
Abstract
Background
Mesenchymal stem cell (MSC) therapy has been shown to be a promising option for liver fibrosis treatment. However, critical factors affecting the efficacy of MSC therapy for liver fibrosis remain unknown. Follistatin-like 1 (FSTL1), a TGF-β-induced matricellular protein, is documented as an intrinsic regulator of proliferation and differentiation in MSCs. In the present study, we characterized the potential role of FSTL1 in MSC-based anti-fibrotic therapy and further elucidated the mechanisms underlying its action.
Methods
Human umbilical cord-derived MSCs were characterized by flow cytometry. FSTL1low MSCs were achieved by FSTL1 siRNA. Migration capacity was evaluated by wound-healing and transwell assay. A murine liver fibrotic model was created by carbon tetrachloride (CCl4) injection, while control MSCs or FSTL1low MSC were transplanted via intravenous injection 12 weeks post CCl4 injection. Histopathology, liver function, fibrosis degree, and inflammation were analysed thereafter. Inflammatory cell infiltration was evaluated by flow cytometry after hepatic nonparenchymal cell isolation. An MSC-macrophage co-culture system was constructed to further confirm the role of FSTL1 in the immunosuppressive capacity of MSCs. RNA sequencing was used to screen target genes of FSTL1.
Results
FSTL1low MSCs had comparable gene expression for surface markers to wildtype but limited differentiation and migration capacity. FSTL1low MSCs failed to alleviate CCl4-induced hepatic fibrosis in a mouse model. Our data indicated that FSTL1 is essential for the immunosuppressive action of MSCs on inflammatory macrophages during liver fibrotic therapy. FSTL1 silencing attenuated this capacity by inhibiting the downstream JAK/STAT1/IDO pathway.
Conclusions
Our data suggest that FSTL1 facilitates the immunosuppression of MSCs on macrophages and that guarantee the anti-fibrotic effect of MSCs in liver fibrosis.
Similar content being viewed by others


Background
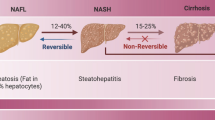
Liver diseases are a global health issue that are associated with high mortality and morbidity worldwide [1, 2]. Liver fibrosis, which occurs as a general response to chronic liver injury, may progress to cirrhosis when not well controlled [3, 4]. Although liver transplantation is the most effective treatment for patients with end-stage cirrhosis, the high costs and shortage of donors limit the clinical application [5]. Recently, following the publication of studies showing the significant efficacy in animal models and preliminary clinical trials, mesenchymal stem cell (MSC) therapy has been shown to be a promising option for liver fibrosis. However, factors regulating the efficacy of MSC therapy for liver fibrosis are not completely understood [6, 7].
MSCs were first isolated and identified by Friedenstein et al. [8] from the bone marrow and have subsequently been isolated from various tissues [9]. The multi-lineage development, immunomodulatory, and trophic properties of MSCs make these cells ideal candidates for clinical applications [10]. During chronic injury, proinflammatory macrophages trigger hepatic stellate cells (HSCs) activation and differentiation into collagen-producing myofibroblasts [11]. Persistent hepatic inflammation activates HSCs to produce extracellular matrix (ECM) and precedes the progression of liver fibrosis [12]. Switching of macrophages from a proinflammatory to an anti-inflammatory phenotype is believed to underlie the therapeutic effects of MSCs in treating liver fibrosis [6]. This function mainly relies on the factors and molecules secreted by MSCs, including Indoleamine 2,3-dioxygenase (IDO), NO, TSG6 and PGE2, which are linked to the immunosuppressive function of MSCs. However, factors which regulate the immunosuppression of MSCs are largely unknown.
Follistatin-like protein 1 (FSTL1), a TGF-β–induced extracellular glycoprotein belonging to the SPARC family of matricellular proteins [13, 14], functions as an important modulator of cell–matrix interactions by integrating signalling networks that regulate essential cell functions. FSTL1 is involved in development, tissue remodelling/repair, and inflammatory processes [15, 16]. Previous studies have shown that FSTL1 could regulate MSC proliferation and chondrogenic differentiation [16,17,18]. Shen et al. [19] and Zhang et al. [20] reported that FSTL1 is an intrinsic cardiokine promoting survival and proliferation of MSC, and facilitates their therapeutic efficacy in a myocardial ischemia model. Kim et al. [21] found that FSTL1, downregulated in the MSC secretome from patients with cerebellar ataxia, could serve as an anti-inflammatory cytokine with suppressive effects on proinflammatory microglial activation. Paradoxically, it is worth noting that FSTL1 has been identified as a novel inflammatory protein that enhances the ability of monocytes/macrophages to respond to inflammatory signals [16, 17, 22]. FSTL1 acts as a pro-fibrotic factor in organs, including the liver, in the case of liver fibrosis [22,23,24,25,26]. Based on these findings, we focused on the role of intrinsic FSTL1 in MSCs for the treatment for liver fibrosis.
In the present study, we knocked down FSTL1 in MSCs and observed the abolishment of MSC-mediated anti-fibrotic therapy in carbon tetrachloride (CCl4)-induced liver fibrosis. Our data suggest that FSTL1 facilitates the immunosuppression of MSCs on macrophages and guarantees the anti-fibrotic effect of MSCs in liver fibrosis.
