
Abstract
Many microbial agents have been implicated as contributors to cancer genesis and development, and the search to identify and characterize new cancer-related organisms is ongoing. Modern developments in methodologies, especially culture-independent approaches, have accelerated and driven this research. Recent work has shed light on the multifaceted role that the community of organisms in and on the human body plays in cancer onset, development, detection, treatment, and outcome. Much remains to be discovered, however, as methodological variation and functional testing of statistical correlations need to be addressed for the field to advance.
Similar content being viewed by others


Introduction
The human microbiome is a significant community, with an estimated ratio of one microbial cell per human cell [1] and nearly 500-fold more microbial genes than host genes [2]. This community is dynamically shaped alongside human development from birth through adolescence. It has coevolved with humans to the degree that it plays an integral role in normal, healthy human functioning [3]. Experiments generating and assessing gnotobiotic and germ-free (GF) mice suggest that, while not a requisite component of physiology, this “hidden organ” provides critical functions that allow for normal metabolic and immune functioning [4]. The role of microbiota as a key functional regulator of metabolic homeostasis [5, 6], drug detoxification and metabolism [7,8,9], and metabolite biosynthesis [10] has been established only recently, and new findings are emerging on a regular basis. Just as microbiota are important in healthy functioning, it has a hand in dysfunction and disorder. Microbial dysbiosis may be loosely described as a human microbiome that does not fulfill all the necessary functions required for health. It has been implicated in metabolic disorders, obesity [5, 11], and immune development, as well as a wide array of disease states [12, 13]. While microbial communities are functionally similar between individuals, they can be wildly dissimilar phylogenetically, a phenomenon that presents unique challenges in studying the microbiome and its role in health and disease [10]. Research on the microbiome has expanded dramatically in the last decade, with increasing interest in microbial community interactions with cancer.
As an emerging field, challenges must be overcome at all facets of research to ensure robust and rigorous science, and these challenges are only exacerbated by the diversity of the human microbiome. Multiple and concerted efforts have been made to identify and provide solutions to these challenges. The MicroBiome Quality Control project (MBQC) attempted to identify the most critical aspects in microbiome studies to improve reproducibility [14], and the International Human Microbiome Standards consortium (IHMS) attempted to address reproducibility concerns by providing standard workflows for microbiome studies [15]. Several reviews have covered issues and solutions for various levels of microbiology research, including fecal DNA extraction [16], 16S rRNA gene analysis and study design [17], and host-microbe multi-omic analyses [18]. These approaches are eminently worthwhile; though it is important to note, they are continuously evolving as the technology and our understanding of the underlying biology improve. In this review, we address current research and issues in targeting cancer as a disease influenced by the microbiome, which includes the issues of microbial studies addressed above but also specific to correlating microbial analyses with cancer pathology or treatment.
Historical relationships between the microbiome and cancer
Various microbial populations have been implicated in cancer. In 2002, 17.8% of all cancers were attributed to microbial action [19]. An early causal relationship between a specific bacterial species and human cancer is Helicobacter pylori and gastric cancer. H. pylori was discovered and later found to be implicated in ulcers by Warren [20]. The development from an H. pylori infection to eventual carcinogenesis has been codified in the Correa pathway. H. pylori can drive chronic inflammation, which leads to atrophic gastritis and eventual dysplasia. CagA-positive H. pylori is especially carcinogenic [21, 22]. More recently, a possible relationship between H. pylori in the gut and increased risk of pancreatic cancer has been explored, although it remains controversial [23]. Curiously, H. pylori may have a protective effect with respect to esophageal adenocarcinomas [24]. Gastroesophageal reflux disease (GERD) can potentially lead to Barrett’s esophagus—that is, a development of scar tissue, cellular dysplasia, and alteration of the cells lining the esophagus from squamous cells to those resembling columnar mucosal cells. These are contributing factors to the development of esophageal adenocarcinoma. There is an inverse correlation between patients with H. pylori infections and Barrett’s esophagus, and thus with esophageal adenocarcinoma, likely due to the reduction in GERD symptoms as a result of H. pylori reducing the local pH in the subregions of the stomach; thereby, the hypothesis goes, reducing the severity of GERD [25]. Thus, a single microbe may have both tumor-suppressing and tumorigenic effects, and deeper research into the host and microbiome relationship is necessary to understand the mechanisms that permit these differing phenotypes.
Transformation-competent viruses have also been shown to cause or be associated with cancer, as was first elucidated through the involvement of Rous sarcoma virus (RSV) in avian sarcoma. RSV is a retrovirus that contains a slightly modified src gene that causes the gene product to be unregulated, which modifies intracellular processes and eventually causes sarcomas in chickens [26]. Human papillomavirus (HPV) has been found to cause cancer by producing the transforming proteins E6 and E7, which prevent Rb from binding E2F and lead to cell cycle dysregulation [27]. Epstein-Barr virus (EBV), a common dsDNA herpesvirus, has been shown to be associated with carcinogenesis, especially Burkitt’s lymphomas. EBV infection alone is not sufficient to cause cancer but may lead to carcinogenesis in tandem with genetic and environmental factors [28]. In the case of breast cancer, there was early suspicion that breast cancer in humans may be driven in part by a mammary tumor virus [29]. While this is a known phenomenon in mice, no such virus has been conclusively identified in humans.
Modern research on the microbiome
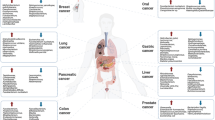
Assessing studies of microbial communities and their interactions with cancer can be difficult as there are many methods to look at these interactions, and, occasionally, the approach used in a given study is not entirely clear. Here, we will describe studies by differentiating the relationship between cancer and microbial communities into three categories: primary, secondary, and tertiary interactions. We are proposing this descriptive nomenclature as a means of clarifying exactly what a given study is assessing, as the relationships can occasionally be unclear. We will define the primary interaction as the interaction between a tumor of interest and the microbiota in the local tumor microenvironment (Fig. 1a). Studies done at this resolution are likely looking for direct mechanistic or causal relationships between the microbiota and the tumor, or therapies within the tumor environment, and often require the use of animal models. Recent mouse studies demonstrating that localized bacteria may modulate chemotherapy efficacy are examples of a primary relationship between the microbiota and tumor [30, 31]. Secondary interactions are defined as those between the microbiota involved with the more general tissue or organ environment and the tumor of interest (Fig. 1b), such as the relationship between the gut microbiota from stool and colorectal cancer (CRC). The distinction between primary and secondary interactions is important because, while studies relying on the primary microbiota may elucidate causal relationships, studies of the secondary microbiota might be less capable in this regard due to the relative dilution of cancer-specific interactions in the more generalized microbial population being evaluated. The secondary microbial communities from these sources may contain some signal in the form of traces and residues from the tumor microenvironment and the primary microbiota, but these signals are inherently noisy since they interact with other tissues besides the neoplasm. However, since samples containing the secondary microbiota are much easier to obtain (e.g., stool), this interaction is critical to study in order to identify biomarkers for disease. Tertiary interactions are those where the effect on a tumor or tumor outcome occurs while the tumor is in an entirely different bodily location than that of the microbial community of interest (Fig. 1c). In the vast majority of cases of tertiary interactions, the microbial community is the gut or stool microbiota and the tumors are those outside the digestive tract—for instance, the interactions seen between breast cancer and the stool microbiota or melanoma and the gut microbiota [32,33,34,35,36]. Tertiary interactions often provide strong clinical implications for treatment options but may also afford insight into systemic relationships between the tumor and a physiologically remote microbial community.

Primary, secondary, and tertiary interactions between the tumor and microbial communities. a Primary interactions are the interactions within the proximal tumor microenvironment. These interactions are important for understanding the mechanisms of microbiome-cancer relationships, such as tumorigenesis through specific microbes or bacterial proliferation in the tumor microenvironment. b Secondary interactions are the interactions between tumors and the microbial community of the tissue or organ system within the same general compartment. These interactions are mostly relevant for discerning potential biomarkers for screening. In this figure, the example is digesta passing by neoplastic tissue in the gut. The digesta may pick up some of the microbes from the tumor, which can be used as a signal of the tumor. Depending on the type and location of the tumor, these interactions may be more or less useful. Generally, an advantage to these interactions is ready access to the material for diagnosis. c Tertiary interactions are interactions between a tumor and a remote microbiome community. Tertiary interactions are less direct than secondary or primary interactions; they include therapeutic modulation by modifying chemotherapy drugs and reduce or increase effectiveness or toxicity, or immune modulation that leads to relevant immune cell differentiation or reaction, or metabolites that regulate hormones or host metabolism that can affect cancer phenotypes or outcomes. In spite of the physical distance and separate organ systems these microbial communities occupy relative to the tumor, they can have a profound effect on the tumor phenotype, treatment, and outcomes
Human tumor microbiota primary interactions
The standard starting place for a study of the microbiota associated with a particular disease state is basic characterization. A first-pass characterization needs to be done to identify the specific taxa that are present in normal and disease states and determine what the potential biomarkers or targets for intervention might be. Thus far, this topic has been studied most commonly through cross-sectional clinical studies, which have identified the microbes or sets of microbes that are differentially present/absent or increased/decreased as a function of the disease once all the potentially confounding patient metadata variables have been accounted for. Most of these types of studies have been restricted to cancers in tissues where there is a resident microbial community. However, one interesting side effect of this line of research is the investigation of tissue sites such as the breast, uterus, prostate, and bladder, among others, that were not previously thought to harbor resident microbial communities [37,38,39,40,41,42,43,44,45].
