
Abstract
Objective
The tarnished plant bug (TPB), Lygus lineolaris (Palisot de Beauvois) (Hemiptera: Miridae), is a pest damaging many cultivated crops in North America. Although partial transcriptome data are available for this pest, a genome assembly was not available for this species. This assembly of a high-quality chromosome-length genome of TPB is aimed to develop the genetic resources that can provide the foundation required for advancing research on this species.
Results
The initial genome of TPB assembled with paired-end nucleotide sequences generated with Illumina technology was scaffolded with Illumina HiseqX reads generated from a proximity ligated (HiC) library to obtain a high-quality genome assembly. The final assembly contained 3963 scaffolds longer than 1 kbp to yield a genome of 599.96 Mbp. The N50 of the TPB genome assembly was 35.64 Mbp and 98.68% of the genome was assembled into 17 scaffolds larger than 1 Mbp. This megabase scaffold number is the same as the number of chromosomes observed in karyotyping of this insect. The TPB genome is known to have high repetitive DNA content, and the reduced assembled genome size compared to flowcytometric estimates of approximately 860 Mbp may be due to the collapsed assembly of highly similar regions.
Similar content being viewed by others

Introduction
The tarnished plant bug (TPB), Lygus lineolaris (Palisot de Beauvois) (Hemiptera: Miridae), has a broad host range exceeding 300 plant species including a large number of cultivated crops in the United States [1, 2]. TPB has five nymphal stages and the ovipositor in the center of abdominal sternites in adult females can distinguish females from males (Supplementary Fig. S1). TPB is present in the continental United States, Canada, and Mexico. This pest causes significant economic damage to a diversity of vegetable crops, fruits, and nursery stock including strawberries, cotton, and seedlings of conifers [3,4,5,6,7]. In 2020, TPB infested more than 4.8 million acres of cotton resulting in an estimated $157 million in control costs and yield losses [8]. Current control of TPB in cotton is carried out almost exclusively using synthetic insecticide sprays. Formulations and mixtures of insecticides including carbamates, organophosphates, nicotinamides, neonicotinoids, and pyrethroids are routinely used along with an insect growth regulator (novaluron) to manage TPB in commercial agriculture. A systemic insecticide (sulfoxaflor) is permitted under special conditions to manage TPB in cotton. Insecticide resistance in the TPB has been reported in the Mississippi Delta since 1995 [9,10,11]. As in the case of many insects, the susceptibility of TPB to different chemicals within three major classes of insecticides (carbamate, organophosphate, and pyrethroid) commonly used for pest control in cotton has varied over the past forty years [12]. Most insecticides that previously provided good control of TPB currently exhibit diminishing effectiveness [13, 14].
Resistance to insecticides may develop by breaking down of the insecticides through a range of mechanisms: by increased levels or enhanced activity of detoxifying enzymes (metabolic resistance), by resisting the binding of the chemical through genetically modified target sites (target-site resistance), by changing the properties of the exoskeleton to reduce the rate of penetration of contact insecticides (penetration resistance), or through behavioral resistance by developing the ability to detect insecticides and avoid exposure [15,16,17,18]. These adaptations reflect shifts in frequencies of alleles responding to changing environmental conditions by substituting genes in populations over time [19, 20]. These shifts in allele frequencies of genes responding to environmental factors can be identified by monitoring the populations using genetic markers. The number of insect population genomic studies has rapidly increased recently due to the availability of genomic data and cost-effective, high throughput sequencing methods used to generate data [reviewed in: 21, 22]. Navel orangeworm, Amyelois transitella [23], brown planthopper, Nilaparvata lugans [24], and Asian tiger mosquito, Aedes albopictus [25], for example, are among the subjects of a growing number of recent population genomic studies. However, lack of genome sequence data has precluded population genomic studies of Lygus species.
Despite TPB being a pest of several economically important crops grown in North America, apart from a few population genetic and transcriptome and gene expression studies, there is a general paucity of research on the genetics of TPB [26,27,28,29,30,31]. Therefore, the development of a comprehensive set of genetic resources including a high-quality genome, full transcriptome with an official gene set that identifies all isoforms, and genetic markers suitable for population genomic and quantitative genetic studies is needed for this species. Community insect genomics initiatives like the i5k consortium [32] and more recently Ag100Pest [33] and AgriVectors [34] have also highlighted the far-reaching consequences and benefits of creating reference-grade genomics resources and building open access tools to make them available [34, 35]. Our goal for sequencing the genome of TPB was to develop these genetic resources that will significantly advance genetics research on TPB. This will allow us to identify the candidate detoxification gene repertoire and genetic polymorphisms required for genetic mapping and ecological genetic studies in the TPB.
