
Abstract
Alzheimer’s disease (AD) represents one of the most common and debilitating neurodegenerative disorders. By the end of 2040, AD patients might reach 11.2 million in the USA, around 70% higher than 2022, with severe consequences on the society. As now, we still need research to find effective methods to treat AD. Most studies focused on the tau and amyloid hypothesis, but many other factors are likely involved in the pathophysiology of AD. In this review, we summarize scientific evidence dealing with the mechanotransduction players in AD to highlight the most relevant mechano-responsive elements that play a role in AD pathophysiology. We focused on the AD-related role of extracellular matrix (ECM), nuclear lamina, nuclear transport and synaptic activity. The literature supports that ECM alteration causes the lamin A increment in the AD patients, leading to the formation of nuclear blebs and invaginations. Nuclear blebs have consequences on the nuclear pore complexes, impairing nucleo-cytoplasmic transport. This may result in tau hyperphosphorylation and its consequent self-aggregation in tangles, which impairs the neurotransmitters transport. It all exacerbates in synaptic transmission impairment, leading to the characteristic AD patient’s memory loss. Here we related for the first time all the evidence associating the mechanotransduction pathway with neurons. In addition, we highlighted the entire pathway influencing neurodegenerative diseases, paving the way for new research perspectives in the context of AD and related pathologies.
Similar content being viewed by others

Introduction
Americans aged 65 and older suffering from Alzheimer’s disease (AD) are estimated to be about 6.5 million [1]. By the end of 2040, U.S. population with dementia might reach 11.2 million of cases, around 70% higher than 2022 (Fig. 1). AD is a neurodegenerative disorder characterized by progressive cognitive impairment with loss of memory and behavioural difficulties [1]. Well-known pathological markers found in AD patients are extracellular aggregates of beta-amyloid (Aβ) and intracellular hyperphosphorylated tau (hyp-tau) deposits. The former lead to the formation of senile plaques, while the latter aggregates are self-organized structures namely tangles, which impair the tau function as stabilizer for microtubules and alter the motor protein-mediated transport [2]. Recently, mechanotransduction has been related to pathological changes occurring during AD progression. Mechanotransduction is the process that converts mechanical stimuli from the extracellular matrix into biochemical signals inside the cell, with consequences on cell structure, gene expression and physiological functions [3]. The extracellular stimuli are propagated into the nucleus via sequential interactions of integrins, F-actins, the Linker of Nucleoskeleton and Cytoskeleton (LINC) complex and the nuclear lamina (Fig. 2) [4,5,6]. Integrins are heterodimeric transmembrane receptors, which mediate the ECM-cytoskeleton microfilaments connection. The ECM-cytoskeleton link is further strengthened by the presence of other key cytosolic proteins, such as talin and vinculin [7]. The cytoskeleton proximal to the plasma membrane is predominantly composed by F-actin microfilaments and supports the cell structure [7]. F-actin microfilaments are connected to the nuclear lamina by the LINC complex, consisting of nesprins and SUN proteins [5]. Nesprin 1 and nesprin 2 pass through outer nuclear membrane, connecting F-actin to the SUN1, a protein crossing the inner nuclear envelope [4, 5]. Among the SUN protein family, SUN1 was reported to interact with the nuclear lamina [4, 5]. As previously stated, this complex transduction network may have a role in AD.


Force transmission pathway from the ECM to the nuclear lamina. Extracellular matrix interacts with integrins that transduce the mechanical stimuli to the cytosolic F-actin through the plasma membrane. The F-actin microfilaments are connected to the nuclear lamina by the LINC complex, consisting of nesprins and SUN proteins. SUN proteins are directly connected to the nuclear lamina
Even tough specific aspects of mechanotransduction in neurons have already been discussed, to the best of our knowledge, there is no work summarizing the entire pathway from ECM to synapse response. To fill this gap, this review summarizes evidence dealing with this hypothesis and explains how the extracellular matrix affects the nuclear lamina and how this may be associated to the impaired synaptic activity affecting AD neurons. To this end, we firstly analyzed literature highlighting the alteration of single components of the mechanotransduction pathway. In particular, we report the changes in ECM, nuclear lamina, nuclear transport and synaptic activity in AD. We then discuss incisive connections between the single players, defining a hypothetic mechanism of the mechanotransduction-driven disease progression.
Extracellular matrix (ECM)
ECM composition is tissue-dependent and collagens, elastin, proteoglycans and glycosaminoglycans (GAGs) are some of the most characterizing fibrous proteins [9]. ECM has a dynamic structure since its organization undergoes repeated modifications in correlation with aging and pathology progression [9, 10]. The extracellular matrix guarantees the ideal environment for cell support, growth, migration, differentiation and survival [11,12,13]. Focusing on the brain, ECM behaves as a three-dimensional network for many physiological processes, including development regulation, tissue homeostasis and neuronal plasticity. ECM structure occupies around 20% of the brain adult volume [14]. The composition reported by Sethi et al. [15] and Hall et al. [9] showed high percentages of proteoglycans and hyaluronan, while a minor proportion is taken by collagens and fibronectin. Using cortex and cerebellum of 24-month-old mice compared to the 4-month-old mice, hyaluronic acid concentration was altered in aging and neurodegenerative diseases [9, 10, 16]. Furthermore, some proteoglycans such as decorin, chondrotin sulfate and heparin sulfate proteoglycans have some effects on neurofibrillary tangles formation and beta amyloid interactions, the two primary elements characterizing Alzheimer’s disease [17,18,19,20,21]. Changes in ECM composition lead to a modification of the mechanical properties (i.e. shear elasticity (μ)) and may be correlated to aging and AD onset. Nowadays the literature on the AD-related ECM features is still dependent on the techniques used. Hiscox et al. [22] used magnetic resonance elastography (MRE) to study the ECM stiffness of 12 healthy young subjects with an age between 19 and 30 (mean age 25.2 ± 3.0 years), gaining a shear stiffness physiological value in the hippocampal region of 2.89 ± 0.32 kPa. According to MRE technique, the ECM softens during aging, resulting in an annual stiffness decrease rate of ∼0.8% (0.015 kPa per year) (Table 1) [22,23,24,25,26,27,28]. MRE is a non-invasive technique, which combines traditional magnetic resonance imaging with acoustic waves, allowing to evaluate viscoelastic properties of soft tissues [29]. However, its resolution is affected by the long time for acquisition (minutes), enlarging the risk of possible patients’ head movements [30]. In line with this, Kalra et al. [28] combined MRE with diffusion tensor imaging (DTI) to deepen the local and directional dependency of brain tissue stiffness and confirmed the stiffness decrement. An anisotropic approach in the brain stiffness study was applied to find more details about shear stress vector orientation on different planes in space. This study reported a decrease in ECM stiffness, in accordance with the results obtained applying only MRE. In contrast to MRE and DTI results, atomic force microscopy (AFM) and indentation techniques showed stiffness increment of ∼20–150% with aging [31, 32]. Unlike MRE and DTI, AFM is an invasive imaging technique that requires the extraction of the tissue to be tested. Shear elasticity is evaluated using Van der Waals interactions forces between the cantilever tip and the tissue, resulting in the deviation of a laser light pointing to the cantilever. Thanks to the laser light reflection, the machinery is able