
Abstract
Background
Mosquitoes (Culicidae) are vectors for most malaria parasites of the Plasmodium species and are required for Plasmodium spp. to complete their life cycle. Despite having 16 species of mosquitoes and the detection of many Plasmodium species in birds, little is known about the role of different mosquito species in the avian malaria life cycle in New Zealand.
Methods
In this study, we used nested polymerase chain reaction (PCR) and real-time PCR to determine Plasmodium spp. prevalence and diversity of mitochondrial cytochrome b gene sequences in wild-caught mosquitoes sampled across ten sites on the North Island of New Zealand during 2012–2014. The mosquitoes were pooled by species and location collected, and the thorax and abdomens were examined separately for Plasmodium spp. DNA. Akaike information criterion (AIC) modeling was used to test whether location, year of sampling, and mosquito species were significant predictors of minimum infection rates (MIR).
Results
We collected 788 unengorged mosquitoes of six species, both native and introduced. The most frequently caught mosquito species were the introduced Aedes notoscriptus and the native Culex pervigilans. Plasmodium sp DNA was detected in 37% of matched thorax and abdomen pools. When considered separately, 33% of abdomen and 23% of thorax pools tested positive by nested PCR. The MIR of the positive thorax pools from introduced mosquito species was 1.79% for Ae. notoscriptus and 0% for Cx. quinquefasciatus, while the MIR for the positive thorax pools of native mosquito species was 4.9% for Cx. pervigilans and 0% for Opifex fuscus. For the overall MIR, site and mosquito species were significant predictors of Plasmodium overall MIR. Aedes notoscriptus and Cx. pervigilans were positive for malaria DNA in the thorax samples, indicating that they may play a role as avian malaria vectors. Four different Plasmodium lineages (SYAT05, LINN1, GRW6, and a new lineage of P (Haemamoeba) sp. AENOT11) were identified in the pooled samples.
Conclusions
This is the first detection of avian Plasmodium DNA extracted from thoraxes of native Culex and introduced Aedes mosquito species in New Zealand and therefore the first study providing an indication of potential vectors in this country.
Graphical Abstract

Similar content being viewed by others

Background
Avian malaria parasites of the genus Plasmodium are common in birds worldwide. Competent insect vectors are compulsory for malaria parasites to complete their life cycle. Following several cycles of schizogony (asexual reproduction) in the vertebrate host, the Plasmodium sexual stages, namely gametocytes or gamonts, start to develop in mature erythrocytes. These sexual stages are acquired by the vector when they feed on bird blood [1] and develop into macro- and micro-gametes in the insect’s gut within the abdomen of the mosquito. After sexual reproduction and one period of asexual reproduction (sporogony), sporozoites travel through the hemocele of the vector and penetrate the salivary glands in the thorax of the mosquito. From there, they are transmitted by the insects to the avian host during blood feeding [2,3,4]. Different Plasmodium parasites can be transmitted by a range of mosquito species. For example, in Hawaii, a culicine mosquito, Culex quinquefasciatus, is the main, but not sole, vector for Plasmodium relictum lineage GRW4, with two other mosquitoes, Aedes albopictus and Wyeomyia mitchellii, also implicated, although transmissibility to confirm vector competence was never tested [5].
Avian malaria parasites of 17 different lineages have been found in 37 different bird species in New Zealand [6, 7]. The lineages most frequently detected infecting native and introduced birds are Plasmodium (Huffia) elongatum GRW6 with the widest host range, Plasmodium matutinum LINN1, and Plasmodium vaughani SYAT05. Other lineages of Plasmodium detected in endemic species include P. relictum lineages GRW4 and SGS1 [6]. The majority of the 17 lineages are thought to have been brought into New Zealand with the many species of introduced passerines from Europe [8, 9]. However, three native Plasmodium lineages have also been reported: P. sp. KOKAKO01/PADOM02 [10], P. (Haemamoeba) lineage NZHIHI [6], and P. sp. BELL01 [9, 11].
Mortality due to Plasmodium species infection in New Zealand has been recorded mostly in captive situations; for example, in New Zealand dotterel (Charadrius obscurus) chicks in 1996 [12], yellowhead/mohua (Mohua ochrocephala) in 2004 [13], brown kiwi (Apteryx mantelli) in 2010/2011 [14], and little penguins (Eudyptula minor) [7]. In addition, there is one confirmed instance of mortality in a reintroduced, wild population of South Island saddlebacks (Philesturnus carunculatus carunculatus) [15]. To date, confirmed mortalities due to Plasmodium spp. in native New Zealand birds have been from infections with the introduced Plasmodium lineages GRW6, LINN1 and a native P. (Haemamoeba) lineage NZHIHI [6].
New Zealand possesses 12 species of native mosquitoes (genus: Culex, Culiseta, Coquillettidia, Opifex, Aedes, Maorigoeldia) and four introduced species (genus: Culex, Aedes) [16, 17], but there is limited information as to which mosquito species are vectors of avian malaria and whether there is any vector-Plasmodium species specificity [8, 18, 19]. A distribution study by Tompkins and Gleeson [8] showed a strong negative correlation between Plasmodium spp. prevalence in birds and longitude, closely matching the known distribution of the invasive Cx. quinquefasciatus, which is thought to have been introduced in the 1880s. However, Gudex-Cross et al. [20] demonstrated that invasive mosquito species were almost exclusively present on the forest edge of a New Zealand regional park, despite a similar Plasmodium spp. infectious prevalence in birds within the forest edge and the interior habitats. These findings, together with the existence of native Plasmodium lineages, indicate a role of native mosquitoes as vectors for Plasmodium species. Only one study to date, by Massey et al. [21], found avian Plasmodium DNA in the abdomen of one engorged female of the native mosquito Cx. pervigilans. However, vector status could not be determined.