Regarding biomarkers, primary tumors are not necessarily a good place to begin a study (i.e., looking at the microbes directly on a tumor to detect if a tumor is present poses some logical challenges). However, there are instances where the primary microbiota can provide useful information. One can discern information about the microbes at the tumor and make predictions about patient outcomes, as is done in parallel research on personalized cancer treatments where a tumor genome is sequenced or specific levels of relevant genes measured as a means of identifying how best to proceed in the clinic. For instance, in some cases, pancreatic cancer can be protected from the immune system by the presence of specific cancer-associated microbial communities and is correlated with patient mortality [46, 47].
Tumors provide a unique hypoxic environment for bacterial growth. In 1955, Malmgren and Flanigan demonstrated in a mouse model that the growth of Clostridium tetani is favored in the tumor microenvironment [48]. Tumors can develop hypoxic conditions due to the outgrowth of oxygen supply as a result of poor vascularization by tumor-stimulated angiogenesis [49, 50]. This hypoxic and necrotic environment allows for the selective growth of anaerobic bacteria, an important characteristic of the tumor microbiome [51, 52].
As established by the H. pylori model, tumorigenesis may occur due to microbial infection. Other microbes have been suggested to play similar roles. The gram-negative genus Fusobacterium is associated with the CRC tumor microenvironment [53, 54]. Fusobacterium nucleatum-derived tumorigenesis is thought to arise through an opportunistic infection followed by chronic inflammation and immunosuppression, establishing F. nucleatum as an opportunistic cancer driver in the microbiome-tumor primary interaction. It also generates bacterial biofilms that increase fitness for tumor-promoting microbial species and complement the hypoxic tumor microenvironment [55,56,57]. Enterotoxigenic Bacteroides fragilis are another gram-negative anaerobic bacterium associated with CRC and is hypothesized to be a driver of tumorigenesis by promoting mutations in host cell genomes. In the tumor microenvironment, besides proliferation under hypoxic conditions, pathogenic B. fragilis may secrete proliferative and proinflammatory signals, thus exacerbating tumorigenesis [58, 59].
The tumor microbiome may play a role in tumor resistance to treatment. Gemcitabine is a cytidine analog used as chemotherapy. In pancreatic ductal adenocarcinoma mouse models, it was found that tumor resistance to gemcitabine is mediated by intratumoral gammaproteobacteria and Bifidobacterium psuedolongum [30, 60].
Microbes as anticancer therapy
A tumor’s hypoxic microenvironment’s selectivity for anaerobic bacteria has been exploited as a method of combating cancer using auxotrophic bacteria to target the tumor in bacteria-mediated cancer therapy (BCT). Early experiments using Clostridium and Salmonella alone have not yielded regression of tumors in clinical trials, in spite of success in mouse models. More recently, in various xenograft models, combination therapy using Clostridium novyi (C. novyi-NT) with chemotherapy, antivascular therapy, or radiation has shown to be more effective than treatment without C. novyi-NT, and auxotrophic Salmonella typhimurium has shown promising effects. These treatments function by eliciting a local inflammatory response to the bacteria via local innate immune recognition of the bacteria, thereby increasing immune effects on the cancer where the bacteria have localized. Challenges in bacteriolytic therapy revolve mostly around successful bacterial growth in the heterogeneous tumor microenvironment and sufficient stimulation of host immunity to effectively attenuate the tumor [61]. Instead of relying on innate immunity to recognize and react to the localized bacteria, an alternative is to use the bacterial localization to deliver anticancer agents to the tumor environment. Normal cells will be minimally affected, as the bacteria will not localize anywhere but the tumor microenvironment. In in vivo models, this method has shown results in delivering cytotoxic chemotherapy and immune-stimulating drugs to the tumor, although it has yet to prove efficacious for long-term effects. However, this may potentially be compensated for in combination with treatment strategies [51, 62].
The treatments above rely on transferring live bacterial communities, but other treatment methods have been developed using bacterial extracts. Bacterial extracts have been used in cancer treatments since the discovery of Coley’s toxins, a concoction of bacterial lysates, in the early twentieth century [61]. More recently, synthetic CpG oligodeoxynucleotides (ODN), small ssDNA segments containing CpG motifs, have been used to stimulate Toll-like receptors (TLRs) on immune cells. They have been classified into three distinct classes: class A stimulates peripheral dendritic cells to secrete type I interferon, class B stimulates B cell maturation, and class C has the effects of both class A and B CpG ODNs [63]. In many models, CpG ODNs are administered intratumorally or locally [64, 65], which can be an issue for administration in some clinical settings. Systemic administration in mice shows promise, however, only when part of combination therapies [66]. At the time of writing, there are multiple ongoing clinical trials for many CpG ODNs, mostly phase 1 or 2 trials for melanomas or lymphomas administered in combination with monoclonal antibody immunotherapies (Table 1) [67, 68].
Using the microbiome as a biomarker—a secondary interaction
Early detection of cancer increases survival rates [69]. In developing biomarkers, it is imperative to keep in mind that the tools must have robust sensitivity and specificity to reduce false positives and false negatives. Since some microbial samples are minimally/non-invasive to collect in routine checkups, such as the microbiome from the skin, mouth, rectal swabs, and urine, they can provide facile sources of investigation to assess as cancer biomarkers.
Microbes involved in the primary tumor microenvironment are capable of becoming biomarkers found in secondary samples. In practice, this depends on a few factors. For one, there is the location of the tumor. Tumors that are in a region of the body exposed to the environment or involved with circulated or excreted substances, such as cancers of the blood or digestive system, have microbial biomarkers that are easier to obtain since they may be detected in the blood, feces, etc. Another important factor is the auxotrophy of the microbes—they may be dependent on the tumor microbiome primary environment and thus not found at detectable levels elsewhere in the body.
Current clinical screening methods for CRC have shortcomings. Some tests that are sensitive are invasive or expensive, and other tests that are non-invasive or inexpensive have low specificity or sensitivity [70]. There remains a need to find non-invasive, effective, and cost-effective screening tools for CRC, and characterization of the microbiome may improve clinical screening and risk assessment. A pilot study has shown that progressive stages of colorectal cancer, labeled as having no malignancy, adenoma, and carcinoma, can be identified based on the relative abundance of microbes in fecal samples. Combining this method with a fecal occult blood test (FOBT) decreased the chance of false positives [71]. Several specific bacteria have been associated with CRC that may be promising targets for developing biomarkers. As mentioned above, Fusobacteria is present in primary colorectal tumor interactions; however, using species-level Fusobacteria taxa as biomarkers has proved challenging. F. nucleatum may be a potential driver of tumorigenesis in the primary interaction, but it is only present in a minority of CRC patients and is also present in the stool of non-CRC individuals [56]. F. nucleatum is also present in the oral microbiome, and unusual levels of this bacterium in the mouth may be a biomarker of CRC, an example of a tertiary interaction as a biomarker for cancer [72].
Pancreatic cancer is one of the most fatal cancers with a 5-year mortality between 91 and 98%. A large contributor to the high mortality is the lack of efficient screening methods [73]. Pancreatic cancer, like many cancers, is largely driven via inflammation. Similar to gastric cancer, H. pylori infection increases the risk of pancreatic cancer. Periodontal disease also leads to an increased risk of pancreatic cancer. Studies thus far have found multiple associations between various oral microbiome perturbations and pancreatic cancer [74, 75].
In addition to those described here, there is a similar clinical need for biomarkers in other cancers. There have been a multitude of studies looking at the microbial communities in secondary compartments for relevant genomic markers, including the head and neck, lung, bladder, prostate, and others [43, 45, 76,77,78]. The stool microbiome has also been used as a tertiary biomarker for distant tumors, including hepatocellular carcinoma, lung cancer, and prostate cancer [78,79,80,81,82].
