
Abstract
Background
Maintaining the structural and functional integrity of the blood–brain barrier (BBB) is vital for neuronal equilibrium and optimal brain function. Disruptions to BBB performance are implicated in the pathology of neurodegenerative diseases.
Main body
Early indicators of multiple neurodegenerative disorders in humans and animal models include impaired BBB stability, regional cerebral blood flow shortfalls, and vascular inflammation associated with BBB dysfunction. Understanding the cellular and molecular mechanisms of BBB dysfunction in brain disorders is crucial for elucidating the sustenance of neural computations under pathological conditions and for developing treatments for these diseases. This paper initially explores the cellular and molecular definition of the BBB, along with the signaling pathways regulating BBB stability, cerebral blood flow, and vascular inflammation. Subsequently, we review current insights into BBB dynamics in Alzheimer’s disease, Parkinson's disease, amyotrophic lateral sclerosis, and multiple sclerosis. The paper concludes by proposing a unified mechanism whereby BBB dysfunction contributes to neurodegenerative disorders, highlights potential BBB-focused therapeutic strategies and targets, and outlines lessons learned and future research directions.
Conclusions
BBB breakdown significantly impacts the development and progression of neurodegenerative diseases, and unraveling the cellular and molecular mechanisms underlying BBB dysfunction is vital to elucidate how neural computations are sustained under pathological conditions and to devise therapeutic approaches.
Similar content being viewed by others

Background
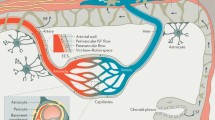
The blood–brain barrier (BBB) serves as a crucial physical barrier separating peripheral blood circulation from the central nervous system (CNS). It safeguards the brain parenchyma by preventing the entry of exogenous pathogens and neurotoxic plasma components. Additionally, the BBB regulates substance transport into and out of the CNS, ensuring the chemical stability of the neuronal milieu and supporting normal neuronal metabolism, which is vital for the optimal functioning of the brain's 86 billion neurons. Impairments to the BBB disrupt the brain's microenvironment homeostasis, ultimately resulting in neuronal injury and functional anomalies, a phenomenon extensively documented in neurodegenerative disease studies [1]. The establishment and preservation of the BBB are intricate cooperative processes, hinging on the synergistic actions among the constituent cells of the neurovascular unit (NVU)—endothelial cells (ECs), pericytes, astrocytes, extracellular matrix, and neurons (Fig. 1). These cells, through complex interactions, guarantee the BBB’s integrity and the normal metabolic state of neurons [2, 3].
To uphold the BBB's integrity and support healthy neuronal metabolism, a continuous and sufficient blood supply is essential for the proper functioning of the brain. Adequate blood supply relies on an efficiently regulated vasculature and the NVU. ECs, pericytes, astrocytes, and neurons, among other cell types, collectively modulate cerebral blood flow (CBF) to cater to the high-energy demands of highly active brain regions [2, 3]. Impaired BBB integrity, such as the loss or degeneration of pericytes, disturbs regional CBF, disrupting the delicate balance required for optimal neural function [4,5,6]. Conversely, abnormal CBF also stresses the BBB, potentially contributing to its dysfunction and exacerbating the challenges faced by the vasculature. This intricate interplay between blood supply and brain health underscores the dual nature of the circulatory system's task: while it serves as a vital conduit for nourishment, it simultaneously manages the influx of potential toxins and mitigates the effects of oxidative stress. The process of nourishing the brain through the peripheral bloodstream inevitably involves the transport of not only essential nutrients but also potentially harmful elements. Simultaneously, the vasculature is confronted with managing oxidative stress products originating from both the peripheral circulation and the metabolic processes within the brain tissue. When confronted with harmful substances such as toxins or byproducts of oxidative stress in the periphery or brain tissue, the vascular system flexibly initiates defensive mechanisms to mitigate potential harm, demonstrating an adaptive response that protects the BBB’s protective function. However, excessive or persistent vascular inflammatory response may cause damage to blood vessels and surrounding tissues, affecting the BBB integrity and thus adversely affecting brain function. This involves immediate responses and refined coordination among various parts of the vasculature, underpinned by intricate signaling pathways and intercellular communication [7, 8]. The BBB's regulation exhibits a high sensitivity and responsiveness to energy requirements and environmental changes. The role of BBB in the pathogenesis of human neurodegenerative disorders highlights the importance of healthy blood vessels [9, 10]. Existing studies on neurodegenerative diseases reveal impairments related to BBB dysfunction, including compromised vascular stability, disrupted CBF, and vascular inflammation. However, in-depth investigations into the pathology of neurodegenerative diseases from the three perspectives, despite being crucial aspects, remain limited. There is an urgent need for a comprehensive review of the mechanisms of the BBB impairment, especially when considering the trio of crucial aspects in the context of various neurodegenerative conditions.
In this review, we initially explore the cellular and molecular mechanisms that underpin the formation of the BBB. Subsequently, our focus shifts to the pivotal pathways that maintain BBB integrity and regulate CBF, as well as vascular inflammation. Further, we investigate the connection between BBB dysfunction and neurodegenerative diseases, predominantly in Alzheimer’s Disease (AD), Parkinson’s Disease (PD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS). Finally, we examine potential BBB-based therapeutic strategies and targets. The review concludes by summarizing key insights and future research directions.
The NVU
EC
Specialized brain ECs construct the primary barrier interface of the BBB, maintaining physiological homeostasis by overseeing transport logistics, regulating vascular permeability, and controlling vascular tone [11]. In contrast to the highly permeable vasculature in peripheral organs, adjacent ECs in the brain are characterized by high trans-endothelial electrical resistance (TEER), low rates of transcytosis, and restricted paracellular permeability, which is primarily governed by junctional molecules [12]. The brain junctional molecules include tight junctions (TJs), adherens junctions (AJs), and gap junctions, which collectively impede the paracellular movement of solutes [13,14,15,16]. TJs, composed of claudins, occludin, junctional adhesion molecules (JAMs), and the zonula occludin (ZO) family, are believed to regulate vascular paracellular transport by forming a high-resistance electrical barrier [17]. Adherens junctions, primarily made up of vascular endothelial (VE)-cadherin, catenin, and platelet endothelial cell adhesion molecule-1 (PECAM-1), are located on the basolateral side of EC TJs [18]. They play a crucial role in trans-endothelial migration of lymphocytes, monocytes, and neutrophils, as well as in cell signaling and transcriptional regulation [19,20,21]. Gap junctions facilitate intercellular communication among ECs through ion currents, second messenger signals, and small metabolites. They are instrumental in vascular angiogenesis and modulating brain endothelial barrier hyperpermeability by influencing TJs. Nonetheless, a more comprehensive understanding of gap junctions concerning the BBB in neurodegenerative diseases is still required.
Transcellular permeability
Given the integrity of cellular structures and junctional proteins between ECs, it is typical for gases and small lipid-soluble molecules with molecular weights under 400 Da to be able to passively diffuse across the BBB [22]. The BBB employs various transport systems, such as carrier-mediated transport (CMT), receptor-mediated transport (RMT), active efflux transport, vesicular trafficking by transcytosis, and ion transport, to facilitate the entry of essential substances into the brain parenchyma and the efflux of metabolic waste and endogenous neurotoxins [19, 23]. The endothelial solute CMT facilitates the crossing of specific solutes across the BBB via substrate-specific transporters, with docosahexaenoic acid (DHA) being transported by major facilitator superfamily domain containing 2a (Mfsd2a) and glucose by glucose transporter 1 (Glut1), ensuring targeted delivery of these vital nutrients to the brain [19, 24,25,26,27]. RMT allows protein movement in and out of the brain, depending on the ligand's binding to specific plasma membrane receptors, such as the interaction between lipoprotein receptors 1 and 2 (LRP1/2) and advanced glycation end products (RAGE) [28, 29]. The ATP-binding cassette (ABC) transporters, including P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), expressed on the luminal endothelial plasma membrane, are pivotal in CNS pharmaco-resistance [24, 30,31,32]. Vesicular trafficking by transcytosis, exemplified by albumin's caveolae-mediated vesicular transport, allows molecules to traverse the BBB based on charged interactions with the ECs' glycocalyx [33]. Ion transporters, such as sodium pumps, calcium transporters, and potassium channels, are essential in regulating neuronal cell electrophysiological activity and maintaining CNS ion concentration gradients [24].