to quantify the height of the cantilever, obtaining the sample rigidity [33, 34]. Similarly, indentation is an invasive procedure consisting in the measurement of the machine tip penetration area on the sample surface. Qian et al. [35] raised some questions about the homogeneity of the results using indentation methods. In case of brain-like soft biomaterials, indentation methods show values with high deviations due to reasons related to structural architecture and heterogeneity of the tissue: (1) the heterogeneity of the biomaterial could lead to difficulties in the test operation. Indeed, a non-flat tissue, which presents numerous asperities (e.g. the brain), shows inaccurate values; (2) in some tissues, the hypothesis of isotropy used in the analytical models for data analysis could be not accurate and this may have a repercussion on the quality of the experiments outputs; (3) a universal protocol for indentation techniques is lacking, allowing user-related variation of the boundary conditions in the experimental setup; (4) the brain stiffness changes according to the tested regions [36]. In vitro AFM and indentation present a technical limitation regarding the small size of the samples that could lead to an erroneous global stiffness measurement [34]. Moreover, it is relevant to highlight that ECM stiffness increase data were obtained only in experiments performed on mice brain samples. This aspect combined with additional data obtained from mouse and bovine models supported the ECM stiffness decrease with aging, showing a direct correlation between myelin concentration and cerebral elasticity [37, 38]. In particular, Weickenmeier et al. [38] found that in bovine brain white matter an increased percentage of myelin leads to a more stiffen tissue. Indeed, a myelin content of 63% showed a stiffness of 0.5 kPa, while a myelin content of 92% matched with a stiffness of 2.5 kPa (Fig. 3). The same Authors confirmed the correlation between myelin content and stiffness in a following study on human brain [39]. Since it has been reported a reduction in the myelin amount during aging [40, 41], it is reasonable to suppose that stiffness decreases with aging. In literature, there is a consensus about the decrement of AD patients’ ECM stiffness compared with the age-matched healthy patients (Table 2). Further experiments on this topic have been conducted on both mice and human brain tissue using different techniques. MRE and multifrequency magnetic resonance elastography (MMRE) have been performed on living subjects [42,43,44,45,46,47,48], while nanoindentation and AFM have been executed in vitro [49, 50]. As an example of stiffness value, in post-mortem human brain tissue has been reported a decrement in stiffness of ∼23.5% for grey matter and ∼27.9% for white matter [49]. These data suggest that ECM stiffness is region-dependent, as this has been further confirmed by experiments on mice brain and on post-mortem human brain samples [36, 51]. Results obtained using in vitro techniques (i.e. nanoindentation and AFM) on the AD hippocampal region were in accordance with the data obtained with non-invasive procedures (i.e. MRE and MMRE). As already discussed, stiffness decrease may be caused by myelin loss occurring both in aging and in AD progression [37, 38, 40, 41, 50]. In line with this, experimental observations in AD exhibited further myelin loss compared to physiological aging [40, 50].

Correlation of myelin percentages with the stiffness of the white matter of a bovine brain. The plot shows the correlation of white matter stiffness in a bovine brain with myelin percentage of the tissue [38]
Nuclear lamina and AD
The nuclear lamina is a nuclear structure that represents the final component of the force transmission pathway from the extracellular matrix to the nucleus. Indeed, the nuclear lamina consists of a nuclear structure that is sensitive to extracellular matrix changes, provides support and a stress-related shield for the inner nuclear membrane. It is a meshwork composed by four intermediate filament proteins (A, B1, B2, C). Nuclear lamina is localized in the proximity of the nuclear inner membrane and it is connected to peripheral chromatin [52]. It is involved in several cell mechanisms and functions such as DNA replication, nuclear and chromatin organization, cell development and differentiation [52]. In physiological conditions, lamin A/C is highly expressed in stiff tissues, whereas it is almost absent in soft tissues such as the brain [53]. Lamin A/C enrichment, which leads to higher nuclear stiffness, may act as a genome protective agent [53, 54]. As for lamin B1, it is necessary for the nuclear shape maintenance, while lamin B2 is important for the neuronal migration during development [55, 56]. In physiological conditions, nuclear lamina is highly dynamic and sensitive to extracellular matrix variations through the mechanotransduction pathways. Indeed, like the ECM, the nuclear lamina showed significant alterations in terms of quantity and thickness during AD progression. In fact, in AD lamin A/C levels increased causing the nuclear envelope stiffening and altering the spatial arrangement of the nuclear scaffold [57, 58]. On the other hand, the lamin B1 reduction leads to a functional and morphological cell nucleus alteration [4, 57, 59]. These data were collected using ex-vivo mice or human brain samples by different techniques, such as immunohistochemistry, immunofluorescence microscopy and Western blotting (Table 3). These immunological techniques are able to evaluate the level of lamin A, lamin B1 and lamin B2 [60,61,62]. It was found that the levels of lamin A and B2 in neurons of AD subjects increased [63, 64], and this variation led to nuclear envelope stiffening, while lamin B2 modifications seemed not to alter the nuclear lamina localizations [55]. In opposition, the elderly and even more AD patients presented a decrease in lamin B1 percentages, suggesting a contribution to nuclear deformation [4]. Due to the lamins changes, AD has been recently considered a laminopathy [4]. A hypothesis on the mechanism leading to the lamin A increment is about the lamin B1 reduction and has been explored in non-neuronal cell types. In order to generate an upregulation of lamin A, cells activate the LMNA gene, which, in the brain tissue, is usually maintained in its silent form inside the condensed heterochromatin [57, 65,66,67]. Chang et al. [68] demonstrated on breast cancer cells that a decrease in lamin B1 levels may lead to heterochromatin decondensation, causing the relocalization of LMNA gene and enabling its transcription. In support to lamins B1-A correlation, Shimi et al. [69] reported that in HeLa cells an increase in lamin A was possible only by silencing lamin B1 gene. Therefore, the Authors suggested that the nucleus could induce a decrease in LMNB1 gene expression levels in order to unfold heterochromatin and increase the expression of the LMNA gene and thus, in lamin A production. These lamins relationship may be favoured by the two lamins spatial disposition. Nmezi et al. [70] conducted a study on HeLa cells, human and mice fibroblasts highlighting the formation of different lamins microdomains. Lamin B1 meshwork was located at inner nuclear membrane periphery, laying on some lamin A districts enabling a continuous interaction with them. Meanwhile, using stochastic optical reconstruction microscopy, the same Authors found lamin A localized in the nucleoplasm inner region. Although the mechanism leading lamins regulation in neurons has still to be identified, lamin A increase and lamin B1 decrease have been recently considered crucial factors in AD onset [4, 57,58,59]. Indeed, lamins reorganization induces nucleocytoplasmic scaffold alterations [4, 55, 65, 71], forming blebs and invaginations on the nuclear envelope [69, 72]. In particular, Matias et al. [72] reported a correlation between lamin B1 loss and invaginations in in vitro hippocampal astrocytes cultures, while Shimi et al. [69] observed lamin A rich blebs in HeLa cells experiments. Frost et al. [4] analyzed post-mortem human AD brains and showed that 60% of analyzed samples had a threefold increase of the invagination number respect to age-matched control brains.
