
Abstract
Endoplasmic reticulum associated degradation (ERAD) is a conserved mechanism to remove misfolded proteins from the ER by targeting them to the proteasome for degradation. To assess the role of ERAD in filamentous fungi, we have examined the consequences of disrupting putative ERAD components in the filamentous fungus Aspergillus niger. Deletion of derA, doaA, hrdC, mifA, or mnsA in A. niger yields viable strains, and with the exception of doaA, no significant growth phenotype is observed when compared to the parental strain. The gene deletion mutants were also made in A. niger strains containing single- or multicopies of a glucoamylase–glucuronidase (GlaGus) gene fusion. The induction of the unfolded protein response (UPR) target genes (bipA and pdiA) was dependent on the copy number of the heterologous gene and the ERAD gene deleted. The highest induction of UPR target genes was observed in ERAD mutants containing multiple copies of the GlaGus gene. Western blot analysis revealed that deletion of the derA gene in the multicopy GlaGus overexpressing strain resulted in a 6-fold increase in the intracellular amount of GlaGus protein detected. Our results suggest that impairing some components of the ERAD pathway in combination with high expression levels of the heterologous protein results in higher intracellular protein levels, indicating a delay in protein degradation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The use of filamentous fungi with the natural property of secreting high amounts of extracellular proteins as cell factories for the production of homologous and heterologous proteins has been extensively exploited for many years. Aspergillus niger, Aspergillus oryzae, and Trichoderma reesei are most often used in industry for the production of proteins. In the search for further improving the properties as protein producer, many attempts and strategies have been employed and optimized such as the knockout of certain genes, the use of strong promoters, mutagenesis, among others (Jeenes et al. 1991; Archer et al. 1994; Punt et al. 1994 Gouka et al. 1997; Nemoto et al. 2009; Nakari-Setälä et al. 2009; Meyer et al. 2010). The recent sequencing of the genomes of these industrially important fungi (Machida et al. 2005; Pel et al. 2007; Martinez et al. 2008; Wortman et al. 2009) provides another starting point to understand and manipulate the outstanding secretion capacities of these fungi (Maeda et al. 2004; Arvas et al. 2006; Guillemette et al. 2007; Gasser et al. 2007; Pel et al. 2007; Jacobs et al. 2009). Several steps occurring during the secretion pathway in filamentous fungi have been pointed out as potential bottlenecks for heterologous protein production (Gouka et al. 1997; Sims et al. 2005). Proteins that enter the secretory pathway begin their journey in the ER, where they are assembled and subjected to a strict quality control (Ellgaard et al. 1999; Lederkremer 2009). The proteins that fail proper folding usually accumulate in the ER leading to the induction of the unfolded protein response (UPR) (Cox et al. 1993), and if UPR is not sufficient to relieve stress, they are eventually targeted to destruction by the ER-associated degradation (ERAD) (Nishikawa et al. 2005). Both the UPR and the ERAD pathways are conserved from yeasts to mammalians (reviewed in Kincaid and Cooper 2007; Anelli and Sitia 2008; Mori 2009); however, apart from a recent publication that studies the effect of deleting A. niger doaA gene (Jacobs et al. 2009), the functional analysis of other putative ERAD-related genes in filamentous fungi has not been reported.
Accumulation of unfolded proteins in the ER lumen results in the dissociation of BiP from Ire1p leading to Ire1p dimerization and thereby the activation of its kinase and endoribonuclease functions (Shamu and Walter 1996; Sidrauski and Walter 1997; Oikawa et al. 2009). In Saccharomyces cerevisiae, Ire1p is responsible for excising a 252-nt intron in Hac1 messenger RNA (mRNA), enabling its translation into an active protein and migration into the nucleus where it binds to UPRE (CANCNTG, Mori et al. 1998) in target genes coding for chaperones and foldases as well as other components of the secretory pathway (Sidrauski et al. 1998; Travers et al. 2000). By homology with the S. cerevisiae model, it is assumed that in A. niger, IreAp is also responsible for the removal of a 20-nt intron in the hacA mRNA. Splicing of the intron leads to the activation of the HacA transcription factor, which in turn controls the expression of genes involved in UPR (Mulder et al. 2004, 2006).
The ER degradation pathway in S. cerevisiae consists of a number of highly conserved proteins. The UPR-induced BiP and disulfide isomerase play important roles in ERAD by preventing misfolded proteins aggregation (Nishikawa et al. 2001) and delivering ERAD substrates to the retrotranslocation machinery (Plemper and Wolf 1999a). Moreover, glycosylation is an important factor in protein folding, and the processing of glycans is indicative of the folding state of the protein (reviewed in Kleizen and Braakman 2004; Lederkremer 2009). If the protein fails to achieve correct conformation, the removal of 1,2 α-mannose units by a specific 1,2 α-mannosidase (mns1) targets the substrate to degradation by ERAD (Gonzalez et al. 1999; Tremblay and Herscovics 1999). When marked for degradation, proteins are retrotranslocated through the Sec61p translocon (Schäfer and Wolf 2009) and/or through Der1p retrotranslocation channel (Goder et al. 2008; Ye et al. 2001a), although the later one only seems to be required for some substrates (Lilley and Ploegh 2004). The Hrd1 complex is involved in the ubiquitination of substrates that contain misfolded luminal domains (Bordallo et al. 1998; Deak and Wolf 2001). In S. cerevisiae, Hrd3p regulates the activity and stability of Hrd1p (Plemper et al. 1999b; Gardner et al. 2000). Together with Sec61p, Hrd1–Hrd3 complex mediates the transfer to the cytosol of proteins targeted for degradation (Plemper and Wolf 1999a). Doa1p forms a complex with Cdc48p allowing the extraction of ubiquitinated substrates via AAA–ATP Cdc48 complex (Ye et al. 2001b; Jarosch et al. 2002; Ogiso et al. 2004; Mullally et al. 2006). Ubiquitinated proteins are degraded by the 26S proteasome in an ATP-dependent manner (Fischer et al. 1994). The translocation of the 26S proteasome from the cytoplasm to the ER membrane seems to be mediated by Mif1p (van Laar et al. 2001). In mammalian systems, the response to ER stress involves four major steps: (1) attenuation of protein synthesis; (2) transcriptional induction of UPR target genes, including chaperones and foldases; (3) transcriptional induction of ERAD components, and in case these three steps are not sufficient, (4) induction of apoptosis (reviewed in Yoshida 2007). For a detailed description of the ERAD pathway, we refer to a recent review by Vembar and Brodsky (2008).
From yeasts to mammals, several elements involved in the recognition and targeting of misfolded proteins for destruction are conserved, allowing the cells to cope with the presence/accumulation of aberrant proteins and their harmful effects. However, not all the processes described in yeast and mammalian system have been established in filamentous fungi (reviewed in van Anken and Braakman 2005a,b).
In this study, we have examined the role of the ERAD pathway in A. niger by disrupting genes that encode proteins suggested to be involved in different parts of ERAD pathway. We have assessed its role both during normal growth conditions, under ER stress-inducing conditions by treatment with dithiothreitol (DTT) or tunicamycin and under conditions when a UPR-inducing heterologous protein is produced. Our results indicate that a functional ERAD pathway is not required for normal growth but that a defective ERAD pathway increases intracellular levels of the UPR-inducing glucoamylase–glucuronidase (GlaGus) protein, indicating that the ERAD pathway is, at least partially, responsible for the degradation of heterologous proteins in A. niger.
