biol Evol. 2014;6:2697–708.
Caro-Quintero A, Konstantinidis KT. Inter-phylum HGT has shaped the metabolism of many mesophilic and anaerobic bacteria. ISME J. 2014;9:958–67.
Stalder T, Press MO, Sullivan S, Liachko I, Top EM. Linking the resistome and plasmidome to the microbiome. ISME J. 2019;13:2437–46.
Kav AB, Sasson G, Jami E, Doron-Faigenboim A, Benhar I, Mizrahi I. Insights into the bovine rumen plasmidome. Proc Natl Acad Sci U S A. 2012;109:5452–7.
Yuan J, Wang X, Shi D, Ge Q, Song X, Hu W, et al. Extensive antimicrobial resistance and plasmid-carrying resistance genes in mcr-1-positive E. coli sampled in swine, in Guangxi, South China. BMC Vet Res. 2021;17:86.
Parsley LC, Consuegra EJ, Kakirde KS, Land AM, Harper WF, Liles MR. Identification of diverse antimicrobial resistance determinants carried on bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. Appl Environ Microbiol. 2010;76:3753–7.
Zhang T, Zhang XX, Ye L. Plasmid metagenome reveals high levels of antibiotic resistance genes and mobile genetic elements in activated sludge. PLoS One. 2011;6:e26041.
Lee K, Kim D-W, Lee D-H, Kim Y-S, Bu J-H, Cha J-H, et al. Mobile resistome of human gut and pathogen drives anthropogenic bloom of antibiotic resistance. Microbiome. 2020;8:2.
Leplae R, Lima-Mendez G, Toussaint A. ACLAME: a CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010;38:D57–61.
Carattoli A, Zankari E, García-Fernández A, Larsen MV, Lund O, Villa L, et al. In silico detection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–903.
Robertson J, Bessonov K, Schonfeld J, Nash JHE. Universal whole-sequence-based plasmid typing and its utility to prediction of host range and epidemiological surveillance. Microb Genom. 2020;6(10):mgen000435.
Ju F, Beck K, Yin X, Maccagnan A, McArdell CS, Singer HP, et al. Wastewater treatment plant resistomes are shaped by bacterial composition, genetic exchange, and upregulated expression in the effluent microbiomes. ISME J. 2019;13:346–60.
Martínez JL. Antibiotics and antibiotic resistance genes in natural environments. Science. 2008;321:365–6.
Martínez JL, Sánchez MB, Martínez-Solano L, Hernandez A, Garmendia L, Fajardo A, et al. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol Rev. 2009;33:430–49.
Aminov RI. The role of antibiotics and antibiotic resistance in nature. Environ Microbiol. 2009;11:2970–88.
Han J, Sahin O, Barton Y-W, Zhang Q. Key role of Mfd in the development of fluoroquinolone resistance in campylobacter jejuni. PLoS Pathog. 2008;4:e1000083.
Beck A, Aanismaa P, Li-Blatter X, Dawson R, Seelig A. SAV1866 from Staphylococcus aureus and P-glycoprotein - similarities and differences in ATPase activity assessed with detergents as Allocrites. Biochemistry. 2013;52:3297–309.
Henderson G, Cox F, Ganesh S, Jonker A, Young W, Global Rumen Census Collaborators, et al. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci Rep. 2015;5:14567. https://doi.org/10.1038/srep14567.
Zhou M, Peng YJ, Chen Y, Klinger CM, Oba M, Liu JX, et al. Assessment of microbiome changes after rumen transfaunation: implications on improving feed efficiency in beef cattle. Microbiome. 2018;6:62.
Li F, Li C, Chen Y, Liu J, Zhang C, Irving B, et al. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome. 2019;7:92.
Patil Y, Gooneratne R, Ju X-H. Interactions between host and gut microbiota in domestic pigs: a review. Gut Microbes. 2020;11:310–34.
Pan D, Yu Z. Intestinal microbiome of poultry and its interaction with host and diet. Gut Microbes. 2014;5:108–19.
Yadav S, Jha R. Strategies to modulate the intestinal microbiota and their effects on nutrient utilization, performance, and health of poultry. J Anim Sci Biotechno. 2019;10:2.
Zeissig S, Blumberg RS. Life at the beginning: perturbation of the microbiota by antibiotics in early life and its role in health and disease. Nat Immunol. 2014;5:307–10.
Cox LM, Blaser MJ. Antibiotics in early life and obesity. Nat Rev Endocrinol. 2015;11:182–90.
Bich VTN, Thanh LV, Thai PD, Phuong TTV, Oomen M, Driessen C, et al. An exploration of the gut and environmental resistome in a community in northern Vietnam in relation to antibiotic use. Antimicrob Resist Infect Control. 2019;8:194.
Pompa C, Rantala M, Greko C, Baptiste KE, Catry B, van Duijkeren E, et al. Public health risk of antimicrobial resistance transfer from companion animals. J Antimicrob Chemother. 2017;72:957–68.
Kim Y, Leung MH, Kwok W, Fournié G, Li J, Lee PKH, et al. Antibiotic resistance gene sharing networks and the effect of dietary nutritional content on the canine and feline gut resistome. Anim Microbiome. 2020;2:4.
Thomson K. Species-specific and indication-based use of antimicrobials in dogs, cats, cattle and horses in Finland: data collected using three different methods. Academic Dissertation. Department of Equine and Small Animal Medicine, Faculty of Veterinary Medicine, University of Helsinki. 2010.
Kauter A, Epping L, Semmler T, Antao E-M, Kannapin D, Stoeckle SD, et al. The gut microbiome of horses: current research on equine enteral microbiota and future perspectives. Anim Microbiome. 2019;1:14.
Álvarez-narváez S, Berghaus LJ, ERA M, Willingham-Lane JM, Slovis NM, Giguere S, et al. A common practice of widespread antimicrobial use in horse production promotes multi-drug resistance. Sci Rep. 2020;10:911.
Stanley D, Geier MS, Chen H, Hughes RJ, Moore RJ. Comparison of fecal and cecal microbiotas reveals qualitative similarities but quantitative differences. BMC Microbiol. 2015;15:51.
Levesque CL, Yu H, Gong J, De Lange CFM. Ileal mucosa-associated—but not ileal digesta—bacterial profiles in grower pigs are influenced by nutrition and use of antibiotics for weaner pigs. J Anim Sci. 2012;90:448–50.
Acknowledgment
The authors thank all members in Tao Ma, Le Luo Guan, and Tim A. McAllister's lab.
Funding
This work was supported by Agricultural Science and Technology Innovation Program of the Chinese Academy of Agricultural Sciences (CAAS-ASTIP-2017-FRI-04), China Agriculture Research System of MOF and MARA, Ministry of Alberta Agriculture and Forestry (2015P008R and 2018F095R), The NSERC Discovery Grant, The One Health Consortium of the University of Calgary - Alberta Government Major Innovation Fund and the International Opportunities Program of Agriculture a Agri-Food Canada.
Author information
Authors and Affiliations
Contributions
TM wrote the manuscript, TAM and LLG structured the manuscript content and contributed to revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, T., McAllister, T.A. & Guan, L.L. A review of the resistome within the digestive tract of livestock. J Animal Sci Biotechnol 12, 121 (2021). https://doi.org/10.1186/s40104-021-00643-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-021-00643-6