Leukocyte adhesion molecules
As vital constituents of the immune system, ECs act as conduits for migrating immune cells. Notably, ECs display considerably reduced levels of intercellular adhesion molecules (ICAMs) in comparison to ECs in peripheral organs, thereby imposing stringent limitations on immune cell ingress into the brain tissue [34, 35]. The traversal of immune cells from the circulation across the BBB is a meticulously orchestrated dance of molecular engagement between leukocytes and ECs. This intricate process, applicable to monocytes and neutrophils, includes initial capture and rolling along the EC lining, followed by integrin activation, adhesion, crawling, and eventual passage either transcellularly or paracellularly [36].
Under inflammatory cues, ECs instigate the expression of P-selectin, causing incoming immune cells to decelerate and roll. Under the orchestration of integrins and their ligands ICAM and VCAM, these cells firmly anchor to the EC surface, undergo polarization, and crawl in preparation for crossing. They then navigate across the endothelial layer either by traversing the TJs or by forming transient pores in the endothelium without compromising the structural integrity of complex TJs. Importantly, monocytes possess the capability to stimulate ECs directly, which may lead to decreased occludin levels and a heightened influx of immune cells into the inflamed site [27].
The leukocyte adhesion molecules located on the apical surface of ECs play a pivotal role in initiating leukocyte attachment, marking the onset of tissue infiltration. In a healthy state, ECs in the CNS exhibit significantly reduced expression of these molecules compared to their peripheral counterparts, further tightly controlling the infiltration of immune cells entry into the brain tissue [34, 35]. Conversely, in pathological conditions, an upsurge in leukocyte adhesion molecule expression on ECs eases the passage of leukocytes across the BBB, culminating in a local buildup of immune cells and intensifying both inflammatory and immune reactions. This amplification exacerbates harm to neurons [37, 38].
Pericyte
Pericytes occupy a central position in the NVU, situated between ECs, astrocytes, and neurons [39]. Embedded in the basement membrane (BM), they are distinguished by their protruding ovoid cell bodies and elongated, slender processes that partly form an incomplete layer over the surface of CNS capillaries [40, 41]. Pericytes are instrumental in regulating several critical neurovascular functions. These include the formation and permeability of the BBB, angiogenesis, CBF, the clearance of toxic cellular metabolites, vascular remodeling, and tone, all of which are essential for the normal functioning of the CNS [39].
Pericytes, responsive to signals from neighboring cells, generate functional responses essential for CNS integrity [39]. During CNS development, pericyte-EC interactions regulate angiogenesis, vascular stability, and remodeling [42, 43]. Pericytes influence vascular permeability by modulating the expression and organization of intercellular junction proteins and vesicular transport [44]. They regulate capillary tone and diameter, thereby controlling CBF, a topic further elaborated in the subsequent section of this review. In multiple neurological disorders, including AD, ALS, and PD, pericyte loss and degeneration are evident [19, 45, 46]. Pericyte deficiency intensifies amyloid-beta (Aβ) and tau pathology and accelerates neuronal loss in the absence of Aβ-precursor protein, leading to cognitive decline [45]. Moreover, pericytes' enhanced expression of adhesion molecules, which guides and facilitates leukocyte activation and transmigration into the CNS, underscores their role in mediating inflammatory signals within the NVU [47]. Neuronal activity drives insulin-like growth factor 2 expression from pericytes to form long-term memory [48].
Astrocyte
Perivascular astrocytic end-feet cover approximately 70–100% of the vascular perimeter [49]. About a third of astrocyte soma directly contacts blood vessels [50]. As integral components of the NVU, perivascular astrocytes maintain BBB characteristics by contributing to basement membrane formation, regulating pericyte differentiation, and modulating EC functions by regulating the expression of TJs such as claudin-5, occludin, and ZO-1 [49, 51,52,53]. Astrocytes, also serving as pivotal relay cells within the NVU, facilitate communication between neurons and cerebral blood vessels [54]. Beyond neurotransmitter-mediated signaling and pericyte involvement, astrocytes regulate CBF through mechanisms linked to increased intracellular Ca2+ levels [7, 54]. Shifts in understanding CBF control will be elaborated upon in the following section. Additionally, heterogeneous astrocytes distributed throughout the brain exacerbate disease progression and compromise the BBB by releasing inflammatory factors and chemokines [55, 56]. Aquaporin-4 (AQP4), predominantly expressed in astrocytic end-feet, regulates the flux of cerebrospinal fluid, clearance of amyloid-beta, and inflammation [57, 58]. In intramural periarterial drainage, astrocytic end-feet are hypothesized to facilitate waste clearance by modulating functional hyperemia and cerebrovascular tone [59, 60]. AQP4 deficiency in mice leads to severe neuroinflammation, increased microglial activity, and neuronal damage [61].
Basement membrane
The BM encompasses the abluminal surface of ECs and pericytes, delineating these cells from neurons and glial cells [62, 63]. BM components form a meshwork that facilitates the diffusion and binding of molecules and serves as an interface for cell adhesion and migration [64]. Comprising a structural matrix of extracellular matrix molecules, including collagens, laminins, fibronectin, heparan sulfate, and proteoglycans, the BM is synthesized and deposited by ECs, astrocytes, and pericytes [41, 65]. Its composition is tissue-specific and varies in response to different neurological diseases. Emerging evidence from human and animal studies of neurological diseases indicates that BBB disruption is associated with changes in BM component synthesis and degradation [64]. In the CNS, laminin deficiency leads to BBB breakdown, characterized by reduced pericyte coverage, leukocyte extravasation, and altered expressions of VE-cadherin, claudin-5, and occludin, underscoring laminin's role in maintaining BBB integrity [51, 66,67,68]. Collagen IV is essential for BBB structural integrity and function during later developmental stages [69]. Perlecan, a prominent heparan sulfate proteoglycan in BM, is crucial in BBB maintenance and repair, particularly in pericyte interactions following brain injuries like ischemic strokes [70,71,72]. Further research into BM's role in neurological disorders is critically necessary.

The NVU (capillary level). The NVU at the capillary level consists of vascular cells (ECs, pericytes, glial cells) and neurons. ECs, situated on the luminal side of blood vessels, are continuously sealed by intercellular junctional molecules like ZO proteins and claudins. The abluminal surface of ECs is enveloped by a basement membrane that incorporates pericytes and their projections. Astrocytic processes culminate in end-feet encircling the abluminal surface of the capillary vessel wall. The BBB functions to preserve neuronal health by preventing the unregulated influx of peripheral cells (such as red blood cells, leukocytes, and lymphocytes) and plasma proteins (e.g., fibrinogen, IgG). It also maintains low paracellular and transcellular permeability for molecules and ions while facilitating the delivery of essential oxygen and nutrients to the brain. Created with bioRender.com
Pathways regulating the BBB stability
The structural stability of the BBB is a prerequisite for its normal functional performance, and anomalies in its structure may impact its selective permeability, thereby influencing the stability of the brain’s internal environment and the health status of neurons. The stability of the BBB largely depends on its vascular integrity and selective permeability. Interactions among ECs, pericytes, and astrocytes are crucial for vascular maintenance. Interventions targeting the processes of survival, proliferation, migration, differentiation, and permeability of vascular components, as well as the signaling pathways governing these processes, all have significant impacts on the stability of BBB. Disruptions to these may result in structural abnormalities, vessel leakage, and BBB functional impairments, consequently affecting brain functioning. The regulation of BBB permeability by various transporters has been extensively discussed elsewhere [31]. Figure 2 provides a summary of the key cellular and molecular pathways underlying BBB stability.