The aim of this study was thus to expand on the limited knowledge of role of mosquitoes in the transmission of avian malaria in New Zealand by assessing the presence of different Plasmodium species separately in their thorax and abdomen. Plasmodium presence in the thorax, where the salivary glands are located, is a stronger indicator of vector competence [43]. We also examined whether location, year of sampling, and mosquito species were significant predictors of infection rates (MIR).
Methods
Sampling locations
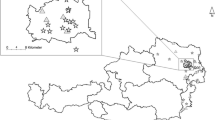
During the period 2012–2014, a total of 13 sampling trips were carried out to catch mosquitoes at the following sites in the North Island of New Zealand from North to South: Whangarei, Hen Island, Cuvier Island, Tiritiri Matangi Island, Auckland/Northshore, Mokoia Island, Cape Kidnappers, Bushy Park, Palmerston North, and Titahi Bay/Porirua (Fig. 1). These sites comprised conservation islands and mainland sites where endangered native species have been translocated, as well as non-conservation public sites in the nearby mainland. Traps were set up along trails and around campsites in sites ranging from uninhabited forested reserves and campgrounds to an urban wetland (Table 1).

Location of sites around the North Island of New Zealand where mosquito species were captured for the detection of Plasmodium spp. DNA
Trapping of mosquitoes
In most cases, sampling was scheduled for summer and/or autumn because mosquitoes caught in summer and autumn are more likely to be infected than those caught in spring [22, 23]. During the summer 2012/2013, dry weather with drought conditions in large parts of the North Island had a negative impact on the number of mosquitoes caught. To increase sample size, sampling was therefore repeated in January/February 2014 (summer) at Mokoia Island, Bushy Park, Titahi Bay, and Palmerston North because these sites were easy to access.
Mosquitoes were trapped using Centres for Disease Control and Prevention (CDC) traps baited with CO2 (dry ice) and UV-diode light such as the BioQuip dry ice traps [24, 25]. The traps were placed approximately 1.5 m high in trees or shrubs. Since most of the mosquito species present in the North Island appear to be crepuscular and/or nocturnal feeders [26], the traps were set approximately 2 hours before sunset (19:00 h) and emptied approximately 2 hours after sunrise (07:00 h) for a total of 12 trapping hours per sampling session. In Titahi Bay, mosquitoes were collected on one afternoon (March 2013) with insect aspirators from around the rock pools. Any female mosquitoes collected in the traps were pooled by date and site location and placed into 1.5 ml microtubes. The mosquitoes were killed by freezing at −20 °C and stored in 80% ethanol until processed for identification and DNA extraction.
DNA extraction
Mosquitoes were identified under a stereomicroscope using the Snell [17] identification keys. Mosquitoes were dissected using a micro-scalpel in 0.9% saline into head, abdomen, and thorax and then separated into species by location and date. Abdomens were dissected first to limit potential contamination by accidental damage to the salivary glands. Next, the heads and thorax were separated. Heads were examined to ensure salivary glands remained in the thorax. The heads were then discarded to avoid introducing PCR inhibitors [27]. Instruments were cleaned with 75% ethanol between individuals and sterilized with the flame of a Bunsen burner to prevent tissue contamination between samples. Not all mosquitoes could be identified to species and genus level as the storage in ethanol caused distortion in some distinguishing features. These unidentified mosquitoes were pooled separately by site and date of collection. Dissected tissues were then pooled by species, ranging from 1 to 20 mosquitoes, depending on the number of each species at each site and sampling period. The insect parts in the species pools were then homogenized with the aid of plastic micro-pestle. The DNA from the abdomen and thorax was extracted separately using the DNeasy Blood and Tissue kit (Qiagen, Düsseldorf, Germany) according to the manufacturer’s instructions with the addition of 30 µl of 100 mg⁄mL dithiothreitol added to the 200 μl of digestion buffer to help dissolve the hard exoskeleton [28] and total DNA was eluted in the final step with 200 µl of elution buffer provided in the kit.
PCR and sequencing
DNA from mosquito abdomen or thorax samples were subjected to an internationally accepted nested PCR for detecting avian Haemosporida parasites using the primer sets HeamNFI/HeamNR2 and HeamF/HeamR2 [29] as previously described [10]. After PCR amplification, all the amplicons were run on a 1% (w/v) ultra-pure agarose gel (Invitrogen, California, USA) containing ethidium bromide and visualized under ultraviolet light on a transilluminator. All positive PCR amplicon samples were purified (PureLink PCR purification kit, Invitrogen, California, USA) and subjected to automatic dye-terminator cycle sequencing with BigDye™ Terminator Version 3.1 Ready Reaction Cycle Sequencing kit and the ABI3730 Genetic Analyzer (Applied Biosystems Inc, California, USA) to confirm genomic sequence. The electropherograms resulting from sequencing were also checked for double nucleotide peaks to infer possible cases of mixed infections of two or more different parasite lineages. The Plasmodium isolate sequences obtained were compared with the MalAvi database [30] and by NCBI Blast to those other published sequences available from GenBank. The resulting sequences were submitted to the GenBank database (OR050956-OR050966).
The 11 mitochondrial cytochrome b sequences from our survey and 18 reference sequences for the MalAvi and GenBank databases, including representatives of the P. (Huffia) elongatum, P. (Haemamoeba) relictum, and P. (Novyella) spp. lineages, as proposed by Valkiunas et al. [31, 32], were trimmed to the same length (450 bases) using Geneious™ (Biomatters, Auckland, New Zealand) and aligned using Clustal W [33]. A phylogenetic tree and sequence divergence was calculated as described by Howe et al. [10].