Materials and methods
Strains, culture conditions, and molecular techniques
A. niger strains used throughout this study are all derivatives of N402 (Bos et al. 1988) (see Table 1 for details). Strains were cultivated in minimal medium (MM) (Bennett and Lasure 1991) containing 1% (w/v) of glucose as a carbon source, 7 mM KCl, 11 mM KH2PO4, 70 mM NaNO3, 2 mM MgSO4, 76 nM ZnSO4, 178 nM H3BO3, 25 nM MnCl2, 18 nM FeSO4, 7.1 nM CoCl2, 6.4 nM CuSO4, 6.2 nM Na2MoO4, and 174 nM EDTA or in complete medium (CM) containing, in addition to MM, 0.1% (w/v) casamino acids and 0.5% (w/v) yeast extract. When using the amdS gene as selection marker, strains were grown in MM in which the 70 mM NaNO3 was replaced with 10 mM acetamide and 15 mM cesium chloride (Meyer et al. 2010). All basic molecular techniques were performed according to standard procedures (Sambrook and Russel 2001). Transformation of A. niger, genomic DNA extraction, screening procedures, Northern analysis, and Southern analysis were conducted as recently described in utmost detail (Meyer et al. 2010).
Phenotypic assays
For plate growth assays, MM or CM was used (as described above) and solidified by the addition of 2% agar. Radial extension rates of the ERAD mutants were determined by inoculating 1 × 104 spores in the centre of a CM and MM plate and growth at 25, 30, 37, and 42°C was followed for 3–4 days. To determine the sensitivity of the ERAD mutants towards ER- and osmotic stress, a 10-fold dilution series of spores (from 1 × 105 until 1 × 101) were spotted on CM and MM plates containing tunicamycin (0.1, 0.5, 1, 5, or 10 mM), DTT (1, 5, 10, or 20 μg/ml) to induce ER stress and containing 0.6 M sorbitol to induce osmotic stress. Sensitivity assays were performed at 25, 30, or 42°C and growth was monitored for 3–5 days. Growth on starch was determined by spotting 10-fold dilution series of spores on CM plates containing 2% starch as the sole carbon source.
Construction of a strain expressing a secreted form of β-glucuronidase
Plasmid pBB19-3 was previously described (Punt et al. 1994). To generate A. niger strains carrying a single copy of this plasmid at a defined position, the pyrG* gene was used (van Gorcom and van den Hondel 1988). The pyrG* was amplified from pAF3 (Damveld et al. 2005) using primers pNC43 and pNC44, where AscI restriction sites were added (Table 2) to facilitate the cloning into pBB19-3. The amplified PCR fragment of 2.2 kb was ligated into pJET1.2 (pJET1.2/blunt cloning vector, Fermentas) to give pJetPyrG*AscI. Finally, the pyrG* fragment was isolated with AscI and cloned into the unique AscI site in pBB19-3 to give pBB19-3pyrG*. This construct was transformed into MA70.15 (ΔkusA, pyrG −, and amdS +), and transformants were purified by repeated streaking of conidia on media without uridine. Transformants were subjected to Southern blot analysis, and MV3.2 was selected as this transformant contains a single copy of the pBB19-3 plasmid at the pyrG locus. The AmdS marker in this strain, which was used to delete the kusA gene, was looped out by selecting fluoroacetamide-resistant colonies by inoculating 2 × 107 spores on MM plates containing 1% (w/v) of glucose as a carbon source, without NaNO3 and supplemented with 0.2% 5′-fluoroacetamide (FAA) and 10 mM urea as additional nitrogen source (for details, see Meyer et al. 2010). Plates were incubated for 1–2 weeks at 30°C, and FAA-resistant mutants were transferred onto fresh FAA-containing plates for purification. Mutants unable to grow on media containing acetamide as sole nitrogen source were subjected to Southern blot analysis, and strain MA99.3, in which the amdS gene was properly looped, out was chosen for further studies.
Construction of ERAD deletion strains
The deletion constructs for the five selected genes involved in the ERAD pathway (derA (An15g00640), doaA (An03g04600), hrdC (An01g12720), mifA (An01g14100), and mnsA (An18g06220) were made using primers listed in Table 2. Briefly, the cloning strategy was as follows: for each individual gene, respective 5′ and 3′ flanking regions and an additional 5′ or 3′ repeat (construct dependent, see Table 2 for details) were amplified using primers where specific restriction enzymes were added and cloned into pGBPEP23 (Jacobs et al. 2009). This vector uses the amdS gene behind the PgpdA promoter as a dominant selection marker. Only in the presence of the amdS gene Aspergillus is able to grow on medium containing acetamide as sole nitrogen source. In general, approximately 1 kb of the 5′ and 3′ sequences flanking the coding regions has been used, and about 500–700 bp repeat of one of the flanks was included to facilitate removal of the amdS marker by homologous recombination forced by growth on FAA. The A. niger doaA deletion strain has been previously described (Jacobs et al. 2009). Deletion constructs were linearized by digestion with NotI and AscI before transformation. To obtain high homologous recombination frequencies to construct ERAD deletion mutants in the multicopy GlaGus strain (AB4-1dglaA36[pBB19-3]#3), the ku70 gene was also deleted in this background using a kusA::amdS deletion construct as previously described (Meyer et al. 2007). Southern blot analysis identified strain MA134.64 as a strain in which the kusA gene was deleted (data not shown). Subsequently, the amdS gene was removed through the FAA loop-out technique and yielded MA135.3 in which the kusA deletion and amdS looped out were confirmed by Southern blot analysis (data not shown). Each ERAD deletion construct was transformed into strains NC5, MA99.3, and MA135.3. Strains MA99.3 and MA135.3 will be referred to as single-copy scGlaGus and multicopy mcGlaGus strains, respectively, in the following sections. All ERAD deletion mutants in the three strain backgrounds were confirmed by Southern analysis (data not shown). All mutants were obtained except for the hrdC deletion strain in the mcGlaGus strain.
Western blot analysis
To analyze the extracellular and intracellular levels of Gus protein, deletion strains and control strains were grown in duplicate for 24 h in 50 ml CM containing 1% glucose as carbon source. All cultures were inoculated with 1 × 106 spores/ml. Mycelium was collected through a myracloth filter, and the supernatant was stored at −20°C before further analysis. Total protein content was extracted by grinding approximately 200 mg frozen mycelium using mortar and pestle in liquid nitrogen. Proteins were extracted using 1 ml extraction buffer [10 mM sodium phosphate buffer, pH 6.0, 2% sodium dodecyl sulfate (SDS). 10 mM EDTA and 1 mM phenylmethylsulfonyl fluoride] and centrifuged twice, collecting the supernatant each time. Protein concentrations of the samples were determined with Bradford assay using bovine serum albumin as standard. For each sample, 10 μg of total protein was mixed with 2× loading buffer (0.5 M HCl, 25% glycerol, 10% SDS, 0.5% bromophenol blue, and 5% β-mercaptoethanol) and boiled for 5 min at 95 °C. Protein samples were loaded on a pre-cast SDS polyacrylamide gel electrophoresis gel (BioRad) and blotted to a nitrocellulose membrane through semi-dry electrotransfer. The membrane was blocked for 1 h with 5% low-fat milk in TTBS (Tris-buffered saline, 0.05% Tween-20), and Gus protein was detected using a Gus-specific antibody (1/5,000) overnight, followed by a goat anti-rabbit horseradish peroxidase secondary antibody (1/20,000) for 1 h. Detection was performed using a chemiluminescence kit (Bio-Rad), according to manufacturer’s instructions. The Gus antibody was kindly provided by Prof. P. Punt (TNO, The Netherlands). Analysis and quantification of band intensities were performed using QuantityOne 1-D Analysis Software (BioRad) and 18S rRNA as loading control.