Results and discussion
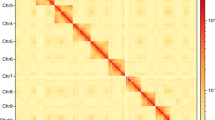
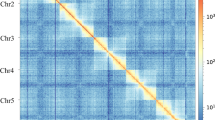
The Meraculous Assembler [36] estimated the genome to be approximately 800 Mb. The scaffolded Illumina-only assembly with two Illumina libraries followed by a round of scaffolding with a third Illumina library was 599.96 Mb with a N50 of 19.8Kb and L50 of 7.1Kb. The total coverage of the L. lineolaris genome by the three Illumina paired-end libraries was 240X. Long range Hi-C scaffolding connected the scaffolds from the Meraculous assembly to create the final assembly with 3963 scaffolds with an N50 of 35.64 Mbp and a total length of 600 Mb (Fig. 1). This assembly contains 80 Mb of Ns with 13.4Kb of Ns per 100Kb of genomic sequence. Accession numbers for genome sequence data are given in the Table 1. This whole genome sequencing project has been deposited at DDBJ/ENA/GenBank under the accession JAEMON000000000. The version described in this paper is version JAEMON010000000.

Linkage density histogram of Lygus lineolaris genome assembly generated from HiC read pairs. The first and second read in a read pair grouped into bins is plotted in the x and y axes, respectively. The intensity of color of each square represents the number of read pairs in each bin. Scaffolds smaller than 1 Mb were not used in this histogram
BUSCO [37] evaluation of the completeness of the Illumina and Hi-C assemblies based on the Hemipteran (2510) and Arthropoda (1013) marker sets indicated that the Hi-C scaffolded assembly improved over the short-read Illumina assembly with an 85.1% of the complete assembly. Only 5.6% of the 1013 Arthropoda BUSCO markers missing (Table 2).
The TPB has 17 chromosome pairs [38] and the 17 largest scaffolds with lengths of more than 1 Mb might represent the 17 chromosomes in the TPB. The GC percentage of 42.7% is higher than the pea aphid (29.6%) and honeybee (38.8%). Flow cytometry analysis of tissue from the heads of male and female TPB resulted in a genome size estimate of 816.6 +/- 2.6 Mb and 869.1 +/- 4.3 Mb, respectively, which is larger than the currently assembled reference assembly (Supplementary Fig. S2).
High repetitive content in the genome may have substantially reduced the genome size by the collapse of repetitive regions during the assembly process. We applied two approaches to identify repeats in the genome. The TPB genome assembly was searched for known repeat families in the order insecta present in the DFAM 2.4 database [39] (Supplementary data Table S1) but this resulted in the annotation of only 3.8% of the genome. RepeatModeler (http://www.repeatmasker.org/) identified 4281 RepeatScout/RECON families and 99 L repeat families with primarily Gypsy/DIRS1 elements. All annotations are available at the AgriVectors portal [34] public database.
Public databases currently list 2,191 and 1,552 nucleotide and protein sequences, respectively, for TPB. In addition, 8 Bioprojects, 21 Biosamples, and 17 population sets are available on the National Center for Bioinformatics (NCBI) database. Four of the eight Bioprojects were submitted by the USDA ARS Southern Insect Management Research Unit, including the TPB genome projects (PRJNA589321 and PRJNA685878) and three transcriptomics projects. We have published RNASeq data from the gut and salivary glands of TPB [26] and two other partial transcriptomes of TPB have been published previously [29, 40]. A high-quality genome with chromosome size scaffolds will facilitate the development of universal markers for mapping genomic loci associated with host selection, insecticide resistance, and population genomic studies. A chromosomal-length genome with annotations from NCBI will provide an official gene set to identify isoforms and study differential gene expression under various physiological conditions such as response to pesticides. The mapping of genomic DNA sequences to the published mitochondrial genome (accession: NC_021975) of TPB from the northern USA identified 34 nucleotide substitutions and three insertions in the protein-coding, rRNA, and tRNA genes of the mitochondrial DNA sequences of TPB from Mississippi. All variant positions, except five single nucleotide variants, were homozygous in southern TPB population.
Filtering of mapped reads identified 842,044 SNPs that were heterozygous in the reads mapped to the largest 18 scaffolds. Flanking sequences, allele-specific primers, and locus-specific primers developed for the manually selected SNPs are shown in the supplementary data Table S2.