Materials and methods
Methods
Animals and cells
Male C57BL/6 J mice (6–8 weeks) were purchased and housed in the animal centre of The Fourth Military Medical University, in a pathogen-free airflow cabinet and food and water were provided ad libitum. All animal study protocols were approved by the Animal Welfare and Ethics Committee of the Fourth Military Medical University (FMMU) and performed in accordance with the ‘Guidelines for the Care and Use of Laboratory Animals.’ The murine liver fibrosis model was established by intraperitoneal injection of 7 ml/kg [20% (v/v)] CCl4 twice a week for 12 weeks. Subsequently, fibrotic mice were randomly divided into groups (n = 6) that received phosphate buffered saline (PBS), 1 × 106 control MSCs, and FSTL1low MSCs (MSCs transfected with FSTL1 siRNA) by tail vein injection. Mice were continually injected with CCl4 during the observation period and were sacrificed at the indicated time for sectioning and analysis.
hMSCs were obtained from the National Engineering Research Centre (Tianjin AmCellGene Engineering Co., Ltd, China) and were passaged in the MSC expansion medium (CCM004, R&D Systems, MN) with 5% CO2 and saturated humidity at 37 °C. hMSCs in the 4th to 10th passages were used in all experiments and for all in vitro evaluation of MSCs. The characterization of hMSCs was performed using flow cytometry. Antibodies (anti-CD29, CD90, CD105, CD73, CD44, CD45, CD34, and HLA-DR) conjugated with PE were purchased from BioLegend (San Diego, CA). Analyses were performed using FACScan (BD Bioscience, CA). hMSCs were cells positive for CD105, CD44, CD29, and CD90, but negative for CD34 and CD45 (Additional file 1: Fig. S1). Osteoblast, adipocyte, and chondrogenic differentiation media were purchased from Cyagen Biosciences (HUXUC-90021, HUXUC-90031, HUXUC-9004, Guangzhou, China) and applied according to the manufacturer’s instructions. Intracellular lipid or calcium deposits were stained using Oil Red O or Alizarin Red S. The presence of proteoglycans, which is indicative of chondrogenic differentiation, was verified by toluidine blue after 21 d of pellet induction in a 15 mL tube. Differentiation assays showed that MSCs could differentiate into adipocytes, osteocytes, and chondrocytes under conditioned medium (Additional file 1: Fig. S1).
Transfection of MSCs with FSTL1-siRNA
The siRNAs against FSTL1 were purchased from Santa Cruz Biotechnology, Inc (USA). One day before transfection, MSCs were seeded in a 6-well plate at a density of 1 × 105 cells/mL. For treatment, 2 μL siRNA was diluted in 120 μL 1 × riboFECT™ CP Buffer. Then, 12 μL riboFECT™ CP Reagent was added and incubated for 15 min. Subsequently, the mixture was diluted in DMEM containing 10% (v/v) serum and added into the wells containing MSCs. After 24–48 h, the cells were harvested for mRNA and protein analysis.
RNA isolation and real-time PCR analysis
Total RNA was extracted using the RNAeasy Plus kit (TaKaRa Biotechnology Co., Ltd., Dalian, China), and reverse transcription was performed using the PrimeScript™ RT Master Mix (RR036A, Takara, Tokyo). Amplification was conducted using TB Green Premix Ex Taq II (DRR820A, Takara, Tokyo) and a CFX96 Touch™ real-time PCR System (Bio-Rad, CA).
Western blot analysis
Proteins were extracted from cells or tissue samples using RIPA lysis buffer (Beyotime biotechnology, China), which was supplemented with proteinase inhibitors and phosphatase inhibitors (Roche, Basel). The Bradford method was applied to quantify the protein samples. Subsequently, 30 μg of protein was loaded onto SDS-PAGE gels before transferring to nitrocellulose membranes (Bio-Rad Biotechnology, America). The membranes were blocked in TBST buffer containing 2.5% skim milk for 30 min. Subsequently, the membranes were incubated with primary antibody at 4 ℃ overnight. The next day, the membranes were washed and incubated with peroxidase-conjugated secondary antibodies at room temperature for 1 h. Blots were imaged using an enhanced chemiluminescence kit.
Wound healing assay
Cells were seeded onto 6-well plates and transfected with siFSTL1 24 h before treatment. The monolayers were scratched with a 200 mL pipette tip and washed with serum-free media to remove the detached cells. The cells were maintained in serum-free media for 12 h. The wound areas were then imaged.
Transwell migration assay
The migration ability of cells was determined using a transwell membrane system comprising an 8 μm pore polycarbonate membrane in a 24-well culture plate. In brief, MSCs were cultured in the upper chamber of a transwell membrane system without serum (1 × 105 cells per well). The lower chamber was filled with 600 μL medium containing 20% serum. Following a 12 h incubation, the cells in the upper chamber were fixed with 4% paraformaldehyde for 30 min, and the non-migrating cells inside the chamber were detached from the membrane. Subsequently, crystal violet was used to stain the migrated cells in the chamber. Finally, the images of stained cells were acquired and counted in five random fields.