Nuclear transport and its impairment in AD
The nucleo-cytoplasmic transport of molecules, such as transcription factors and mRNA, is essential for cell survival and function [65, 74,75,76]. The nuclear pore complex (NPC) is a protein-based structure, which connects the inner and outer nuclear membranes, playing a key role in macromolecular transport from nucleoplasm to cytoplasm and vice versa [77]. NPCs are responsible for the correct maintenance of proteostasis, a process regulating the proper transport and distribution of proteins between nucleoplasm and cytoplasm. Based on electron microscopy acquisitions, its structure appears to remind a cylindrical shape with a diameter of ∼30 nm and a length of ∼50 nm [77]. NPC function is defined by the interactions between NPC binding sites and some of its constituent proteins named nucleoporins (NUPs). The NPC role in nuclear-cytoplasmic transport is dependent to the proper NUPs positioning [77, 78]. Two types of nucleo-cytoplasmic transport can be distinguished: (1) small molecules (typically up to ∼5 nm radius) diffusion by passive transport; (2) larger cargoes (> 15 nm) facilitated transport by carrier proteins [79]. Aging and AD have been correlated with the loss or alteration of essential NUPs. For instance, NUP93 is damaged and lost in aging conditions [74], while NUP98 is mislocalized in AD and contributes to tau tangles formation [80]. Interestingly, the loss or dysregulation of essential NUPs has been associated to the reduction in the number of nuclear pore complexes with consequences on the nucleo-cytoplasmic transport [80]. Eftekharzadeh et al. [80] conducted a study on AD human hippocampal sections and a study on AD mice brain samples. Combining immunostaining and electron microscopy, they found a reduction of around 300 NPCs (about 45%) in AD cases respect to the healthy cases. Moreover, nuclear blebs and invaginations occurring in AD are known to disrupt the nucleoskeleton morphology, leading to the occlusion of the NPCs [65]. Taken together, the reduction in NPCs number and the NPCs closure interfere with the nucleo-cytoplasmic molecular transport [76]. In addition, using in situ hybridization with β-actin in the hippocampal region of AD human brain samples, it was reported that protein phosphatase 2A (PP2A) subunit mRNA and β-actin mRNA decreased, revealing that PP2A mRNA could be limited in its transport through the nuclear membrane [75, 81]. Interestingly, the downregulation of PP2A in AD neurons impairs the physiological phosphorylation process of tau, involved in tangles generation and better detailed below [82, 83].
Tau dynamics and aggregation in relation to nuclear pore transport
Alternative splicing of tau leads to the production of six isoforms with a molecular weight ranging from 37 to 46 kDa and N- and C-termini are very close when tau is unbound in the cytoplasm [84]. To stabilize microtubules, tau binds the filaments with the C-terminus region, while the N-terminus remains far from both the tau C-terminus and the microtubule [84]. Through this molecular interaction, tau indirectly regulates the nucleo-cytoplasmic transport, which is strongly influenced by the microtubule stabilization [85, 86]. Indeed, microtubules act as rails for molecular transport and kinesin, which is the motor protein allowing the motion of vesicles rich of neurotransmitters throughout the cell [80, 85, 87]. As stabilizer, tau can influence microtubules dynamics, acting simultaneously with other proteins [88]. In neurons, tau is physiologically localized in the cytoplasm of the axon region [84, 89]. Hochmair et al. [90] conducted an experiment on AD human brain samples, showing tau accumulation localized throughout the whole cell. Tau was observed in cytoplasmic inclusions localized in proximity to the nucleus (∼1.5 μm distance to the nuclear envelope) and in low chromatin density areas of the nucleus, where tau had a spherical geometry. Tau was also observed close to the NPC at the nuclear side, composing a fine irregular layer of small granules in the nucleoplasm. The three different localizations were due to the alteration of tau phosphorylation level [90]. In physiological conditions, this process is balanced by activity and concentration of kinase and phosphatase. The former oversees the adding of a phosphate group to a protein, while the latter can remove a phosphate group [84, 89, 91]. PP2A and glycogen synthase kinase-3β (GSK-3β) are the enzymes responsible for tau dephosphorylation and phosphorylation, respectively [84, 89, 91]. While dephosphorylated tau strongly binds the microtubules guaranteeing their stability, phosphorylated tau shows a lower binding affinity to microtubules, resulting in its delocalization in the cytoplasm [84, 92]. During AD onset, PP2A activity decreases by almost 50%, GSK-3β increases and, thus, phospho-tau increases too [81, 83, 84, 91, 93]. Tau hyperphosphorylation was found to increase the molecular affinity to self-aggregate and to generate intracellular tangles [2, 55, 84, 94]. Studies using surface plasmon resonance measurements showed that hyp-tau interacts with the NPC, leading to NUP98 delocalization in the cytoplasm [80]. NUP98 delocalization contributes to the destabilization of the NPC arrangement and thus to the alteration of nucleo-cytoplasmic trafficking. Moreover, cytoplasmic NUP98 attracts tau molecules, promoting tau tangles agglomeration [80]. In AD, tau tangles are located in microtubules proximity and affect the kinesin-mediated transport (Fig. 4). Indeed, kinesin in proximity of tau agglomerations dissociates instantaneously from microtubules, releasing transported cargoes into the cytoplasm [2]. Stokin et al. demonstrated that βAPP, Aβ precursor, is one of the possible proteins carried by kinesin [95]. The kinesin detachment from microtubules could promote the βAPP proteolysis, leading to Aβ accumulation in the cytoplasm [96, 97]. The proteolysis process could also be influenced by the disruption of tau and βAPP interaction [98].