Modulation of therapeutic drug efficacy by the microbiome—a tertiary interaction
Chemotherapy, a common and effective means of treating many cancers, involves using drugs that preferably destroy transformed cells with minimal damage to normal cells, although side effects may be common. Bacteria in the microbiome have been shown to modulate chemotherapy in various capacities. The most obvious effect is the microbial effect on orally administered drug metabolism. Microbial species lining the intestinal mucosa can perform various metabolic reactions, such as reduction, hydrolysis, ring opening, and functional group removal. These alterations can activate or deactivate drugs, or affect drug toxicity [83]. Alternatively, these microbes can affect intestinal cell gene expression to modulate host cell metabolism of the drugs, which has implications for drug absorption and efficacy over time [84, 85] The direct clinical relevance of gut bacterial modulation of drugs is apparent in the case of irinotecan (CPT-11) treatment, where the presence of bacteria in the gut expressing β-glucuronidase increases the CPT-11 side effect of severe diarrhea [85, 86].
The microbiome has modulatory effects in other therapies. For allogeneic hematopoietic cell transplantation (allo-HCT), a treatment for hematologic cancers, the gut microbiome may be predictive for both risk for treatment and chances of relapse post-treatment [87, 88]. In the case of inflammation-based immunotherapy, such as intratumoral administration of CpG ODN described above, an intact gut microbiome was found to significantly increase treatment efficacy in mice [89]. TLR response-based therapeutics are connected to innate immunity, which may generally be affected by the host microbial communities, although the specific mechanisms remain unknown. Other therapeutic agents that do not rely on innate immunity have also been studied. Cisplatin and oxaliplatin, platinum-based chemotherapy that forms intrastrand adducts and interstrand crosslinks to prevent polymerase activity, were found to have reduced efficacy when combined with common antibiotics in mice [89]. Cyclophosphamide (CTX) is a chemotherapy drug that alkylates and crosslinks DNA and similarly prevents polymerase activity. CTX is known to be immunogenic, meaning through cell lysis via CTX, tumor cells release immune-stimulating signals that allow dendritic cells (DC) to target the tumor, thus increasing CTX efficacy [90]. In mice, Viaud et al. found that CTX also leads to increased numbers of pathogenic TH17 and increased CD4+ T cell differentiation to TH1/TH17 by modulating the gut microbiota. However, the use of antibiotics along with CTX was found to decrease CTX efficacy [91].
The efficacy of the checkpoint inhibitors PD-1, PD-L1, and CTLA-4 is modulated by the gut microbiota. In multiple melanoma GF mouse models, the presence of specific microbial taxa along with checkpoint inhibitors leads to an increased presence of relevant immune-activating CD4+ and CD8+ T cells in the tumor microenvironment and DC IL-12 secretion [92,93,94]. These studies can have significant clinical relevance, as the gut microbial composition is a likely determinant of patient response or non-response to anti-PD-1 and anti-PD-L1 therapy [32, 95]. While microbial-mediated immune stimulation can have a positive effect on the tertiary tumor, it can also have a negative effect by driving immune checkpoint inhibitor (ICI)-associated colitis. Colitis has been shown in mouse models to drive primary interactions that can allow genotoxic bacteria to ingraft and eventually lead to dysplasia [96]. This issue is manageable, however, given that patients who develop or did not develop colitis after anti-CTLA-4 therapy have distinct microbial communities [35], IL-10−/− mouse models show Bifidobacteria may prevent colitis after anti-CTLA-4 therapy [97], and FMT may prove effective [98]. Thus, analysis of a patient’s gut microbiota may provide insight into the checkpoint inhibitor treatment efficacy for a remote tumor and risk assessment for ICI-associated colitis.
In general, dysbiosis or altered states of microbial communities may result from a patient’s genotype, antibiotic exposures, or chemotherapy, and altering or restoring microbiota may lead to increased immune responses via innate immune signaling (TLR) or specific immune cell activation or differentiation, e.g., TH1/TH17, CD8+ T cells, or DC [51]. There is emerging interest and effort being put forth to model the interactions between the immune system, therapeutic interactions, and cancer outcomes with the microbial communities in the host as the key variable [91, 99,100,101,102,103].
Future directions
Culture-independent approaches have become the forefront of research on the cancer microbiome. As sequencing has become much less expensive and the 16S rRNA gene databases have expanded, 16S rRNA gene sequencing has become an efficient and high-throughput method for identifying bacteria present in a microbial community [104, 105]. Issues with the resolution of 16S rRNA gene sequencing at the species and strain levels remain a challenge [105, 106]. With the development of second-generation and third-generation sequencing, one method to overcome the shortfalls of 16S rRNA gene sequencing is to simply sequence the entire metagenome. Whole-genome shotgun (WGS) sequencing, especially WGS techniques that provide long reads, has the ability to overcome the 16S rRNA gene sequencing limitations [104, 107, 108]. By analyzing the entire genome of all the microbial populations present, greater microbial diversity can be represented with greater accuracy than 16S rRNA gene sequencing. Viral and fungal species can be represented as well [109]. While advances in these technologies are making WGS sequencing become more affordable, the cost to accurately sequence and analyze all the bacteria in a microbial population with sufficient coverage remains expensive, and the data analysis is laborious and intensive [107, 109] Progress is being made on these fronts as well, with researchers leveraging advances in the algorithms used to identify species- and strain-level details to allow shallow WGS to be used to gain the advantage of the resolution of WGS with the economic benefit of 16S rRNA gene sequencing [110].
Generally speaking, microbiome studies each come with their own caveats and challenges. The Human Microbiome Project was the first large-scale effort to catalog the microbial communities present across multiple body sites [10, 111,112,113,114]. These studies, along with other, similar efforts [115], have acted as the cornerstone of human microbiome work as they allow researchers to compare and contrast their own work on the cancer microbiome with the research generated by others. However, while conceptually simple, in practice, these comparisons are plagued with confounding variables that hamper clear comparisons (Fig. 2 and Table 2).

Studies of primary interactions between tumors/precancerous lesions and bacteria are diverse with respect to the sequencing platforms used and the 16S rRNA gene variable regions targeted for PCR and sequencing. This Sankey plot highlights that among 40 different studies of the primary microbial communities found at the site of a lesion, there are 5 different sequencing technologies that have been used and 13 different sets of variable regions targeted
The work of Sinha et al. has evaluated many experimental factors that influence the evaluation of microbial communities [14]. Their work is part of a larger effort to identify and iterate upon “best practices” in the microbiome research field. For example, the specific microbial taxa detected in samples vary as a function of which region of the 16S rRNA gene is targeted for PCR amplification and sequencing (e.g., V1–V3 vs. V3–V5) [10, 14, 124]. Subsequent research by other groups has also uncovered many additional variables that can influence the results of 16S rRNA gene sequencing studies, including the use of sample preservatives [116,117,118,119,120,121,122], sample storage [117,118,119, 129, 132,133,134,135,136,137,138,139,140,141,142], sample lysis protocol [143,144,145, 149, 150], and DNA purification technique [116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156]. The choice of PCR primers, even for the same variable region; the DNA polymerase used; the PCR cycling conditions; and the analysis platform can influence the outcome of 16S rRNA gene sequencing studies [129, 157,158,159]. These variables, combined with the additional factors such as bioinformatic pipelines used for analysis, data quality control tuning, software parameters, and reference database choices, raise many questions related to the value of comparing the final results of these studies to one another, when none of these factors are controlled for. One would hope that the results from one bioinformatic analysis platform to another would be robust and adequately reflect the biological reality of the experimental system; however, this has been demonstrated by several groups to not necessarily be the case [159,160,161,162,163,164,165]. The development of new tools and the constant updating of existing tools are proceeding rapidly, with a healthy amount of back and forth discussion between different research groups (e.g., see Edgar [166] and the associated pre-publication comment by the QIIME development team).