Presence of several different lineages in one sample
Samples that were positive on the standard nested PCR were reexamined using a real-time PCR (qPCR) protocol using the primers HRMF [34] and HaemR2 [29] amplifying a 127 bp product of the cytochrome b gene as described by Schoener et al. [35]. This qPCR method can discern between the most common Plasmodium lineages in New Zealand [P. elongatum GRW6 (GenBank MK652238), P. matutinum LINN1 (GenBank MT912106), and P. vaughani SYAT05 (GenBank MT912207) as well as P. relictum GRW4 (GenBank AY099041)] to give an indication of possible presence of several different lineages within the same sample.
Statistical analysis
The minimum infection rate (MIR) of each mosquito species was calculated as described by White et al. [36] to evaluate the infection rate of the collected mosquitoes. If a mosquito pool was positive for Plasmodium DNA on PCR, it was assumed that the pool contained at least one positive individual. Minimum infection rate was calculated for positive abdomen + thorax and thorax only pools. Therefore, MIR (percentage) was calculated as follows:
The small sample size and the unbalanced dataset, due to the nature of our sampling, limited the number of potentially meaningful statistical models that could be used. For this reason, we used model simplification [37] to compare, using the Akaike information criterion (AIC), a set of generalized linear models (GLM). The full model examined the predictive value of mosquito species (two most common species: Aedes notoscriptus and Culex pervigilans), site (Auckland, Bushy Park, Cape Kidnappers, Cuvier, Hen, Mokoia, Palmerston North, Titahi Bay, and Whangarei), and year of sampling (2012, 2013, and 2014) on overall MIR and thorax MIR. The number of mosquito pools was used as a covariate in the models. We accepted the model with the lowest AIC as the one that best predicted the MIR data. When this model and the next model were within two AIC points, this model was also highlighted (Greenwood 2023-8.13.1—https://libretexts.org/—book downloaded 10-01-2024).
Owing to the low sample sizes of certain mosquito species, we did not test for differences in the parasite prevalence among mosquito species. Tests were carried out in IBM SPSS statistics (Version 28.0.0.0, Armonk, New York).
Results
Mosquito Fauna
We collected a total of 788 mosquitoes of six species at nine sites (Table 2). All mosquitoes were unengorged. No mosquitoes were caught on Tiritiri Matangi. In Titahi Bay, only the endemic Opifex fuscus was collected. The most common mosquito species captured was the introduced Ae. notoscriptus with 70.5% (556/788) of the total individuals caught (Table 2). This species was found at all sites where CO2-baited traps were used but it was most common in urban areas where it dominated the collected community, with 100% of the caught mosquitoes in Whangarei (9/9) and Auckland (21/21) as well as 94% (442/472) in Palmerston North being of this species (Table 2). The native Cx. pervigilans was the second most common mosquito found, with 12.9% (102/788) of the individuals caught, but was only present at 4/10 sites. This mosquito was more prominent in nature reserves and predominated in March 2012 on Mokoia Island (49.15%, 29/59) and in February 2013 in Bushy Park (61.61%, 61/99). Other species identified included the native Ae. (Ochlerotatus) antipodeus (34 individuals) and Cx. astelidae (1 individual), the introduced Cx. quinquefasciatus (11 individuals), two individuals which could only be identified at the genus level (Culex spp.), and 39 individuals that could not be identified at the genus level due to loss of key features while in ethanol storage (Table 2).
There were differences in mosquito species composition at the two sites where several sampling trips were conducted in consecutive years. On Mokoia Island, in March 2012, Ae. notoscriptus was most prevalent, while in February 2013 no mosquitoes were caught, and in February 2014 Cx. pervigilans was the most common species. In Bushy Park, Ae. notoscriptus was predominant in February 2013, while 1 year later in February 2014, the native Cx. pervigilans was more common (Table 2).
Potential avian malaria vectors
When the thorax and abdomen pools were matched and results combined, there was a minimal Plasmodium sp DNA prevalence of 37% (28/75), where a matched pool was considered positive if either the thorax or abdomen pool was PCR positive, but not counted twice if both were positive. A total of 42 (28%) of 150 (75 of each abdomen and thorax) were positive for Plasmodium spp. DNA (Table 3). Of these, 25/75 (33%) abdomen and 17/75 (23%) thorax pools tested positive. Avian malaria parasite DNA was found in four different mosquito species. However, of these four species, only two, the native Cx. pervigilans and introduced Ae. notoscriptus, had both thorax and abdomen pools that tested positive (Table 3).
The Plasmodium MIR of the thorax positive pools was highest for Cx. pervigilans (4.9%), followed by Ae. notoscriptus (1.79%). Although abdominal pools for both Cx. quinquefasciatus and Opifex fuscus were positive, the thorax pools were negative, resulting in an 0% thorax MIR (Table 4). When the simplified AIC models were considered, the two models including only site and mosquito species + site were significant predictors of Plasmodium overall MIR for the two most captured mosquito species sampled (Table 5). For the thorax MIR, the model with only mosquito species and the model with only year as fixed factors were the best for predicting its values (Table 5).
Plasmodium lineages
A total of 42 mosquito species pools were positive for the presence of Plasmodium spp. However, only 30 of these could be sequenced with conclusive results (Tables 6 and 7). Of these, analysis of the electropherograms from direct sequencing or the qPCR results revealed that 16 (53.33%) pools carried more than one Plasmodium lineage (Tables 6 and 7). Of note, only four of these mixed pools were identified by both the nested PCR and qPCR.
