
Abstract
Liver disease is prevalent worldwide. When it reaches the end stage, mortality rises to 50% or more. Although liver transplantation has emerged as the most efficient treatment for end-stage liver disease, its application has been limited by the scarcity of donor livers. The lack of acceptable donor organs implies that patients are at high risk while waiting for suitable livers. In this scenario, cell therapy has emerged as a promising treatment approach. Most of the time, transplanted cells can replace host hepatocytes and remodel the hepatic microenvironment. For instance, hepatocytes derived from donor livers or stem cells colonize and proliferate in the liver, can replace host hepatocytes, and restore liver function. Other cellular therapy candidates, such as macrophages and mesenchymal stem cells, can remodel the hepatic microenvironment, thereby repairing the damaged liver. In recent years, cell therapy has transitioned from animal research to early human studies. In this review, we will discuss cell therapy in end-stage liver disease treatment, especially focusing on various cell types utilized for cell transplantation, and elucidate the processes involved. Furthermore, we will also summarize the practical obstacles of cell therapy and offer potential solutions.
Similar content being viewed by others


Introduction
The liver, a vital organ for survival, is responsible for bile production, nutrient metabolism, toxin removal, blood purification, and inflammation [1]. The prevalence of liver diseases, such as non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH) and metabolic liver diseases, has increased and may ultimately progress to end-stage liver disease such as liver failure and liver cirrhosis [2, 3]. The only efficient treatment for these diseases is orthotopic liver transplantation (OLT); however, suitable organ donors are insufficient to meet clinical demand [4]. Due to the shortage of healthy donor livers, a wide gap exists between the number of patients on the waiting list and the number of organs available, and those waiting for organ donation have a high mortality rate [5, 6]. Despite the emergence of novel surgical transplantation procedures such as split liver transplantation, the problem of donor liver scarcity has not been satisfactorily addressed [7, 8]. Fortunately, cell therapy, an increasingly popular strategy for treating end-stage liver disease, can effectively address the shortage of donor livers and reduce the need for invasive surgical procedures.
Cell therapy involves using cells of various types to remodel or replace damaged organs or tissues. The transformed cells may be injected into the liver locally or intravenously to restore liver function or encourage liver regeneration [9]. The therapeutic effect of cell therapy has been extensively studied in animal models [10, 11]. The first study on cell therapy was conducted in 1976 by Najarian, who transplanted allogeneic hepatocytes into a rat model of congenital enzyme deficiency disease via the portal vein [12]. Later, Mito et al. attempted to transplant hepatocytes into the spleen, suggesting that the spleen may be used as an ectopic liver [13]. In 1992, researchers successfully restored the liver function of a patient with hepatic encephalopathy and severe ascites by transplanting hepatocytes into the spleen [14]. Subsequently, Strom et al. proved the viability of human hepatocyte trans-splenial artery transplantation in patients with end-stage liver disease, and the spleens of transplanted patients displayed a characteristic hepatic cord structure [15]. Of the five patients who received treatment, three made a full recovery and were successfully bridged to OLT, while in specific individuals with acute liver failure, cell therapy has also been effective in bridging to OLT [16]. Since then, hundreds of clinical hepatocyte transplants have been recorded worldwide.
Compared with OLT, cell therapy offers many benefits among surgical techniques. From an operational standpoint, cell therapy is more feasible than OLT or split liver transplantation because (1) multiple recipients can receive hepatocytes from a single donor; (2) the procedure is less invasive and simpler; (3) donor cells can be cryopreserved and accessed as needed; (4) the recipient liver is not removed and can continue functioning normally if the treatment is unsuccessful; and (5) the cost is lower [17]. While cell therapy shows great promise, some significant issues remain unresolved. Transplanted hepatocytes do not proliferate in patients with metabolic illnesses when the liver is undamaged and entirely healthy, such as Crigler–Najjar syndrome or familial hypercholesterolemia, because the liver does not need such proliferation during physiological processes. To allow transplanted cells to proliferate, the liver must be pretreated to boost the proliferative advantage of donor cells [18, 19]. Furthermore, immunological rejection of transplanted cells in the liver presents a significant obstacle.
Overall, cell therapy is an effective and promising approach for end-stage liver disease, and much research has been performed in this field. Before OLT, cell therapy can be used as a bridge treatment. However, it also has limitations in cell acquisition engraftment, proliferation and delivery. The solutions usually include cytokine stimulation, immune regulation, organoids, hyaluronan matrix, reprogramming media with antioxidants, intrasplenic cell infusion and peritoneal delivery. In this review, we summarize the latest progress in end-stage liver disease using cell therapy.
Diverse cell sources and therapeutic sites for cell therapy
The hepatic lobule has a basic hexagonal shape, with the portal triad (the portal vein, bile duct, and hepatic artery) located at the lobule's periphery and the central vein in the middle. Hepatocytes, as primary parenchymal cells of the liver, perform most of the liver's physiological functions and maintain a certain liver-to-body ratio owing to the powerful regenerative capacity of the liver [20]. Thus, hepatocytes can be implanted into the liver to boost liver regeneration, meaning that transplanted hepatocytes multiplied and replaced host hepatocytes (Fig. 1). However, the acquisition and preservation of hepatocytes is a common problem. To obtain more hepatocytes, some teams gradually began to use stem cells or cells with stem cell properties that can transdifferentiate or differentiate into hepatocytes for cell transplantation (Fig. 1). Bile duct epithelial cells (BECs), for instance, can transdifferentiate into hepatocytes, which we believe is due to cell plasticity [21, 22]. Labelling BECs revealed that these cells can be converted to hepatocytes in the event of severe liver injury [21], implying that they are probably accessible as a source of transplantable cells. In recent studies, extrahepatic organoids derived from cholangiocytes have been demonstrated to preserve plasticity within the human biliary tree after transplantation and can repair human intrahepatic bile ducts [23]. This has piqued researchers' curiosity, and some have used human bile duct epithelial cells (hBECs) from discarded donor livers to rescue bile duct structure and function in a mouse model of biliary disease [24]. These studies showed that BECs may be an alternative cell source for cell transplantation. In further studies, pluripotent stem cells (PSCs) and foetal liver progenitor cells may differentiate in vitro into hepatocytes or hepatocyte-like cells for cell therapy, which will be described in detail later (Fig. 1).

Cell sources and entry routes for cell therapy. Replacement and remodelling are the two primary purposes of cells deployed in cell therapy. Before cell transplantation, bile duct epithelial cells, pluripotent stem cells, and foetal liver progenitor cells must be differentiated into hepatocytes in vitro. Following transplantation, these cells proliferate within the liver and replace the host hepatocytes. Additionally, mesenchymal stem cells, macrophages, and hematopoietic progenitor cells can remodel the hepatic microenvironment. The liver, spleen, and peritoneum are all potential sites for cell transplantation
In addition to replacing host hepatocytes and expanding within the liver, transplanted cells may remodel the hepatic microenvironment (Fig. 1). For example, transplanted cells can exert immunomodulatory, anti-inflammatory and antifibrotic effects on end-stage liver disease [25, 26]. Additionally, it has been shown that bone marrow transplanted cells, when abandoning transdifferentiation, may stimulate hepatocyte proliferation and restore liver function [27, 28]. In conclusion, transplanted cells can treat liver diseases by replacing host hepatocytes and remodelling the hepatic microenvironment (Fig. 1). Moreover, the feasibility of hepatocyte acquisition is also improved due to the plasticity of the cells [29].
In addition to the liver, some extrahepatic organs may serve as cell therapy sites. Currently, the more reliable transplantation sites are the spleen and peritoneum (Fig. 1). The transplanted cells may colonize these organs and perform the liver's functions [15, 30, 31]. In conclusion, the flexible cell therapy technique enables the selection of suitable transplanted cells and delivery organs according to the status of the specific patient.
Replacing the damaged liver
Hepatocytes
Adult hepatocytes have been shown to expand in vivo, similar to haematopoietic stem cells [32]. Several successful genetic mouse models have been used to validate the use of cell transplantation to treat inborn errors of metabolism (IEM) of the human liver, including albumin-uPA transgenic mice, mice with alpha-1-antitrypsin deficiency, and those with fumaryl acetoacetate hydrolase deficiency (familial tyrosinemia) [33,34,35]. Within a few weeks, the transplanted cells effectively replaced most of the host hepatocytes in these immunodeficient mouse models, resulting in the formation of chimeric tissues. According to a case report in the New England Journal of Medicine, a 10-year-old girl with Crigler–Najjar syndrome type I and severe unconjugated hyperbilirubinemia was effectively treated by 7.5 × 109 hepatocytes infused into the portal vein [36], and the therapeutic benefits for the patient persisted for up to 11 months and permitted a bridge to OLT [16]. Additionally, hepatocyte transplantation (HT) has been utilized to treat paediatric patients with liver metabolic abnormalities [37,38,39], and several investigations are being conducted to enhance the availability and safety of cells [40]. Even a small amount of host hepatocytes replaced by cell therapy produces significant clinical effects in treating IEM [41]. The efficacy of cell therapy has been shown in several illnesses, including phenylketonuria, Crigler–Najjar syndrome and propionic acidemia [42,43,44]. Most patients finally underwent OLT, and the greatly extended life before liver transplantation enhanced the likelihood that the patient would obtain a suitable donor liver. On the other hand, hepatocyte suspension can be safely administered into the portal vein, proving that cell therapy has a lower surgical risk than OLT. Strom et al. have shown the safety and therapeutic effectiveness of hepatocyte splenic artery infusion in patients with chronic end-stage liver disease [15]. However, the transplanted cells in some individuals do not dramatically proliferate but are progressively eliminated over several months. This occurs because the proliferation of transplanted cells in the liver necessitates a high proliferative advantage relative to the host cells and proper immunological microenvironment [45]. Therefore, a hypothesis can be put forth: halting the patient's current therapy may enable the patient's liver to have an environment beneficial to the engraftment of the transplanted cells.
