
Abstract
Background
Kluyveromyces marxianus is a promising cell factory for producing bioethanol and that raised a demand for a high yield of heterologous proteins in this species. Expressions of heterologous proteins usually lead to the accumulation of misfolded or unfolded proteins in the lumen of the endoplasmic reticulum (ER) and then cause ER stress. To cope with this problem, a group of ER stress response target genes (ESRTs) are induced, mainly through a signaling network called unfolded protein response (UPR). Characterization and modulation of ESRTs direct the optimization of heterologous expressions. However, ESRTs in K. marxianus have not been identified so far.
Results
In this study, we characterized the ER stress response in K. marxianus for the first time, by using two ER stress-inducing reagents, dithiothreitol (DTT) and tunicamycin (TM). Results showed that the Kar2–Ire1–Hac1 pathway of UPR is well conserved in K. marxianus. About 15% and 6% of genes were upregulated during treatment of DTT and TM, respectively. A total of 115 upregulated genes were characterized as ESRTs, among which 97 genes were identified as UPR target genes and 37 UPR target genes contained UPR elements in their promoters. Genes related to carbohydrate metabolic process and actin filament organization were identified as new types of UPR target genes. A total of 102 ESRTs were overexpressed separately in plasmids and their effects on productions of two different lignocellulolytic enzymes were systematically evaluated. Overexpressing genes involved in carbohydrate metabolism, including PDC1, PGK and VID28, overexpressing a chaperone gene CAJ1 or overexpressing a reductase gene MET13 substantially improved secretion expressions of heterologous proteins. Meanwhile, overexpressing a novel gene, KLMA_50479 (named ESR1), as well as overexpressing genes involved in ER-associated protein degradation (ERAD), including HRD3, USA1 andYET3, reduced the secretory expressions. ESR1 and the aforementioned ERAD genes were deleted from the genome. Resultant mutants, except the yet3Δ mutant, substantially improved secretions of three different heterologous proteins. During the fed-batch fermentation, extracellular activities of an endoxylanase and a glucanase in hrd3Δ cells improved by 43% and 28%, respectively, compared to those in wild-type cells.
Conclusions
Our results unveil the transcriptional scope of the ER stress response in K. marxianus and suggest efficient ways to improve productions of heterologous proteins by manipulating expressions of ESRTs.
Similar content being viewed by others

Background
Protein secretion is initiated from the rough endoplasmic reticulum (ER) lumen, where nascent polypeptides are bound by ER-resident proteins for correct folding and processing. Only properly folded and assembled proteins are exported from the ER to the Golgi for further modification, before being transported to the extracellular space, vacuoles or other organelles [1]. When the ER encounters a high flux of heterologous proteins, its folding capacity could be transiently saturated, thus leading to the accumulation of misfolded or unfolded proteins and causing ER stress [2]. ER stress causes substantial transcriptomic changes. For instance, 7.8% of genes in Neurospora crassa and 6.8% of genes in Saccharomyces cerevisiae were upregulated by both dithiothreitol (DTT) and tunicamycin (TM), while 6.8% of genes in Komagataella phaffii were upregulated by both DTT and the overexpression of HAC1 [3,4,5]. ER stress response target genes (ESRTs) were defined as genes induced upon ER stress and were involved in the response to cope with the stress. A proportion of ESRTs was induced through a signaling network called unfolded protein response (UPR), which is one of the best-characterized pathways to deal with ER stress [6]. Meanwhile, some ESRTs were induced independently of UPR, as reported in N. crassa and S. cerevisiae [4, 7].
The UPR network is composed of stress sensors, transcriptional activators and downstream target genes. Three branches of UPR were identified and named after sensors, including IRE1 (inositol-requiring enzyme 1), PERK (protein kinase RNA-like endoplasmic reticulum kinase) and ATF6 (activating transcription factor 6) [6]. In yeast, UPR regulation solely depends on the most conserved IRE1 branch. In S. cerevisiae, Ire1 may be dissociated from the ER-luminal resident chaperone Kar2 (Bip) upon ER stress. The released Ire1 recognizes misfolded proteins, triggers Ire1 oligomerization and then activates themselves to splice the HAC1 mRNA into the translationally competent HAC1 mRNA [8]. Then, the spliced HAC1 mRNA is translated into a transcriptional activator that recognizes the specific DNA sequences, called UPR elements (UPRE), to induce transcriptions of UPR target genes [9]. UPR target genes function from protein folding, phospholipid synthesis, protein translocation, glycosylation and vacuolar transport to ER-associated degradation (ERAD) [3].
Naturally, components of the UPR network can serve as targets for optimization to improve the secretory expressions of heterologous proteins. In Aspergillus niger var. awamori, overexpression of the activated UPR transcription factor HacA constitutively induced the UPR pathway and enhanced productions of both Trametes versicolor laccase and bovine preprochymosin [10]. ER-resident chaperones, including Kar2, Lhs1 and Jem1, and protein disulfide isomerase Pdi1, are classic UPR targets [3]. The introduction of an extra copy of KAR2 into S. cerevisiae caused more than a 20-fold increase in the amount of extracellular prochymosin [11]. Similarly, overexpression of KAR2 in K. phaffii increased the secretion of a single-chain antibody fragment [12]. In S. cerevisiae, overexpression of LHS1 or JEM1 significantly increased the secretory expression of recombinant human albumin [13]. Overexpressing S. cerevisiae disulfide isomerase Pdi1 in K. phaffii increased the secretory yield of human parathyroid hormone even though it does not contain any cysteine residue [14]. In addition, UPR-independent ESRTs can also be modified to improve secretion. RES-1 encodes an N. crassa transcription factor that responds to intracellular calcium disturbances induced by the ER stress. RES-1 was induced independently of Ire1 or Hac1 and deletion of RES-1 elevated the amount of secreted cellulase by 50% [4].
Kluyveromyces marxianus is a homothallic hemiascomycetous yeast species commonly isolated in dairy products, grape, papaya and Mexican fermented corn dough [15]. It is the fastest-growing eukaryote known so far, and can assimilate inulin, lactose and pentose (e.g., xylose and arabinose) that cannot be utilized by the traditional ethanologenic yeast S. cerevisiae. It is noteworthy that K. marxianus exhibits weak glucose repression that is preferable for the fermentation of mixed sugars such as hemicellulose hydrolysate [16]. Beyond that, K. marxianus is highly thermotolerant, as it can grow at a temperature up to 52 °C. Given its desirable traits, K. marxianus is considered a promising host for the productions of heterologous proteins and bioethanol [17].
So far, ER stress response and UPR pathway have not been characterized in K. marxianus, which hinders the improvement of heterologous proteins expressions by modulating ESRTs. In this study, we showed that the Kar2–Ire1–Hac1 pathway of UPR was well conserved in K. marxianus. During treatments of ER stress-inducing reagents DTT and TM, 15.1% and 6.4% of genes were upregulated, respectively. A total of 115 upregulated genes were identified as ESRTs, among which 97 genes were characterized as UPR target genes. Effects of the overexpression or deletion of ESRT on productions of heterologous proteins were systematically evaluated. Overexpression of genes involved in carbohydrate metabolism, chaperone gene and reductase gene improved the secretory expression. Meanwhile, deletions of genes involved in ERAD improved the secretory expressions. Our results revealed the transcriptional scope of ER stress response in K. marxianus and identified valuable target genes to be engineered to improve expressions of heterologous proteins.
Results
Upstream components of UPR pathway in K. marxianus
In the upstream processes of the UPR pathway, Kar2, Ire1 and Hac1 cooperate to sense unfolded or misfolded proteins, to trigger unfolded protein response and maintain the homeostasis in the endoplasmic reticulum [18]. Orthologs of these three proteins were identified in K. marxianus based on sequence similarity (Fig. 1a). Hac1 shares poor sequence identity with its ortholog in S. cerevisiae, while Kar2 is relatively well conserved.