Biochemical analysis and histological staining
The serum of mice was obtained at each time point. The levels of alanine aminotransferase (ALT) were analysed using an automatic biochemistry analyser in Xijing Hospital. The liver tissue samples were prepared as paraffin-embedded sections by YiKE Biotechnology, China. Subsequently, the sections were stained with haematoxylin and eosin (HE) for routine histological examination and Sirius Red for fibrosis evaluation. Further quantification and analysis of the collagen fibres were performed using Image-Pro Plus software (v6.0, Media Cybernetics Inc.).
Immunohistochemistry staining
The liver tissue samples were prepared as paraffin-embedded sections by YiKE Biotechnology, China. Subsequently, the sections were dewaxed and sequentially rehydrated by in xylol and alcohol in sequence. Antigen retrieval was achieved by microwave using sodium citrate solution with pH 6.0. Subsequently, 1% H2O2 was used to block endogenous peroxidase activity. The slides were further blocked in goat serum for 30 min. Anti-iNOS and anti-CD206 antibodies were incubated with the slides at 4 °C overnight. The next day, after washing with PBS, the secondary antibodies were added and diaminobenzidine (DAB) was applied to visualise the images. Further quantification and analysis of the positive areas were performed using Image-Pro Plus software (v6.0, Media Cybernetics Inc.).
Flow cytometry analysis of liver leukocyte subsets
Hepatic mononuclear cells were isolated from the liver by using collagenase IV (Sigma) and 40% Percoll (GE Healthcare). Single-cell suspensions were first incubated with anti-mouse FcR blocking reagent (BioLegend) and then labelled with mixed fluorochrome-conjugated antibodies, (APC/Cyanine 7 anti-mouse CD45.2, PE anti-mouse F4/80, Brilliant Violet 421 anti-mouse CD206 (MMR), APC anti-mouse CD86, FITC anti-mouse/human CD11b, PerCP/Cyanine5.5 anti-mouse CD11c, and APC anti-mouse I-A/I-E). Flow cytometric data were acquired on a FACSVerse flow cytometer (BD Bioscience) and analysed using FlowJo software (TreeStar, Ashland, OR, US).
RNA sequencing (RNA-seq) analysis
MSCs were transfected with FSTL1 siRNA for 24 h, and then treated with or without FSTL1 (100 ng/ml) for 24 h. Cells were washed with PBS for 3 times. Total RNA was isolated from control MSCs, MSCs transfected with siRNA treated with or without FSTL1 using Trizol (Invitrogen, USA) according to the manual instruction. RNA sequencing libraries were generated with an insert size ranging from 370 to 420 bp, and sequenced using the Illumina NovaSeq 6000 platform (Novogene-Tianjin, China). The image data measured by the high-throughput sequencer are converted into sequence data (reads) by CASAVA base recognition. Raw data (raw reads) of fastq format were firstly processed through in-house perl scripts. In this step, clean data (clean reads) were obtained by removing reads containing adapter, reads containing N base and low quality reads from raw data. At the same time, Q20, Q30 and GC content of the clean data were calculated. All the downstream analyses were based on the clean data. Each sample produced 15.0 G data on average. The clean reads were mapped to the reference genome using HISAT2 (v2.0.5) software. Data processing and analysis were performed using the R programming language. Quantification of gene expression level featureCounts (v1.5.0-p3) was used to count the reads numbers mapped to each gene. And then FPKM of each gene was calculated based on the length of the gene and reads count mapped to this gene. FPKM, expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced. (For DESeq2 with biological replicates) Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq2 R package (1.20.0). DESeq2 provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting P-values were adjusted using the Benjamini and Hochberg’s approach for controlling the false discovery rate. padj < = 0.05 and |log2(foldchange)|> = 1 were set as the threshold for significantly differential expression. Differentially expressed genes (DEGs) were included for further functional analysis based on GO and KEGG databases. The details of all the identified genes were listed in Additional file 2: Table 2.
Statistical analysis
Data were expressed as the mean values ± SD. One-way analysis of variance and a t-test were performed to identify the significant differences. A P value of < 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, CA).
Results
Generation of FSTL1low MSCs
As FSTL1 expression is robust in most types of mesenchymal cells, we evaluated FSTL1 mRNA in subsequent passages in human umbilical cord-derived MSCs (hMSCs). We found that Fstl1 expression was different in MSCs from different origin (Fig. 1A). And it was generally downregulated in MSCs after growth for over ten passages (Fig. 1B, Additional file 1: Fig.S2A). To study the function of FSTL1 on the therapeutic effect of MSCs, FSTL1low MSCs were generated using RNA interference in MSCs to knock down endogenous FSTL1 (Fig. 1C, D). FSTL1low MSCs showed no obvious change in expression of CD surface-markers compared with controls (Fig. 1E), but they significantly inhibited osteogenesis and chondrogenesis as previously reported [27] (Fig. 1F, Additional file 1: Fig. S2B). Further, we observed that FSTL1low MSCs had attenuated migration capacity in wound healing (Additional file 1: Fig. S2C) and transwell (Fig. 1G) assays. These results indicate that silencing of FSTL1 does not affect MSC identity, but inhibits their differentiation and migration capacity.

