
Abstract
Background
Comparative knowledge of microbiomes and resistomes across environmental interfaces between animal production systems and urban settings is lacking. In this study, we executed a comparative analysis of the microbiota and resistomes of metagenomes from cattle feces, catch basin water, manured agricultural soil and urban sewage.
Results
Metagenomic DNA from composite fecal samples (FC; n = 12) collected from penned cattle at four feedlots in Alberta, Canada, along with water from adjacent catchment basins (CB; n = 13), soil (n = 4) from fields in the vicinity of one of the feedlots and urban sewage influent (SI; n = 6) from two municipalities were subjected to Illumina HiSeq2000 sequencing. Firmicutes exhibited the highest prevalence (40%) in FC, whereas Proteobacteria were most abundant in CB (64%), soil (60%) and SI (83%). Among sample types, SI had the highest diversity of antimicrobial resistance (AMR), and metal and biocide resistance (MBR) classes (13 & 15) followed by FC (10 & 8), CB (8 & 4), and soil (6 & 1). The highest antimicrobial resistant (AMR) gene (ARG) abundance was harboured by FC, whereas soil samples had a very small, but unique resistome which did not overlap with FC & CB resistomes. In the beef production system, tetracycline resistance predominated followed by macrolide resistance. The SI resistome harboured β-lactam, macrolide, tetracycline, aminoglycoside, fluoroquinolone and fosfomycin resistance determinants. Metal and biocide resistance accounted for 26% of the SI resistome with a predominance of mercury resistance.
Conclusions
This study demonstrates an increasing divergence in the nature of the microbiome and resistome as the distance from the feedlot increases. Consistent with antimicrobial use, tetracycline and macrolide resistance genes were predominant in the beef production system. One of the feedlots contributed both conventional (raised with antibiotics) and natural (raised without antibiotics) pens samples. Although natural pen samples exhibited a microbiota composition that was similar to samples from conventional pens, their resistome was less complex. Similarly, the SI resistome was indicative of drug classes used in humans and the greater abundance of mercury resistance may be associated with contamination of municipal water with household and industrial products.
Similar content being viewed by others


Background
Antimicrobials have played an important role in controlling bacterial infectious diseases in both humans and animals. In livestock, antimicrobials are used mainly for the treatment and prevention of disease as label claims for their use at sub-therapeutic levels to promote growth are being removed [1]. The worldwide consumption of antimicrobials in food animal production has been reported at ≥57 million kg with a projected increase to ≥95 million kilogram by 2030 [2]. In North American beef feedlots, a number of antimicrobials are administered to cattle, with macrolides and tetracyclines accounting for the majority of antimicrobial use (AMU) [3]. Bacteria residing in the bovine gastrointestinal tract may become resistant to these antibiotics and, once released into the environment, they may transfer antimicrobial resistance (AMR) genes (ARGs) to other bacteria including potential human pathogens [4, 5]. Furthermore, residual antibiotics may enter the environment through runoff from manure, where they may select for antimicrobial resistant bacteria [6, 7]. Consequently, it is not surprising that for almost every livestock-associated bacterial pathogen, resistance to at least one antimicrobial from each antimicrobial class has been reported [8].
Antimicrobials are not fully metabolized when administered to either humans or livestock. Gao et al. [9] estimated that up to 90% of many of the antibiotics used in livestock are excreted in urine or feces. Sewage treatment plants (STP) receive waste streams that contain a mixture of nutrients, metals, antibiotics, and industrial/household chemicals from a variety of sources [10]. Antimicrobials, antimicrobial resistant bacteria (ARB) and ARGs are frequently detected in STP [11, 12] and as a result these facilities have been identified as a potential hotspot for antibiotic resistance, where ARGs spread among bacteria via horizontal gene transfer. These biological pollutants are also released into the environment in STP effluent [13,14,15].
Knowledge of the microbiome and resistome within and between the environmental interface between animal production systems and urban centres is lacking. Information gained from an understanding of this interface could help support more prudent use of antimicrobials in livestock, more specifically, in defining targeted treatment options and distinguishing between essential and non-essential AMU to ensure safer food production practices.
Culture independent techniques, such as next generation sequencing (NGS) can be used to quantitatively assess the microbiota composition and its associated resistome. Advances in high-throughput NGS technologies have enabled rapid understanding of overall microbial ecology as well as occurrence and diversity of ARGs from diverse environments. Whole-metagenome shotgun analyses are accomplished by unrestricted sequencing of the genomes of most microorganisms present in a sample, including currently uncultured organisms. The present study describes the microbial metagenomes and resistomes of a variety of environmental samples from beef production to human-associated wastes (urban sewage). We utilize a NGS approach to inform surveillance as well as to improve the current understanding of the microbial community structure, the prevalence of ARGs within these microbial communities and to investigate overlaps between various components of the environmental spectrum.
Results and discussion
All 35 samples (FC = 12, CB = 13, soil = 4 and SI = 6) were sequenced to an average of ~ 54 million reads per sample. This sequencing depth was found appropriate, as indicated by the saturation of novel taxa and ARGs in our previous study which investigated the microbiota and resistome of bovine fecal samples [16]. The average read quality score for samples in the present study ranged from 33 to 37, indicative of high quality reads. Of the total number of reads generated, 94–97% survived quality filtering and trimming across all datasets.
Each sampling group exhibited distinct composition of microbiota
Across all samples 5.9% of total reads aligned to bacterial and archaeal species, representing 816 genera and 35 phyla. The proportion of prokaryote-associated (bacteria and archaea) raw (trimmed and quality filtered) reads arising from the total metagenomic raw reads varied among various sample types. Sewage influent (SI) had the highest number of prokaryote-associated reads, followed by soil, catch-basin (CB) water, and bovine feces (FC). For SI, 24.5% of the sequence reads were associated with bacteria and archaea, whereas soil, CB and FC had a much smaller proportion of prokaryote-associated reads (3.4, 4.5 and 2.1%, respectively), as revealed by the taxonomic classification via Kraken. The majority of remaining read fractions in these samples were uncharacterized, most likely originating from uncharacterized prokaryotes as well as eukaryotic organisms including algae, plants, small eukaryotes, avian or mammalian sources that are absent from the Kraken database. The comparatively high proportion of prokaryote-associated reads in SI is reflective of the very high density (2–10 g dry weight/L) of microorganisms within sewage [17]. Comparison of normalized data across all samples also supported the largest abundance of microbial taxa reads in SI, being 6.2, 6.7, and 2.4 fold higher than in FC, CB and soil, respectively (Fig. 1).

Abundance and relative proportion of microbial phyla in sample types. Abundance (a) is a measure of read counts aligning to various phyla (indicated by different colors) and normalized across samples whereas proportion (b) indicates percentage of each phylum in a sample type. The category ‘Other’ includes the rest of the low abundance phyla for each sample type
In FC, Firmicutes were the most abundant (39%) followed by Bacteroidetes (32%), Proteobacteria (11%), Actinobacteria (7%), Spirochaetes (5%) and Euryarchaeota (4%). These six phyla constituted 98.5% of the FC microbial community with Firmicutes and Bacteroidetes accounting for over 70% of the community. Predominance of Firmicutes and Bacteroidetes in livestock gastrointestinal tract microbiota is in agreement with other studies [18,19,20,21]. The most abundant classes of Firmicutes and Bacteroidetes in FC included Clostridia and Bacteroidia, respectively (Fig. 2) corresponding to 59% of prokaryotic reads, whereas Bacteroidaceae, Prevotellaceae, Methanobacteriaceae, Flavobacteriaceae, Clostridiaceae, Enterobacteriaceae were among the most abundant families (relative abundance range 12.6–7.5%). The five most predominant bacterial genera included Prevotella, Bacteroides, Treponema, Bifidobacterium and Clostridium (Table 1). Methanobrevibacter was the most prevalent genus from the archaeal phylum Euryarchaeota (Table 1). This genus has been previously characterized as hydrogenotrophic rumen methanogens [22]. Methanobrevibacter accounts for 80–85% of all methanobacterial reads in the cattle fecal methanogenic community [21, 23]; it is also the dominant methanogen in the rumen [24, 25].