Identification of upstream components of the UPR pathway in K. marxianus. a Identities of Kar2, Ire1 and Hac1 in K. marxianus to their orthologs in S. cerevisiae (Sc). b Sensitivity of W303-1a, FIM-1ΔU, ire1Δ and hac1Δ mutants to ER stress-inducing reagents. Cells were diluted fivefold and dilutions were spotted onto YPD containing different concentrations of DTT or TM. c Induction of KAR2 upon treatment of DTT and TM. Exponentially growing cells were treated with DTT and TM for indicated times. mRNA level of KAR2 relative to 18s rDNA was measured. The value represented mean ± SD (n = 3). d Splicing of HAC1 upon treatment of DTT and TM. Sizes of the product representing unspliced and spliced forms of HAC1 were 443 bp and 117 bp, respectively. Cells were treated as in c. SWC4 was detected as a loading control. e Schematic representation of HAC1 introns in K. marxianus, K. lactis and S. cerevisiae
DTT and TM are two classic ER stress-inducing reagents. DTT blocks disulfide-bond formation and TM inhibits N-linked glycosylation, both leading to the disruption of protein folding in the ER [19]. As shown in Fig. 1b, 25 mM DTT caused severe growth defect of a wild-type S. cerevisiae strain W303-1a, while wild-type K. marxianus strain FIM-1ΔU strain was able to grow in the presence of 30 mM DTT. The result suggests K. marxianus is more tolerant to DTT stress than S. cerevisiae. The sensitivity of K. marxianus and S. cerevisiae to TM were similar, as FIM-1ΔU and W303-1a cells exhibited growth defects in the presence of 0.5 μg/mL TM (Fig. 1b). To verify the roles of Ire1 and Hac1 in ER stress in K. marxianus, IRE1 and HAC1 were knocked out by CRISPR separately and growth of these two mutants was examined in the medium containing DTT or TM. As shown in Fig. 1b, the growth of ire1Δ and hac1Δ mutants was significantly impaired in the presence of 0.1 μg/mL TM. The ire1Δ mutant was more sensitive to DTT than the hac1Δ mutant, as ire1Δ and hac1Δ cells exhibited severe growth defects in the presence of 25 mM and 30 mM DTT, respectively. The result suggests that Ire1 and Hac1 maintain homeostasis of the ER to reduce deleterious effects caused by the ER stress in K. marxianus.
In S. cerevisiae, KAR2 is an essential gene and transcription of KAR2 is rapidly induced upon DTT or TM treatment, which is a hallmark event of UPR response [20, 21]. We failed to obtain the null mutant of KAR2 in K. marxianus, suggesting that KAR2 is also an essential gene in K. marxianus. Similar inductions of KAR2 after being treated with ER stress-inducing reagents were observed in K. marxianus. In the presence of 10 mM DTT or 0.5 μg/mL TM, the increases of the KAR2 mRNA levels were quite similar, which were about 50-fold after 30 min and 75-fold after 60 min. After 120 min, the level of KAR2 induced by DTT increased by 150-fold, which was twice that induced by TM (Fig. 1c). The results suggest prolonged incubation with DTT induces an acuter ER stress than with TM.
In S. cerevisiae, ER stress triggers Ire1-mediated splicing of HAC1 mRNA [8]. In K. marxianus, the ORF of HAC1 is 912-bp in length. Few spliced HAC1 mRNA was detected in cells in normal conditions (Fig. 1d). In contrast, splicings of HAC1 mRNA were rapidly induced after adding DTT or TM, leading to the appearance of a small fragment in the RT-PCR assay (Fig. 1d). Sequence analyses of both the unspliced and spliced forms of HAC1 revealed that a 325-bp intron was removed. As shown in Fig. 1e, cleavage motifs before and after exon–intron junctions were the same among K. marxianus, K. lactis and S. cerevisiae, suggesting that a conserved HAC1-splicing mechanism exists within Saccharomycetaceae.
Transcriptomic analysis of K. marxianus during ER stress
To analyze the transcriptional scope of ER stress in K. marxianus, wild-type FIM-1ΔU cells were grown in the presence of 10 mM DTT or 0.5 µg/mL TM for 15, 30 and 60 min before they were collected and subjected to RNAseq (Additional file 1: Table S1). The expression level of a gene in the treated sample at each time point was compared with that in the untreated control. As shown in Fig. 2a–c, several classic UPR target genes, including HAC1, KAR2, PDI1, ERO1 and HRD3, were substantially upregulated during the treatment of DTT or TM. The result suggests that ER stress and UPR in K. marxianus are invoked after adding DTT or TM.

Identification of differentially expressed genes during ER stress. Comparison of ER stress induced by DTT and TM. Exponentially growing cells were treated with DTT or TM for 15 (a), 30 (b) and 60 min (c). Induction by ER stress was calculated by dividing the average abundance of a gene in cells treated with DTT or TM by that in cells without treatment. At the indicated time point, fold change (Log2) of a certain gene upon DTT treatment was plotted against that upon TM treatment in the chart. Points representing ESRTs were red. Points corresponding to several known UPR target genes were blue. Points representing other genes were green. The size of the point reflected the mean of FPKM value of a certain gene after treatment of DTT and TM. d, f, h Venn diagrams of upregulated and downregulated genes after TM and DTT treatment. Genes significantly upregulated (Fc > 2, q value < 0.1) or downregulated (Fc < 0.5, q value < 0.1) upon drug treatment were counted. The number of upregulated or downregulated genes in indicated conditions is shown in the parenthesis. e, g, i GO enrichment analysis of differentially expressed genes during ER stress. The enrichment analysis was based on the biological process (BP). Top-ranked terms containing 10 ~ 50 genes and with an adjusted P-value < 0.05 are listed in e, g. Top-ranked terms containing 10 ~ 100 genes and with an adjusted P-value < 0.05 are listed in i. The size of a point represented the adjusted P-value. A full list of enriched GO terms is shown in Additional file 3: Table S3
FIM-1ΔU strain harbors 5202 genes in total [22]. After a 15-min treatment of DTT, 60 genes were significantly upregulated (Fold change > 2, q value < 0.1), among which genes involved in the glycolytic process were enriched (Fig. 2d, e, Additional file 2: Table S2), while 77 genes were significantly downregulated, among which genes involved in the oxidative phosphorylation were enriched (Fig. 2f, g, Additional file 2: Table S2). The results suggest a shift from respiration to glycolysis during the initial response to DTT. After a 30-min treatment of DTT, 695 genes were significantly upregulated, among which genes involved in ERAD and retrograde protein transport were enriched (Fig. 2d, e). Meanwhile, 687 genes were significantly downregulated, among which genes involved in rRNA processing were enriched, suggesting the repression of translation (Fig. 2f, g). Reduction of the rRNA processing probably is not a specific response to DTT, as a transient reduction in transcripts for the translation apparatus was observed in S. cerevisiae during various environmental stresses, including heat shock, acid, alkali, H2O2, salt and sorbitol [23]. After a 60-min treatment of DTT, 301 and 212 genes were upregulated and downregulated, respectively (Fig. 2d, f). At this time point, cells kept the upregulation of genes involved in ERAD and retrograde protein transport, while genes involved in respiration, TCA cycles and arginine metabolic process were downregulated (Fig. 2e, g).
During the treatment of TM, 48, 170 and 243 genes were significantly upregulated after 15, 30 and 60 min, respectively (Fig. 2d, Additional file 2: Table S2). Among upregulated genes, genes involved in the processes of ERAD and protein targeting to ER were enriched (Fig. 2e). Interestingly, genes involved in the regulation of actin filament polymerization were upregulated after 60 min (Fig. 2e), composing a new type of genes induced upon ER stress. During the treatment of TM, 10, 43 and 126 genes were significantly downregulated after 15, 30 and 60 min, respectively (Fig. 2f). Among the downregulated genes, genes involved in iron ion transport, arginine and histidine biosynthesis were enriched (Fig. 2g, Additional file 2: Table S2). Downregulated iron ion transport might reduce the metabolic and respiratory activity of mitochondria [24], leading to the repression of respiration as shown in cells treated with DTT. Downregulation of amino acid biosynthesis might be related to the repression of the translation of certain target proteins [25].
In general, 15.2% of genes and 14.9% of genes were upregulated and downregulated, respectively, after DTT treatment, while 6.4% and 3.1% of genes were upregulated and downregulated after TM treatment. As shown in Additional file 11: Fig. S1, 15-min and 30-min DTT treatment caused a larger magnitude of changes in the expressed transcripts than 15-min, 30-min and 60-min TM treatment. These results indicated that in the current concentrations of both chemicals, DTT induced an acuter ER stress than TM. In total, 16.9% and 15.9% of genes were upregulated and downregulated, respectively, after treatment of at least one ER stress-inducing reagents, suggesting that ER stress leads to a profound transcriptomic change in K. marxianus. A total of 240 genes (4.6% of total genes) were upregulated by both DTT and TM. Since DTT and TM caused ER stress through different mechanisms, these genes were likely to be induced by misfolding of proteins in ER rather than other effects (Fig. 2h and Additional file 2: Table S2). Among the co-induced genes, genes involved in ERAD, ER protein transport and localization, and actin filament organization were enriched (Fig. 2i). A total of 103 genes were downregulated by both DTT and TM, among which genes involved in rRNA processing, organic acid transport and arginine biosynthesis were enriched (Fig. 2i).
