
Abstract
Microplitis manilae Ashmead (Hymenoptera: Braconidae) is an important parasitoid of agricultural pests in lepidopteran species. So far, two extant genome assembles from the genus Microplitis are fragmented. Here, we offered a high-quality genome assembly of M. manilae at the chromosome level with high accuracy and contiguity, assembled by ONT long-read, MGI-SEQ short-read, and Hi-C sequencing methods. The final assembled genome size was 282.85 Mb, with 268.17 Mb assigned to 11 pseudochromosomes. The scaffold N50 length was 25.23 Mb, and the complete BUSCO score was 98.61%. The genome contained 152.37 Mb of repetitive elements, representing 53.87% of the total genome size. We predicted 15,689 protein-coding genes, of which 13,580 genes were annotated functionally. Gene family evolution investigations of M. manilae revealed 615 expanded and 635 contracted gene families. The high-quality genome of M. manilae reported in this paper will be a useful genomic resource for research on parasitoid wasps in the future.
Similar content being viewed by others


Background & Summary
Microplitis manilae Ahmead (Hymenoptera: Braconidae: Microgastrinae) is a solitary endoparasitoid wasp and is primarily distributed in the Asia-Pacific region1. It attacks several lepidopteran species, with Spodoptera species being its preferred target, including S. frugiperda, S. exigua and S. litura2, which of them are the world’s most significant agricultural pests3. M. manilae is thought to be an ideal biological control agent for Spodoptera spp.
So far, it has approximately 200 species have been recognized within Microplitis1, and some of them, i.e. M. croceipes, M. demolitor and M. mediator, have been widely used in biological pest control4,5,6. The virulence factors of Microplitis wasps that act to suppress or circumvent host immunity are primarily composed of polydnaviruses (PDV), venom, and teratocytes7,8. In recent years, the biology, ecology, and interaction with the host of Microplitis have been studied9,10. The study of the interactions between parasitoid wasps and their host insects, particularly the regulation of host immunity and development by parasitoid wasps, has great potential for increasing the use of parasitoid wasps in sustainable pest management in agriculture. To further understand the complex relationship between parasitoids and their hosts, high quality genome data would play an important role. The genome at the chromosome level may shed light on the evolution of parasites, the mechanisms of parasitism, the potential for developing new techniques for biological control and utilizing natural enemies as resources. However, only two fragmented genomes from the genus Microplitis (M. demolitor and M. mediator) are currently available in NCBI and a chromosome-level genome assembly for Microplitis spp. has not been published.
In this study, we used MGI short-read, ONT long-read and Hi-C sequencing technologies to assemble the M. manilae chromosome-level genome. The final genome size was 282.85 Mb with a scaffold N50 length of 25.23 Mb, and 268.17 Mb assembled genome sequences were successfully anchored on 11 chromosomes. In total, 15,689 protein-coding genes were identified, and 13,580 of them were functionally annotated.
Methods
Insect collection and rearing
The wasps Microplitis manilae were collected from maize fields in Dongfang City, Hainan Province, China (18.86°N, 108.72°E) in November 2020 and reared using their host Spodoptera frugiperda under laboratory conditions of 26 ± 1 °C, 65 ± 5% RH, and a 14 h light: 10 h dark photoperiod.
Sequencing
The extraction of DNA and RNA was performed on newly emerged male individuals that had been raised for five or more generations. Genomic DNA was obtained using the Blood & Cell Culture DNA Mini Kit (Qiagen, Hilden, Germany) for both long-read and short-read whole genome sequencing. RNA was isolated using the TRlzol reagent (Vazyme, Nanjing, China). The Hi-C library was generated using the restriction endonuclease DpnII. Long-read sequencing was carried out using the Nanopore PromethION platform (Oxford Nanopore Technologies, UK), with an insert size of approximately 20 kb. Short-read and transcriptome sequencing were performed using libraries with an insert size of 350 bp and sequenced on the MGISEQ. 2000 platform. The total data generated from the long-read sequencing was 76.31 Gb, while the total data generated from the short-read sequencing was 82.60 Gb (Table 1).
Genome size estimation and assembly
The raw reads obtained from the MGISEQ. 2000 platform were subjected to quality control using fastp v0.21.011 to filter adapter sequences and low-quality reads. The remaining reads of MGI library were then used to estimate the genome size of M. manilae by GenomeScope v1.0.012 and analyze the 17-mer distribution with Jellyfish v2.3.013. The final genome size was estimated to be 297.29 Mb through K-mer analysis.