Generation of human umbilical cord-derived FSTL1 low MSCs. A-B: FSTL1 level was evaluated by qPCR in human umbilical cord-derived MSCs. FSTL1 expression in MSCs in passage 3 from different origin (A). FSTL1 expression in MSCs after grow for eight passages (B). C-E: MSCs were transfected with siFSTL1 for 12 h prior to RNA analysis, or 24 h for protein or flow cytometry (FCM) analysis. FSTL1 mRNA levels were determined with qRT-PCR (C, n ≥ 5, **, P < 0.01). Western blot analysis of FSTL1 in cell extracts (D). Relative density of FSTL1-normalised β-actin is represented. FCM analysis of surface markers after FSTL1 silencing (E). F-G: MSCs were transfected with siFSTL1 for 24 h, followed by differentiation or migration capacity evaluation. The ability of MSCs to differentiate into the chondrogenic was confirmed by Alcian blue staining (F). The migration ability of MSCs was evaluated by transwell assays (G). ***, P < 0.001; **, P < 0.01; *, P < 0.05. Bars, 100 µm. All experiments were performed three times. Error bars indicate the mean ± SEM
FSTL1low MSCs had impaired treatment efficacy in liver fibrosis
To further assess the role of intrinsic FSTL1 in MSCs for their anti-fibrotic efficacy, we transplanted FSTL1low MSCs via tail vein into mice with CCl4-induced liver fibrosis (Fig. 2A). Histological analyses of liver tissues showed that mice transplanted with control MSCs exhibited remarkably reduced fibrosis, while those transplanted with FSTL1low MSCs did not show any pronounced amelioration at 4 weeks post transplantation (Fig. 2B). The area of Sirius Red staining (Fig. 2C) and serum parameters (alanine aminotransferase, ALT) that indicated hepatic function restoration (Fig. 2D) was barely altered at 4 weeks post FSTL1low MSC transplantation. Immunofluorescence of α-SMA and Col1, both markers of activated myofibroblasts, revealed a significant decrease in the MSC-transplanted group, but no obvious changes in the FSTL1low MSC-transplanted group (Fig. 3E). Consistent with these results, FSTL1low MSC treatment did not inhibit the transcript-level expression of fibrotic genes (Col1al, Col3a1,α-SMA,, TGF-β1,) in the liver tissue, while the expression of genes in the control group was markedly decreased (Fig. 3F–I). These findings suggested the indispensable role of FSTL1 in MSC antifibrotic therapy in the context of liver fibrosis.

FSTL1low MSCs showed impaired treatment capacity in liver fibrosis. A Schematic illustration of the establishment of an in vivo hepatic fibrosis model and the MSC-based treatment strategy. MSC transplantation was performed at 12 weeks post CCl4 injury, livers were harvested at 4 weeks post transplantation for subsequent analysis. B Histological analysis and Sirius red staining of collagen in liver sections. Representative images of the staining are shown (n = 4). C Liver fibrosis score analysis of Sirius red-stained liver sections. The fibrotic area is presented as a percentage. D Serum ALT changes after MSC treatment. E–G: Immunofluorescent staining (E) and qRT-PCR analyses (F–I) of fibrotic markers. Representative images of the staining are shown (n = 4). **, P < 0.01; *, P < 0.05. Bars, 100 µm. All experiments were performed three times. Error bars indicate the mean ± SEM

FSTL1low MSCs lose their immunosuppressive capacity on inflammatory macrophages in fibrotic mouse model. A and B: Infiltrating inflammatory macrophages and cytokines in liver tissue were evaluated 1 weeks after MSCs transplantation. Pro-inflammatory macrophages (iNOS +) and anti-inflammatory macrophages (CD206 +) were analysed using IHC staining (A). Cytokines in liver tissues related to inflammatory macrophages were evaluated by qRT-PCR analyses (B). C–E: Non-parenchymal cells in livers were isolated and analysed at 1 week post MSC transplantation for subsequent analyses. Total number of macrophages (F4/80 + CD11b +) in the fibrotic liver (C and D, n = 4). (E) Mean fluorescence intensity (MFI) of inflammatory surface markers on macrophages (CD11c, CD206, CD86) after MSC treatment (n = 4). **, P < 0.01; *, P < 0.05. Bars, 100 µm. All experiments were performed in triplicate. Error bars indicate the mean ± SEM
FSTL1low MSCs lose their immunosuppressive capacity on inflammatory macrophages in liver fibrosis mouse model
With a crucial role in the pathogenesis of chronic liver injury, hepatic macrophages have been proposed as potential therapeutic targets in fibrosis [11]. It has further been found that liver macrophage depletion ameliorates the effect of MSC transplantation, indicating the importance of macrophages for MSC therapy [28]. As previously reported [29], MSC transplantation significantly decreased the amount of iNOS-positive pro-inflammatory macrophages and increased the number of CD206-positive anti-inflammatory macrophages in liver Sects. 1 week post MSCs transplantation (Fig. 3A). However, the same improvement was not observed in the FSTL1low MSC-transplanted group. Furthermore, consistent with these findings, in the FSTL1low MSC-transplanted group, expression of genes related to the pro-inflammatory function of macrophages (TNF-α, iNOS and IL-6) showed no obvious downregulation and there was no increase in the anti-inflammatory function of macrophages (indicated by CD206 and Arg1 expression) (Fig. 3B). Flow cytometry also showed that FSTL1low MSCs did not sufficiently inhibit macrophage infiltration (Fig. 3C, D). Further, FSTL1low MSCs exerted no obvious suppression of pro-inflammatory markers (CD86, CD11c) or elevation of anti-inflammatory markers (CD206), as was observed in the MSC-transplanted group (Fig. 3E). FSTL1low MSCs partially restore their immunosuppressive capacity after FSTL1 addition in culture system. These data suggest that FSTL1 is essential for MSC immunosuppression on pro-inflammatory macrophages during liver fibrosis therapy.
