
Abstract
Background
Identifying the parasites transmitted by a particular vector and the factors that render this vector susceptible to the parasite are key steps to understanding disease transmission. Although avian malaria has become a model system for the investigation of the ecological and evolutionary dynamics of Plasmodium parasites, little is still known about the field prevalence, diversity and distribution of avian Plasmodium species within the vectors, or about the extrinsic factors affecting Plasmodium population dynamics in the wild.
Methods
We examined changes in avian malaria prevalence and Plasmodium lineage composition in female Culex pipiens caught throughout one field season in 2006, across four sampling sites in southern France. Using site occupancy models, we correct the naive estimates of Plasmodium prevalence to account for PCR-based imperfect detection. To establish the importance of different factors that may bear on the prevalence and diversity of avian Plasmodium in field mosquitoes, we focus on Wolbachia and filarial parasite co-infections, as well as on the insecticide resistance status of the mosquito.
Results
Plasmodium prevalence in Cx. pipiens increased from February (0%) to October (15.8%) and did not vary significantly among the four sampling sites. The application of site occupancy models leads to a 4% increase in this initial (naive) estimate of prevalence. The parasite community was composed of 15 different haemosporidian lineages, 13 of which belonged to the Plasmodium genus, and 2 to the Haemoproteus genus. Neither the presence of different Wolbachia types and of filarial parasites co-infecting the mosquitoes, nor their insecticide resistance status were found to affect the Plasmodium prevalence and diversity.
Conclusion
We found that haemosporidian parasites are common and diverse in wild-caught Cx. pipiens mosquitoes in Southern France. The prevalence of the infection in mosquitoes is unaffected by Wolbachia and filarial co-infections as well as the insecticide resistant status of the vector. These factors may thus have a negligible impact on the transmission of avian malaria. In contrast, the steady increase in prevalence from February to October indicates that the dynamics of avian malaria is driven by seasonality and supports that infected birds are the reservoir of a diverse community of lineages in southern France.
Similar content being viewed by others

Background
In the last few decades, avian malaria has become a model system for the investigation of the ecological and evolutionary dynamics of Plasmodium parasites in the wild [1–4]. These studies have allowed the identification of more than 900 lineages (as defined by their cytochrome-b sequence) in over 600 bird species distributed all over the world [5]. Some of these lineages are extremely prevalent in particular geographical areas (upwards of 90% [3, 6]) and able to infect a wide range of hosts, while others are rarer and confined to a particular species or family [5, 7]. Comparatively little is known about the prevalence and distribution of avian Plasmodium species within the vectors in the field [4, 6–13]. Only a fraction of the known avian Plasmodium lineages have been matched to a putative vector and, thus far, only ca. 20 mosquito vector species have been identified (MalAvi Database). Vectors, however, play a key role in dynamics and epidemiology of the disease. Vector populations fluctuate temporally and spatially, and these processes generate variability in the host biting rate, which ultimately bear on the parasite prevalence and population dynamics of the infection [14]. In addition, vectors play a key role in structuring host-parasite relationships by, for example, restricting the access of certain parasites to a particular subset of hosts (but see [2, 8]) or by limiting or blocking the transmission of parasites to which the vector is less susceptible [15, 16]. Identifying the parasites transmitted by a particular vector and the factors that render this vector susceptible to the parasite are therefore key steps to understanding the epidemiology of the disease.
The aim of the present study is two-fold. The first aim is to establish whether there are variations across space and time in the prevalence and diversity of avian malaria infections in Culex pipiens mosquitoes, the main vector of avian malaria in Europe [6, 7, 10]. For this purpose, we sampled Cx. pipiens mosquitoes for ten consecutive months on four different locations within the Rhone delta (France) and we looked for differences in prevalence and diversity of avian malaria infections across space and time. One pervasive, but rarely acknowledged, problem of parasite prevalence studies in vectors is imperfect detection. Even with modern and a priori more sensitive PCR-based detection techniques, the probability of detection of parasites is strongly correlated with parasite load. Low parasite loads can be easily missed, leading to an underestimation of the parasite’s prevalence [17, 18]. This problem is particularly acute for vectors that feed on birds because of interference between the parasite’s DNA and the DNA from the nucleated erythrocytes. To address this issue, our prevalence estimates were corrected by using site occupancy models. These models are based on the repeated sampling of each “site” (in our case, a site corresponds to a single mosquito) in order to obtain an estimate of the probability of detection of the parasite (p), which is then used to correct the observed prevalence [17, 19]. Site-occupancy models are commonly used in ecology to estimate the density and range of species distributions (e.g. [20–22]) but are still seldom used in the field of host-parasite interactions [17, 18] and have, to our knowledge, never been applied to malaria vectors.
The second aim is to establish the importance of different factors that may bear on the prevalence of avian Plasmodium in Cx. pipiens mosquitoes in the field. In the wild, vectors are rarely infected by a single parasite. More often than not, a suite of microorganisms ranging from virus and bacteria to protozoan, and even metazoan, parasites can be found competing with each other for space and nutritional resources within the vector [23–26]. As a result, recent years have seen a growing interest on the role of such co-infections in shaping the epidemiology of vector-transmitted diseases. Here, we focus specifically on the presence of Wolbachia and filarial parasite co-infections on avian Plasmodium prevalence and diversity in the field. Wolbachia pipientis, a maternally inherited intracellular bacterium, is the most common microorganism in insects. In recent years there has been a plethora of studies showing that Wolbachia interferes with the development of a wide range of pathogens [27, 28]. Studies conducted on Plasmodium, however, suggest that the outcome of the co-infection is largely dependent on the particular Wolbachia-Plasmodium combination used: some combinations seem to inhibit [29–33] while others facilitate [32, 34, 35] the parasite’s development. To our knowledge, however, no study has investigated the role that Wolbachia infections may play in structuring Plasmodium infections in field-caught mosquitoes. Wolbachia infections are near to or at fixation in Cx. pipiens populations worldwide [36, 37], where it is responsible for complex patterns of cytoplasmic incompatibility, a type of conditional sterility between hosts harboring incompatible infections [38]. In the Montpellier region, Cx. pipiens populations harbor a considerable diversity of Wolbachia strains belonging to three different phylogenetic groups: w Pip-I, w Pip-II and w Pip-III [37, 39, 40]. Therefore, although no Wolbachia- uninfected Cx. pipiens mosquitoes exist in nature that would allow testing the effect of Wolbachia presence/absence on the probability of being infected with Plasmodium, this system provides an interesting opportunity to investigate the role of Wolbachia diversity on Plasmodium prevalence in the field. Filarial infections have also been shown to influence the prevalence and intensity of infection of Plasmodium in mosquitoes [23, 41]. In Southern Europe, Cx. pipiens is the main vector of Dirofilaria immitis, a filarial parasite of humans and cannids [42–44], but there is a paucity of data on the prevalence and co-occurrence of Plasmodium and filarial infections in this mosquito species.
The region where the study took place has been repeatedly treated with organophosphate insecticides for the last 40 years. As a result, the prevalence of insecticide resistance in Cx. pipiens mosquitoes in the Montpellier region is high [45, 46]. It has been suggested that the evolution of insecticide resistance in mosquitoes entails a series of drastic physiological and immunological changes that may potentially alter their ability to transmit diseases [47]. Using data obtained both in the laboratory and in the field, McCarroll et al.[48, 49] have indeed shown that insecticide resistant Cx. quinquefasciatus mosquitoes are less likely to transmit the filarial parasite Wuchereria bancrofti. The limited evidence available from Plasmodium is, however, contradictory and comes exclusively from the laboratory: while one study found no effect of insecticide resistance on Plasmodium prevalence or intensity [50], a later study found that insecticide resistance increases the susceptibility of mosquitoes to Plasmodium[51]. Our sampling provided an unparalleled opportunity to investigate whether the insecticide resistance status of mosquitoes bears on the prevalence or diversity of Plasmodium in wild-caught mosquitoes. For this purpose, we typed mosquitoes for the two main types of insecticide resistance present in the area: target site resistance (through the modification of the acetylcholinesterase [52]) and metabolic resistance (through the overproduction of detoxifying carboxylesterases [53]).
By following avian malaria infections in Cx. pipiens mosquitoes for ten consecutive months and across four different sites, our study aimed to address several relevant but hitherto seldom explored determinants of avian malaria prevalence and diversity in wild populations of mosquitoes, namely: 1) Does Plasmodium prevalence and diversity vary across space and time in the area of study? 2) Can site occupancy models be used to detect and correct a bias in the estimation of Plasmodium prevalence in mosquitoes? 3) Do mosquitoes bearing a particular strain of Wolbachia have a higher probability of being infected by avian malaria? 4) What is the prevalence of filarial infections in the area and are these infections correlated with the prevalence of Plasmodium? and 5) Are insecticide resistant mosquitoes more or less likely to transmit avian malaria than their susceptible counterparts and, if so, is this correlation associated with a particular insecticide resistant mechanism (detoxification vs target site modification)?