Combined genomic and transcriptomic data (RNASeq + gDNA + BAC = 3,335,989,518 reads) will facilitate identifying non-transcribed genomic regions and regulatory sequences influencing gene expression. In addition, minor effect genes that are coregulated with major effect genes can be identified using expression profiles and gene coregulatory network analysis [41].
Methods
TPB collected from field locations in Stoneville, MS were mated as single pairs to obtain progeny that were used to establish a colony inbred for five generations. DNA extracted from adult females from the inbred colony was submitted to Dovetail Genomics (Scotts Valley, CA) for library construction and genome sequencing. Illumina paired-end short reads (2 × 150 bp) were generated from a Chicago library made from TPB genomic DNA. Sequencing adapters and low-quality reads were removed before assembly using Trimmomatic [42]. All bases with quality scores lower than Q20 were removed from the leading and trailing ends and the middle of the reads.
A Dovetail Omni-C library was prepared as described in Saha et al. 2022 [43]. Briefly, chromatin was fixed in the nucleus by immersing the tissues in formaldehyde. Ends of DNAse I digested chromatin were repaired followed by ligation to a biotinylated bridge adapter. The adapter containing ends were proximity ligated and the crosslinks were reversed before the DNA was purified. Biotin not internal to ligated fragments were removed and the sequencing libraries containing Illumina-compatible adapters were generated using NEBNext Ultra reagents. Streptavidin beads were used to isolate biotin-containing DNA fragments and each library was PCR enriched. Illumina HiSeqX platform was used to sequence the libraries to approximately 30x coverage. HiRise, a pipeline specifically designed to scaffold initial genome assemblies using proximity ligation sequence data was used to generate final scaffolds using initial assembly and OmniC reads [44].
BUSCO version 5.2.2 was used to evaluate genome completeness [37]. Dfam TE tools docker container (version 1.4) of the RepeatModeler (https://github.com/Dfam-consortium/TETools) was used to annotate repeats. RepeatMasker and RepeatClassifier Version 2.0.2 (http://www.repeatmasker.org/) was used to classify the repeat types in the TPB genome. Dfam 3.4 database was used for repeat classification [39].
A published mitochondrial genome of TPB (accession: NC_021975) [45] was used as the reference to map 2,723,838,186 Illumina short reads generated by sequencing initial shotgun libraries and the Hi-C library using CLC Genome WorkBench (Qiagen, Redwood City, CA, USA). Variant analysis was performed on the mapped reads to identify single nucleotide polymorphisms and indels between the reference and the reads. Single nucleotide polymorphisms (SNP) were identified by filtering variants in Illumina reads mapped to the eighteen largest scaffolds using the variant filtering function in CLC Genome Workbench. SNPs with at least 60 mapped reads with greater than 30% heterozygosity and coverage greater than 200 were filtered and exported. A set of 96 SNPs representing 18 largest scaffolds were manually selected to develop an SNP assay panel.
Limitations: Proprietary methods developed by a service provider to prepare Genomic DNA library preparation and assembly are not publicly available. Difference between the genome size estimated by flow cytometry and the size of the assembled genome may needs to be corrected using long read technology.
Data Availability
All raw sequencing data and assemblies have been submitted to NCBI BioProject: PRJNA685878.
References
Capinera JL. Handbook of vegetable pests. San Diego, CA. 729 pp.: Academic Press; 2001. 729 p.
Kelton LA. The Lygus bugs (genus Lygus Hahn) of North America (Heteroptera: Miridae). Mem Entomol Soc Can. 1975;107:5–101.
Mailloux G, Bostanian N. Economic injury level model for tarnished plant bug, Lygus lineolaris (Palisot de Beauvois)(Hemiptera: Miridae), in strawberry fields. Env Entomol. 1988;17:581–6.
Handley DT. Strawberry fruit development and the effects of feeding by the tarnished plant bug (Lygus lineolaris). In:. The strawberry into the 21st century Proceedings of the Third North American Strawberry Conference Houston, Texas 14–16 February 1990. Timber Press; 1991.
Layton M. Biology and damage of the tarnished plant bug, Lygus lineolaris, in cotton. Southwest Entomol. 2000:7–20.
Fleischer S, Gaylor M, Hue N. Dispersal of Lygus lineolaris (Heteroptera: Miridae) adults through cotton following nursery host destruction. Env Entomol. 1988;17:533–41.
Wold SJ, Hutchison WD. Comparison of economic and plant phenology-based thresholds for management of Lygus lineolaris (Hemiptera: Miridae) in Minnesota strawberries. J Econ Entomol. 2003;96:1500–9.
Cook DR, Threet M. Cotton insect losses – 2019. 2019. https://www.biochemistry.msstate.edu/resources/2019loss.php. Accessed March 31, 2020, 2020.