Identification of UPR target genes and UPRE in K. marxianus
To determine ESRT and UPR target genes, homologues of 12 classic UPR target genes were selected as reference genes [19], including genes encoding chaperone proteins (KAR2, LHS1), genes involved in the disulfide bond formation (PDI1, ERO1, MPD1), genes involved in the vesicle transport (SFB3, SEC24, SEC12) and genes involved in ERAD (DER1, HRD1, HRD3, UBC7). All reference genes were induced both by DTT and TM. Analyses of the transcriptional profiles of the 12 reference genes revealed four different patterns of induction during the treatment of DTT and TM. Each pattern was represented by a group of reference genes. A given gene displaying a similar transcriptional pattern to those of representative reference genes of a certain group was identified as an ESRT and classified into the corresponding group. By analyzing the transcriptomic data of hac1Δ cells, ESRTs whose inductions were dependent on Hac1 were identified as UPR target genes (see detail in “Methods” section).
Genes in the first group were rapidly induced upon DTT treatment. Reference genes were DER1, HRD1 and UBC7. Der1 and Hrd1 are the subunits of the Hrd1 complex, an essential component of ERAD, while Ubc7 is an E2 interacting with the Hrd1 complex [26]. Expression levels of genes in this group reached the peak after 15 min upon DTT treatment and then gradually went down at the following time points. A total of 41 ESRTs, including 34 UPR target genes, were included in this group. Since all three reference genes were related to ERAD, it was expected to obtain the enrichment of terms associated with ubiquitin-dependent protein catabolic process. 9 genes involved in the carbohydrate metabolic process were enriched in this group, which included 4 genes (VID28, VID30, FYV10 and GID8) encoding subunits of the glucose-induced degradation deficient (GID) complex (Fig. 3a). The GID complex is responsible for the repression of gluconeogenesis [27]. Among the 9 genes, GPM3 was not designated as a UPR target gene and RAG5 (homolog of ScHXK2) was previously identified as a UPR target gene in S. cerevisiae [3], while the rest 7 genes were linked with UPR for the first time. UBC7, a reference gene of this group, did not pass the statistical criterion of UPR target genes (P = 0.71). The upregulation of UBC7 by 15-min DTT treatment was slightly but not significantly reduced in hac1Δ cells (Additional file 1: Table S1). A similar situation was reported in S. cerevisiae, as UBC7 was just below the criterion of the UPR target gene in a transcriptomic analysis [19]. However, UBC7 was still identified as a UPR target gene in the same report, probably due to its role in ERAD [19]. In another report, Hac1-dependent upregulation of UBC7 was proved experimentally in S. cerevisiae [28], suggesting UBC7 was a false negative in the transcriptomic analysis. Therefore, UBC7 was designated as a UPR target gene of K. marxianus.

ESRTs clustered in 4 groups. a-d Significantly upregulated genes during DTT or TM treatment were clustered in 4 groups. Characteristics of transcriptional patterns in each group were defined by reference genes (marked by asterisks). Genes displaying similar transcriptional patterns to those of reference genes were clustered together. Genes rapidly induced in DTT are shown in a. Genes gradually induced in DTT are shown in b. Genes gradually induced in TM are shown in c. Genes induced late in TM are shown in d. UPR target genes are labeled in red. GO enrichment of genes in each group are shown below the cluster. The colour of a point represented the ontology. BP stood for biological process, CC for cellular component and MF for molecular function. The size of a point represented the adjusted P-value
Genes of the second group were induced gradually upon treatment of DTT. KAR2, LHS1, ERO1, MPD1 and PDI1 were reference genes of this group. A total of 55 genes fit the criterion, including 47 UPR target genes. GO analysis revealed that 7 genes involved in the actin filament organization were enriched in this group (Fig. 3b). None of these 7 genes was directly linked with ER stress or UPR before. However, genes related to actin filament organization, including RDI1, were overexpressed in an ethanol-tolerant S. cerevisiae strain during ethanol stress, while UPR target genes, including HAC1, ERO1 and KAR2 were also upregulated [29]. Secretory vesicles are transported on actin filaments [30]. Therefore, the induction of genes involved in actin filament organization might contribute to the relief of ER stress by promoting secretory vesicles transport.
During the treatment of TM, 39 genes, including reference genes KAR2, ERO1 and PDI1, were gradually induced. A total of 35 UPR target genes were included in this group. Among 39 ESRTs, genes encoding ER proteins and genes involved in ERAD were enriched. In S. cerevisiae and A. niger, ER stress triggered the removal of unfolded proteins by the ERAD system [3, 31]. Our results suggest a conserved relationship between ER stress response and the ERAD in K. marxianus.
The fourth group represented genes induced late upon TM treatment. The levels of four reference genes, including SEC12, SEC24, SFB3 and HRD3 remained constant at 15 and 30 min and then were upregulated at 60 min. Another 4 ESRTs, including FYV8, MNR2, SPF1 and ORM1, were included in this group. All 8 genes were identified as UPR target genes. FYV8 was a gene with an unknown function. MNR2 encodes a vacuolar membrane protein required for magnesium homeostasis [32]. SPF1 encodes an ion transporter of the ER membrane [33]. ORM1 encodes an ER membrane protein that mediates sphingolipid homeostasis [34]. Three reference genes, including SEC12 [35], SEC24 [36] and SFB3 [37], encode proteins involved in the COP II-coated vesicle formation from the ER membrane. The other reference gene, HRD3 encodes a subunit of the Hrd1 complex located in the ER membrane [38]. Since MNR2, SPF1 and four reference genes were related to ER membrane, it was not surprising that the GO term of ER membrane was enriched in this group (Fig. 3d).
There was an overlapping of ESRTs and UPR targets genes from the four groups discussed above (Fig. 4a, b). In total, 115 ESRTs and 97 UPR target genes were included in the four groups (Additional file 4: Table S4). Among 115 ESRTs, 67 genes were related to UPR or ER stress previously. The remaining 48 genes were linked with ER stress for the first time (Additional file 4: Table S4). Among 97 UPR targets, 62 genes were linked with UPR for the first time (Additional file 4: Table S4). Novel ESRTs and UPR target genes might underlie species-specific characteristics of the ER stress response and UPR network in K. marxianus.

Identification of UPREs in the promoters of UPR target genes. a The Venn diagram of the overlapping of ESRTs and UPR target genes. The numbers of ESRTs and UPR target genes in each subset were labeled in black and red, respectively. b Distribution of UPRE-1 and UPRE-2 motifs in the UPR targets genes from different groups. c, d Alignment of promoters of UPR target genes that contain UPRE-1 (c) or UPRE-2 motifs (d). The consensus is listed below the alignment. Flanking nucleotides are indicated in light grey. Coordinates are relative to the start codon of the respective gene
In S. cerevisiae, Hac1 binds to UPRE motifs to initiate the expression of UPR target genes [39]. The promoters of UPR target genes in K. marxianus were analyzed to identify potential Hac1-binding sites. A 7-bp consensus (5ʹ-CASNGKD-3ʹ) resembled the core UPRE-1 motif (5ʹ-CAGNGTG-3ʹ) in S. cerevisiae (Fig. 4c). Another 7-bp consensus (5ʹ-ACGTGKY-3ʹ) exhibited high similarity to the UPRE-2 motif (5ʹ-TACGTG-3ʹ) in S. cerevisiae (Fig. 4d). In terms of sequence similarity, the UPRE-2 motif was more conserved than the UPRE-1 motif. Among 97 UPR target genes identified in this study, 14 genes contain a single UPRE-1 motif and 16 genes contain a single UPRE-2 motif in their promoters, suggesting these genes are induced by direct binding of Hac1 (Fig. 4b and Additional file 4: Table S4). Meanwhile, 7 genes contain both UPRE motifs (Fig. 4b and Additional file 4: Table S4). Notably, among homologues of the 7 genes in S. cerevisiae, ERO1, LHS1 and KAR2 contained UPRE-1 and UPRE-2 motifs. The result suggests that the dual-site recognition of ERO1, LHS1 and KAR2 by Hac1 may be conserved in S. cerevisiae and K. marxianus [39].