Results
The level of induction of the unfolded protein response pathway by heterologous protein expression is protein specific
In order to study the effects of deleting ERAD components on heterologous protein production in A. niger, we started our research by choosing a suitable heterologous protein reporter through an inventory of A. niger strains expressing/overexpressing different heterologous proteins (Table 1). Each strain was cultured under identical conditions and UPR and ERAD responses were determined by examining the expression of UPR and ERAD marker genes in these strains. As markers for UPR induction, we have chosen bipA and pdiA, as an increase in the expression levels of these genes has been observed in strains expressing heterologous proteins (Punt et al. 1998; Kauffman et al. 2002; Guillemette et al. 2007). The ERAD markers (derA and hrdC) were chosen based on S. cerevisiae studies in which induction of these genes was observed after protein folding stress (Knop et al. 1996; Travers et al. 2000). To confirm that derA and hrdC in A. niger were also induced under ER stress conditions, we grew N402 in the presence of increasing concentrations of DTT and tunicamycin to induce ER stress (Fig. 1). Northern analysis and blot quantification revealed a high induction of both genes in the presence of DTT. In the case of growing in the presence of tunicamycin, an increase in derA and hrdC was observed at the higher concentrations tested (5 and 10 μg/ml). Having established good marker genes for UPR and ERAD responses, we then studied the induction of these pathways in strains expressing different heterologous proteins (Fig. 2). Results in Fig. 2 visibly show different gene expression levels depending on the heterologous protein expressed. Although we see an increase in expression of UPR target genes in most of the strains bearing heterologous proteins in relation to N402, both UPR and ERAD responses were more boosted when A. niger strains expressed tPA (D15) and GlaGus (AB4-1dglaA36#3) heterologous proteins. In S. cerevisiae, a link between UPR and ERAD pathways has been established (Travers et al. 2000; Friedlander et al. 2000), and the co-induction of both UPR genes (bipA and pdiA) and ERAD genes (derA and hrdC) in A. niger in response to the expression of the heterologous GlaGus protein as observed in Fig. 2 suggests a similar link between these two pathways in A. niger. For reasons of availability of activity assays and antibodies against β-glucuronidase, the heterologous fusion protein GlaGus was then chosen as a reporter to study the fate of heterologous proteins under ERAD-deficient conditions.

Induction of two genes involved in the ERAD pathway (derA and hrdC) by the presence of increasing concentrations of DTT or tunicamycin stress agents. Samples for Northern analysis were collect after 16 h growth on liquid CM (1% glucose) at 30°C. On the y-axis is the relative expression of derA and hrdC in arbitrary units, normalized for loading differences by comparison with 18S ribosomal RNA probe

Relative expression (arbitrary units) of ERAD (derA and hrdC) and UPR (bipA and pdiA) reporter genes in strains expressing different heterologous proteins (see Table 1 for details). Samples for Northern blot analysis were collected from these strains grown for 16 h at 30°C in liquid CM. Values were normalized for loading differences. The gene expression levels were normalized using the N402 values as reference
The level of GlaGus expression affects UPR induction
To express a secreted form of the bacterial β-glucuronidase in A. niger, plasmid pBB19-3 was used (Punt et al. 1994). This plasmid contains the bacterial uidA gene (encoding β-glucuronidase), which is fused to the glucoamylase gene. Plasmid (pBB19-3pyrG*) was constructed to generate strain MV3.2, which contains a single-copy integration of the GlaGus construct at the pyrG locus (data not shown). We will refer to this strain as the single-copy GlaGus (scGlaGus) strain in the remaining of the paper. Strain AB4.1Δgla#A36#3 has been reported to contain multiple copies of the pBB19-3 plasmid (Punt et al. 1994; 1998).
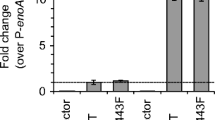
To determine the number of copies of GlaGus gene present in the AB4.1Δgla36#3 strain, to which we will refer to as the multicopy GlaGus (mcGlaGus) strain, we performed Southern blot analysis (Fig. 3a). After correcting for loading differences, we determined about eight copies of the glaA gene in the mcGlaGus strain. Additionally, Western blot analysis using a Gus-specific antibody was performed on a total protein extract on these two strains and N402, where we observe that the difference in the number of copies between them relates to the amounts of Gus protein detected (or absence in the case of N402), as band intensity in mcGlaGus is higher than in scGlaGus (Fig. 3b). Western analysis of medium samples from both the scGlaGus or mcGlaGus strains failed to detect the GlaGus protein in the medium, using Gus antiserum. To determine UPR induction in these strains, we examined the mRNA expression levels of bipA, pdiA, and hacA (Fig. 4a). By comparison with N402, quantification of the mRNA levels shows an induction of bipA and pdiA in the mcGlaGus strain but not in the scGlaGus strain (Fig. 4b), demonstrating that the copy number of this heterologous protein affects the UPR response.

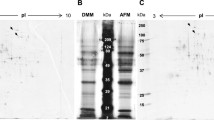
a Southern blot analysis of the GlaGus copy number in mcGlaGus strain. Genomic DNA was digested with NcoI and probed with a probe annealing within the glucoamylase ORF. Expected band size for endogenous glucoamylase is 4.8 kb; for the scGlaGus strain, 7.1 and 4.8 kb bands are expected. Ectopic integration of pBB19-3 in the mcGlaGus strain does not allow band size predictions; however, the band(s) observed at 9.4 kb indicate that the plasmids have been tandemly integrated. Loading differences were corrected using a gel stained with ethidium bromide. b Western analysis of GlaGus amounts on total protein of mycelium samples of scGlaGus and mcGlaGus strains; N402 was used as a control for Gus antibody specificity. Samples were grown in CM for 24 h at 30°C. The protein content was extracted; 10 μg of total protein were separated by gel electrophoresis and immunodetected with an anti-Gus antibody. Detection was carried out through a chemiluminescence reaction for 5 min

a Northern blot analysis of mRNA levels of UPR target genes on strains containing either a single- or multicopy GlaGus genes in comparison to N402. Total RNA was extracted from mycelia grown for 24 h at 30°C in CM. b The UPR target genes expression levels were normalized using N402 as reference
Construction and analysis of ERAD deletion strains
Misfolded proteins that become destined to be degraded are taken by the ERAD pathway, which involves many components that recognize aberrant proteins and activate their retrotranslocation to the cytosol for proteasome-mediated degradation. Among these many components, we have selected five genes indicated to be involved in different parts of the ERAD system to assess the effects of having a compromised ERAD in different A. niger backgrounds. We have deleted derA, doaA, hrdC, mifA, and mnsA in the control strain (NC5; ΔkusA, amdS −), the scGlaGus strain (MA99.3; ΔkusA, amdS −, and scpBB19-3pyrG*) and the mcGlaGus strain (MA135.3 (ΔkusA, amdS −, mcpBB19-3). Transformants for each strain were purified on media containing acetamide and further examined by Southern blot analysis (data not shown). All five ERAD genes were successfully deleted in both NC5 and scGlaGus (MA99.3) backgrounds. In the mcGlaGus background, four ERAD genes were successfully disrupted, but obtaining a deletion mutant of the hrdC gene was unsuccessful, although over 140 putative transformants were screened. It should be noted that the inability to obtain this disruptant was not caused because the disruption was lethal as also no heterokaryons were obtained on primary transformation plates. For unknown reasons the frequency of getting a homologous recombination in the hrdC locus is very low, even in the ku70 mutant background. During the process of making deletion mutants in hrdC in the other two strain backgrounds, we also noticed a low homologous recombination frequency to obtain the knockout strains. Thus, in total, 14 disruptions strains have been generated (Table 1). To further confirm the deletions and to examine whether the deletion of any of the ERAD-related genes has an effect on the expression of ERAD itself, Northern blot analysis was performed. Figure 5 depicts an example of one of the Northern analysis and shows the effect of deleting ERAD-related genes on the expression of the other ERAD genes in the scGlaGus strain background. First, the Northern blot analysis confirmed the Southern blot data, and no mRNA was detected when using probes corresponding to the respective gene deletion mutant. In addition, the hybridization (Fig. 5) and subsequent blot quantifications (data not shown) revealed no apparent increase or decrease in expression of any of the ERAD genes tested among the different strains, suggesting that deletion of a single component of the ERAD pathway does not affect the expression of other components of this pathway. Furthermore, probes against glaA and gus were used as an indication of the transcription of the fusion gene in the scGlaGus and mcGlaGus background, or its absence in the case of N402 (Fig. 5).

