
Abstract
Hymenoptera is an order accounting for a large proportion of species in Insecta, among which Chalcidoidea contains many parasitoid species of biocontrol significance. Currently, some species genomes in Chalcidoidea have been assembled, but the chromosome-level genomes of Aphelinidae are not yet available. Using Illumina, PacBio HiFi and Hi-C technologies, we assembled a genome assembly of Eretmocerus hayati (Aphelinidae, Hymenoptera), a worldwide biocontrol agent of whiteflies, at the chromosome level. The assembled genome size is 692.1 Mb with a contig N50 of 7.96 Mb. After Hi-C scaffolding, the contigs was assembled onto four chromosomes with a mapping rate of > 98%. The scaffold N50 length is 192.5 Mb, and Benchmarking Universal Single-Copy Orthologues (BUSCO) value is 95.9%. The genome contains 370.8 Mb repeat sequences and total of 24471 protein coding genes. P450 gene families were identified and analyzed. In conclusion, our chromosome-level genome assembly provides valuable support for future research on the evolution of parasitoid wasps and the interaction between hosts and parasitoid wasps.
Similar content being viewed by others

Background & Summary
Hymenoptera is a mega-diverse insect order with >169,000 described species, and a large proportion of species in this order are parasitoid wasps, such as the species of Chalcidoidea, that play an important role in the biological control of insect pests. Derived from the same ancestor that lived in the Permian or Triassic, parasitoid wasps have evolved numerous branches with unique characteristics to adapt to different living environments1.
Within Hymenoptera, over 30 species genomes in Chalcidoidea have been assembled hitherto, and some of them were assembled at the chromosome level, including Aphelinus atriplicis (Aphelinidae), Aphelinus certus (Aphelinidae)2, Muscidifurax raptorellus (Pteromalidae)3, Nasonia vitripennis (Pteromalidae)4,5, Pteromalus puparum (Pteromalidae)6, Theocolax elegans (Pteromalidae)7, Valisia javana (Agaonidae)8, Anastatus japonicus (Eupelmidae), Anastatus fulloi (Eupelmidae)9, Megastigmus duclouxiana (Megastigmidae) and Megastigmus sabinae (Megastigmidae)10. Among these, the chromosome-level assemblies of A. atriplicis and A. certus were generated according to a genetic map of hybrids between each other2.
Eretmocerus hayati (Aphelinidae, Hymenoptera) is an obligate parasitoid of the global prominent pest whitefly Bemisia tabaci (Aleyrodidae, Hemiptera)11,12,13. The release of this parasitoid wasp can effectively control the outbreak of whitefly14,15,16. The parasitoid wasps of the genus Eretmocerus have unique parasitic habits. With a curved tip on their ovipositor, they can lay eggs between the host nymphs and plant leaves instead of directly into host body17. The hatched first instar larva breaks through the abdomen of host, but still lives outside the host body, and does not burrow into the host body to complete the subsequent development until the host has developed to the last nymphal stage18,19. The first and second instar larvae are wrapped by the capsule structure produced by the host, and will not fully contact the host tissue until the third instar18.
Here, we report a chromosome-level genome assembly of E. hayati using combined Illumina, PacBio and Hi-C sequencing technology. The assembly is 692.1 Mb in length with a scaffold N50 of 192.5 Mb and BUSCO completeness 97.4%. 53.58% repeat sequences and 24471 protein coding sequences were identified. We also identified 74 P450 genes and discovered the expression patterns at different development stages and sex. This assembly provides a valuable resource for evolutionary and host-parasitoid interaction studies in parasitoid wasps as well as biological control application of E. hayati.
Methods
Sampling and genome sequencing
A strain of E. hayati was initially imported by the Chinese Academy of Agricultural Sciences in 2008 and had been reared on Gossypium hirsutum plants carrying whitefly nymphs in our laboratory (26 ± 1°C, 14 L:10D, 70 ± 10%RH) over seven years. To collect the newly emerged parasitoid wasps, the pupae of E. hayati were first collected in a petri dish, then a triangle funnel of appropriate size was inversely placed on the petri dish with a 1.5 ml plastic sampling tube on the outlet of the funnel neck (Fig. 1a,b). We anesthetized the emerged parasitoid wasps on ice and collected approximately 3000 newly emerged males for DNA extraction using QIAamp DNA Mini Kit (QIAGEN) (Fig. 1c,d). After extraction, the DNA purity, concentration and integrity were detected using NanoDrop 2000&8000, Qubit fluorescence photometer and Agilent 4200 Bioanalyzer, respectively.