Effects of overexpressing ESRTs on secretory expressions of lignocellulolytic enzymes
To study the role of ESRTs in regulating secretory expression, two strains constitutively secreting heterologous lignocellulolytic enzymes were constructed. RuCelA is a bifunctional xylanase/endoglucanase from yak rumen microorganisms that can simultaneously produce xylo-oligosaccharides and cello-oligosaccharides from lignocellulose [40]. AnFaeA is a feruloyl esterase from A. niger, which is a part of the hemicellulase complex that acts collectively and synergistically to completely hydrolyze feruloyl-polysaccharide. Genes encoding RuCelA and AnFaeA were integrated into the INU1 loci of a T1 strain to obtain LHP1021 and LHP643, respectively. The T1 strain was derived from FIM-1ΔU that improved the yield of heterologous proteins by attenuating autophagy [41]. In LHP1021 and LHP643, RuCelA and AnFaeA were expressed by a strong INU1 promoter and their secretions were directed by an alpha factor signal peptide from S. cerevisiae. RuCelA is a 532 aa protein containing 5 cysteine residues, while AnFaeA is a 282 aa protein containing 7 cysteine residues. Overexpression of these two heterologous enzymes might cause different types of ER stress, which provided a good system to evaluate the versatility of ESRTs in handling ER stress.
The ESRTs were inserted behind a strong TEF promoter separately on a multi-copy plasmid. The resultant plasmids were separately transformed into LHP1021 and LHP643. Except for 13 ESRTs that might cause cytotoxicity to K. marxianus, thus failing to obtain transformants, a total of 102 ESRTs were successfully overexpressed and effects on the secretory expressions of both AnFaeA and RuCelA were measured. As shown in Fig. 5a and b, overexpressing 16 and 11 ESRTs significantly increased the extracellular activities of AnFaeA and RuCelA, respectively. Overexpressing 10 and 9 ESRTs significantly reduced the secretory expressions of AnFaeA and RuCelA, respectively. Representative ESRTs that affected secretory expressions are listed in Fig. 5c. Due to the limited fundamental research in K. marxianus, functions of these ESRTs were mainly predicted by their orthologs in S. cerevisiae. PDC1, PGK and VID28 are involved in carbohydrate metabolism. PDC1 encodes a pyruvate decarboxylase isozymes that decarboxylates pyruvate to acetaldehyde [42]. Overexpression of PDC1 caused the highest improvement for the secretory expression of AnFaeA. PGK encodes phosphoglycerate kinase [43]. Vid28 is a subunit of the GID complex, which is a highly conserved ubiquitin ligase complex targeting key enzymes of gluconeogenesis for degradation [27]. Overexpressing PGK and VID28 significantly improved the expression of RuCelA and AnFaeA. Met13 is involved in the tetrahydrofolate interconversion pathway [44]. Overexpressing MET13 significantly improved the expression of RuCelA. Caj1 is a chaperone that regulates the stability or turnover of plasma membrane proteins [45]. Out of the 102 ESRTs overexpressed in this study, CAJ1 was the only gene that improved the secretory expressions of both AnFaeA and RuCelA. The result suggests that only a few ESRTs, when overexpressed, displayed positive effects on the productions of heterologous proteins in K. marxianus.

Effects of overexpressing ESRTs on secretory expressions of lignocellulolytic enzymes. Effects of overexpressing ESRTs on secretory expressions of AnFaeA (a) and RuCelA (b). AnFaeA or RuCelA was integrated at INU1 loci in the genome. Overexpression of the ESRT was driven by a TEF promoter on a multi-copy plasmid. The activity of AnFaeA or RuCelA in the supernatant of cells transformed with a void vector was designated as unit 1. The column corresponding to the void vector control is in red. Value represented mean ± SD (n = 3) (*P < 0.05, **P < 0.01, ***P < 0.001). ESRTs that significantly improved and reduced extracellular activities are in red and blue, respectively. c Representative ESRTs that displayed significant effects on secretory expressions of lignocellulolytic enzymes (*P < 0.05, **P < 0.01, *** P < 0.001). d Effects of known UPR target genes on secretory expressions of lignocellulolytic enzymes
On the other hand, overexpressing several genes involved in ERAD significantly reduced the secretory expressions of AnFaeA or RuCelA. Yet3 is a homolog of human Bap31p which plays a role in targeting the misfolded protein to ERAD [46]. Secretory expressions of RuCelA and AnFaeA were reduced upon overexpressing YET3. Hrd3 and Usa1 belong to the Hrd1 complex which is responsible for the ubiquitination of ERAD-L substrates [47]. Overexpressing HRD3 and USA1 significantly reduced the expression of RuCelA.
In addition, overexpression of an unknown gene, KLMA_50479 increased the extracellular expression of RuCelA but reduced the expression of AnFaeA. Since KLMA_50479 was induced by ER stress and might play a role in regulating secretion, KLMA_50479 was named ESR1 (ER stress response gene 1).
In previous reports, overexpressing KAR2, PDI1, ERO1, JEM1 or LHS1, promoted the productions of heterologous proteins [10, 12,13,14]. However, overexpressing these genes, as well as four other conserved UPR target genes, MCD4, MPD1, OST2 and SEC12, exhibited no significant or very mild effects on the expressions of AnFaeA and RuCelA (Fig. 5d). The failure of improving secretion by overexpressing these genes in K. marxianus might be because choices of promoter and host in this study were different from those in previous studies, including AOX1 promoter in K. phaffii [12, 14], glaA promoter in A. niger [10], ADH1 and PGK1 promoters in S. cerevisiae [13]. In addition, the properties of an esterase (AnFaeA) and a glycosidase (RuCelA) aimed for secretion in this study were different from those of proteins used previously, including protease, oxidoreductase [10], antibody fragment [12], human albumin and hormone [13, 14].
Deletions of ESR1 and ERAD genes improved secretory expressions
As shown in Fig. 5c, overexpressing some ESRTs significantly reduced the secretory expressions of AnFaeA or RuCelA. To investigate whether their deletions can promote secretion, ERS1, HRD3, YET3 and USA1, were deleted in T1, respectively, to obtain LHP1027 ~ LHP1030 (Additional file 5: Table S5). Besides RuCelA and AnFaeA, an endo-1,4-β-endoxylanase Xyn-CDBFV was also overexpressed by a pKD1-based multi-copy plasmid. Xyn-CDBFV undergoes heavy glycosylation in K. marxianus [48]. Plasmids expressing RuCelA, AnFaeA or Xyn-CDBFV were transformed into LHP1027 ~ LHP1030 separately. Transformants were grown in flasks and extracellular activities in the supernatant were measured. As shown in Fig. 6a–d, deletion of USA1, HRD3 or ESR1 significantly improved the expression of RuCelA, AnFaeA and Xyn-CDBFV. To investigate the relationships between ESR1, YET3 and USA1, double-deletion mutants, including usa1Δyet3Δ, usa1Δesr1Δ and yet3Δesr1Δ were constructed. Expressions of AnFaeA and RuCelA in a combined mutant were measured and compared with those in a single mutant (Fig. 6e, f). Deletions of USA1 and YET3 showed weak negative interaction, suggesting both genes affect the secretion in partially overlapped pathways. This was expected because both USA1 and YET3 play roles in ERAD [46, 47]. Deletions of ESR1 and YET3 showed additive interaction, suggesting two genes mediated secretory expressions through different pathways. The deletion of USA1 showed relatively strong negative interactions with the deletion of ESR1, suggesting two genes used overlapped pathways to affect secretory expressions. Therefore, ESR1 might be functionally related to USA1. The esr1Δ, hrd3Δ, usa1Δ mutants were genomically stable and were suitable to serve as chassis for the expression of heterologous proteins and construction of consolidated bioprocessing (CBP) strains for bioethanol production.