Expression analysis of different genes in the five ERAD deletion strains on the scGlaGus background, scGlaGus parental strain, mcGlaGus and N402. Total RNA was extracted from mycelia grown for 24 h at 30°C in CM. RNA (5 μg) was separated by agarose gel electrophoresis, blotted and hybridized with 32P-labeled probes specific for the genes indicated. 18S rRNA was used as loading control
The morphological and growth effects of the disruption of these ERAD genes in the 14 A. niger strains were analyzed on CM and MM agar plates and compared to the growth phenotype of its corresponding wild type. We performed a drop dilution test on solid MM and monitored growth at 25, 30, and 42°C (Fig. 6). At 25 and 30°C, strains are able to grow, and only ΔdoaA revealed a different phenotype. This mutant strain showed irregular colony morphology, slower growth, and reduced sporulation. At 42°C and at the lower spore concentrations, the mcGlaGus and respective ERAD deletions are no longer able to form colonies, unlike N402 and the other strains tested. As the sensitivity toward high temperature is already observed in the mcGlaGus parental strain, we can conclude that is the expression of a high copy number of this heterologous protein that confers this growth defect and not a defective ERAD. Furthermore, at this temperature, ΔderA shows a more apparent growth defect than the other deletions.

Growth assay at different temperatures of parental strains NC5, scGlaGus and mcGlaGus, and respective ERAD deletions. Spore serial dilutions were spotted onto solid MM and incubated under the given conditions. Growth was monitored for 3 days
Subsequently, the sensitivity of the ΔERAD strains towards a chemical that disturbs the ER homeostasis was tested by spotting 104 spores per 10 μl on solid MM containing increasing concentrations of DTT and incubating at 25, 30, and 42°C for 3 days (Fig. 7). In general, at the temperatures 25 and 30°C and either absence or in the presence of increasing concentrations of the DTT, deletions strains grew like their parental strain. The growth of the ΔdoaA mutant was affected in the absence of DTT, but the ΔdoaA did not seem to be more sensitive toward DTT in this spot assay in comparison to the other ERAD deletions. As the growth phenotype of the doaA deletion strain was observed in all the different backgrounds, we attribute this growth phenotype to the absence of the doaA gene and not to the expression of the heterologous protein. On the other hand, at 42°C, the mcGlaGus strain not only shows a reduction in colony size compared to NC5 and scGlaGus but also reveals an increased sensitivity toward 5 mM DTT (Fig. 7). At the concentration of 10 mM DTT, the growth of both scGlaGus and mcGlaGus strains is almost completely abolished, whereas the NC5 wild-type and ERAD deletion mutants are able to grow. A 20-mM concentration of DTT abolishes growth of all the strains and indicated that none of the ERAD Δstrains become more resistant toward DTT. As also observed in Fig. 6, the mcGlaGusΔderA strain displays a reduced growth and sporulation phenotype at 42°C (Fig. 7). Thus, the strains expressing the GlaGus protein are more sensitive to DTT compared to the respective parental strain that does not express the GlaGus protein, and high levels of GlaGus expression is correlated with a higher sensitivity to DTT. We further conclude that the disruption of ERAD component had no further effect on the growth and/or the sensitivity toward DTT. Additional growth tests such as on simple/complex carbon sources (glucose vs starch) or under osmotic stress conditions (1 M NaCl, 0.6 M sorbitol) with those deletion mutants (besides ΔdoaA) resulted in no significant differences (data not shown). As none of the ERAD genes described revealed to be essential to A. niger, we decided to test for synthetic lethality by the combination of deleting any two of the five genes in study. The double ERAD knockout mutants were made by deleting an additional gene in the existing single knockouts strains in the NC5 background (data not shown) after looping out the amdS marker used to disrupt the first ERAD gene. Then, the ERAD deletion constructs were transformed into these new five ERAD deletions (amdS −) strains obtained in order to get the ten possible double deletion combinations. All the double knockout (KO) transformants were purified on acetamide media and confirmed by Southern blot (data not shown; see Table 1). All the double KO mutants were subjected to the phenotypic tests as described above, but no additional differences or effects on morphology and growth were found by having any of the double ERAD deletions compared to the single mutants. The combination of doaA deletion with any of the other deletions did not give extra phenotypic features than the ones observed for the single doaA KO in any of the background strains (data not shown).

Comparison of colony morphology of parental strains and respective ERAD deletion mutants incubated at different temperatures (indicated on the right) and in the presence of increasing concentrations of the stress agent DTT (indicated at the bottom). 104 spores per 10 μl of each strain were spotted on solid MM and growth was monitored for 3 days. NA not available
Activation of the UPR by strains expressing/overexpressing the GlaGus protein in combination with a defective ERAD pathway
To investigate whether deletion of ERAD components in combination with expression or overexpression of GlaGus has an effect on the UPR, the ERAD mutants were analyzed for the expression of UPR target genes (hacA, bipA, and pdiA), and their expression was compared to their corresponding parental strain. Figure 8 shows the Northern blot results and quantified mRNA levels of UPR target genes in the ΔERAD strains not expressing the GlaGus protein (NC5 background; Fig. 8a, b), the ΔERAD strains in the scGlaGus background (Fig. 8c, d), and the ΔERAD strains in the mcGlaGus background (Fig. 8e, f). In the case of having a deficient ERAD pathway but no expression of heterologous protein (NC5 background), the UPR pathway is not induced (Fig. 8a, b). In the ΔdoaA, ΔmifA, and ΔmnsA strains in the scGlaGus background, no increase in the expression levels of the UPR target genes is observed (Fig. 8c, d). However, the bipA expression level in derA and hrdC deletion strains are 1.8- and 2-fold higher, respectively, in comparison to the scGlaGus parental strain. Hence, there seems to be specific induction of bipA expression upon deletion of derA or hrdC. As depicted in Fig. 8e and f, the combination of overexpression of GlaGus with the deletion of any of ERAD components tested further induces the transcription of the UPR reporter genes. As shown in Figs. 2 and 5, the multicopy expression of the GlaGus protein induces the expression of UPR and ERAD target genes. This induction of the UPR target genes is further enhanced by deleting ERAD components as deletion of derA, mifA, and mnsA genes lead to an induction of pdiA and bipA mRNAs of more than 2-fold higher compared to the mcGlaGus parental strain (Fig. 8e, f). In the case of ΔdoaA, the values of pdiA and bipA are induced 1.6- and 1.8-fold, respectively. The values of hacA mRNA levels do not show a noteworthy change. In summary, we have shown that in the mcGlaGus, the UPR and ERAD-related genes are induced and deletion of ERAD genes in the mcGlaGus background further induces UPR. This hyperactivation of UPR might be explained by an increasing accumulation of GlaGus protein in the ER in the absence of a functional ERAD system.