Heat map of prevalent taxonomic classes across all samples grouped by phyla. As described in methods section, fecal composite (FC) samples were obtained from four feedlots a, b, c and d. The subscript letters C and N denote conventional and natural practices, respectively
The catch basin water community was dominated by Proteobacteria (67.4%), Actinobacteria (9.3%), Firmicutes (7.9%), Bacteroidetes (5.9%), Euryarchaeota (3.3%) and Spirochaetes (3.3%), accounting for 97% of prokaryotic microbiota reads (Fig. 1). Bacterial classes ɣ-proteobacteria and β-proteobacteria were abundant (Fig. 2) and constituted 45% of the prokaryotic reads, while Rhodocyclaceae and Moraxellaceae were the most abundant families in CB. Within these families, Thauera and Psychrobacter were the most abundant Proteobacterial genera in catch basin samples (Table 1). Psychrobacter are salt-tolerant, chemoheterotrophic, cold-adapted bacteria, which oxidize ammonia in high concentration under saline conditions [26]. Species from genus Thauera are frequently found in wet soil and polluted freshwater and have been considered important for industrial wastewater treatment systems as they play a key role in refractory aromatic hydrocarbon (e.g., indole and toluene) degradation under anaerobic and denitrifying conditions [26, 27]. Thauera were also observed in sewage influent. Occurrence of species from this genus in these polluted waters indicates the potential presence of aromatic hydrocarbons in these environments and as a result these functional species are of great significance for wastewater management.
The soil microbial community was predominated by Proteobacteria (60.3%) and Actinobacteria (35.2%), constituting 95.5% of the prokaryotic microbiota (Fig. 1). North American and European agroecosystems studies have also identified a high abundance of Proteobacteria and Actinobacteria associated with rhizosphere and rhizoplane [28, 29]. Wang et al. [30] have reported a 27 and 14% abundance of these two phyla respectively, in Chinese soils, followed by Acidobacteria (14%), Chloroflexi (8%) and Firmicutes (6%). In our soil samples, Bacteroidetes was the third most abundant phylum (1.6%), whereas Acidobacteria, Chloroflexi and Firmicutes were only present at 0.45, 0.41 and 0.13%, respectively. Lower abundance of Acidobacteria, and higher abundance of Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes has been associated with healthy agricultural soils with higher available phosphorus content [30]. Soil microbial communities can be highly diverse due to heterogeneity of soils, manure application as well as the nature of the rhizosphere [31]. In our soil samples, plant-associated species belonging to family Rhizobeaceae (α-Proteobacteria) were most prevalent (Table 1). Healthy soils generally have higher abundances of beneficial microbes including nitrogen-fixing and plant growth-promoting bacteria [32]. Interestingly, in present study, the soil collected 6 months after manure application had a higher number of Bacteroidetes (> 5 fold) and Euryarchaeota (> 3 fold) compared to non-manured and not recently manured fields. This likely reflects the presence of residual fecal bacteria from manure. Lupwayi et al. [33] also reported a higher proportion of Bacteroidetes in soils receiving composted beef feedlot manure in southern Alberta. While acknowledging the low number of soil samples originating from two agricultural fields in the vicinity of feedlot C over two years, inclusion of these samples in the analysis presents a snapshot of the influence of the feedlot manure on the soil microbiota and resistome.
Proteobacteria (83.5%), Bacteroidetes (10.4%) and Firmicutes (3.8%) represented the majority of sewage microbes with Acinetobacter (29%) and Aeromonas (16%) being the most abundant of the Proteobacteria. Others have found Proteobacteria to be among the most abundant bacteria in urban wastewater followed by Bacteroidetes and Firmicutes [34]. Acinetobacter johnsii and Acinetobacter baumannii accounted for the majority of the Acinetobacter identified. The former species rarely causes human infections, whereas the latter is an emerging hospital pathogen. In addition to being frequently recovered from patients during hospital outbreaks, A. baumannii have been reported in untreated as well as in biologically or chemically treated hospital and municipal wastewaters [35,36,37,38]. Our normalized species richness data indicated that SI harbored on average 2000 or more A. baumannii sequence reads as compared to FC, CB and soil (only 4, 15 and 1 respectively; Additional file 1) This suggests that the risk to human health from A. baumannii is far greater with SI than with the other environmental samples examined. In addition to Acinetobacter spp., the most abundant bacterial taxa detected in SI by others are Campylobacteraceae (Arcobacter spp.), Aeromonadaceae and Carnobacteriaceae [39,40,41,42]. Consistent with these studies Arcobacter and Aeromonas were among the most abundant genera in SI samples in our study, followed by Acinetobacter. Among Aeromonas spp. A. hydrophila, A. media, A. veronii, A. salmonicida, and A. schubertii were prevalent in SI. Most of these species are emerging human pathogens and have been associated with gastroenteritis, wound and soft tissue infections, necrotizing fasciitis, urinary tract infections, pulmonary infections in cystic fibrosis, and septicemia [43, 44]. Aeromonas spp. produce an array of virulence factors including cytolytic toxins with hemolytic activity and enterotoxins. Prevalence of these pathogens in FC, CB and soil was negligibly low as compared to SI.
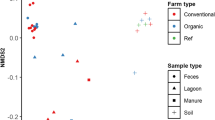
Although 793 of the total 816 prokaryotic genera detected across all samples were represented in all sample types, their relative distribution was very unique between matrices (Fig. 2; Additional file 1). The non-metric multidimensional scaling (NMDS) plot formed distinct sample type-specific clusters (Fig. 3) with significant separation at all taxa levels (ANOSIM R: 0.9–0.98, P < 0.05; Fig. 3). As expected, the distinct microbial composition of each sample matrix appears to be a reflection of the unique composition of nutrients, physical, physicochemical and other biotic and abiotic factors within each niche.

Comparative microbiota and resistome compositions of various sample types. Non-metric multidimensional scaling (NMDS) ordination plot indicate microbiota and resistome composition differences between fecal, catch basin, soil and wastewater (SI) samples at (a) genus (ANOSIM P = 0.001, ANOSIM R = 0.9804) and (b) AMR group (ANOSIM P = 0.001, ANOSIM R = 0. 9767) levels
The SI microbiome exhibited the highest richness of microbial genera as indicated by the number of unique taxonomic (genus) assignments corresponding to discovery of new species, but the lowest α-diversity and evenness as depicted by low inverse Simpson and Pielou’s evenness indexes respectively, across all sample types (Fig. 4). Wastewater biosolids are a rich source of nitrogen, phosphorus, potassium and organic matter as well as micro-nutrients [45]. This nutrient-rich environment may allow certain resident bacteria to thrive and therefore promotes richness over diversity. Although the median α-diversity of phyla was higher for fecal samples than for any other matrices, soil had the largest (p < 0.05) median α-diversity at the lower taxonomic ranks.

Quantitative comparisons of microbiota between various sample types. Richness (a) as indicated by number of unique taxa (genus discovery) assignments, α-diversity (b) as measured through inverse Simpson index, and evenness (c) of microbiota as Pielou’s evenness index at the genus level among various sample matrices are depicted by box-and-whisker plots. Boxes represent the interquartile ranges (upper line is the 75% quantile, and the lower line is the 25% quantile), the lines inside the boxes are the medians, the whiskers span the range of the 25% quantile or the 75% quantile plus 1.5 times the interquartile range, and dots are outliers
Distinct resistome composition of each sample matrix with predominance of tetracycline resistance in the beef production system
Across all samples, ~ 0.12% of total reads aligned to 35 mechanisms of antimicrobial resistance (AMR), coding resistance to 15 classes of antimicrobials, and ~ 0.04% of all reads corresponded to 15 classes of metal and biocide resistance (MBR) spanning 32 mechanisms. The proportion of AMR-MBR associated raw reads to the corresponding total reads was highest in conventional FC (0.25%) followed by SI (0.12%), CB (0.03%) and soil (0.002%), indicating a high prevalence of resistance genes in bovine feces. The proportion of AMR-MBR associated reads to the corresponding prokaryote-microbial reads was highest in conventional FC (11.3%) followed by CB (0.8%), SI (0.5%) and soil (0.07%) indicating that a higher fraction of bacteria and archaea in bovine feces harboured ARGs compared to other sample types. Comparison of normalized data across all samples also supported the larger abundance of ARG-associated reads in FC compared to soil, CB and SI (Fig. 5).

Abundance and relative proportion of antimicrobial resistance gene (ARG) and metal and biocide resistant gene (MBRG) classes in different sample types. Abundance (a) is a measure of read counts aligning to ARG-MBRG database and normalized across samples whereas proportion (b) indicates percentage of each class in a sample type. The category ‘Other’ includes the rest of the low abundance ARG/MBRG classes for each sample type
At the class level, tetracycline resistance was the most prevalent (82%) in FC followed by macrolide (14%), aminoglycoside (2.2%) and β-lactams (1.3%), respectively. Sequence reads aligned to 120 ARG and MBR gene (MBRG) groups collectively, belonging to 41 mechanisms within 18 classes. The tetracycline resistance ribosomal protection protein mechanism was most abundant (81%) predominantly represented by: TETQ,> TETW,> TET40,> TETO,> TET32 > TET44. Other tetracycline resistance genes including TET(X, M, A, B, G, 36, Z) were also present, but in lower abundance. Resistance to macrolides followed tetracycline resistance abundance, conferring lincosamide nucleotidyltransferases and efflux pump genes belonging to the LNUC and MEFA groups, respectively (Additional file 1). Previous studies reported a high prevalence of tetracycline resistance genes in cattle feces, with ~ 98% of reads aligning to ribosomal protection proteins represented in TETQ and TETW groups [46,47,48].