Four different introduced Plasmodium lineages were identified, namely P. matutinum LINN1 (18/30, 99.6–100% sequence homology to GenBank MT912106), P. vaughani SYAT05 (15/30, 99.8–100% sequence homology to GenBank MT912207), P. (Huffia) elongatum GRW6 (8/30, 100% sequence homology to GenBank MK652238), and one case of a previously undescribed P. (Haemamoeba) isolate (lineage AENOT11) (Table 6). Both Ae. notoscriptus and Cx. pervigilans carried the Plasmodium lineages LINN1, SYAT05, and GRW6 in both the abdomen and thorax. A single pool from the abdomen of Ae. notoscriptus was found to carry the novel lineage AENOT11 (Table 6).
Three of the Plasmodium lineages LINN1, SYAT05, and GRW6 had widespread prevalence and were found on three of the six Plasmodium spp. positive sites around the North Island (Table 7). For the three additional positive sites, Cape Kidnappers had only Plasmodium lineages SYAT05 and GRW6 detected, Hen Island had only P. vaughani SYAT05 detected, and one positive mosquito pool from Titahi Bay could not be genotyped. The novel P. lineage AENOT11 was only detected in Palmerston North (Table 7), which was the largest sample of mosquitoes obtained. There was no significant difference in lineage diversity between sites (χ2 = 7.27, df = 10, P > 0.05).
To further characterize the new P. (Haemamoeba) lineage AENOT11, a phylogenetic tree was constructed with known reference sequences (Fig. 2). As expected, the mosquito isolates identified as Plasmodium lineages LINN1, SYAT05, and GRW6 all clustered with high sequence homology (100%, 99–78-100%, and 100%, respectively) with their relevant reference sequences. Similar isolates have been identified in various avian species in New Zealand including isolates of P. matutinum LINN1 and P. vaughani SYAT05 from New Zealand blackbirds (NZTM2 GenBank HQ454002; NZTM2 GenBank HQ453997) and P. elongatum GRW6 isolate from a South Island saddleback (SISB GenBank GU552449)(Fig. 2). The new P. relictum isolate AENOT11 clustered with reference sequences generally considered part of the P. relictum GRW4 (GenBank AY099041) cluster rather than the closely related P. relictum SGS1 (GenBank MZ465355) or New Zealand native isolate KOKAKO01 (GenBank JQ905573). The P. relictum GRW4 group includes isolates from around the world, including Madagascar (GenBank MF442547), Japan (GenBank LC230050), USA (GenBank KX867058), South Africa (GenBank KU375974), PADOM10 (MalAvi database), and a New Zealand isolate HIHI01 (GenBank HQ453996). Sequence homology between the new isolate P. (Haemamoeba) lineage AENOT11 and the P. relictum cluster varied slightly between 99.3 and 99.8%, with the P. relictum isolates KOKAKO01 and SGS1 sharing 98.4% and 97.7% homology, respectively, with P. relictum AENOT11.

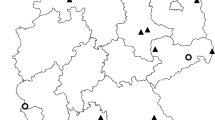
Bayesian phylogenetic analysis and comparison of 11 Plasmodium spp. isolates (bold) from Aedes notoscriptus ( ), Culex pervigilans (
), Culex pervigilans ( ), and previously published Plasmodium spp. sequences present in the GenBank and/or MalAvi database. Avian icons represent previous reports of Plasmodium lineages identified in New Zealand avifauna, including kiwi (
), and previously published Plasmodium spp. sequences present in the GenBank and/or MalAvi database. Avian icons represent previous reports of Plasmodium lineages identified in New Zealand avifauna, including kiwi ( ), raptor species (
), raptor species ( ), penguins (
), penguins ( ), and a variety of Passeriformes (
), and a variety of Passeriformes ( ). Plasmodium falciparum used as an outgroup. Numbers above or below branch nodes indicate bootstrap support based on 1000 replicates. Names of the lineages (when available) and GenBank accession numbers of the sequences are given after the species names of the parasites. The branch lengths are drawn proportionally to the amount of changes (scale bar is shown)
). Plasmodium falciparum used as an outgroup. Numbers above or below branch nodes indicate bootstrap support based on 1000 replicates. Names of the lineages (when available) and GenBank accession numbers of the sequences are given after the species names of the parasites. The branch lengths are drawn proportionally to the amount of changes (scale bar is shown)
Discussion
This study is the first detection of avian Plasmodium DNA from mosquito thoraxes in New Zealand. Plasmodium lineages LINN1, SYAT05, and GRW6 were found in both the abdomen and thorax of the introduced Ae. notoscriptus mosquito and the native Cx. pervigilans mosquito, suggesting that both are competent vectors for Plasmodium spp. Aedes notoscriptus (with by far the largest sample collected) was also found to carry a novel P. (Haemamoeba) lineage AENOT11 in the abdomen. This study also confirmed the findings made by Gudex-Cross et al. [20] that native mosquitoes outnumber introduced mosquitoes at uninhabited conservation sites, while the introduced species Cx. quinquefasciatus and Ae. notoscriptus are known to prefer habitats modified by humans and are therefore often found in urban and semi-urban areas [18, 38, 39]. As observed here, Ae. notoscriptus was also the most abundant introduced species recorded by Gudex-Cross et al. [20] during a study in the northern North Island of New Zealand.
Sampling methods used to capture mosquitoes are known to influence the species and individual numbers caught. Gudex-Cross et al. [20] emphasized the placement of traps both on the ground and canopy to discern vertical distribution patterns of each species that may be related to feeding patterns and host preference. Different kinds of traps also influence the outcome of a study. Carlson et al. [40] even suggest that conclusions made on the role of vectors by examinations using only a single trapping method should be viewed with caution. While CO2-baited light traps such as the ones used in this study collect host-seeking mosquitoes that may feed on a variety of animals, traps baited with readily available birds such as chickens or canaries will accomplish a more specific collection [41]. The possibility of trap bias should be addressed in future studies by using a variety of trapping techniques to consider the specific biology of different New Zealand mosquito species.