Tau tangles-driven impairment of vesicle transport on microtubules. Kinesin is the motor protein responsible for vesicles transport along the microtubules. In the presence of tau tangles, kinesin dissociates from microtubules releasing the cargo into the cytoplasm
Alzheimer’s disease from ECM alteration to synapse loss
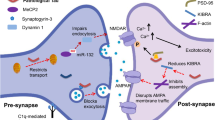
As above stated, AD is one of the most common forms of dementia clinically characterized by memory impairment and underlining neurodegeneration with an increasing trend in prevalence [1]. Effective treatments are recently emerging [99], but they are under further development and in the meanwhile it appears relevant to continue deepening the key elements of AD aetiology and progression. Even though the brain has been considered a static organ for long time, recent studies, showing that the brain is a perfused organ, introduced the relevant role of the ECM-lamina interactions and mechanotransduction in neurodegenerative diseases (Fig. 5, A) [3, 4, 71, 100]. In the context of brain perfusion, the glymphatic system (GS) is rising interest. GS is a physiological mechanism that regulates interstitial flow throughout the cerebral parenchyma [101, 102] and it may transmit the flow-related stimuli to the neural nuclei. The aquaporin-4 (AQP4) is a key water channel in the GS, since it regulates the flow of water in and out the astrocytes that are key components of the GS. AD is characterized by the AQP4 differential expression or localization with consequent interstitial pressure increment on the softened ECM [100, 103,104,105,106,107]. Indeed, MRE-based studies showed the softening of ECM in aging [22,23,24,25,26,27,28] and a further remarkable ECM stiffness decrease in AD [42,43,44,45,46,47,48,49,50]. Considering the high adaptability of the nuclear lamina to external substrates [53], it is reasonable to suppose that ECM changes affect lamins variations. Indeed, lamin B1 concentration was found to decrease while, unexpectedly, lamin A levels had a notable increase [4, 57,58,59] (Table 4). This inverse correlation is also observed in other experiments conducted on HeLa cells and breast cancer cells [4, 57,58,59, 68, 69]. A possible explanation of lamin A increment in response to ECM stiffness decrease may be found by applying extrinsic forces on the HeLa cells and fibroblast cytoskeleton using magnetic tweezers [68,69,70]. When subjected to extrinsic forces, nuclei isolated from their ECM reorganized their lamin-based structure to modulate their stiffness. This highlights that mechanotransduction involves both the ECM and the nucleus, even if the latter keeps a plastic adaptive response when isolated [57, 106]. Even though more studies are required to verify this hypothesis (dashed arrow), the increment of pressure induced by GS combined with the decrement of ECM stiffness in AD neurons support the possibility that lamin A level increases in order to protect the neuronal genome [54, 55, 57, 106] (Fig. 5, B). Lamin variation induces nucleocytoplasmic scaffold alterations [4, 55, 65, 71], forming swellings and sinkings on the nuclear envelope called blebs and invaginations, respectively [69, 72] (Fig. 5, C). Nuclear scaffold modifications cause an impaired NPC opening due to their spatial localization on the nuclear membrane. Indeed, undergoing blebs to an imaging technique, a percentage of NPC were not visible on their surfaces [69], as declared in several works [55, 71, 74, 80] (Fig. 5, D). Because of the steric hindrance of some NPCs, some proteins remained entrapped without being able to move from the nucleus to the cytoplasm and vice versa [108] (Fig. 5, E). Indeed, protein tau exchange between nucleoplasm and cytoplasm could be altered with possible consequent cytoplasmic tau accumulation [65, 74, 76] (Fig. 5, F). Further studies will better clarify this aspect (dashed arrow). Hyp-tau also contributes to the NPC closure by promoting the dissociation of NUP98 from NPC and thus inducing their destabilization [80] (Fig. 5, G). Moreover, since blebs formation impairs NPCs, mRNA transport is also locally hindered and proteins translation is reduced [74, 75] (Fig. 5, H). For instance, in AD, this mechanism may involve PP2A protein whose expression is actually reduced [81, 93] (Fig. 5, I). The downregulation of the phosphatase PP2A levels within the AD neurons seems to be coupled with an increase in kinase quantity between the controls and the AD cases [91]. It seems that GSK-3β activity increases and phosphatase quantity decreases when tau phosphorylation is unbalanced [83, 84, 89] (Fig. 5, J). Indeed, in AD protein tau, which remains entrapped in the cytoplasm, undergoes hyperphosphorylation and self-aggregation in structures called tangles [2, 55, 82, 84, 94, 109, 110] (Fig. 5, K). Hyp-tau affects synaptic signalling by impairing neurotransmitter release from the axonal terminal. In the physiological condition, the vesicles containing the neurotransmitters migrate to the pre-synaptic terminal and they fuse with the pre-synaptic membrane. Then the neurotransmitters are released in the synaptic space by exocytosis [111, 112]. In AD, since hyp-tau loses its affinity with microtubules, it partially relocalizes in pre-synaptic terminal proximity [92]. At this point, hyp-tau interacts stably with synaptic vesicles impairing their fusion with the plasma membrane and thus the release of the neurotransmitters [111, 112]. Furthermore, a work by Jiwon Choi et al. [113] on the development of synapses suggested that in AD tau tangles impair neurotransmission not only by affecting neurotransmitter release but also contributing to synapses loss (Fig. 5, L). Meanwhile, cytosolic tangles positioned in proximity of microtubules obstacle kinesin-mediated transport, exacerbating the βAPP intraneuronal release (Fig. 4). βAPP scission generates Aβ40 and Aβ42 peptides [2, 87, 95]. Aβ oligomers in the extracellular environment tend to self-aggregate in extracellular structures named senile plaques or amyloid plaques [114] (Fig. 5, M). It is of particular interest to notice that pathological Aβ42 quantity increment compromises the viability of oligodendrocytes, which are glial cells responsible for myelin production [115, 116]. In line with these data, in AD patients it has been observed a further reduction in myelin levels in comparison with healthy elderly subjects [40, 50]. As previously highlighted, the reduction in myelin levels support the data about the ECM stiffness decrement in AD [37, 38] (Fig. 5, N). AD-related intracellular and extracellular Aβ accumulation has two main effects: α-tubulin polymerization reduction and long-term potentiation decrease. The former leads to microtubules decrement in quantity and length, as α-tubulin is their fundamental component [117]. In fact, in physiological conditions, microtubule positive terminal undergoes repetitive polymerization and depolymerization, characterizing the mature neurons dendritic spines [117,118,119] Changes in cell microenvironment, such as the decrease of α-tubulin polymerization could lead to axonal stretching and thus neurotransmitter axonal transport and neuronal function impairment [118, 120, 121]. Accordingly, dynamic microtubules quantity and length reduction prevent them from polymerizing along the dendrites, obstructing the proper synaptic signal transmission [117] (Fig. 5, O). On the other hand, the increment of Aβ leads to a reduction of long-term potentiation, a continuous excitatory impulse that fortifies synaptic connections [2, 122]. The impairment of long-term potentiation alters hippocampal neurons activity, leading to synapses loss and progressive memory impairment [123] (Fig. 5, P). All combined, the reduction of the network of microtubules, the long-term potentiation and the neurotransmitter release contribute to the damage of the synaptic function and the consequent memory loss, typical features of AD.