Northern blot analysis of ERAD deletion strains in NC5 (a), scGlaGus (c), and mcGlaGus (e). Total RNA was extracted from mycelia grown for 24 h at 30°C in CM. RNA (5 μg) was separated, blotted, and hybridized with 32P-labeled probes specific for the genes indicated. b, d, f Quantification of the mRNA expression levels of hacA, pdiA, and bipA. Signals were corrected for loading differences using 18S. The UPR target genes expression levels were then normalized using the respective parental strain values as reference
Effects of the deletion of ERAD genes on protein production
To examine the effects of an impaired ERAD pathway on the GlaGus protein production or accumulation, we performed Western blot analysis on medium samples and intracellular protein samples collected from scGlaGus and mcGlaGus strains. In the medium samples, no Gus activity was detected, and no GlaGus protein could be detected using a Gus-specific antibody (data not shown), indicating that secretion levels are low. At this stage, we cannot exclude the possibility that some secreted GlaGus protein is degraded by extracellular proteases. To examine the effect of the ERAD deletion mutant on the intracellular pool of GlaGus protein, total protein content was extracted from fungal biomass as described in “Materials and methods.” For each set of experiments, two gels were run in parallel; one of them was immunoblotted and probed with an antibody against Gus (Fig. 9a, d), and the other gel was stained with Coomassie blue to be used as loading control (Fig. 9b, e). The relative amount of protein present in each deletion strain was determined in relation to the amount of protein detected in the parental strain (Fig. 9c, f). Using the Gus antibody, we were able to detect a band corresponding to the GlaGus fusion protein (around 140 kDa), as well as smaller bands (Fig. 9a, d), which might represent truncated versions of the protein most likely caused by proteolytic activity. The amount of fusion protein detected in the scGlaGus background strains (Fig. 9c) is the highest in the ΔderA and ΔhrdC strains. Subsequently, we determined the amounts of fusion protein present in ERAD deletion strains in the mcGlaGus background by Western blot analysis (Fig. 9f). Both the Western blot and the quantified data clearly indicate higher amounts of fusion protein for all the deletions when compared to mcGlaGus parental strain. The deletion of derA had the most significant effect as a 6-fold increase in GlaGus protein levels was detected. The results indicate that a defective ERAD leads to the accumulation of intracellular GlaGus, but this does not result in detectable GlaGus production in the culture medium.

Effect of deletion of ERAD components on the amount of GlaGus fusion protein in total protein extracts. Western analysis of GlaGus amounts in total protein of mycelium samples of scGlaGus (a) and mcGlaGus (d) ERAD deletion strains. Samples were grown in CM for 24 h at 30°C. Ten micrograms of total protein was separated by gel electrophoresis and immunodetected with an anti-Gus antibody. Detection was carried out through a chemiluminescence reaction for 5 min. As a positive and negative control, 50 ng of purified Gus and a total protein extract from N402 were loaded. The arrow indicates the band corresponding to the GlaGus fusion protein (≈140 kDa). The relative amounts of protein were normalized for loading differences by comparison with a “twin” gel stained with Coomassie blue (b, e). c, f Relative amount of GlaGus fusion protein detected in total protein extracts of strains with impaired ERAD and respective parental strain. Bars indicate standard deviations from two independent experiments
Discussion
Different heterologous proteins, distinctive bottlenecks?
In filamentous fungi, the levels of heterologous protein production are often low (Gouka et al. 1997). Possible processes and mechanisms involved in protein degradation, especially those related to the ERAD pathway, are poorly described or understood in these fungi. As A. niger has such an outstanding capacity as a cell factory, the understanding of these mechanisms becomes crucial to improve heterologous protein production. We started out our study by comparing the effect of expressing different heterologous proteins in A. niger. In our analysis, we have included proteins from bacterial origin, β-glucuronidase, which has successfully used as a reporter in gene expression in innumerable cell systems (Punt et al. 1994, 1998; Gilissen et al. 1998; Ayra-Pardo et al. 1999); the metazoan green fluorescent protein, widely used as a fluorescent marker; the human proteins tPA and IL6 with valuable medical applicability (Upshall et al. 1987; Broekhuijsen et al. 1993; Punt et al. 1998; Wiebe et al. 2001); and basidiomycetes enzymes with wide biotechnological applications manganese peroxidase and laccase (Conesa et al. 2000; Weenink et al. 2006; Elisashvili and Kachlishvili 2009). Expression of all the heterologous proteins result in relative low production levels compared to the production of glucoamylase expressed form the same promoters (Archer et al. 1994; Gouka et al. 1997). Several potential bottlenecks for the production of proteins have been evaluated and discussed over the last decade, and a potential bottleneck for efficient secretion in folding of the heterologous proteins in the ER has been considered as a major issue. BipA and PdiA, encoding a chaperone and a foldase, respectively, have been identified as reliable reporter genes as indicated for ER stress in filamentous fungi (Punt et al. 1998; Kauffman et al. 2002; Guillemette et al. 2007). Comparison of the different bipA and pdiA mRNA levels in the strains expressing the different heterologous proteins revealed that not all heterologous proteins induce a strong UPR response (Fig. 2) despite the fact that the production levels of, for example, the Human IL6 protein, are low. Clearly, the lack of a strong UPR response in some strains producing low levels of heterologous proteins strongly suggests that also non-UPR mediated bottlenecks exist in A. niger that hamper efficient secretion. Two heterologous proteins, human tissue plasminogen activator (t-PA) and the bacterial glucuronidase (Gus) displayed a strong induction of the bipA and pdiA reporters, indicating that these two proteins induce a strong UPR response (Fig. 2). Interestingly, the expression of two genes involved in ER associated degradation pathway (derA and hrdC) was also induced, suggesting that t-PA and Gus might be targeted for proteolytic degradation via the ERAD system. Besides the protein specific issues, we also show that the induction of the UPR pathway is dependent on the level of expression (Fig. 4). The UPR-inducing property of the glucuronidase is only observed when the protein is highly expressed. Apparently, under relative low-expressing conditions, A. niger is capable of dealing with the protein in such a way that the protein does not induce the UPR. Our results suggest that in the case of having a single-copy of GlaGus, the basal protein folding and quality control machinery are able to cope with the heterologous protein, and only high levels of GlaGus protein in the ER induces ER stress.
Expression of the bacterial glucuronidase results in increased thermo and DTT sensitivity
The growth of transformants containing single- or multicopy insertions of the GlaGus construct at different temperatures was compared to the parental strains (Fig. 6). Growth of the mcGlaGus strain was severely impaired at 42°C, suggesting that the temperature stress (42°C is above the optimal growth temperature of A. niger) in combination with the presence of misfolded GlaGus protein in the ER also affects the processing of endogenous cargo resulting in a growth retardation. Environmental factors have an influence on cells productivity, and it has been recently shown in Pichia pastoris that cultivating this fungus below its optimal growth temperature results in a more efficient secretion of heterologous proteins due to a general decrease of folding stress at lower temperatures (Dragosits et al. 2009). Furthermore, we reasoned that an additional ER stress-inducing condition might further aggravate this phenotype, and therefore, the strains were also growth impaired in the presence of increasing concentrations of DTT (Fig. 7). The results clearly indicate that high temperature, the presence of DTT, and the expression of the GlaGus protein act additionally and interfere with growth. In the case of expressing high levels of GlaGus, a concentration of 5 mM DTT was enough to prevent growth at 42°C, whereas growth of the single-copy GlaGus strain was inhibited at 10 mM DTT, a condition that still allowed growth of the strain lacking this heterologous protein.
ERADication of misfolded proteins in A. niger
The function of the ERAD pathway during normal vegetative growth and its possible involvement in the degradation of misfolded proteins in the ER were analyzed by disrupting putative ERAD components in a wild-type background and in backgrounds expressing the glucoamylase-β-glucuronidase (GlaGus) fusion protein as a reporter. Five genes (derA, doaA, hrdC, mifA, and mnsA) involved in different aspects in the ERAD pathway were selected and identified in the A. niger genome to establish whether this pathway has an important role during the degradation of the GlaGus protein. The systematic analysis of these five genes either as single deletions or as double mutants clearly showed that the effect of the gene deletion on growth as well as on the faith of the heterologous protein was limited. Phenotypic assays performed on the ERAD deletion strains showed that, except for ΔdoaA, the deletion of ERAD components does not result in an apparent phenotype (Fig. 6). Moreover, deletion of the ERAD genes did not increase the sensitivity of the ERAD mutants in comparison to the respective parental strains toward tunicamycin (data not shown) or DTT (Fig. 7). It has been reported that the deletion of DER1 and HRD3 in the yeast S. cerevisiae does not lead to a detectable growth phenotype although the ERAD pathway is strongly affected (Knop et al. 1996; Travers et al. 2000). However, this lack of phenotype has been explained as a result of compensatory effects of the UPR induction (Travers et al. 2000), as deletion of DER1 only becomes lethal when combined with the deletion of IRE1 and at the restrictive temperature of 37°C (Mori et al. 1993; Travers et al. 2000). The deletion of the DER1 and HDR3 homologues in A. niger does not result in a phenotype different from the wild-type strain, but contrary to Travers et al. (2000), under normal growth conditions, there is no evidence for activation of UPR in the A. niger strains lacking derA or hrdC (Fig. 8a, b). The doaA deletion was the only mutant showing a growth defect in all the strains tested, translated into an irregular morphology and reduced sporulation (Figs. 6 and 7). In S. cerevisiae, Doa1p is known to play an important role in the ubiquitin-dependent protein degradation by a direct interaction with Cdc48p (a member of the AAA-ATPase family of molecular chaperones; Ye et al. 2001b; Ogiso et al. 2004; Mullally et al. 2006). In fission yeast, the deletion of the doa1 homologue (lub1) results in a defective ubiquitin/proteasome-dependent proteolysis, causing increased cell sensitivity to several stress conditions (Ogiso et al. 2004). Although there is no evidence for protein accumulation either in scGlaGus or mcGlaGus ΔdoaA background strains (Fig. 9), we might hypothesize that in A. niger, the lack of ubiquitination fails to target proteins to ERAD-mediated destruction, inducing another degradation pathway that could impair the fungal growth.