Major signaling pathways regulating BBB stability. Pericyte-EC communications. PDGF-BB-PDGFRβ pathways regulate various pericyte functions (survival, migration, proliferation, differentiation) by activating phosphoinositide 3-Kinase (PI3K) and Src homology-2 domain-containing protein tyrosine phosphatase-2 (SHP-2). In ECs, TGF-β–TGFβR2 activates different cascades: Alk5-Smad2/3/4 complex boosts differentiation, curbs proliferation, Alk1-Smad1/5/8 complex enhances proliferation; and Alk1-PI3K–Akt pathway fortifies survival and BBB stability. In pericytes, Smad2/3 activation restrains proliferation and migration, promoting differentiation. Notch3 receptor engagement by Notch ligands strengthens BBB stability through Notch-regulated ankyrin repeat protein (Nrarp) or Notch intracellular domain (NICD) pathways when binding to Notch1 or Notch4. Excessive VEGF-A from pericytes and astrocytes weakens BBB by interacting with EC's VEGFR2. Ang1 binding to Tie2 receptors activates PI3K–Akt and inhibits β-catenin, supporting BBB stability, whereas Ang2 antagonizes this effect. Astrocyte-Pericyte/ EC communications. Astrocytes secrete APOE2 and APOE3 (not APOE4), which bind to pericyte LRP1 receptors, inhibiting the CypA–NFκB–MMP-9 pathway and bolstering BBB stability. Moreover, astrocytic Shh binds to PTCH1 receptors on ECs, playing a critical role in BBB stabilization. Astrocyte-derived laminin significantly supports BBB integrity. Neuron-EC communications. Neurons release Wnt, which upon binding to frizzled (FZD) receptors on ECs stimulates their differentiation. Microglia-EC Communications. Microglia release TNF-α and IL-1β, which cumulatively diminish the stability of BBB. Other factors influencing BBB integrity. Additional factors include MFSD2A's role in BBB formation and hyperglycemia-induced pericyte apoptosis via ROS
Platelet-derived growth factor-BB (PDGF-BB), upon binding to its receptor PDGF receptor-β (PDGFRβ), triggers receptor dimerization, autophosphorylation, and activation [39]. The PDGF-BB–PDGFRβ pathway and its downstream signaling mechanisms regulate BBB maintenance by promoting pericyte survival, proliferation, migration, differentiation, adherence to the vascular wall, and leukocyte trafficking [42, 73]. Endothelial-derived impaired PDGFRβ signaling results in microvascular reductions, decreased CBF, and accumulation of neurotoxic molecules from the blood, contributing to secondary neurodegeneration [45, 74]. Endothelium-derived Notch ligands binding to pericyte Notch3 receptors promote pericyte survival. Pericyte-originated Notch ligands attaching to endothelial Notch1/4 receptors enhance N-cadherin expression and regulate PDGF-BB in ECs, reinforcing BBB integrity [39]. The vascular endothelial growth factor-A (VEGF-A)–VEGFR2 signaling cascade is critical for cell survival, angiogenesis, and vascular permeability [42, 75]. However, aberrant activation of this pathway disrupts TJs, causing increased BBB leakage in CNS diseases [76, 77]. The transforming growth factor-β (TGF-β)–TGF-β receptor 2 (TGFβR2) signaling pathway regulates differentiation, maturation, proliferation, migration, and attachment of both ECs and pericytes [78, 79]. The angiopoietin-1 (Ang1)–Tie2 and Ang2–Tie2 signaling pathways, involved in angiogenesis and BBB permeability regulation, have varying effects on EC functions.
Transporters in ECs play a pivotal role in maintaining BBB stability. For example, Mfsd2a, an essential omega-3 fatty acid transporter in ECs vital for BBB formation, maintains BBB integrity by suppressing caveolae-mediated transcytosis [80, 81]. The brain's uptake of glucose is facilitated by Glut1. Significantly, deletion of a single allele encoding Glut1 markedly reduces glucose uptake, leading to BBB disruption, a decline in CBF, and ultimately, neuronal death and loss [27].
Beyond the previously mentioned pathways, perivascular microglia and astrocytes are critical in sustaining BBB stability. These cells release cytokines that interact with the BBB. Proinflammatory cytokines compromise ECs, stimulate the release of ICAM, and enhance the adhesion and infiltration of immune cells. This process amplifies inflammatory responses, contributing to the onset of CNS disorders [82]. Moreover, activated microglia phagocytose astrocytic end-feet, lead to the loss of astrocyte-vascular connections and subsequent BBB disruption [83, 84].
Pathways regulating the CBF
The BBB’s primary role is to regulate the exchange of substances between blood and brain tissues, ensuring that essential nutrients reach neurons while excluding potential toxins and pathogens. CBF delivers the necessary oxygen and nutrients to the brain cells, and its regulation is dynamically adjusted according to the metabolic needs of the neurons. The integrity of the BBB is fundamental to this progress since it affects the vascular response and the overall efficiency of blood delivery. The human brain's blood supply is managed through a vascular network of arteries, arterioles, capillaries, and venules [26]. Given the predominance of capillaries in the brain's vasculature, their significant role in vascular resistance, and the extensive review of CBF control by smooth muscle cells (SMCs) elsewhere [54], this discussion focuses on capillary-based CBF regulation [26]. The NVU upholds BBB integrity and modulates CBF. Disruptions in the NVU, leading to BBB dysfunction and reduced CBF, are early events in various neurodegenerative diseases, including AD, PD, and ALS [85]. Indeed, the cellular components of the NVU play a crucial role in physiologically regulating CBF. This review explores how the brain modulates CBF in response to different information-processing tasks. Figure 3 presents the key cellular and molecular pathways involved in capillary-based CBF regulation.

Major signaling pathways regulating CBF (capillary level). Increased CBF. Neuronal ATP and glutamate activate P2X receptors and metabotropic glutamate receptors (mGluRs) on astrocytes respectively, triggering Ca2+ influx. Elevated cytoplasmic Ca2+ stimulates COX-2 activity, leading to the conversion of AA into prostaglandin E2 (PGE2). PGE2 induces pericyte relaxation by binding to its receptor EP4 (EP4R), causing pericyte hyperpolarization. Similarly, acetylcholine released by neurons and adenosine from neurons and astrocytes target pericytes and ECs through muscarinic receptors and A2ARs respectively, resulting in hyperpolarization and relaxation, thereby increasing CBF. Furthermore, high extracellular K+ from neurons activates VGCCs, causing pericyte depolarization and contraction. Conversely, activation of channels like the inward rectifier potassium channel and KCa or KATP channels leads to K+ efflux and pericyte hyperpolarization, reducing Ca2+ entry through VGCCs. ACh or high extracellular K+ can stimulate KIR or KCa channels on ECs, causing endothelial hyperpolarization that propagates via gap junctions between ECs, enhancing CBF in a retrograde manner. Decreased CBF. Neurogenic ATP or NA binds to P2X or α2A receptors, respectively, on pericytes, resulting in cell depolarization and constriction, thereby reducing CBF. In astrocytes, AA is synthesized and then enters smooth muscle cells and pericytes where it transforms into 20-HETE, leading to pericyte depolarization, contraction, and a subsequent decrease in CBF. In AD models, excess Aβ triggers ROS generation in pericytes, upping ET-1 levels and inducing pericyte contraction and capillary constriction via ET-1 type A receptor (ETAR). Concurrently, EC-released PDGFB and blood-borne IGF1 interact with PDGFRβ and IGF1R, respectively, causing Ca2+ influx and pericyte depolarization. Moreover, brain pericyte AT1 receptors (AT1R) respond to neurogenic angiotensin II, amplifying contractile ability
Neuron-mediated regulation of CBF
Neurons release chemical signals that directly or indirectly influence receptors on pericytes and astrocytes, resulting in either decreased CBF due to depolarization and cell contraction or increased CBF through hyperpolarization and cell relaxation. Earlier studies posited that CBF is contingent on the energy demands of local neuronal activities. Following the generation of synaptic and action potentials, neurons use ATP released by glutamate to restore ion gradients, creating a metabolic signal that elevates CBF to meet energy requirements [86]. Recent research indicates that neuronal ATP and adenosine interact with their respective receptors-P2X and P2Y for ATP, and adenosine A2A receptors (A2AR) for adenosine-leading to pericyte contraction, which reduces CBF, or membrane hyperpolarization, which increases CBF [26, 87,88,89]. Additionally, neuronal noradrenaline (NA) and angiotensin II act on α2-adrenergic receptors (α2A) for NA and AT1 for angiotensin II on pericytes. This action causes local Ca2+ elevations in pericytes, leading to depolarization and reduced CBF [90, 91]. Acetylcholine (ACh) released from neurons activates muscarinic ACh receptors (MRs) on ECs, increasing nitric oxide (NO) production, EC hyperpolarization, and consequently, elevated CBF [92]. NO released by neurons influences pericytes, causing hyperpolarization and exerting vasodilatory effects, further impacting CBF [93].