Acute liver failure (ALF) is a rare acute disease with a high mortality rate [46]. An "indeterminate" aetiology is a reasonably frequent cause that often lacks a transparent causal element and affects people who have never had liver disease. OLT is the only effective treatment option [47]. Many studies have shown that primary hepatocyte transplantation may rescue animals from ALF and improve survival in a rat model where successful donor chimaerism occurs after transplantation [48,49,50]. The liver and spleen are the most reliable transplantation locations (Fig. 1) because their distinct vascular and extracellular matrix structures are ideal places for transplanted hepatocytes to stay and proliferate [50]. Alternatively, hepatocytes may be transplanted into the peritoneum [51]. Cell therapy is more suited for ALF patients with a typical liver structure. At several research centres, clinical hepatocyte transplantation studies have been performed. Strom et al. described five patients who underwent hepatocyte transplantation after perfusion of a mixture of 107–109 freshly isolated and cryopreserved hepatocytes through the splenic artery [52]. The four control individuals and all five test participants developed multisystem organ failure and grade IV hepatic encephalopathy. Among them, five participants were successfully bridged to OLT while maintaining normal cerebral perfusion and cardiac stability, whereas all four control subjects died within three days. At 20 months of follow-up, three of the five patients who successfully received liver transplantation were still alive and in good physical health. It remains challenging to use HT to provide consistent therapeutic benefits. After the transplanted cells are fully assimilated into the body, macrophages are activated and begin to phagocytose diseased cells and other debris. They also secrete transforming growth factor-β (TGF-β), which causes hepatocytes in the liver to undergo replicative senescence, which is then transmitted to the transplanted cells [53]. Smad2/3 is phosphorylated by the TGF-β signalling pathway, which activates downstream molecules such as extracellular matrix (ECM) genes and causes collagen deposition [54]. This causes the transplanted cells to separate from the recipient, thus hindering the regeneration process. Extrahepatic HT, such as in the spleen and peritoneum (Fig. 1), may be considered a solution to this issue. Seven patients from India received peritoneal infusions of foetal liver cells from 26–34-week-old unborn babies. The results revealed that HT patients had a 10% higher overall survival rate than controls [55]. Birir et al. described five patients with ALF who underwent intrasplenic HT [56]. At 48 h post-transplantation, three of the five patients showed significant improvements in encephalopathy scores, blood ammonia levels, prothrombin time, and overall survival after HT. In other studies, sodium alginate microencapsulation has been used to encapsulate hepatocytes in microspheres, protecting them against the body's immunological response and enabling molecular exchange [31]. Of course, HT also has adverse effects in specific clinical situations. During hepatocyte infusion in the portal vein, intrapleural, or peritoneal, some patients developed deadly mesenteric vein thrombosis [30] and non-lethal splenic vein thrombosis [57]. This shows that there is still room for improvement in hepatocyte delivery.
Acute-on-chronic liver failure (ACLF) is characterized by acute decompensation (AD), organ failure, and high short-term mortality [58, 59]. The development of ACLF frequently occurs due to an unanticipated occurrence, such as bacterial infection, acute alcohol-associated hepatitis, drug-induced hepatitis and viral hepatitis, with a 28-day death rate of 30%. Depending on the grade, regular treatment enables only 16–51% of patients to reverse ACLF, leaving a sizable percentage of patients with ACLF stable or progressing [60]. In a clinical experiment by Wang et al. on patients with ACLF, one patient was bridged to OLT, three restored liver function, and another three died after intrasplenic injection of 4.2–6.0 × 1010 hepatocytes [61].
Overall, the application of HT has shown great therapeutic potential for end-stage liver disease. However, this approach has limitations. One of the main challenges is the limited availability and engraftment efficiency of hepatocytes, as well as manufacturing difficulties. These obstacles include difficulties in isolating high-quality cells from donor livers, mechanistic limitations in cryopreserving liver cells while maintaining viability, low levels of engraftment and proliferation in transplanted liver cells, and issues related to cell delivery. It has been shown that long-term proliferation of human hepatocytes (lc-ProliHH) can induce activation of dedifferentiation-associated inflammatory factors (DAIF), thus inducing increased macrophage activation. Blockage of the innate immune response by dexamethasone improved engraftment and repopulation [62]. The strategy of coculturing hepatocytes with IL-6, epidermal growth factor (EGF) and hepatocyte growth factor (HGF) can promote long-term expansion of primary hepatocytes (> 30 passages in ~ 150 days with theoretical expansion of ~ 1035 times) and repopulate livers [63]. Recent advances in liver/cholangiocyte organoids have provided a promising solution to address these challenges. Organoids often have higher viability and efficiency in engraftment and proliferation in vivo [64]. Moreover, the delivery issues associated with HT may result from alternative strategies such as intrasplenic cell infusion and peritoneal delivery (Table 1).
Foetal liver progenitor cells
In vitro, foetal liver progenitor cells (FLPs) may differentiate into hepatocytes (Fig. 1). It has been shown that foetal hepatocytes from humans and other primates may proliferate and mature in the livers of immunodeficient mice, repopulating 10% of the organ [65, 66]. Furthermore, it has been shown that FLPs can differentiate into functional human endothelial cells and that no tumour development was observed within nine months after transplantation [67]. Additionally, it was demonstrated that liver fibrosis was reduced in an animal model that received transplants of human FLPs and that the majority of the human hepatocytes still present in the mouse livers were from proliferation rather than the original transplant [68]. In an immunodeficient animal model, FLPs have a poorer proliferative ability than adult hepatocytes, despite results showing that FLPs can differentiate into hepatocytes [68, 69]. It is possible that the liver could not provide adequate signals to promote the proliferation and engraftment of FLPs. The issues of inefficient engraftment remain a significant challenge. Some researchers have shown that direct intrahepatic injection of cells within a hyaluronan matrix may significantly improve engraftment [64]. Furthermore, the development and use of FLPs for treating liver disease have also been constrained by ethical debate over the acquisition of foetal tissue.
Pluripotent stem cells
Pluripotent stem cells (PSCs), which need to be differentiated into hepatocytes before cell transplantation, are regarded as an alternative cell source for HT (Fig. 1) [70]. To distinguish between embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), several techniques have been developed [71,72,73,74,75]. ESCs are derived from pluripotent stem cells that form in the blastocyst cluster of cells in humans 4–5 days after fertilization. Because ESCs have been researched for a long time, some protocols employ cell development signals to promote the differentiation of ESCs into hepatocyte-like cells (HLCs). Bone morphogenetic protein (BMP) and fibroblast growth factor (FGF) promote hepatocyte development, whereas activin A and WNT3 signalling favour endoderm differentiation [71, 76, 77]. Despite many attempts, the phenotype of HLCs derived from ESCs fails to match that of adult hepatocytes [78]. In addition, immune rejection after ESC transplantation and ethical aspects of ESC acquisition are inevitable issues. In 2013, many research groups used nuclear transplantation to effectively create ESCs from the somatic cells of patients, although the process is still technically challenging [79].
Through the induction of adult fibroblasts, Takahashi's team created iPSCs in 2007 [80]. In addition to human fibroblasts, other cells may be reprogrammed into iPSCs, including keratinocytes, lymphocytes, endothelial colony-forming cells, and mesenchymal stem cells [81,82,83,84]. iPSCs are comparable to ESCs in terms of pluripotency, infinite proliferative ability, and plasticity. The benefits of iPSCs for hepatocyte differentiation and organoid culture in vitro have been shown in several studies [75, 81, 85]. Most earlier investigations have shown that HTs and hepatocytes produced from iPSCs have similar transplant efficacy [86]. Nevertheless, research by Carpentier's team showed that by employing iPSCs for implantation in urokinase-type plasminogen activator (uPA) immunodeficient mice, 15% liver regeneration (LR) could be attained [87]. Furthermore, cell transplantation using HLCs differentiated from iPSCs is also associated with a risk of carcinogenesis resulting from genetic mutations. Merkle et al. found that PSCs accumulated single nucleotide variants (SNVs) in cancer-related genes, including the tumour suppressor gene TP53 [88]. Moreover, poor engraftment and immune-mediated loss are regarded as crucial factors [89]. In an attempt to overcome these challenges, a previous study has shown that supplementing the reprogramming media with antioxidants is attributed to the reduction of genomic aberrations in iPSCs [90]. Some studies have used activin A, dimethyl sulfoxide, hepatocyte growth factor, oncostatin M, and dexamethasone to induce hepatocyte maturation [91]. These strategies provide solutions for the engraftment and repopulation of iPSCs. Clinical translation will be substantially accelerated by creating iPSCs with minimal immunogenicity and improved differentiation ability. In conclusion, many modifications are still needed for application of iPSCs in clinical translation.
Organoids
The application of organoids has increasingly aroused researchers’ interest. “Organoid” is defined as a three-dimensional (3D) structure that is grown from stem cells or adult cells. Compared with two-dimensional (2D) cell lines, organoids have the advantages of organ structure and genetic stability. Appropriate organoids are considered an important source of available cell therapy [92]. Previous studies have successfully constructed liver organoids from Lgr5+ cells and iPSCs that can be applied to treat liver failure in mice [93,94,95]. Takebe et al. reported that after transplantation, liver organoids can reconstruct functional blood vessels and save mice from drug-induced liver damage [96]. Forbes et al. rescued bile duct structure and function in a mouse model of biliary disease with organoids constructed from purified human biliary epithelial cells (hBECs) [24]. A study from the University of Cambridge found that cholangiocyte organoids can repair bile duct injury after transplantation in the human liver [23].
In summary, organoids show great promise in cell transplantation. Future research could apply liver/cholangiocyte organoids to treat end-stage liver disease and improve the quality of expanded criteria donors in expanded liver donor pools.
Remodelling the hepatic microenvironment
Anti-inflammatory and immunomodulatory effects
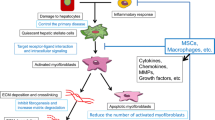
In addition to replacing host hepatocytes, transplanted cells promote LR by remodelling the liver microenvironment. One of the important factors is the immunomodulatory effect of transplanted cells. Mesenchymal stem cells (MSCs) are often employed in the clinic due to their simplicity in isolation, growth, and preservation. MSCs were first isolated in bone marrow [97] and can also be obtained from umbilical cords and adipose tissue (Fig. 2). Nevertheless, MSCs vary in their characteristics depending on the origin of cells and tissues as well as culture conditions [98]. MSCs are in vitro progenitor cells that exhibit plastic adhesion capabilities and a fibroblast-like appearance. MSCs express specific cell surface markers such as CD105, CD73, and CD90 [99]. Studies on MSCs have shown that they can effectively improve immune response [100]. MSCs were discovered in one investigation to increase CCL18, which in turn promoted monocyte survival [101]. Furthermore, by interacting with dendritic cells and decreasing their expression of inflammatory molecules such as IL12, TNF-α, and IFN-γ while increasing their secretion of IL-10 (Fig. 2), MSCs can differentiate antigen-presenting cells towards monocytes, which may lead to an increase in the number of regulatory T cells [102]. Furthermore, MSCs are able to suppress the proliferation and pro-inflammatory cytokine secretion of immune cells through the secretion of anti-inflammatory factors. MSCs improve the anti-inflammatory effects of macrophages by secreting prostaglandin E2 (PGE2), stimulated gene/protein 6, and indoleamine 2,3-dioxygenase (IDO) [103]. Pro-inflammatory cytokines, such as IL-1β and IL-6, were also inhibited by MSCs [104]. Moreover, MSCs inhibit antibody production and secretion and the proliferation of activated B lymphocytes in an IDO-dependent manner [105, 106]. In summary, the anti-inflammatory and immunomodulatory effects of MSCs can be exploited to promote liver regeneration.