The body of microbiome studies on cancer as a whole manifests as one might expect a decentralized, multidisciplinary research endeavor would: there is a profound lack of consistency regarding the analyses. Figure 2 is a Sankey plot that summarizes the tumor types, sequencing technologies, and the 16S rRNA gene variable region targeted across 40 different studies of the primary interactions between tumors and their associated bacteria [39, 40, 54, 167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203]. While not shown, these studies also vary with respect to the data analysis tools, pipelines, and databases used. Of these, more than half (26/40) did not provide sufficient information in the methods section or supplementary materials to ascertain what the bioinformatic approaches used were in sufficient detail to be able to replicate their analyses. Of the others that indicated the tools and approaches used, at least seven different tools or combinations of tools were used. Furthermore, the specifics of the parameters applied were not sufficiently reported. The studies included in Fig. 2 are also quite varied with respect to the availability of the raw sequence data. From an assessment of the manuscripts and their accompanying supplementary materials, as well as a cold search of the NCBI Sequence Read Archive (SRA) for the studies themselves, 27 of the 40 have deposited their data in the SRA, in some form or other, 1 of the 40 deposited their data in the European Nucleotide Archive (ENA) and 12 of the 40 include a statement that data are available upon request or do not mention data availability at all. One study has the data deposited in a public database, but not the associated metadata that would allow other researchers to replicate their findings, specifically due to the manner in which the IRB protocol for this study was written. In order to improve the ability of researchers to replicate published findings, journals have begun to require that data underlying figures as well as the code used to generate statistical models and visualizations be made available as supplementary information, though even with published code and tools, there is a broad spectrum of usability [204]. Code generated and used in publications is often made available on GitHub or SourceForge. The journal eLife has made code persistently available by forking the authors' published version of code directly to the eLife GitHub account. Challenges remain regarding software and database availability, however. There are efforts in the field to provide bioinformatic virtual environments in the form of virtual machine (VM) images or dockerized containers that would allow interested researchers to be able to easily repeat the analyses shown in a publication, as these environments contain the exact software and database versions used at the time—QIIME 2, a popular metagenomic analysis suite, is provided in a preconfigured VM [205]. Even in these best-case scenarios, this distribution model is suboptimal when the tools or databases used are proprietary or closed source and not distributable. Despite these challenges, steps are being taken in the field to develop guidelines for computational tool development and publication [206].
The field of microbiome research is still new, and it is understandable that there would be diversity in data analysis approaches and reporting. Unfortunately, as the field of biomedical research struggles with a reproducibility crisis, it is important that these deviations are addressed [207]. To deal with these challenges, several groups have provided guidelines that researchers can utilize to improve the utility and robustness of their findings, including MIMARKS and MIMAG for reporting sequencing data from microbes [208, 209], STROBE and STROME-ID for molecular data tied to epidemiological projects [210, 211], and REMARK for data used as tumor markers [212, 213]. Marques and colleagues, working in the field of hypertension, have published a set of general guidelines and checklists that researchers and publishers can use when designing, executing, and reporting microbiome-related studies of disease in either animal models or humans that could easily be adapted to cancer research [214]. Other groups, including the International Human Microbiome Standards Project and the Microbiome Quality Control project, have proposed research and reporting guidelines for experimental design when assessing the microbiome in humans and model systems [14, 215, 216].
The many variables related to research on cancer-associated microbiota have led other research groups to attempt to integrate findings across studies in meta-analyses, typically focused on CRC and other gut-related diseases [198, 217,218,219], while other reviews on the topic have attempted to integrate the non-gut-related microbial changes [220]. In the case of the former, a recent meta-analysis that was performed on the colorectal cancer microbiome [219] found that when integrating data from multiple projects, many of the significant factors that were reported in individual studies were discovered to not be generalizable (i.e., the relationship between the increased abundance of Fusobacterium ssp. in CRC patients). In the case of the latter, the findings documented in these reviews are intriguing—however, as the comparisons are massively confounded by the indicated variables mentioned above, they are challenging to rely upon as a basis for future work.
As the field generates more and more studies of the microbial communities relevant to cancer biology, it is common (and quite reasonable) for researchers and reviewers to want to contextualize findings in light of what has already been done on the topic. At the moment, this is done ad hoc either by comparing the final results of other projects to one’s own work or by more quantitatively integrating the work of other researchers into the analyses using either raw sequencing data or finalized OTU tables, when available. Standardizing methods and protocols across the field of cancer, microbiome research will prove useful in the short run, but since tools will change and evolve, robust controls, logical study design, and sparing no effort to document and report on research materials will be vital to the field.
Conclusions
The promise of the microbiome in cancer research is tremendous. The field is rapidly expanding in scope along with our understanding of how microbial communities that live in and on us shape our behavior and influence our health. These breakthroughs in microbiome research have only been made possible by the advances in genomics—these advances have allowed us to collect vast amounts of metagenomic and microbial marker gene sequences. Studies have now characterized the essential role of the microbiota in tumor formation and fitness. We have highlighted several areas that show prospective benefits for cancer patients in the clinic. Characterization of the tumor microenvironment led to the use of microbes that are selective for the said microenvironment in therapies. Current trials are exploring the use of bacterial extracts in combination with checkpoint inhibitor immunotherapies. A deeper understanding of the microbiome in the context of secondary interactions brought about research into useful diagnosis and detection of cancer early in the process. We have also shown that continued, efficient research in the emerging field of the microbiome in the context of cancer is predicated on identifying best practices surrounding the design, execution, bioinformatic analysis, interpretation, and reporting of studies. Due to the lack of applied common experimental and analytical approaches for research methods and subsequent extreme variability in study design, outlining overarching, interpretable themes between multiple studies even within the same cancer type, much less across cancer types, is challenging to say the least. We propose an update to the manner in which the interactions between cancer and microbial communities are described to rectify these inconsistencies and allow for larger-scale interpretations from multiple studies. Our hope is that the temporary shortcomings in microbiome research will be recognized and accounted for in order to provide the research community, clinicians, and patients with the best possible information and outcomes possible.
References
Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 2016;14:e1002533.
Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–41.
Dominguez-Bello MG, Godoy-Vitorino F, Knight R, Blaser MJ. Role of the microbiome in human development. Gut. 2019;68:1108–14.
Kennedy EA, King KY, Baldridge MT. Mouse microbiota models: comparing germ-free mice and antibiotics treatment as tools for modifying gut bacteria. Front Physiol. 2018;9:1534.
Bäckhed F. Programming of host metabolism by the gut microbiota. Ann Nutr Metab. 2011;58(Suppl 2):44–52.
Dobrijevic D, Abraham A-L, Jamet A, Maguin E, van de Guchte M. Functional comparison of bacteria from the human gut and closely related non-gut bacteria reveals the importance of conjugation and a paucity of motility and chemotaxis functions in the gut environment. PLoS One. 2016;11:e0159030.
Björkholm B, Bok CM, Lundin A, Rafter J, Hibberd ML, Pettersson S. Intestinal microbiota regulate xenobiotic metabolism in the liver. PLoS One. 2009;4:e6958.
Coryell M, McAlpine M, Pinkham NV, McDermott TR, Walk ST. The gut microbiome is required for full protection against acute arsenic toxicity in mouse models. Nat Commun. 2018;9:5424.
Selwyn FP, Cheng SL, Bammler TK, Prasad B, Vrana M, Klaassen C, et al. Developmental regulation of drug-processing genes in livers of germ-free mice. Toxicol Sci. 2015;147:84–103.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–14.
Weiss GA, Hennet T. Mechanisms and consequences of intestinal dysbiosis. Cell Mol Life Sci. 2017;74:2959–77.
Gensollen T, Iyer SS, Kasper DL, Blumberg RS. How colonization by microbiota in early life shapes the immune system. Science. 2016;352:539–44.
Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. 2017;474:1823–36.
Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol. 2017;35:1077–86.
Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35:1069–76.
Greathouse KL, Sinha R, Vogtmann E. DNA extraction for human microbiome studies: the issue of standardization. Genome Biol. 2019;20:212.
Debelius J, Song SJ, Vazquez-Baeza Y, Xu ZZ, Gonzalez A, Knight R. Tiny microbes, enormous impacts: what matters in gut microbiome studies? Genome Biol. 2016;17:217.
Mallick H, Ma S, Franzosa EA, Vatanen T, Morgan XC, Huttenhower C. Experimental design and quantitative analysis of microbial community multiomics. Genome Biol. 2017;18:228.
Parkin DM. The global health burden of infection-associated cancers in the year 2002. Int J Cancer. 2006;118:3030–44.
Warren JR. Gastric pathology associated with Helicobacter pylori. Gastroenterol Clin N Am. 2000;29:705–51.
Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–9.
Karimi P, Islami F, Anandasabapathy S, Freedman ND, Kamangar F. Gastric cancer: descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol Biomark Prev. 2014;23:700–13.
Chen J, Domingue JC, Sears CL. Microbiota dysbiosis in select human cancers: evidence of association and causality. Semin Immunol. 2017;32:25–34.
Polyzos SA, Zeglinas C, Artemaki F, Doulberis M, Kazakos E, Katsinelos P, et al. Helicobacter pylori infection and esophageal adenocarcinoma: a review and a personal view. Ann Gastroenterol Hepatol. 2018;31:8–13.