During the summer 2012/2013, dry weather with drought conditions in large parts of the North Island negatively impacted the number of mosquitoes caught, particularly at Tiritiri Matangi Island and Mokoia Island in late summer (February 2013), where no mosquitoes were caught. Other similar studies have caught between 40 and 100 individuals per sampling period per site in favorable climatic conditions [23, 42, 43]. Unfortunately, very few mosquitoes were collected on the offshore islands in this study (Hen Island, Cuvier Island, and Tiritiri Matangi Island) during the sampling period. These sites require permits, financial effort, extended logistics, and trips that must be arranged months in advance. As a result, a second trip was not feasible to further examine vector populations on these islands.
To conserve resources and limit expense, mosquitoes of each species were pooled for analysis. As each pool of thorax or abdomen could contain between 1 and 20 individuals, a direct parasite prevalence in each species could not be determined. Therefore, a MIR was calculated for each mosquito species with a PCR-positive thorax and abdomen pool and for a positive thorax pool only. When 23% of total thorax pools is considered, the MIR ranged from 0% to 4.90% depending on the mosquito species examined. Previous studies have shown that the parasite prevalence in thorax samples for other members of the Culex species of mosquitoes is variable, and can range widely within species depending on season, year, and location. For example, two studies in Switzerland examining the prevalence in individual thorax samples of Culex pipiens found an overall Plasmodium spp. prevalence of 6.6% (n = 394) in 2006/2007 [44] and 16.3% in 2011, but noted prevalence varied from 1.5% to 20.3% depending on the month sampled [23]. This temporal variation was also noted in similar studies in the USA, Europe, and Japan [43, 45, 46] and should be further investigated in the New Zealand context.
One limitation with only studying vector thoraxes with molecular methods is the possibility that sporozoites, the parasite life stage before transmission to the vertebrate host, can occur in the hemocele during their travel from the midgut to the salivary glands [47]. If the vector is not fully competent, sporozoites may never reach and fully develop in the salivary glands. Consequently, the amplified parasite DNA from mosquito thoraxes may come from noninfectious parasite stages, and therefore non-competent mosquitoes. In addition, to be able to fully confirm the competent vector status of different mosquito species in New Zealand, future studies will have to involve microscopic detection of oocytes in the midgut and sporozoites in the salivary glands of the mosquitoes, followed by experimental transmission studies [44, 48].
With this limitation considered, the results of this study show that the two models including site and mosquito species were significant predictors of Plasmodium overall MIR for the two most captured mosquito species sampled, indicating that conditions associated with location and mosquito species are as important in New Zealand as they are in other countries [49,50,51,52]. The results for overall and thorax MIR suggest that Cx. pervigilans, our native mosquito, with higher MIR, is more likely to transmit avian malaria than Ae. notoscriptus. Culex pervigilans has long been suspected to be a competent vector for avian malaria in New Zealand [17, 53] and mosquitoes of the genus Culex are the most common vectors for these parasites worldwide [5, 41, 44]. However, a similar vector role of other mosquito species in New Zealand cannot be ruled out.
While not all avian Plasmodium lineages have a putative vector, members of the Culex pipiens complex, which includes the notorious and established vector Cx. quinquefasciatus [5], are often featured. For example, the three most common Plasmodium lineages found in this study have also been reported in Cx. pipiens in several studies on mosquito vectors throughout Europe [23, 44, 45, 54] and in Cx. pipiens and Cx. theileri in Portugal [55]. To date, mosquito studies in the Pacific region have been limited to Japan, however, the results are consistent with those in Europe with Plasmodium lineage SGS1 and PADOM02 detected in the thorax of Cx. pipiens and the abdomens of Aedes albopictus and Tripterodie bambusa [56].
The Plasmodium lineages found in mosquitoes during this study are Plasmodium (Huffia) elongatum lineage GRW06, Plasmodium sp. LINN1, P. (Novyella) spp. lineage SYAT05 and a novel isolate of a P. (Haemamoeba) lineage, AENOT11. This is consistent with the most common Plasmodium lineages that have been found in New Zealand raptors (Falco sp., Circus sp., and Ninox sp.), little penguins (Eudyptula minor), kiwi (Apteryx sp.), and a variety of Passeriformes [6, 7, 57, 58]. Both A. notoscriptus and C. pervigilans carried the Plasmodium lineages LINN1, SYAT05, and GRW6 in both the abdomen and the thorax and may be competent vectors for these lineages.
Compared with the number of lineages identified in avian species in New Zealand, diversity appeared to be low during this examination of mosquito vectors. The Plasmodium lineages LINN1, SYAT05, and GRW6 were widespread and found on all positive sites around the North Island, except for Cape Kidnappers (only SYAT05 and GRW6) and Hen Island (only SYAT05). The Plasmodium (Haemamoeba) lineage AENOT11 was only identified in Palmerston North, which was most likely due to low sample sizes at other locations, especially from the island sites. In contrast, an examination of Plasmodium spp. diversity among introduced birds with overlapped ranges with the threatened North Island Saddleback (Philesturnus carunculatus rufusater) revealed six distinct avian Plasmodium lineages, including the three cosmopolitan lineages SYAT05, LINN1, and GRW6 at many of the same locations as the present study [59].
In addition, the low diversity of collected lineages might also be connected to sampling only being performed during late summer and early autumn. Lalubin et al. [23] have reported seasonal changes in lineage composition throughout the year, where SYAT05 decreased from spring to summer in favor of three lineages of the P. relictum group (SGS1, GRW11, and PADOM02). Similar seasonal changes have also been observed by Kim and Tsuda [60] in Japan, where they found a temporal association with a high prevalence of Plasmodium lineages infecting the two dominant mosquito species and the transmission season. In future studies in New Zealand, a wider range of seasons should be considered to mitigate possible seasonal variations in Plasmodium lineage composition.