Vascular inflammation. A Changes in TJs. Immune cells and molecules like LPS, TNF-α, IL-1β, ROS, and MMPs break TJs, while VEGF from astrocytes weakens them via PLCγ/PKCα/eNOS or PI3K/Akt/eNOS pathways when interacting with VEGFR on ECs. B Inflammation affects ECs. ROS disrupts the cytoskeleton, impairs Ca2+ influx, and damages membrane proteins, causing EC dysfunction. LPS harms ECs by inhibiting P-gp, promoting prostaglandin E2 synthesis, and triggering EC apoptosis through the MAPK pathway alongside TNF. Microglia recruited by CCL5-CCR5 signaling phagocytose ECs under chronic inflammation, worsening BBB breakdown. Activated T cells target ECs for apoptosis via the Fas pathway. C Modifications of transport pathways and receptors. Peripheral inflammation significantly impacts various transport pathways. Efflux transporters (e.g., P-gp) decrease, while influx transporters for insulin, monoamines, and lysosomal enzymes increase. Endothelium displays heightened IL-1, IL-6, RAGE, and TNF-α receptors, and reduced LRP, P-gp, and glutamate transporters. Pericytes downregulate LRP but upregulate PDGFβ and TLR. D Activation of Astrocyte and microglia. Astrocytes and microglia react to inflammation differently. Microglia polarize into pro-inflammatory M1 or anti-inflammatory M2 types, affecting synaptic function and neurogenesis. Reactive astrocytes triggered by M1 microglia cytokines lose their regulatory control over microglia, while M2 microglia-secreted cytokines dampen inflammation. Inflammatory cytokines released by microglia induce AQP4 upregulation, causing astrocytic end-feet swelling. E Infiltration of immune cells. Immune cell infiltration follows a multistep process involving capture, rolling, adhesion, crawling, and migration. Once infiltrated, lymphocytes adopt pro- or anti-inflammatory roles based on secreted cytokines
Astrocyte-mediated regulation of CBF
Perivascular astrocytes, located near synapses, are stimulated by neuronal activity, while their end-feet envelop blood vessels and communicate with SMCs and pericytes, which regulate CBF. The strategic positioning of perivascular astrocytes enables their function as relay cells in the NVU [54]. These astrocytes respond to neuronal glutamate via metabotropic glutamate receptors (mGluR), leading to an increase in intracellular Ca2+. This elevation triggers the synthesis of arachidonic acid (AA) in astrocytes. AA then enters SMCs and pericytes and is converted to 20-hydroxy-eicosatetraenoic acid (20-HETE), causing pericyte depolarization and contraction, ultimately reducing CBF [94]. Additionally, elevated extracellular Ca2+ and K+ levels, either from neuronal activities or astrocytic end-feet, can activate Ca2+-gated K+ channels, leading to intracellular K+ efflux. A moderate increase in extracellular K+ concentration (above 10 mM) results in pericyte depolarization and vasoconstriction [8]. AQP4 on astrocytic end-feet relocates towards the soma, causing astrocyte swelling, capillary compression, and decreased CBF [95,96,97]. The deletion of AQP4 has been shown to improve CBF distribution in cases of brain edema, highlighting its significance in CBF dynamics [98].

BBB dysfunction in AD. BBB breakdown in AD is characterized by TJ disruption, pericyte degeneration, and imbalanced Aβ clearance/production, impacting neuronal activity. ECs express receptors (LRP1, RAGE, FCRN) and transporters (P-gp, ABCA1) facilitating Aβ transcytosis across the BBB. Aβ complexes with APOE or IgG for enhanced recognition and clearance, with Aβ-APOE4-VLDLR interaction slowing endocytosis. Reduced Aβ clearance due to downregulated LRP1/P-gp and upregulated RAGE coincides with lower Glut1 expression, suggesting EC clearance impairment. Loss of AQP4 in perivascular astrocytes is associated with Aβ accumulation. Astrocytes, neutrophils, and BACE1 regulate Aβ secretion, while APOE4, ASC, fibrinogen, and Tregs contribute to Aβ plaque formation. Microglia cluster around plaques forming a protective barrier, influenced by fibrinogen, which activates EC IL-8. Infiltrated fibrinogen/IgG trigger microglia/astrocyte proliferation and release of various factors, including APOE, C1q, TNF-α, ROS, NO, and TGFβ, with IL-1β potentially phosphorylating tau. Neutrophils infiltrating the brain cause neurotoxicity via IL-17, NETs, and MPO. Aβ-specific CD4+ T cells exacerbate Aβ accumulation, microgliosis, inflammation, and cognition decline. Hemosiderin deposits from microhemorrhages boost ROS and vascular permeability, attracting inflammatory monocytes to amyloid deposits, and contributing to CAA. Pericyte-derived TNF-α/IL-1β reduces claudin-5, amplifies infiltration of vasoconstrictors Ang-2/EDN1, and constricts pericytes. Infiltration of Ang-2/EDN1, EC-derived Willebrand Factor (VWF), and pericyte loss reduce CBF. APOE4, mainly from astrocytes/pericytes, weakens APOE2/3 binding to LRP1 and activates CypA–MMP9 pathway, exacerbating BBB instability
Pericytes-mediated regulation of CBF
Brain pericytes express contractile and cytoskeletal proteins that can modify CBF by adjusting their contractile tone and diameter [39, 99]. The degree of pericyte loss or degeneration directly correlates with the magnitude of functional changes in the vasculature, underscoring their critical role in preserving cerebral homeostasis. Selective optical ablation of individual capillary pericytes leads to sustained local capillary dilation and a twofold increase in blood cell flux until pericyte contact is regained [5, 4]. Pericyte degeneration reduces both global and individual capillary CBF responses to neuronal and astrocytic stimuli, resulting in abnormal blood perfusion [4, 100]. Pericyte ablation thoroughly leads to acute BBB breakdown and severe loss of CBF [101].