Mesenchymal stem cell and macrophage reduce ECM deposition in the liver. MSCs, which are acquired from bone marrow, umbilical cord, and adipose tissue, can reduce ECM deposition by promoting the polariton of macrophages from M1 to M2 and inhibiting HSCs. Macrophage M1 secretes IL-12, PDGF, and TGF-β to exacerbate ECM deposition. In contrast, macrophages M2 secrete TNF-α, IL-6, and IL-10 to accelerate ECM degradation. Meanwhile, damaged hepatocytes and cholangiocytes secrete DAMP and ROS to activate Kupffer cells in the hepatic blood sinusoids. Activated Kupffer cells could recruit Ly-6Chi monocytes by secreting IL-1, TNF-α, CCL2, and CCL5 and activate HSCs by secreting TGF-β. In contrast, Ly-6Chi monocytes recruit CXCR6+ natural killer T cells to damage hepatocytes. Meanwhile, Ly-6Chi monocytes further differentiate into Ly-6Clo monocytes, which can accelerate ECM degradation by secreting MMP-9, MMP-12, MMP-13, IL-10, IGF-1, VEGF-α, and CSF-1
Liver sinusoidal endothelial cells (LSECs) constitute the vascular wall of the hepatic sinusoids and are considered key regulators of LR. Loss of LSECs exacerbates inflammation in liver disease [107]. Fang et al. found that LESCs could improve NASH by alleviating inflammation [108]. The underlying cause may be a decrease in NO secretion. LSEC-derived NO can effectively play an immunomodulatory role in the liver. Targeting LSECs for treatment has been shown to be effective [109]. Therefore, it may be a good choice to use LSECs to remodel the liver microenvironment in liver disease.
Anti-fibrosis
Fibrosis is one of the main manifestations of end-stage liver disease. MSCs and macrophages can effectively alleviate liver fibrosis and ECM deposition. The primary functions of MSCs are to release numerous trophic factors to suppress hepatic stellate cell (HSC) activation and ECM formation during LR, thus decreasing fibrosis, apoptosis, and inflammation via immunomodulation of T cells, B cells, and macrophages (Fig. 2). Liver disease and fibrosis are closely connected processes. In liver fibrosis and fibrosis regression, macrophages are crucial [110, 111]. Furthermore, macrophages secreting pro-fibrotic factors, including TGF-β and platelet-derived growth factor (PDGF) (Fig. 2), may worsen liver fibrosis when in the M1 state [110,111,112]. However, the ability of MSCs to polarize macrophages to the M2 state increases the possibility that they may alter the cytokine profile of activated macrophages biologically and reduce fibrosis [113, 114]. The antifibrotic ability of MSCs was further validated by coculture with HSCs in vitro and an animal model of liver fibrosis. Wang et al. reported that when MSCs and HSCs were cocultured, the expression of liver fibrosis markers was considerably decreased [115]. By boosting caspase3/7 activity (Fig. 2), another work by Lin et al. showed that MSCs may prevent the proliferation of activated HSCs and promote their death [116]. Certain clinical trials have confirmed the therapeutic benefits of MSC transplantation in patients with chronic liver disease [117,118,119]. According to a randomized controlled trial from Lin et al., peripheral infusion of allogeneic bone marrow-derived MSCs was safe and practical for patients with HBV-associated ACLF. By enhancing liver function and lowering the risk of serious infections, this treatment significantly increased 24-week survival [119]. Additionally, it has been shown that MSCs administered by hepatic arterial infusion considerably decreased liver fibrosis in patients and resulted in a notable improvement in Child–Pugh scores [117]. Nevertheless, relative to one transplant, liver fibrosis did not deteriorate after two MSC transplants. For these reasons, MSCs are considered a potential cell source for cell therapy.
Hepatic macrophages are one of the critical cells in the pathophysiology of chronic liver damage and have recently been highlighted as possible antifibrosis targets [120]. Macrophages undergo substantial phenotypic and functional changes after tissue damage and are essential for the initiation, maintenance, and resolution phases of tissue recovery. By changing their phenotype in response to cues from the liver microenvironment, hepatic macrophages may perform several tasks [121]. The typical categorization of macrophages as either "pro-inflammatory" type M1 or "pro-repair" type M2 does not accurately represent how they operate [122]. According to rodent liver damage models, circulating monocytes are mainly categorized as Ly-6C low (Ly-6Clo) monocytes and Ly-6C high (Ly-6Chi) monocytes (Fig. 2) [123]. The former display patrolling behaviour and express more clearance receptors in the liver, while the latter express "inflammatory" chemokine receptors such as CCR2, pattern recognition receptors, and cytokines [124,125,126,127,128]. The origin of hepatic macrophage subpopulations has a significant impact on how well they function in liver disease [123]. Ly-6Chi monocytes are mainly derived from bone marrow [129], and Ly-6Clo monocytes are mainly derived from the spleen [130]. Kupffer cells (KCs), mainly located in the blood sinusoids of the liver, act as the body's first line of defence against pathogens by effectively identifying and eliminating blood-borne germs, particularly gram-positive germs [131].
Kupffer cells (KCs) are rapidly lost during the damage period after liver injury [132, 133]. Reactive oxygen species (ROS) and damage-associated molecular patterns (DAMPs), such as high mobility group Box 1 (HMGB1), mitochondrial DNA (mtDNA), and ATP, are released by injured hepatocytes or cholangiocytes, and these molecules activate resident KCs on the luminal side of the hepatic sinusoidal endothelium (Fig. 2). After that, KCs quickly release pro-inflammatory cytokines and chemokines such as IL-1β, TNF-α, CCL2, and CCL5, which stimulate hepatocytes to secrete protective or apoptotic signalling pathways and recruit Ly-6Chi monocytes. Ly-6Chi monocytes then boost liver damage by recruiting CXCR6+ natural killer T (NKT) cells (Fig. 2) [134, 135]. However, there is evidence that KCs may activate HSCs through a paracrine mechanism and encourage their transdifferentiation into myofibroblasts, worsening liver fibrosis [136, 137]. Macrophages, as one of the primary producers of matrix metalloproteinases (MMPs), may breakdown various ECM proteins, but specific matrix metalloproteinases can also accelerate liver fibrosis [138]. The antifibrotic, pro-ECM degradation and pro-wound healing functions of macrophages are also regulated. For instance, MMPs released by macrophages may accelerate fibrosis regression. Homologous mature macrophages injected into a CCl4-treated animal model raised the levels of the antifibrotic cytokine IL-10 and the regional growth factors IGF-1, VEGF, and CSF-1 by delivering MMP-13 and MMP-9 to the liver scar to prevent the progression of liver scars [139]. In a mouse model of ALF, Kupffer cells recruit Ly-6Chi monocytes to the damaged area and display pro-inflammatory (TNF-α, IL-1, IL-6, CCL2, and CCL5) signals. Subsequently, they directly activate HSCs in a TGF-β-dependent manner while displaying pro-fibrotic (IL-13) phenotypes [137, 140,141,142,143]. Ly-6Clo monocytes, on the other hand, have antifibrotic properties (Fig. 2) [138]. In a mouse model of liver fibrosis, CCL2 inhibitors prevented Ly-6Chi monocytes from entering the liver, thus indirectly increasing Ly-6Clo monocytes, and liver fibrosis was shown to significantly decrease [139]. In another study, the conversion of Ly-6Chi to Ly-6Clo by injecting liposomes sped up liver fibrosis recovery [143]. These studies show that while distinct phenotypes of macrophages may carry out opposing tasks, they mostly contribute to fibrosis regression in the case of liver fibrosis (Fig. 2).
Given the critical role of recruited monocytes, specifically Ly-6Clo, in promoting the regression of liver fibrosis, the use of exogenously differentiated macrophages in vitro for treating liver illness may be a promising strategy. By injecting bone marrow-derived macrophages (BMDMs) into a mouse model of CCL4-induced liver fibrosis, researchers were able to drastically lower ECM deposition, the number of myofibroblasts (activated HSCs), and MMP-9 levels [139]. Nevertheless, individuals with liver disease, portal hypertension, and soft coagulation may not be candidates for infusion. According to later research, the delivery of primary human monocyte-derived macrophages (MDM) into the spleen of mice with liver fibrosis also has an antifibrotic ability [144]. Qin et al. discovered that the infusion of active macrophages into animals with liver fibrosis via the tail vein could efficiently breakdown collagen in the liver [145]. Recently, patients with liver cirrhosis have been investigated for response to autologous macrophage infusion treatment. Forbes et al. administered autologous macrophage treatment to nine persons with cirrhosis and an MELD score of 10 to 16 (ISRCTN 10,368,050). Each group of three subjects received a single peripheral infusion of cells at 107, 108, or up to 109 [146, 147]. Within a year, all individuals were still alive and transplant-free, meeting the study's primary goal of safety and viability. This work provides a theoretical foundation for cell treatment for cirrhosis and other fibrotic disorders.
In conclusion, MSCs and macrophages are critical to the regeneration of the liver and may serve as a source of cell therapy.
Tissue repair and regeneration
Another function of cell therapy is to promote tissue repair and regeneration. Haematopoietic progenitor cells play a major role in this process. Bone marrow (BM) is an alternate source of hepatocytes in the context of liver damage [148,149,150,151]. Previous research has shown that BM can generate a range of adult stem cells that express biomarkers for non-haematopoietic progenitor cells [152,153,154]. Although they may produce hepatocytes under tissue stress, BM cells have little impact on parenchymal replenishment in liver damage [27, 155,156,157,158]. The current hypothesis holds that the therapeutic improvement after haematopoietic progenitor cell therapy for damaged liver is primarily mediated by stimulating endogenous progenitor cells through paracrine signalling between the donor and host cells, which provides cytokines and growth factors [158,159,160]. Studies have shown that following partial hepatectomy in mice and humans, haematopoietic progenitor cells may reduce IL-1-mediated inflammation and boost liver regeneration in a CD39-dependent manner [161]. CD34 and CD133 are frequent markers for collecting haematopoietic progenitor cells [162, 163]. However, human CD34 cells have been shown to have little activity after implantation [164]. According to follow-up research, this may be connected to the activation-dependent expression of CD34, which may have clone-forming properties in multilineage progenitor cells [165, 166]. The metabolic marker aldehyde dehydrogenase (ALDH), which is linked to increased stem cell activity in vivo [165, 166], is found in early immature cells. Currently, a popular strategy is to extract haematopoietic progenitor cells based on vigorous ALDH activity in conjunction with markers such as CD34 or CD133 [168, 169]. Granulocyte colony-stimulating factor (G-CSF), a haematopoietic growth factor, promotes the mobilization of haematopoietic progenitor cells to peripheral circulation [170]. In acute and chronic liver damage models, G-CSF-induced proliferation of haematopoietic progenitor cells has been demonstrated to aid liver regeneration [171, 172]. In recent clinical research, patients with alcoholic cirrhosis who had haematopoietic progenitor cell transplantation achieved long-lasting therapeutic results [172, 173]. E. Yannaki et al. demonstrated that G-CSF hastened recovery and enhanced survival in a model of acute liver damage. In a large randomized controlled clinical study, the combination of G-CSF with the infusion of autologous haematopoietic progenitor cells via the portal vein dramatically boosted survival and liver function (as measured by the Child–Pugh score and liver biochemistry) [174]. However, individuals with liver cirrhosis who received G-CSF or an infusion of G-CSF with autologous CD133 + cells did not have appreciable effectiveness [175]. The diversity of these clinical studies may result from various infusion protocols, infusion techniques, and doses.