The mechanotransduction pathway from ECM to synapses failure in Alzheimer’s disease. Grey arrows “↑” and “↓” indicate increase and decrement of quantities; the continuous arrows refer to data from literature; dashed arrows stand for hypothesis; blue arrows represent the connections from literature; the green arrows are the retroaction effects. A ECM stiffness decreases in Alzheimer’s disease. Stimuli from ECM reach the nuclear lamina by integrins, F-actin, nesprins and SUN proteins. B Under mechanical stimuli, the nucleus requires an increase in lamin A quantity to protect the genome (hypothesis not yet verified). C The increase of lamin A exacerbates in blebs and invaginations formation, leading to the nuclear scaffold deformation and bringing to the nuclear pore complexes closure (D). E It causes the impairment of nucleo-cytoplasmic transport of proteins, resulting in proteostasis interruption. F Tau nuclear-cytoplasmic transport is impaired and remains entrapped in the cytoplasmic compartment, causing a pathological accumulation (hypothesis not yet verified). G Hyp-tau localizes also near NPCs, releasing NUP98 in the cytoplasm and further compromising NPCs function. The release of NUP98, accelerates hyp-tau aggregation. H NPCs closure also causes the damage of mRNA transport, which induces PP2A gene translation impairment, a decrement of phosphatase concentration (I) and an increase in tau hyperphosphorylation (J). K The high quantity of tau protein in the cytoplasm combined with tau hyperphosphorylation leads to tau self-aggregation in tangles. Tau tangles interacts stably with pre-synaptic vesicles, impeding the neurotransmitter release into the synaptic space (L). M When tangles affect the motor protein kinesin-mediated transport, kinesin detaches from microtubules and releases the vesicles containing Aβ precursor, resulting in Aβ precursor accumulation. Aβ accumulation compromises oligodendrocytes viability, hindering their production of myelin and contributing to further ECM softening (N). Furthermore, Aβ accumulation reduces tubulin polymerization, leading to dendritic spines signalling loss (O) and alters the physiological long-term potentiation contributing to the synapses loss and progressive memory impairment (P)
Conclusion
In summary, this review brings evidence on how the mechanotransduction pathway from ECM to the nuclear lamina may be a player in the dynamics of AD molecular markers and vice versa (Fig. 5). In AD context, changes in physio-chemical properties of ECM affect the nuclear envelope by forming nuclear envelope-related blebs and obstructing nuclear pore complexes, leading to PP2A concentration decrease. All these dysfunctional events lead to the hyperphosphorylation of the cytosolic tau and its self-aggregation into tau tangles which impair both pre-synaptic exocytosis and microtubule-mediated transport. Therefore, the neurotransmitter release in the synaptic space and the cytosolic kinesin-mediated transport of βAPP are altered, inducing microtubules reduction in quantity and length and synaptic signal transmission impairment. Overall, the mechanotransduction pathway seems relevant in the AD context that includes the nuclear scaffold deformation and role of AD molecular markers, such as hyp-tau and Aβ. In line with this, specific aspects of mechanotransduction in neurons have been extensively discussed since 1998 (Table 5) but, to the best of our knowledge, there is no work showing the entire pathway from ECM to synapse response. To fill this gap, we have resumed all the scientific evidence supporting the whole mechanotransduction pathway from the extracellular environment to the neurons. We also focused on its implication in AD, paving the way for innovative therapeutic targets (e.g. ECM and the NL) to fight this disorder. Although most of the reported connections have already been described in the literature, the processes that induce the increase in lamin A through mechanotransduction and the correlation between proteostasis interruption and tau protein accumulation have yet to be clearly confirmed (Fig. 5, dashed arrows).
In conclusion, for the first time we have broadly collected current evidence correlating ECM to synapses, identifying nuclear lamina, NPCs and tau-protein as key elements in maintaining the physiological behaviour of neurons.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- AFM:
-
Atomic force microscopy
- AQP4:
-
Aquaporin-4
- Aβ:
-
Beta-amyloid
- DTI:
-
Diffusion tensor imaging
- ECM:
-
Extracellular matrix
- GAGs:
-
Glycosaminoglycans
- GS:
-
Glymphatic system
- GSK-3β:
-
Glycogen synthase kinase-3β
- hyp-tau:
-
Hyperphosphorylated tau
- LINC:
-
Linker of nucleoskeleton and cytoskeleton
- MMRE:
-
Multifrequency magnetic resonance elastography
- MRE:
-
Magnetic resonance elastography
- NPC:
-
Nuclear pore complex
- NUP:
-
Nucleoporin
- PP2A:
-
Protein phosphatase 2A
References
Alzheimer’s Association. 2022 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2022;18(4):700–89. https://doi.org/10.1002/alz.12638.
Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathologica. 2020;140(4):417–47. https://doi.org/10.1007/s00401-020-02196-w. Springer Science and Business Media Deutschland GmbH.
Osmanagic-Myers S, Dechat T, Foisner R. Lamins at the crossroads of mechanosignaling. Genes Dev. 2015;29(3):225–37. https://doi.org/10.1101/gad.255968.114. Cold Spring Harbor Laboratory Press.
Frost B. Alzheimer’s disease: An acquired neurodegenerative laminopathy. Taylor & Francis. 2016https://doi.org/10.1080/19491034.2016.1183859
Wang N, Tytell JD, and Ingber DE. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat Rev Mol Cell Biol. 2009. https://doi.org/10.1038/nrm2594. [Online]. Available: www.nature.com/reviews/molcellbio.
Khilan AA, Al-Maslamani NA, Horn HF. Cell stretchers and the LINC complex in mechanotransduction. Arch Biochem Biophys. 2021;702:108829. https://doi.org/10.1016/J.ABB.2021.108829.
Sun Z, Guo SS, Fässler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215(4):445–56. https://doi.org/10.1083/jcb.201609037. Rockefeller University Press.
Alzheimer’s Association. 2014 Alzheimer’s disease facts and figures. Alzheimers Dement. 2014;10(2). https://doi.org/10.1016/j.jalz.2014.02.001.
Hall CM, Moeendarbary E and Graham Sheridan CK. Mechanobiology of the brain in ageing and Alzheimer’s disease. Wiley Online Library. 2020. https://doi.org/10.1111/ejn.14766.
Reed MJ, Damodarasamy M, Pathan JL, Erickson MA, Banks WA, Vernon RB. The effects of normal aging on regional accumulation of hyaluronan and chondroitin sulfate proteoglycans in the mouse brain. J Histochem Cytochem. 2018;66(10):697–707. https://doi.org/10.1369/0022155418774779.
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27. https://doi.org/10.1016/J.ADDR.2015.11.001.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(123):4195–200. https://doi.org/10.1242/jcs.023820.
Clause KC, Barker TH. Extracellular matrix signaling in morphogenesis and repair. Curr Opin Biotechnol. 2013. https://doi.org/10.1016/j.copbio.2013.04.011.
Nicholson C, Syková E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21(5):207–15. https://doi.org/10.1016/S0166-2236(98)01261-2.
Sethi MK, Zaia J. Extracellular matrix proteomics in schizophrenia and Alzheimer’s disease. Anal Bioanal Chem. 2016. https://doi.org/10.1007/s00216-016-9900-6.
Sun Y, et al. Role of the Extracellular Matrix in Alzheimer’s Disease. Front Aging Neurosci. 2021. https://doi.org/10.3389/fnagi.2021.707466.
Snow AD, Mar H, Nochlin D, Kresse H, and Wight TN. Peripheral Distribution of Dermatan Sulfate Proteoglycans (Decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of alzheimer’s disease. 1992.
Dewitt DA, Silver J, Canning DR and Perry G. Chondroitin sulfate proteoglycans are associated with the lesions of Alzheimer’s disease. 1993. [Online]. Available: https://digitalcommons.liberty.edu/bio_chem_fac_pubs/15
Perry G. et al. Association of Heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s Disease. 1991.
Zhu Y. et al. Heparan sulfate proteoglycans in tauopathy. Biomolecules. 2022;12(12). https://doi.org/10.3390/biom12121792. MDPI.
Snow AD, Cummings JA and Lake T. The unifying hypothesis of Alzheimer’s Disease: Heparan Sulfate Proteoglycans/Glycosaminoglycans are key as first hypothesized over 30 years ago. Front Aging Neurosci. 2021;13. https://doi.org/10.3389/fnagi.2021.710683. Frontiers Media S.A.