BBB dysfunction in PD. BBB breakdown and dysfunction exist in the basal ganglia of PD patients. In ECs, excessive α-Syn PFFs induce the upregulation of LRP1-ICD, which, in turn, exacerbates αSyn PFF accumulation, and reduces the expression of endothelial TJs such as ZO-1 and occludin, and P-gp. Additionally, endothelial LRP1-ICD increases the expression of protease-activated receptor (PAR), leading to the release of proBDNF, which affects dopaminergic neurons and contributes to early neuronal apoptosis. Enhanced expression of the COL4A2 gene, encoding a subunit of type IV collagen, may alter the morphology and function of the basement membrane. Activated astrocytes secrete VEGFA and NO, causing downregulation of TJs. αSyn PFFs activate pericytes, microglia, and astrocytes, promoting the release of cytokines (IL-1-α, IL-1-β, IL-6, TNF-α, IFN-γ), MMP-9, and ROS, triggering inflammatory reactions and cellular degeneration in ECs, thereby increasing BBB permeability. Upregulated pro-inflammatory chemokines (CCL2, CCL10, CCL20, CXCL2) released by astrocytes attract immune cells from the peripheral circulation into the CNS, exacerbating the inflammatory response. Microglia activated by αSyn PFFs exhibit elevated expression of chemokine receptor CXCR4 and its ligand CXCL12, promoting dopaminergic neuron apoptosis. Furthermore, activated microglia release ROS, leading to pericyte necrosis. The disruption of the BBB allows the entry of plasma fibrinogen, RBCs, and other substances into the brain parenchyma, aggravating pathological processes
The brain capillary pericytes respond to neuronal and astrocytic signals by regulating intracellular Ca2+ and K+, leading to either hyperpolarization or depolarization, thereby influencing CBF. Neuronal transmitters such as ACh and adenosine bind to their respective receptors on pericytes—MRs for ACh and adenosine A2A receptors for adenosine—causing pericyte hyperpolarization and increased CBF. Activation of large-conductance (BKCa) and small-conductance (KCa) K+ channels, as well as ATP-sensitive K+ (KATP) channels, induces K+ efflux, reducing Ca2+ entry through voltage-gated calcium channels (VGCCs), leading to pericyte hyperpolarization and increased CBF [93]. Increases in extracellular K+ mediated by neuronal activities, along with vasoconstrictors such as endothelin 1 (ET1), PDGFB, and blood-derived insulin-like growth factor 1 (IGF1), trigger Ca2+ entry into pericytes through VGCCs, causing depolarization and contraction [7, 93]. Depolarization of pericytes can also result from neuronal transmitters like NA, ATP, and Ang2 binding to their corresponding receptors on pericytes, as well as from astrocytic AA entering pericytes [102, 103]. Excessive Ca2+ entry into the cytosol leads to pericyte calcium overload and contraction, causing microvascular constriction. The resulting death of pericytes leaves the microvasculature continuously constricted, leading to microcirculatory failure and long-term cerebral hypoperfusion due to the contractile apparatus remaining in rigor upon pericyte death, thus reducing capillary diameter [94, 104].

BBB dysfunction in ALS. Hallmark features of ALS include vascular dysfunction and motor neuron degeneration. The BBB breakdown in ALS is characterized by the loss of TJs including ZO-1, claudin-5, and occludin, along with degeneration of pericytes and ECs. This degeneration also involves swelling and detachment of astrocytic end-feet from vessels, elevated levels of BM components such as collagen IV and fibrin deposits, leading to the infiltration of blood-borne cells (red blood cells, neutrophils, monocytes, and mast cells), and plasma-derived proteins (fibrinogen, IgG, and thrombin) into the CNS. In ALS patients, augmented expression and activity of P-gp and BCRP on ECs have been observed. Decreased expression of the potassium channel Kir4.1 and an increased level of AQP4 in astrocytic end-feet are implicated in the swelling of these structures and detachment from the endothelium. Upregulation of TDP-43 in astrocytes promotes microgliosis through the NF-κB pathway. Activated microglia secrete IL-1β, which in turn stimulates astrocytes to release VEGF and pro-inflammatory chemokines including CXCL2, CCL2, and CCL20, resulting in BBB breakdown. Activated microglia contribute to oxidative stress in neurons through the release of ROS, NO, and pro-inflammatory cytokines (TNF-α, IL-6, and IL-1β), leading to motor neuron degeneration. COX-2 and iNOS released by astrocytes surrounding motor neurons, as well as free iron (Fe2+) and subsequent ROS production induced by extravasated RBCs, also contribute to this degeneration. Inflammatory reactive pericytes promote neutrophil transmigration via the release of IL-8 and MMP-9. Astrocytes secrete MCP-1, mediating monocyte migration into the CNS. Additionally, activated microglia release IL-6, CCL5, and TNF-α, which lead to the activation and infiltration of mast cells into the CNS
Endothelium-mediated regulation of CBF
ECs receive signals from the blood and perivascular cells, including neurons, astrocytes, and pericytes, and play a critical role in regulating CBF. When ACh released from neurons or blood activates endothelial MRs, it triggers an increase in NO production in ECs, leading to cell hyperpolarization and relaxation. ECs contain two types of K+ channels: Ca2+-activated K+ channels (KCa) and inward-rectifier K+ channels from the Kir2 family (KIR). KCa channels respond to signals from activated MRs, facilitating K+ influx. On the other hand, KIR channels, activated by elevated external K+ during neural activity, primarily mediate K+ influx without causing cellular hyperpolarization [105]. Instead, they initiate a rapidly propagating retrograde hyperpolarization, transmitting electrical signals to upstream small arteries. This process results in the dilation of these arteries, consequently increasing blood supply to the capillary bed [26, 106].
Vascular inflammation
The vasculature serves as a vital conduit for nourishment, it inevitably involves the transportation of not only essential nutrients but also potentially harmful substances. Concurrently, the vascular system also grapples with managing the byproducts of oxidative stress generated from both the peripheral circulation and metabolic processes within the brain tissue. An instant vascular inflammatory response, though initially protective against such threats, can become detrimental if excessive or chronic. For example, preconditioning with low doses of lipopolysaccharides (LPS), a component of bacteria cell walls known to trigger an immune response, has demonstrated neuroprotective effects including enhancing cellular resilience, supporting neuronal health, and promoting a monocyte phenotype that aids in tissue repair rather than causing damage [27, 107, 108]. The BBB can resist leakage during systemic inflammation, especially at low levels, which is partly attributed to its robust efflux system, which plays a crucial role in maintaining the brain’s protected environment [109, 110]. These observations suggest that mild inflammatory stimuli can activate endogenous defense mechanisms that strengthen the BBB and the overall CNS resilience. However, when inflammation escalates beyond a certain threshold, vascular inflammation can compromise the structure and function of the BBB, playing a pathological role in multiple CNS diseases [110]. The following section will examine how vascular inflammation induced by BBB disruption manifests in various CNS disorders (Fig. 4).
Inflammation adversely affects the BBB and neural transmission by escalating “inflammatory” signals such as cytokines and chemokines, and by mobilizing “inflammatory” cells including macrophages, neutrophils, and lymphocytes. These elements disrupt homeostasis and exert inflammatory effects on ECs. As the primary barrier, ECs are pivotal targets for peripheral inflammatory factors. These factors, including lipopolysaccharide (LPS), tumor necrosis factor-alpha (TNF-α), and interleukin-1 beta (IL-1β), may impede P-gp activity, activate the mitogen-activated protein kinase (MAPK) signaling pathway, and increase nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) expression in ECs. This cascade induces endoplasmic reticulum stress and mitochondrial damage in ECs, leading to apoptosis [111, 112]. Additionally, ECs can respond to IL-1β in the bloodstream by synthesizing and releasing prostaglandin E2, which influences prostaglandin receptors on neurons and glial cells, potentially leading to CNS diseases [113]. Enhanced BBB permeability can be seen as a result of peripheral inflammation, mediated by alterations in the expression of TJs and transporters. “Inflammatory” signals, including IL-1β, IL-6, IL-9, IL-17, interferon-gamma (IFN-γ), TNF-α, and chemokine ligand 2 (CCL2), contribute to diminished TJs expression or misallocation [114,115,116,117,118,119]. Additionally, there are indirect factors influencing TJs, such as matrix metalloproteinases (MMPs), NO, ROS, Rho-associated protein kinase (ROCK), and NF-κB signaling pathways [120,121,122,123,124]. In addition to ECs, pericytes and astrocytes are also activated in response to peripheral inflammation, significantly impacting BBB permeability. These “inflammatory” signals further affect BBB transport systems, which regulate molecular exchanges between the blood and the brain. Specifically, certain efflux transporters like P-gp on astrocytic end-feet, as well as transporters for anions, amino acids, and β-amyloid, are downregulated. Conversely, influx transporters for insulin, monoamines, and lysosomal enzymes undergo upregulation [125]. Additionally, inflammatory signals lead to increased expression of adhesive molecules on ECs, including vascular cell adhesion molecule 1 (VCAM-1), intercellular adhesion molecule 1 (ICAM-1), and E-selectin. This elevation facilitates the entry of peripheral immune cells into the CNS, a process observed in aging and chronic inflammation [126,127,128,129]. Another adverse effect involves the leakage of toxic blood substances such as albumin, fibrinogen, thrombin, and immunoglobulin G (IgG) into the brain. This extravasation alters the neural environment, potentially triggering CNS diseases. For instance, blood-borne fibrin is known to activate neurotoxic innate immune responses in multiple sclerosis mice and to stimulate neurotoxic microglial programs in AD mice [130].