Wang Z, Shaheen NJ, Whiteman DC, Anderson LA, Vaughan TL, Corley DA, et al. Helicobacter pylori infection is associated with reduced risk of Barrett’s esophagus: an analysis of the Barrett’s and Esophageal Adenocarcinoma Consortium. Am J Gastroenterol. 2018;113:1148–55.
Prakash O, Bardot SF, Cole JT. Chicken sarcoma to human cancers: a lesson in molecular therapeutics. Ochsner J. 2007;7:61–4.
Harden ME, Munger K. Human papillomavirus molecular biology. Mutat Res - Rev Mut Res. 2017;772:3–12.
Hau PM, Tsao SW. Epstein-Barr virus hijacks DNA damage response transducers to orchestrate its life cycle. Viruses. 2017;9. Available from: https://doi.org/10.3390/v9110341.
Amarante MK, de Sousa PN, Vitiello GAF, Watanabe MAE. Involvement of a mouse mammary tumor virus (MMTV) homologue in human breast cancer: evidence for, against and possible causes of controversies. Microb Pathog. 2019;130:283–94.
Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science. 2017;357:1156–60.
Lehouritis P, Cummins J, Stanton M, Murphy CT, McCarthy FO, Reid G, et al. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci Rep. 2015;5:14554.
Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103.
Kwa M, Plottel CS, Blaser MJ, Adams S. The intestinal microbiome and estrogen receptor-positive female breast cancer. J Natl Cancer Inst. 2016;108. Available from: https://doi.org/10.1093/jnci/djw029.
Yang J, Tan Q, Fu Q, Zhou Y, Hu Y, Tang S, et al. Gastrointestinal microbiome and breast cancer: correlations, mechanisms and potential clinical implications. Breast Cancer. 2017;24:220–8.
Dubin K, Callahan MK, Ren B, Khanin R, Viale A, Ling L, et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat Commun. 2016;7:10391.
Frankel AE, Coughlin LA, Kim J, Froehlich TW, Xie Y, Frenkel EP, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. 2017;19:848–55.
Cavarretta I, Ferrarese R, Cazzaniga W, Saita D, Lucianò R, Ceresola ER, et al. The microbiome of the prostate tumor microenvironment. Eur Urol. 2017;72:625–31.
Banerjee S, Alwine JC, Wei Z, Tian T, Shih N, Sperling C, et al. Microbiome signatures in prostate cancer. Carcinogenesis. 2019;40:749–64.
Urbaniak C, Gloor GB, Brackstone M, Scott L, Tangney M, Reid G. The microbiota of breast tissue and its association with breast cancer. Appl Environ Microbiol. 2016;82:5039–48.
Hieken TJ, Chen J, Hoskin TL, Walther-Antonio M, Johnson S, Ramaker S, et al. The microbiome of aseptically collected human breast tissue in benign and malignant disease. Sci Rep. 2016;6:30751.
Cicinelli E, De Ziegler D, Nicoletti R, Colafiglio G, Saliani N, Resta L, et al. Chronic endometritis: correlation among hysteroscopic, histologic, and bacteriologic findings in a prospective trial with 2190 consecutive office hysteroscopies. Fertil Steril. 2008;89:677–84.
Walther-António MRS, Chen J, Multinu F, Hokenstad A, Distad TJ, Cheek EH, et al. Potential contribution of the uterine microbiome in the development of endometrial cancer. Genome Med. 2016;8:122.
Wu P, Zhang G, Zhao J, Chen J, Chen Y, Huang W, et al. Profiling the urinary microbiota in male patients with bladder cancer in China. Front Cell Infect Microbiol. 2018;8:167.
Bajic P, Wolfe AJ, Gupta GN. The urinary microbiome: implications in bladder cancer pathogenesis and therapeutics. Urology. 2019;126:10–5.
Bučević Popović V, Šitum M, Chow C-ET, Chan LS, Roje B, Terzić J. The urinary microbiome associated with bladder cancer. Sci Rep. 2018;8:12157.
Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018;8:403–16.
Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806 e12.
Malmgren RA, Flanigan CC. Localization of the vegetative form of Clostridium tetani in mouse tumors following intravenous spore administration. Cancer Res. 1955;15:473–8.
Wouters BG, Koritzinsky M, Chiu RK, Theys J, Buijsen J, Lambin P. Modulation of cell death in the tumor microenvironment. Semin Radiat Oncol. 2003;13:31–41.
Zu C, Wang J. Tumor-colonizing bacteria: a potential tumor targeting therapy. Crit Rev Microbiol. 2014;40:225–35.
Bashiardes S, Tuganbaev T, Federici S, Elinav E. The microbiome in anti-cancer therapy. Semin Immunol. 2017;32:74–81.
Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B. Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A. 2001;98:15155–60.
Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012;22:299–306.
Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–8.
Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal cancer. Annu Rev Microbiol. 2016;70:395–411.
Shang F-M, Liu H-L. Fusobacterium nucleatum and colorectal cancer: a review. World J Gastrointest Oncol. 2018;10:71–81.
Li S, Konstantinov SR, Smits R, Peppelenbosch MP. Bacterial biofilms in colorectal cancer initiation and progression. Trends Mol Med. 2017;23:18–30.
Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60:208–15.
Keenan JI, Aitchison A, Purcell RV, Greenlees R, Pearson JF, Frizelle FA. Screening for enterotoxigenic Bacteroides fragilis in stool samples. Anaerobe. 2016;40:50–3.
Wei M-Y, Shi S, Liang C, Meng Q-C, Hua J, Zhang Y-Y, et al. The microbiota and microbiome in pancreatic cancer: more influential than expected. Mol Cancer. 2019;18:97.
Wong S, Slavcev RA. Treating cancer with infection: a review on bacterial cancer therapy. Lett Appl Microbiol. 2015;61:107–12.
Din MO, Danino T, Prindle A, Skalak M, Selimkhanov J, Allen K, et al. Synchronized cycles of bacterial lysis for in vivo delivery. Nature. 2016;536:81–5.
Martinson JA, Tenorio AR, Montoya CJ, Al-Harthi L, Gichinga CN, Krieg AM, et al. Impact of class A, B and C CpG-oligodeoxynucleotides on in vitro activation of innate immune cells in human immunodeficiency virus-1 infected individuals. Immunology. 2007;120:526–35.
Makkouk A, Joshi VB, Wongrakpanich A, Lemke CD, Gross BP, Salem AK, et al. Biodegradable microparticles loaded with doxorubicin and CpG ODN for in situ immunization against cancer. AAPS J. 2015;17:184–93.
Czerwinski DK, Frank MJ, Shree T, Khodadoust MS, Long SR, Levy R. Intratumoral injection of CpG-ODN plus systemic ibrutinib induces an anti-tumor immune response affecting T cell subsets in the microenvironment of both injected and non-injected tumor sites in patients with low-grade lymphoma. Blood Am Soc Hematol. 2018;132:1612.
Appelbe OK, Moynihan KD, Flor A, Rymut N, Irvine DJ, Kron SJ. Radiation-enhanced delivery of systemically administered amphiphilic-CpG oligodeoxynucleotide. J Control Release. 2017;266:248–55.
Levy R, Reagan PM, Friedberg JW, Bartlett NL, Gordon LI, Leung A, et al. SD-101, a novel class C CpG-oligodeoxynucleotide (ODN) Toll-like receptor 9 (TLR9) agonist, given with low dose radiation for untreated low grade B-cell lymphoma: interim results of a phase 1/2 trial. Blood American Society of Hematology. 2016;128:2974.
Search of: CpG ODN | Recruiting, not yet recruiting, active, not recruiting, enrolling by invitation studies | Cancer - List Results - ClinicalTrials.gov. [cited 2019 Jul 11]. Available from: https://clinicaltrials.gov/ct2/results?term=CpG+ODN&cond=Cancer&recrs=b&recrs=a&recrs=f&recrs=d&age_v=&gndr=&type=&rslt=&Search=Apply.
Kent A. Cancer screening and survival. Rev Obstet Gynecol. 2011;4:129–30.
Simon K. Colorectal cancer development and advances in screening. Clin Interv Aging. 2016;11:967–76.
Zackular JP, Rogers MAM, Ruffin MT 4th, Schloss PD. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res. 2014;7:1112–21.
Flemer B, Warren RD, Barrett MP, Cisek K, Das A, Jeffery IB, et al. The oral microbiota in colorectal cancer is distinctive and predictive. Gut. 2018;67:1454-63.
Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22:9694–705.