Methods
Study areas and mosquito sampling
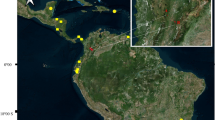
The study was carried out in four sample sites in the Rhône delta, along a North-West to South-East transect (43°42’07”- 43°30’20”N, 4°00’33”- 4°47’29” E, Figure 1) which mostly consisted of wetlands (ponds, marshes, paddies, reed beds and swamps). Sample sites are roughly 23 km away from each other: the Sussargues site (SUS: 43°42’07” N, 4°00’33”E, elevation 50 m) contains Mediterranean forest, scrubland, and stone quarries, with few habitations; Tour Carbonnière (TC: 43°36’28” N, 4°13’49”E, elev. 0 m) is located close to the village of Saint Laurent d’Aigouze where rice fields, ponds and reed beds dominate; in Méjanes (MEJ: 43°34’13”N, 4°30’02”E, elev. 3 m), rice fields, reed beds and marshes with meadows and horses are abundant; finally, Marais du Vigueirat is a natural bird reserve (MDV: 43°30’20”N, 4°47’29” E, elev. 0 m) and consists of marshes, swamps, paddies and reed beds. Culex pipiens females were trapped every fortnight, for two consecutive nights (from 18:00 to 10:00), over a 10 month period (February 20th to December 1st) in 2006. Two pigeon-baited traps hung on trees (protected from sunlight and wind exposure) were used per site, one in the canopy (5–10 meters height, depending on the vegetation) and the other one close to the ground. A detailed description of the pigeon-baited traps used here is given in L’Ambert et al. [54]. This method allows the sampling of host-seeking mosquitoes and maximizes the chances of collecting non-blood-fed females. As a precaution, however, the absence of remnant bird blood in the digestive tract was controlled for by observing under a binocular microscope (mosquitoes with blood meal were removed from further analyses). Eliminating blood fed mosquitoes reduced the chances of false positives because of parasites in the blood meal inside the gut. A total of 1156 unfed Cx. pipiens mosquitoes (identified using morphological characteristics [55]) were collected in this way.

Map showing sampling sites (crosses) where Culex pipiens mosquitoes were collected in the Rhône delta in South of France. Map: GOOGLE EARTH - Data SIO, NOAA, U.S. Navy, NGA, GEBCO, © 2013 Google Landsat Image.
Detection of avian malaria parasites
Total DNA was extracted from each individual mosquito (n = 1156) using the QIAGEN protocol and materials (DNeasy 96 Tissue Kit, Qiagen NV, Venlo, The Netherlands) and total DNA was eluted in the final step with 80 μL RNase free water (Qiagen). The DNA quality was systematically tested using a PCR amplification of a fragment of the Cx. pipiens cytb as described in [56]. Avian malaria parasites were detected for each mosquito sampled by using the nested PCR method developed by [57], which amplifies a 448 bp fragment of the haemosporidia cytb gene. Infected-positive individuals were used as positive controls in each PCR assay. This technique allows the detection of haemosporidian parasites belonging to the genus Plasmodium but also Haemoproteus. This method detected haemosporidian parasites in 98 individual mosquitoes (henceforth “haemosporidian pool”) across a set of geographic locations and collection dates (see Figures 1 and 2). In order to obtain a non-infected “control pool”, we randomly sampled (using RANDOM.ORG; http://www.random.org/) 140 haemosporidian- negative individuals uniformly distributed across the same geographic locations and collection sites.

Prevalence of haemosporidian infection over time. Naïve mosquito prevalence over the ten month sampling period is plotted as gray-filled circles, corrected prevalence (calculated using site-occupancy models) as empty circles. Gray and dashed lines indicate the predicted values for naïve and estimated infection prevalence respectively using a GLM models with a logit link (binomial errors). Numbers between brackets indicate the total number of mosquitoes sampled at each time point.
Estimation of avian malaria parasite prevalence
Haemosporidian detection rates have been shown by qPCR to be strongly dependent on host parasite load: low parasite loads can be easily mistaken for lack of infection leading to an underestimation of prevalence [17, 18]. To correct for false negatives and thereby obtain a more accurate estimate of prevalence, we applied a site-occupancy modeling framework to the dataset [19, 58]. Site occupancy models are based on the repeated sampling of each individual “site” (in our case a “site” corresponds to an individual mosquito) in order to obtain an estimate of the probability of detection or, in other words, an estimate of the sensitivity of the test (i.e. false negatives, see [17] for more conceptual and methodological information). For this purpose, we repeated the nested PCRs an average of 6 times for the individuals from the “haemosporidian pool” (n = 98), and an average of 8 times for the individuals from the “control pool” (n = 140). In the “haemosporidian pool”, 66.3% of the samples always gave a positive result across the 6 PCRs, 13.3% showed variable patterns of positive/negative results, and 20.4% were positive only once. After this intense sampling effort, 12 individuals originally assigned to the “control pool” (n = 128 after reassignment) were re-assigned to the “haemosporidian pool” (n = 110 after reassignment). False positive diagnoses were assumed not to occur (the nested PCRs systematically used a negative control). This method gave us what we call ‘naïve prevalence’ hereafter.
We fitted models using the software PRESENCE version 3.1 [59] and we used the Akaike Information Criterium (AIC; see [60]), to select the best fit model (“1 group constant probability”). The number of sampling occasions was set to 12 (the maximum number of PCRs carried out on a single individual) since the program controls for missing data. The individual detection probabilities given by the model were used to correct the prevalence of haemosporidians per location and sampling month: called ‘estimated prevalence’ hereafter.
Phylogenetic analysis of avian Plasmodium parasites
Molecular identification and phylogenetic analysis of avian malaria parasites was carried out using 96 samples randomly picked from the “ haemosporidian pool” (96 is the number of wells in a standard PCR plate). Fragments (at least 448-bp-long) of the cytb gene were sequenced in forward sense, using the internally nested primer HAEMF from products of the initial PCR [57]. Sequences were edited and aligned using the program ClustalW included in the software MEGA version 5.1 beta [61] with additional manual editing. Mitochondrial DNA lineages were blasted against known avian malaria lineage sequences available in the MalAvi database [5] and in GenBank. The chromatograms were also checked for double nucleotide peaks to infer possible cases of mixed infections of two different parasite lineages. The sequences were assigned to an already described lineage only if they were identical to a reference sequence present in the MalAvi database. For the lineages differing by one nucleotide from the known lineage the term “-like” was appended to the name (e.g. “SGS1-like”) for the phylogenetic analyses but, for simplicity, they were considered to be the same lineage for all other analyses. New sequences have been deposited in GenBank (see Table 1) and whenever the information was available, lineages were then assigned to a given morphospecies using the MalAvi database (see Table 1).
We compared 448-bp-long fragments of the sequences obtained from this study with five published sequences that have been reliably identified to Plasmodium morphospecies level and/or that are as close as possible to the new sequences obtained in this study: CXQUI01, GRW04, MANSON01, PADOM05 and SYAT03 [GenBank: AB308051, AF254975, AB308052, EU708328 and AY831752, respectively]. We rooted our tree with four mammalian malaria parasites sequences [78]. The program jModelTest version 0.1.1 [79] indicated that the most likely model of sequence evolution was TIM2 + G. We used Maximum-Likelihood analysis implemented in PhyML (v.3.0) [80] to reconstruct a phylogeny using these parameters and the software FigTree v1.3.1 was used to draw the phylogenetic tree. Node supports in the resulting phylogeny were tested using 5000 bootstrap replications.
Molecular identification of Wolbachia and filarial nematode
Wolbachia and filarial genotyping were performed on 100 randomly chosen mosquitoes from the “haemosporidian pool” and 92 randomly chosen mosquitoes from the “control pool”. Wolbachia genotyping was performed by analyzing the polymorphism of two genes encoding proteins with ankyrin domains, ank2 and pk1, and one gene putative secreted protein gene, GP15, following the method of [40, 81]. Polymorphism of ank2 and pk1 markers was analyzed using RFLP analyses as described by [81], while polymorphism of GP15 was examined through direct sequencing of PCR products. Examination of the allelic profile of these 3 markers allows the assignment of each individual infection to one of the five known Wolbachia groups in Cx. pipiens (named w Pip-I to w Pip-V [40]).
Filarial nematodes were detected using a PCR assay amplifying a fragment of the cytochrome c oxidase 1 gene (CO1) using the generalist primers COlintF and COlintR as described in [82]. DNA from a rodent filarial nematodes, Litomosoides sigmodontis, was used as positive control in each PCR assay. DNA sequencing of the obtained products was performed in reverse sense using COlintR and sequences have been deposited in GenBank.