Snodgrass G. Glass-vial bioassay to estimate insecticide resistance in adult tarnished plant bugs (Heteroptera: Miridae). J Econ Entomol. 1996;89:1053–9.
Snodgrass G, Elzen G. Insecticide resistance in a tarnished plant bug population in cotton in the Mississippi Delta. Southwest Entomol. 1995;20:317–232.
Gore J, Catchot A, Musser F, Greene J, Leonard BR, Cook DR, et al. Development of a plant-based threshold for tarnished plant bug (Hemiptera: Miridae) in cotton. J Econ Entomol. 2012;105:2007–14.
Parys KA, Luttrell RG, Snodgrass GL, Portilla M, Copes JT. Longitudinal measurements of tarnished plant bug (Hemiptera: Miridae) susceptibility to Insecticides in Arkansas, Louisiana, and Mississippi: Associations with Insecticide Use and Insect Control Recommendations. Insects. 2017;8.
Dorman SJ, Gross AD, Musser FR, Catchot BD, Smith RH, Reisig DD, et al. Resistance monitoring to four insecticides and mechanisms of resistance in Lygus lineolaris Palisot de Beauvois (Hemiptera: Miridae) populations of southeastern USA cotton. Pest Manag Sci. 2020;76:3935–44.
Gore J, Snodgrass G, Jackson R, Catchot A, editors. Managing difficult to control tarnished plant bugs in the Mid-South. Memphis, TN: Proc Beltwide Cotton Conf, National Cotton Council of America; 2007.
Roush RT, McKenzie JA. Ecological genetics of insecticide and acaricide resistance. Ann Rev Entomol. 1987;32:361–80.
Li X, Schuler MA, Berenbaum MR. Molecular mechanisms of metabolic resistance to synthetic and natural xenobiotics. Ann Rev Entomol. 2007;52:231–53.
Guedes NM, Guedes RN, Ferreira GH, Silva LB. Flight take-off and walking behavior of insecticide-susceptible and -resistant strains of Sitophilus zeamais exposed to deltamethrin. Bull Entomol Res. 2009;99:393–400.
Antonio-Nkondjio C, Sonhafouo-Chiana N, Ngadjeu C, Doumbe-Belisse P, Talipouo A, Djamouko-Djonkam L, et al. Review of the evolution of insecticide resistance in main malaria vectors in Cameroon from 1990 to 2017. Parasites Vectors. 2017;10:1–14.
Black WC, Baer CF, Antolin MF, DuTeau NM. Population genomics: genome-wide sampling of insect populations. Annu Rev Entomol. 2001;46:441–69.
Fenton B, Margaritopoulos JT, Malloch GL, Foster SP. Micro-evolutionary change in relation to insecticide resistance in the peach-potato aphid, Myzus persicae. Ecol Entomol. 2010;35:131–46.
Pelissie B, Crossley MS, Cohen ZP, Schoville SD. Rapid evolution in insect pests: the importance of space and time in population genomics studies. Curr Opin insect Sci. 2018;26:8–16.
Webster MT, Beaurepaire A, Neumann P, Stolle E. Population Genomics for Insect Conservation. Annu Rev Anim Biosci. 2023;11:115–40.
Calla B, Demkovich M, Siegel JP, Viana JPG, Walden KK, Robertson HM, et al. Selective sweeps in a nutshell: the genomic footprint of rapid insecticide resistance evolution in the almond agroecosystem. Genome Biol Evol. 2021;13:evaa234.
Hereward JP, Cai X, Matias AMA, Walter GH, Xu C, Wang Y. Migration dynamics of an important rice pest: the brown planthopper (Nilaparvata lugens) across Asia—Insights from population genomics. Evol Appl. 2020;13:2449–59.
Schmidt TL, Swan T, Chung J, Karl S, Demok S, Yang Q, et al. Spatial population genomics of a recent mosquito invasion. Mol Ecol. 2021;30:1174–89.
Perera OP, Shelby KS, Pierce CA, Snodgrass GL. Expression profiles of Digestive genes in the gut and salivary glands of tarnished plant bug (Hemiptera: Miridae). J Insect Sci. 2021;21.
Perera O, Snodgrass G, Scheffler B, Gore J, Abel C. Characterization of eight polymorphic microsatellite markers in the tarnished plant bug, Lygus lineolaris (Palisot de Beauvois). Mol Ecol Notes. 2007;7:987–9.