Conclusions
This study identified four Plasmodium lineages, LINN1, SYAT05, GRW6, and a new P. (Haemamoeba) lineage AENOT11 in the mosquitoes tested, with the first three in both abdomens and thoraxes of introduced A. notoscriptus and native C. pervigilans. These mosquitoes are therefore likely competent vectors for avian malaria in New Zealand and found in high abundance at all sampled sites. With site and mosquito species being significant predictors of Plasmodium overall MIR, our findings support the hypothesis that at least one native and one introduced mosquito species are competent vectors for introduced Plasmodium lineages in New Zealand. This study provides the first step to improving our understanding of mosquito transmission of Plasmodium species in New Zealand and will lead to improving our understanding of risk for native avifauna.
Availability of data and materials
The Plasmodium species cytochrome b gene sequences were submitted to the GenBank database under the reference nos. OR050956-OR050966.
Abbreviations
- MIR:
-
Minimum infection rate
- AIC:
-
Akaike information criterion
- GLM:
-
Generalized linear models
- PCR:
-
Polymerase chain reaction
- qPCR:
-
Real-time PCR
References
Valkiūnas G. Avian malaria parasites and other haemosporidia. USA: CRC Press; 2004.
Atkinson CT, van Riper IIIC. Pathogenicity and epizootiology of avian haematozoa: Plasmodium, Leucocytozoon, and Haemoproteus. In: Loye JE, Zuk M, editors. Bird-parasite interactions: ecology, evolution, and behavior. Oxford: Oxford University Press; 1991. p. 19–48.
Ritchie BW, Harrison GJ, Harrison LR. Avian medicine: principles and application. Lake Worth: Wingers Publishing Inc; 1994.
Valkiūnas G, Liutkevicius G, Iezhova TA. Complete development of three species of Haemoproteus (Haemosporida, Haemoproteidae) in the biting midge Culicoides impunctatus (Diptera, Ceratopogonidae). J Parasitol. 2002;88:864–8.
Lapointe DA, Goff ML, Atkinson CT. Comparative susceptibility of introduced forest dwelling mosquitoes in Hawai’I to avian malaria Plasmodium relictum. J Parasitol. 2005;91:843–9.
Schoener ER, Banda M, Howe L, Castro IC, Alley MR. Avian malaria in New Zealand. New Zeal Vet J. 2014;62:189–98.
Sijbranda DC, Hunter S, Howe L, Lenting B, Argilla L, Gartrell BD. Cases of mortality in little penguins (Eudyptula minor) in New Zealand associated with avian malaria. New Zeal Vet J. 2017;65:332–7. https://doi.org/10.1080/00480169.2017.1359124.
Tompkins DM, Massey B, Sturrock H, Gleeson D. Avian malaria in native New Zealand birds? Kararehe Kino. 2008;13:8–9.
Ewen JG, Bensch S, Blackburn TM, Bonneaud C, Brown R, Cassey P, et al. Establishment of exotic parasites: the origins and characteristics of an avian malaria community in an isolated island avifauna. Ecol Lett. 2012;15:1112–9.
Howe L, Castro IC, Schoener ER, Hunter S, Barraclough RK, Alley MR. Malaria parasites (Plasmodium spp.) infecting introduced, native and endemic New Zealand birds. Parasitol Res. 2012;110:913–23.
Baillie SM, Brunton DH. Diversity, distribution and biogeographical origins of Plasmodium parasites from the New Zealand bellbird (Anthornis melanura). Parasitol. 2011;38:1843–51.
Tompkins DM, Gleeson DM. Relationship between avian malaria distribution and an exotic invasive mosquito in New Zealand. J Roy Soc N Z. 2006;36:51–62.
Alley MR, Fairley RA, Martin DG, Howe L, Atkinson T. An outbreak of avian malaria in captive yellowheads/mohua (Mohoua ochrocephala). New Zeal Vet J. 2008;56:247–51.
Banda ME, Howe L, Gartrell BD, Mcinnes K, Hunter S, French NP. A cluster of avian malaria cases in a kiwi management programme. New Zeal Vet J. 2013;61:121–6.
Alley MR, Hale KA, Cash W, Ha HJ, Howe L. Concurrent avian malaria and avipox virus infection in translocated South Island saddlebacks (Philesturnus carunculatus carunculatus). New Zeal Vet J. 2010;58:218–23.
Derraik JGBA. Survey of the mosquito (Diptera: Culicidae) fauna of the Auckland zoological park. NZ Entomol. 2004;27:51–5.
Snell AE. Identification keys to larval and adult female mosquitoes (Diptera: Culicidae) of New Zealand. New Zeal J Zoo. 2005;32:99–110.
Derraik JGB. Exotic mosquitoes in New Zealand: a review of species intercepted, their pathways and ports of entry. Aust NZ J Publ Heal. 2004;28:433–44.
Derraik JGB, Slaney D. Anthropogenic environmental change, mosquito-borne diseases and human health in New Zealand. EcoHealth. 2007;4:72–81.
Gudex-Cross D, Barraclough RK, Brunton DH, Derraik JGB. Mosquito communities and avian malaria prevalence in silvereyes (Zosterops lateralis) within forest edge and interior habitats in a New Zealand regional park. EcoHealth. 2015;12:432–40. https://doi.org/10.1007/s10393-015-1039-y.
Massey B, Gleeson DM, Slaney D, Tompkins DM. PCR detection of Plasmodium and blood meal identification in a native New Zealand mosquito. J Vector Ecol. 2007;32:154–6.