LSECs can activate intracellular pathways, including the Notch1 signalling pathway, activation of the transcription factor KLF2, and expression of CD44, vascular cell adhesion molecule 1 (VCAM-1), c-myc and c-jun [176,177,178]. These molecules have been shown to play a crucial role in LR. Meanwhile, LSECs secrete HGF, Wnt2, NO, IL-6 and TNF-α to promote LR [179, 180]. HGF stimulates hepatocyte proliferation through c-Met [181]. LR failed in a mouse model in which HGF was specifically knocked out in LSECs [182]. Id1 knockout mice have reduced HGF and Wnt2 expression, but the injection of allogeneic LSECs can restore hepatic angiogenesis [180]. LESCs also recruit monocytes to stimulate LR. In CD11b knockout mice, cellular crosstalk between monocytes and LESCs was disrupted, resulting in decreased angiogenesis and survival [183]. In conclusion, LSECs promote liver regeneration by releasing angiocrine factors. Transplanting LSECs to promote LR is a promising therapeutic strategy.
Optimizing the operation of cell therapy
Cell delivery
The liver is highly vascularized; therefore, transplanted cells may be administered by several different pathways, of which the portal vein or hepatic artery are the most common (Fig. 3). The transplanted cells should be delivered to the hepatic sinusoids, where they need to subsequently integrate with the liver parenchyma [184]. The most frequent clinical technique is cell infusion via the portal vein. Three options are available for adults: an intrahepatic splenic vein tributary puncture, an intrahepatic portal vein tributary puncture, and an intrahepatic portal shunt via the jugular vein through the hepatic venous system [185,186,187]. For newborns, access is via umbilical vein cannulation. In older children, laparoscopic or minimum dissection surgery or an incisional method may be used to place a central venous cannula into the portal vein stent. Depending on the size of the cell, hepatocytes cross-sinusoidal veins, temporarily occluding the periportal vascular area, and normal blood flow is subsequently restored via vascular permeability [49, 188]. Nevertheless, all of these treatment options have a risk of bleeding, particularly for patients with liver illness who often have portal hypertension, collateral circulation formation, and coagulation disorders. The transplanted cells may also reach the pulmonary capillaries via the hepatic veins and produce thrombi there, causing pulmonary infarction. The high portal pressure in individuals with portal hypertension makes it difficult for cells to reach the hepatic sinusoids. Within 24 h, macrophages eliminated all remaining hepatocytes from the portal veins (Fig. 3). Therefore, there are two possible solutions for managing this issue: (1) continuous Doppler ultrasonography monitoring to ensure that the portal pressure does not exceed 12 mmHg [89] and (2) hepatic artery administration of transplanted cells (Fig. 3). In contrast to the portal vein approach, the hepatic artery is often employed as a conduit for internal radiation treatment and arterial chemoembolization and may serve as a more effective way to deliver cells [189]. Other research has suggested alternative strategies, such as intrasplenic cell infusion for cirrhosis patients [52] and peritoneal delivery methods for ALF patients [55, 190]. The advantage of these strategies is that they permit functional liver recovery while preventing hepatic immune rejection.

Optimization of hepatocyte transplantation. Hepatocyte transplantation through the hepatic artery or the spleen may result in better colonization. And hepatocytes may be rejected by macrophages after reaching the liver. Macrophages phagocytose donor hepatocytes and secrete TGF-β to induce senescence. In this case, macrophages can avoid phagocytosis by encasing the donor hepatocytes in a hydrogel. On the other hand, the proliferative advantage of donor hepatocytes can be enhanced by irradiating the liver or injecting vascular epithelial growth factors
Cell recovery and proliferation
The activity and proliferation capacity of donor cells after transplantation play a major role in the effectiveness of cell therapy. Hepatocyte preservation has always been a serious issue. Cryopreservation significantly harms hepatocytes, reducing ATP generation and downregulating integrin-β1 and E-cadherin [191,192,193]. Hepatocyte acquisition and preservation procedures have considerably improved in recent years, and the activity of rewarmed hepatocytes is comparable to that of fresh hepatocytes [193, 194]. The techniques developed at the University of Wisconsin in the late 1980s are still used in most modern preservation techniques for donor organs and suspended cells [193, 194]. Many laboratories are also beginning to accept newer hepatocyte preservation media, such as Hypothermosol-FRS (HTS-FRS) and Institut Georges Lopez 1 (IGL-1), due to their better quality and lower cost [197,198,199]. Meanwhile, hepatocyte activity and function have been improved using apoptosis inhibitors and cystathionine inhibitors [200, 201].
Through the blood sinusoids, the transplanted cells enter the liver and attach to the liver parenchyma. It takes 1–5 days for transplanted cells to bind to recipient hepatocytes, and a gap junction, as well as a bile duct network formed by the two types of cells, can be seen in rats [202, 203]. During this process, HSCs are activated, while the implanted hepatocytes express the genes of the host hepatocytes at their location and undergo proliferative activity [204, 205]. Compared to the spleen and peritoneum, the liver exhibits much higher levels of proliferation [206]. This implies that the liver is a better candidate for the transplant location. Another crucial element in the effectiveness of cell therapy is intentionally controlling how tightly transplanted cells adhere to the recipient’s liver. The growth of transplanted cells in rats may be aided by pretreatment techniques based on the production of liver damage [18]. These techniques include boosting the release of advantageous compounds such as vascular endothelial growth factor (VEGF) (Fig. 3) from HSCs and rupturing the endothelial barrier that separates the parenchyma and hepatic blood sinusoids [207,208,209,210]. VEGF may also be externally supplied. Additionally, by causing liver damage, irradiation of the LSECs and a portion of the liver lobe (Fig. 3) may also improve the competitive advantage of transplanted cell proliferation [43, 211]. Immune rejection issues are avoided, and the effectiveness of transplantation is increased by coating the transplanted cells with hydrogel in material rich in growth factors and matrix proteins (Fig. 3) [212].
Conclusions and perspectives
As the demand for liver transplantation grows worldwide, more research is required to bridge the gap between donor livers and waiting list patients, as well as enhance the long-term prognosis of liver transplant recipients. Over the last several decades, cell therapy has significantly advanced, proving its effectiveness and safety. Before it can be utilized in clinical trials, a number of challenges must be resolved, including isolating high-quality cells from donor livers, enhancing cell implantation to reduce immune reactions, and improving engraftment and long-term outcomes. Additionally, stem cells or cells with stem cell qualities may be employed to address the challenges associated with hepatocyte acquisition. HLCs derived from iPSCs and organoids of various cell origins (iPSCs, progenitor cells, cholangiocytes) provide sufficient cell sources for cell therapy. Furthermore, iPSCs and organoids always have higher viability and efficiency in engraftment in vivo. Organoids with genetic stability and highly expandable properties could be a promising strategy for end-stage liver disease. Different cells in the liver have a role in replacing and remodelling the liver microenvironment; thus, combining various cell treatments may be the future tendency of cell therapy. The most significant advantage of cell therapy is a high degree of flexibility in selecting the best cells and transplantation sites for treatment catering to the patient’s needs.
Availability of data and materials
Not applicable.
Abbreviations
- NAFLD:
-
Non-alcoholic fatty liver disease
- NASH:
-
Non-alcoholic steatohepatitis
- OLT:
-
Orthotopic liver transplantation
- BEC:
-
Bile duct epithelial cell
- hBEC:
-
Human bile duct epithelial cell
- PSC:
-
Pluripotent stem cell
- MSC:
-
Mesenchymal stem cell
- LR:
-
Liver regeneration
- HSC:
-
Hepatic stellate cell
- ECM:
-
Extracellular matrix
- MMPs:
-
Matrix metalloproteinases
- IEM:
-
Inborn errors of metabolism
- HT:
-
Hepatocyte transplantation
- ALF:
-
Acute liver failure
- TGF-β:
-
Transforming growth factor-β
- ACLF:
-
Acute-on-chronic liver failure
- AD:
-
Acute decompensation
- FLP:
-
Foetal liver progenitor cell
- ESC:
-
Embryonic stem cell
- iPSC:
-
Induced pluripotent stem cell
- HLC:
-
Hepatocyte-like cell
- BMP:
-
Bone morphogenetic protein
- FGF:
-
Fibroblast growth factor
- uPA:
-
Urokinase-type plasminogen activator
- SNV:
-
Single nucleotide variants
- Ly-6Clo :
-
Ly-6C low
- Ly-6Chi :
-
Ly-6C high
- KC:
-
Kupffer cell
- ROS:
-
Reactive oxygen species
- DAMP:
-
Damage-associated molecular patterns
- HMGB1:
-
High mobility group box 1
- mtDNA:
-
Mitochondrial DNA
- LSEC:
-
Liver sinusoidal endothelial cell
- NKT:
-
Natural killer T
- BMDM:
-
Bone marrow-derived macrophage
- MDM:
-
Monocyte-derived macrophage
- BM:
-
Bone marrow
- ALDH:
-
Aldehyde dehydrogenase
- G-CSF:
-
Granulocyte colony-stimulating factor
- HTS-FRS:
-
Hypothermosol-FRS
- IGL-1:
-
Institut Georges Lopez 1
- VEGF:
-
Vascular endothelial growth factor
- PDGF:
-
Platelet-derived growth factor
- MDM:
-
Monocyte-derived macrophages
- VCAM-1:
-
Vascular cell adhesion molecule 1
- HGF:
-
Hepatocyte growth factor
- hBEC:
-
Human biliary epithelial cell
- lc-ProliHH:
-
Long-term proliferation human hepatocytes
- DAIF:
-
Dedifferentiation-associated inflammatory factors
- EGF:
-
Epidermal growth factor
- PGE2:
-
Prostaglandin E2
- IDO:
-
Indoleamine 2,3-dioxygenase
References
Wang J, Sun M, Liu W, Li Y, Li M. Stem cell-based therapies for liver diseases: an overview and update. Tissue Eng Regen Med. 2019;16(2):107–18.
Huang H, Guo S, Chen Y-Q, Liu Y-X, Jin J-Y, Liang Y, et al. Increased RTN3 phenocopies nonalcoholic fatty liver disease by inhibiting the AMPK-IDH2 pathway. MedCommun. 2023;4(2):e226.