Leaky vascular signals and immune cells serve as potential activators of the innate immune response in CNS diseases. Innate immune cells, including microglia, effectively integrate and rapidly respond to environmental cues. Microglia possess the capability to detect microorganisms, leukocytes, and their metabolites, such as LPS, granulocyte–macrophage colony-stimulating factor (GM-CSF), and can swiftly initiate an immune response by secreting IL-1β, IFN-γ, and TNF-α. These cells are drawn towards blood vessels by the chemokine CCL5, released by ECs, which also plays a role in the production of claudin-5. Activation of glial cells leads to the release of inflammatory cytokines, including IL-1β, IL-6, and TNF-α. This process induces the upregulation of AQP4 and causes swelling in the astrocytic end-feet [131]. Furthermore, IL-1α, TNF-α, and complement component subunit 1q (C1q) encourage the transformation of astrocytes into reactive astrocytes (A1), subsequently impairing the astrocytic capacity to regulate microglial activity [132]. Following this, activated microglial cells adhere to ECs, a process mediated by macrophage-1 antigen (Mac-1). The release of IL-1β and TNF-α can lead to a reduction in the expression of TJs and facilitate the engulfment of ECs by microglial cells [133, 134]. In the later stages of stroke, microglial cells contribute to the remodeling of the ECM by secreting MMPs such as MMP-3, MMP-9, and MMP-19 [135]. The breakdown of the BBB allows infiltrating leukocytes, including monocytes, B cells, T cells, and neutrophils, to be recruited to the CNS [136]. These leukocytes secrete various cytokines that can either exacerbate or mitigate inflammatory responses.
BBB dysfunction in neurodegenerative diseases
AD
In the early stages of AD, there is clear evidence of both disruption and dysfunction within the BBB [26]. Disruption of the BBB including compromised BBB integrity and subsequent leakage of plasma constituents into the brain parenchyma, and impaired BBB dysfunction consisting of compromised transport system, reduced CBF, and heightened vascular inflammatory response at the BBB level, which are closely interlinked with the accumulation of Aβ and the presence of neurofibrillary tangles [137]. Emerging evidence suggests that declines in CBF and vascular dysfunction may precede other established AD biomarkers, including Aβ, amyloid, and tau proteins (Fig. 5) [26, 138].
Impaired BBB integrity
Postmortem studies of AD patients and animal models have revealed cellular infiltration, including red blood cells [139], macrophages [140], and neutrophils [141], as well as capillary leakage of blood-derived components such as fibrin(ogen), IgG, albumin, thrombin, and hemosiderin [142,143,144,145]. These findings indicate compromised integrity of the BBB. Before obvious clinical symptoms appear in AD patients, increased BBB permeability occurs in many brain regions, including the hippocampus, cortex, deep gray matter, and white matter by Dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) [146,147,148,149]. The deterioration of BBB integrity is further correlated with pericyte degeneration and apoptosis, as evidenced by the presence of CSF PDGFRβ, a pericyte marker, and the detachment of PDGFR-β from pericytes [146]. As a significant genetic risk factor for sporadic AD, the Apolipoprotein E (APOE4) allele demonstrates a reduced affinity for LRP1 in pericytes. This reduction activates the Cyclophilin A (CypA)-MMP-9 pathway, which is responsible for the breakdown of the BBB, degeneration of pericytes, and loss of CBF [143, 150,151,152]. Furthermore, APOE4 is associated with a decrease in the basal membrane area and an increase in thrombin concentrations in the vessel walls [144, 153].
Impaired transport system
Impaired glucose metabolism is a prominent characteristic of AD [154, 155]. A reduction in Glut in the ECs of AD patients highlights the compromised function of the BBB [156,157,158]. Patients with mild cognitive impairment (MCI) exhibit decreased glucose absorption [159]. Fluorodeoxyglucose-PET imaging shows characteristic hypo-metabolism in the posterior cingulate cortex and precuneus in the early stages of Alzheimer's disease, with temporoparietal hypo-metabolism manifesting before structural changes [160]. As AD progresses, this diminished uptake of glucose becomes increasingly apparent in regions such as the hippocampus, parietal cortex, cingulate cortex, and temporal cortex [161, 162]. Glut1deficiency suppresses the expression of low-density LRP1 and reduces CBF, suggesting a connection between CBF, Aβ clearance, and glucose metabolism disturbances [163]. While phosphorylated tau (p-tau) does not directly affect Glut1, it triggers widespread neuronal death, leading to a decline in D-glucose transport in p25 mice [164]. Tau-induced activation of glial cells increases the expression of EC adhesion molecules, facilitating leukocyte migration across the BBB [165]. Although a specific transporter for p-tau has yet to be identified, the involvement of tau pathology in BBB dysfunction necessitates further investigation.
Aβ is primarily cleared through transport-mediated processes, notably via the low-density LRP [166]. Apolipoprotein J (APOJ) binds to Aβ, facilitating its transport across the BBB through the LRP2 receptor [167]. P-gp not only exports Aβ from BBB endothelium but also plays a role in clearing it from neurons [168]. Accumulated Aβ peptides can disrupt these clearance processes by reducing P-gp expression and activity [169,170,171]. Additionally, Aβ can be eliminated through interstitial fluid (ISF) flow in perivascular spaces [172]. However, Aβ plaques in these spaces can lead to reduced expression of AQP4 at the astrocytic end-feet, adversely affecting Aβ efflux with the ISF and exacerbating intraparenchymal Aβ deposition [58, 173, 174]. Other factors that can hinder Aβ clearance include extracellular proprotein convertase subtilisin/kexin type 9 (PCSK9) impacting LRP1 on ECs and altered expression of β-site amyloid precursor protein cleaving enzyme 1 (BACE1) [175, 176].
CBF reductions
Individuals and animals with AD exhibit reductions in CBF and impaired cerebrovascular reactivity, particularly in the hippocampus, thalamus, frontal, and occipital cortices, as assessed by arterial spin labeling magnetic resonance imaging [157, 177, 178]. Decreased CBF precedes the detectable changes of classical Aβ and tau biomarkers, as well as brain atrophy, suggesting that reduced CBF could be a valuable biomarker in preclinical AD [138, 179]. Pericyte degeneration, which can diminish capillary CBF responses to neuronal stimulation, has been reported. Furthermore, pericyte degeneration and apoptosis, as evidenced by the detachment of PDGFR-β from pericytes, are induced by accumulated Aβ and impaired Aβ clearance, leading to reduced CBF [45, 74, 146, 180]. In human brain cortex slices from AD patients, excessive Aβ triggers the production of ROS, consequently elevating intracellular ET1 levels. This elevation activates the endothelin-A receptor on pericytes, causing pericyte contraction and capillary constriction, which ultimately leads to decreased CBF [15]. Aβ also stimulates the activation of NADPH oxidases (NOXs), integrin Iib-3, and ROS, promoting platelet adhesion, increasing levels of the endogenous inhibitors plasminogen activator inhibitor-1 and endothelin-1 (ET-1), and resulting in thrombosis, capillary constriction, and impaired blood flow [99, 181, 182]. There is evidence of a negative correlation between tau protein expression and CBF [183]. Blood vessels in AD exhibit structural irregularities, such as twisting and varying diameters, and may develop cerebral amyloid angiopathy (CAA), significantly contributing to BBB disruption, diminished CBF, and cognitive deficits [19, 94, 184,185,186]. Furthermore, chronic overproduction of TGFβ1 from astrocytes and microglia leads to decreased pericyte coverage, facilitating vascular remodeling and suboptimal brain perfusion [187,188,189].