Hiscox LV, et al. High-resolution magnetic resonance elastography reveals differences in subcortical gray matter viscoelasticity between young and healthy older adults. Neurobiol Aging. 2018;65:158–67. https://doi.org/10.1016/J.NEUROBIOLAGING.2018.01.010.
Sack I, et al. The impact of aging and gender on brain viscoelasticity. Neuroimage. 2009;46(3):652–7. https://doi.org/10.1016/J.NEUROIMAGE.2009.02.040.
Sack I, Streitberger KJ, Krefting D, Paul F and Braun J. The influence of physiological aging and atrophy on brain viscoelastic properties in humans. PLoS One. 2011;6(9). https://doi.org/10.1371/journal.pone.0023451.
Arani A, et al. Measuring the effects of aging and sex on regional brain stiffness with MR elastography in healthy older adults. Neuroimage. 2015;111:59–64. https://doi.org/10.1016/j.neuroimage.2015.02.016.
Takamura T, et al. Influence of age on global and regional brain stiffness in young and middle-aged adults. J Magn Reson Imaging. 2020;51(3):727–33. https://doi.org/10.1002/jmri.26881.
Delgorio PL, et al. Effect of aging on the viscoelastic properties of hippocampal subfields assessed with high-resolution MR elastography. Cereb Cortex. 2021;31(6):2799–811. https://doi.org/10.1093/cercor/bhaa388.
Kalra P, Raterman B, Mo X, Kolipaka A. Magnetic resonance elastography of brain: comparison between anisotropic and isotropic stiffness and its correlation to age. Magn Reson Med. 2019;82(2):671–9. https://doi.org/10.1002/mrm.27757.
Hiscox LV, Schwarb H, McGarry MDJ, and Johnson CL. Aging brain mechanics: Progress and promise of magnetic resonance elastography. NeuroImage. 2021;232. https://doi.org/10.1016/j.neuroimage.2021.117889. Academic Press Inc.
MacÉ E, Cohen I, Montaldo G, Miles R, Fink M, Tanter M. In vivo mapping of brain elasticity in small animals using shear wave imaging. IEEE Trans Med Imaging. 2011;30(3):550–8. https://doi.org/10.1109/TMI.2010.2079940.
Ryu Y, Iwashita M, Lee W, Uchimura K, and Kosodo Y. A shift in tissue stiffness during hippocampal maturation correlates to the pattern of neurogenesis and composition of the extracellular matrix. Front Aging Neurosci, 2021;13. https://doi.org/10.3389/fnagi.2021.709620.
Antonovaite N, Hulshof LA, Hol EM, Wadman WJ, Iannuzzi D. Viscoelastic mapping of mouse brain tissue: Relation to structure and age. J Mech Behav Biomed Mater. 2021;113:104159. https://doi.org/10.1016/J.JMBBM.2020.104159.
Allison DP, Hinterdorfer P, Han W. Biomolecular force measurements and the atomic force microscope. Curr Opin Biotechnol. 2002;13(1):47–51. https://doi.org/10.1016/S0958-1669(02)00283-5.
Stylianou A, Kontomaris SV, Grant C and Alexandratou E. Atomic force microscopy on biological materials related to pathological conditions. Scanning. 2019;2019. https://doi.org/10.1155/2019/8452851.
Qian L, Zhao H. Nanoindentation of soft biological materials. Micromachines (Basel). 2018. https://doi.org/10.3390/mi9120654.
Dauth S, et al. Extracellular matrix protein expression is brain region dependent. J Compar Neurol. 2016;524(7):1309–36. https://doi.org/10.1002/cne.23965.
Schregel K, et al. Demyelination reduces brain parenchymal stiffness quantified in vivo by magnetic resonance elastography. Proc Natl Acad Sci U S A. 2012;109(17):6650–5. https://doi.org/10.1073/pnas.1200151109.
Weickenmeier J, de Rooij R, Budday S, Steinmann P, Ovaert TC, Kuhl E. Brain stiffness increases with myelin content. Acta Biomater. 2016;42:265–72. https://doi.org/10.1016/j.actbio.2016.07.040.
Budday S, et al. Towards microstructure-informed material models for human brain tissue. Acta Biomater. 2020;104:53–65. https://doi.org/10.1016/j.actbio.2019.12.030.
Bouhrara M, et al. Evidence of demyelination in mild cognitive impairment and dementia using a direct and specific magnetic resonance imaging measure of myelin content. Alzheimer’s Dement. 2018;14(8):998–1004. https://doi.org/10.1016/j.jalz.2018.03.007.
Wang F, et al. Myelin degeneration and diminished myelin renewal contribute to age-related deficits in memory. Nat Neurosci. 2020;23(4):481–6. https://doi.org/10.1038/s41593-020-0588-8.
Murphy MC, et al. Magnetic resonance elastography of the brain in a mouse model of Alzheimer’s disease: Initial results. Magn Reson Imaging. 2012;30(4):535–9. https://doi.org/10.1016/j.mri.2011.12.019.
Munder T, et al. MR elastography detection of early viscoelastic response of the murine hippocampus to amyloid β accumulation and neuronal cell loss due to Alzheimer’s disease. J Magn Reson Imaging. 2018;47(1):105–14. https://doi.org/10.1002/jmri.25741.
Murphy MC, et al. Decreased brain stiffness in Alzheimer’s disease determined by magnetic resonance elastography. J Magn Reson Imaging. 2011;34:494–8. https://doi.org/10.1002/jmri.22707.
Murphy MC, et al. Regional brain stiffness changes across the Alzheimer’s disease spectrum. Neuroimage Clin. 2016;10:283–90. https://doi.org/10.1016/j.nicl.2015.12.007.
ElSheikh M, et al. MR elastography demonstrates unique regional brain stiffness patterns in dementias. Am J Roentgenol. 2017;209(2):403–8. https://doi.org/10.2214/AJR.16.17455.
Hiscox LV. et al. Mechanical property alterations across the cerebral cortex due to Alzheimer’s disease. Brain Commun. 2020;2(1). https://doi.org/10.1093/braincomms/fcz049.
Gerischer LM, et al. Combining viscoelasticity, diffusivity and volume of the hippocampus for the diagnosis of Alzheimer’s disease based on magnetic resonance imaging. Neuroimage Clin. 2018;18:485–93. https://doi.org/10.1016/j.nicl.2017.12.023.
Park K, Lonsberry GE, Gearing M, Levey AI, Desai JP. Viscoelastic properties of human autopsy brain tissues as biomarkers for Alzheimer’s diseases. IEEE Trans Biomed Eng. 2019;66(6):1705–13. https://doi.org/10.1109/TBME.2018.2878555.
Menal MJ. et al. Alzheimer’s disease mutant mice exhibit reduced brain tissue stiffness compared to wild-type mice in both normoxia and following intermittent hypoxia mimicking sleep apnea. Front Neurol. 2018;9. https://doi.org/10.3389/fneur.2018.00001.
Budday S, et al. Mechanical characterization of human brain tissue. Acta Biomater. 2017;48:319–40. https://doi.org/10.1016/j.actbio.2016.10.036.