Overall, the CB resistome was represented by 84 ARG and MBRG groups. Similar to FC, in the CB resistome tetracycline resistance (59%) was the most abundant followed by resistance to macrolide (17.5%), aminoglycosides (7.2%) β-lactams (4.2%), sulfonamides (3.3%), mercury (2.8%) and multidrug resistance (MDR; 2.8%) (Fig. 5). This likely reflects the surface runoff of manure-associated tetracycline resistant ARB from feedlot pen floors into the catch basins. Miller et al. [49] quantified a runoff depth of 54 mm during a major rainfall event at a southern Alberta feedlot. Feedlots A, B, C and D shared 24, 31, 28 and 38 ARG groups between FC and their associated CB, respectively. The shared ARG groups were members of the tetracycline, macrolide and aminoglycoside resistance classes (Additional file 2). Among the tetracycline resistance groups, TETQ, TETM, TETW, TET36, TETT and TET44 were most prevalent. However, the relative abundance profile of these ARG classes differed between CB and FC reflecting the niche specificity of bacteria harboring these ARGs, considering that Proteobacteria were predominant in the CB microbial community as compared to Firmicutes and Bacteroidetes in FC. Among macrolide resistance ARG groups, MEFA, MEFB and MSR were more abundant in CB. Interestingly, MEFB was not detected in FC, but was present in SI samples. This gene has been found to be generally hosted by Proteobacteria [50], whereas MEFA and MSR genes have been associated with a wide variety of enteric bacterial phyla including Proteobacteria, Bacteroidetes, Actinobacteria and Firmicutes [51]. The high relative abundance of these genes could reflect their common presence in enteric bacteria, and/or due to co-selection with other ARGs as many tetracycline ARGs are linked to macrolide ARGs through common mobile genetic elements [52].
In North America, the use of in-feed tetracycline and macrolides to prevent liver abscesses and other bacterial diseases is a common management strategy in beef cattle production. Macrolides are also used to treat and manage Bovine Respiratory Disease (BRD). Conventional feedlots in the present study administered ionophores in combination with chlortetracycline or tylosin in-feed on a daily basis throughout the feeding period. Occasionally, therapeutic doses of antimicrobials were also administered to clinically ill cattle within a pen. It is acknowledged that the physical presence of a resistance gene may not always be interpretable as functional presence in the absence of gene expression data. However, the presence of an abundant gene is generally associated with some degree of its functional expression within a particular environment. The high prevalence of both tetracycline and macrolide resistance gene classes in FC and CB, therefore is likely a reflection of the ubiquitous use of these antibiotics in beef production [53, 54].
Soil samples originating from agricultural fields adjacent to feedlot C had a small and unique resistome with only 9 ARG groups belonging to 6 classes and did not align with the feedlot resistome (Fig. 5; Additional file 1). Tetracycline ARG TETL was only found in recently manured soil. Compared to soil, this ARG group had a 9–17 times lower prevalence in FC and CB and was completely absent in SI. It may be that TETL harboring bacterial species from manure survived better in soil compared to other tetracycline ARG carrying bacteria. Tetracycline was the most widely used antibiotic class in the feedlots enrolled in this study. Glycopeptide resistance associated genes were present across all soil samples, but were absent from any other sample type. Specifically, VanO-type regulators (VANRO) [55] were the only glycopeptide-related genes detected in soil samples. The vanO operon initially identified in Rhodococcus equi [55], harbors a vanHOX resistance gene cluster transcribed convergent to that of the vanS-vanR two-component regulatory system. The vanO locus in Rhodococcus equi exhibits similarity to genera Amycolatopsis and the nitrogen fixing, root nodule-forming Frankia [55] and to the teicoplanin producer Actinoplanes teichomyceticus [56]. The Amycolatopsis and Actinoplanes were among the most prevalent genera in soil samples from our study (Table 1). Other than vanO-type regulators no other vancomycin resistance operon-associated reads (Vancomycin D-alanyl-D-alanine dipeptidase and/or ligase etc.) were detected, which may be due to low homology or absence of the vanO operon associated genes in soil bacteria. The second most abundant ARGs in soil were multidrug resistance (MDR) efflux pump coding genes. The organisms with the largest number of MDR pumps are in fact found in the soil or in association with plants [57]. Along with their potential roles as multidrug efflux pumps, these are important for detoxification of intracellular metabolites, bacterial virulence in both animal and plant hosts, cell homeostasis, and intercellular signal trafficking [58]. Therefore, bacteria harboring MDR pumps are not always associated only with high antibiotic load environments.
The SI from two urban municipalities in Southern Alberta exhibited similar resistome composition. Across all sample matrices SI had the largest number of ARG groups (229) belonging to 28 classes of ARGs and MBRGs. The most prevalent resistance classes in SI included multi-drug resistance (28%), β-lactam (15.28%), mercury (11.83%), tetracycline (11.16%) macrolide (10.72%) and aminoglycoside resistance (5.78%) (Fig. 5). Historically, mercury contamination of wastewater occurs from a variety of sources including dental practice wastes, lawn fertilisers, landfill leachate, paints, domestic waste inputs, groundwater infiltration and storm water drainage. Of the 2000 tonnes per year of global atmospheric mercury that is discharged into the air and water from anthropogenic sources, Canada’s atmospheric mercury share account for <0.5% of the world’s emissions (https://www.canada.ca/en/environment-climate-change/services/pollutants/mercury-environment.html).
Among β-lactam ARGs, cephalosporin resistance groups OXA and CTX were predominant, with 8 fold more richness of OXA in SI compared to CB, and its complete absence in FC and soil. Conversely, CTX was 71 fold more abundant in SI compared to FC and absent in CB and soil (Additional file 1). QNRD, a plasmid-mediated quinolone resistance (PMQR) gene group was only present in SI, likely reflecting its use in human medicine. Among all sample types, only the SI resistome contained a large variety of metal and biocide resistance genes (Additional file 1). Recently, Gupta et al. [42] reported a similar relative abundance of ARGs and a high prevalence of heavy metal resistance genes (HMRGs) in samples from a wastewater treatment plant.
Thirty four ARG groups belonging to tetracycline (TET32, TET40, TET36, TETA, TETG, TETM, TETO, TETS, TETQ, TETW, TETX,), macrolide (ERMA, ERMB, ERMF, ERMG, LNUC, MEFA, MEL, MPHB, MPHE, MSR, MSRD), aminoglycoside (ANT6, ANT9, APH3’ APH3”, APH6, SAT, ANT3”), class A beta lactamase (CFX & CARB), sulfonamide (SULII), mercury resistance (MERA), and drug and biocide small multidrug resistance (SMR) efflux pump (qacEΔ1) were shared in FC & CB and SI sample groups at varying abundances (Additional file 1). For tetracycline resistance TETQ, TETW, TET40, TETO, TET32 and TET44 were among the most abundant tetracycline ARG groups in beef production. TETQ, TETW, TET40, TETO and TET32 have also been identified among the most prevalent groups in fecal samples collected from humans in China, Denmark and Spain [59, 60], suggesting their high abundance in both cattle and human microbiota. Studies across diverse agricultural ecosystems have also demonstrated the ubiquity of tetracycline resistance genes [61, 62].
Sewage wastewater is an effective source of fecal bacteria and provides a unique opportunity to monitor fecal microbes from large human populations without compromising privacy [63]. Wastewater treatment plants are considered hotspots of ARB and ARGs [15, 64, 65], as they receive wastewater from households and hospitals where antimicrobials are administered. The persistent selective pressure posed by sub-inhibitory concentrations of antimicrobial residues in wastewater combined with the high density [17] and diversity [66] of microorganisms could promote horizontal transfer of ARGs and HMRGs [67,68,69]. Co-selection of ARGs and HMRGs in SI [70, 71] is favoured when these genes are carried on the same mobile genetic element [72]. Furthermore, leachate from wastewater sludge disposed of in landfills may promote the spread of ARGs into sub-soils and ground water [73].