Induced BipA levels correlate with increased levels of intracellular GlaGus
To examine possible UPR induction in the strains with an impaired ERAD pathway in combination with the GlaGus protein expressed, we analyzed mRNA expression levels of hacA, bipA, and pdiA. Results in Fig. 8a and b clearly show that under normal growth conditions, in the wild-type background, the absence of any of the ERAD genes in study does not lead to induction of the UPR pathway. Overall, in the scGlaGus background (Fig. 8c, d), the deletion of ERAD components does not seem to trigger the UPR as values of hacA, bipA, and pdiA are maintained relatively constant. Only in the derA and hrdC deletion strains, an increase of about 2-fold of bipA mRNA is observed. In parallel, we observe in scGlaGus derA and hrdC deletions the highest accumulation of GlaGus intracellularly, indicating that high levels of bipA mRNA are correlated with higher levels of GlaGus protein (Fig. 9c). Our results suggest that in the absence of ERAD proteins DerA and HrdC, GlaGus might be retained in the ER longer, which might be responsible for triggering the induction of bipA levels. In the mcGlaGus background, an increase in the levels bipA and pdiA mRNA levels was observed for all the ERAD gene disruptions (Fig. 8f). Hence, not only the high levels of GlaGus produced triggers a UPR (Fig. 4a), but also the combination with the deletion of any of the ERAD genes and consequent accumulation of intracellular GlaGus might stimulate it even further (Fig. 8f). Again, the highest level of induction of bipA mRNA is correlated with the highest levels of GlaGus protein. In the mcGlaGus background, the deletion of derA had the most significant effect on the amount of intracellular protein detected and resulted in a 6-fold increase in GlaGus levels.
A general observation concerning the UPR induction throughout our study is the constant values of hacA mRNA itself. Mulder et al. (2004) have shown that upon UPR induction, hacA is able to up-regulate its own transcription via HacA binding sites in the HacA promoter region (Mulder et al. 2004, 2009). Examining the hacA expression levels in our studies showed that the levels of hacA mRNA were not induced in response to the expression of the GlaGus protein (Figs. 8 and 9). In the studies of Mulder et al., in which the induced expression levels of HacA are reported, the cells were suddenly exposed to ER-stress inducing chemicals, whereas in our case, the strains might have been adapted to the conditions of expressing/overexpressing the heterologous protein and therefore the hacA induction is not evident.
Alternative mechanisms of degradation
Our research revealed a surprisingly modest effect on the deletion of ERAD functions in A. niger, even under ER stress conditions. It was anticipated that the inability to remove misfolded proteins from the ER by deleting ERAD components would result in severe ER stress situation and, by analogy to metazoans, might induce apoptosis-like phenotypes (see for review Rasheva and Domingos 2009). Therefore, other mechanisms besides ERAD might be of importance in the clearance of misfolded proteins and help the cells cope with the stress. The lack of proteins in the medium together with the observed degradation in our Western analysis (Fig. 9) suggests alternative pathways to remove misfolded proteins, such as the presence of proteases directly in the ER, as it has been shown for mammalian systems (Evnouchidou et al. 2009), Sec61p-DerA/HrdC-independent transport to the cytosol, or mechanisms of direct targeting of misfolded proteins to the vacuole. The GlaGus reporter strains used in this study allow non-biased genetic screens to identify mutants involved in these alternative protein degradation pathways.
References
Anelli T, Sitia R (2008) Protein quality control in the early secretory pathway. EMBO J 27(2):315–327
Archer DB, Jeenes DJ, Mackenzie DA (1994) Strategies for improving heterologous protein production from filamentous fungi. Antonie Leeuwenhoek 65(3):245–250
Arvas M, Pakula T, Lanthaler K, Saloheimo M, Valkonen M, Suortti T, Robson G, Penttilä M (2006) Common features and interesting differences in transcriptional responses to secretion stress in the fungi Trichoderma reesei and Saccharomyces cerevisiae. BMC Genomics 7:32
Ayra-Pardo C, Montejo-Sierra IL, Vázquez-Padrón RI, García-Martínez C (1999) beta-D-glucuronidase gene from Escherichia coli is a functional reporter in the methylotrophic yeast Pichia pastoris. Lett Appl Microbiol 29(5):278–283
Bennett JW, Lasure LL (1991) More gene manipulations in fungi. Academic, San Diego
Bordallo J, Plemper RK, Finger A, Wolf DH (1998) Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell 9(1):209–222
Bos CJ, Debets AJ, Swart K, Huybers A, Kobus G, Slakhorst SM (1988) Genetic analysis and the construction of master strains for assignment of genes to six linkage groups in Aspergillus niger. Curr Genet 14:437–443
Broekhuijsen MP, Mattern IE, Contreras R, Kinghorn JR, van den Hondel CA (1993) Secretion of heterologous proteins by Aspergillus niger: production of active human interleukin-6 in a protease-deficient mutant by KEX2-like processing of a glucoamylase-hIL6 fusion protein. J Biotechnol 31(2):135–145
Carvalho ND, Arentshorst M, Jin Kwon M, Meyer V, Ram AF (2010) Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol 87(4):1463–1473
Conesa A, van den Hondel CA, Punt PJ (2000) Studies on the production of fungal peroxidases in Aspergillus niger. Appl Environ Microbiol 66(7):3016–3023
Cox JS, Shamu CE, Walter P (1993) Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73(6):1197–1206
Damveld RA, Arentshorst M, Franken A, vanKuyk PA, Klis FM, van den Hondel CA, Ram AF (2005) The Aspergillus niger MADS-box transcription factor RlmA is required for cell wall reinforcement in response to cell wall stress. Mol Microbiol 58(1):305–319
Deak PM, Wolf DH (2001) Membrane topology and function of Der3/Hrd1p as a ubiquitin- protein ligase (E3) involved in endoplasmic reticulum degradation. J Biol Chem 276(14):10663–10669
Dragosits M, Stadlmann J, Albiol J, Baumann K, Maurer M, Gasser B, Sauer M, Altmann F, Ferrer P, Mattanovich D (2009) The effect of temperature on the proteome of recombinant Pichia pastoris. J Proteome Res 8(3):1380–1392
Elisashvili V, Kachlishvili E (2009) Physiological regulation of laccase and manganese peroxidase production by white-rot Basidiomycetes. J Biotechnol 144(1):37–42
Ellgaard L, Molinari M, Helenius A (1999) Setting the standards: quality control in the secretory pathway. Science 286(5446):1882–1888
Evnouchidou I, Papakyriakou A, Stratikos E (2009) A new role for Zn(II) aminopeptidases: antigenic peptide generation and destruction. Curr Pharm Des 15(31):3656–3670
Fischer M, Hilt W, Richter-Ruoff B, Gonen H, Ciechanover A, Wolf DH (1994) The 26S proteasome of the yeast Saccharomyces cerevisiae. FEBS Lett 355(1):69–75
Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T (2000) A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol 2(7):379–384
Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY (2000) Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol 151(1):69–82
Gasser B, Maurer M, Rautio J, Sauer M, Bhattacharyya A, Saloheimo M, Penttilä M, Mattanovich D (2007) Monitoring of transcriptional regulation in Pichia pastoris under protein production conditions. BMC Genomics 8:179
Gilissen LJ, Metz PL, Stiekema WJ, Nap JP (1998) Biosafety of E. coli beta-glucuronidase (GUS) in plants. Transgenic Res 7(3):157–163
Goder V, Carvalho P, Rapoport TA (2008) The ER-associated degradation component Der1p and its homolog Dfm1p are contained in complexes with distinct cofactors of the ATPase Cdc48p. FEBS Lett 582(11):1575–1580
Gonzalez DS, Karaveg K, Vandersall-Nairn AS, Lal A, Moremen KW (1999) Identification, expression, and characterization of a cDNA encoding human endoplasmic reticulum mannosidase I, the enzyme that catalyzes the first mannose trimming step in mammalian Asn-linked oligosaccharide biosynthesis. J Biol Chem 274(30):21375–21386
Gordon CL, Archer DB, Jeenes DJ, Doonan JH, Wells B, Trinci AP, Robson GD (2000) A glucoamylase::GFP gene fusion to study protein secretion by individual hyphae of Aspergillus niger. J Microbiol Methods 42(1):39–48
Gouka RJ, Punt PJ, van den Hondel CA (1997) Efficient production of secreted proteins by Aspergillus: progress, limitations and prospects. Appl Microbiol Biotechnol 47(1):1–11
Guillemette T, van Peij NN, Goosen T, Lanthaler K, Robson GD, van den Hondel CA, Stam H, Archer DB (2007) Genomic analysis of the secretion stress response in the enzyme-producing cell factory Aspergillus niger. BMC Genomics 8:158
Jacobs DI, Olsthoorn MM, Maillet I, Akeroyd M, Breestraat S, Donkers S, van der Hoeven RA, van den Hondel CA, Kooistra R, Lapointe T, Menke H, Meulenberg R, Misset M, Müller WH, van Peij NN, Ram A, Rodriguez S, Roelofs MS, Roubos JA, van Tilborg MW, Verkleij AJ, Pel HJ, Stam H, Sagt CM (2009) Effective lead selection for improved protein production in Aspergillus niger based on integrated genomics. Fungal Genet Biol 46(Suppl 1):S141–S152
Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, Sommer T (2002) Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat Cell Biol 4(2):134–139
Jeenes DJ, Mackenzie DA, Roberts IN, Archer DB (1991) Heterologous protein production by filamentous fungi. Biotechnol Genet Eng Rev 9:327–367
Kauffman KJ, Pridgen EM, Doyle FJ 3rd, Dhurjati PS, Robinson AS (2002) Decreased protein expression and intermittent recoveries in BiP levels result from cellular stress during heterologous protein expression in Saccharomyces cerevisiae. Biotechnol Prog 18(5):942–950
Kincaid MM, Cooper AA (2007) ERADicate ER stress or die trying. Antioxid Redox Signal 9(12):2373–2387
Kleizen B, Braakman I (2004) Protein folding and quality control in the endoplasmic reticulum. Curr Opin Cell Biol 16(4):343–349
Knop M, Finger A, Braun T, Hellmuth K, Wolf DH (1996) Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J 15(4):753–763
Lederkremer GZ (2009) Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol 19(5):515–523
Lilley BN, Ploegh HL (2004) A membrane protein required for dislocation of misfolded proteins from the ER. Nature 429(6994):834–840
Machida M, Asai K, Sano M, Tanaka T, Kumagai T, Terai G, Kusumoto K, Arima T, Akita O, Kashiwagi Y, Abe K, Gomi K, Horiuchi H, Kitamoto K, Kobayashi T, Takeuchi M, Denning DW, Galagan JE, Nierman WC, Yu J, Archer DB, Bennett JW, Bhatnagar D, Cleveland TE, Fedorova ND, Gotoh O, Horikawa H, Hosoyama A, Ichinomiya M, Igarashi R, Iwashita K, Juvvadi PR, Kato M, Kato Y, Kin T, Kokubun A, Maeda H, Maeyama N, Maruyama J, Nagasaki H, Nakajima T, Oda K, Okada K, Paulsen I, Sakamoto K, Sawano T, Takahashi M, Takase K, Terabayashi Y, Wortman JR, Yamada O, Yamagata Y, Anazawa H, Hata Y, Koide Y, Komori T, Koyama Y, Minetoki T, Suharnan S, Tanaka A, Isono K, Kuhara S, Ogasawara N, Kikuchi H (2005) Genome sequencing and analysis of Aspergillus oryzae. Nature 438(7071):1157–1161
Maeda H, Sano M, Maruyama Y, Tanno T, Akao T, Totsuka Y, Endo M, Sakurada R, Yamagata Y, Machida M, Akita O, Hasegawa F, Abe K, Gomi K, Nakajima T, Iguchi Y (2004) Transcriptional analysis of genes for energy catabolism and hydrolytic enzymes in the filamentous fungus Aspergillus oryzae using cDNA microarrays and expressed sequence tags. Appl Microbiol Biotechnol 65(1):74–83
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol 26(5):553–560
Meyer V, Arentshorst M, El-Ghezal A, Drews AC, Kooistra R, van den Hondel CA, Ram AF (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128(4):770–775
Meyer V, Ram AF, Punt PJ (2010) Genetics, genetic manipulation and approaches to strain improvement of filamentous fungi. In: Demain AL, Davis J (eds) Manual of industrial microbiology and biotechnology, 3rd edn. Wiley, New York, pp 318–329
Mori K (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146(6):743–750
Mori K, Ma W, Gething MJ, Sambrook J (1993) A transmembrane protein with a cdc2+/CDC28− related kinase activity is required for signaling from the ER to the nucleus. Cell 74(4):743–756
Mori K, Ogawa N, Kawahara T, Yanagi H, Yura T (1998) Palindrome with spacer of one nucleotide is characteristic of the cis-acting unfolded protein response element in Saccharomyces cerevisiae. J Biol Chem 273(16):9912–9920
Mulder HJ, Nikolaev I (2009) HacA-dependent transcriptional switch releases hacA mRNA from a translational block upon endoplasmic reticulum stress. Eukaryot Cell 8(4):665–675
Mulder HJ, Saloheimo M, Penttilä M, Madrid SM (2004) The transcription factor HACA mediates the unfolded protein response in Aspergillus niger, and up-regulates its own transcription. Mol Genet Genomics 271(2):130–140
Mulder HJ, Nicolaev I, Madrid SM (2006) HACA, the transcriptional activator of the unfolded protein response (UPR) in Aspergillus niger, binds to partly palindromic UPR elements of the consensus sequence 5′-CAN(G/A)NTGT/GCCT-3′. Fungal Genet Biol 43(8):560–572
Mullally JE, Chernova T, Wilkinson KD (2006) Doa1 is a Cdc48 adapter that possesses a novel ubiquitin binding domain. Mol Cell Biol 26(3):822–830
Nakari-Setälä T, Paloheimo M, Kallio J, Vehmaanperä J, Penttilä M, Saloheimo M (2009) Genetic modification of carbon catabolite repression in Trichoderma reesei for improved protein production. Appl Environ Microbiol 75(14):4853–4860
Nemoto T, Maruyama J, Kitamoto K (2009) Improvement of heterologous protein production in Aspergillus oryzae by RNA interference with alpha-amylase genes. Biosci Biotechnol Biochem 73(11):2370–2373
Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, Endo T (2001) Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J Cell Biol 153(5):1061–1070
Nishikawa SI, Brodsky JL, Nakatsukasa K (2005) Roles of molecular chaperones in endoplasmic reticulum (ER) quality control and ER-associated degradation (ERAD). J Biochem 137(5):551–555