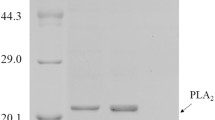
Secretory expressions of lignocellulolytic enzymes in the cells with deletion of ESRTs. Secretory expression of AnFaeA (a), RuCelA (b) or Xyn-CDBFV (c) in the cells with deletion of ESRTs. ESR1, HRD3, YET3 or USA1 was deleted. A multi-copy plasmid overexpressing AnFaeA, RuCelA or XynCDB was transformed into deletion mutants. Extracellular activity in the supernatant was measured. The activity of AnFaeA, RuCelA or Xyn-CDBFV expressed by the wild-type strain was designated as unit 1. Value represented mean ± SD (n = 3) (*P < 0.05, **P < 0.01, ***P < 0.001). d Summary of effects of deletion of ESRTs. Data were extracted from (a ~ c) (*P < 0.05, **P < 0.01, ***P < 0.001). e Extracellular activities of AnFaeA and RuCelA in double-deletion mutants. The activity of AnFaeA or RuCelA expressed by the wild-type strain was designated as unit 1. Value represented mean ± SD (n = 3) (*P < 0.05, **P < 0.01). f Interaction between deletion of ESRTs. Trait value (T) was represented by the relative activity in the double or single deletion mutant compared with that in wild-type cells. The interaction value was calculated as TAB-TA-TB, where TAB represented the trait value of a double-deletion mutant, while TA and TB represented values of single mutants. Interaction values are shown in the chart. Values closing to zero indicated additive interactions and negative values indicated negative interactions. g–j Expression of Xyn-CDBFV or AnFaeA by wild-type and hrd3Δ cells in a 5-L fermentor. Cells were grown in the fermentor for 72 h. Curves of enzymatic activity and OD600 were plotted for cells expressing Xyn-CDBFV (g) and AnFaeA (i). SDS-PAGE analysis of 8 ul supernatant collected at indicated time point is shown in h, j. The arrow in j indicated the position of inulinase, which was a host protein secreted by K. marxianus [63]
To evaluate the industrial potential of ESRT mutants for producing lignocellulose-degrading enzymes, episomal plasmids expressing Xyn-CDBFV or AnFaeA were transformed into the hrd3Δ mutant and transformants were grown in a 5-L fermentor for 72 h. A synthetic medium was used in the fermentation to reduce cost. During the 72 h fermentation, the OD600 curve of hrd3Δ cells resembled those of wild-type cells, suggesting the mutation did not affect the growth of cells (Fig. 6g, i). The extracellular amounts of Xyn-CDBFV and AnFaeA in hrd3Δ cells were 43% and 28% higher than those of wild-type cells, with the activities of 48,970 U/mL and 14,570 U/mL at 72 h, respectively (Fig. 6g, i). Improved secretions of enzymes in the hrd3Δ mutant were confirmed by the SDS-PAGE analysis (Fig. 6 h, j). Based on the specific activities of Xyn-CDBFV (4000 U/mg) and AnFaeA (18,000 U/mg), the protein concentrations of Xyn-CDBFV and AnFaeA in the culture of hrd3Δ cells were predicted to be 12.2 g/L and 6.16 g/L, respectively, which were the highest concentrations of endoxylanase and feruloyl esterase produced by K. marxianus so far.
Discussion
In this study, we characterized the transcriptional scope of ER stress in K. marxianus for the first time. A total of 892 genes were significantly upregulated upon treatment of DTT or TM. Among upregulated genes, 115 genes were identified as ESRTs and 97 genes as UPR target genes. 58% of ESRTs and 53% of UPR target genes were previously linked with UPR or ER stress, suggesting the majority of the ER response and UPR network was conserved in yeast and fungi. Meanwhile, the rest genes were novel ESRTs and UPR target genes, which provides valuable information for a better understanding of ER response and UPR network in K. marxianus. Among the newly identified UPR target genes, four genes encoding subunits of the GID complex, including VID28, VID30, FYV10 and GID8, were induced rapidly upon DTT treatment. VID30 and FYV10 promoters contain UPRE-2 motifs (Additional file 4: Table S4), which are not found in the promoters of their orthologs in S. cerevisiae. GID complex is required for the degradation of Fbp1, a key enzyme in gluconeogenesis, and that leads to the metabolic switch from gluconeogenesis to glycolysis [27]. Consistently, glycolytic gene PGK, fermentation genes PDC1 and ADH1 were induced following the induction of genes encoding GID subunits (Fig. 3b, Additional file 1: Table S1). Gluconeogenesis hydrolyzes four ATPs and two GTPs to direct the process of glucose formation [49]. In contrast, glycolysis and fermentation produce two ATP per glucose with a high rate and low yield, compared to ATP production with a low rate and high yield in respiration [50]. Therefore, one direct outcome of the switch from gluconeogenesis to glycolysis and fermentation is to provide ATP quickly, suggesting ER stress raises an urgent demand for ATP in K. marxianus. Overexpressing genes encoding GID subunits and glycolytic enzymes might fasten the flux to glycolysis and help cells to relieve the ER stress more quickly. Consistent with this idea, overexpression of VID28, PDC1 or PGK improved expressions of lignocellulolytic enzymes (Fig. 5c). Increased expression of genes involved in glycolysis was in company with decreased expressions of genes involved in cellular respiration, as shown by the downregulation of oxidative phosphorylation genes after 15-min DTT treatment (Fig. 2e, g). The result suggests a preference for the high rate of ATP production during the initial response to ER stress. Interestingly, a shift from respiration to fermentation was observed in a set of S. cerevisiae mutants displaying improved secretory expressions of α-amylase [25]. This suggests promoting a flux to glycolysis and fermentation is an effective strategy to improve secretory expressions in yeast.
ERAD is a noteworthy pathway that was related to the secretory expression in this study. In the ERAD-L system, which is best characterized in S. cerevisiae, misfolded proteins in the ER lumen are processed, recognized and then transferred to the Hrd1 complex. Hrd1 complex, composed of multispanning ubiquitin ligase Hrd1 and four additional proteins (Hrd3, Der1, Usa1, Yos9), is responsible for the retrotranslocation-coupled ubiquitination of the ERAD substrates. Following retrotranslocation to the cytoplasm, ubiquitinated substrates are transferred to the 26S proteasome via the Cdc48 complex and are degraded afterwards [26]. In this study, genes encoding subunits of the Hrd1 complex, including HRD1, HRD3, DER1 and USA1, were identified as UPR target genes. Deletion of USA1 and HRD3 promoted the secretory expressions of three lignocellulolytic enzymes in the flask using a rich medium. Improved secretory expression in hrd3Δ cells was reproduced in a 5-L fermentor. In S. cerevisiae, deletion of Hrd1, Hrd3 or Yos9 decreased the secretory expression of a human IgG [51]. In A. niger, deletion of Der1 or Hrd3 improved the intracellular amount of GalGus [31]. Therefore, deletions of the Hrd1 complex subunits displayed species-specific effects on the productions of heterologous proteins. Hrd3 is responsible for the initial recognition of glycosylated substrates [38]. Usa1 is required for optimal function and regulation of Hrd1 [52]. Deletion of HRD3 and USA1 severely impaired the degradation of ERAD substrates [47]. In K. marxianus, the degradation of unfolded or misfolded heterologous proteins via the ERAD system might be interfered in hrd3Δ and usa1Δ cells, and heterologous proteins are cleared from the cells through the alternative secretion route, which improves the secretion of heterologous proteins. Disruption of the Hrd1 complex might serve as a new strategy to improve the secretion expression of heterologous proteins in K. marxianus.
In this study, a novel gene, ESR1, was identified. There is no ortholog of Esr1 in S. cerevisiae and Esr1 in K. marxianus shares only 66% identity with its ortholog in the sister species K. lactis, suggesting it is a newly evolved protein. ESR1 was an ESRT but not a UPR target gene. Deletion of ESR1 promoted secretory production of three lignocellulolytic enzymes (Fig. 6d). Deletion of ESR1 displayed a negative interaction with deletion of USA1 and an additive interaction with deletion of YET3 in improving secretory expressions (Fig. 6e). ESR1 were induced by both DTT and TM (Fig. 3b, Additional file 2: Table S2). Genes displaying similar transcriptional patterns to ESR1 were enriched in the GO terms of ‘membrane trafficking’ and ‘protein processing in the endoplasmic reticulum’. Moreover, Esr1 was predicted by OrthoDB to be an integral component of the membrane. Taken together, Esr1 might be a protein localized in the ER membrane involved in protein secretion.
Overexpressing CAJ1 improved the extracellular activities of both AnFaeA and RuCelA. Caj1 belongs to the Hsp40/DnaJ family. DnaJ/Hsp40-like genes were identified as UPR target genes in human [53]. In S. cerevisiae, Caj1 colocalizes with the plasma membrane and overexpressing Caj1 stabilizes plasma membrane proteins, including amino acid permeases [45]. Overexpression of Caj1 may stabilize specific plasma membrane proteins required for the export of proteins through the membrane, which leads to improved secretion in K. marxianus.