FSTL1low MSCs lose their immunosuppressive function towards macrophages in vitro
To further explore the effect of intrinsic FSTL1 of MSCs in their immunoregulatory function, we performed an in vitro MSC-macrophage co-culture assay. Raw264.7, a mouse monocyte/macrophage cell line, co-cultured with hMSCs. Flow cytometry of macrophage populations indicated an increase in scavenger receptor CD206-expressing cells in the MSC-macrophage co-culture system. However, macrophages from FSTL1low-MSC co-cultures showed no significant induction of the population of CD206-positive cells (Fig. 4A, B). Then, Raw264.7 was treated with pro-inflammatory macrophage (M1) stimuli (LPS and IFN-γ). Flow cytometry of macrophage populations indicated a reduction in co-stimulatory molecule CD86-expressing cells in this MSC-macrophage co-culture system (Fig. 4C, D). In contrast, macrophages from FSTL1low-MSC co-cultures showed no significant suppression of the population of CD86-positive cells. Moreover, FSTL1low MSCs were inefficient with respect to inducing the expression of anti-inflammatory cytokines, Mrc-1, Arg-1 and interleukin (IL)-10, and incapable of attenuating the expression of prototypic pro-inflammatory cytokines, TNF-α and iNOS (Fig. 4E). FSTL1low MSCs partially restore their immunosuppressive capacity after FSTL1 addition in culture system in both situations. These results further demonstrated that FSTL1 silencing attenuates the immunosuppressive capacity of MSCs against inflammatory macrophages.

FSTL1low MSCs lose their immunosuppressive function on macrophages in vitro. A-B: MSC transfected with siFSTL1treated with or without FSTL1 (100 ng/ml) were co-cultured with Raw264.7 cells. The proportions of CD206-positive macrophages were determined after 24 h co-culture with MSCs (A). The mean percentages of CD206-positive cells in total macrophages were summarised (B). C, D: MSCs transfected with siFSTL1 treated with or without FSTL1 (100 ng/ml) were co-cultured with Raw264.7 cells after 24 h IFN-γand LPS treatment. The proportions of CD86-positive cells were subsequently determined (C). Mean percentages of CD86-positive cells in total macrophages were summarised after 24 h co-culture with MSCs (D). E Expression of differential markers in macrophages evaluated by qPCR under different conditions after 12 h co-culture with MSCs. **, P < 0.01; *, P < 0.05. All experiments were performed in triplicate. Error bars indicate the mean ± SEM
FSTL1 downregulation induces transcriptional reprogramming in MSCs
To elucidate whether the expression of FSTL1 influences the gene expression profiles of MSCs, we performed RNA-seq analysis on FSTL1WT and FSTL1low MSCs. In total, we identified 1410 differentially expressed genes (DEGs) after FSTL1 silencing (600 upregulated genes and 810 downregulated genes) (Fig. 5A). With respect to functional analysis based on the gene ontology (GO) database, the DEGs were annotated into multiple subdivisions within the three GO categories, ‘Biological process,’ ‘Cellular component,’ and ‘Molecular function,’ implying distinct transcriptional changes induced by FSTL1 silencing (Fig. 5B).

FSTL1 downregulation induces transcriptional reprogramming in MSCs. A Volcano plot showing DEGs in FSTL1low MSCs compared to FSTL1WT MSCs. The blue and red dots indicate downregulated and upregulated genes, respectively. Fold change > 1.5 and P value < 0.05) B Gene ontology (GO) analysis of the DEGs categorised as ‘Cellular component,’ ‘Molecular function,’ and ‘Biological process.’ C KEGG pathway enrichment analysis of the DEGs. The Y-axis represents KEGG terms, and the X-axis represents the rich factor. The colour of the bubble represents the enrichment significance, and the size of the bubble represents the number of DEGs. D The top downregulated enriched terms in KEGG pathway are presented as a bubble chart. E Representative differentially expressed genes (DEGs) between FSTL1WT MSC and FSTL1low MSC
Enrichment analysis revealed that FSTL1 silencing resulted in increased expression of genes associated with cell cycle (histone-serine phosphorylation, DNA binding and phospholipase activity) (Additional file 1: Fig. S3), and a decreased expression of genes associated with signalling transduction (cytokine receptor binding, cytokine activity, receptor complex, receptor regulator activity) (Additional file 1: Fig. S4). KEGG pathway analysis further revealed an obvious enrichment of DEGs associated with multiple inflammatory-related pathways, including “JAK/STAT signalling pathway”, “TNF signalling pathway,” and “MAPK signalling pathway”, “NOD-like receptor signalling pathway”, indicating that FSTL1 functions as a modulator of MSC behaviours and functions by integrating signalling networks (Fig. 5C). And genes associated with JAK/STAT signalling pathway were downregulated after FSTL1 silencing (Fig. 5D), which had been reported to empower the immunoregulation capacity of MSCs after cytokines stimuli. Representative DEGs that changed after FSTL1 downregulation are shown in Fig. 5E. These data indicate that silencing of FSTL1 induces the transcriptional reprogramming of MSCs.