Ertz-Archambault N, Keim P, Von Hoff D. Microbiome and pancreatic cancer: a comprehensive topic review of literature. World J Gastroenterol. 2017;23:1899–908.
Zambirinis CP, Pushalkar S, Saxena D, Miller G. Pancreatic cancer, inflammation, and microbiome. Cancer J. 2014;20:195–202.
Vesty A, Gear K, Biswas K, Radcliff FJ, Taylor MW, Douglas RG. Microbial and inflammatory-based salivary biomarkers of head and neck squamous cell carcinoma. Clin Exp Dent Res. 2018;4:255–62.
Yang J, Mu X, Wang Y, Zhu D, Zhang J, Liang C, et al. Dysbiosis of the salivary microbiome is associated with non-smoking female lung cancer and correlated with immunocytochemistry markers. Front Oncol. 2018;8:520.
Golombos DM, Ayangbesan A, O’Malley P, Lewicki P, Barlow L, Barbieri CE, et al. The role of gut microbiome in the pathogenesis of prostate cancer: a prospective. Pilot Study Urology. 2018;111:122–8.
Zhang W-Q, Zhao S-K, Luo J-W, Dong X-P, Hao Y-T, Li H, et al. Alterations of fecal bacterial communities in patients with lung cancer. Am J Transl Res. 2018;10:3171–85.
Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, et al. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 2019;68:1014–23.
Amirian ES, Petrosino JF, Ajami NJ, Liu Y, Mims MP, Scheurer ME. Potential role of gastrointestinal microbiota composition in prostate cancer risk. Infect Agent Cancer. 2013;8:42.
Stanisławska IJ, Granica S, Piwowarski JP, Szawkało J, Wiązecki K, Czarnocki Z, et al. The activity of urolithin A and M4 valerolactone, colonic microbiota metabolites of polyphenols, in a prostate cancer in vitro model. Planta Med. 2019;85:118–25.
Li H, He J, Jia W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2016;12:31–40.
Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell. 2013;152:39–50.
Wilkinson EM, Ilhan ZE, Herbst-Kralovetz MM. Microbiota-drug interactions: impact on metabolism and efficacy of therapeutics. Maturitas. 2018;112:53–63.
Garrett WS. Cancer and the microbiota. Science. 2015;348:80–6.
Taur Y, Jenq RR, Perales M-A, Littmann ER, Morjaria S, Ling L, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood. 2014;124:1174–82.
Peled JU, Devlin SM, Staffas A, Lumish M, Khanin R, Littmann ER, et al. Intestinal microbiota and relapse after hematopoietic-cell transplantation. J Clin Oncol. 2017;35:1650–9.
Iida N, Dzutsev A, Stewart CA, Smith L, Bouladoux N, Weingarten RA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342:967–70.
Schiavoni G, Sistigu A, Valentini M, Mattei F, Sestili P, Spadaro F, et al. Cyclophosphamide synergizes with type I interferons through systemic dendritic cell reactivation and induction of immunogenic tumor apoptosis. Cancer Res. 2011;71:768–78.
Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342:971–6.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–84.
Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–9.
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre M-L, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–8.
Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T-J, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–3.
Wang F, Yin Q, Chen L, Davis MM. Bifidobacterium can mitigate intestinal immunopathology in the context of CTLA-4 blockade. Proc Natl Acad Sci U S A. 2018;115:157–61.
Wang Y, Wiesnoski DH, Helmink BA, Gopalakrishnan V, Choi K, DuPont HL, et al. Fecal microbiota transplantation for refractory immune checkpoint inhibitor-associated colitis. Nat Med. 2018;24:1804–8.
Zimmermann M, Zimmermann-Kogadeeva M, Wegmann R, Goodman AL. Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature. 2019;570:462–7.
Panebianco C, Andriulli A, Pazienza V. Pharmacomicrobiomics: exploiting the drug-microbiota interactions in anticancer therapies. Microbiome. 2018;6:92.
Koppel N, Maini Rekdal V, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science. 2017;356.
Jiang P, Sellers WR, Liu XS. Big data approaches for modeling response and resistance to cancer drugs. Annu Rev Biomed Data Sci. 2018;1:1–27.
Ma W, Mao Q, Xia W, Dong G, Yu C, Jiang F. Gut microbiota shapes the efficiency of cancer therapy. Front Microbiol. 2019;10:1050.
Land M, Hauser L, Jun S-R, Nookaew I, Leuze MR, Ahn T-H, et al. Insights from 20 years of bacterial genome sequencing. Funct Integr Genomics. 2015;15:141–61.
Janda JM, Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol. 2007;45:2761–4.
Fox GE, Wisotzkey JD, Jurtshuk P Jr. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Bacteriol. 1992;42:166–70.
Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell. 2019;177:70–84.
Luo C, Knight R, Siljander H, Knip M, Xavier RJ, Gevers D. ConStrains identifies microbial strains in metagenomic datasets. Nat Biotechnol. 2015;33:1045–52.
Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL. Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun. 2016;469:967–77.
Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, et al. Evaluating the information content of shallow shotgun metagenomics [Internet]. bioRxiv. 2018 [cited 2019 Aug 26]. p. 320986. Available from: https://www.biorxiv.org/content/10.1101/320986v1.
NIH HMP Working Group, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–2323.
Human Microbiome Project Consortium. A framework for human microbiome research. Nature. 2012;486:215–21.
Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Brantley Hall A, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. Nature Research; 2017;550:61-66.
Proctor LM. The human microbiome project in 2011 and beyond. Cell Host Microbe. 2011;10:287–91.
Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science. 2009;326:1694–7.
Hallmaier-Wacker LK, Lueert S, Roos C, Knauf S. The impact of storage buffer, DNA extraction method, and polymerase on microbial analysis. Sci Rep. 2018;8:6292.
Kerckhof F-M, Courtens ENP, Geirnaert A, Hoefman S, Ho A, Vilchez-Vargas R, et al. Optimized cryopreservation of mixed microbial communities for conserved functionality and diversity. PLoS One. 2014;9:e99517.
Gorzelak MA, Gill SK, Tasnim N, Ahmadi-Vand Z, Jay M, Gibson DL. Methods for improving human gut microbiome data by reducing variability through sample processing and storage of stool. PLoS One. 2015;10:e0134802.
Choo JM, Leong LEX, Rogers GB. Sample storage conditions significantly influence faecal microbiome profiles. Sci Rep. 2015;5:16350.
Song SJ, Amir A, Metcalf JL, Amato KR, Xu ZZ, Humphrey G, et al. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSystems. 2016;1:e00021–16.
McKain N, Genc B, Snelling TJ, Wallace RJ. Differential recovery of bacterial and archaeal 16S rRNA genes from ruminal digesta in response to glycerol as cryoprotectant. J Microbiol Methods. 2013;95:381–3.
Gray MA, Pratte ZA, Kellogg CA. Comparison of DNA preservation methods for environmental bacterial community samples. FEMS Microbiol Ecol. 2013;83:468–77.
Al V, et al. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015;16:73.
Rintala A, Pietilä S, Munukka E, Eerola E, Pursiheimo J-P, Laiho A, et al. Gut microbiota analysis results are highly dependent on the 16S rRNA gene target region, whereas the impact of DNA extraction is minor. J Biomol Tech. 2017;28:19–30.
Al H, et al. Multicenter quality assessment of 16S ribosomal DNA-sequencing for microbiome analyses reveals high inter-center variability. Int J Med Microbiol. 2016;306:334–42.
Al H, et al. Comparison of brush and biopsy sampling methods of the ileal pouch for assessment of mucosa-associated microbiota of human subjects. Microbiome. 2014;2:5.
Al H, et al. 16S rRNA gene pyrosequencing of reference and clinical samples and investigation of the temperature stability of microbiome profiles. Microbiome. 2014;2:31.
Al S, et al. Collecting fecal samples for microbiome analyses in epidemiology studies. Cancer Epidemiol Biomark Prev. 2015;25:407–16.
Panek M, Čipčić Paljetak H, Barešić A, Perić M, Matijašić M, Lojkić I, et al. Methodology challenges in studying human gut microbiota - effects of collection, storage, DNA extraction and next generation sequencing technologies. Sci Rep. 2018;8:5143.
Burbach K, Seifert J, Pieper DH, Camarinha-Silva A. Evaluation of DNA extraction kits and phylogenetic diversity of the porcine gastrointestinal tract based on Illumina sequencing of two hypervariable regions. Microbiologyopen. 2016;5:70–82.
Al F, et al. Comparison of self-collected and physician-collected vaginal swabs for microbiome analysis. J Clin Microbiol. 2010;48:1741–8.