Ferraguti M, Martinez-De La Puente J, Munoz J, Roiz D, Ruiz S, Soriguer R. Avian Plasmodium in Culex and Ochlerotatus mosquitoes from southern Spain: effects of season and host-feeding source on parasite dynamics. PLoS ONE. 2013; 8, e66237 https://doi.org/10.1371/journal.pone.0066237
Lalubin F, Deledevant A, Glaizot O, Christe P. Temporal changes in mosquito abundance (Culex pipiens), avian malaria prevalence and lineage composition. Parasite Vector. 2013;25:307. https://doi.org/10.1186/1756-3305-6-307.
Russell CB, Hunter FF. A modified centers for disease control and prevention gravid trap for easier mosquito collection. J Am Mosquito Contr. 2010;2010:119–20.
Panella NA, Crockett RJK, Biggerstaff BJ, Komar N. The Centres for Disease Control and Prevention resting trap: a novel device for collecting resting mosquitoes. J Am Mosquito Contr. 2011; 27:323-325
Derraik JGB, Snell AE, Slaney D. An investigation into the circadian response of adult mosquitoes (Diptera: Culicidae) to host-cues in West Auckland. NZ Entomol. 2005;28:89–94.
Arez P, Lopes D, Pinto J, Franco AS, Nounou GS, do Rosario VE. Plasmodium sp.: Optimal protocols for PCR detection of low parasite numbers from mosquito (Anopheles sp) samples. Exp Parasitol. 2000;94:269–272.
Cooper A. DNA from museum specimens. In: Herrmann BHS, editor. Ancient DNA, recovery and analysis of genetic material from paleontological, archaeological, museum, medical and forensic specimens. New York: Springer; 1994. p. 149–65.
Hellgren O, Waldenstrom J, Bensch S. A new PCR assay for simultaneous studies of Leucocytozoon, Plasmodium, and Haemoproteus from avian blood. J Parasitol. 2004;90:797–802.
Bensch S, Hellgren O, Perez-Tris J. MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Resour. 2009;9:1353–8.
Valkiunas G, Zehtindjiev P, Dimitrov D, Krizanauskiene A, Iezhova TA, Bensch S. Polymerase chain reaction-based identification of Plasmodium (Huffia) elongatum, with remarks on species identity of haemosporidian lineages deposited in GenBank. Parasitol Res. 2008;102:1185–93.
Valkiunas G, Iezhova TA, Loiseau C, Smith TB, Sehgal RNM. New malaria parasites of the subgenus Novyella in African rainforest birds, with remarks on their high prevalence, classification and diagnostics. Parasitol Res. 2009;104:1061–77.
Higgins DG, Thompson JD, Gibson TJ. ClustalW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties, and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80.
Njabo KY, Cornel AJ, Bonneaud C, Toffelmier E, Sehgal RNM, Valkiūnas G, et al. Nonspecific patterns of vector, host and avian malaria parasite associations in a central African rainforest. Mol Ecol. 2011;20:1049–61.
Schoener ER, Hunter S, Howe L. Development of a rapid HRM qPCR for the diagnosis of the four most prevalent Plasmodium lineages in New Zealand. Parasitol Res. 2017;116:1831–41. https://doi.org/10.1007/s00436-017-5452-8.
White BJ, Andrew DR, Mans NZ, Ohajuruka OA, Garvin MC. West Nile virus in mosquitoes of northern Ohio, 2003. Am J Trop Med Hyg. 2006;75:346–9.
Harrison XA, Donaldson L, Correa-Cano ME, Evans J, Fisher DN, Goodwin CE, et al. A brief introduction to mixed effects modelling and multi-model inference in ecology. PeerJ. 2018;6:e4794.
Knoeckel J, Molina-Cruz A, Fischer E, Muratova O, Haile A, Barillas-Mury C, et al. An impossible journey? The development of Plasmodium falciparum NF54 in Culex quinquefasciatus. PLoS ONE. 2013;8:e63387.
Molina-Cruz A, Lehmann T, Knoeckel J. Could culicine mosquitoes transmit human malaria? Trends Parasitol. 2013;29:530–7.
Carlson JS, Walther E, Troutfryxell R, Staley S, Tell LA, Sehgal RNM, et al. Identifying avian malaria vectors: sampling methods influence outcomes. Parasite Vector. 2015;8:365–365.
Kimura M, Darbro JM, Harrington LC. Avian malaria parasites share congeneric mosquito vectors. J Parasitol. 2010;96:144–51.
Okanga S, Cumming GS, Hockey PAR. Avian malaria prevalence and mosquito abundance in the Western Cape. South Africa Malaria J. 2013. https://doi.org/10.1186/1475-2875-12-370.
Fryxell RTT, Lewis TT, Peace H, Hendricks BBM, Paulsen D. Identification of avian malaria (Plasmodium sp.) and canine heartworm (Dirofilaria immitis) in the mosquitoes of Tennessee. J Parasitol. 2014;100:455–62.
Glaizot O, Fumagalli L, Iritano K, Lalubin F, Van Rooyen J, Christe P. High prevalence and lineage diversity of avian malaria in wild populations of great tits (Parus major) and mosquitoes (Culex pipiens). PLoS ONE. 2012;7:e34964. https://doi.org/10.1371/journal.pone.0034964.
Zele F, Vezilier J, Lambert G, Nicot A, Gandon S, Rivero A, et al. Dynamics of prevalence and diversity of avian malaria infections in wild Culex pipiens mosquitoes: the effects of Wolbachia, filarial nematodes and insecticide resistance. Parasite Vector. 2014;7:437.