Gruenbaum Y, et al. The Nuclear Lamina and Its Functions in the Nucleus. Int Rev Cytol. 2003;226:1–62. https://doi.org/10.1016/S0074-7696(03)01001-5.
Swift J. et al. Nuclear Lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science (1979). 2013. https://doi.org/10.1126/science.1240104.
Ovsiannikova NL, Lavrushkina SV, Ivanova AV, Mazina LM, Zhironkina OA, Kireev II. Lamin A as a determinant of mechanical properties of the cell nucleus in health and disease. Biochemistry. 2021;86(10):1288–300. https://doi.org/10.1134/S0006297921100102. Pleiades Journals.
Gil L, Niño SA, Capdeville G, Jiménez-Capdeville ME. Aging and Alzheimer’s disease connection: Nuclear Tau and lamin A. Neurosci Lett. 2021;749:135741. https://doi.org/10.1016/J.NEULET.2021.135741.
Coffinier C. et al. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. PNAS. 2010;107. https://doi.org/10.1073/pnas.0908790107.
Gil L. et al. Perinuclear lamin a and nucleoplasmic lamin B2 characterize two types of hippocampal neurons through Alzheimer’s disease progression. Int J Mol Sci. 2020;21(5). https://doi.org/10.3390/ijms21051841.
Méndez-López I. et al. Hippocampal LMNA gene expression is increased in late-stage alzheimer’s disease. Int J Mol Sci. 2019;20(4). https://doi.org/10.3390/ijms20040878.
Islam I. et al. Regulatory role of cathepsin L in induction of nuclear laminopathy in Alzheimer’s disease. Aging Cell. 2022;21. https://doi.org/10.1111/acel.13531.
Im K, Mareninov S, Fernando M, Diaz P and Yong WH. An Introduction to Performing Immunofluorescence Staining.Springer. 2019https://doi.org/10.1007/978-1-4939-8935-5_26
Magaki S, Hojat SA, Wei B, So A, Yong WH. An introduction to the performance of immunohistochemistry.Springer. 2019.https://doi.org/10.1007/978-1-4939-8935-5_25
Kartheek G and Kodeeswaran P. Western Blot. 2022.
Rober RA, Weber K and Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: a developmental study. 1989. https://doi.org/10.1242/dev.105.2.365.
Takamori Y. et al. Differential expression of nuclear lamin, the major component of nuclear lamina, during neurogenesis in two germinal regions of adult rat brain.Wiley Online Library. 2007.https://doi.org/10.1111/j.1460-9568.2007.05450.x
Frost B, Bardai FH, Feany MB. Lamin dysfunction mediates neurodegeneration in Tauopathies. Curr Biol. 2016;26(1):129–36. https://doi.org/10.1016/j.cub.2015.11.039.
Camozzi D. et al. Diverse lamin-dependent mechanisms interact to control chromatin dynamics Focus on laminopathies. Taylor & Francis. 2014https://doi.org/10.4161/nucl.36289
Morrison O and Thakur J. Molecular complexes at euchromatin, heterochromatin and centromeric chromatin. Int J Mol Sci. 2021;22(13). https://doi.org/10.3390/ijms22136922.
Chang L, et al. Nuclear peripheral chromatin-lamin B1 interaction is required for global integrity of chromatin architecture and dynamics in human cells. Protein Cell. 2022;13(4):258–80. https://doi.org/10.1007/s13238-020-00794-8.
Shimi T, et al. The A- and B-type nuclear lamin networks: Microdomains involved in chromatin organization and transcription. Genes Dev. 2008;22(24):3409–21. https://doi.org/10.1101/gad.1735208.
Nmezi B, et al. Concentric organization of A-and B-type lamins predicts their distinct roles in the spatial organization and stability of the nuclear lamina. PNAS. 2019. https://doi.org/10.1073/pnas.1810070116.
Donnaloja F, Carnevali F, Jacchetti E, and Raimondi MT. Lamin A/C Mechanotransduction in Laminopathies. Cells. 2020;9(5). https://doi.org/10.3390/cells9051306. NLM (Medline).
Matias I. et al. Loss of lamin-B1 and defective nuclear morphology are hallmarks of astrocyte senescence in vitro and in the aging human hippocampus. Aging Cell. 2022;21. https://doi.org/10.1111/acel.13521.
Noguchi A, et al. Decreased Lamin B1 levels affect gene positioning and expression in postmitotic neurons. Neurosci Res. 2021;173:22–33. https://doi.org/10.1016/J.NEURES.2021.05.011.
Hachiya N. et al. Nuclear envelope and nuclear pore complexes in neurodegenerative diseases-new perspectives for therapeutic interventions.Springer. 2020.https://doi.org/10.1007/s12035-020-02168-x/Published
de Magistris P, Lim R, Zilman A, Veenhoff L. The great escape: mRNA export through the nuclear pore complex. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms222111767.
Almendáriz-Palacios C, et al. The nuclear lamina: protein accumulation and disease. Biomedicines. 2020. https://doi.org/10.3390/biomedicines8070188.
Wente SR and Rout MP. The nuclear pore complex and nuclear transport. Cold Spring Harbor Perspectives Cell Biology. 2010. https://doi.org/10.1101/cshperspect.a000562.
Daniel H, Hoelz A. The Structure of the Nuclear Pore Complex (An Update). Annu Rev Biochem. 2019. https://doi.org/10.1146/annurev-biochem-062917-011901.
Paci G, Caria J, Lemke EA. Cargo transport through the nuclear pore complex at a glance. J Cell Sci. 2021. https://doi.org/10.1242/jcs.247874.
Eftekharzadeh B, et al. Tau protein disrupts nucleocytoplasmic transport in alzheimer’s disease. Neuron. 2018;99(5):925-940.e7. https://doi.org/10.1016/j.neuron.2018.07.039.
Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VMY. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol. 2001;168(2):402–12. https://doi.org/10.1006/exnr.2001.7630.
Lauretti E, Praticò D. Alzheimer’s disease: phenotypic approaches using disease models and the targeting of tau protein. Exp Opin Therapeut Targets. 2020;24(4):319–30. https://doi.org/10.1080/14728222.2020.1737012. Taylor and Francis Ltd.
Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. J Biol Chem. 2000;275(8):5535–44. https://doi.org/10.1074/jbc.275.8.5535.
Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665–704. https://doi.org/10.1007/s00401-017-1707-9.
Combs B, Mueller RL, Morfini G, Brady ST and Kanaan NM. Tau and Axonal Transport Misregulation in Tauopathies. 2019. https://doi.org/10.1007/978-981-32-9358-8_7.
Avila J, Lucas JJ, Perez M, Hernandez F. Role of Tau protein in both physiological and pathological conditions. 2004. https://doi.org/10.1152/physrev.00024.2003.-The.
Dietrich KA, Sindelar CV, Brewer PD, Downing KH, Cremo CR, and Rice SE. The kinesin-1 motor protein is regulated by a direct interaction of its head and tail. PNAS. 2008, [Online]. Available: www.pnas.org/cgi/content/full/
Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G. Tau protein modifications and interactions: their role in function and dysfunction. OPEN ACCESS Int J Mol Sci. 2014;15:15. https://doi.org/10.3390/ijms15034671.