Maukonen J, Simões C, Saarela M. The currently used commercial DNA-extraction methods give different results of clostridial and actinobacterial populations derived from human fecal samples. FEMS Microbiol Ecol. 2012;79:697–708.
Fouhy F, Deane J, Rea MC, O’Sullivan Ó, Ross RP, O’Callaghan G, et al. The effects of freezing on faecal microbiota as determined using MiSeq sequencing and culture-based investigations. PLoS One. 2015;10:e0119355.
Tedjo DI, Jonkers DMAE, Savelkoul PH, Masclee AA, van Best N, Pierik MJ, et al. The effect of sampling and storage on the fecal microbiota composition in healthy and diseased subjects. PLoS One 2015;10:e0126685.
Shaw AG, Sim K, Powell E, Cornwell E, Cramer T, McClure ZE, et al. Latitude in sample handling and storage for infant faecal microbiota studies: the elephant in the room? Microbiome. 2016;4:40.
Lewis ZT, Davis JCC, Smilowitz JT, German JB, Lebrilla CB, Mills DA. The impact of freeze-drying infant fecal samples on measures of their bacterial community profiles and milk-derived oligosaccharide content. PeerJ. 2016;4:e1612.
Al C, et al. Storage conditions of intestinal microbiota matter in metagenomic analysis. BMC Microbiol. 2012;12:158.
Al N, et al. High stability of faecal microbiome composition in guanidine thiocyanate solution at room temperature and robustness during colonoscopy. Gut. 2016;65:1574–5.
Al R, et al. Influence of fecal sample storage on bacterial community diversity. Open Microbiol J. 2009;3:40–6.
Bahl MI, Bergström A, Licht TR. Freezing fecal samples prior to DNA extraction affects the Firmicutes to Bacteroidetes ratio determined by downstream quantitative PCR analysis. FEMS Microbiol Lett. 2012;329:193–7.
Lauber CL, Zhou N, Gordon JI, Knight R, Fierer N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol Lett. 2010;307:80–6.
Hale VL, Tan CL, Knight R, Amato KR. Effect of preservation method on spider monkey (Ateles geoffroyi) fecal microbiota over 8 weeks. J Microbiol Methods. 2015;113:16–26.
Ariefdjohan MW, Savaiano DA, Nakatsu CH. Comparison of DNA extraction kits for PCR-DGGE analysis of human intestinal microbial communities from fecal specimens. Nutr J. 2010;9:23.
Lim MY, Song E-J, Kim SH, Lee J, Nam Y-D. Comparison of DNA extraction methods for human gut microbial community profiling. Syst Appl Microbiol. 2018;41:151–7.
Knudsen BE, Bergmark L, Munk P, Lukjancenko O, Priemé A, Aarestrup FM, et al. Impact of sample type and DNA isolation procedure on genomic inference of microbiome composition. mSystems [Internet]. 2016;1. Available from: https://doi.org/10.1128/mSystems.00095-16.
Eriksson P, Mourkas E, González-Acuna D, Olsen B, Ellström P. Evaluation and optimization of microbial DNA extraction from fecal samples of wild Antarctic bird species. Infect Ecol Epidemiol. 2017;7:1386536.
Kashinskaya EN, Andree KB, Simonov EP, Solovyev MM. DNA extraction protocols may influence biodiversity detected in the intestinal microbiome: a case study from wild Prussian carp, Carassius gibelio. FEMS Microbiol Ecol 2017;93. Available from: https://doi.org/10.1093/femsec/fiw240.
Esteves AIS, Amer N, Nguyen M, Thomas T. Sample processing impacts the viability and cultivability of the sponge microbiome. Front Microbiol. 2016;7:499.
Bag S, Saha B, Mehta O, Anbumani D, Kumar N, Dayal M, et al. An improved method for high quality metagenomics DNA extraction from human and environmental samples. Sci Rep. 2016;6:26775.
Santiago A, Panda S, Mengels G, Martinez X, Azpiroz F, Dore J, et al. Processing faecal samples: a step forward for standards in microbial community analysis. BMC Microbiol. 2014;14:112.
Gill C, van de Wijgert JHHM, Blow F, Darby AC. Evaluation of lysis methods for the extraction of bacterial DNA for analysis of the vaginal microbiota. PLoS One. 2016;11:e0163148.
Couch RD, Navarro K, Sikaroodi M, Gillevet P, Forsyth CB, Mutlu E, et al. The approach to sample acquisition and its impact on the derived human fecal microbiome and VOC metabolome. PLoS One. 2013;8:e81163.
Shobar RM, Velineni S, Keshavarzian A, Swanson G, DeMeo MT, Melson JE, et al. The effects of bowel preparation on microbiota-related metrics differ in health and in inflammatory bowel disease and for the mucosal and luminal microbiota compartments. Clin Transl Gastroenterol. 2016;7:e143.
Al K, et al. Heterogeneity of vaginal microbial communities within individuals. J Clin Microbiol. 2009;47:1181–9.
Al G, et al. Direct sampling of cystic fibrosis lungs indicates that DNA-based analyses of upper-airway specimens can misrepresent lung microbiota. Proc Natl Acad Sci U S A. 2012;109:13769–74.
Al G-G, et al. A new method for extracting skin microbes allows metagenomic analysis of whole-deep skin. PLoS One. 2013;8:e74914.
Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, et al. Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol. 2016;34:942–9.
D’Amore R, Ijaz UZ, Schirmer M, Kenny JG, Gregory R, Darby AC, et al. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genomics. 2016;17:55.
Allali I, Arnold JW, Roach J, Cadenas MB, Butz N, Hassan HM, et al. A comparison of sequencing platforms and bioinformatics pipelines for compositional analysis of the gut microbiome. BMC Microbiol. 2017;17:194.
Majaneva M, Hyytiäinen K, Varvio SL, Nagai S, Blomster J. Bioinformatic amplicon read processing strategies strongly affect eukaryotic diversity and the taxonomic composition of communities. PLoS One. 2015;10:e0130035.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Nearing JT, Douglas GM, Comeau AM, Langille MGI. Denoising the denoisers: an independent evaluation of microbiome sequence error-correction methods [Internet]. PeerJ Preprints; 2018 Feb. Report No.: e26566v1. Available from: https://peerj.com/preprints/26566/.
Nilakanta H, Drews KL, Firrell S, Foulkes MA, Jablonski KA. A review of software for analyzing molecular sequences. BMC Res Notes. 2014;7:830.
Kopylova E, Navas-Molina JA, Mercier C, Xu ZZ, Mahé F, He Y, et al. Open-source sequence clustering methods improve the state of the art. mSystems [Internet]. 2016;1. Available from: https://doi.org/10.1128/mSystems.00003-15.
Xue Z, Kable ME, Marco ML. Impact of DNA sequencing and analysis methods on 16S rRNA gene bacterial community analysis in dairy products [Internet]. bioRxiv. 2018 [cited 2018 Jun 11]. p. 305078. Available from: https://www.biorxiv.org/content/early/2018/04/20/305078.
Edgar RC. Accuracy of microbial community diversity estimated by closed- and open-reference OTUs. PeerJ. 2017;5:e3889.
Wang H, Funchain P, Bebek G, Altemus J, Zhang H, Niazi F, et al. Microbiomic differences in tumor and paired-normal tissue in head and neck squamous cell carcinomas. Genome Med. 2017;9:14.
Yang L, Lu X, Nossa CW, Francois F, Peek RM, Pei Z. Inflammation and intestinal metaplasia of the distal esophagus are associated with alterations in the microbiome. Gastroenterology. 2009;137:588–97.
McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8:e53653.
Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog. 2013;5:2.
Gong H-L, Shi Y, Zhou L, Wu C-P, Cao P-Y, Tao L, et al. The composition of microbiome in larynx and the throat biodiversity between laryngeal squamous cell carcinoma patients and control population. PLoS One. 2013;8:e66476.
Seo S-S, Oh HY, Lee J-K, Kong J-S, Lee DO, Kim MK. Combined effect of diet and cervical microbiome on the risk of cervical intraepithelial neoplasia. Clin Nutr. 2016;35:1434–41.
Allali I, Delgado S, Marron PI, Astudillo A, Yeh JJ, Ghazal H, et al. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes. 2015;6:161–72.
Nasrollahzadeh D, Malekzadeh R, Ploner A, Shakeri R, Sotoudeh M, Fahimi S, et al. Variations of gastric corpus microbiota are associated with early esophageal squamous cell carcinoma and squamous dysplasia. Sci Rep. 2015;5:8820.