The draft genome is obtained by first assembling long reads and then polishing the results with short reads, which has been widely used in genome assembly research for different organisms recently14,15,16,17,18. NextDenovo v2.5.0 (https://github.com/Nextomics/NextDenovo) was used to assemble the initial assembly with ONT sequences. NextPolish v1.4.019 was then applied to polish the draft genome assembly using MGISEQ sequences. Juicer v1.6.220 was used to align Hi-C reads to the draft assembly and subject them to quality control. 3D-DNA21 was used to anchor primary contigs into chromosomes, then corrected the possible errors manually with Juicebox v1.11.0822. The final genome assembly of M. manilae was 282.85 Mb with a scaffold N50 of 25.23 Mb. The Hi-C analyses scaffolded 11 pseudomolecules (Fig. 1), anchoring 94.81% (268.17 Mb) of the genome assembly of M. manilae. The average GC content of M. manilae genome assembly was 31.26% (Table 2, Fig. 2).

Heat map of Hi-C assembly of Microplitis manilae. The scale bar represents the interaction frequency of Hi-C links.

Genome characteristics of Microplitis manilae. (1) Pseudo-chromosomes; (2) gene distribution; (3) GC content; (4) repeat distribution; (5) ncRNA distribution.
The genome completeness was evaluated with the BUSCO v4.1.4 pipeline23, searching against the insect_odb10 database24. The analysis identified 98.61% (single-copied genes: 97.88%, duplicated genes: 0.73%), 0.44%, and 0.95% of the 1,367 predicted genes in this genome as complete, fragmented, and missing sequences, respectively. These results suggested the assembled genome is highly complete.
Genome annotation
The genome of M. manilae was annotated for repetitive elements, non-coding RNAs (ncRNAs), and protein-coding genes (PCGs). The Extensive de novo TE Annotator (EDTA) pipeline25 was used to build TE libraries for repeat annotation initially. Non-LTR retrotransposons and any unclassified TEs missed by the TE annotators mentioned above were then identified by RepeatModeler v2.0.226. A comprehensive non-redundant TE library was generated combining with above results and Dfam3.227. RepeatMasker v4.1.2 (http://www.repeatmasker.org) was subsequently used to search for known and novel TEs. In the genomic sequences, a total of 152.37 Mb repetitive elements were identified, constituting 53.87% of the total. The most abundant repeating element was DNA transposons (13.54%), followed by long terminal repeats (LTR, 10.43%) and long interspersed nuclear elements (LINEs, 1.75%), while unclassified repeats made up 27.43% of the total (Table 3, Fig. 2). Infernal 1.1.228 was used to identify rRNAs, snRNAs, and miRNAs based on the alignment with the Rfam library29. tRNAscan-SE v2.0.630 was used to predict tRNAs. Finally, 1,894 noncoding RNAs were predicted, including 1,269 transfer RNAs (tRNAs), 194 ribosomal RNAs (rRNAs), 74 micro-RNAs (miRNAs), 63 small nuclear RNAs (snRNAs), and 294 others (Table S1, Fig. 2).
Three different strategies were applied for the annotation of PCGs: transcriptome-based prediction, de novo gene prediction, and homology-based prediction. In transcriptome-based prediction, the transcriptome was assembled from RNA-seq alignments by HISAT2 v2.2.131 and the candidate coding region was identified by PASA pipeline v2.4.1 (https://github.com/PASApipeline/PASApipeline). The repeat-masked genome was analyzed using AUGUSTUS v3.3.332 and SNAP v2006-07-2833 for de novo gene prediction. The protein sequences of hymenopteran species were downloaded from the NCBI Database as references for homology-based prediction. Exonerate v2.4.034 was utilized to align the reference proteins to the genome assembly and predict gene structures. Finally, a consensus gene set was created by integrating the genes predicted by the aforementioned three methods using EVidenceModeler v1.1.135. We predicted 15,689 protein-coding genes for the M. manilae genome by combining the evidences from the transcriptome, ab initio, and homology-based predictions. The average length of the predicted gene was 8,718 base pairs, while that of a protein-coding region was 1,575 bp. Exon and intron lengths on average were 319 and 1,814 bp, respectively. There were 4.9 exons on average per gene (Table 4).