Cerebrovascular reactivity is indicative of the cerebrovascular system's capacity for contraction and dilatation in response to stimuli, such as variations in cerebral perfusion pressure and arterial CO2 partial pressure levels. This reactivity reflects the overall functionality of the cerebrovascular system [190]. The reduction in CBF and the diminished vasodilatory response of cerebral vessels to CO2, due to the accumulation of Aβ, suggest compromised cerebrovascular reactivity examined by transcranial Doppler and the blood oxygenation level-dependent (BOLD) functional MRI (fMRI) [191,192,193]. MRI research has disclosed a diminished CBF response to visual stimulation and during memory encoding processes specifically in the hippocampal region of AD patients [194, 195]. Additionally, pathogenic soluble tau proteins impair endothelium-dependent CBF responses, leading to microvascular deficits. These deficits are mediated by EC senescence and inhibited activation of endothelial nitric oxide synthase (eNOS), which is exacerbated by the internalization of soluble tau [196].
Vascular inflammatory response
The disruption of the BBB can lead to inflammation that exhibits a dual nature. In acute inflammation phases, this disruption may offer protection in microglial cells, but it becomes detrimental during chronic responses [84]. In the early stages of AD, heightened immune responses facilitate the clearance of Aβ [197, 198]. However, prolonged inflammation impairs the microglial cells' ability to bind and phagocytose Aβ, diminishes the activity of Aβ-degrading enzymes, and weakens their capacity to disintegrate Aβ plaques [199, 200]. Peripheral macrophages, attracted to Aβ plaque deposition sites by CCL2, assist in Aβ clearance, but CCL2 simultaneously exacerbates NVU injury [201,202,203]. M1 microglia release cytokines such as CCL2, CXCL10, TNF-α, IL-1β, and IL-6, which lead to a decreased expression of TJs in blood vessels [203,204,205,206]. While TNF-α does not affect P-gp protein levels, it inhibits its efflux transport activity [207, 208].
Extravasated plasma components act as potent stimulators of inflammatory chemokines, triggering the activation of microglia and astrocytes. Conversely, they damage the BBB and exacerbate the pathology [209,210,211,212]. Fibrinogen infiltration, co-localizing with Aβ, promotes IL-8 release by ECs. This alters EC properties through the production of a broad spectrum of proteases and coagulation factors, which disrupt NVU cells and contribute to BBB dysfunction, leading to prolonged inflammation [211, 213,214,215]. The complexities of how BBB dysfunction is linked with extravasated plasma components, including fibrinogen, IgG, albumin, thrombin, and hemosiderin, remain to be fully elucidated [216].
PD
PD is primarily attributed to the degeneration and death of dopaminergic neurons in the substantia nigra pars compacta, accompanied by the accumulation of Lewy bodies and Lewy neurites, predominantly comprising α-synuclein, in degenerating neurons [217]. Vascular risk factors have been identified as accelerating the onset and severity of motor and cognitive impairments in PD [19]. Clinical investigations using DCE-MRI reveal a stark contrast in BBB performance between healthy individuals and patients diagnosed with PD. PD patients exhibit pronounced BBB dysfunction, evidenced by heightened leakage, notably in the substantia nigra and striatum pivotal to the disease's pathology [218, 219]. This BBB dysfunction in PD is linked to the degeneration of dopaminergic neurons, which release alpha-synuclein (αSyn) into the brain parenchyma, exacerbating BBB dysfunction (Fig. 6) [220].
In patients with PD, ECs in the basal ganglia undergo degeneration, accompanied by a reduction in the expression of TJs including occludin, and ZO-1 [221]. Decreased expression of P-gp, which participates in the clearance of toxic substances, has been observed in PD patients’ ECs, with dysfunction linked to membrane LRP1 [222, 223]. In PD mouse models, the intracellular domain of LRP1 (LRP1-ICD), released through γ-secretase and matrix metalloproteinase cleavage, accumulates within cells. This leads to the entry and aggregation of alpha-synuclein-preformed fibrils (αSyn PFFs), resulting in the loss of occludin and decreased P-gp expression [224]. Under the influence of αSyn PFFs, LRP1-ICD interacts with poly (adenosine 5'-diphosphate-ribose) polymerase 1 (PARP1), upregulating PAR expression and promoting the secretion of pro-brain-derived neurotrophic factor (proBDNF) by ECs into dopaminergic neurons. This interaction accelerates the damage caused by αSyn PFFs to both ECs and neurons [225, 226]. As an important mechanism of PD neurogenesis, LRP1-ICD/PARP1 provides a vascular perspective for PD treatment. Pericytes activated by αSyn in PD patients express high levels of IL-1β, IL-6, TNF-α, and MMP-9, exacerbating BBB disruption [227,228,229]. Alpha-synuclein induces activated astrocytes to release VEGF-A and NO, leading to vascular damage and the downregulation of TJs [228, 229]. Elevated expression levels of collagen IV in the BM have been identified in the post-mortem brains of PD patients. Early-stage PD patients and 6-hydroxydopamine (6-OHDA)-treated rodent models exhibit aberrant angiogenesis [230, 231]. This correlates with the significant disruption of the capillary network observed in the substantia nigra [232]. The BBB disruption, indicated by microhemorrhages [26], allows the entry of fibrinogen, plasma proteins, RBC, and other toxic substances into the brain parenchyma, further damaging neurons [233]. Furthermore, MRI studies have revealed microbleeds in both gray and white matter regions of PD patients [234]. Clinical research has demonstrated that a reduction in CBF occurs during the initial phases of the disease, suggesting a connection to αSyn pathology and the aberrant activation of pericytes [27, 235, 236].
The aggregation of αSyn activates microglia and astrocytes, promoting the release of inflammatory mediators and contributing to the degeneration of ECs and neurons. Direct injection of αSyn into the substantia nigra of model rats induces microglial activation, upregulates inflammatory mediators, and increases the expression of chemokine receptor CXCR4 and its ligand CXCL12. This CXCR4-CXCL12 signaling pathway activates Caspase-3, leading to apoptotic neuronal death [237]. Additionally, activated microglia promote pericyte apoptosis through the production of ROS [238]. In the astrocytes of patients with PD, αSyn predominantly accumulates in astrocytic end-feet, particularly within lysosomes. These activated astrocytes secrete numerous inflammatory factors, including TNF-α, IL-1β, and IL-6 [188]. In response to inflammatory mediators, ECs produce pro-inflammatory cytokines and chemokines such as IL-1β, IL-6, CCL2, and CXCL1, and synthesize ICAM-1 and VCAM-1, facilitating the infiltration of immune cells [239].
ALS
ALS is characterized by the degeneration of motor neurons in the spinal cord, brainstem, and brain, progressively weakening voluntary skeletal muscles and ultimately leading to paralysis [240]. Disrupted BBB and the blood-cerebrospinal fluid barrier are evident in both sporadic and familial ALS, occurring before the onset of motor neuron degeneration (Fig. 7). Research involving ALS patients and model mice has revealed EC degeneration, characterized by cellular damage, swelling, and cytoplasmic vacuolization, along with diminished levels of TJs such as ZO-1, claudin-V, and occluding [241]. ALS patients exhibit increased expression of BM components like collagen IV, whereas model mice show a reduction in collagen IV [241, 242]. The BBB disruption is associated with leakage of blood-derived proteins, including IgG, thrombin, hemoglobin, hemosiderin, and fibrinogen into the parenchyma, and increased IgG, albumin, and complement protein C3a levels in the CSF of ALS patients [46, 240]. MRI examinations have uncovered microbleeds in the deep layers of the motor cortex in ALS patients, while the precise mechanisms driving this BBB impairment in ALS remain elusive [243]. Iron (Fe2+) derived from hemoglobin metabolism and immune responses, implicated in early ALS pathogenesis via oxidative stress, contributes to motor neuron damage, exacerbating BBB permeability changes [244, 245].
Significant alterations in the BBB transport systems have been observed in ALS. Increased expression and activity of P-gp in ECs and BCRP in reactive astrocytes are evident in the motor cortex of ALS patients, potentially contributing to multidrug resistance [246]. Notably, a reduction in the potassium channel Kir4.1 and an increase in AQP4 in the astrocyte end-feet of ALS model rats indicate challenges in maintaining water and potassium balance, further compromising BBB stability [247].