Insecticide resistance status
The insecticide resistant or susceptible status of the mosquitoes was carried out on the same random sample of individuals as above (n = 100 from the “haemosporidian pool” and n = 92 from the “control pool”). This was analyzed using RFLP analysis as described in [50]. This technique allows us to distinguish between 4 insecticide resistance status in Cx. pipiens: S (fully susceptible), E (resistant through the overproduction of the carboxylesterase, Ester gene), A (resistant through the modification of the acetylcholinesterase gene, encoded by ace-1) or AE (resistant through both acetylcholinesterase modification and esterase overproduction).
Statistical analyses
Analyses were carried out using the R statistical package (v. 3.0.2). The prevalence of infected mosquitoes was analyzed on the whole dataset (n = 1156 mosquitoes) using GLM models with a binomial error structure, fitting mosquito sampling location (site), sampling time (month) and their interaction as fixed explanatory variables. The significant effect of month on mosquitoes infection prevalence was further confirmed by a mixed effect model approach in order to account for i) the nested structure of the dataset (month within site); and ii) potential temporal auto-correlations [83]. Parasite prevalence (calculated for each site x month combination) was arcsine square root transformed [83] prior to fitting in a lme model (nlme package) using month as a fixed effect, site as a random term, and adding a temporal autocorrelation structure (month within site, corAR1 function) to our model as described in [84]. All other analyses were performed on the subsample of 192 mosquitoes belonging either to the “haemosporidian” or “control” pools. Haemosporidian lineage richness was calculated as the total number of lineages encountered, while lineage diversity was calculated using the Shannon-Weaver index (vegan package). Richness and diversity were calculated for each site x month combination. GLM models with a normal error structure were used to test the effect of the following four explanatory variables on both richness and diversity: site, month, sample size (number of mosquitoes captured), and infection prevalence (proportion of Haemosporidian-infected mosquitoes). Sample size and infection prevalence were also estimated for each site x month combination. As there were not enough individuals of each insecticide resistant category to warrant separate analyses for each of them, all insecticide resistant mosquitoes were grouped within a single insecticide resistant class (giving a binomial IR response variable with two levels: resistant and susceptible). Similarly, only two Wolbachia subgroups were found in mosquitoes giving a binomial response variable (Wolb). The effect of site and month on the probability of being insecticide resistant (IR) or belonging to a given Wolbachia subgroup (Wolb) were therefore analyzed using GLM models with a binomial error structure. The effect of haemosporidian infection on IR or Wolb was analyzed using a mixed model procedure (lmer, lme4 package) with a binomial error structure, fitting infection as a fixed explanatory variable and site as a random explanatory variable.
The general procedure for building all statistical models was as follows. Maximal models were built including all high order interactions and were simplified by sequentially eliminating non-significant terms and interactions to establish a minimal model [83]. The significance of the explanatory variables was established using F-tests or χ2 tests for models with normal error structure or binomial error structure respectively. P = 0.05 was used as a cut-off p-value. The significant F or χ2 values given in the text are for the minimal model, whereas non-significant values correspond to those obtained before the deletion of the variable from the model. In models using a binomial error structure we systematically checked for over dispersion by calculating that the ratio of residual deviance over residual degrees of freedom was <2 [83]. All our models satisfied this premise. The full data-set is given in the Additional file 1.
Results
Infection prevalence
Of the 1156 Cx. pipiens mosquitoes that were captured at the four sampling sites (Figure 1), 9.52 ± 0.86% were found to be infected with haemosporidian parasites by nested PCR (henceforth ‘naive’ prevalence, see Methods and Table 2 for details). When we accounted for imperfect PCR detection (occurrence of false negatives) using site occupancy models, the estimated infection prevalence was found to be somewhat higher: 13.82 ± 0.82%. Only a single infected mosquito was collected in November so this time period was not included in subsequent analyses. The proportion of infected mosquitoes fluctuated across months, showing a distinctive “humped” pattern from April to August. Over the 9-month sampling period, however, the overall trend was that of a significant increase in prevalence: from 0% in February to 15.8% in October, (main month effect, χ21 = 13.23, p < 0.001, see Figure 2). This effect remained significant when accounting for potential temporal autocorrelation patterns or when nesting sampling time within sampling sites (main month effect, χ21 = 9.74, p = 0.002, autocorrelation term, χ21 = 1.62, p = 0.20). These fluctuations in parasite prevalence were similar across the four sampling sites (main site effect, χ23 = 0.63, p = 0.89; site * month, χ23 = 4.84, p = 0.18).
Richness and diversity of haemosporidian lineages
Of the 96 haemosporidian–infected mosquitoes whose cytb gene was sequenced, 92 contained single infections. In the four remaining cases, chromatograms showed double nucleotide peaks indicating mixed infections by different hemosporidian lineages. In two of these cases, it was possible to identify the peaks as being a mixture of SGS1 and DELURB5. In the other two cases, however, it was not possible to identify the combination of lineages and they were therefore taken out from the analyses.
Overall, the analysis led to the identification of 15 different haemosporidian lineages, 13 of which belonged to the Plasmodium genus and two to the Haemoproteus genus (see Table 2, Figure 3). Thirteen of the haemosporidian lineages found in this study clustered with previously known lineages (see Table 1). Three other lineages were found for the first time and could not be assigned to a given morphospecies, though two of them unambiguously fell within the Plasmodium genus and the third one within the Haemoproteus genus. The first one, which we named CXPIPS2 is close to several lineages within the P. relictum morphospecies (e.g. 97% identity at the nucleotide level with SGS1, GRW11 and COLL1; cf. Table 1 and Figure 3 for GenBank accession nos), while the second one, CXPIPS1, is closest to CXQUI01 (99% identity) and MANSON01 (99% identity) lineages which have been isolated in Japanese mosquitoes (Cx. quinquefasciatus and Mansonia sp. respectively [72]). Finally, the CXPIPS3 sequence matches that of several Haemoproteus lineages such as H. pallidulus lineage SYAT03 (97% identity), and H. passeris lineage PADOM05 (95% identity).

Phylogenetic relationships of haemosporidian parasites isolated from mosquitoes (●) and other reference malaria parasites (○ ) based on the cytochrome b (448 bp). Haemosporidian sequences from Cx. pipiens mosquitoes sampled in this study are shown in bold. Branches having bootstrap support (5000 replicates) with values under 60% are omitted. Scale bar indicates number of nucleotide substitutions per site. “like” means that the strain isolated in this study differs only by 1 bp from the reference sequence. Morphospecies or parasite genuses are given for lineages referenced in GeneBank and using the MalAvi database.
There was a considerable fluctuation in the number of lineages (lineage richness) present in mosquitoes across the 10-month study (see Figure 4, Table 2) with the maximum richness (7–8 lineages) happening in late spring (May) and summer (August). This fluctuation across time, which was independent of the sampling site, was only marginally non-significant (month effect: F1,25 = 3.94, p = 0.058, site effect: F3,23 = 0.32, p = 0.81). The diversity of these lineages (estimated by the Shannon-Weaver index), on the other hand, was not dependent on either the time (F1,20 = 0.75, p = 0.40) or the location of sampling (F3,18 = 0.68, p = 0.57). Both haemosporidian richness and diversity were best predicted by the interaction between the number of mosquitoes captured and the prevalence of infected mosquitoes (significant prevalence x sample size interaction for lineage richness: F1,24 = 8.12, p = 0.009; for lineage diversity: F1,19 = 4.89, p = 0.040).

Number of mosquitoes and haemosporidian lineage richness over time. The number of mosquitoes over the ten-month sampling period is plotted as empty circles; the number of haemosporidian lineages (lineage richness) is plotted as black-filled circles over the same sampling period.
Effect of Insecticide resistance, Wolbachia and filarial nematodes on Plasmodium prevalence
Of the 192 mosquitoes used for this analysis, 86.6% were found to be insecticide resistant through known mutations either at the ace-1 locus (A: 0.5%), the Ester locus (E: 73.8%), or both loci at the same time (AE: 12.3%; Table 2). The site at which mosquitoes were captured was a significant predictor of insecticide resistance status (χ23 = 11.83, p = 0.008, see Figure 5A) while sampling time had no effect (χ21 = 0.02, p = 0.89). Haemosporidian infection in mosquitoes was not correlated with their insecticide resistance status (χ21 = 0.63, p = 0.43, see Figure 6A).

Sampling site effect on insecticide resistance and Wolbachia -infection. (A) Barplot of the proportion of insecticide susceptible (S) or resistant Culex pipiens mosquitoes (E, A or AE) in each site; (B) Barplot of the proportion of Culex pipiens mosquitoes infected with either w Pip II or w Pip III Wolbachia in each of the 4 sampling sites (Sus: Sussargues, TC: Tour Charbonnière, Méj: Méjanes, MDV: Marais du Vigueirat).