Guerrero-Preston R, Godoy-Vitorino F, Jedlicka A, Rodríguez-Hilario A, González H, Bondy J, et al. 16S rRNA amplicon sequencing identifies microbiota associated with oral cancer, human papilloma virus infection and surgical treatment. Oncotarget. 2016;7:51320–34.
Wang H, Altemus J, Niazi F, Green H, Calhoun BC, Sturgis C, et al. Breast tissue, oral and urinary microbiomes in breast cancer. Oncotarget. 2017;8:88122–38.
Li TH, Qin Y, Sham PC, Lau KS, Chu K-M, Leung WK. Alterations in gastric microbiota after H. pylori eradication and in different histological stages of gastric carcinogenesis. Sci Rep. 2017;7:44935.
Yoon H, Kim N, Park JH, Kim YS, Lee J, Kim HW, et al. Comparisons of gut microbiota among healthy control, patients with conventional adenoma, sessile serrated adenoma, and colorectal cancer. J Cancer Prev. 2017;22:108–14.
Flemer B, Herlihy M, O’Riordain M, Shanahan F, O’Toole PW. Tumour-associated and non-tumour-associated microbiota: addendum. Gut Microbes. 2018;9:369-73.
Kinross J, Mirnezami R, Alexander J, Brown R, Scott A, Galea D, et al. A prospective analysis of mucosal microbiome-metabonome interactions in colorectal cancer using a combined MAS 1HNMR and metataxonomic strategy. Sci Rep. 2017;7:8979.
Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, Sieling PA, et al. Microbial dysbiosis is associated with human breast cancer. PLoS One. 2014;9:e83744.
Schmidt BL, Kuczynski J, Bhattacharya A, Huey B, Corby PM, Queiroz ELS, et al. Changes in abundance of oral microbiota associated with oral cancer. PLoS One. 2014;9:e98741.
Oh HY, Kim B-S, Seo S-S, Kong J-S, Lee J-K, Park S-Y, et al. The association of uterine cervical microbiota with an increased risk for cervical intraepithelial neoplasia in Korea. Clin Microbiol Infect. 2015;21:674 e1–9.
Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol Syst Biol. 2014;10:766.
Gong H, Shi Y, Zhou X, Wu C, Cao P, Xu C, et al. Microbiota in the throat and risk factors for laryngeal carcinoma. Appl Environ Microbiol Am Soc Microbiol. 2014;80:7356.
Eun CS, Kim BK, Han DS, Kim SY, Kim KM, Choi BY, et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter. 2014;19:407–16.
Gao Z, Guo B, Gao R, Zhu Q, Wu W, Qin H. Probiotics modify human intestinal mucosa-associated microbiota in patients with colorectal cancer. Mol Med Rep. 2015;12:6119–27.
Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A. 2014;111:18321–6.
Jo HJ, Kim J, Kim N, Park JH, Nam RH, Seok Y-J, et al. Analysis of gastric microbiota by pyrosequencing: minor role of bacteria other than Helicobacter pylori in the gastric carcinogenesis. Helicobacter. 2016;21:364–74.
Al-Hebshi NN, Nasher AT, Idris AM, Chen T. Robust species taxonomy assignment algorithm for 16S rRNA NGS reads: application to oral carcinoma samples. J Oral Microbiol. 2015;7:28934.
Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun. 2015;6:8727.
Burns M, Lynch J, Starr T, Knights D, Blekhman R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med. 2015;7:55.
Wang L, Zhou J, Xin Y, Geng C, Tian Z, Yu X, et al. Bacterial overgrowth and diversification of microbiota in gastric cancer. Eur J Gastroenterol Hepatol. 2016;28:261–6.
Chng KR, Chan SH, Ng AHQ, Li C, Jusakul A, Bertrand D, et al. Tissue microbiome profiling identifies an enrichment of specific enteric bacteria in Opisthorchis viverrini associated cholangiocarcinoma. EBioMedicine. 2016;8:195–202.
Lu Y, Chen J, Zheng J, Hu G, Wang J, Huang C, et al. Mucosal adherent bacterial dysbiosis in patients with colorectal adenomas. Sci Rep. 2016;6:26337.
Audirac-Chalifour A, Torres-Poveda K, Bahena-Román M, Téllez-Sosa J, Martínez-Barnetche J, Cortina-Ceballos B, et al. Cervical microbiome and cytokine profile at various stages of cervical cancer: a pilot study. PLoS One. 2016;11:e0153274.
Zhang C, Liu Y, Gao W, Pan Y, Gao Y, Shen J, et al. The direct and indirect association of cervical microbiota with the risk of cervical intraepithelial neoplasia. Cancer Med. 2018;7:2172–9.
Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, et al. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes. 2017;3:34.
Thomas AM, Jesus EC, Lopes A, Aguiar S Jr, Begnami MD, Rocha RM, et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling. Front Cell Infect Microbiol. 2016;6:179.
Zhao H, Chu M, Huang Z, Yang X, Ran S, Hu B, et al. Variations in oral microbiota associated with oral cancer. Sci Rep. 2017;7:11773.
Gao R, Kong C, Huang L, Li H, Qu X, Liu Z, et al. Mucosa-associated microbiota signature in colorectal cancer. Eur J Clin Microbiol Infect Dis. 2017;36:2073-83.
Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012;6:1858–68.
Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743.
Mangul S, Martin LS, Eskin E, Blekhman R. Improving the usability and archival stability of bioinformatics software. Genome Biol. 2019;20:47.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
List M, Ebert P, Albrecht F. Ten simple rules for developing usable software in computational biology. PLoS Comput Biol. 2017;13:e1005265.
Stupple A, Singerman D, Celi LA. The reproducibility crisis in the age of digital medicine. NPJ Digit Med. 2019;2:2.
Yilmaz P, Kottmann R, Field D, Knight R, Cole JR, Amaral-Zettler L, et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat Biotechnol. 2011;29:415–20.
Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 2017;35:725–31.
von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. PLoS Med. 2007;4:e296.
Field N, Cohen T, Struelens MJ, Palm D, Cookson B, Glynn JR, et al. Strengthening the Reporting of Molecular Epidemiology for Infectious Diseases (STROME-ID): an extension of the STROBE statement. Lancet Infect Dis. 2014;14:341–52.
Altman DG, McShane LM, Sauerbrei W, Taube SE. Reporting recommendations for tumor marker prognostic studies (REMARK): explanation and elaboration. PLoS Med. 2012;9:e1001216.
Sauerbrei W, Taube SE, McShane LM, Cavenagh MM, Altman DG. Reporting recommendations for tumor marker prognostic studies (REMARK): an abridged explanation and elaboration. J Natl Cancer Inst. 2018;110:803–11.
Marques FZ, Jama HA, Tsyganov K, Gill PA, Rhys-Jones D, Muralitharan RR, et al. Guidelines for transparency on gut microbiome studies in essential and experimental hypertension. Hypertension. 2019;74:1279–93.
Laukens D, Brinkman BM, Raes J, De Vos M, Vandenabeele P. Heterogeneity of the gut microbiome in mice: guidelines for optimizing experimental design. FEMS Microbiol Rev. 2016;40:117–32.
Raising standards in microbiome research. Nat Microbiol. 2016;1:16112.
Duvallet C, Gibbons SM, Gurry T, Irizarry RA, Alm EJ. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun. 2017;8:1784.
Shah MS, DeSantis TZ, Weinmaier T, McMurdie PJ, Cope JL, Altrichter A, et al. Leveraging sequence-based faecal microbial community survey data to identify a composite biomarker for colorectal cancer. Gut. 2018;67:882–91.
Sze MA, Schloss PD. Leveraging existing 16S rRNA gene surveys to identify reproducible biomarkers in individuals with colorectal tumors. MBio. 2018;9. Available from: https://doi.org/10.1128/mBio.00630-18.
Pevsner-Fischer M, Tuganbaev T, Meijer M, Zhang S-H, Zeng Z-R, Chen M-H, et al. Role of the microbiome in non-gastrointestinal cancers. World J Clin Oncol. 2016;7:200–13.
Peer review information
Kevin Pang was the primary editor of this article and managed its editorial and peer review in collaboration with the rest of the editorial team.
Review history
The review history is available as Additional file 1.
Funding
None applicable.
Author information
Authors and Affiliations
Contributions
MBB and SSM co-wrote the first draft of the manuscript. AMD substantially revised the manuscript and coordinated with SSM to generate the tables. All authors contributed to the writing, revision, and preparation of the review. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Competing interests
The authors affirm that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Review history.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Manzoor, S.S., Doedens, A. & Burns, M.B. The promise and challenge of cancer microbiome research. Genome Biol 21, 131 (2020). https://doi.org/10.1186/s13059-020-02037-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13059-020-02037-9