High-level secretory expressions of heterologous cellulases and glycosidases might promote the efficiency of CBP. For example, a semi-industrial S. cerevisiae strain displaying improved expressions of heterologous α-amylase and glucoamylase from multi-copy plasmids achieved a 70% increase in the production of ethanol, compared with a laboratory strain expressing the same enzymes [54]. The improved extracellular activity of an integrated inulinase in S. cerevisiae increased ethanol production from inulin and Jerusalem artichoke tuber powder in 24 h [55]. In this study, the deletion or overexpression of specific ESRTs of K. marxianus improved the secretion of lignocellulolytic enzymes expressed from episomal plasmids or integrated loci. So far, genes encoding heterologous cellulolytic enzymes were integrated into the genome of K. marxianus for CBP [56,57,58]. In the future, it will be intriguing to investigate the effect of modulating ESRTs on the productions of enzymes and the CBP efficiency in these strains.
Methods
Strains and plasmids
Strains used in this study are listed in Additional file 5: Table S5. Plasmids are listed in Additional file 6: Table S6. Relevant primers are listed in Additional file 7: Table S7. FIM-1ΔU strain was used as a wild-type strain for RNAseq [48]. T1 strain was used for expressions of lignocellulolytic enzymes [41].
LHZ765 served as a backbone for overexpressing ESRTs. INU1 promoter and terminator in pUKDN132 were replaced by TEF promoter and TEF terminator, respectively, to obtain LHZ765. The ORF of ESRTs were inserted between SacII and PacI sites of LHZ765 separately to obtain LHZ767 ~ LHZ880. The ORF of AnFaeA was inserted between SacII and PacI sites of LHZ765 to obtain LHZ766. LHZ442 (pZP52) overexpressing RuCelA and LHZ443 (pZP46) overexpressing Xyn-CDBFV were described previously [48].
Deletion of a gene in K. marxianus was performed by homologous recombination with the aid of a CRISPR plasmid. Three CRISPR vectors, LHZ296, LHZ301 and LHZ531, were constructed and used as backbones to build CRISPR plasmids (Additional files 8, 9, 10). pUKD-N122-AUC contains two SapI sites for the insertion of a target sequence in front of gRNA [41]. The ARS7 fragment in pUKD-N122-AUC was replaced by a pKD1 fragment [59], to obtain LHZ296. A second gRNA expression cassette containing two AarI sites for the insertion of the target sequence was cloned upstream of KmURA3 in LHZ296 to construct LHZ301. The pKD1 fragment in LHZ296 was replaced by ARS1/CEN [60], to construct LHZ531. Primers containing 20 bp target sequence were annealed in pairs and inserted into SapI or AarI sites of LHZ296, LHZ301 and LHZ531. Details of resultant CRISPR plasmids are listed in Additional file 6: Table S6.
To delete IRE1 in FIM-1ΔU, IRE1 with the flanking sequence was amplified and ligated with pMD18-T. The ORF of IRE1 in the resulting plasmid was removed by mutagenesis PCR to obtain LHZ748. Donor sequence was amplified from LHZ748 and co-transformed with CRISPR plasmid LHZ747 into FIM-1ΔU to obtain LHP1019. Similarly, HAC1 was deleted in FIM-1ΔU to obtain LHP1020. ESR1, HRD3, YET3 and USA1 were deleted in T1, respectively, to obtain LHP1027 ~ LHZ1030. ESR1 was deleted in LHP1029 and LHP1030 to obtain LHP1031 and LHP1032, respectively. YET3 was deleted in LHP1030 to obtain LHP1033.
To replace INU1 with a gene of interest, INU1 with the flanking sequence was amplified and ligated with pMD18-T. The ORF of INU1 in the resulting plasmid was removed by mutagenesis PCR to obtain LHZ424. To replace INU1 with AnFaeA, the ORF of AnFaeA was amplified and inserted between SpeI and NotI sites of LHZ424. The donor sequence was amplified from the resultant plasmid and co-transformed with LHZ759 into T1 to obtain LHP643. Similarly, INU1 in T1 was replaced by RuCelA to obtain LHP1021.
For enzymatic assays, plasmids overexpressing different ESRT (LHZ767 ~ LHZ880) was transformed into LHP643 and LHP1021. The plasmid overexpressing Xyn-CDBFV (LHZ443) was transformed into LHP1027 ~ LHP1031. The plasmid overexpressing RuCelA (LHZ442) or AnFaeA (LHZ766) was transformed into LHP1027 ~ LHP1033. Transformants were selected on Sc-Ura medium.
Media
K. marxianus cells were cultivated at 30°. C. YPD medium (2% peptone, 1% yeast extract, 2% agar for plates), synthetic complete minus uracil medium (SC-Ura) and synthetic dextrose minimal medium (SD) were prepared as described before [61]. YG liquid medium (4% glucose, 2% yeast extract) was prepared for expressions of AnFaeA, RuCelA and Xyn-CDBFV in flasks.
Spot assay
Fresh FIM-1ΔU, LHP1019 or LHP1020 cells were grown in YPD liquid medium overnight. Cells were collected and adjusted to an OD600 of 1.0. Then, fivefold serial dilutions were performed and 5 µL dilutions were spotted onto YPD containing 10 ~ 30 mM DTT (D8220, Solarbio, China) or 0.1 ~ 0.5 µg/mL TM (T8480, Solarbio). The plates were incubated at 30 °C for 2 days.
RNA extraction and qPCR
Fresh FIM-1ΔU and LHP1020 cells were grown in YPD liquid medium overnight. Cells were diluted into a fresh YPD liquid medium to start at an OD600 of 0.2 and were grown for 3 ~ 5 h until the exponential phase (OD600 of 0.6–0.8). The cultures were supplemented with DTT (a final concentration of 10 mM) or TM (a final concentration of 0.5 µg/mL). FIM-1ΔU cells were harvested at 0, 15, 30 and 60 min after treatment. LHP1020 cells were harvested at 15 and 60 min after treatment. Cells were frozen at − 80 °C. Three biological repeats were collected at each time point. RNA was extracted from frozen cells using a Quick-RNA Fungal/Bacterial Miniprep kit (R2010, Zymo Research, USA) and were reverse transcribed using a PrimeScript RT Reagent Kit (RR037A, Takara, China). The qPCR was performed using TB Green Premix Ex Taq (RR820A, Takara). Primers used in qPCR are listed in Additional file 7: Table S7.
RNA sequencing
RNA was extracted as described above. Samples were reversed transcribed and sequenced by Illumina HiSeq X Ten system at Biomedical Big Data Center, Shanghai Institutes for Biological Sciences, or by Illumina NovaSeq 6000 system at Mingma Biotechnology. A total of 5.6 million reads and 40 million reads were obtained on average for samples of FIM-1ΔU and LHP1020, respectively. Raw sequencing data were uploaded to NCBI. Reads were aligned, assembled and analyzed as previously described [62], by using the genome of K. marxianus DMKU 3–1042 as a reference (GenBank assembly accession GCA_001417885.1). A q-value threshold of 0.05 was set in terms of altered expression. At an indicated time point, the relative transcript abundance of a gene was calculated by dividing the Fragments Per Kilobase of exon model per Million mapped fragments (FPKM) value in the cells treated with DTT or TM by the FPKM value in the cell without treatment. The relative transcript abundances of genes are listed in Additional file 1: Table S1.
Identification of ESRTs and UPR target genes
Orthologs of classic UPR target genes that displayed specific transcriptional patterns during ER stress were selected as reference genes. DER1, HRD1 and UBC7 were references for the group of genes induced rapidly upon DTT treatment (Group A). KAR2, LHS1, ERO1, MPD1 and PDI1 were references for the group of genes gradually induced upon DTT treatment (Group B). KAR2, ERO1 and PDI1 were references for the group of genes gradually induced upon TM treatment (Group C). SEC12, SEC24, SFB3 and HRD3 were references for the group of genes induced late upon TM treatment (Group D). In each group, the difference of the relative transcript abundance (log2) of a reference gene between adjacent time points (15 min-0 min, 30 min-15 min or 60 min-30 min) was calculated. Mean (\({\mu }_{i}\)) and variance (\({\nu }_{i}\)) of the difference between reference genes were calculated for each group. Chi-square tests were performed to identify potential ESRT genes based on the formula:
\({x}_{i}\) represented the difference of the relative transcript abundance (log2) of a gene of interest between adjacent time points. \({\chi }^{2}\) statistic was calculated by using \({\mu }_{i}\) and \({\nu }_{i}\) of each group with the assumption that the distribution of observation \({x}_{i}\) had a normal distribution. Under the null hypothesis, the \({\chi }^{2}\) statistic follows a central Chi-square distribution with a degree of freedom 3. P-value was obtained according to \({\chi }^{2}\). Because DTT induced an acuter ER stress than TM, a high threshold of P-value was applied to identify ESRTs induced by DTT in group A and B, while a low threshold of P-value was applied to identify UPR targets genes induced by TM in group C and D. A gene with a P-value > 0.1 and a fold change > 4 at 15 min during DTT treatment was identified as an ESRT of group A. A gene with a P-value > 0.1 was identified as an ESRT of group B. A gene with a P-value > 0.05 was identified as an ESRT of group C or D. Two-tailed T-tests were performed to determine if there is a significant difference between the means of relative transcript abundance of a given ESRT in FMI1ΔU cells and hac1Δ cells. A timepoint displaying the highest relative transcript abundance in each group was chosen for the T-test, including 15-min DTT treatment in group A, 60-min DTT treatment in group B, 60-min TM treatment in group C and D. An ESRT with a P-value < 0.05 was designated as a UPR target gene. A full list of ESRTs and UPR target genes is shown in Additional file 4: Table S4.