Haemosporidian infection, Wolbachia infection and insecticide resistance. (A) Barplot of the proportion of insecticide susceptible (S) or resistant Culex pipiens mosquitoes (E, A or AE) in Haemosporidia- infected and -uninfected mosquitoes; (B) Barplot of the proportion of Culex pipiens mosquitoes infected with either w Pip II or w Pip III Wolbachia in Haemosporidia- infected and -uninfected mosquitoes.
Wolbachia infection was detected in the 192 examined specimens, as expected from results of previous studies showing that infection is fixed in Cx. pipiens populations [36, 37, 85]. The analysis of allelic profiles of three diagnostic Wolbachia genes revealed that 47% of these mosquitoes were infected with bacteria from the w Pip-II group while the remaining 53% were infected with the w Pip-III group (Table 2). As observed for insecticide resistance, the only significant predictor of w Pip group was the site at which mosquitoes were captured (χ23 = 13.91, p = 0.003, see Figure 5B) while sampling time had no effect (χ21 = 0.23, p = 0.63). Haemosporidian infection in mosquitoes was also not correlated with the w Pip group they were infected with (χ21 = 0.12, p = 0.73, see Figure 6B).
Infection by filarial nematodes was rare: it concerned only two of 192 individuals (1%), both of which were in the Plasmodium-infected group (coinfections with the DELURB5 and CXPIPS2 lineages). The two CO1 filiarial sequences were strictly identical and clearly match with sequences from members of the Onchocercidae family. The CO1 sequences obtained in this study [GenBank: KJ612514] were however not identical to sequences available in GenBank, preventing us from identifying the species infecting our Cx. pipiens samples. The most closely related sequence (90.4% identity at the nucleotide level) found in GenBank is from the avian filarial nematode Chandlerella quiscali [GenBank: HM773029], which is naturally found in several species of Passeriformes.
Discussion
Infection prevalence and diversity
The high prevalence of Plasmodium spp. found in wild-caught Cx. pipiens in this study confirms the important role of this mosquito species as the main vector of avian malaria in the European continent [4, 6, 7, 10], although definite proof of its role requires further experimental efforts. The prevalence of Plasmodium spp found in Cx. pipiens (9.5% naive and 13.8% estimated), is roughly of the same magnitude as that observed in a Swiss forest in the same period (6.6-16.6%; [4, 6]). Remarkably, this also falls within the range observed in wild-caught Anopheles mosquitoes infected with human Plasmodium parasites (ca. 10-15% of infected mosquitoes, e.g. [86, 87]), suggesting that different malaria parasites may be ultimately driven by similar biological constraints [88].
We observed a marked seasonal variation in avian malaria parasite prevalence in mosquitoes, as has also been documented in birds [73, 89], and, recently, in mosquitoes from Switzerland [4] and Spain [11]. Avian parasites were not observed in February-March, but from April to October the prevalence increased from ca. 3.9% to ca. 15.8%. This is consistent with the biology of European populations of Cx. pipiens which are known to go into diapause in winter and to cease blood feeding from October to March [90]. The mosquitoes collected at the beginning of the season (February, March) were thus either old overwintered mosquitoes, or their first descendants. Either way, the lack of avian Plasmodium infections in mosquitoes at the beginning of the season proves that the parasite’s winter reservoir is the bird and not the mosquito host [73, 91]. Plasmodium relictum is indeed unlikely to survive the overwintering period within mosquitoes as prolonged exposures to low (ca. 12-15°C) temperatures are shown to be lethal for the parasite developing within mosquitoes [92, 93]. In contrast, infections in the bird are characterized by an initial (acute) phase with high parasitaemias, followed by a low level (chronic) phase which can last for months or even years [94–96]. It therefore follows that the rise in malaria prevalence observed from April onwards must be the result of de novo infections following the first infected blood meals at the beginning of the season. From here on, prevalence increases almost linearly until October. Worthy of note is that although Cx. pipiens constituted over 90% of the mosquitoes collected in our sampling, other mosquito species were also present [54], some of which may play a role in the transmission of avian haemosporidians [12] and thus in the overall dynamics of infections.
Our results reveal that avian malaria parasites are very diverse in Cx. pipiens females, as also recently pointed out by recent studies [1, 4, 6]. Fifteen different lineages were obtained amongst the 96 mosquitoes that tested Plasmodium-positive, all of which have been described as being either partially or exclusively parasites of Passeriform birds (Table 1). The most prevalent lineage was SGS1 (44.90% of the lineages found), confirming its status as the most abundant haemosporidian lineage in Europe [4, 6, 95], followed by DELURB4 (21.43%) and DELURB5 (10.20%). All lineages identified here are known to infect both migratory and sedentary Passeriforms (Table 1), except DELURB5, which has, to date, only been sampled from a migratory bird, the common house martin (Delichon urbicum), in Spain [66]. Interestingly, the highest richness in terms of the number of lineages happens in the summer, coinciding with both the period of high vector abundance, a common pattern in haemosporidia [97], and with the breeding period of migratory birds in the study area.
The high diversity of lineages in Culex pipiens mosquitoes suggests that such generalist vectors [62, 98, 99] may play an important role in the high frequency of host switching that characterizes avian malaria, and which can sometimes take place across great host taxonomic distances [100]. Yet the factors allowing the maintenance of the coexistence between multiple malaria lineages must be considered. For instance, migratory birds may also play a role in the maintenance of this diversity [63, 101, 102]. For instance, Waldenstrom et al.[63] have evidenced that resident African birds can exchange African haemosporidian local lineages with European migrant birds, highlighting the role of bird migration in Plasmodium distribution. A recurrent problem of parasite prevalence estimations from wild caught mosquito samples is the potential for either overestimating or underestimating the proportion of infectious mosquitoes in the population. Overestimation comes about from the assumption that all PCR-positive samples translate into a vector-competent mosquito. Parasites present in the blood meal may however fail to establish a viable infection in mosquitoes and may be subsequently eliminated. Leftovers from previous infected blood meals containing parasite DNA residues may lead to PCR amplifications [13]. In addition, even when parasites are seen to be infecting certain vector tissues (e.g. oocysts in the midgut), this does not necessarily imply that the parasite will complete its intrinsic incubation cycle all the way to the transmissible (sporozoite) stages in the salivary glands [103]. Examples of such ectopic parasite development have been already reported in the literature [104, 105]. To avoid these pitfalls, we sampled our mosquitoes using bird-baited traps, under the assumption that mosquitoes searching for a blood meal have entirely digested previous blood meals, an assumption confirmed by the visual inspection of the mosquitoes’ abdomens prior to freezing. We cannot, however, exclude the possibility that some haemosporidian lineages detected in this study are not transmitted by Cx. pipiens. Such may be the case for the two Haemoproteus lineages (CXPIPS3 and GAGLA03) found in our samples. Other studies have also detected Haemoproteus parasites in field-caught mosquitoes [9, 10, 13]. Haemoproteus parasites are, however, currently thought to be transmitted exclusively by biting midges [106]. Experimental evidence available, albeit limited, indeed seems to exclude mosquitoes as vectors [105, 107].
Underestimation, on the other hand, constitutes a more pervasive problem as it comes about through difficulties in diagnosing haemosporidian infection using the nested-PCR method. Indeed, because detection rates are strongly dependent on the relative concentrations of parasites and total DNA within the extract [17, 18], low parasite loads go through undetected (i.e. false negatives). Although seldom used in the context of host-parasite interactions, site-occupancy modelling is an efficient means to examine the accuracy of naive estimates of organismal prevalence and for determining potential sources of detection bias [17]. Gomez-Diaz et al. [17] advocate the inclusion of such models for all pathogen survey techniques. Here, the application of site occupancy models leads us to increase our initial (“naive”) estimate of Plasmodium prevalence in mosquitoes by over 4%. An accurate estimation of the proportion of infectious mosquitoes in a population is essential to estimate the level of exposure of hosts to the parasite within a population (the “entomological inoculation rate”), a key epidemiological tool to estimate malaria endemicity and transmission intensity within a host population [108].
Effect of Wolbachia co-infections on Plasmodium prevalence, richness and diversity
Although an enormous amount of effort has gone into investigating the interaction between Wolbachia endosymbionts and a range of parasites, our study is, to our knowledge, the first investigation of Wolbachia-Plasmodium interactions in field-caught mosquitoes. Wolbachia infections are fixed in wild Cx. pipiens mosquito populations worldwide [36, 37], so it is not possible to compare the prevalence of Plasmodium in Wolbachia-infected and uninfected mosquitoes. However, Wolbachia infections are polymorphic within Cx. pipiens populations, with individual mosquitoes being infected by one of the five known Wolbachia w Pip groups [40, 81]. In our sampling area, we identified two of these groups: w Pip-II and w Pip-III, both of which are commonly found in the Northern hemisphere [40, 81]. The frequency of these two w Pip groups varied between the four study sites, with w Pip-II being more frequent in the western populations (76% in Sussargues) and less frequent in eastern populations (roughly 34% in Méjanes or in Marais du Vigueirat, Figure 5B). This result agrees with previous work done in this region, showing geographic differences in the frequency of the different Wolbachia groups [37, 39]. While in other mosquito species previous work has shown that the effect of Wolbachia on Plasmodium development is strain-specific [29–35], here, neither Plasmodium prevalence nor diversity was affected by the w Pip group co-infecting Cx. pipiens females. This result thus suggests that these two groups of Wolbachia have either no effect on Plasmodium development [109] or act in the same way (i.e. inhibit [29–33] or facilitate [32, 34, 35] the parasite’s development). Although a Wolbachia strain from the wPip-III group has been shown to facilitate P. relictum SGS1 infection in Cx. pipiens[35], further experimental work, using strains from both Wolbachia groups and different Plasmodium lineages would be necessary in order to generalize this pattern.