A heat map of prevalent ARGs groups across all samples grouped by AMR classes (Fig. 6) indicated that the majority of AMR/MBR classes represented in FC, CB and SI resistome were absent in soil. Tetracycline, β-lactam and multidrug efflux ARGs were present among all sample types, whereas ARGs for fluoroquinolones, fosfomycin and metronidazole were only present in SI (Additional file 1), suggesting that use of these antimicrobials in humans selected for these genes. The NMDS analysis showed that the resistome from different sample types differed at the AMR gene group (ANOSIM P = 0.001, ANOSIM R = 0. 98) level (Fig. 3B) and all other levels of ARG categories (ANOSIM P < 0.05, R: 0.92–0.98) confirming the uniqueness of resistome in each sample type. Across sample types, 5, 9, 98 and 5 resistance gene groups were uniquely present in FC, CB, SI and soil respectively (Fig. 6; Additional file 2). In addition to the microbial source and the microbial niche specificity in different environments the distinct resistome composition of each sample matrix could also be a reflection of the specific antimicrobial residues in each environment. Recent studies have identified a link between community structure and antibiotic resistance gene dynamics [74]. Future metagenomics-based microbiome and resistome studies that include bacterial genome assemblies from deep metagenomics sequencing data will shed light on the association of ARGs with their host bacteria.

Heat map of prevalent antimicrobial resistant gene groups across all samples grouped by antimicrobial resistance class. As described in methods section, fecal composite samples were obtained from 4 feedlots a, b, c and d. The subscript letters C and N denote conventional and natural practices, respectively
The SI wastewater resistome exhibited the highest richness of ARG mechanism types among sample types (Fig. 7). In addition to having high richness, SI contained the most diverse and even resistome among all sample types as indicated by high inverse Simpson index of α-diversity and Pielou’s evenness index (Fig. 7B), which reflects the diverse classes of antimicrobials used in human medicine [75] as compared to those used in cattle. After ionophores, tetracycline and macrolides are among the most frequently used antimicrobials in livestock [76, 77].

Quantitative comparisons of resistome between various sample types. Richness (a) as indicated by number of unique gene groups (gene group discovery) assignments, α-diversity (b) as measured through inverse Simpson index, and evenness (c) of resistome as Pielou’s evenness index at the resistance gene group level among various sample matrices are depicted by box-and-whisker plots. Boxes represent the interquartile ranges (upper line is the 75% quantile, and the lower line is the 25% quantile), the lines inside the boxes are the medians, the whiskers span the range of the 25% quantile or the 75% quantile plus 1.5 times the interquartile range, and dots are outliers
Natural feedlot FC samples harboured relatively similar microbiota but smaller resistome compared to conventional samples
The microbial composition of fecal samples from ‘natural’ and ‘conventional’ beef production systems had comparable richness, diversity, and similar prevalence of microbial phyla. The exception was that the composition of natural FC microbiota had a lower abundance of two bacterial (Bacteroidetes, Spirochaetes; log FC values − 0.7 and − 2.3 respectively; p < 0.05) and one archaeal (Euryarchaeota; log FC value − 3.8; p < 0.001) phyla in natural, compared with conventional FC. A 17-fold increase in the methanogenic archaeal genus Methanobrevibacter (Phylum Euryarchaeota) was observed in the samples originating from conventional pens as compared to the natural pens (Additional file 1). Considering that the animal diets between the natural and conventional feedlot practices were similar, these differences in fecal microbiota may be related to antimicrobial use. Given the small number of samples compared between natural and conventional feedlots, further studies are needed to more thoroughly investigate this phenomenon.
The proportion of AMR-MBR associated raw reads to the corresponding total reads for feedlot D conventional FC samples was higher (0.23%) compared to natural FC samples (0.09%) indicating high prevalence of resistance genes in bovine feces. The average number of ARG-associated reads identified was higher for the conventional FC compared to natural FC (Fig. 8). This trend was observed across the top three abundant ARG classes including tetracycline, macrolide and aminoglycoside (p < 0.05). Regardless of higher ARG abundance in conventional samples, diversity of resistomes between natural and conventional pen samples was similar (Additional file 1). Prior studies have concluded no correlation between the presence of antimicrobial resistance genes in the gut microbiota and the administration of antibiotic feed additives [78,79,80,81]. However, in contrast to our study, most of these studies either did not quantify comparative prevalence of ARGs in production systems managed with and without using antimicrobials or their comparative investigation was limited to a few bacterial species and ARGs. Single-colony subcultures do not recover the actual AMR reservoir of a microbial community.

Comparisons of microbiota and resistome between samples from conventional and natural feedlot pens. Relative abundance of microbial phyla (a) and antimicrobial resistance classes (b) among fecal composite samples from conventional and natural feedlot pens are indicated as read counts on the X-axis
Phenicol and sulfonamide were the only resistance classes absent in the natural samples. Other groups belonging to tetracycline (TETA, TETB, TET32, TETW, TET40, TET44, TETO, TETQ, TETX), macrolide (MEFA, LNUC), aminoglycoside (APH3’, ANT6) and β-lactams (CFX, ACI) resistance were present in both natural and conventional FC, whereas tetracycline (TETH, TET36, TETZ, TETS, TETT), macrolide (APH6, MPHE, MPHB, MSRD ERMA, MPHE, MEL, ERMR, ERMC, ERMT), aminoglycoside (ANT3”), β-lactamase (CARB), phenicol (FLOR, CMXAB) and sulfonamide (SULII) were absent in natural samples, but were present in at least one of three conventional samples. The ARG groups MSR and TETM belong to macrolide and tetracycline drug classes respectively, and were present in all conventional FC pen samples from feedlot D, but were absent in all natural pen samples. Assuming that the presence of a gene means that it is being expressed, their presence may be associated with the use of these drug classes in the conventional feedlot. Genes belonging to this family have been shown to be associated with transposons and integrative conjugative elements [82, 83], which may contribute to their ubiquitous prevalence through intra- and inter-species mobility under the added selective pressure of antimicrobial use. Considering that ARGs are ancient [84] their diverse presence in natural production systems is not surprising. The occurrence of certain ARGs within bacterial populations is likely a reflection of their association with fitness traits that enable bacteria to persist within a particular environment. While antibiotic resistance and its spread by horizontal gene transfer are ancient mechanisms, the rate at which these processes occur and the proliferation of certain ARG-harboring bacteria has increased tremendously over the last decades due to the selective pressure exerted through anthropogenic administration of antimicrobials. We argue that a holistic approach of identifying ARGs and microbiota, and quantitating their prevalence as undertaken in our study is necessary for informing surveillance and to understand the evolution and transmission of AMR in an environmental spectrum.
Conclusions
Consistent with its abundant use in feedlots, tetracycline resistance was predominant in the beef production system followed by macrolide resistance. Regardless of possessing a comparable composition of microbiota, fecal samples collected from cattle raised without antibiotics exhibited a smaller resistome as compared to fecal samples collected from conventionally raised cattle. This study enhances our understanding of the microbial composition and the occurrence of ARGs and identifies common elements between those components of the environmental spectrum and indicates a distinct separation of associated microbial communities. The specific resistance profiles across various sample matrices were dependent upon the microbial community composition as well as differences in the nature and prevalence of drug, metal and biocide contaminants.
Methods
Sample collection, DNA isolation, quantitation and quality assessment
Composite fecal samples analyzed in this study (n = 12) were collected from four different beef cattle feedlots (A, B, C, D) within the province of Alberta Canada (sampling locations in Additional file 6: Fig. S1). Feedlot sampling was conducted from April – June 2014. The feedlots had operating capacities of ∼15,000–30,000 head of cattle. Production conditions were typical of western Canadian commercial feedlots, with animals housed in open-air, clay-floor pens arranged side-by-side with central feed alleys. Feedlot D had two separate wings for hosting natural (raised without antibiotics) and conventional (with antibiotics) cattle pens. Samples in Feedlot D were collected from both natural (n = 3) and conventional (n = 3) pens. The rest of the fecal composite samples (n = 6 of a total of 12) originated from conventional feedlots A, B and C (Supplementary data_3), where antimicrobials were used in a routine manner similar to the conventional wing in Feedlot D. Within a feedlot, samples were collected on the same day from pens containing 150–300 animals. Sampling procedures were reviewed and approved by the Lethbridge Research Centre Animal Care and Use Committee (AC# 14–0029), and were conducted according to the Canadian Council of Animal Care Guidelines. Each composite fecal sample comprised ~ 20 g aliquots collected from 20 individual fresh fecal pats within each pen. Fecal samples were thoroughly mixed, placed in 532 mL Whirl-Pak bags, flash frozen in liquid nitrogen and stored at -80 °C. The antimicrobials used in the sampled conventional feedlots are listed in Additional file 4. The in-feed antimicrobials (ionophores, chlortetracycline or tylosin) were administered to all cattle in the conventional feedlot throughout the feeding period with the therapeutic parenteral drugs administered to clinically ill cattle as required.