Gene functions were annotated using BLASTP v2.9.036 (-evalue 1e-5) to search against UniProtKB (Swiss-Prot + TrEMBL)37, and InterProScan 5.52-86.038 to search against the Pfam39, CDD40, Gene3D41, Smart42, and Superfamily43 databases. The eggnog-mapper v2.1.444 was used to predict conserved sequences and domains, GO terms, and KEGG pathways against the eggnog v5.0 database45. A total of 13,580 (86.56%) genes were functionally annotated against the UniProtKB database. In integrating with InterProScan and eggnog annotation results, 13,227 (84.31%) protein-coding genes with protein domains were identified, which were assigned 11,276 COG Functional Categories genes, 9,489 Reactome pathways, 7,819 MetaCyc, 7,722 GO terms, 7,324 KEGG KO terms, and 4,274 KEGG pathways, respectively.
Data Records
The MGI, ONT, RNA-seq and Hi-C sequencing data used for the genome assembly have been deposited in the NCBI Sequence Read Archive (SRA) database with accession numbers SRR2135882846, SRR2135882747, SRR2135882948 and SRR2135882649, respectively, under the BioProject accession number PRJNA872950. The chromosomal assembly has been deposited at GenBank with accession number JAPFQK00000000050. Genome annotation information has been deposited in the Figshare database51.
Technical Validation
Evaluating the quality of the genome assembly
The quality of M. manilae genome assembly was evaluated using two approaches. Firstly, sequencing data were mapped to the genome to verify the accuracy, yielding mapping rates of 99.52% for MGI, 94.40% for RNA-seq, and 98.52% for ONT data. Secondly, BUSCO analysis found 98.6% of the 1,367 single-copy orthologues (in the insects_odb10 database) to be complete (97.9% single-copied genes and 0.7% duplicated genes), 0.4% fragmented, and 1.0% missing.
Chromosome synteny
Chromosome synteny between M. manilae and Cotesia congregata was detected by MCScanX52 with default parameters. The genome assembly of C. congregata53 was retrieved from NCBI with accession number GCA_905319865.3. The visual diagram was generated using TBtools54. The synteny of the M. manilae assembly was compared to that of C. congregata, a closely related species of the subfamily Microgastrinae. The results showed a low level of synteny between M. manilae and C. congregata (Fig. 3). A number of fusion and fission events were detected between these two wasps. For instance, Chr11 and a part of Chr5 of M. manilae were syntenic to Chr4 of C. congregata, whereas Chr1 of M. manilae was syntenic to a portion of Chr2 and Chr3 of C. congregata. Low genome synteny was also identified between Nasonia vitripennis and Pteromalus puparum, both of which are members of the same family Pteromalidae55.

Chromosomal synteny between Microplitis manilae and Cotesia congregata genomes.
Gene annotation validation
OrthoFinder v2.5.456 was utilized to infer sequence orthology, based on protein annotation sequences of 11 additional hymenopteran organisms retrieved from NCBI, including Apis mellifera, Athalia rosae, Bombus terrestris, Chelonus insularis, Diachasma alloeum, Fopius arisanus, M. demolitor, Nasonia vitripennis, Orussus abietinus, Polistes dominula, and Venturia canescens (Table S2). A total of 132,122 genes were assigned to 12,544 gene families. Among them, 4,910 gene families were presented in all the species genomes, with 3,780 single-copy and 1,130 multicopy gene families. In the 15,689 predicted genes of M. manilae, 14,822 (94.47%) were grouped into 9,725 families. There were 1,295 genes in 241 families unique to M. manilae (Fig. 4, Table S3).

Distribution of genes in different Hymenoptera species. “1:1:1” represents shared single-copy genes, “N:N:N” as multicopy genes shared by all species, “others” as unclassified orthologs, “unassigned” as orthologs which cannot be assigned into any gene families (orthogroups).