Effect of filarial co-infections on Plasmodium prevalence and diversity
While Culex mosquitoes are vectors of a wide range of filarial parasites [42–44, 110], little is known about the outcome of concomitant filarial-Plasmodium infections within the vector. Remarkably, previous studies on non-Culex species have shown that simultaneous transmission of the two parasites is particularly rare in the field, suggesting that competition is likely to be a common outcome in nature [111, 112]. Here, we only found two mosquitoes (1%) infected with an unknown avian filaria parasite, even though birds are known to be often infected with multiple filarial species [113–115]. The observed avian filarial parasite is closely related to Chanderella quiscalli, a species known to parasitize the brain of various Passeriform birds. Both of these filaria-positive mosquitoes were found in the Plasmodium-positive pool, one in co-infection with DELURB5 (and with the w Pip3 Wolbachia group) and the other one with CSPIPS2 (and with w Pip2). These results agree with data obtained from Cx. pipiens mosquitoes in Germany, where an unidentified avian filarial parasite, very close to C. quiscalli, was also found at very low prevalence [110]. Overall, our data suggest that filarial parasites may be not common enough to have a significant effect on the population dynamics of avian Plasmodium parasites in this region.
Effect of insecticide resistance on Plasmodium prevalence and diversity
Despite extensive knowledge on the intimate physiological relationships existing between Plasmodium and mosquitoes on the one hand, and on the physiological consequences of insecticide resistance for the mosquito on the other, these two questions have, surprisingly, rarely been put together to ask whether the evolution of insecticide resistance can affect the transmission of Plasmodium (but see [50, 51, 116]). Insecticide resistance could interfere with Plasmodium development in at least two ways [47]. First, the physiological modifications that accompany the deployment of insecticide resistance mechanisms may render the vector toxic to parasites. Second, insecticide resistance could affect vector immunity. In one of the few studies to have explicitly investigated the connection between insecticide resistance and disease transmission, McCarroll and collaborators showed that the development of the filaria Wuchereria bancrofti larvae was arrested in insecticide-resistant Cx. quinquefasciatus mosquitoes [48, 49]. Exactly what rendered the insecticide-resistant mosquito toxic to the parasite is not known, but it was hypothesised that the overproduction of carboxylesterases in these mosquitoes resulted in a change in the redox potential of the tissues hosting the parasite, which led to the death of the larvae. Experimental infections carried out in the laboratory have rendered contradictory results [50, 116]. To our knowledge, no study exists that investigates the impact of insecticide resistance on Plasmodium prevalence in naturally infected wild-caught mosquitoes. Following repeated treatments of larval sites with organophosphate insecticides (initiated 40 years ago), it is therefore not surprising that insecticide resistance was found in the Cx. pipiens populations examined. However, that the overwhelming majority of the mosquitoes sampled (86.6%) were found to be insecticide resistant was somewhat of a surprise, given that in this region classic (organophosphate) insecticides were substituted in 2006 by Bti (Bacillus thuringiensis israelensis[117]). The consequences of these high resistance levels for the circulation of pathogens such as West Nile virus, which has been known to cause episodic cases in both humans and animals in the region, need further study [118]. The high prevalence of insecticide resistance through carboxylesterase overproduction (a general detoxification mechanism) may be the result of pollutant transfer from neighbouring agrosystems. We found no effect of insecticide resistance on avian malaria prevalence or diversity. However, the low frequency of insecticide-susceptible mosquitoes in the sample considerably reduced the statistical power to detect differences between resistant and susceptible mosquitoes. Further work should consider sampling Cx. pipiens populations along several North–south transects spanning populations with low (North) and high (South) recorded frequencies of insecticide resistant genes [119].
Conclusions
In conclusion, we found that haemosporidian parasites are common and diverse in wild-caught Cx. pipiens mosquitoes in Southern France, but that their prevalence is independent of the co-infection and insecticide resistant status of the vector. A correct estimation of the prevalence of infected mosquitoes in a population is essential in order to understand the epidemiology of the disease. The application of site occupancy models leads to a considerable increase in our estimates of Plasmodium prevalence in mosquitoes, suggesting that imperfect detection should be taken into account in further studies. Our study fails to detect spatial variations in prevalence among sampling sites. Yet, we confirm the existence of a temporal pattern where malaria prevalence increases throughout the season. This temporal trend strongly suggests that the bird population is used as a reservoir of avian malaria during the winter. The fact that multiple sedentary or migratory bird species may host Plasmodium parasites during the winter may explain the maintenance of the diversity of malaria lineages in the Southern France.
References
Kim KS, Tsuda Y: Seasonal changes in the feeding pattern of Culex pipiens pallens govern the transmission dynamics of multiple lineages of avian malaria parasites in Japanese wild bird community. Mol Ecol. 2010, 19 (24): 5545-5554.
Medeiros MCI, Hamer GL, Ricklefs RE: Host compatibility rather than vector-host-encounter rate determines the host range of avian Plasmodium parasites. Proc R Soc Lond B Biol Sci. 2013, 280 (1760): 20122947-
Rooyen J, Lalubin F, Glaizot O, Christe P: Altitudinal variation in haemosporidian parasite distribution in great tit populations. Parasit Vectors. 2013, 6: 139-139.
Lalubin F, Deledevant A, Glaizot O, Christe P: Temporal changes in mosquito abundance (Culex pipiens), avian malaria prevalence and lineage composition. Parasit Vectors. 2013, 6: 307-
Bensch S, Hellgren O, Perez-Tris J: MalAvi: a public database of malaria parasites and related haemosporidians in avian hosts based on mitochondrial cytochrome b lineages. Mol Ecol Res. 2009, 9 (5): 1353-1358.
Glaizot O, Fumagalli L, Iritano K, Lalubin F, Van Rooyen J, Christe P: High prevalence and lineage diversity of avian malaria in wild populations of great tits (Parus major) and mosquitoes (Culex pipiens). PLoS One. 2012, 7 (4): e34964-
Ventim R, Ramos J, Osório H, Lopes R, Pérez-Tris J, Mendes L: Avian malaria infections in western European mosquitoes. Parasitol Res. 2012, 111 (2): 637-645.
Gager AB, Loaiza JDR, Dearborn DC, Bermingham E: Do mosquitoes filter the access of Plasmodium cytochrome b lineages to an avian host?. Mol Ecol. 2008, 17 (10): 2552-2561.
Ishtiaq F, Guillaumot L, Clegg SM, Phillimore AB, Black RA, Owens IPF, Mundy NI, Sheldon BC: Avian haematozoan parasites and their associations with mosquitoes across Southwest Pacific Islands. Mol Ecol. 2008, 17 (20): 4545-4555.
Inci A, Yildirim A, Njabo KY, Duzlu O, Biskin Z, Ciloglu A: Detection and molecular characterization of avian Plasmodium from mosquitoes in central Turkey. Vet Parasitol. 2012, 188 (1–2): 179-184.
Ferraguti M, Martinez-de la Puente J, Munoz J, Roiz D, Ruiz S, Soriguer R, Figuerola J: Avian Plasmodium in Culex and Ochlerotatus mosquitoes from Southern Spain: Effects of season and host-feeding source on parasite dynamics. PLoS One. 2013, 8 (6): e66237-
Santiago-Alarcon D, Palinauskas V, Schaefer HM: Diptera vectors of avian Haemosporidian parasites: untangling parasite life cycles and their taxonomy. Biol Rev. 2012, 87 (4): 928-964.
Njabo KY, Cornel AJ, Bonneaud C, Toffelmier E, Sehgal RNM, Valkiunas G, Russell AF, Smith TB: Nonspecific patterns of vector, host and avian malaria parasite associations in a central African rainforest. Mol Ecol. 2011, 20 (5): 1049-1061.
Smith DL, Dushoff J, McKenzie FE: The risk of a mosquito-borne infection in a heterogeneous environment. PLoS Biol. 2004, 2 (11): e368-
Michalski ML, Erickson SM, Bartholomay LC, Christensen BM: Midgut barrier imparts selective resistance to filarial worm infection in Culex pipiens pipiens. PLoS Negl Trop Dis. 2010, 4 (11): e875-
Molina-Cruz A, Lehmann T, Knoeckel J: Could culicine mosquitoes transmit human malaria?. Trends Parasitol. 2013, 29 (11): 530-537.