Device and process for collecting adult male wasps. (a) Pupae of E. hayati. Male pupae are on the left and two female pupae on the right. The sides of the small square are 1.0 mm in length. (b) Device for collecting newly emerged parasitoid wasps. Pupae were collected and placed in the petri dish, and the newly emerged parasitoid wasps would move upward into the sampling tube. (c) Device for distinguishing the sex of parasitoid wasps and collecting male wasps. Newly emerged wasps were placed in a petri dish sitting on ice and were anesthetized for examination and collection. (d) Tubes for resuscitating parasitoid wasps. Anesthetized male wasps were collected and placed in these tubes for resuscitation.
A short-read sequencing library with an insert size of 350 bp was generated using Universal DNA Fragmentase kit V2.0 (Annoroad) and Universal DNA Library Prep Kit V2.0 (Annoroad) with 0.5 ug DNA as input material. Hiseq x ten platform was used to sequence the library and generate 47.65 Gb paired-end clean reads after removing adapters and low-quality reads.
For long-read library, the SMRT bell express template prep kit 2.0 (Pacific Biosciences) was used to construct a SMRT bell library using 8 ug sheared DNA. After size-selection, primer annealing and binding the SMRT bell templates to polymerases, the library was sequenced on the Pacific Bioscience Sequel II platform in Annoroad Gene Technology company (TianJin). Single SMRT cells were processed, generating 481.17 Gb of subreads data.
Estimation of genome size
Flow cytometry was performed as described below to estimate the genome size. The heads of E. hayati were cut off with a dissecting needle after parasitoid wasps were anesthetized on ice. Next, we collected 300 heads as one sample in a 1.5 mL centrifuge tube and ground them with an abrasive rod and 200 μL Galbraith’s nuclear dissociation solution20. Then the fully ground tissue solution was filtered with a 38 μm pore size nylon mesh to obtain the nuclear suspension. Then, the cells were centrifuged at 1000 r/min for 5 min at 4 °C.
Next the precipitated nuclei were resuspended with 400 μL 1 × PBS buffer by lightly scratching at the bottom of the tube. Finally, we added PI solution (final concentration was 50 μg/mL) and RNase A solution (final concentration was 20 μg/mL) to the nuclei, and stained the samples at 4 °C in dark for 5–20 min. Heads of Drosophila melanogaster (genome size is 176.4 Mb) were collected as a reference to calculate the size of the genome.
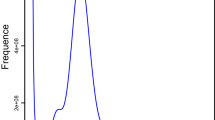
Flow cytometry results show that the DNA content in the nucleus of the female E. hayati was four times that of the female Drosophila melanogaster, indicating that the estimated genome size was about 756.227 Mb (Fig. 2a, Table 1).

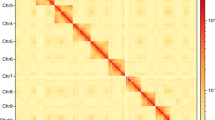
Genome assembly of Eretmocerus hayati. (a) Flow cytometry results of 4 samples. The ordinate is the number of nuclei, and the abscissa is the fluorescence of the nucleus. Drosophila melanogaster was used as reference sample. (b) Contig distribution on genome chromosomes. Grey bars represent different lengths of the corresponding chromosome. Rectangles of other colors represent contigs of different lengths loading on the chromosomes. (c) Genome-wide all by all Hi-C interaction heatmap of E. hayati (4 chromosomes, resolution 100 kb). The intensity of chromosomal interactions is shown on the right shading gradient. Intrachromosomal interactions (red blocks in the diagonal) are much stronger than interchromosomal interactions (light yellow blocks). (d) Chromosome-level genome assembly results information circle plot (window size 50 kb). A: chromosome information, B: gene density, C: GC content, D: second-generation sequencing depth, E: third-generation sequencing depth, F: heterozygous SNP distribution (outer) and homozygous SNP distribution (inner), G: heterozygous Indel distribution (outer) and homozygous Indel distribution (inner), H: single copy (outer) and multiple copy (inner) BUSCO genes distribution.