Ling S, Jiang G, Que Q, Xu S, Chen J, Xu X. Liver transplantation in patients with liver failure: twenty years of experience from China. Liver Int. 2022;42(9):2110–6.
Dwyer BJ, Macmillan MT, Brennan PN, Forbes SJ. Cell therapy for advanced liver diseases: repair or rebuild. J Hepatol. 2021;74(1):185–99.
Wahid NA, Rosenblatt R, Brown RS. A review of the current state of liver transplantation disparities. Liver Transpl. 2021;27(3):434–43.
Zhou J, Chen J, Wei Q, Saeb-Parsy K, Xu X. The role of ischemia/reperfusion injury in early hepatic allograft dysfunction. Liver Transpl. 2020;26(8):1034–48.
Wang Z, Gao W, Dong C, Sun C, Wang K, Zhang W, et al. Outcome of split-liver transplantation from pediatric donors weighing 25 kg or less. Liver Transpl. 2022;5:417.
Zheng S-S, Yang Z, Wu Y-C. Liver transplantation for intrahepatic and perihilar cholangiocarcinoma: current and future. Hepatobiliary Pancreat Dis Int. 2020;19(2):101–2.
Okano H, Sipp D. New trends in cellular therapy. Development. 2020;147(18):471.
Zheng X, Zhou X, Ma G, Yu J, Zhang M, Yang C, et al. Endogenous Follistatin-like 1 guarantees the immunomodulatory properties of mesenchymal stem cells during liver fibrotic therapy. Stem Cell Res Ther. 2022;13(1):403.
Margiana R, Markov A, Zekiy AO, Hamza MU, Al-Dabbagh KA, Al-Zubaidi SH, et al. Clinical application of mesenchymal stem cell in regenerative medicine: a narrative review. Stem Cell Res Ther. 2022;13(1):366.
Bram Y, Nguyen D-HT, Gupta V, Park J, Richardson C, Chandar V, et al. Cell and tissue therapy for the treatment of chronic liver disease. Annu Rev Biomed Eng. 2021;23:517–46.
Tatsumi K, Okano T. Hepatocyte transplantation: cell sheet technology for liver cell transplantation. Curr Transpl Rep. 2017;4(3):184–92.
Li T-T, Wang Z-R, Yao W-Q, Linghu E-Q, Wang F-S, Shi L. Stem cell therapies for chronic liver diseases: progress and challenges. Stem Cells Transl Med. 2022;11(9):900–11.
Strom SC, Bruzzone P, Cai H, Ellis E, Lehmann T, Mitamura K, et al. Hepatocyte transplantation: clinical experience and potential for future use. Cell Transpl. 2006;15(Suppl 1):S105–10.
Hansel MC, Gramignoli R, Skvorak KJ, Dorko K, Marongiu F, Blake W, et al. The history and use of human hepatocytes for the treatment of liver diseases: the first 100 patients. Curr Protoc Toxicol. 2014;62:14–1223.
Forbes SJ, Gupta S, Dhawan A. Cell therapy for liver disease: from liver transplantation to cell factory. J Hepatol. 2015;62(1 Suppl):S157–69.
Shafritz DA, Oertel M. Model systems and experimental conditions that lead to effective repopulation of the liver by transplanted cells. Int J Biochem Cell Biol. 2011;43(2):198–213.
Karnezis AN, Dorokhov M, Grompe M, Zhu L. Loss of p27(Kip1) enhances the transplantation efficiency of hepatocytes transferred into diseased livers. J Clin Invest. 2001;108(3):383–90.
Pu W, Zhou B. Hepatocyte generation in liver homeostasis, repair, and regeneration. Cell Regen. 2022;11(1):2.
Deng X, Zhang X, Li W, Feng R-X, Li L, Yi G-R, et al. Chronic liver injury induces conversion of biliary epithelial cells into hepatocytes. Cell Stem Cell. 2018;23(1):4587.
Raven A, Lu W-Y, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature. 2017;547(7663):350–4.
Sampaziotis F, Muraro D, Tysoe OC, Sawiak S, Beach TE, Godfrey EM, et al. Cholangiocyte organoids can repair bile ducts after transplantation in the human liver. Science. 2021;371(6531):839–46.
Hallett JM, Ferreira-Gonzalez S, Man TY, Kilpatrick AM, Esser H, Thirlwell K, et al. Human biliary epithelial cells from discarded donor livers rescue bile duct structure and function in a mouse model of biliary disease. Cell Stem Cell. 2022;29(3):589.
Tsuchiya A, Takeuchi S, Watanabe T, Yoshida T, Nojiri S, Ogawa M, et al. Mesenchymal stem cell therapies for liver cirrhosis: MSCs as “conducting cells” for improvement of liver fibrosis and regeneration. Inflamm Regen. 2019;39:18.
Alfaifi M, Eom YW, Newsome PN, Baik SK. Mesenchymal stromal cell therapy for liver diseases. J Hepatol. 2018;68(6):1272–85.
Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, et al. Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature. 2003;422(6934):897–901.
Vassilopoulos G, Wang P-R, Russell DW. Transplanted bone marrow regenerates liver by cell fusion. Nature. 2003;422(6934):901–4.
Li W, Li L, Hui L. Cell plasticity in liver regeneration. Trends Cell Biol. 2020;30(4):329–38.
Li B, Wang X, Wang Y, Gou W, Yuan X, Peng J, et al. Past, present, and future of microcarrier-based tissue engineering. J Orthop Translat. 2015;3(2):51–7.
Dhawan A, Chaijitraruch N, Fitzpatrick E, Bansal S, Filippi C, Lehec SC, et al. Alginate microencapsulated human hepatocytes for the treatment of acute liver failure in children. J Hepatol. 2020;72(5):877–84.
Merrell AJ, Peng T, Li J, Sun K, Li B, Katsuda T, et al. Dynamic transcriptional and epigenetic changes drive cellular plasticity in the liver. Hepatology. 2021;74(1):444–57.
Ding J, Yannam GR, Roy-Chowdhury N, Hidvegi T, Basma H, Rennard SI, et al. Spontaneous hepatic repopulation in transgenic mice expressing mutant human α1-antitrypsin by wild-type donor hepatocytes. J Clin Invest. 2011;121(5):1930–4.
Liu Y, Maya S, Ploss A. Animal models of hepatitis B virus infection-success, challenges, and future directions. Viruses. 2021;13(5):4589.
Azuma H, Paulk N, Ranade A, Dorrell C, Al-Dhalimy M, Ellis E, et al. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat Biotechnol. 2007;25(8):903–10.
Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, et al. Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med. 1998;338(20):1422–6.
Dhawan A, Puppi J, Hughes RD, Mitry RR. Human hepatocyte transplantation: current experience and future challenges. Nat Rev Gastroenterol Hepatol. 2010;7(5):288–98.
Soltys KA, Soto-Gutiérrez A, Nagaya M, Baskin KM, Deutsch M, Ito R, et al. Barriers to the successful treatment of liver disease by hepatocyte transplantation. J Hepatol. 2010;53(4):769–74.
Enosawa S, Horikawa R, Yamamoto A, Sakamoto S, Shigeta T, Nosaka S, et al. Hepatocyte transplantation using a living donor reduced graft in a baby with ornithine transcarbamylase deficiency: a novel source of hepatocytes. Liver Transpl. 2014;20(3):391–3.
Alwahsh SM, Rashidi H, Hay DC. Liver cell therapy: is this the end of the beginning? Cell Mol Life Sci. 2018;75(8):1307–24.
Esrefoglu M. Role of stem cells in repair of liver injury: experimental and clinical benefit of transferred stem cells on liver failure. World J Gastroenterol. 2013;19(40):6757–73.
Stéphenne X, Debray FG, Smets F, Jazouli N, Sana G, Tondreau T, et al. Hepatocyte transplantation using the domino concept in a child with tetrabiopterin nonresponsive phenylketonuria. Cell Transpl. 2012;21(12):2765–70.
Soltys KA, Setoyama K, Tafaleng EN, Soto Gutiérrez A, Fong J, Fukumitsu K, et al. Host conditioning and rejection monitoring in hepatocyte transplantation in humans. J Hepatol. 2017;66(5):890.
Celik N, Squires JE, Soltys K, Vockley J, Shellmer DA, Chang W, et al. Domino liver transplantation for select metabolic disorders: expanding the living donor pool. JIMD Rep. 2019;48(1):83–9.
Peng WC, Kraaier LJ, Kluiver TA. Hepatocyte organoids and cell transplantation: what the future holds. Exp Mol Med. 2021;53(10):1512–28.
Kolodziejczyk AA, Federici S, Zmora N, Mohapatra G, Dori-Bachash M, Hornstein S, et al. Acute liver failure is regulated by MYC- and microbiome-dependent programs. Nat Med. 2020;26(12):1899–911.
Brennan PN, Donnelly MC, Simpson KJ. Systematic review: non A-E, seronegative or indeterminate hepatitis; what is this deadly disease? Aliment Pharmacol Ther. 2018;47(8):1079–91.
Patel P, Okoronkwo N, Pyrsopoulos NT. Future approaches and therapeutic modalities for acute liver failure. Clin Liver Dis. 2018;22(2):419–27.
Nguyen MP, Jain V, Iansante V, Mitry RR, Filippi C, Dhawan A. Clinical application of hepatocyte transplantation: current status, applicability, limitations, and future outlook. Expert Rev Gastroenterol Hepatol. 2020;14(3):185–96.
Liu J, Yuan Z, Wang Q. Pluripotent stem cell-derived strategies to treat acute liver failure: current status and future directions. J Clin Transl Hepatol. 2022;10(4):692–9.
Kawai T, Ito M, Hayashi C, Yamamoto N, Asano Y, Arakawa S, et al. Novel strategy for hepatocyte transplantation using resected organ with hepatocellular carcinoma or cholangiocarcinoma after hepatectomy. Fujita Med J. 2020;6(1):790.
Zimmerer JM, Ringwald BA, Chaudhari SR, Han J, Peterson CM, Warren RT, et al. Invariant NKT cells promote the development of highly cytotoxic multipotent CXCR3CCR4CD8 T cells that mediate rapid hepatocyte allograft rejection. J Immunol. 2021;207(12):3107–21.
Bird TG, Müller M, Boulter L, Vincent DF, Ridgway RA, Lopez-Guadamillas E, et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med. 2018;10(454):2145.
Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;127(10):3770–83.
Nicolas CT, Kaiser RA, Hickey RD, Allen KL, Du Z, VanLith CJ, et al. Cell therapy by ectopic hepatocyte transplantation treats the porcine tyrosinemia model of acute liver failure. Mol Ther Methods Clin Dev. 2020;18:738–50.
Bilir BM, Guinette D, Karrer F, Kumpe DA, Krysl J, Stephens J, et al. Hepatocyte transplantation in acute liver failure. Liver Transpl. 2000;6(1):32–40.