Natural resources legislation in Alberta stipulates that feedlots must have catch basins (also known as retention or runoff holding ponds) for containment of surface runoff water from pens or manure storage areas generated by rainfall or snowmelt. At each feedlot, surface water was sampled from a catchment basin adjacent to the sampled feedlot pens. Water samples (2, 3, 4 and 4 samples were collected from catch basins at feedlots A, B, C, and D respectively, n = 13) (Additional file 3). One liter of water was collected at a depth of 0.5 m into a 1.3 L polyethylene bottle attached to a telescopic pole. Water was collected from four different locations within the catchment basin and the samples were combined to generate a single composite sample which was immediately transferred to the lab on ice. To complement the cattle production and associated environmental sampling, two wastewater treatment plants in Southern Alberta (Additional file 1: Fig. S1) provided sewage influent samples (n = 6) to represent the urban element of the environmental spectrum. One liter of sewage influent water was collected from post-grit tanks of the wastewater treatment facility.
Catchment basin or sewage influent water samples (n = 13, up to 100 mL each) were filtered through 0.45 μm pore size nylon filters (MilliporeSigma, Etobicoke, ON, Canada) using a water filtration manifold and membrane filtration units (Pall Corporation Ltd. Mississauga, Canada). The membrane filter was aseptically removed from the filter base using sterile forceps and stored at − 20 °C in a sterile 5 ml OMNI Bead Ruptor tube (Cole-Parmer, Montreal, Canada) for later DNA extraction. If the membrane filter became plugged, samples were centrifuged at 10,000 x g in 50 mL tube to obtain a pelleted biomass for DNA extraction.
Composite core soil samples (n = 4) were collected from agricultural fields adjacent to feedlot C and included the following sample types: field with no history of manure application, from the same field as above but ~ 6 months after manure application, and from a field with a continuous history of manure application, but not within 1–2 year prior to sampling. Soil samples were collected twice over two years (see Additional file 3 for details). Soil sampling was carried out using a soil coring kit (5 cm diameter) to a depth of 10 cm and samples at 10 points along a 100 m transect were collected and pooled for each field to constitute a composite sample.
Metagenomic DNA isolation from the bovine fecal samples was performed as previously described [16]. The DNA was extracted from soil and pelleted biomass from water samples in a manner similar to feces, with the nylon filters subject to bead-beating and incubation steps at 70 °C [16]. The DNA concentrations were measured using the Quant-iT™ PicoGreen (Thermo Fisher Scientific, Mississauga,ON, Canada) and the DNA purity was determined by measuring the ratios of absorbance at 260/280 and 260/230 using a NanoDrop spectrophotometer (Thermo Fisher Scientific). The DNA extracts with a 260/280 ratio between 1.8–2.0 and a 260/230 ratio between 2.0–2.2 were regarded as pure. The presence of PCR-inhibitors was also evaluated by amplifying the 16S rRNA gene using the universal 16S rRNA gene primers 27F and 1492R [85] with undiluted and diluted samples [16].
Metagenomic DNA sequencing and data processing
All library preparations, next generation sequencing and quality control steps were performed by the McGill University and Genome Quebec Innovation Centre (Montréal, QC, Canada). TruSeq DNA libraries were prepared and samples were run on an Illumina HiSeq2000 platform, with 4 samples multiplexed per sequencing lane to generate 2 × 100 base paired-end (PE) sequences [16]. As a quality control for cluster generation and sequencing, each HiSeq2000 sequencing lane was spiked with the PhiX174 sensu lato virus genomic DNA library at ~ 1% concentration of the total DNA loaded per lane.
Trimmomatic version 0.36 [86] was used to remove adapter contamination and low quality reads using the following parameters: trimming leading and the trailing low quality or N bases (below quality 3) from sequence reads; performing quality score filtering using a sliding window at every four bases with a minimum Phred score of 15; discarding sequences with < 36 nucleotides; removing adapters supplied in the TruSeq3 adapter sequence file using a maximum of 2 mismatches in the initial seed, and clipping the adapter if a match score of 30 was reached. Singleton reads, whereby the other pair was discarded were also included in downstream analysis.
Determination of the taxonomic and ARG composition of microbiota
Taxonomic classification of microbiota and determination of AGR assignments for resistome analysis of the sequence data were performed using previous methods and parameters [16]]via a Galaxy Web server instance (https://galaxyproject.org/) supported by the National Microbiology Laboratory, Public Health Agency of Canada (PHAC NML Galaxy). The Kraken taxonomic classification tools (version 0.10.5 beta) and the resistome analysis tools were integrated in a workflow to obtain output for both the resistome and microbiome analyses (workflow details in Additional file 6: Fig. S2).
In that workflow, the trimmed paired reads that passed the quality assessment criteria from the pre-processing step with Trimmomatic were aligned to the genome of the enterobacteria phage phiX174 (GenBank accession NC_001422.1) using the minimum exact match (MEM) algorithm of the Burrows-Wheeler aligner (BWA) [87]. The sorted alignments were then processed with samtools [88] to filter out reads that did not map to the PhiX 174 bacteriophage genome. This was done using a flag value of 4 to extract the unmapped reads in binary alignment map (BAM) format. The paired reads that did not map to PhiX 174 bacteriophage were then extracted from the alignment using the bamToFastq tool of BEDTools [89]. The PhiX-filtered reads were then classified with Kraken v 1.2.3 [90] using the custom Kraken database bvfpa [16]. Kraken results were filtered using a confidence threshold of 0.05 to select for taxonomic assignments with high precision and sensitivity and thus high accuracy at the genus rank [http://ccb.jhu.edu/software/kraken/MANUAL.html; 16]. Resistome analysis was conducted in parallel with the taxonomic classification as follows: Trimmed paired reads were mapped to the ARG sequences in the MEGAREs database v1.01 [91] combined with a custom metal and biocide resistance (MBR) database (MegaBio; P.S. Morley’s lab; Additional file 5) using BWA-MEM v 0.7.17.1 [87] alignments in BAM format followed by conversion to sequence alignment map (SAM) format and post-processing with the Coverage Sampler tool (https://github.com/cdeanj/coveragesampler) using a 75% gene fraction threshold and other parameters [15].
Data analyses
The microbiome and resistome data reports from individual samples were aggregated into corresponding matrices using R for downstream analyses. Microbiome and resistome matrices were normalized using the data-driven approach of Cumulative Sum Scaling normalization (CSS) with the metagenomeSeqR package [92]. This method calculates a scaling threshold that is the quantile after which the distribution of raw counts among samples is invariant. The method calculates the sum, up to and including that quantile threshold for re-scaling. In this study, a CSS normalization quantile threshold of 0.5 (the median) was used. The cumulative sum scaling method has been previously reported for normalization of comparative metagenomic sequencing data from various environments [93]. CSS has greater sensitivity and specificity compared to other normalization methods and it corrects the bias in the assessment of differential abundance introduced by total-sum normalization therefore improving sample clustering [94]. Other methods such as rarefaction analysis can lead to higher false discovery rate while comparing differentially abundant genes [95]. The exploratory analyses performed in this study included: relative abundance analysis for microbiome and resistome for all sample matrix types, assessment of α-diversity and richness for all sample types, ordination using nonmetric multidimensional scaling (NMDS), and comparative visualization of data with heatmaps and barplots. Observed richness, the Shannon’s and Inverse Simpson’s α -diversity indices, and Pielou’s evenness were calculated using functions of the vegan package version 2.5.1 [96] and their distributions were plotted for each sample type as box-and-whisker plots using ggplot2 [97]. Heatmaps were constructed using the log2 transformed CSS-normalized counts which were plotted using white to orange gradient scale.
A zero-inflated Gaussian (ZIG) mixture model was applied to evaluate differentially abundant features in the resistomes and microbiomes between sample matrix types. This model has been reported to increase sensitivity and specificity when working with datasets with high sparsity (abundance of zero counts). Ordination plots were generated using NMDS and statistical inference was made using the analysis of similarity (ANOSIM) with the vegan R package version 2.5.1 [96]. ANOSIM R-values ranged from 0 (total similarity) to 1 (total dissimilarity). The Kruskal–Wallis test [98] was performed to compare the distributions of richness and the Inverse Simpson’s indices of α--diversity for both ARGs and microbial taxa among the various sample types. Nemenyi post-hoc comparisons [99] were conducted for incidences where differences were declared significant at P < 0.05 as per the Kruskal-Wallis analysis. The R code for the data analysis is available at https://github.com/ropolomx/one_health_continuum.
Availability of data and materials
All Illumina sequence read data from current study have been deposited to the NCBI database as Short Read Archive (SRA) under BioProject IDs PRJNA420682, PRJNA529711, PRJNA507800 and PRJNA482680. These data are publically available at https://www.ncbi.nlm.nih.gov/bioproject/.