We also used k-mer distribution analysis21 to estimate the genome size and characteristics of E. hayati. For k-mer analyze, 47.65 Gb clean reads from the short-read library were used with K-mer size of 21. The genome size of E. hayati was estimated to be 694.3 Mb by 21 k-mer analysis, with a heterozygosity of about 0.01% and a repeat sequence ratio of 40.94% (Table 2).
Genome assembly
We used HiFiasm v0.15.1 to preliminarily assemble the E. hayati genome, which could resolve near-identical repeats and segmental duplications to generate better haplotype assemblies22. The HiFiasm outputs a primary assembly after performing all-versus-all read overlap alignment and correcting sequencing errors.
Purge Haplotigs software was used to complete genome de-redundancy after initial assembly and error corrected, and the redundant heterozygous contigs were identified and removed according to reads depth distribution and sequence similarity23.
The hybrid set with 47.65 Gb Illumina short read sequencing (67-fold coverage) and 481.17 Gb PacBio sequencing reads (693-fold coverage) (Table 3) was used to obtain a de novo genome assembly for E. hayati with total length of 763.42 Mb and contig N50 length of 7.96 Mb (Table 4).
Hi-C scaffolding
To obtain the genome at the chromosomal level, Hi-C technology was applied24. We commissioned Annoroad (TianJin) to construct a Hi-C sequencing library using more than 1500 adult male wasps. Firstly, DNA containing biotin was captured under the adsorption of avidin magnetic beads and terminal repair of DNA fragments was performed. Then poly A was added to the end of the DNA fragment, the joint was connected, the number of PCR amplification cycles was evaluated, and the final purification library was built. The library was sequenced on the Illumina platform and generated 70 Gb sequencing data (98-fold coverage), which was used to construct the genome at the chromosome level.
We used bowtie 2 (v2.2.3)25 to map the paired-end reads to the preliminary assembly. Then, HiC-Pro (v2.7.8)26 was used to detect the ligation site of unmapped reads, which were mainly composed of the chimeric regions spanning across the ligation junction. The 5’ fractions of unmapped reads were aligned back onto the genome. Next, in order to generate a high quality alignment file, we discarded the low mapping quality, multiple hitting reads and singletons according to the mapping results. To obtain the chromosome-scale scaffolds from the primary contig-level assembly, we used 3d-DNA27 and LACHESIS28 to treat the valid interaction pairs and generate the interaction matrices. According to the results of the karyotype study of Chalcidoidea, the subfamily Aphelininae harbored parasitoids with n = 4–529. We consulted the literature and the output results of 3D-DNA to assess the number of chromosomes, which appears to be four. To ascertain the precision of the outcomes, we built the interaction graph with Juicer30 and performed manual visual error correction using JuiceBox (v2.13.07) (https://github.com/aidenlab/Juicebox). After that, we cut the pseudochromosomes into equal 100 kb length bins and used Hic-Pro to constructe the heatmap showing the significant linkage clusters on diagonal.
The 244 contigs were divided, anchored, sorted, oriented, and merged into 4 chromosomes using LACHESIS and corrected by JuiceBox (Fig. 2b), with 13 sequences unanchored. The chromosomal heat map showed good collinearity on the diagonal (Fig. 2c), which confirms the high quality of scaffolding. The final genome assembly was 692.1 Mb with a scaffold N50 of 192.5 Mb (Table 4, Fig. 2d).
Genome completeness and quality assessment
We used three methods to assess the completeness and quality of the assembly. First, the genome sequence was split into 1000 bp fragments, and then these fragments were BLASTN against the NCBI nucleotide database (NT library). Second, we used BWA (v0.7.17)31 and Minimap2 (v2.24)32 to compare the second and third generation sequencing data with the genome, and calculated the comparison rate and sequence coverage. Third, BUSCO (v5.3.2)33 was used to estimate the gene space of chromosome-level genome assembly by searching the 1367 BUSCO genes in insecta_odb10.
The best five hits of BLASTN again NCBI nt database were from Nasonia, Gossypium, Eretmocerus, Ceratosolen and Copidosoma (Table 5). 96.09% of the Illumina short reads and 100% of the PacBio HiFi long reads were mapped to the genome assembly successfully. Moreover, we compared the insect_odb10 database using BUSCO. The assessment showed 95.9% of BUSCO genes were successfully detected, of which 94.6% were single copy and 1.3% were duplicated (Table 6). The results of these evaluations indicate that the genome assembly has a high level of completeness and accuracy.