GO analysis
Gene ontology (GO) analyses were performed in each group of genes, respectively, via R (version 4.0.5) with R packages and RSQLite (version 2.2.5), clusterProfiler (version 3.18.1), Rgraphviz (version 2.34.0), pathview (version 1.30.1) and org.Sc.sgd.db (version 3.12.0).
Enzyme activity assay
Transformants that contained the plasmid overexpressing ESRT, Xyn-CDBFV, RuCelA or AnFaeA were grown on the SC-Ura medium for one day. Then, fresh cells were grown in a YG medium for 72 h. The activities of RuCelA, AnFaeA and Xyn-CDBFV in the supernatant were measured as described previously [48].
Fed-batch fermentation
Plasmid overexpressing RuCelA (LHZ442) or Xyn-CDBFV (LHZ443) was transformed into LHP1028. Seed culture was prepared by growing fresh transformant in SD liquid medium for 16 h. Fed-batch fermentations were carried out in a 5-L bioreactor (BXBIO, Shanghai, China) as described previously [48]. The supernatant was collected every 24 h. Samples were subjected to the SDS-PAGE analysis and enzymatic assay.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the NCBI (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA729880; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA777750).
Abbreviations
- Bp:
-
Base pair
- CBP:
-
Consolidated bioprocessing
- DTT:
-
Dithiothreitol
- ER:
-
Endoplasmic reticulum
- ERAD:
-
ER-associated protein degradation
- ERS1:
-
ER stress responsible gene 1
- ESRTs:
-
ER stress response target genes
- TM:
-
Tunicamycin
- ORF:
-
Open reading frame
- UPR:
-
Unfolded protein response
- UPRE:
-
Unfolded protein response element
References
Idiris A, Tohda H, Kumagai H, Takegawa K. Engineering of protein secretion in yeast: strategies and impact on protein production. Appl Microbiol Biotechnol. 2010;86(2):403–17.
Gasser B, Saloheimo M, Rinas U, Dragosits M, Rodriguez-Carmona E, Baumann K, Giuliani M, Parrilli E, Branduardi P, Lang C, et al. Protein folding and conformational stress in microbial cells producing recombinant proteins: a host comparative overview. Microb Cell Fact. 2008;7:11.
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–58.
Fan F, Ma G, Li J, Liu Q, Benz JP, Tian C, Ma Y. Genome-wide analysis of the endoplasmic reticulum stress response during lignocellulase production in Neurospora crassa. Biotechnol Biofuels. 2015;8:66.
Graf A, Gasser B, Dragosits M, Sauer M, Leparc GG, Tuchler T, Kreil DP, Mattanovich D. Novel insights into the unfolded protein response using Pichia pastoris specific DNA microarrays. BMC Genomics. 2008;9:390.
Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13(2):89–102.
Pincus D, Aranda-Diaz A, Zuleta IA, Walter P, El-Samad H. Delayed Ras/PKA signaling augments the unfolded protein response. Proc Natl Acad Sci U S A. 2014;111(41):14800–5.
Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457(7230):687–93.
Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87(3):391–404.
Valkonen M, Ward M, Wang H, Penttila M, Saloheimo M. Improvement of foreign-protein production in Aspergillus niger var. awamori by constitutive induction of the unfolded-protein response. Appl Environ Microbiol. 2003;69(12):6979–86.
Harmsen MM, Bruyne MI, Raue HA, Maat J. Overexpression of binding protein and disruption of the PMR1 gene synergistically stimulate secretion of bovine prochymosin but not plant thaumatin in yeast. Appl Microbiol Biotechnol. 1996;46(4):365–70.
Damasceno LM, Anderson KA, Ritter G, Cregg JM, Old LJ, Batt CA. Cooverexpression of chaperones for enhanced secretion of a single-chain antibody fragment in Pichia pastoris. Appl Microbiol Biotechnol. 2007;74(2):381–9.
Payne T, Finnis C, Evans LR, Mead DJ, Avery SV, Archer DB, Sleep D. Modulation of chaperone gene expression in mutagenized Saccharomyces cerevisiae strains developed for recombinant human albumin production results in increased production of multiple heterologous proteins. Appl Environ Microbiol. 2008;74(24):7759–66.
Vad R, Nafstad E, Dahl LA, Gabrielsen OS. Engineering of a Pichia pastoris expression system for secretion of high amounts of intact human parathyroid hormone. J Biotechnol. 2005;116(3):251–60.
Phister TG, O’Sullivan DJ, McKay LL. Identification of bacilysin, chlorotetaine, and iturin a produced by Bacillus sp. strain CS93 isolated from pozol, a Mexican fermented maize dough. Appl Environ Microbiol. 2004;70(1):631–4.
dos Santos VC, Braganca CR, Passos FJ, Passos FM. Kinetics of growth and ethanol formation from a mix of glucose/xylose substrate by Kluyveromyces marxianus UFV-3. Antonie Van Leeuwenhoek. 2013;103(1):153–61.
Gombert AK, Madeira JV Jr, Cerdan ME, Gonzalez-Siso MI. Kluyveromyces marxianus as a host for heterologous protein synthesis. Appl Microbiol Biotechnol. 2016;100(14):6193–208.
Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Dissociation of Kar2p/BiP from an ER sensory molecule, Ire1p, triggers the unfolded protein response in yeast. Biochem Biophys Res Commun. 2000;279(2):445–50.
Kimata Y, Ishiwata-Kimata Y, Yamada S, Kohno K. Yeast unfolded protein response pathway regulates expression of genes for anti-oxidative stress and for cell surface proteins. Genes Cells. 2006;11(1):59–69.
Normington K, Kohno K, Kozutsumi Y, Gething MJ, Sambrook J. S cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell. 1989;57(7):1223–36.
Kohno K, Normington K, Sambrook J, Gething MJ, Mori K. The promoter region of the yeast KAR2 (BiP) gene contains a regulatory domain that responds to the presence of unfolded proteins in the endoplasmic reticulum. Mol Cell Biol. 1993;13(2):877–90.
Yu Y, Mo W, Ren H, Yang X, Lu W, Luo T, Zeng J, Zhou J, Qi J, Lu H. Comparative Genomic and Transcriptomic Analysis Reveals Specific Features of Gene Regulation in Kluyveromyces marxianus. Front Microbiol. 2021;12:598060.
Causton HC, Ren B, Koh SS, Harbison CT, Kanin E, Jennings EG, Lee TI, True HL, Lander ES, Young RA. Remodeling of yeast genome expression in response to environmental changes. Mol Biol Cell. 2001;12(2):323–37.
Lange H, Kispal G, Lill R. Mechanism of iron transport to the site of heme synthesis inside yeast mitochondria. J Biol Chem. 1999;274(27):18989–96.
Huang M, Bao J, Hallstrom BM, Petranovic D, Nielsen J. Efficient protein production by yeast requires global tuning of metabolism. Nat Commun. 2017;8(1):1131.
Sun Z, Brodsky JL. Protein quality control in the secretory pathway. J Cell Biol. 2019;218(10):3171–87.
Liu H, Pfirrmann T. The Gid-complex: an emerging player in the ubiquitin ligase league. Biol Chem. 2019;400(11):1429–41.
Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2(7):379–84.
Navarro-Tapia E, Nana RK, Querol A, Perez-Torrado R. Ethanol Cellular Defense Induce Unfolded Protein Response in Yeast. Front Microbiol. 2016;7:189.
DePina AS, Langford GM. Vesicle transport: the role of actin filaments and myosin motors. Microsc Res Tech. 1999;47(2):93–106.