Gomez-Diaz E, Doherty PF, Duneau D, McCoy KD: Cryptic vector divergence masks vector-specific patterns of infection: an example from the marine cycle of Lyme borreliosis. Evol Appl. 2010, 3 (4): 391-401.
Lachish S, Gopalaswamy AM, Knowles SCL, Sheldon BC: Site-occupancy modelling as a novel framework for assessing test sensitivity and estimating wildlife disease prevalence from imperfect diagnostic tests. Methods Ecol Evol. 2012, 3 (2): 339-348.
MacKenzie DI, Nichols JD, Lachman GB, Droege S, Royle JA, Langtimm CA: Estimating site occupancy rates when detection probabilities are less than one. Ecology. 2002, 83 (8): 2248-2255.
Bailey LL, Simons TR, Pollock KH: Estimating site occupancy and species detection probability parameters for terrestrial salamanders. Ecol Appl. 2004, 14 (3): 692-702.
Altwegg R, Wheeler M, Erni B: Climate and the range dynamics of species with imperfect detection. Biol Letters. 2008, 4 (5): 581-584.
Weller TJ: Using occupancy estimation to assess the effectiveness of a regional multiple-species conservation plan: Bats in the Pacific Northwest. Biol Cons. 2008, 141 (9): 2279-2289.
Aliota MT, Chen CC, Dagoro H, Fuchs JF, Christensen BM: Filarial worms reduce Plasmodium infectivity in mosquitoes. PLoS Negl Trop Dis. 2011, 5 (2): e963-
Zouache K, Michelland RJ, Failloux A-B, Grundmann GL, Mavingui P: Chikungunya virus impacts the diversity of symbiotic bacteria in mosquito vector. Mol Ecol. 2012, 21 (9): 2297-2309.
Chahar HS, Bharaj P, Dar L, Guleria R, Kabra SK, Broor S: Co-infections with chikungunya virus and dengue virus in Delhi. India Emerg Infect Dis. 2009, 15 (7): 1077-1080.
Nakazawa S, Marchand RP, Quang NT, Culleton R, Manh ND, Maeno Y: Anopheles dirus co-infection with human and monkey malaria parasites in Vietnam. Int J Parasitol. 2009, 39 (14): 1533-1537.
Cook PE, McGraw EA: Wolbachia pipientis: an expanding bag of tricks to explore for disease control. Trends Parasitol. 2010, 26 (8): 373-375.
Eleftherianos I, Atri J, Accetta J, Castillo JC: Endosymbiotic bacteria in insects: guardians of the immune system?. Front Physiol. 2013, 4: 46-
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu GJ, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE, Johnson KN, Kay BH, McGraw EA, van den Hurk AF, Ryan PA, O'Neill SL: A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell. 2009, 139 (7): 1268-1278.
Kambris Z, Blagborough AM, Pinto SB, Blagrove MSC, Godfray HCJ, Sinden RE, Sinkins SP: Wolbachia stimulates immune gene expression and inhibits Plasmodium development in Anopheles gambiae. PLoS Pathog. 2010, 6 (10): e1001143-
Hughes GL, Koga R, Xue P, Fukatsu T, Rasgon JL: Wolbachia infections are virulent and inhibit the human malaria parasite Plasmodium falciparum in Anopheles gambiae. PLoS Pathog. 2011, 7 (5): e1002043-
Hughes GL, Vega-Rodriguez J, Xue P, Rasgon JL: Wolbachia strain w AlbB enhances infection by the rodent malaria parasite Plasmodium berghei in Anopheles gambiae mosquitoes. Appl Environ Microbiol. 2012, 78 (5): 1491-1495.
Bian G, Joshi D, Dong Y, Lu P, Zhou G, Pan X, Xu Y, Dimopoulos G, Xi Z: Wolbachia invades Anopheles stephensi populations and induces refractoriness to Plasmodium infection. Science. 2013, 340: 748-751.
Baton LA, Pacidonio EC, Goncalves DS, Moreira LA: w Flu: Characterization and evaluation of a native Wolbachia from the mosquito Aedes fluviatilis as a potential vector control agent. PLoS One. 2013, 8 (3): e59619-e59619.
Zélé F, Nicot A, Berthomieu A, Weill M, Duron O, Rivero A: Wolbachia increases susceptibility to Plasmodium infection in a natural system. Proc R Soc Lond B Biol Sci. 2014, 281 (1779): 20132837-
Duron O, Lagnel J, Raymond M, Bourtzis K, Fort P, Weill M: Transposable element polymorphism of Wolbachia in the mosquito Culex pipiens: evidence of genetic diversity, superinfection and recombination. Mol Ecol. 2005, 14 (5): 1561-1573.
Duron O, Raymond M, Weill M: Many compatible Wolbachia strains coexist within natural populations of Culex pipiens mosquito. Heredity. 2011, 106 (6): 986-993.
Werren JH, Baldo L, Clark ME: Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008, 6 (10): 741-751.
Duron O, Fort P, Weill M: Hypervariable prophage WO sequences describe an unexpected high number of Wolbachia variants in the mosquito Culex pipiens. Proc R Soc Lond B Biol Sci. 2006, 273 (1585): 495-502.
Atyame CM, Delsuc F, Pasteur N, Weill M, Duron O: Diversification of Wolbachia endosymbiont in the Culex pipiens mosquito. Mol Biol Evol. 2011, 28 (10): 2761-2772.
Albuquerque CMR, Ham PJ: Concomitant malaria (Plasmodium gallinaceum) and filaria (Brugia pahangi) infections in Aedes aegypti effect on parasite development. Parasitology. 1995, 110: 1-6.
Cancrini G, Scaramozzino P, Gabrielli S, Di Paolo M, Toma L, Romi R: Aedes albopictus and Culex pipiens implicated as natural vectors of Dirofilaria repens in central Italy. J Med Entomol. 2007, 44 (6): 1064-1066.
Morchon R, Bargues MD, Latorre JM, Melero-Alcibar R, Pou-Barreto C, Mas-Coma S, Simon F: Haplotype H1 of Culex pipiens implicated as natural vector of Dirofilaria immitis in an endemic area of Western Spain. Vector Borne Zoonotic Dis. 2007, 7 (4): 653-658.
Capelli G, di Regalbono AF, Simonato G, Cassini R, Cazzin S, Cancrini G, Otranto D, Pietrobelli M: Risk of canine and human exposure to Dirofilaria immitis infected mosquitoes in endemic areas of Italy. Parasit Vectors. 2013, 6: 60-
Labbe P, Berticat C, Berthomieu A, Unal S, Bernard C, Weill M, Lenormand T: Forty years of erratic insecticide resistance evolution in the mosquito Culex pipiens. Plos Genet. 2007, 3 (11): 2190-2199.
Labbe P, Sidos N, Raymond M, Lenormand T: Resistance gene replacement in the mosquito Culex pipiens: Fitness estimation from long-term cline series. Genetics. 2009, 182 (1): 303-312.
Rivero A, Vézilier J, Weill M, Read AF, Gandon S: Insecticide control of vector-borne diseases: When is insecticide resistance a problem?. PLoS Pathog. 2010, 6 (8): e1001000-
McCarroll L, Hemingway J: Can insecticide resistance status affect parasite transmission in mosquitoes?. Insect Biochem Mol Biol. 2002, 32 (10): 1345-1351.
McCarroll L, Paton MG, Karunaratne S, Jayasuryia HTR, Kalpage KSP, Hemingway J: Insecticides and mosquito-borne disease. Nature. 2000, 407 (6807): 961-962.
Vézilier J, Nicot A, Gandon S, Rivero A: Insecticide resistance and malaria transmission: infection rate and oocyst burden in Culex pipiens mosquitoes infected with Plasmodium relictum. Malar J. 2010, 9: 379-
Alout H, Ndam NT, Sandeu MM, Djegbe I, Chandre F, Dabire RK, Djogbenou LS, Corbel V, Cohuet A: Insecticide resistance alleles affect vector competence of Anopheles gambiae s.s. for Plasmodium falciparum field isolates. PLoS One. 2013, 8 (5): e63849-
Weill M, Lutfalla G, Mogensen K, Chandre F, Berthomieu A, Berticat C, Pasteur N, Philips A, Fort P, Raymond M: Comparative genomics: Insecticide resistance in mosquito vectors. Nature. 2003, 423: 136-137.
Raymond M, Chevillon C, Guillemaud T, Lenormand T, Pasteur N: An overview of the evolution of overproduced esterases in the mosquito Culex pipiens. Philos Trans R Soc Lond B Biol Sci. 1998, 353 (1376): 1707-1711.