In addition, the alignment results of second generation sequencing data were used to identify SNP and INDELS using GATK (v4.4.0.0)34 with hard filtering parameters “–filter-expression “QD < 2.0 || FS > 60.0 || SOR > 3.0 || MQRankSum < −12.5 || ReadPosRankSum < −8.0””. VCFtools (0.1.16)35 was used to calculate the density of SNP and INDELS on the assembly, and window size was 50,000 bp.
RNA sequencing and analysis
A total of 17 RNA-seq libraries were constructed, including four different development stages and various tissues of E. hayati. We extracted total RNA from egg (<post-deposition, 2000 individuals, single sample), 1st instar larva (1-2 d after hatching and 5–6 d after the eggs were first deposited, still attached to the leaf, 1000 individuals, three replicates), later instar larva (7–8 d after hatching and 10–11 d after the eggs were first deposited, mostly 3rd-4th instar within the body of whitefly nymphs and sampled by dissecting whitefly nymphs, 100 individuals, three replicates), female adult (<12 h post emergence and without exposure to male wasps, 150 individuals, three replicates), male adult (<12 h post emergence and without exposure to female wasps, 150 individuals, three replicates), female head (anaesthetized and dissected on ice with the method described above, 600 individuals, single sample), and male head (anaesthetized and dissected on ice with the method described above, 600 individuals, single sample). Total RNA was used for library preparations, which were sequenced on an Illumina Novaseq platform. All 150 bp paired-end raw reads were processed and a total of 98.5 Gb (approximately 14 Gb per sample) clean reads data were obtained. Then the clean reads were used to annotate the genome.
Repeat sequence annotation
We identified repeat sequences and transposable elements (TEs) using the methods of de novo assembly and homologous prediction. First, we used RepeatModeler (v2.0.3) (https://github.com/Dfam-consortium/RepeatModeler) to predict the repeat sequence with default parameters. Then RepBase database36 and RepeatMasker (v4.1.0) (https://github.com/rmhubley/RepeatMasker) were used to annotate the sequence homologous.
The results showed that 370.8 Mb are repeat sequences, accounting for 53.58% of the E. hayati genome. Among these repeat sequences, most (32.39%) are unclassified elements, followed by 8% of long terminal repeats (LTRs), 7.27% of DNA elements, 5.63% of long interspersed nuclear elements (LINEs) and only 0.29% of short interspersed nuclear elements (SINEs) (Table 7).
Gene prediction and function assignment
We annotated protein coding genes in the E. hayati genome using a pipeline that combines de novo prediction, homology searching and transcriptome evidence37. The de novo prediction was implemented with BRAKER (v2)38. For details, we randomly selected 10000 intact E. hayati genes from the transcriptomes to generate prediction parameters using AUGUSTUS (v3.1)39, GeneMark-ET Suite (v4.21)40, and SNAP (v2006-07-28)41. After that, the newly trained parameters were applied to the repeat sequence masked genome to predict genes following the BRAKER pipeline. For homology searching, the protein sequences of Nasonia vitripennis5 and Encarsia Formosa were TBLASTN (v2.8.1) (E-value < 1e-5) against the E. hayati genome42. Then, the gene models were defined by aligning the genome sequences to the matched protein using GeneWise (v2.4.1)43. For transcriptome evidence, we aligned the RNA-seq reads to the E. hayati genome by Hisat2 (v2.2.1)44 to identify gene information such as candidate exon regions, donor, and acceptor sites. StringTie45 was used to assemble the alignments into transcripts. Finally, the above three lines of gene prediction evidence were integrated with EVidenceModeler (EVM)46 to generate a consensus gene set. Specifically, the weight for different methods were “4”, “5” and “10” for de novo prediction, homology searching, and transcriptome evidence, respectively.
We used several convention methods to annotate the function of predicted protein coding genes. First, gene ontology (GO) annotation was generated by using both PANNZER247 web server and BLAST2GO (v5)48 with default parameters. Second, online service of BLASTKOALA (v2.2)49 was used to map the protein sequences of E. hayati to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways with database parameter “family_eukaryotes”. InterProScan (v5.57-90.0)50 was used to obtain motifs and domains information of protein sequences by searching the default available database (e.g. CDD, Gene3D, PANTHER, Pfam and SMART). To obtain function prediction of protein sequences, we combined the results of eggNOG-mapper (v2)51 and blastp against SwissProt and Trembl database (E-value < 1e-5).