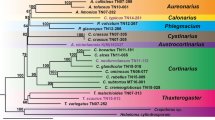
All single-copy protein sequences were concatenated into one data matrix after being aligned with MAFFT v7.42757. The phylogenetic tree was constructed using IQ-TREE v2.0.558 with the best model (JTT + F + R7) estimated by ModelFinder59. Statistical support for the phylogenetic trees was evaluated by Ultrafast bootstrap60 analysis using 1000 replicates. The phylogenetic tree reconstructed by IQ-TREE had high bootstrap support values. The topology of the phylogeny was consistent with that of the previous study61. The MCMCTree package in PAML v4.9j62 was used to estimate divergence times. Based on a previous study, five calibration time points were used: root holometabolous: <300 million years ago (mya); Orussoidea + Apocrita: 211–289 mya; Apocrita: 203–276 mya; Aculeata: 160–224 mya; and Ichneumonoidea: 151–218 mya61. As expected, our analysis revealed that M. manilae was closely related to M. demolitor and these two species diverged approximately 7.6 mya (Fig. 5). CAFE v4.2.163 was used to estimate gene family expansions and contractions with a p value of 0.01. Finally, we found 615 and 635 gene families experienced expansions and contractions in M. manilae, respectively, and 395 (310 expanded and 85 contracted) of them were rapidly evolved (Fig. 5).

Phylogenetic and gene family evolution analyses of Microplitis manilae and 11 other Hymenoptera species. The bootstrap values of all nodes are supported at 100/100. Node values indicate the number of gene families showing expansion, contraction, and rapid evolution. The scale at the bottom of the figure represents the divergence time.
Code availability
This work did not utilize a custom script. Data processing was carried out using the protocols and manuals of the relevant bioinformatics software.
References
Fernandez-Triana, J., Shaw, M. R., Boudreault, C., Beaudin, M. & Broad, G. R. Annotated and illustrated world checklist of Microgastrinae parasitoid wasps (Hymenoptera, Braconidae). Zookeys, 1–1089 (2020).
Gupta, A. Revision of the Indian Microplitis Foerster (Hymenoptera: Braconidae: Microgastrinae), with description of one new species. Zootaxa 3620, 429–452 (2013).
Huang, S.-H. et al. Insecticidal activity of pogostone against Spodoptera litura and Spodoptera exigua (Lepidoptera: Noctuidae). Pest Manag. Sci. 70, 510–516 (2014).
Powell, J. E. & King, E. G. Behavior of adult Microplitis croceipes (Hymenoptera: Braconidae) and parasitism of Heliothis spp. (Lepidoptera: Noctuidae) host larvae in cotton. Environ. Entomol. 13, 272–277 (1984).
Shepard, M., Powell, J. E. & Jones, W. A. Biology of Microplitis demolitor (Hymenoptera: Braconidae), an Imported Parasitoid of Heliothis (Lepidoptera: Noctuidae) spp. and the Soybean Looper, Pseudoplusia includens (Lepidoptera: Noctuidae). Environ. Entomol. 12, 641–645 (1983).
Yu, H. et al. Electrophysiological and Behavioral Responses of Microplitis mediator (Hymenoptera: Braconidae) to Caterpillar-Induced Volatiles From Cotton. Environ. Entomol. 39, 600–609 (2010).
Lin, Z. et al. Insights into the venom protein components of Microplitis mediator, an endoparasitoid wasp. Insect Biochem. Mol. Biol. 105, 33–42 (2019).
Burke, G. R., Simmonds, T. J., Thomas, S. A. & Strand, M. R. Microplitis demolitor bracovirus proviral loci and clustered replication genes exhibit distinct DNA amplification patterns during replication. J. Virol. 89, 9511–9523 (2015).
Tang, C. K. et al. MicroRNAs from Snellenius manilae bracovirus regulate innate and cellular immune responses of its host Spodoptera litura. Commun. Biol. 4, 11 (2021).
Qiu, B., Zhou, Z.-S., Luo, S.-P. & Xu, Z.-F. Effect of temperature on development, survival, and fecundity of Microplitis manilae (Hymenoptera: Braconidae). Environ. Entomol. 41, 657–664 (2012).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204 (2017).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Hu, Y. et al. Genome assembly and population genomic analysis provide insights into the evolution of modern sweet corn. Nat. Commun. 12, 1227 (2021).
Karimi, K. et al. A chromosome-level genome assembly reveals genomic characteristics of the American mink (Neogale vison). Commun. Biol. 5, 1381 (2022).
Li, R., Zhang, M., Cha, M., Xiang, J. & Yi, X. Chromosome-level genome assembly of the Siberian chipmunk (Tamias sibiricus). Sci. Data 9, 783 (2022).