FSTL1 guarantees MSC immunosuppressive function by regulating IDO expression
IDO, the rate-limiting enzyme in the kynurenine pathway of tryptophan degradation, plays an important role in inflammation. It has been reported that IDO can not only affect the infiltration of immune cells in many pathological disease conditions, including liver fibrosis [30, 31], but also further inhibits T cell proliferation and promotes anti-inflammatory macrophage differentiation during MSC therapy [29, 32]. Based on the results of our RNA-seq analysis, we hypothesized that IDO was one of the target genes of FSTL1. Using qPCR and Western blot, we further confirmed that silencing of FSTL1 downregulated IDO at both the mRNA (Fig. 6A) and protein levels (Fig. 6B). We found that JAK/STAT1 signalling was downregulated following FSTL1 silencing. Most importantly, we found that FSTL1 promotes MSC immunosuppressive functions by activating the downstream JAK/STAT1/IDO pathway. FSTL1low MSCs partially restore IDO expression and STAT1 phosphorylation after FSTL1 addition in culture system (Fig. 6C, D). 2-(1,8-naphthyridin-2-yl)-Phenol (2-NP), a STAT1 transcriptional activator, could rescue JAK/STAT1/IDO axis in FSTL1low MSCs (Fig. 6E, F). Besides, we also found that 2-NP could significantly enhance immunosuppression of FSTL1low MSC after co-cultured with macrophage (Fig. 6G–I). These data indicate FSTL1 promotes MSC immunosuppressive functions by activating the downstream JAK/STAT1/IDO pathway.

FSTL1 guarantees immunosuppressive function of MSCs on macrophages by regulating JAK/STAT1/IDO pathway. MSCs were transfected with siFSTL1. RNA expression of FSTL1 and IDO was determined by qRT-PCR at 12 h post transfection (A). Protein level of FSTL1, IDO, and pSTAT1 was determined by western blotting at 24 h post transfection (B). C–D: MSC transfected with siFSTL1treated with or without FSTL1 (100 ng/ml). IDO mRNA was determined by qRT-PCR at 12 h post FSTL1 treatment (C). Protein level of FSTL1, IDO, and pSTAT1 was determined by western blotting at 24 h post FSTL1 treatment (D). E–F: MSC transfected with siFSTL1treated with or without 2-NP (15 ug/ml). IDO mRNA was determined by qRT-PCR at 12 h post 2-NP treatment (E). Protein level of FSTL1, IDO, and pSTAT1 was determined by western blotting at 24 h post 2-NP treatment (F). G-I: MSC transfected with siFSTL1treated with or without 2-NP treatment were co-cultured with Raw264.7 cells. The proportions of CD206-positive macrophages were determined after 24 h co-culture with MSCs (G). The mean percentages of CD206-positive cells in total macrophages were summarised (H). Expression of differential markers in macrophages evaluated by qPCR under different conditions after 12 h co-culture with MSCs (I). ***, P < 0.001; **, P < 0.01; *, P < 0.05. All experiments were performed in triplicate. Error bars indicate the mean ± SEM
Discussion
Liver disease is associated with high mortality and morbidity, with an increasing incidence worldwide. Recently, MSC transplantation is generally believed to be a promising therapeutic strategy for patients with end-stage liver disease [33]. However, it is still not recognized as a reproducible, predictable, and standardized therapeutic approach in the clinical setting [34]. Thus, factors affecting the efficacy of MSC therapy for liver fibrosis need to be fully characterized. These results provide evidence at the cellular, molecular, and animal levels to support a regulatory role of FSTL1 with respect to the immunosuppressive effect of MSCs on inflammatory macrophages, which affects the therapeutic outcome.
MSCs have a self-renewal ability and multi-directional differentiation potential. FSTL1, a multifunctional secreted protein mainly expressed in interstitial cells, has a role in maintaining MSC stemness [18]. It has been reported to participate in cell survival, proliferation, differentiation, migration, and organ development. Our work here was consistent with previous findings that FSTL1 could regulate the chondrogenic differentiation of MSCs [18]. It has been shown that Follistatin, another SPARC family member, promotes MSC migration in vitro [35]. We also found that FSTL1 could facilitate MSC migration capacity, which potentially enhances the rate of MSC homing to injured sites.
Our work further illustrates the role of endogenous FSTL1 in MSC therapy. During MSC therapy for myocardial infarction, Han et al. showed that overexpression of FSTL1 in MSCs can significantly reduce inflammation and enhance neovascularization [19]. However, as FSTL1 can be used as a pro-survival cytokine to inhibit the death of cardiomyocytes [27], it is hard to distinguish whether the therapeutic improvement in FSTL1-overexpressed MSCs depends on more activated MSCs or the molecular function of FSTL1 alone. Here, we tried to determine the role of FSTL1 in MSC therapy and investigate how it affects the treatment outcome in liver fibrosis. The data indicated that endogenous FSTL1 expression in MSCs is indispensable for MSC treatment outcomes in liver fibrosis without a tendency to promote fibrogenesis.