Kim KS, Tsuda Y. Seasonal changes in the feeding pattern of Culex pipiens pallens govern the transmission dynamics of multiple lineages of avian malaria parasites in Japanese wild bird community. Mol Ecol. 2010;19:5545–54.
Valkiūnas G. Haemosporidian vector research: marriage of molecular and microscopical approaches is essential. Mol Ecol. 2011;20:3084–6.
Kim KS, Tsuda Y. Sporogony and sporozoite rates of avian malaria parasites in wild Culex pipiens pallens and C. inatomii in Japan. Parasit Vectors. 2015;8:633. https://doi.org/10.1186/s13071-015-1251-1.
Yaladanda N, Mopuri R, Vavilala H, Vavilala H, Bhimala KR, Gouda KC, et al. The synergistic effect of climatic factors on malaria transmission: a predictive approach for northeastern states of India. Environ Sci Pollut Res. 2023;30:59194–211.
Otambo WO, Onyango PO, Wang C, Olumeh J, Ondeto BM, Lee MC, et al. Influence of landscape heterogeneity on entomological and parasitological indices of malaria in Kisumu. Western Kenya Parasites Vectors. 2022;15:340.
Keyel AC, Timm O, Backenson PB, Prussing C, Quinones S, McDonough KA, et al. Seasonal temperatures and hydrological conditions improve the prediction of West Nile virus infection rates in Culex mosquitoes and human case counts in New York and Connecticut. PLoS ONE. 2019;14:e0217854.
Yé Y, Hoshen M, Kyobutungi C, Louis VR, Sauerborn R. Local scale prediction of Plasmodium falciparum malaria transmission in an endemic region using temperature and rainfall. Glob Health Action. 2009;2:1923.
Derraik JGB, Tompkins DM, Alley MR, Holder P, Atkinson T. Epidemiology of an avian malaria outbreak in a native bird species (Mohoua ochrocephala) in New Zealand. J Roy Soc New Zeal. 2008;38:237–42.
Iurescia M, Romiti F, Cocumelli C, Diaconu EL, Stravino F, Onorati R, et al. Plasmodium matutinum transmitted by Culex pipiens as a cause of avian malaria in captive African penguins (Spheniscus demersus) in Italy. Front Vet Sci. 2021. https://doi.org/10.3389/fvets.2021.621974.
Ventim R, Ramos JA, Osorio H, Lopes RJ, Perez-Tris J, Mendes L. Avian malaria infections in western European mosquitoes. Parasitol Res. 2012;111:637–45.
Odagawa T, Inumaru M, Sato Y, Murata K, Higa Y, Tsuda Y. A long-term field study on mosquito vectors of avian malaria parasites in Japan. J Vet Med Sci. 2022;4:1391–8. https://doi.org/10.1292/jvms.22-0211.
Mirza V, Burrows EB, Gils S, Hunter S, Gartrell BD, Howe L. A retrospective survey into the presence of Plasmodium spp. and Toxoplasma gondii in archived tissue samples from New Zealand raptors: New Zealand falcons (Falco novaeseelandiae), Australasian harriers (Circus approximans) and moreporks (Ninox novaeseelandiae). Parasitol Res. 2017;116:2283–9. https://doi.org/10.1007/s00436-017-5536-5.
Gulliver E, Hunter S, Howe L, Castillo-Alcala F. The pathology of fatal avian malaria due to Plasmodium elongatum (GRW6) and Plasmodium matutinum (LINN1) infection in New Zealand kiwi (Apteryx spp.). Animals (Basel). 2022;12:3376. https://doi.org/10.3390/ani12233376.
Schoener ER, Tompkins DM, Parker KA, Howe L, Castro IC. Presence and diversity of mixed avian Plasmodium spp. infections in introduced birds whose distribution overlapped with threatened New Zealand endemic birds. New Zeal Vet J. 2020;68:101–6. https://doi.org/10.1080/00480169.2019.1680326.
Kim KS, Tsuda Y. Avian Plasmodium lineages found in spot surveys of mosquitoes from 2007 to 2010 at Sakata wetland, Japan: do dominant lineages persist for multiple years? Mol Ecol. 2012;21:5374–85.
Acknowledgement
The authors would like to thank Bethany Jackson for assisting with mosquito sampling on Tiritiri Matangi Island, the Bushy Park Trust for access, logistical support and accommodation at Bushy Park, and the team of Cape Sanctuary (Cape Kidnappers/ Hawkes Bay) for access to the sanctuary. The authors would also like to thank the following iwi for access to their ancestral land to collect samples- Te Arawa, Ngati Wai and Ngati Whatua. Finally, we would like to thank our funders for their financial support.
Funding
This project received funding from the Morris Animal Foundation study grant ID no. D13ZO-811: Do Translocations for Species Restoration Cause Pathogen Pollution? Further funding was received from Julie Alley Bursary, Marion Cunningham memorial fund, Forest and Bird J S Watson Trust, Massey University (IAE), and the WILDLIFE SOCIETY OF THE NEW ZEALAND Veterinary Association.
Author information
Authors and Affiliations
Contributions
Project inception IC with help from DT. Data collection: ERS (2012–2014 all sites); IC Mokoia Island. Laboratory work ERS with important contributions from LH. Data analyses: IC (stats) and LH (phylogenetics). Manuscript writing: ERS with important contributions from IC and LH and comments from DT.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal ethics protocol for this study was approved by the Animal Ethics Committee at Massey University, MUAEC Protocol 11/59.
Competing interests
The authors declare no competing interests.
Consent for Publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Schoener, E.R., Tompkins, D.M., Howe, L. et al. New insight into avian malaria vectors in New Zealand. Parasites Vectors 17, 150 (2024). https://doi.org/10.1186/s13071-024-06196-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-024-06196-7