Abbreviations
- AMR:
-
Antimicrobial resistance
- AMU:
-
Antimicrobial use
- ANOSIM:
-
Analysis of similarity
- ARB:
-
Antimicrobial resistant bacteria
- ARG:
-
Antimicrobial resistance gene
- BAM:
-
Binary alignment map
- CB:
-
Feedlot catchment basin
- CSS:
-
Cumulative sum scaling normalization
- FC:
-
Composite fecal sample
- HMRGs:
-
Heavy metal resistance genes
- MBR:
-
Metal and biocide resistance
- MBRG:
-
Metal and biocide resistance gene
- MDR:
-
Multidrug resistance
- MEM:
-
Minimum exact match
- NGS:
-
Next generation sequencing
- NMDS:
-
Nonmetric multidimensional scaling
- SI:
-
Urban sewage influent
- STP:
-
Sewage treatment plant
References
Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010;74(3):417–33. https://doi.org/10.1128/MMBR.00016-10.
Van Boeckel TP, Brower C, Gilbert M, Grenfell BT, Levin SA, Robinson TP, et al. Global trends in antimicrobial use in food animals. Proc Natl Acad Sci U S A. 2015;112:5649–54. https://doi.org/10.1073/pnas.1503141112.
Silbergel EK, Graham J, Price LB. Industrial food animal production, antimicrobial resistance, and human health. Annu Rev Public Health. 2008;29:151–69. https://doi.org/10.1146/annurev.publhealth.29.020907.090904.
Andremont A. Commensal flora may play key role in spreading antibiotic resistance. ASM News. 2003;69:601–7.
Marshall BM, Ochieng DJ, Levy SB. Commensals: underappreciated reservoirs of antibiotic resistance. Microbe. 2009;4:231–8.
Campagnolo ER, Johnson KR, Karpati A, Rubin CS, Kolpin DW, Meyer MT, et al. Antimicrobial residues in animal waste and water resources proximal to large-scale swine and poultry feeding operations. Sci Total Environ. 2002;299:89–95.
Heuer H, Solehati Q, Zimmerling U, Kleineidam K, Schloter M, Müller T, et al. Accumulation of sulfonamide resistance genes in arable soils due to repeated application of manure containing sulfadiazine. Appl Environ Microbiol. 2011;77(7):2527–30. https://doi.org/10.1128/AEM.02577-10.
Cameron A, McAllister TA. Antimicrobial usage and resistance in beef production. J Anim Sci Biotech. 2016;7:68. https://doi.org/10.1186/s40104-016-0127-3.
Gao P, Munir M, Xagoraraki I. Correlation of tetracycline and sulfonamide antibiotics with corresponding resistance genes and resistant bacteria in a conventional municipal wastewater treatment plant. Sci Total Environ. 2012;421-422:173–83. https://doi.org/10.1016/j.scitotenv.2012.01.061.
Naquin A, Shrestha A, Sherpa M, Nathaniel R, Boopathy R. Presence of antibiotic resistance genes in a sewage treatment plant in Thibodaux, Louisiana, USA. Bioresour Technol. 2015;188:79–83. https://doi.org/10.1016/j.biortech.2015.01.052.
Szczepanowski R, Linke B, Krahn I, Gartemann KH, Gützkow T, Eichler W, et al. Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiology. 2009;155:2306–19. https://doi.org/10.1099/mic.0.028233-0.
Devarajan N, Laffite A, Mulaji CK, Otamonga JP, Mpiana PT, Mubedi JI, et al. Occurrence of antibiotic resistance genes and bacterial markers in a tropical river receiving hospital and urban wastewaters. PLoS One. 2016;11:e0149211. https://doi.org/10.1371/journal.pone.0149211.
Giger W, Alder AC, Golet EM, Kohler HPE, McArdell CS, Molnar E, et al. Occurrence and fate of antibiotics as trace contaminants in wastewaters, sewage sludges, and surface waters. CHIMIA Int J Chem. 2003;57:485–91. https://doi.org/10.2533/000942903777679064.
Hijosa-Valsero M, Fink G, Schlüsener MP, Sidrach-Cardona R, Martín-Villacorta J, Ternes T, et al. Removal of antibiotics from urban wastewater by constructed wetland optimization. Chemosphere. 2011;83:713–9. https://doi.org/10.1016/j.chemosphere.2011.02.004.
Rizzo L, Manaia C, Merlin C, Schwartz T, Dagot C, Ploy MC, et al. Urban wastewater treatment plants as hotspots for antibiotic resistant bacteria and genes spread into the environment: a review. Sci Total Environ. 2013;447:345–60. https://doi.org/10.1016/j.scitotenv.2013.01.032.
Zaheer R, Noyes N, Ortega Polo R, Cook SR, Marinier E, Van Domselaar G, et al. Impact of sequencing depth on the characterization of the microbiome and Resistome. Sci Rep. 2018;8:5890. https://doi.org/10.1038/s41598-018-24280-8.
Grady CPL Jr, Daigger GT, Love NG, Filipe CD. Biological wastewater treatment; 3rd edition. New York: IWA Publishing; 2011. ISBN13:9781843393429
Durso LM, Harhay GP, Smith TPL, Bono JL, DeSantis TZ, Harhay DM, et al. Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol. 2010;76:4858–62. https://doi.org/10.1128/AEM.00207-10.
Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, et al. Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol. 2011;77:2992–3001. https://doi.org/10.1128/AEM.02988-10.
Xu Y, Dugat-Bony E, Zaheer R, Selinger L, Barbieri R, Munns K, et al. Escherichia coli O157:H7 super-shedder and non-shedder feedlot steers harbour distinct fecal bacterial communities. PLoS One. 2014;9:e98115. https://doi.org/10.1371/journal.pone.0098115.
Ozbayram EG, Ince O, Ince B, Harms H, Kleinsteuber S. Comparison of rumen and manure microbiomes and implications for the inoculation of anaerobic digesters. Microorganisms. 2018;6. https://doi.org/10.3390/microorganisms6010015.
Bayané A, Guiot SR. Animal digestive strategies versus anaerobic digestion bioprocesses for biogas production from lignocellulosic biomass. Rev Environ Sci Biotechnol. 2011;10:43–62.
Seedorf H, Kittelmann S, Janssen PH. Few highly abundant operational taxonomic units dominate within rumen methanogenic archaeal species in New Zealand sheep and cattle. Appl Environ Microbiol. 2015;81:986–95. https://doi.org/10.1128/AEM.03018-14.
McSweeney C, Mackie R. Micro-organisms and ruminant digestion: state of knowledge, trends and future prospects. Comm Genet Resour Food Agric. 2012; Background Study Paper No. 61. Available online: http://www.fao.org/docrep/016/me992e/me992e/pdf. Accessed 8 Aug 2018.
King EE, Smith RP, St-Pierre B, Wright AD. Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen. Appl Environ Microbiol. 2011;77:5682–7. https://doi.org/10.1128/AEM.05130-11.
Alfiansah YR, Hassenrück C, Kunzmann A, Taslihan A, Harder J, Gärdes A. Bacterial abundance and community composition in pond water from shrimp aquaculture systems with different stocking densities. Front Microbiol. 2018;9:2457. https://doi.org/10.3389/fmicb.2018.02457.
Mao YJ, Zhang XJ, Xia X, Zhong HH, Zhao LP. Versatile aromatic compound-degrading capacity and microdiversity of Thauera strains isolated from a coking wastewater treatment bioreactor. J Ind Microbiol Biotechnol. 2010;37(9):927–34. https://doi.org/10.1007/s10295-010-0740-7.
Edwards JA, Santos-Medellín CM, Liechty ZS, Nguyen B, Lurie E, Eason S, et al. Compositional shifts in root-associated bacterial and archaeal microbiota track the plant life cycle in field-grown rice. PLoS Biol. 2018;16:e2003862. https://doi.org/10.1371/journal.pbio.2003862.
Hartman K, van der Heijden MGA, Wittwer RA, Banerjee S, Walser JC, Schlaeppi K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming [published correction appears in microbiome]. Microbiome. 2018;6:14. https://doi.org/10.1186/s40168-017-0389-9.
Wang R, Zhang H, Sun L, Qi G, Chen S, Zhao X. Microbial community composition is related to soil biological and chemical properties and bacterial wilt outbreak. Sci Rep. 2017;7:343. https://doi.org/10.1038/s41598-017-00472-6.
Fierer N. Embracing the unknown: disentangling the complexities of the soil microbiome. Nat Rev Microbiol. 2017;5:579–90. https://doi.org/10.1038/nrmicro.2017.87.
Raaijmakers JM, Paulitz TC, Steinberg C, Alabouvette C, Moënne-Loccoz Y. The rhizosphere: a playground and battlefield for soilborne pathogens and beneficial microorganisms. Plant Soil. 2009;321:341–61.
Lupwayi NZ, Larney FJ, Blackshaw RE, Kanashiro DA, Pearson DC, Petri RM. Pyrosequencing reveals profiles of soil bacterial communities after 12 years of conservation management on irrigated crop rotations. Appl Soil Ecol. 2017;121:65–73.