Tapia-Rojas C, Cabezas-Opazo F, Deaton CA, Vergara EH, Johnson GVW, Quintanilla RA. It’s all about tau. Progress Neurobiol. 2019;175:54–76. https://doi.org/10.1016/j.pneurobio.2018.12.005. Elsevier Ltd.
Hochmair J. et al. Molecular crowding and RNA synergize to promote phase separation, microtubule interaction, and seeding of Tau condensates. EMBO J. 2022;41(11). https://doi.org/10.15252/embj.2021108882.
Leroy K, Yilmaz Z, Brion JP. Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol. 2007;33(1):43–55. https://doi.org/10.1111/j.1365-2990.2006.00795.x.
Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discovery. 2007;6(6):464–79. https://doi.org/10.1038/nrd2111.
Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer’s disease. Prog Mol Biol Transl Sci. 2012;106:343–79. https://doi.org/10.1016/B978-0-12-396456-4.00012-2. Elsevier B.V.
Mondragón-Rodríguez S, et al. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. Int J Exp Pathol. 2008;89(2):81–90. https://doi.org/10.1111/j.1365-2613.2007.00568.x.
Stokin GB, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307(5713):1279–82. https://doi.org/10.1126/science.1105681.
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J. Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci. 2006. https://doi.org/10.1523/JNEUROSCI.1450-06.2006.
Zheng H and Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegen. 2006;1(1). https://doi.org/10.1186/1750-1326-1-5.
Barbato C, et al. Interaction of Tau with Fe65 links tau to APP. Neurobiol Dis. 2005;18(2):399–408. https://doi.org/10.1016/J.NBD.2004.10.011.
Decourt B, Noorda K, Noorda K, Shi J, Sabbagh MN. Review of advanced drug trials focusing on the reduction of brain beta-amyloid to prevent and treat dementia. J Exper Pharmacol. 2022;14:331–52. https://doi.org/10.2147/JEP.S265626. Dove Medical Press Ltd.
Silva I, Silva J, Ferreira R, and Trigo D. Glymphatic system, AQP4, and their implications in Alzheimer’s disease. Neurol Res Pract. 2021;3(1). https://doi.org/10.1186/s42466-021-00102-7.
Hablitz LM, Nedergaard M. The glymphatic system: A novel component of fundamental neurobiology. J Neurosci. 2021;41(37):7698–711. https://doi.org/10.1523/JNEUROSCI.0619-21.2021. Society for Neuroscience.
Nycz B and Mandera M. The features of the glymphatic system. 2021.
Peng W, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2016;93:215–25. https://doi.org/10.1016/j.nbd.2016.05.015.
Mestre H, et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife. 2018. https://doi.org/10.7554/eLife.40070.
Bloch O, Auguste KI, Manley GT, Verkman AS. Accelerated progression of kaolin-induced hydrocephalus in aquaporin-4-deficient mice. J Cereb Blood Flow Metab. 2006;26:1527–37. https://doi.org/10.1038/sj.jcbfm.9600306.
Guilluy C, et al. Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat Cell Biol. 2014;16(4):376–81. https://doi.org/10.1038/NCB2927.
Tarasoff-Conway JM, et al. Clearance systems in the brain - implications for Alzheimer disease. Nat Rev Neurol. 2015;11(8):457–70. https://doi.org/10.1038/nrneurol.2015.119. Nature Publishing Group.
Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475(7356):324–32. https://doi.org/10.1038/nature10317.
HY Chang, TK Sang and AS Chiang. Untangling the Tauopathy for Alzheimer’s disease and parkinsonism. J Biomed Sci. 2018;25(1). https://doi.org/10.1186/s12929-018-0457-x. BioMed Central Ltd
Bretteville A. et al. Hypothermia-induced hyperphosphorylation: a new model to study tau kinase inhibitors. Sci Rep. 2012;2. https://doi.org/10.1038/srep00480.
Zhou L. et al. Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun. 2017;8. https://doi.org/10.1038/ncomms15295.
Naseri NN, Wang H, Guo J, Sharma M, Luo W. The complexity of tau in Alzheimer’s disease. Neurosci Lett. 2019;705:183–94. https://doi.org/10.1016/J.NEULET.2019.04.022.
Choi BJ, et al. Miniature neurotransmission regulates drosophila synaptic structural maturation. Neuron. 2014;82(3):618–34. https://doi.org/10.1016/j.neuron.2014.03.012.
Gouras GK, Olsson TT, Hansson O. β-amyloid peptides and amyloid plaques in alzheimer’s disease. Neurotherapeutics. 2015;12(1):3–118. https://doi.org/10.1007/s13311-014-0313-y. Springer New York LLC.
Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177(3):1422–35. https://doi.org/10.2353/ajpath.2010.100087.
Roth AD, Ramírez G, Alarcón R, von Bernhardi R. Oligodendrocytes damage in Alzheimer’s disease: Beta amyloid toxicity and inflammation. Biol Res. 2005;38(4):381–7. https://doi.org/10.4067/S0716-97602005000400011.
Pchitskaya EI, Zhemkov VA, Bezprozvanny IB. Dynamic Microtubules in Alzheimer’s Disease: association with Dendritic spine pathology. Biochemistry. 2018;83(9):1068–74. https://doi.org/10.1134/S0006297918090080. Pleiades Journals.
Tian J, et al. A mechanoelectrical coupling model of neurons under stretching. J Mech Behav Biomed Mater. 2019;93:213–21. https://doi.org/10.1016/J.JMBBM.2019.02.007.
Parato J and Bartolini F. The microtubule cytoskeleton at the synapse. Neurosci Lett. 2021;753. https://doi.org/10.1016/j.neulet.2021.135850.
de Rooij R, Kuhl E. Microtubule polymerization and cross-link dynamics explain axonal stiffness and damage. Biophys J. 2018;114(1):201–12. https://doi.org/10.1016/j.bpj.2017.11.010.
de Rooij R, Miller KE, Kuhl E. Modeling molecular mechanisms in the axon. Springer. 2017;59:523–37. https://doi.org/10.1007/s00466-016-1359-y.
Walsh DM. et al. Naturally secreted oligomers of amyloid b protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002. [Online]. Available: www.nature.com
Cobar LF, Yuan L, Tashiro A. Place cells and long-term potentiation in the hippocampus. Neurobiol Learn Mem. 2017;138:206–14. https://doi.org/10.1016/j.nlm.2016.10.010.
Acknowledgements
N/A
Funding
European Research Council project BEACONSANDEGG; G.A. nr 101053122. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the ERC. Neither the European Union nor the granting authority can be held responsible for them.
Author information
Authors and Affiliations
Contributions
M. T. Raimondi, D. Albani supervised the redaction of the article. F. Donnaloja conceptualized the project and contributed to the writing of the article. E. Limonta, C. Mancosu, F. Morandi, L. Boeri contributed in equal parts to the writing of the article. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved the manuscript and gave their consent for submission and publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Donnaloja, F., Limonta, E., Mancosu, C. et al. Unravelling the mechanotransduction pathways in Alzheimer’s disease. J Biol Eng 17, 22 (2023). https://doi.org/10.1186/s13036-023-00336-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13036-023-00336-w