L’Ambert G, Ferré J-B, Schaffner F, Fontenille D: Comparison of different trapping methods for surveillance of mosquito vectors of West Nile virus in Rhône Delta. France J Vector Ecol. 2012, 37 (2): 269-275.
Schaffner F, Angel G, Geoffroy B, Hervy JP, Rhaeim A, Brunhes J: The mosquitoes of Europe / Les moustiques d’Europe. 2001, Paris, France: Programme d’identification et d’enseignement
Zélé F, Nicot A, Duron O, Rivero A: Infection with Wolbachia protects mosquitoes against Plasmodium-induced mortality in a natural system. J Evol Biol. 2012, 25 (7): 1243-1252.
Waldenstrom J, Bensch S, Hasselquist D, Ostman O: A new nested polymerase chain reaction method very efficient in detecting Plasmodium and Haemoproteus infections from avian blood. J Parasitol. 2004, 90 (1): 191-194.
MacKenzie DI: Modeling the probability of resource use: The effect of, and dealing with, detecting a species imperfectly. J Wildl Manage. 2006, 70 (2): 367-374.
MacKenzie DI, Nichols JD, Royle JA, Pollock KH, Bailey LL, Hines JE: Occupancy Estimation and Modeling - Inferring Patterns and Dynamics of Species Occurrence. 2005, Burlington, MA: Elsevier Publishing
Burnham KP, White GC: Evaluation of some random effects methodology applicable to bird ringing data. J Appl Stat. 2002, 29 (1–4): 245-264.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739.
Kim KS, Tsuda Y, Yamada A: Bloodmeal identification and detection of avian malaria parasite from mosquitoes (Diptera: Culicidae) inhabiting coastal areas of Tokyo bay. Japan J Med Entomol. 2009, 46 (5): 1230-1234.
Waldenstrom J, Bensch S, Kiboi S, Hasselquist D, Ottosson U: Cross-species infection of blood parasites between resident and migratory songbirds in Africa. Mol Ecol. 2002, 11 (8): 1545-1554.
Ejiri H, Sato Y, Sawai R, Sasaki E, Matsumoto R, Ueda M, Higa Y, Tsuda Y, Omori S, Murata K, Yukawa M: Prevalence of avian malaria parasite in mosquitoes collected at a zoological garden in Japan. Parasitol Res. 2009, 105 (3): 629-633.
Kim KS, Tsuda Y, Sasaki T, Kobayashi M, Hirota Y: Mosquito blood-meal analysis for avian malaria study in wild bird communities: laboratory verification and application to Culex sasai (Diptera: Culicidae) collected in Tokyo. Japan Parasitol Res. 2009, 105 (5): 1351-1357.
Marzal A, Bensch S, Reviriego M, Balbontin J, de Lope F: Effects of malaria double infection in birds: one plus one is not two. J Evol Biol. 2008, 21 (4): 979-987.
Bonneaud C, Perez-Tris J, Federici P, Chastel O, Sorci G: Major histocompatibility alleles associated with local resistance to malaria in a passerine. Evolution. 2006, 60 (2): 383-389.
Valkiunas G, Atkinson CT, Bensch S, Sehga RNM, Ricklefs RE: Parasite misidentifications in GenBank: how to minimize their number?. Trends Parasitol. 2008, 24 (6): 247-248.
Perez-Tris J, Hellgren O, Krizanauskiene A, Waldenstrom J, Secondi J, Bonneaud C, Fjeldsa J, Hasselquist D, Bensch S: Within-host speciation of malaria parasites. PLoS One. 2007, 2 (2): e235-
Kimura M, Darbro JM, Harrington LC: Avian malaria parasites share congeneric mosquito vectors. J Parasitol. 2010, 96 (1): 144-151.
Perez-Tris J, Bensch S: Dispersal increases local transmission of avian malarial parasites. Ecol Lett. 2005, 8 (8): 838-845.
Ejiri H, Sato Y, Sasaki E, Sumiyama D, Tsuda Y, Sawabe K, Matsui S, Horie S, Akatani K, Takagi M, Omori S, Murata K, Yukawa M: Detection of avian Plasmodium spp. DNA sequences from mosquitoes captured in Minami Daito Island of Japan. J Vet Med Sci. 2008, 70 (11): 1205-1210.
Cosgrove CL, Wood MJ, Day KP, Sheldon BC: Seasonal variation in Plasmodium prevalence in a population of blue tits Cyanistes caeruleus. J Anim Ecol. 2008, 77 (3): 540-548.
Knowles SCL, Wood MJ, Sheldon BC: Context-dependent effects of parental effort on malaria infection in a wild bird population, and their role in reproductive trade-offs. Oecologia. 2010, 164 (1): 87-97.
Wood MJ, Cosgrove CL, Wilkin TA, Knowles SCL, Day KP, Sheldon BC: Within-population variation in prevalence and lineage distribution of avian malaria in blue tits. Cyanistes Caeruleus Mol Ecol. 2007, 16 (15): 3263-3273.
Bentz S, Rigaud T, Barroca M, Martin-Laurent F, Bru D, Moreau J, Faivre B: Sensitive measure of prevalence and parasitaemia of Haemosporidia from European blackbird (Turdus merula) populations: value of PCR-RFLP and quantitative PCR. Parasitology. 2006, 133 (6): 685-692.
Dimitrov D, Zehtindjiev P, Bensch S: Genetic diversity of avian blood parasites in SE Europe: cytochrome b lineages of the genera Plasmodium and Haemoproteus (Haemosporida) from Bulgaria. Acta Parasitol. 2010, 55 (3): 201-209.
Escalante AA, Freeland DE, Collins WE, Lal AA: The evolution of primate malaria parasites based on the gene encoding cytochrome b from the linear mitochondrial genome. Proc Natl Acad Sci U S A. 1998, 95 (14): 8124-8129.
Posada D: jModelTest: phylogenetic model averaging. Mol Biol Evol. 2008, 25 (7): 1253-1256.
Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O: New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010, 59 (3): 307-321.
Dumas E, Atyame CM, Milesi P, Fonseca DM, Shaikevich EV, Unal S, Makoundou P, Weill M, Duron O: Population structure of Wolbachia and cytoplasmic introgression in a complex of mosquito species. BMC Evol Biol. 2013, 13: 181-
Casiraghi M, Anderson TJC, Bandi C, Bazzocchi C, Genchi C: A phylogenetic analysis of filarial nematodes: comparison with the phylogeny of Wolbachia endosymbionts. Parasitology. 2001, 122: 93-103.
Crawley MJ: The R Book. 2007, Chichester, England: John Wiley & Sons, Ltd
Pollitt LC, Reece SE, Mideo N, Nussey DH, Colegrave N: The problem of auto-correlation in parasitology. PLoS Pathog. 2012, 8 (4): e1002590-
Rasgon JL, Cornel AJ, Scott TW: Evolutionary history of a mosquito endosymbiont revealed through mitochondrial hitchhiking. Proc R Soc Lond B Biol Sci. 2006, 273 (1594): 1603-1611.
Fryxell RTT, Nieman CC, Fofana A, Lee Y, Traore SF, Cornel AJ, Luckhart S, Lanzaro GC: Differential Plasmodium falciparum infection of Anopheles gambiae s.s. molecular and chromosomal forms in Mali. Malar J. 2012, 11: 133-
Dhiman S, Bhola RK, Goswami D, Rabha B, Kumar D, Baruah I, Singh L: Polymerase chain reaction detection of human host preference and Plasmodium parasite infections in field collected potential malaria vectors. Pathog Glob Health. 2012, 106 (3): 177-180.
Sinden RE, Carter R, Drakeley C, Leroy D: The biology of sexual development of Plasmodium: the design and implementation of transmission-blocking strategies. Malar J. 2012, 11: 70-
Grillo EL, Fithian RC, Cross H, Wallace C, Viverette C, Reilly R, Mayer DCG: Presence of Plasmodium and Haemoproteus in breeding prothonotary warblers (Protonotaria citrea: Parulidae): temporal and spatial trends in infection prevalence. J Parasitol. 2012, 98 (1): 93-102.
Gazave E, Chevillon C, Lenormand T, Marquine M, Raymond M: Dissecting the cost of insecticide resistance genes during the overwintering period of the mosquito Culex pipiens. Heredity. 2001, 87 (4): 441-448.
Applegate JE, Beaudoin RL, Seeley DC: The effect of spring relapse in English sparrows on infectivity of malaria to mosquitoes. J Wildl Dis. 1971, 7 (2): 91-92.
Chao J, Ball GH: Effect of low temperature on Plasmodium relictum in Culex tarsalis. J Parasitol. 1962, 48 (2): 252-
LaPointe DA, Goff ML, Atkinson CT: Thermal constraints to the sporogonic development and altitudinal distribution of avian malaria Plasmodium relictum in Hawaï. J Parasitol. 2010, 96 (2): 318-324.