Lee S, Geller JT, Torok T, Wu CH, Singer M, Reid FC, et al. Characterization of wastewater treatment plant microbial communities and the effects of carbon sources on diversity in laboratory models. PLoS One. 2014;9:e105689. https://doi.org/10.1371/journal.pone.0105689.
Ferreira AE, Marchetti DP, De Oliveira LM, Gusatti CS, Fuentefria DB, Corção G. Presence of OXA-23-producing isolates of Acinetobacter baumannii in wastewater from hospitals in southern Brazil. Microb Drug Resist. 2011;17:221–7. https://doi.org/10.1089/mdr.2010.0013.
Zhang C, Qiu S, Wang Y, Qi L, Hao R, Liu X, et al. Higher isolation of NDM-1 producing Acinetobacter baumannii from the sewage of the hospitals in Beijing. PLoS One. 2013;8:e64857. https://doi.org/10.1371/journal.pone.0064857.
Seruga MM, Hrenovic J, Goic-Barisic I, Hunjak B, Skoric D, Ivankovic T. Emission of extensively-drug resistant Acinetobacter baumannii from hospital settings to the natural environment. J Hosp Infect. 2017;96:323–7. https://doi.org/10.1016/j.jhin.2017.04.005.
Higgins PG, Hrenovic J, Seifert H, Dekic S. Characterization of Acinetobacter baumannii from water and sludge line of secondary wastewater treatment plant. Water Res. 2018;140:261–7. https://doi.org/10.1016/j.watres.2018.04.057.
Moreno Y, Botella S, Alonso JL, Ferrus MA, Hernandez M, Hernandez J. Specific detection of Arcobacter and campylobacter strains in water and sewage by PCR and fluorescent in situ hybridization. Appl Environ Microbiol. 2003;69:1181–6.
Vandewalle JL, Goetz GW, Huse SM, Morrison HG, Sogin ML, Hoffmann RG, et al. Acinetobacter, Aeromonas and Trichococcus populations dominate the microbial community within urban sewer infrastructure. Environ Microbiol. 2012;14:2538–52. https://doi.org/10.1111/j.1462-2920.2012.02757.x.
Shanks OC, Newton RJ, Kelty CA, Huse SM, Sogin ML, McLellan SL. Comparison of the microbial community structures of untreated wastewaters from different geographic locales. Appl Environ Microbiol. 2013;79:2906–13. https://doi.org/10.1128/AEM.03448-12.
Gupta SK, Shin H, Han D, Hur HG, Unno T. Metagenomic analysis reveals the prevalence and persistence of antibiotic- and heavy metal-resistance genes in wastewater treatment plant. J Microbiol. 2018;56:408–15. https://doi.org/10.1007/s12275-018-8195-z.
Wu C-J, Chen P-L, Hsueh P-R Chang MC, Tsai PJ, Shih HI, Wang HC, Chou PH, Ko WC. Clinical implications of species identification in monomicrobial Aeromonas bacteremia. PloS ONE 2015;10: e0117821. doi:https://doi.org/10.1371/journal.pone.0117821.
Persson S, Al-Shuweli S, Yapici S, Jensen JN, Olsen KE. Identification of clinical Aeromonas species by rpoB and gyrB sequencing and development of a multiplex PCR method for detection of Aeromonas hydrophila, A. caviae, A. veronii, and A. media. J Clin Microbiol. 2015;53:653–6. https://doi.org/10.1128/JCM.01963-14.
Kirchmann H, Börjesson G, Kätterer T, Cohen Y. From agricultural use of sewage sludge to nutrient extraction: a soil science outlook. Ambio. 2016;46:143–54.
Chambers L, Yang Y, Littier H, Ray P, Zhang T, Pruden A, et al. Metagenomic analysis of antibiotic resistance genes in dairy cow feces following therapeutic Administration of Third Generation Cephalosporin. PLoS One. 2015;10:e0133764. https://doi.org/10.1371/journal.pone.0133764.
Noyes NR, Yang X, Linke LM, Magnuson RJ, Cook SR, Zaheer R, et al. Characterization of the resistome in manure, soil and wastewater from dairy and beef production systems. Sci Rep. 2016a;6:24645. https://doi.org/10.1038/srep24645.
Noyes NR, Yang X, Linke LM, Magnuson RJ, Dettenwanger A, Cook S, et al. Resistome diversity in cattle and the environment decreases during beef production. Elife. 2016b;5:e13195. https://doi.org/10.7554/eLife.13195.
Miller JJ, Handerek BP, Beasley BW, Olson ECS, Yanke LJ, Larney FJ, et al. Quantity and quality of runoff from a beef cattle feedlot in southern Alberta. J Environ Qual. 2004;33:1088–97.
Liu J, Keelan P, Bennett PM, Enne VI. Characterization of a novel macrolide efflux gene, mef(B), found linked to sul3 in porcine Escherichia coli. J Antimicrob Chemother. 2009;63:423–6. https://doi.org/10.1093/jac/dkn523.
Roberts MC. Resistance to macrolide, lincosamides, streptogramin, ketolide, and oxazolidinone antibiotics. Mol Biotechnol. 2004;28:47–62.
Chung WO, Young K, Leng Z, Roberts MC. Mobile elements carrying ermF and tetQ genes in gram-positive and gram-negative bacteria. J Antimicob Chemother. 1999;44:329–35.
Alexander TW, Yanke JL, Reuter T, Topp E, Read RR, Selinger BL, et al. Longitudinal characterization of antimicrobial resistance genes in feces shed from cattle fed different subtherapeutic antibiotics. BMC Microbiol. 2011;11:19. https://doi.org/10.1186/1471-2180-11-19.
Peak KCW, Yang RK, Hanfelt MM, Smith MS, Aga DS, Graham DW. Abundance of six tetracycline resistance genes in wastewater lagoons at cattle feedlots with different antibiotic use strategies. Environ Microbiol. 2007;9:143–51.
Gudeta DD, Moodley A, Bortolaia V, Guardabassi L. vanO, a new Glycopeptide resistance operon in environmental Rhodococcus equi isolates. Antimicrob Agents Chemother. 2014;58:1768–70. https://doi.org/10.1128/AAC.01880-13.
Serina S, Radice F, Maffioli S, Donadio S, Sosio M. Glycopeptide resistance determinants from the teicoplanin producer Actinoplanes teichomyceticus. FEMS Microbiol Lett. 2004;240:69–74.
Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci U S A. 2004;101:3160–5.
Martinez JL, Sánchez MB, Martínez-Solano L, Hernandez A, Garmendia L, Fajardo A, et al. Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol Rev. 2009;33:430–49. https://doi.org/10.1111/j.1574-6976.2008.00157.x.
Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, et al. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013;23:1163–9. https://doi.org/10.1101/gr.155465.113.
Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013;4:2151. https://doi.org/10.1038/ncomms3151.
Schmitt H, Stoob K, Hamscher G, Smit E, Seinen W. Tetracyclines and tetracycline resistance in agricultural soils: microcosm and field studies. Microb Ecol. 2006;51:267–76.
Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. mBio. 2012;3. https://doi.org/10.1128/mBio.00377-12.
Newton RJ, McLellan SL, Dila DK, Vineis JH, Morrison HG, Eren AM, et al. Sewage reflects the microbiomes of human populations. mBio. 2015;6:e02574–14. https://doi.org/10.1128/mBio.02574-14.
Gatica J, Cytryn E. Impact of treated wastewater irrigation on antibiotic resistance in the soil microbiome. Environ Sci Pollut Res Int. 2013;20:3529–38. https://doi.org/10.1007/s11356-013-1505-4.
Bouki C, Venieri D, Diamadopoulos E. Detection and fate of antibiotic resistant bacteria in wastewater treatment plants: a review. Ecotoxicol Environ Saf. 2013;91:1–9. https://doi.org/10.1016/j.ecoenv.2013.01.016.
Zhang T, Shao MF, Ye L. 454 pyrosequencing reveals bacterial diversity of activated sludge from 14 sewage treatment plants. ISME J. 2012;6:1137–47. https://doi.org/10.1038/ismej.2011.188.
Yang Y, Li B, Zou S, Fang HHP, Zhang T. Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach. Water Res. 2014;62:97–106. https://doi.org/10.1016/j.watres.2014.05.019.
Li A-D, Li L-G, Zhang T. Exploring antibiotic resistance genes and metal resistance genes in plasmid metagenomes from wastewater treatment plants. Front Microbiol. 2015;6:1025. https://doi.org/10.3389/fmicb.2015.01025.
Guo J, Ni B-J, Han X, Chen X, Bond P, Peng Y, et al. Data on metagenomic profiles of activated sludge from a full-scale wastewater treatment plant. Data Brief. 2017;15:833–9. https://doi.org/10.1016/j.dib.2017.10.048.