A total of 24,471 protein coding genes were annotated following the pipeline combined with above-mentioned three methods. In total, 7,803 genes could be assigned with GO terms and 7,159 genes with KEGG ID. In addition, 16808 genes have BLASTN hits in NCBI non-redundant database, 11,556 genes contain Pfam domains (Table 8). Finally, a total number of 23974 genes were annotated by at least one database. Gene density was generated by TBtools (v1.120)52.
Cytochrome P450 genes
P450 genes are key genes closely related to insect stress resistance and environmental adaptation. E. hayati shows many unique and interesting features in association with host. For example, its eggs and first instar larvae survive in vitro, and the subsequent stages survive inside the host body. We hope to infer the mechanism of its unique parasitic habits through the identification and expression pattern analysis of P450 genes. To identify the P450 genes in the E. hayati genome, we first downloaded P450 protein sequences from UniPortKB, GeneBank and FlyBase to build a reference database. All predicted protein sequences of E. hayati were used as query to BLASTP against the P450 reference sequences. The Blast hits were retained for further domain analysis using InterProScan. Finally, genes with P450 conservative domain pf00047 were retained and classified according to the BLASTP analysis results.
To reconstruct the phylogeny tree of P450 genes from E. hayati, E. Formosa and N. vitripennis, we used MAFFT (v7.471)53 with the parameter “-auto” to align P450 sequences. The alignment was filtered by TRIMAL (v1.4.rev22)54 with the parameter “-automated1”,and IQ-tree (v2.1.2)55 with parameters “-m MFP -bb 1000” was used to construct the maximum likelihood (ML) phylogenetic tree with the best model (JTT + F + R8) estimated by ModelFinder56. RSEM (v1.3.3)57 was used to map the transcriptome clean data to the P450 sequences to estimate expression level of P450 genes in various tissues.
In total, we annotated 74 P450 genes in E. hayati, among which 6 genes belong to CYP2 clan, 42 belong to CYP3 clan, 19 belong to CYP4 clan, and 7 belong to Mito clan (Table 9). Chromosome mapping results show that the P450 genes are distributed on all four chromosomes, with more on chromosome 1, 2 and 4, and fewer on chromosome 3. There are six gene clusters containing three or more P450 genes on four chromosomes (Fig. 3a). The results of the phylogenetic tree show that the P450 genes from E. hayati, E. formosa and N. vitripennis expanded in the CYP3 and CYP4 branches (Fig. 3b).

Chromosomal location, classification and expression of P450 genes. (a) The location of the P450 genes on chromosomes. The density of the chromosomal genes is displayed by stripes of different colors. The superfamily type of the P450 gene is represented by the text color of the gene name, orange represents CYP4, blue represents CYP3, green represents CYP2, and purple represents Mito. (b) The maximum-likelihood phylogenetic tree of P450 genes derived from E. hayati, E. formosa and N. vitripennis using IQ-TREE software. The branch colors indicate different P450 superfamilies, consistent with the above description. (c) FPKM values (log2 transformed) of P450 genes in different tissues. Gene superfamilies are marked on the left using colored squares and tissue types are marked below (E: eggs; F: 1st instar larvae; T: later instar larvae; FA: female adults; MA: male adults; numbers represent biological replicates).
The expression patterns of P450 genes differ significantly among various tissues. Some CYP3 clan genes are expressed in all stages, but the expression levels are significantly higher in adult male wasps than in other stages (e.g. Ehay097930.1, Ehay252140.1, and Ehay264040.1). Some CYP3 clan genes are expressed only in the adult stage and are expressed at higher levels in male than in females (e.g. Ehay163240.1, Ehay288700.1, and Ehay270900.1). Some P450 genes show relatively higher expression levels in the 1st instar larvae (e.g. Ehay260430.1 and Ehay170850.1), and others show higher expression levels in later instar larvae (e.g. Ehay202870.1 and Ehay269780.1) (Fig. 3c).
P450 genes are highly related to responses to external stimuli and stress, and whether the different expression levels of P450 in different stages of E. hayati are related to the parasitic behavior of the wasp in vivo and in vitro deserves further study.