Snounou G, Jarra W, Preiser PR: Malaria multigene families: The price of chronicity. Parasitol Today. 2000, 16 (1): 28-30.
Valkiūnas G: Avian Malaria Parasites and Other Haemosporidia. 2005, Boca Raton, Florida: CRC Press
van Rooyen J, Lalubin F, Glaizot O, Christe P: Avian haemosporidian persistence and co-infection in great tits at the individual level. Malar J. 2013, 12: 40-
Santiago-Alarcon D, Havelka P, Pineda E, Segelbacher G, Schaefer HM: Urban forests as hubs for novel zoonosis: blood meal analysis, seasonal variation in Culicoides (Diptera: Ceratopogonidae) vectors, and avian haemosporidians. Parasitology. 2013, 140 (14): 1799-1810.
Ziegyte R, Bernotiene R, Bukauskaite D, Palinauskas V, Iezhova T, Valkiunas G: Complete sporogony of Plasmodium relictum (lineages pSGS1 and pGRW11) in mosquito Culex pipiens pipiens form molestus, with implications to avian malaria epidemiology. J Parasitol. 2014, in press
Jansen CC, Webb CE, Graham GC, Craig SB, Zborowski P, Ritchie SA, Russell RC, van den Hurk AF: Blood sources of mosquitoes collected from urban and peri-urban environments in Eastern Australia with species-specific molecular analysis of avian blood meals. Am J Trop Med Hyg. 2009, 81 (5): 849-857.
Ricklefs RE, Fallon SM, Bermingham E: Evolutionary relationships, cospeciation, and host switching in avian malaria parasites. Syst Biol. 2004, 53 (1): 111-119.
Levin II, Zwiers P, Deem SL, Geest EA, Higashiguchi JM, Iezhova TA, Jimenez-Uzcategui G, Kim DH, Morton JP, Perlut NG, Renfrew RB, Sari EHR, Valkiunas G, Parker PG: Multiple lineages of avian malaria parasites (Plasmodium) in the Galapagos Islands and evidence for arrival via migratory birds. Conserv Biol. 2013, 27 (6): 1366-1377.
Hellgren O, Waldenstrom J, Perez-Tris J, Szollosi E, Hasselquist D, Krizanauskiene A, Ottosson U, Bensch S: Detecting shifts of transmission areas in avian blood parasites - a phylogenetic approach. Mol Ecol. 2007, 16 (6): 1281-1290.
Valkiunas G: Haemosporidian vector research: marriage of molecular and microscopical approaches is essential. Mol Ecol. 2011, 20 (15): 3084-3086.
Schneider D, Shahabuddin M: Malaria parasite development in a Drosophila model. Science. 2000, 288 (5475): 2376-2379.
Valkiunas G, Kazlauskiene R, Bernotiene R, Palinauskas V, Iezhova TA: Abortive long-lasting sporogony of two Haemoproteus species (Haemosporida, Haemoproteidae) in the mosquito Ochlerotatus cantans, with perspectives on haemosporidian vector research. Parasitol Res. 2013, 112 (6): 2159-2169.
Martinsen ES, Perkins SL, Schall JJ: A three-genome phylogeny of malaria parasites (Plasmodium and closely related genera): Evolution of life-history traits and host switches. Mol Phylogenet Evol. 2008, 47 (1): 261-273.
Valkiunas G, Kazlauskiene R, Bernotiene R, Bukauskaite D, Palinauskas V, Iezhova TA: Haemoproteus infections (Haemosporida, Haemoproteidae) kill bird-biting mosquitoes. Parasitol Res. 2014, 113 (3): 1011-1018.
Smith DL, McKenzie FE: Statics and dynamics of malaria infection in Anopheles mosquitoes. Malar J. 2004, 3: 13-
Murdock CC, Blanford S, Hughes GL, Rasgon JL, Thomas MB: Temperature alters Plasmodium blocking by Wolbachia. Sci Rep. 2014, 4: 3932-
Czajka C, Becker N, Poppert S, Jost H, Schmidt-Chanasit J, Kruger A: Molecular detection of Setaria tundra (Nematoda: Filarioidea) and an unidentified filarial species in mosquitoes in Germany. Parasit Vectors. 2012, 5: 14-
Kelly-Hope LA, Diggle PJ, Rowlingson BS, Gyapong JO, Kyelem D, Coleman M, Thomson MC, Obsomer V, Lindsay SW, Hemingway J, Molyneux DH: Short communication: negative spatial association between lymphatic filariasis and malaria in West Africa. Trop Med Int Health. 2006, 11 (2): 129-135.
Muturi EJ, Mbogo CM, Mwangangi JM, Ng’ang’a ZW, Kabiru EW, Mwandawiro C, Beier JC: Concomitant infections of Plasmodium falciparum and Wuchereria bancrofti on the Kenyan coast. Filaria J. 2006, 5: 8-
Bedin M, Petterino C, Gallo E, Selleri P, Morgante M: Clinical pathological endings in an owl (Athene noctua) with microfilaraemia in Italy. J Vet Med Ser A - Physiol Pathol Clin Med. 2007, 54 (3): 128-130.
Acosta I, Hernandez S, Gutierrez PN, Martinez-Cruz MS, Hernandez E, Buffoni L, Javier Martinez-Moreno F: Acuaroid nematodes in the common kestrel (Falco tinnunculus) in the south of Spain. Vet J. 2010, 183 (2): 234-237.
Larrat S, Dallaire AD, Lair S: Emaciation and larval filarioid nematode infection in boreal owls (Aegolius funereus). Avian Pathol. 2012, 41 (4): 345-349.
Vontas JG, McCarroll L, Karunaratne S, Louis C, Hurd H, Hemingway J: Does environmental stress affect insect-vectored parasite transmission?. Physiol Entomol. 2004, 29 (3): 210-213.
Abdul-Ghani R, Al-Mekhlafi AM, Alabsi MS: Microbial control of malaria: Biological warfare against the parasite and its vector. Acta Trop. 2012, 121 (2): 71-84.
Del Giudice P, Schuffenecker I, Vandenbos F, Counillon E, Zeller H: Human West Nile Virus. France Emerg Infect Dis. 2004, 10 (10): 1885-1886.
Lenormand T, Bourguet D, Guillemaud T, Raymond M: Tracking the evolution of insecticide resistance in the mosquito Culex pipiens. Nature. 1999, 400 (6747): 861-864.
Acknowledgments
We are very grateful to Karen McCoy for her help with the site occupancy models, to Muriel Dietrich for her help with the phylogenetic analyses, to Valérie Noel, Emilie Dumas and the Mylène Weill’s group for laboratory assistance and useful advice. This study was funded by the French ANR program (ANR “IRMAL”) to AR. AN and JV were funded by an ERC Starting Grant 243054 to SG, FZ was funded by a PhD grant from the CNRS and the Languedoc-Roussillon Region. This is contribution ISEM 2014–127 of the Institut des Sciences de l’Evolution de Montpellier (UMR 5554 CNRS – Université Montpellier 2).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Experimental conception and design: FZ GL SG AR OD; mosquito sampling: GL; molecular analyses: FZ AN; site occupancy modeling: FZ; phylogenetic analyses: FZ; statistical analyses: JV; paper writing: FZ JV SG AR OD. All authors read and approved the final version of the manuscript.
Ana Rivero and Olivier Duron contributed equally to this work.
Electronic supplementary material
13071_2014_1612_MOESM1_ESM.xlsx
Additional file 1: Complete dataset. Haemosporidian naïve prevalence (0 or 1), corrected prevalence (based on repeated PCRs results, 0,0549 to 1), lineage identity and the occurrence of co-infections are given for Culex pipiens mosquitoes collected between February and December 2006 in 4 different sampling sites (Sus: Sussargues, TC: Tour Charbonnière, Méj: Méjanes, MDV: Marais du Vigueirat). The “pool” columns indicate the data subset used for (a) phylogenetic analyses, (b) diversity index and species richness calculation, and (c) the insecticide resistance and Wolbachia subgroup prevalence according to different covariates (site, month and haemosporidian prevalence, see Methods). Wolbachia groups (wolb, wPip2 or wPip3) and insecticide resistance status (S: fully susceptible, E: overproduction of carboxylesterase, A: acetylcholinesterase modification, AE: both acetylcholinesterase modification and esterase overproduction) are indicated for all mosquitoes that were included in these analyses. Other abbreviations used in the table are ‘ - ’ (not studied), ‘ 0 ’ (absence), ‘ 1 ’ (presence), and ‘ na ’ (failed). (XLSX 224 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zélé, F., Vézilier, J., L’Ambert, G. et al. Dynamics of prevalence and diversity of avian malaria infections in wild Culex pipiens mosquitoes: the effects of Wolbachia, filarial nematodes and insecticide resistance. Parasites Vectors 7, 437 (2014). https://doi.org/10.1186/1756-3305-7-437
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-7-437