It was widely accepted that the anti-fibrotic effect in liver fibrosis mainly depended on the immunomodulatory capacity of MSCs, especially when they were administered via intravenous infusion [36]. MSCs have been reported to induce a shift in the polarization of pro-inflammatory macrophages (M1) towards alternative macrophages (M2) both in vivo and in vitro. This polarization is driven by the ability of MSCs to secrete soluble factors, such as IL-10, IL-1Ra, and PGE2 by enhancing the number of M2 macrophages [6]. IDO, an IFN-γ-inducible intracellular enzyme, catalyses the first step in tryptophan degradation along the kynurenine pathway, which was reported to regulate macrophage recruitment and polarization both in humans [29] and mice [31]. We identified IDO as the most significantly varied gene in FSTL1 silencing MSCs using RNA-seq, a result that we further confirmed at the transcriptional and translational levels.
As previously reported, FSTL1 regulates several other pathways controlling chondrocyte proliferation and differentiation, including those regulated by BMP4, IGF, and Wnt signalling [18]. FSTL1 enhances the activity of various intermediaries of the TGF-β pathway, which include the canonical Smad3, as well as alternative p38 MAPK and Akt [18]. Our data show that FSTL1 silencing downregulates JAK/STAT1 signalling, which directly regulates IDO transcriptional activity. Moreover, although FSTL1 silencing suppresses the activation of several signalling pathways, we found that most upregulated genes enriched in RNA-seq were those related to DNA replication, translation, and transcription. Genes related to non-coding RNA were also significantly upregulated after FSTL1 downregulation, the mechanism of which needs further work to illustrate. Above all, FSTL1 plays a critical role in the extensive crosstalk between the numerous signalling pathways that are activated in MSCs in response to stimuli. Further research to determine how FSTL1 is integrated into the signal-transduction network maintaining MSC stemness and immunoregulatory function is important to elucidate the mechanisms underlying MSC therapy.
Conclusion
In summary, we have described that FSTL1 silencing attenuates the immunosuppressive capacity of MSCs on inflammatory macrophages by inhibiting downstream JAK/STAT1/IDO. Our data suggest that FSTL1 facilitates the immunosuppression of MSCs on macrophages and guarantee the anti-fibrotic effect of MSCs in liver fibrosis.
Availability of data and materials
The datasets supporting the results of this article are included within the article and its additional files.
Abbreviations
- MSC:
-
Mesenchymal stem cell
- FSTL1:
-
Follistatin-like 1
- TGF-β:
-
Ttransforming growth factor β
- CCl4:
-
Carbon tetrachloride
- JAK:
-
Janus kinase
- STAT:
-
Signal transducer and activator of transcription
- IDO:
-
Indoleamine 2,3-dioxygenase
- HSC:
-
Hepatic stellate cells
- ECM:
-
Extracellular matrix
- RNA:
-
Ribonucleic Acid
- DNA:
-
Deoxyribonucleic acid
- a-SMA:
-
Alpha-smooth muscle actin
- Col1al:
-
Collagen I
- Col3a1:
-
Collagen III
- iNOS:
-
Inducible nitric oxide synthase
- TNF-α:
-
Tumor Necrosis Factor Alpha
- LPS:
-
Lipopolysaccharides
- IFN-γ:
-
Interferon (IFN)-γ
- DEGs:
-
Differentially expressed genes
- GO:
-
Gene ontology
- MAPK:
-
MAP kinase
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- PGE2:
-
Prostaglandin E2
- IGF:
-
Insulin like growth factor
- 2-NP:
-
2-(1,8-Naphthyridin-2-yl)-phenol
References
Collaborators GBDC. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(3):245–66.
Cheemerla S, Balakrishnan M. Global epidemiology of chronic liver disease. Clin Liver Dis (Hoboken). 2021;17(5):365–70.
Sarin SK, et al. Liver diseases in the Asia-Pacific region: a Lancet Gastroenterology & Hepatology Commission. Lancet Gastroenterol Hepatol. 2020;5(2):167–228.
Ramachandran P, Henderson NC. Antifibrotics in chronic liver disease: tractable targets and translational challenges. Lancet Gastroenterol Hepatol. 2016;1(4):328–40.
Zhang Z, et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J Gastroenterol Hepatol. 2012;27(Suppl 2):112–20.
Alfaifi M, et al. Mesenchymal stromal cell therapy for liver diseases. J Hepatol. 2018;68(6):1272–85.
Dwyer BJ, et al. Cell therapy for advanced liver diseases: repair or rebuild. J Hepatol. 2021;74(1):185–99.
Friedenstein AJ, Piatetzky S II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16(3):381–90.
Yang Y, et al. The application of mesenchymal stem cells in the treatment of liver diseases: mechanism, efficacy, and safety issues. Front Med. 2021;8: 655268.
Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell. 2013;13(4):392–402.
Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59(5):2034–42.
Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127(1):55–64.
Shibanuma M, et al. Cloning from a mouse osteoblastic cell line of a set of transforming-growth-factor-beta 1-regulated genes, one of which seems to encode a follistatin-related polypeptide. Eur J Biochem. 1993;217(1):13–9.
Zheng X, et al. TGF-beta1 induces Fstl1 via the Smad3-c-Jun pathway in lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L240–51.
Sylva M, Moorman AF, van den Hoff MJ. Follistatin-like 1 in vertebrate development. Birth Defects Res C Embryo Today. 2013;99(1):61–9.