Carvalho ND, Arentshorst M, Kooistra R, Stam H, Sagt CM, van den Hondel CA, Ram AF. Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger. Appl Microbiol Biotechnol. 2011;89(2):357–73.
Pisat NP, Pandey A, Macdiarmid CW. MNR2 regulates intracellular magnesium storage in Saccharomyces cerevisiae. Genetics. 2009;183(3):873–84.
Cronin SR, Rao R, Hampton RY. Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol. 2002;157(6):1017–28.
Han S, Lone MA, Schneiter R, Chang A. Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc Natl Acad Sci U S A. 2010;107(13):5851–6.
Sato K, Nakano A. Mechanisms of COPII vesicle formation and protein sorting. FEBS Lett. 2007;581(11):2076–82.
Miller E, Antonny B, Hamamoto S, Schekman R. Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 2002;21(22):6105–13.
Shimoni Y, Kurihara T, Ravazzola M, Amherdt M, Orci L, Schekman R. Lst1p and Sec24p cooperate in sorting of the plasma membrane ATPase into COPII vesicles in Saccharomyces cerevisiae. J Cell Biol. 2000;151(5):973–84.
Wu X, Siggel M, Ovchinnikov S, Mi W, Svetlov V, Nudler E, Liao M, Hummer G, Rapoport TA. Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science. 2020;368:6489.
Fordyce PM, Pincus D, Kimmig P, Nelson CS, El-Samad H, Walter P, DeRisi JL. Basic leucine zipper transcription factor Hac1 binds DNA in two distinct modes as revealed by microfluidic analyses. Proc Natl Acad Sci U S A. 2012;109(45):E3084-3093.
Chang L, Ding M, Bao L, Chen Y, Zhou J, Lu H. Characterization of a bifunctional xylanase/endoglucanase from yak rumen microorganisms. Appl Microbiol Biotechnol. 2011;90(6):1933–42.
Liu Y, Mo WJ, Shi TF, Wang MZ, Zhou JG, Yu Y, Yew WS, Lu H. Mutational Mtc6p attenuates autophagy and improves secretory expression of heterologous proteins in Kluyveromyces marxianus. Microb Cell Fact. 2018;17(1):144.
Schmitt HD, Zimmermann FK. Genetic analysis of the pyruvate decarboxylase reaction in yeast glycolysis. J Bacteriol. 1982;151(3):1146–52.
Watson HC, Walker NP, Shaw PJ, Bryant TN, Wendell PL, Fothergill LA, Perkins RE, Conroy SC, Dobson MJ, Tuite MF, et al. Sequence and structure of yeast phosphoglycerate kinase. EMBO J. 1982;1(12):1635–40.
Shan X, Wang L, Hoffmaster R, Kruger WD. Functional characterization of human methylenetetrahydrofolate reductase in Saccharomyces cerevisiae. J Biol Chem. 1999;274(46):32613–8.
Dobriyal N, Sagarika P, Shrivastava A, Verma AK, Islam Z, Gupta P, Mochizuki T, Abe F, Sahi C. Over-expression of Caj1, a plasma membrane associated J-domain protein in Saccharomyces cerevisiae, stabilizes amino acid permeases. Biochim Biophys Acta Biomembr. 2020;1862(11):183435.
Wakana Y, Takai S, Nakajima K, Tani K, Yamamoto A, Watson P, Stephens DJ, Hauri HP, Tagaya M. Bap31 is an itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a juxtanuclear compartment related to ER-associated Degradation. Mol Biol Cell. 2008;19(5):1825–36.
Vashistha N, Neal SE, Singh A, Carroll SM, Hampton RY. Direct and essential function for Hrd3 in ER-associated degradation. Proc Natl Acad Sci U S A. 2016;113(21):5934–9.
Zhou J, Zhu P, Hu X, Lu H, Yu Y. Improved secretory expression of lignocellulolytic enzymes in Kluyveromyces marxianus by promoter and signal sequence engineering. Biotechnol Biofuels. 2018;11:235.
Jeremy M, Berg JLT, Lubert S. Biochemistry, Fifth Edition: International Version (hardcover). New York: WH Freeman; 2002.
Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504–7.
de Ruijter JC, Frey AD. Analysis of antibody production in Saccharomyces cerevisiae: effects of ER protein quality control disruption. Appl Microbiol Biotechnol. 2015;99(21):9061–71.
Carroll SM, Hampton RY. Usa1p is required for optimal function and regulation of the Hrd1p endoplasmic reticulum-associated degradation ubiquitin ligase. J Biol Chem. 2010;285(8):5146–56.
Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–59.
Viktor MJ, Rose SH, van Zyl WH, Viljoen-Bloom M. Raw starch conversion by Saccharomyces cerevisiae expressing Aspergillus tubingensis amylases. Biotechnol Biofuels. 2013;6(1):167.
Wang D, Li FL, Wang SA. Engineering a natural Saccharomyces cerevisiae strain for ethanol production from inulin by consolidated bioprocessing. Biotechnol Biofuels. 2016;9:96.
Chang JJ, Ho FJ, Ho CY, Wu YC, Hou YH, Huang CC, Shih MC, Li WH. Assembling a cellulase cocktail and a cellodextrin transporter into a yeast host for CBP ethanol production. Biotechnol Biofuels. 2013;6(1):19.
Anandharaj M, Lin YJ, Rani RP, Nadendla EK, Ho MC, Huang CC, Cheng JF, Chang JJ, Li WH. Constructing a yeast to express the largest cellulosome complex on the cell surface. Proc Natl Acad Sci U S A. 2020;117(5):2385–94.
Yanase S, Hasunuma T, Yamada R, Tanaka T, Ogino C, Fukuda H, Kondo A. Direct ethanol production from cellulosic materials at high temperature using the thermotolerant yeast Kluyveromyces marxianus displaying cellulolytic enzymes. Appl Microbiol Biotechnol. 2010;88(1):381–8.
Chen XJ, Saliola M, Falcone C, Bianchi MM, Fukuhara H. Sequence organization of the circular plasmid pKD1 from the yeast Kluyveromyces drosophilarum. Nucleic Acids Res. 1986;14(11):4471–81.
Lee KS, Kim JS, Heo P, Yang TJ, Sung YJ, Cheon Y, Koo HM, Yu BJ, Seo JH, Jin YS, et al. Characterization of Saccharomyces cerevisiae promoters for heterologous gene expression in Kluyveromyces marxianus. Appl Microbiol Biotechnol. 2013;97(5):2029–41.
Amberg DC, Burke D, Strathern JN, Burke D. Methods in yeast genetics : a Cold Spring Harbor Laboratory course manual. 2005th ed. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2005.
Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT StringTie and Ballgown. Nat Protoc. 2016;11(9):1650–67.
Rouwenhorst RJ, Hensing M, Verbakel J, Scheffers WA, van Duken JP. Structure and properties of the extracellular inulinase of Kluyveromyces marxianus CBS 6556. Appl Environ Microbiol. 1990;56(11):3337–45.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (31771384 to YY), Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-KJGG-006), National Natural Science Foundation of China (91731310 to HY), Science and Technology Research Program of Shanghai (19DZ2282100 to HL) and the Open Fund of State Key Laboratory of Genetic Engineering.
Author information
Authors and Affiliations
Contributions
TS, JZ and YY performed most of the experiments and analyzed the data. AX and YH analyzed the transcriptomic data. HL, YH and YY conceived and supervised the project. TS, JZ and YY wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Changes of gene expressions during treatment of DTT or TM.
Additional file 2: Table S2.
Differentially expressed genes upon treatment of DTT or TM.
Additional file 3: Table S3.
GO analysis of differentially expressed genes upon treatment of DTT or TM.
Additional file 4: Table S4.
ESRTs and UPR target genes.
Additional file 5: Table S5.
Strains used in this study.
Additional file 6: Table S6.
Plasmids used in this study.
Additional file 7: Table S7.
Primers used in this study.
Additional file 8.
Sequence of LHZ296 in genbank format.
Additional file 9.
Sequence of LHZ301 in genbank format.
Additional file 10.
Sequence of LHZ531 in genbank format.
Additional file 11: Figure S1.
Box plot of distributions of fold changes of transcript abundance under different treatment conditions.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shi, T., Zhou, J., Xue, A. et al. Characterization and modulation of endoplasmic reticulum stress response target genes in Kluyveromyces marxianus to improve secretory expressions of heterologous proteins. Biotechnol Biofuels 14, 236 (2021). https://doi.org/10.1186/s13068-021-02086-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-021-02086-7