Zhu YC, Luttrell R. Altered gene regulation and potential association with metabolic resistance development to imidacloprid in the tarnished plant bug, Lygus lineolaris. Pest Manag Sci. 2015;71:40–57.
Showmaker KC, Bednářová A, Gresham C, Hsu C-Y, Peterson DG, Krishnan N. Insight into the salivary gland transcriptome of Lygus lineolaris (Palisot de Beauvois). PLoS ONE. 2016;11:e0147197.
Perera OP, Gore J, Snodgrass GL, Jackson RE, Allen KC, Abel CA, et al. Temporal and spatial genetic variability among tarnished plant bug (Hemiptera: Miridae) populations in a small geographic area. Ann Entomol Soc Am. 2015;108:181–92.
Hull JJ, Perera OP, Wang MX. Molecular cloning and comparative analysis of transcripts encoding chemosensory proteins from two plant bugs, Lygus lineolaris and Lygus hesperus. Insect Sci. 2020;27:404–24.
i5K-Consortium. The i5K Initiative: advancing arthropod genomics for knowledge, human health, agriculture, and the environment. J Hered. 2013;104:595–600.
Childers AK, Geib SM, Sim SB, Poelchau MF, Coates BS, Simmonds TJ, et al. The USDA-ARS Ag100Pest Initiative: High-Quality Genome Assemblies for Agricultural Pest Arthropod Research. Insects. 2021;12:626.
Saha S, Cooper WR, Hunter W, Mueller L, Consortium A. An Open Access Resource Portal for Arthropod Vectors and Agricultural Pathosystems: AgriVectors. Org. 1st Int Electron Conf Entomol. 2021. https://doi.org/10.3390/IECE-10576.
Giraldo-Calderón GI, Emrich SJ, MacCallum RM, Maslen G, Dialynas E, Topalis P, et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 2015;43:D707–13.
Chapman JA, Ho I, Sunkara S, Luo S, Schroth GP, Rokhsar DS. Meraculous: de novo genome assembly with short paired-end reads. PLoS ONE. 2011;6:e23501.
Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021;38:4647–54.
Akingbohungbe A. Chromosome numbers of some north american mirids (Heteroptera: Miridae). Can J Genet Cytol. 1974;16:251–6.
Storer J, Hubley R, Rosen J, Wheeler TJ, Smit AF. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA. 2021;12:2.
Magalhaes LC, van Kretschmar JB, Donohue KV, Roe M. Pyrosequencing of the adult tarnished plant bug, Lygus lineolaris, and characterization of messages important in metabolism and development. Entomol Exp Appl. 2013;146:364–78.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Saha S, Allen KC, Mueller LA, Reddy GV, Perera OP. Chromosome length genome assembly of the redbanded stink bug, Piezodorus guildinii (Westwood). BMC Res Notes. 2022;15:1–5.
Putnam NH, O’Connell BL, Stites JC, Rice BJ, Blanchette M, Calef R, et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016;26:342–50.
Roehrdanz R, Cameron SL, Toutges M, Wichmann SS. The complete mitochondrial genome of the tarnished plant bug, Lygus lineolaris (Heteroptera: Miridae). Mitochondrial DNA A. 2016;27:48–9.
Acknowledgements
We thank Calvin Pierce (USDA ARS SIMRU) for assistance with TPB colony maintenance and sample preparation. The use or mention of a trademark or proprietary product does not constitute an endorsement, guarantee, or warranty. It does not imply its approval to exclude other suitable products by the U.S. Department of Agriculture, an equal opportunity employer.
Funding
This research was funded by USDA Agricultural Research Service in house research project 6066-22000-091-00D of the Southern Insect Management Research Unit. Cotton Incorporated provided grants 12–349 and 15–105 to OPP provided partial funding.
Author information
Authors and Affiliations
Contributions
Conceptualization, OPP; methodology, OPP, SS, JPG, SG, JSJ, MD, TS; formal analysis, OPP, SG, JSJ, SS, TS; investigation, OPP, SS, JPG, KAP, KCA, SG, JSJ, MD, TS, GVPR; resources, OPP, GVPR, RK; data curation, OPP, SS, MD, TS; writing-original draft preparation, OPP, SS; writing-review and editing, OPP, SS, KCA, JPG, KAP, KCA, RK, JSJ, GVPR; project administration, OPP. All authors have read and agreed to the published version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Perera, O.P., Saha, S., Glover, J. et al. A chromosome scale assembly of the tarnished plant bug, Lygus lineolaris (Palisot de Beauvois), genome. BMC Res Notes 16, 125 (2023). https://doi.org/10.1186/s13104-023-06408-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13104-023-06408-w