Cardinale V, Carpino G, Overi D, Safarikia S, Zhang W, Kanke M, et al. Human duodenal submucosal glands contain a defined stem/progenitor subpopulation with liver-specific regenerative potential. J Hepatol. 2022;5:2147.
Arroyo V, Jalan R. Acute-on-chronic liver failure: definition, diagnosis, and clinical characteristics. Semin Liver Dis. 2016;36(2):109–16.
Chen P, Wang Y-Y, Chen C, Guan J, Zhu H-H, Chen Z. The immunological roles in acute-on-chronic liver failure: an update. Hepatobiliary Pancreat Dis Int. 2019;18(5):403–11.
Arroyo V, Moreau R, Kamath PS, Jalan R, Ginès P, Nevens F, et al. Acute-on-chronic liver failure in cirrhosis. Nat Rev Dis Primers. 2016;2:16041.
Wang F, Zhou L, Ma X, Ma W, Wang C, Lu Y, et al. Monitoring of intrasplenic hepatocyte transplantation for acute-on-chronic liver failure: a prospective five-year follow-up study. Transpl Proc. 2014;46(1):192–8.
Wang C, Zhang L, Sun Z, Yuan X, Wu B, Cen J, et al. Dedifferentiation-associated inflammatory factors of long-term expanded human hepatocytes exacerbate their elimination by macrophages during liver engraftment. Hepatology. 2022;76(6):1690–705.
Guo R, Jiang M, Wang G, Li B, Jia X, Ai Y, et al. IL6 supports long-term expansion of hepatocytes in vitro. Nat Commun. 2022;13(1):7345.
Jalan-Sakrikar N, Brevini T, Huebert RC, Sampaziotis F. Organoids and regenerative hepatology. Hepatology. 2023;77(1):305–22.
Mahieu-Caputo D, Allain J-E, Branger J, Coulomb A, Delgado J-P, Andreoletti M, et al. Repopulation of athymic mouse liver by cryopreserved early human fetal hepatoblasts. Hum Gene Ther. 2004;15(12):1219–28.
Weber A, Delgado J-P, Parouchev A, Branger J, Mainot S, Coulomb A, et al. Primate hepatic foetal progenitor cells and their therapeutic potential. Pathol Biol (Paris). 2006;54(2):58–63.
Irudayaswamy A, Muthiah M, Zhou L, Hung H, Jumat NHB, Haque J, et al. Long-term fate of human fetal liver progenitor cells transplanted in injured mouse livers. Stem Cells. 2018;36(1):103–13.
Hu H, Gehart H, Artegiani B, Löpez-Iglesias C, Dekkers F, Basak O, et al. Long-term expansion of functional mouse and human hepatocytes as 3D organoids. Cell. 2018;175(6):478.
Haridass D, Yuan Q, Becker PD, Cantz T, Iken M, Rothe M, et al. Repopulation efficiencies of adult hepatocytes, fetal liver progenitor cells, and embryonic stem cell-derived hepatic cells in albumin-promoter-enhancer urokinase-type plasminogen activator mice. Am J Pathol. 2009;175(4):1483–92.
Luce E, Messina A, Duclos-Vallée J-C, Dubart-Kupperschmitt A. Advanced techniques and awaited clinical applications for human pluripotent stem cell differentiation into hepatocytes. Hepatology. 2021;74(2):1101–16.
Hay DC, Zhao D, Fletcher J, Hewitt ZA, McLean D, Urruticoechea-Uriguen A, et al. Efficient differentiation of hepatocytes from human embryonic stem cells exhibiting markers recapitulating liver development in vivo. Stem Cells. 2008;26(4):894–902.
Yuan L, Zhang Y, Liu X, Chen Y, Zhang L, Cao J, et al. Agonist c-met monoclonal antibody augments the proliferation of hiPSC-derived hepatocyte-like cells and improves cell transplantation therapy for liver failure in mice. Theranostics. 2019;9(7):2115–28.
Guo J, Duan L, He X, Li S, Wu Y, Xiang G, et al. A combined model of human iPSC-derived liver organoids and hepatocytes reveals ferroptosis in DGUOK mutant mtDNA depletion syndrome. Adv Sci (Weinh). 2021;8(10):2004680.
Olgasi C, Cucci A, Follenzi A. iPSC-derived liver organoids: a journey from drug screening, to disease modeling, arriving to regenerative medicine. Int J Mol Sci. 2020;21(17):247.
Wang S, Wang X, Tan Z, Su Y, Liu J, Chang M, et al. Human ESC-derived expandable hepatic organoids enable therapeutic liver repopulation and pathophysiological modeling of alcoholic liver injury. Cell Res. 2019;29(12):1009–26.
Hay DC, Fletcher J, Payne C, Terrace JD, Gallagher RCJ, Snoeys J, et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci U S A. 2008;105(34):12301–6.
Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26(5):1117–27.
Baxter M, Withey S, Harrison S, Segeritz C-P, Zhang F, Atkinson-Dell R, et al. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J Hepatol. 2015;62(3):581–9.
Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, et al. Human embryonic stem cells derived by somatic cell nuclear transfer. Cell. 2013;153(6):1228–38.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Dianat N, Steichen C, Vallier L, Weber A, Dubart-Kupperschmitt A. Human pluripotent stem cells for modelling human liver diseases and cell therapy. Curr Gene Ther. 2013;13(2):120–32.
Seki T, Yuasa S, Fukuda K. Derivation of induced pluripotent stem cells from human peripheral circulating T cells. Curr Protoc Stem Cell Biol. 2011;4:3.
Geti I, Ormiston ML, Rouhani F, Toshner M, Movassagh M, Nichols J, et al. A practical and efficient cellular substrate for the generation of induced pluripotent stem cells from adults: blood-derived endothelial progenitor cells. Stem Cells Transl Med. 2012;1(12):855–65.
Yan X, Qin H, Qu C, Tuan RS, Shi S, Huang GTJ. iPS cells reprogrammed from human mesenchymal-like stem/progenitor cells of dental tissue origin. Stem Cells Dev. 2010;19(4):469–80.
Hannoun Z, Steichen C, Dianat N, Weber A, Dubart-Kupperschmitt A. The potential of induced pluripotent stem cell derived hepatocytes. J Hepatol. 2016;65(1):182–99.
Asgari S, Moslem M, Bagheri-Lankarani K, Pournasr B, Miryounesi M, Baharvand H. Differentiation and transplantation of human induced pluripotent stem cell-derived hepatocyte-like cells. Stem Cell Rev Rep. 2013;9(4):493–504.
Carpentier A, Tesfaye A, Chu V, Nimgaonkar I, Zhang F, Lee SB, et al. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J Clin Invest. 2014;124(11):4953–64.
Morizane A, Kikuchi T, Hayashi T, Mizuma H, Takara S, Doi H, et al. MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nat Commun. 2017;8(1):385.
Dhawan A. Clinical human hepatocyte transplantation: current status and challenges. Liver Transpl. 2015;21(Suppl 1):S39–44.
Ji J, Sharma V, Qi S, Guarch ME, Zhao P, Luo Z, et al. Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Rep. 2014;2(1):44–51.
Kondo Y, Iwao T, Nakamura K, Sasaki T, Takahashi S, Kamada N, et al. An efficient method for differentiation of human induced pluripotent stem cells into hepatocyte-like cells retaining drug metabolizing activity. Drug Metab Pharmacokinet. 2014;29(3):237–43.
Rossi G, Manfrin A, Lutolf MP. Progress and potential in organoid research. Nat Rev Genet. 2018;19(11):671–87.
Huch M, Dorrell C, Boj SF, van Es JH, Li VSW, van de Wetering M, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature. 2013;494(7436):247–50.
Takebe T, Zhang R-R, Koike H, Kimura M, Yoshizawa E, Enomura M, et al. Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc. 2014;9(2):396–409.
Choi J, Shin E, Lee JDS, Kim D, Shin JH, et al. Light-stimulated insulin secretion from pancreatic islet-like organoids derived from human pluripotent stem cells. Mol Ther. 2023;5:489.
Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499(7459):481–4.
Friedenstein AJ, Chailakhjan RK, Lalykina KS. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell Tissue Kinet. 1970;3(4):393–403.
Han H-T, Jin W-L, Li X. Mesenchymal stem cells-based therapy in liver diseases. Mol Biomed. 2022;3(1):23.
Hu C, Wu Z, Li L. Mesenchymal stromal cells promote liver regeneration through regulation of immune cells. Int J Biol Sci. 2020;16(5):893–903.
Wang M-Y, Zhou T-Y, Zhang Z-D, Liu H-Y, Zheng Z-Y, Xie H-Q. Current therapeutic strategies for respiratory diseases using mesenchymal stem cells. MedComm. 2021;2(3):351–80.
Hu C, Zhao L, Zhang L, Bao Q, Li L. Mesenchymal stem cell-based cell-free strategies: safe and effective treatments for liver injury. Stem Cell Res Ther. 2020;11(1):377.
Takeuchi S, Tsuchiya A, Iwasawa T, Nojiri S, Watanabe T, Ogawa M, et al. Small extracellular vesicles derived from interferon-γ pre-conditioned mesenchymal stromal cells effectively treat liver fibrosis. NPJ Regen Med. 2021;6(1):19.
Yang X, Li Q, Liu W, Zong C, Wei L, Shi Y, et al. Mesenchymal stromal cells in hepatic fibrosis/cirrhosis: from pathogenesis to treatment. Cell Mol Immunol. 2023;5:448.
Cheng H, Huang H, Guo Z, Chang Y, Li Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics. 2021;11(18):8836–54.
Su W, Yu S, Yin Y, Li B, Xue J, Wang J, et al. Diabetic microenvironment preconditioning of adipose tissue-derived mesenchymal stem cells enhances their anti-diabetic, anti-long-term complications, and anti-inflammatory effects in type 2 diabetic rats. Stem Cell Res Ther. 2022;13(1):422.
Volarevic V, Arsenijevic N, Lukic ML, Stojkovic M. Concise review: mesenchymal stem cell treatment of the complications of diabetes mellitus. Stem Cells. 2011;29(1):5540.
Hammoutene A, Biquard L, Lasselin J, Kheloufi M, Tanguy M, Vion A-C, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72(3):528–38.
Fang Z-Q, Ruan B, Liu J-J, Duan J-L, Yue Z-S, Song P, et al. Notch-triggered maladaptation of liver sinusoidal endothelium aggravates nonalcoholic steatohepatitis through endothelial nitric oxide synthase. Hepatology. 2022;76(3):742–58.
Wang X, Walkey CJ, Maretti-Mira AC, Wang L, Johnson DL, DeLeve LD. Susceptibility of rat steatotic liver to ischemia-reperfusion is treatable with liver-selective matrix metalloproteinase inhibition. Hepatology. 2020;72(5):1771–85.