Data Records
Illumina, PacBio and Hi-C data for E. hayati genome sequencing have been deposited in the NCBI Sequence Read Archive with accession number SRR24094107, SRR24099399 and SRR24094118 under BioProject accession number PRJNA95147858.
Illumina transcriptome data for egg (SRR24094058, SRR24094059, SRR24094060), 1st instar larva (SRR24094061, SRR24094062, SRR24094063), later instar larva (SRR24094064, SRR24094050, SRR24094051), female adult (SRR24094052, SRR24094053, SRR24094054), male adult (SRR24094055, SRR24094056, SRR24094057), female head (SRR24094065) and male head (SRR24094066) are available under Bioproject PRJNA95147858.
This Whole Genome Shotgun project has been deposited at GenBank under the accession JARUXH00000000059. The version described in this paper is version JARUXH010000000.
Technical Validation
After extraction, the DNA purity, concentration and integrity were detected using NanoDrop 2000&8000, Qubit fluorescence photometer, and Agilent 4200 Bioanalyzer (Agilent Technologies, CA, USA), respectively. RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA). High-quality DNA and RNA were used for sequencing.
Code availability
All software and scripts were executed according to user manual, and default parameters were applied if not mentioned in the Methods described above.
References
Peters, R. S. et al. Evolutionary history of the hymenoptera. Curr. Biol. 27, 1013–1018 (2017).
Wittmeyer, K. T., Oppenheim, S. J. & Hopper, K. R. Assemblies of the genomes of parasitic wasps using meta-assembly and scaffolding with genetic linkage. G3 (Bethesda, Md.) 12 (2022).
Xiong, X. et al. Long-read assembly and annotation of the parasitoid wasp Muscidifurax raptorellus, a biological control agent for filth flies. Front. Genet. 12, 748135 (2021).
Werren, J. H. et al. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343–348 (2010).
Dalla Benetta, E. et al. Genome elimination mediated by gene expression from a selfish chromosome. Sci Adv 6, eaaz9808 (2020).
Ye, X. H. et al. A chromosome‐level genome assembly of the parasitoid wasp Pteromalus puparum. Mol. Ecol. Resour. 20, 1384–1402 (2020).
Xiao, S. et al. Genome assembly of the ectoparasitoid wasp Theocolax elegans. Sci. Data 10, 159 (2023).
Chen, L. et al. A chromosome-level genome assembly of the pollinating fig wasp Valisia javana. DNA Res. 29 (2022).
Ye, X. H. et al. Genomic signatures associated with maintenance of genome stability and venom turnover in two parasitoid wasps. Nat. Commun. 13, 6417 (2022).
Kuang, J. G. et al. Chromosome-level de novo genome assembly of two conifer-parasitic wasps, Megastigmus duclouxiana and Megastigmus sabinae, reveals genomic imprints of adaptation to hosts. Mol Ecol Resour 23, 1142–1154 (2023).
De Barro, P. J., Liu, S. S., Boykin, L. M. & Dinsdale, A. B. Bemisia tabaci: a statement of species status. Annu. Rev. Entomol. 56, 1–19 (2011).
Rose, M. & Zolnerowich, G. Eretmocerus Haldeman (Hymenoptera: Aphelinidae) in the United States, with descriptions of new species attacking Bemisia (tabaci complex) (Homoptera: Aleyrodidae). Proc. Entomol. Soc. Wash. 99, 1–27 (1997).
Zolnerowich, G. & Rose, M. Eretmocerus Haldeman (Hymenoptera: Aphelinidae) imported and released in the United States for control of Bemisia (tabaci complex)(Homoptera: Aleyrodidae). Proc. Entomol. Soc. Wash. 100, 310–323 (1998).
Abd-Rabou, S. Biological control of Bemisia tabaci biotype “B” (Homoptera: Aleyrodidae) by introduction, release and establishment of Eretmocerus hayati (Hymenoptera: Aphelinidae). J. Pest Sci. 77, 91–94 (2004).
De Barro, P. J. & Coombs, M. T. Post-release evaluation of Eretmocerus hayati Zolnerowich and Rose in Australia. Bull. Entomol. Res. 99, 193–206 (2009).
Liu, T. X., Stansly, P. A. & Gerling, D. Whitefly parasitoids: distribution, life history, bionomics, and utilization. Annu. Rev. Entomol. 60, 273–292 (2015).