Ji X, Shen Q, Liu F, Ma J, Xu G, Wang Y, et al. Antibiotic resistance gene abundances associated with antibiotics and heavy metals in animal manures and agricultural soils adjacent to feedlots in Shanghai; China. J Hazard Mater. 2012;235-236:178–85. https://doi.org/10.1016/j.jhazmat.2012.07.040.
Seiler C, Berendonk TU. Heavy metal driven co-selection of antibiotic resistance in soil and water bodies impacted by agriculture and aquaculture. Front Microbiol. 2012;3:399. https://doi.org/10.3389/fmicb.2012.00399.
Pal C, Bengtsson-Palme J, Kristiansson E, Larsson DGJ. Co-occurrence of resistance genes to antibiotics, biocides and metals reveals novel insights into their co-selection potential. BMC Genomics. 2015;16:964. https://doi.org/10.1186/s12864-015-2153-5.
Threedeach S, Chiemchaisri W, Watanabe T, Chiemchaisri C, Honda R, Yamamoto K. Antibiotic resistance of Escherichia coli in leachates from municipal solid waste landfills: comparison between semi-aerobic and anaerobic operations. Bioresour Technol. 2012;113:253–8. https://doi.org/10.1016/j.biortech.2012.01.086.
Dunivin TK, Shade A. Community structure explains antibiotic resistance gene dynamics over a temperature gradient in soil. FEMS Microbiol Ecol. 2018;94:fiy016. https://doi.org/10.1093/femsec/fiy016.
Gow S. Antimicrobial resistance, prudent use, and the Canadian integrated program for antimicrobial resistance surveillance (CIPARS). Large Anim Vet Rounds. 2005;5:1–6. https://doi.org/10.1111/j.1863-2378.2010.01356.x.
Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. 2001;65:232–60.
Canadian Integrated Program for Antimicrobial Surveillance (CIPARS). Annual report; 2013. http://publications.gc.ca/collections/collection_2015/aspc-phac/HP2-4-2013-2-eng.pdf
Thomas M, Webb M, Ghimire S, Blair A, Olson K, Fenske GJ, et al. Metagenomic characterization of the effect of feed additives on the gut microbiome and antibiotic resistome of feedlot cattle. Sci Rep. 2017;7:12257. https://doi.org/10.1038/s41598-017-12481-6.
Harvey R, Funk J, Wittum TE, Hoet AE. A metagenomic approach for determining prevalence of tetracycline resistance genes in the fecal flora of conventionally raised feedlot steers and feedlot steers raised without antimicrobials. Am J Vet Res. 2009;70:198–202. https://doi.org/10.2460/ajvr.70.2.198.
Santamaria J, Lopez L, Soto CY. Detection and diversity evaluation of tetracycline resistance genes in grassland-based production systems in Colombia, South America. Front Microbiol. 2011;2:252.
Vikram A, Rovira P, Agga GE, Arthur TM, Bosilevac JM, Wheeler TL, et al. Impact of "raised without antibiotics" beef cattle production practices on occurrences of antimicrobial resistance. Appl Environ Microbiol. 2017pii: AEM.01682–17. https://doi.org/10.1128/AEM.01682-17.
Gillings MR, Labbate M, Sajjad A, Giguere NJ, Holley MP, Stokes HW. Mobilization of a Tn402-like class 1 integron with a novel cassette array via flanking miniature inverted-repeat transposable element-like structures. Appl Environ Microbiol. 2009;75:6002–4.
Ciric L, Jasni A, ElviradeVries L, Agerso Y, Mullany P, Roberts AP. The Tn916/Tn1545 family of conjugative transposons. In: Roberts AP, Mullany P, editors. Bacterial and integrative mobile genetic elements. Austin: Landes Bioscience; 2013. http://www.ncbi.nlm.nih.gov/books/NBK63531/.
D’Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, et al. Antibiotic resistance is ancient. Nature. 2011;477:457–61.
Lane DJ. 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. New York: Wiley; 1991. p. 115–76.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM; 2013. bioRxiv:arXiv:1303.3997 [q-bio.GN]
Li HB, Handsaker AW, Fennell T, Ruan J, Homer N, Marth G, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Quinlan AR. BEDTools: the Swiss-Army tool for genome feature analysis. In: Current protocols in bioinformatics. John Wiley & Sons, Inc; 2002. http://onlinelibrary.wiley.com/doi/10.1002/0471250953.bi1112s47/abstract.
Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014;15:R46. https://doi.org/10.1186/gb-2014-15-3-r46.
Lakin SM, Dean C, Noyes NR, Dettenwanger A, Ross AS, Doster E, et al. MEGARes: an antimicrobial resistance database for high throughput sequencing. Nucleic Acids Res. 2017;45(D1):D574–80. https://doi.org/10.1093/nar/gkw1009.
Paulson JN, Talukder H, Pop M, Bravo HC. metagenomeSeq: statistical analysis for sparse high-throughput sequencing; 2017. Bioconductor package: 1.18.0. http://cbcb.umd.edu/software/metagenomeSeq
Doster E, Rovira P, Noyes NR, et al. Investigating effects of Tulathromycin Metaphylaxis on the fecal Resistome and microbiome of commercial feedlot cattle early in the feeding period. Front Microbiol. 2018;9:1715. https://doi.org/10.3389/fmicb.2018.01715.
Paulson JN, Stine OC, Bravo HC, Pop M. Differential abundance analysis for microbial marker-gene surveys. Nat Methods. 2013;10(12):1200–2. https://doi.org/10.1038/nmeth.2658.
McMurdie PJ, Holmes S. Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput Biol. 2014;10(4):e1003531. https://doi.org/10.1371/journal.pcbi.1003531.
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan: community ecology PackageR package version 2.5–2; 2018. https://CRAN.R-project.org/package=vegan
Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621 and errata, ibid. 48:907–911.
Nemenyi P. Distribution-free multiple comparisons: Ph.D. thesis, Princeton University; 1963.
Acknowledgements
The authors would like to thank the NML bioinformatics lab staff, especially Philip Mabon, Shane Thiessen and Paul Williams for help with software and tool installation in the NML computing cluster. All metagenomic sequencing was done at McGill University and Génome Québec Innovation Centre, Montréal.
Funding
This study was supported by funding from the Beef Cattle Research Council (BCRC Project FOS 10.13) – Agriculture and Agri-Food Canada’s Science Cluster and the Genomic Research and Development Initiative (GRDI)-AMR program of the Federal Government of Canada. Funding agencies had no role in the design of the study, sample collection, analysis, interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
RZ, PSM, CB and TAM designed the study. SH and FJL arranged feedlot and soil samples and metadata collection; RZ and SRC isolated metagenomic DNA; RZ and ROP analyzed the data. SL wrote the initial version of the R code for data analysis, which ROP modified and expanded. GVD provided bioinformatics cluster facility; RZ wrote the manuscript; ROP, SRC, FJL, RRR and TAM edited the manuscript; TAM, RRR and PSM provided funding. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Sampling procedures were reviewed and approved by the Lethbridge Research Centre Animal Care and Use Committee (AC# 14–0029), and were conducted according to the Canadian Council of Animal Care Guidelines. Feedlots were recruited for participation by Feedlot Health Management Services (FHMS), Okotoks, Alberta, Canada, and the owners of the participating feedlots provided explicit permission in accordance with provincial agriculture operation practices to work on the premises and to collect sample from feedlots that were enrolled in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any competing interest or commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Microbiota and AMR stats – Ten Excel sheets describing raw and normalized read count data for microbiome and resistome of studied samples. (XLSX 3121 kb)
Additional file 2:
Shared and Unique ARG groups – Two Excel sheets with lists of shared and unique ARG groups among various sample types. (XLSX 17 kb)
Additional file 3:
Sample metadata and sequencing stats – Two excel sheets describing details of sample collection metadata and Illumina HiSeq read counts of studied samples. (XLSX 12 kb)
Additional file 4:
Antibiotics used in feedlots – Excel sheet describing antibiotics used in the feedlots enrolled in present study and their mode of administration. (XLSX 9 kb)
Additional file 5:
Excel file describing accession and annotation details of genes included in metal and biocide database, MegaBio. (XLSX 28 kb)
Additional file 6:
Figure S1. Sampling locations in the province of Alberta, Canada. Figure S2. Galaxy workflow for antimicrobial resistance (AMR) and taxonomic profiling of metagenomics sequencing read data. (DOCX 1067 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zaheer, R., Lakin, S.M., Polo, R.O. et al. Comparative diversity of microbiomes and Resistomes in beef feedlots, downstream environments and urban sewage influent. BMC Microbiol 19, 197 (2019). https://doi.org/10.1186/s12866-019-1548-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-019-1548-x