Liao, N. et al. Chromosome-level genome assembly of bunching onion illuminates genome evolution and flavor formation in Allium crops. Nat. Commun. 13, 6690 (2022).
Liu, Z. et al. Chromosome-level genome assembly and population genomic analyses provide insights into adaptive evolution of the red turpentine beetle, Dendroctonus valens. BMC Biol. 20, 190 (2022).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Zdobnov, E. M. et al. OrthoDB in 2020: evolutionary and functional annotations of orthologs. Nucleic Acids. Res. 49, D389–D393 (2021).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457 (2020).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids. Res. 44, D81–D89 (2016).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids. Res. 49, D192–D200 (2021).
Chan, P. P., Lin, B. Y., Mak, A. J. & Lowe, T. M. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids. Res. 49, 9077–9096 (2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Stanke, M., Diekhans, M., Baertsch, R. & Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644 (2008).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Slater, G. S. & Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Camacho, C. et al. BLAST plus: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Bateman, A. et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids. Res. 49, D480–D489 (2021).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Mistry, J. et al. Pfam: the protein families database in 2021. Nucleic Acids. Res. 49, D412–D419 (2021).
Lu, S. et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids. Res. 48, D265–D268 (2020).
Lees, J. et al. Gene3D: a domain-based resource for comparative genomics, functional annotation and protein network analysis. Nucleic Acids. Res. 40, D465–D471 (2012).
Letunic, I., Khedkar, S. & Bork, P. SMART: recent updates, new developments and status in 2020. Nucleic Acids. Res. 49, D458–D460 (2021).
Wilson, D. et al. SUPERFAMILY-sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids. Res. 37, D380–D386 (2009).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids. Res. 47, D309–D314 (2019).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21358828 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21358827 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21358829 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR21358826 (2022).
NCBI GenBank https://identifiers.org/nucleotide:JAPFQK000000000 (2022).
Shu, X. H., Tang, P. & Chen, X. X. Chromosome-level genome assembly of Microplitis manilae Ashmead, 1904 (Hymenoptera: Braconidae). figshare https://doi.org/10.6084/m9.figshare.21971738 (2023).
Wang, Y. et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids. Res. 40, e49–e49 (2012).
Gauthier, J. et al. Chromosomal scale assembly of parasitic wasp genome reveals symbiotic virus colonization. Commun. Biol. 4, 104 (2021).
Chen, C. et al. TBtools: an Integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202 (2020).
Ye, X. et al. A chromosome-level genome assembly of the parasitoid wasp Pteromalus puparum. Mol. Ecol. Resour. 20, 1384–1402 (2020).
Emms, D. M. & Kelly, S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238 (2019).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Minh, B. Q. et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589 (2017).
Diep Thi, H., Chernomor, O., von Haeseler, A., Minh, B. Q. & Le Sy, V. UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522 (2018).
Peters, R. S. et al. Evolutionary history of the Hymenoptera. Curr. Biol. 27, 1013–1018 (2017).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007).
Han, M. V., Thomas, G. W. C., Lugo-Martinez, J. & Hahn, M. W. Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol. Biol. Evol. 30, 1987–1997 (2013).
Acknowledgements
This work was supported by the Key Project of Laboratory of Lingnan Modern Agriculture (NT2021003); the Key International Joint Research Program of National Natural Science Foundation of China (31920103005); the National Natural Science Foundation of China (32070467, 31901942); the Provincial Key Research and Development Plan of Zhejiang (2020C02003, 2021C02045); and the Fundamental Research Funds for the Central Universities (2021FZZX001-31).
Author information
Authors and Affiliations
Contributions
Conceptualization, supervision and funding acquisition, X.X.C. and P.T.; Resources, X.H.S. and P.T.; Software, X.Q.Y. and B.Y.Z.; Investigation, X.H.S., R.Z.Y, Z.Z.W.; Visualization, X.H.S. and R.Z.Y.; Writing, X.X.C., P.T. and X.H.S. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shu, X., Yuan, R., Zheng, B. et al. Chromosome-level genome assembly of Microplitis manilae Ashmead, 1904 (Hymenoptera: Braconidae). Sci Data 10, 266 (2023). https://doi.org/10.1038/s41597-023-02190-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02190-3
- Springer Nature Limited