Gerling, D., Quicke, D. L. J. & Orion, T. Oviposition mechanisms in the whitefly parasitoids Encarsia transvena and Eretmocerus mundus. Biocontrol 43, 289–297 (1998).
Gerling, D. & Blackburn, M. B. Immature development of Eretmocerus mundus (Hymenoptera: Aphelinidae). Arthropod. Struct. Dev. 42, 309–314 (2013).
Gerling, D., Orion, T. & Delarea, Y. Eretmocerus penetration and immature development: a novel-approach to overcome host immunity. Arch. Insect Biochem. Physiol. 13, 247–253 (1990).
Galbraith, D. W. et al. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220, 1049–1051 (1983).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H. & Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Meth. 18, 170–175 (2021).
Roach, M. J., Schmidt, S. A. & Borneman, A. R. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics 19, 460 (2018).
van Berkum, N. L. et al. Hi-C: a method to study the three-dimensional architecture of genomes. J. Vis. Exp. (2010).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 9, 357–359 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125 (2013).
Gokhman, V. E. Chromosomes of parasitic wasps of the superfamily Chalcidoidea (Hymenoptera): An overview. Comparative Cytogenetics 14, 399–416 (2020).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Systems 3, 95–98 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Manni, M., Berkeley, M. R., Seppey, M., Simao, F. A. & Zdobnov, E. M. BUSCO Update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of Eukaryotic, Prokaryotic, and Viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11 (2015).
Mei, Y. et al. InsectBase 2.0: a comprehensive gene resource for insects. Nucleic Acids Res. 50, D1040–D1045 (2022).
Bruna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics and Bioinformatics 3, lqaa108 (2021).
Stanke, M. et al. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–439 (2006).
Lomsadze, A., Burns, P. D. & Borodovsky, M. Integration of mapped RNA-Seq reads into automatic training of eukaryotic gene finding algorithm. Nucleic Acids Res. 42, e119 (2014).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinformatics 10, 421 (2009).
Birney, E. & Durbin, R. Using GeneWise in the Drosophila annotation experiment. Genome Res. 10, 547–548 (2000).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Meth. 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 9, R7 (2008).
Toronen, P., Medlar, A. & Holm, L. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 46, W84–W88 (2018).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731 (2016).
Jones, P. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: Functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Chen, C. et al. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 13, 1194–1202 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Capella-Gutierrez, S., Silla-Martinez, J. M. & Gabaldon, T. TrimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A. & Jermiin, L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Meth. 14, 587–589 (2017).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP431306 (2023).
Zhong, Y. W., Liu, S. S. & Liu, Y. Q. Eretmocerus hayati isolate ZJU_SS_LIU_2023, whole genome shotgun sequencing project. GenBank https://identifiers.org/ncbi/insdc:JARUXH000000000 (2023).
Zhong, Y. W. Annotation files of Eretmocerus hayati. figshare https://doi.org/10.6084/m9.figshare.22709485 (2023).
Zhong, Y. W. Repeat sequence analyze result of Eretmocerus hayati. figshare https://doi.org/10.6084/m9.figshare.22709494 (2023).
Acknowledgements
This work was supported by the Key R&D Program of Zhejiang Province (Grant No. 2021C02045) and the earmarked fund for China Agriculture Research System (CARS-23-C05).
Author information
Authors and Affiliations
Contributions
S.S.L. supervised the project. F.L., S.S.L. and Y.Q.L. planned and coordinated the project. Y.W.Z. and Y.Y.F. prepared the samples for PacBio, Illumina and Hi-C sequencing as well as tissue samples for transcriptome sequencing. Y.W.Z. and R.G.S. prepared and conducted flow cytometry experiments. Z.Q.Z. and Y.W.Z. performed the genome annotation. J.B.L. provided the annotation of Encarsia formosa. Y.W. Z performed P450 gene analysis. Y.W.Z., Y.Y.F., Y.Q.L., F.L. and S.S.L. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhong, YW., Fan, YY., Zuo, ZQ. et al. A chromosome-level genome assembly of the parasitoid wasp Eretmocerus hayati. Sci Data 10, 585 (2023). https://doi.org/10.1038/s41597-023-02450-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02450-2
- Springer Nature Limited