Nuciforo S, Heim MH. Organoids to model liver disease. JHEP Rep. 2021;3(1):100198.
Kim G, Huh JH, Lee KJ, Kim MY, Shim KY, Baik SK. Relative adrenal insufficiency in patients with cirrhosis: a systematic review and meta-analysis. Dig Dis Sci. 2017;62(4):1067–79.
El Agha E, Kramann R, Schneider RK, Li X, Seeger W, Humphreys BD, et al. Mesenchymal stem cells in fibrotic disease. Cell Stem Cell. 2017;21(2):166–77.
Nevens F, van der Merwe S. Mesenchymal stem cell transplantation in liver diseases. Semin Liver Dis. 2022;42(3):283–92.
Manka P, Zeller A, Syn W-K. Fibrosis in chronic liver disease: an update on diagnostic and treatment modalities. Drugs. 2019;79(9):903–27.
Wang P-p, Xie D-y, Liang X-J, Peng L, Zhang G-l, Ye Y-n, et al. HGF and direct mesenchymal stem cells contact synergize to inhibit hepatic stellate cells activation through TLR4/NF-kB pathway. PLoS One. 2012;7(8):e43408.
Lin N, Hu K, Chen S, Xie S, Tang Z, Lin J, et al. Nerve growth factor-mediated paracrine regulation of hepatic stellate cells by multipotent mesenchymal stromal cells. Life Sci. 2009;85(7–8):291–5.
Suk KT, Yoon J-H, Kim MY, Kim CW, Kim JK, Park H, et al. Transplantation with autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: phase 2 trial. Hepatology. 2016;64(6):2185–97.
Jang YO, Kim YJ, Baik SK, Kim MY, Eom YW, Cho MY, et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: a pilot study. Liver Int. 2014;34(1):33–41.
Lin B-L, Chen J-F, Qiu W-H, Wang K-W, Xie D-Y, Chen X-Y, et al. Allogeneic bone marrow-derived mesenchymal stromal cells for hepatitis B virus-related acute-on-chronic liver failure: a randomized controlled trial. Hepatology. 2017;66(1):209–19.
Schuppan D, Kim YO. Evolving therapies for liver fibrosis. J Clin Invest. 2013;123(5):1887–901.
Tacke F, Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60(5):1090–6.
Morris G, Gevezova M, Sarafian V, Maes M. Redox regulation of the immune response. Cell Mol Immunol. 2022;41:789.
Zimmermann HW, Trautwein C, Tacke F. Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol. 2012;3:56.
Flores-Costa R, Duran-Güell M, Casulleras M, López-Vicario C, Alcaraz-Quiles J, Diaz A, et al. Stimulation of soluble guanylate cyclase exerts antiinflammatory actions in the liver through a VASP/NF-κB/NLRP3 inflammasome circuit. Proc Natl Acad Sci U S A. 2020;117(45):28263–74.
Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016;64(5):1667–82.
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62(1):279–91.
David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. 2016;151(6):1176–91.
Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317(5838):666–70.
Shi K, Li H, Chang T, He W, Kong Y, Qi C, et al. Bone marrow hematopoiesis drives multiple sclerosis progression. Cell. 2022;185(13):748.
Lu W, Cao F, Feng L, Song G, Chang Y, Chu Y, et al. LncRNA Snhg6 regulates the differentiation of MDSCs by regulating the ubiquitination of EZH2. J Hematol Oncol. 2021;14(1):196.
Zeng Z, Surewaard BGJ, Wong CHY, Geoghegan JA, Jenne CN, Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe. 2016;20(1):418.
Borst K, Frenz T, Spanier J, Tegtmeyer P-K, Chhatbar C, Skerra J, et al. Type I interferon receptor signaling delays Kupffer cell replenishment during acute fulminant viral hepatitis. J Hepatol. 2018;68(4):682–90.
Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 2015;42(1):145–58.
Luedde T, Schwabe RF. NF-κB in the liver–linking injury, fibrosis and hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2011;8(2):108–18.
Wehr A, Baeck C, Heymann F, Niemietz PM, Hammerich L, Martin C, et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J Immunol. 2013;190(10):5226–36.
De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, et al. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology. 2007;132(5):1937–46.
Pradere J-P, Kluwe J, De Minicis S, Jiao J-J, Gwak G-Y, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–73.
Madala SK, Pesce JT, Ramalingam TR, Wilson MS, Minnicozzi S, Cheever AW, et al. Matrix metalloproteinase 12-deficiency augments extracellular matrix degrading metalloproteinases and attenuates IL-13-dependent fibrosis. J Immunol. 2010;184(7):3955–63.
Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53(6):2003–15.
Heymann F, Tacke F. Immunology in the liver–from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):214.
Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol. 2016;13(3):316–27.
Liaskou E, Zimmermann HW, Li K-K, Oo YH, Suresh S, Stamataki Z, et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology. 2013;57(1):385–98.
Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A. 2012;109(46):E3186–95.
Moore JK, Mackinnon AC, Wojtacha D, Pope C, Fraser AR, Burgoyne P, et al. Phenotypic and functional characterization of macrophages with therapeutic potential generated from human cirrhotic monocytes in a cohort study. Cytotherapy. 2015;17(11):1604–16.
Ma P-F, Gao C-C, Yi J, Zhao J-L, Liang S-Q, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatol. 2017;67(4):770–9.
Moroni F, Dwyer BJ, Graham C, Pass C, Bailey L, Ritchie L, et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat Med. 2019;25(10):1560–5.
Brennan PN, MacMillan M, Manship T, Moroni F, Glover A, Graham C, et al. Study protocol: a multicentre, open-label, parallel-group, phase 2, randomised controlled trial of autologous macrophage therapy for liver cirrhosis (MATCH). BMJ Open. 2021;11(11):e053190.
Siapati EK, Roubelakis MG, Vassilopoulos G. Liver regeneration by hematopoietic stem cells: have we reached the end of the road? Cells. 2022;11(15):514.
Nikokiraki C, Psaraki A, Roubelakis MG. The potential clinical use of stem/progenitor cells and organoids in liver diseases. Cells. 2022;11(9):745.
Tsolaki E, Athanasiou E, Gounari E, Zogas N, Siotou E, Yiangou M, et al. Hematopoietic stem cells and liver regeneration: differentially acting hematopoietic stem cell mobilization agents reverse induced chronic liver injury. Blood Cells Mol Dis. 2014;53(3):124–32.
Okabayashi T, Shima Y, Sumiyoshi T, Kozuki A, Iiyama T, Tokumaru T, et al. Extrahepatic stem cells mobilized from the bone marrow by the supplementation of branched-chain amino acids ameliorate liver regeneration in an animal model. J Gastroenterol Hepatol. 2014;29(4):870–7.
Araújo AB, Soares TB, Schmalfuss T, Angeli MH, Furlan JM, Salton GD, et al. Non-cryopreserved peripheral blood stem cells as a safe and effective alternative for autologous transplantation in multiple myeloma. Transfusion. 2022;2:558.
Lenaerts A, Kucinski I, Deboutte W, Derecka M, Cauchy P, Manke T, et al. EBF1 primes B-lymphoid enhancers and limits the myeloid bias in murine multipotent progenitors. J Exp Med. 2022;219(11):7489.
Perriot S, Canales M, Mathias A, Du Pasquier R. Generation of transgene-free human induced pluripotent stem cells from erythroblasts in feeder-free conditions. STAR Protoc. 2022;3(3):101620.
Fu X, He Q, Tao Y, Wang M, Wang W, Wang Y, et al. Recent advances in tissue stem cells. Sci China Life Sci. 2021;64(12):1998–2029.
Gao S, Shi Q, Zhang Y, Liang G, Kang Z, Huang B, et al. Identification of HSC/MPP expansion units in fetal liver by single-cell spatiotemporal transcriptomics. Cell Res. 2022;32(1):38–53.
Vig P, Russo FP, Edwards RJ, Tadrous PJ, Wright NA, Thomas HC, et al. The sources of parenchymal regeneration after chronic hepatocellular liver injury in mice. Hepatology. 2006;43(2):316–24.
Thorgeirsson SS, Grisham JW. Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology. 2006;43(1):2–8.
Chen Y, Pu Q, Ma Y, Zhang H, Ye T, Zhao C, et al. Aging reprograms the hematopoietic-vascular niche to impede regeneration and promote fibrosis. Cell Metab. 2021;33(2):214.
Shido K, Chavez D, Cao Z, Ko J, Rafii S, Ding B-S. Platelets prime hematopoietic and vascular niche to drive angiocrine-mediated liver regeneration. Signal Transduct Target Ther. 2017;2:2147.
Hirata Y, Furuhashi K, Ishii H, Li HW, Pinho S, Ding L, et al. CD150 bone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell. 2018;22(3):478.
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, et al. Successful transfer of umbilical cord blood CD34 hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res. 2017;23(15):4107–18.
Calvanese V, Capellera-Garcia S, Ma F, Fares I, Liebscher S, Ng ES, et al. Mapping human haematopoietic stem cells from haemogenic endothelium to birth. Nature. 2022;604(7906):534–40.
Yong KSM, Keng CT, Tan SQ, Loh E, Chang KT, Tan TC, et al. Human CD34(lo)CD133(lo) fetal liver cells support the expansion of human CD34(hi)CD133(hi) hematopoietic stem cells. Cell Mol Immunol. 2016;13(5):605–14.
Morcos MNF, Zerjatke T, Glauche I, Munz CM, Ge Y, Petzold A, et al. Continuous mitotic activity of primitive hematopoietic stem cells in adult mice. J Exp Med. 2020;217(6):2147.
Hess DA, Karanu FN, Levac K, Gallacher L, Bhatia M. Coculture and transplant of purified CD34(+)Lin(-) and CD34(-)Lin(-) cells reveals functional interaction between repopulating hematopoietic stem cells. Leukemia. 2003;17(8):1613–25.
Cooper TT, Sherman SE, Kuljanin M, Bell GI, Lajoie GA, Hess DA. Inhibition of aldehyde dehydrogenase-activity expands multipotent myeloid progenitor cells with vascular regenerative function. Stem Cells. 2018;36(5):723–36.
Hess DA, Meyerrose TE, Wirthlin L, Craft TP, Herrbrich PE, Creer MH, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood. 2004;104(6):1648–55.
Zimran E, Papa L, Djedaini M, Patel A, Iancu-Rubin C, Hoffman R. Expansion and preservation of the functional activity of adult hematopoietic stem cells cultured ex vivo with a histone deacetylase inhibitor. Stem Cells Transl Med. 2020;9(4):531–42.
Metcalf D. The molecular control of cell division, differentiation commitment and maturation in haemopoietic cells. Nature. 1989;339(6219):27–30.
Bhardwaj R, Kumar L, Chhabra D, Mehra NK, Sharma A, Mohanty S, et al. In vitro expansion of fetal liver hematopoietic stem cells. Sci Rep. 2021;11(1):11879.