Ogiso Y, Sugiura R, Kamo T, Yanagiya S, Lu Y, Okazaki K, Shuntoh H, Kuno T (2004) Lub1 participates in ubiquitin homeostasis and stress response via maintenance of cellular ubiquitin contents in fission yeast. Mol Cell Biol 24(6):2324–2331
Oikawa D, Kimata Y, Kohno K, Iwawaki T (2009) Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res 315(15):2496–2504
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JA, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EG, Debets AJ, Dekker P, van Dijck PW, van Dijk A, Dijkhuizen L, Driessen AJ, d’Enfert C, Geysens S, Goosen C, Groot GS, de Groot PW, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JP, van den Hondel CA, van der Heijden RT, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk PA, Lauber J, Lu X, van der Maarel MJ, Meulenberg R, Menke H, Mortimer MA, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NN, Ram AF, Rinas U, Roubos JA, Sagt CM, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJ, Wedler H, Wösten HA, Zeng AP, van Ooyen AJ, Visser J, Stam H (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25(2):221–231
Plemper RK, Wolf DH (1999a) Endoplasmic reticulum degradation. Reverse protein transport and its end in the proteasome. Mol Biol Rep 26(1–2):125–130
Plemper RK, Bordallo J, Deak PM, Taxis C, Hitt R, Wolf DH (1999b) Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J Cell Sci 112(Pt 22):4123–4134
Punt PJ, Veldhuisen G, van den Hondel CA (1994) Protein targeting and secretion in filamentous fungi. A progress report. Antonie Van Leeuwenhoek 65(3):211–216
Punt PJ, van Gemeren IA, Drint-Kuijvenhoven J, Hessing JG, van Muijlwijk-Harteveld GM, Beijersbergen A, Verrips CT, van den Hondel CA (1998) Analysis of the role of the gene bipA, encoding the major endoplasmic reticulum chaperone protein in the secretion of homologous and heterologous proteins in black Aspergilli. Appl Microbiol Biotechnol 50(4):447–454
Rasheva VI, Domingos PM (2009) Cellular responses to endoplasmic reticulum stress and apoptosis. Apoptosis 14(8):996–1007
Sambrook J, Russel DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbour Laboratory Press, New York
Schäfer A, Wolf DH (2009) Sec61p is part of the endoplasmic reticulum-associated degradation machinery. EMBO J 28(19):2874–2884
Shamu CE, Walter P (1996) Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J 15(12):3028–3039
Sidrauski C, Walter P (1997) The transmembrane kinase Ire1p Is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 90:1031–1039
Sidrauski C, Chapman R, Walter P (1998) The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol 8(6):245–249
Sims AH, Gent ME, Lanthaler K, Dunn-Coleman NS, Oliver SG, Robson GD (2005) Transcriptome analysis of recombinant protein secretion by Aspergillus nidulans and the unfolded-protein response in vivo. Appl Environ Microbiol 71(5):2737–2747
Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101(3):249–258
Tremblay LO, Herscovics A (1999) Cloning and expression of a specific human alpha 1, 2-mannosidase that trims Man9GlcNAc2 to Man8GlcNAc2 isomer B during N-glycan biosynthesis. Glycobiology 9(10):1073–1078
Upshall A, Kumar AA, Bailey MC, Parker MD, Favreau MA, Lewison KP, Joseph ML, Maraganore JM, McKnight GL (1987) Secretion of active human tissue plasminogen activator from the filamentous fungus Aspergillus nidulans. Biotechnology 5:1301–1304
van Anken E, Braakman I (2005a) Endoplasmic reticulum stress and the making of a professional secretory cell. Crit Rev Biochem Mol Biol 40(5):269–283
van Anken E, Braakman I (2005b) Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol 40(4):191–228
van Gorcom RF, van den Hondel CA (1988) Expression analysis vectors for Aspergillus niger. Nucleic Acids Res 16(18):9052
van Laar T, van der Eb AJ, Terleth C (2001) Mif1: a missing link between the unfolded protein response pathway and ER-associated protein degradation? Curr Protein Pept Sci 2(2):169–190
Vembar SS, Brodsky JL (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat Rev Mol Cell Biol 9(12):944–957
Verdoes JC, Punt PJ, Schrickx JM, van Verseveld HW, Stouthamer AH, van den Hondel CA (1993) Glucoamylase overexpression in Aspergillus niger: molecular genetic analysis of strains containing multiple copies of the glaA gene. Transgenic Res 2(2):84–92
Weenink XO, Punt PJ, van den Hondel CA, Ram AF (2006) A new method for screening and isolation of hypersecretion mutants in Aspergillus niger. Appl Microbiol Biotechnol 269(6):711–717
Wiebe MG, Karandikar A, Robson GD, Trinci AP, Candia JL, Trappe S, Wallis G, Rinas U, Derkx PM, Madrid SM, Sisniega H, Faus I, Montijn R, van den Hondel CA, Punt PJ (2001) Production of tissue plasminogen activator (t-PA) in Aspergillus niger. Biotechnol Bioeng 76(2):164–174
Wortman JR, Gilsenan JM, Joardar V, Deegan J, Clutterbuck J, Andersen MR, Archer D, Bencina M, Braus G, Coutinho P, von Döhren H, Doonan J, Driessen AJ, Durek P, Espeso E, Fekete E, Flipphi M, Estrada CG, Geysens S, Goldman G, de Groot PW, Hansen K, Harris SD, Heinekamp T, Helmstaedt K, Henrissat B, Hofmann G, Homan T, Horio T, Horiuchi H, James S, Jones M, Karaffa L, Karányi Z, Kato M, Keller N, Kelly DE, Kiel JA, Kim JM, van der Klei IJ, Klis FM, Kovalchuk A, Krasevec N, Kubicek CP, Liu B, Maccabe A, Meyer V, Mirabito P, Miskei M, Mos M, Mullins J, Nelson DR, Nielsen J, Oakley BR, Osmani SA, Pakula T, Paszewski A, Paulsen I, Pilsyk S, Pócsi I, Punt PJ, Ram AF, Ren Q, Robellet X, Robson G, Seiboth B, van Solingen P, Specht T, Sun J, Taheri-Talesh N, Takeshita N, Ussery D, van Kuyk PA, Visser H, van de Vondervoort PJ, de Vries RP, Walton J, Xiang X, Xiong Y, Zeng AP, Brandt BW, MJ C ornell, van den Hondel CA, Visser J, Oliver SG, Turner G (2009) The 2008 update of the Aspergillus nidulans genome annotation: a community effort. Fungal Genet Biol 1(46):S2–S13
Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA (2001a) A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 429(6994):841–847
Ye Y, Meyer HH, Rapoport TA (2001b) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414(6864):652–656
Yoshida H (2007) ER stress and diseases. FEBS J 274(3):630–658
Acknowledgements
We thank Lucienne Vonk, Noortje Klaver, Mathijs van Vliet, and Renske van Andel for helpful contributions to this project. We thank Peter Punt for materials and helpful discussions. This project was carried out within the research programme of the Kluyver Centre for Genomics of Industrial Fermentation, which is part of The Netherlands Genomics Initiative/Netherlands Organization for Scientific Research. Part of this work was supported by SENTER (BTS project BTS00010, TSGE 3012).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Carvalho, N.D.S.P., Arentshorst, M., Kooistra, R. et al. Effects of a defective ERAD pathway on growth and heterologous protein production in Aspergillus niger . Appl Microbiol Biotechnol 89, 357–373 (2011). https://doi.org/10.1007/s00253-010-2916-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2916-5