Chaly Y, et al. Follistatin-like protein 1 and its role in inflammation and inflammatory diseases. Immunol Res. 2014;59(1–3):266–72.
Chaly Y, et al. FSTL1 promotes arthritis in mice by enhancing inflammatory cytokine/chemokine expression. Arthritis Rheum. 2012;64(4):1082–8.
Chaly Y, et al. Follistatin-like protein 1 regulates chondrocyte proliferation and chondrogenic differentiation of mesenchymal stem cells. Ann Rheum Dis. 2015;74(7):1467–73.
Shen H, et al. Follistatin-like 1 protects mesenchymal stem cells from hypoxic damage and enhances their therapeutic efficacy in a mouse myocardial infarction model. Stem Cell Res Ther. 2019;10(1):17.
Zhang MQ, et al. Follistatin-like 1-engineered mesenchymal stem cells prevent myocardial ischemic reperfusion injury. Zhonghua Yi Xue Za Zhi. 2016;96(25):2017–22.
Kim JH, et al. Characterization of mesenchymal stem cells derived from patients with cerebellar ataxia: downregulation of the anti-inflammatory secretome profile. Cells. 2020;9(1):212.
Rao J, et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut. 2022. https://doi.org/10.1136/gutjnl-2021-325150.
Xu XY, et al. Targeting Follistatin like 1 ameliorates liver fibrosis induced by carbon tetrachloride through TGF-beta1-miR29a in mice. Cell Commun Signal. 2020;18(1):151.
Vollmann EH, et al. Identification of Novel Fibrosis Modifiers by In Vivo siRNA Silencing. Mol Ther Nucleic Acids. 2017;7:314–23.
Ramnath D, et al. Hepatic expression profiling identifies steatosis-independent and steatosis-driven advanced fibrosis genes. JCI Insight. 2018. https://doi.org/10.1172/jci.insight.120274.
Dong Y, et al. Blocking follistatin-like 1 attenuates bleomycin-induced pulmonary fibrosis in mice. J Exp Med. 2015;212(2):235–52.
Oshima Y, et al. Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117(24):3099–108.
Ghanem LY, et al. Liver macrophage depletion ameliorates the effect of mesenchymal stem cell transplantation in a murine model of injured liver. Sci Rep. 2019;9(1):35.
Francois M, et al. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol Ther. 2012;20(1):187–95.
Mo C, et al. Indoleamine 2,3-dioxygenase 1 limits hepatic inflammatory cells recruitment and promotes bile duct ligation-induced liver fibrosis. Cell Death Dis. 2021;12(1):16.
Jiang N, et al. Indoleamine 2,3-dioxygenase regulates macrophage recruitment, polarization and phagocytosis in aspergillus fumigatus keratitis. Invest Ophthalmol Vis Sci. 2020;61(8):28.
Wang XF, et al. The role of indoleamine 2,3-dioxygenase (IDO) in immune tolerance: focus on macrophage polarization of THP-1 cells. Cell Immunol. 2014;289(1–2):42–8.
Lee CW, et al. Historical perspectives and advances in mesenchymal stem cell research for the treatment of liver diseases. Gastroenterology. 2018;154(1):46–56.
Krampera M, Le Blanc K. Mesenchymal stromal cells: putative microenvironmental modulators become cell therapy. Cell Stem Cell. 2021;28(10):1708–25.
Fahmy-Garcia S, et al. Follistatin effects in migration, vascularization, and osteogenesis in vitro and bone repair in vivo. Front Bioeng Biotechnol. 2019;7:38.
Li YH, et al. Mesenchymal stem cells attenuate liver fibrosis by targeting Ly6C(hi/lo) macrophages through activating the cytokine-paracrine and apoptotic pathways. Cell Death Discov. 2021;7(1):239.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key R&D Program of China, 2020YFA0710803 (to J.W.), 2017YFA0105704 (to Y.H.), National Natural Science Foundation of China (NSFC) grants 81900570 (to X.Z.), 81870421 (to X.Z.) 81700569 (to Y.H.), 82030001(to W.N.), and 81873562 (to Z.H.), the Funds for International Cooperation and Exchange of the National Natural Science Foundation of China 81820108005 (to Y.H.), the China Postdoctoral Science Foundation 2019M653966 (to X.Z.).
Author information
Authors and Affiliations
Contributions
Study concept and design: Y.H., J.W., X.Z.; Acquisition of data: X.Z., X.Z., G.M., J.Y., M.Z., C.Y., Y.H., S.M.; Analysis and interpretation of data: Z.H., W.N., B.J.; Drafting of the manuscript: X.Z., X.Z., G.M., Y.H., J.W., X.Z.; Statistical analysis: X.Z, X.Z., G.M.; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
All animal study protocols were approved by the Animal Welfare and Ethics Committee of the Fourth Military Medical University (FMMU) and performed in accordance with the ‘Guidelines for the Care and Use of Laboratory Animals.’
Consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest with respect to the authorship and publication of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplemental material 1.
Additional file 2
. Supplemental material 2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zheng, X., Zhou, X., Ma, G. et al. Endogenous Follistatin-like 1 guarantees the immunomodulatory properties of mesenchymal stem cells during liver fibrotic therapy. Stem Cell Res Ther 13, 403 (2022). https://doi.org/10.1186/s13287-022-03042-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-022-03042-4