Yannaki E, Athanasiou E, Xagorari A, Constantinou V, Batsis I, Kaloyannidis P, et al. G-CSF-primed hematopoietic stem cells or G-CSF per se accelerate recovery and improve survival after liver injury, predominantly by promoting endogenous repair programs. Exp Hematol. 2005;33(1):108–19.
Sharma M, Kulkarni A, Sasikala M, Kumar P, Jaggaiahgari S, Pondugala K, et al. Long-term outcome of autologous hematopoietic stem cell infusion in cirrhosis: waning effect over time. J Clin Transl Hepatol. 2020;8(4):385–90.
Salama H, Zekri A-RN, Bahnassy AA, Medhat E, Halim HA, Ahmed OS, et al. Autologous CD34+ and CD133+ stem cells transplantation in patients with end stage liver disease. World J Gastroenterol. 2010;16(42):5297–305.
Newsome PN, Fox R, King AL, Barton D, Than N-N, Moore J, et al. Granulocyte colony-stimulating factor and autologous CD133-positive stem-cell therapy in liver cirrhosis (REALISTIC): an open-label, randomised, controlled phase 2 trial. Lancet Gastroenterol Hepatol. 2018;3(1):25–36.
De Rudder M, Dili A, Stärkel P, Leclercq IA. Critical role of LSEC in post-hepatectomy liver regeneration and failure. Int J Mol Sci. 2021;22(15):247.
Guixé-Muntet S, de Mesquita FC, Vila S, Hernández-Gea V, Peralta C, García-Pagán JC, et al. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J Hepatol. 2017;66(1):86–94.
Díaz-Juárez JA, Hernández-Muñoz R. Rat liver enzyme release depends on blood flow-bearing physical forces acting in endothelium glycocalyx rather than on liver damage. Oxid Med Cell Longev. 2017;2017:1360565.
Michalopoulos GK, Bhushan B. Liver regeneration: biological and pathological mechanisms and implications. Nat Rev Gastroenterol Hepatol. 2021;18(1):40–55.
Ding B-S, Nolan DJ, Butler JM, James D, Babazadeh AO, Rosenwaks Z, et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature. 2010;468(7321):310–5.
Zhao Y, Ye W, Wang Y-D, Chen W-D. HGF/c-met: a key promoter in liver regeneration. Front Pharmacol. 2022;13:808855.
Zhang X-J, Olsavszky V, Yin Y, Wang B, Engleitner T, Öllinger R, et al. Angiocrine hepatocyte growth factor signaling controls physiological organ and body size and dynamic hepatocyte proliferation to prevent liver damage during regeneration. Am J Pathol. 2020;190(2):358–71.
Yap KK, Gerrand Y-W, Dingle AM, Yeoh GC, Morrison WA, Mitchell GM. Liver sinusoidal endothelial cells promote the differentiation and survival of mouse vascularised hepatobiliary organoids. Biomaterials. 2020;251:120091.
Demaret T, Evraerts J, Ravau J, Roumain M, Muccioli GG, Najimi M, et al. High dose versus low dose syngeneic hepatocyte transplantation in -G844D NMRI mouse model is safe but does not achieve long term engraftment. Cells. 2020;10(1):189.
Haddad MM, Fleming CJ, Thompson SM, Reisenauer CJ, Parvinian A, Frey G, et al. Comparison of bleeding complications between transplenic versus transhepatic access of the portal venous system. J Vasc Interv Radiol. 2018;29(10):1383–91.
Dissegna D, Sponza M, Falleti E, Fabris C, Vit A, Angeli P, et al. Morbidity and mortality after transjugular intrahepatic portosystemic shunt placement in patients with cirrhosis. Eur J Gastroenterol Hepatol. 2019;31(5):626–32.
Saad WEA, Madoff DC. Percutaneous portal vein access and transhepatic tract hemostasis. Semin Intervent Radiol. 2012;29(2):71–80.
Takano C, Grubbs BH, Ishige M, Ogawa E, Morioka I, Hayakawa S, et al. Clinical perspective on the use of human amniotic epithelial cells to treat congenital metabolic diseases with a focus on maple syrup urine disease. Stem Cells Transl Med. 2021;10(6):829–35.
Sun A, Gao W, Xiao T. Autologous bone marrow stem cell transplantation via the hepatic artery for the treatment of hepatitis B virus-related cirrhosis: a PRISMA-compliant meta-analysis based on the Chinese population. Stem Cell Res Ther. 2020;11(1):104.
Amaral RJFC, Arcanjo KD, El-Cheikh MC, de Oliveira FL. The peritoneum: health, disease, and perspectives regarding tissue engineering and cell therapies. Cells Tissues Organs. 2017;204:211–7.
Patrono D, Zanierato M, Avolio AW, Romagnoli R. Different timing of cholangiocyte and hepatocyte damage in liver preservation: time to implement donor interventions and new preservation techniques to prevent ischemic-type biliary lesions. Transplantation. 2021;105(7):e77–8.
Mun SJ, Ryu J-S, Lee M-O, Son YS, Oh SJ, Cho H-S, et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J Hepatol. 2019;71(5):970–85.
Matsumura M, Imura T, Inagaki A, Ogasawara H, Miyagi S, Ohashi K, et al. Use of perfluorohexyloctane for preservation of rat liver after circulatory death and a prolonged cold preservation model for hepatocyte transplantation. Transpl Int. 2022;35:10659.
Li AP. Human hepatocytes: isolation, cryopreservation and applications in drug development. Chem Biol Interact. 2007;168(1):16–29.
Gramignoli R, Dorko K, Tahan V, Skvorak KJ, Ellis E, Jorns C, et al. Hypothermic storage of human hepatocytes for transplantation. Cell Transplant. 2014;23(9):1143–51.
Tsutsui K, Monfredi OJ, Sirenko-Tagirova SG, Maltseva LA, Bychkov R, Kim MS, et al. A coupled-clock system drives the automaticity of human sinoatrial nodal pacemaker cells. Sci Signal. 2018;11(534):2104.
Ben Abdennebi H, Elrassi Z, Scoazec J-Y, Steghens J-P, Ramella-Virieux S, Boillot O. Evaluation of IGL-1 preservation solution using an orthotopic liver transplantation model. World J Gastroenterol. 2006;12(33):5326–30.
Dondéro F, Paugam-Burtz C, Danjou F, Stocco J, Durand F, Belghiti J. A randomized study comparing IGL-1 to the University of Wisconsin preservation solution in liver transplantation. Ann Transpl. 2010;15(4):2147.
Mathew AJ, Baust JM, Van Buskirk RG, Baust JG. Cell preservation in reparative and regenerative medicine: evolution of individualized solution composition. Tissue Eng. 2004;10(11–12):1662–71.
Ölander M, Wiśniewski JR, Flörkemeier I, Handin N, Urdzik J, Artursson P. A simple approach for restoration of differentiation and function in cryopreserved human hepatocytes. Arch Toxicol. 2019;93(3):819–29.
Jitraruch S, Dhawan A, Hughes RD, Filippi C, Lehec SC, Glover L, et al. Cryopreservation of hepatocyte microbeads for clinical transplantation. Cell Transpl. 2017;26(8):1341–54.
Barahman M, Zhang W, Harris HY, Aiyer A, Kabarriti R, Kinkhabwala M, et al. Radiation-primed hepatocyte transplantation in murine monogeneic dyslipidemia normalizes cholesterol and prevents atherosclerosis. J Hepatol. 2019;70(6):1170–9.
Deep A, Alexander EC, Bulut Y, Fitzpatrick E, Grazioli S, Heaton N, et al. Advances in medical management of acute liver failure in children: promoting native liver survival. Lancet Child Adolesc Health. 2022;6(10):725–37.
Ko S, Russell JO, Molina LM, Monga SP. Liver progenitors and adult cell plasticity in hepatic injury and repair: knowns and unknowns. Annu Rev Pathol. 2020;15:23–50.
Barahman M, Asp P, Roy-Chowdhury N, Kinkhabwala M, Roy-Chowdhury J, Kabarriti R, et al. Hepatocyte transplantation: quo vadis? Int J Radiat Oncol Biol Phys. 2019;103(4):922–34.
Hoppo T, Komori J, Manohar R, Stolz DB, Lagasse E. Rescue of lethal hepatic failure by hepatized lymph nodes in mice. Gastroenterology. 2011;140(2):24457.
Enami Y, Bandi S, Kapoor S, Krohn N, Joseph B, Gupta S. Hepatic stellate cells promote hepatocyte engraftment in rat liver after prostaglandin-endoperoxide synthase inhibition. Gastroenterology. 2009;136(7):2356–64.
Krohn N, Kapoor S, Enami Y, Follenzi A, Bandi S, Joseph B, et al. Hepatocyte transplantation-induced liver inflammation is driven by cytokines-chemokines associated with neutrophils and Kupffer cells. Gastroenterology. 2009;136(5):1806–17.
Bahde R, Kapoor S, Viswanathan P, Spiegel H-U, Gupta S. Endothelin-1 receptor A blocker darusentan decreases hepatic changes and improves liver repopulation after cell transplantation in rats. Hepatology. 2014;59(3):1107–17.
Bahde R, Kapoor S, Bandi S, Bhargava KK, Palestro CJ, Gupta S. Directly acting drugs prostacyclin or nitroglycerine and endothelin receptor blocker bosentan improve cell engraftment in rodent liver. Hepatology. 2013;57(1):320–30.
Yamanouchi K, Zhou H, Roy-Chowdhury N, Macaluso F, Liu L, Yamamoto T, et al. Hepatic irradiation augments engraftment of donor cells following hepatocyte transplantation. Hepatology. 2009;49(1):258–67.
Nevi L, Carpino G, Costantini D, Cardinale V, Riccioni O, Di Matteo S, et al. Hyaluronan coating improves liver engraftment of transplanted human biliary tree stem/progenitor cells. Stem Cell Res Ther. 2017;8(1):68.
Acknowledgements
All the figures were created with BioRender.com.
Funding
This research was funded by the National Key Research and Development Program of China (No. 2021YFA1100500), the Key Program, National Natural Science Foundation of China (No. 81930016) and the Key Research & Development Plan of Zhejiang Province (No. 2021C03118). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed to conceptualization; X.X. and J.C. contributed to resources; X.H.H. and L.C. were involved in writing—original draft preparation; X.H.H. and Q.M.Z. assisted in writing—review and editing; Y.B.T. and H.W. contributed to the visualization; X.Y.W. helped in the supervision; X.H.H. and Q.H. were involved in the project administration; X.X. and Q.W. acquired the funding. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval and Consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing financial interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, XH., Chen, L., Wu, H. et al. Cell therapy in end-stage liver disease: replace and remodel. Stem Cell Res Ther 14, 141 (2023). https://doi.org/10.1186/s13287-023-03370-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-023-03370-z