Astrocytes surrounding degenerating motor neurons show increased expression of inflammatory markers, notably cyclooxygenase-2 (COX-2) and inducible nitric oxide synthase (iNOS) [248]. TDP-43, accumulating in glial and neuronal cytoplasm, is associated with immune and neuroinflammatory processes [249]. In cultured astrocytes, TDP-43 elevates IL-1β, IL-6, and TNF-α levels, mirroring ALS patient blood profiles and stimulating microglial proliferation via NF-κB activation [250, 251]. Overexpression of TDP-43 in ALS results in the infiltration of immune cells such as CD3+ T cells, CD4+ T cells, and monocytes, along with IgG leakage, activation of EC and pericyte [240, 252, 253]. Activated microglia release IL-1β, which stimulates astrocytes to secrete VEGF and proinflammatory chemokines like CXCL2, CCL2, and CCL20, further exacerbating BBB dysfunction [240]. Additionally, activated microglia contribute to oxidative stress through the release of ROS, NO, and pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, inducing motor neuron death [254].
Peripheral immune cells, initially activated within the peripheral immune system, subsequently transition to the CNS, facilitating the proinflammatory milieu together with the CNS resident immune cells [255]. Beyond the astrocytic activation by TDP-43, pericytes, astrocytes, and microglia regulate the infiltration of peripheral immune cells. Inflammatory reactive pericytes promote neutrophil transmigration through the release of IL-8 and MMP-9 [256]. Astrocytes secrete monocyte chemoattractant protein-1 (MCP-1), mediating monocyte migration and amplifying neuroinflammation [257]. Activated microglia release IL-6, CCL5, and TNF-α contributing to the activation and infiltration of mast cells [258]. Interactions between resident immune cells in the CNS and peripheral immune cells through immune molecules render inflammation systemic, ultimately leading to the dysfunction of neuromuscular junctions, damage to motor neuron axons, and motor neuron death [255].
In conclusion, impairment of the BBB in ALS involves reduced TJs, degeneration of pericytes and ECs, infiltration of peripheral immune cells, and accumulation of plasma-derived toxic proteins. These changes often precede and exacerbate ALS symptoms.
MS
Multiple sclerosis is an autoimmune, chronic inflammatory disease of the CNS, hallmarked by demyelination and inflammation due to immune cell invasion into the brain parenchyma via a disrupted BBB [259]. Characteristic hyperintense lesions within white matter, discernible on T2-weighted MRI scans, signify core pathological markers of MS [260]. Gadolinium-enhanced neuroimaging has illuminated that BBB dysfunction is a foundational event in MS pathogenesis, occurring early in the disease process [261]. Intriguingly, MRI evidence of BBB disruption has been noted in seemingly normal white matter even before the emergence of enhancing lesions, as well as in non-enhancing regions, implying that BBB impairment precedes both clinical symptoms and other MRI-evident changes in the disease course.
Indicative markers of BBB breakdown, including elevated albumin quotient (Qalb), increased MMP-9 activity in CSF and serum, and a higher CSF leukocyte count, have been documented across various stages of MS [19]. Inflammation is a constant feature across all MS stages, with immune cell-mediated inflammation being particularly pronounced during the initial acute phases. The infiltration of leukocytes across the BBB necessitates intricate molecular interactions between these cells and ECs [19]. T and B lymphocytes, along with monocytes, are activated in the peripheral compartment before infiltrating the CNS [19, 259]. Recent research employing radiolabeled MMP PET has revealed that heightened MMP activity is localized around active lesions, which are linked to leukocyte infiltration [262]. The transmigration of immune cells across the BBB involves interactions with ECs, changes in endothelial transcellular and paracellular transport, and morphological alterations of the immune cells. Upon entry into the parenchyma, these cells initiate a secondary immune response, releasing inflammatory mediators such as cytokines, chemokines, proteases, and toxic substances, aggravating myelin and axonal damage [263]. Resident glial cells become activated, aiding in antigen presentation to T helper cells. Macrophages, upon activation by Th1 cells, release various cytokines and inflammatory molecules, including TNF-α, proteases, NO, and oxygen-free radicals, causing myelin damage [264]. Th17 and Th1 cells influence astrocyte functions, downregulating neurotrophic factors and upregulating inflammatory cytokines and chemokines [265]. Furthermore, TH17 cells impede the maturation and survival of oligodendrocytes (OLs), as well as induce their apoptosis [266, 267]. CD8+ cytotoxic T lymphocytes, activated by antigens presented by oligodendrocytes and neurons, are implicated in demyelination and axonal/neuronal damage [268]. Lymphocytes activate ECs and affect BBB permeability through cytokines like IL-6, IL-17, IL-22, and IFN-γ [266]. Pericytes secrete inflammatory factors and physically detach from ECs, thereby creating a local inflammatory milieu that facilitates the recruitment of peripheral immune cells across the BBB [269]. In the early stages of experimental autoimmune encephalomyelitis (EAE), astrocytes within demyelinating lesions become activated before leukocyte infiltration. They release various factors including TNF-α, IL-1β, IL-6, glutamate, and NO, contributing to oligodendrocyte injury and axonal degeneration. IL-17-mediated signaling in astrocytes promotes the secretion of MMP-3 and MMP-9, further compromising BBB integrity. Activated microglia inflict damage on oligodendrocytes through the release of pro-inflammatory cytokines such as IL-1, IL-6, TNF-α, and interferons, alongside phagocytic activity, and antigen presentation to CD4+ T cells via MHC class II [270]. Furthermore, neurons are directly impaired by reactive oxygen and nitrogen species (ROS/NRs) and inflammatory cytokines [270].
MRI studies have revealed global cerebral hypoperfusion at all stages of MS, suggesting a link to hypoxic conditions [271]. Imaging studies using SPECT, PET, and ASL have shown reduced CBF in MS patients across all major subtypes, without reaching ischemic severity [271, 272]. The mechanisms behind this hypoperfusion may involve astrocyte energy metabolism compromise due to lacking β2-adrenergic receptors, and increased vasoconstriction from elevated ET-1 [273,274,275]. Excess NO production and associated cellular desensitization, along with vascular inflammation and structural changes, also contribute to the complex vasculature-related pathology of MS [276, 277]. Oligodendrocytes are notably susceptible to hypoxia [278], and the oxidative stress resulting from chronic hypoxia is a critical mechanism in neurodegeneration and axonal loss in multiple sclerosis. Extravasation of plasma albumin, occurring before cellular inflammation and clinical symptoms, contributes to damage in myelin and oligodendrocytes [279]. Albumin triggers inflammatory responses through astrocytic and microglial expression of IL-1β, CXCL3, and NO. Additionally, albumin’s interaction with TGF-β receptors on astrocytes affects calcium concentrations [280] and leads to the downregulation of Kir4.1. Albumin uptake in neurons heightens NMDA receptor excitability, provoking glutamate excitotoxicity that exacerbates myelin and oligodendrocyte injury, culminating in neuronal apoptosis [281, 282]. Glutamate accumulation, caused by release from activated microglia and lymphocytes and impaired clearance by resident cells like astrocytes, further exacerbates neuronal damage [283]. In the CNS, zinc released from nerve endings activates the MAPK pathway, inducing neuronal apoptosis, MMP-9-dependent BBB disruption, and microglial activation, which in turn releases proinflammatory cytokines, ultimately damaging the myelin sheath [284].
Mechanisms affecting BBB to neurodegeneration
Neurodegenerative diseases such as AD, PD, ALS, and MS, while distinct in clinical presentation, share a convergence on impaired BBB stability, CBF reduction, and vascular inflammation, reflecting parallel paths of progressive neurodegeneration distinct from acute brain injuries.
Impaired BBB stability
DCE-MRI has exposed increased regional BBB permeability in the cortex and hippocampus in AD, basal ganglia in PD, and white matter in MS. Increased BBB permeability suggests loss of BBB integrity evidenced by EC degeneration, TJs decrease, and pericyte deterioration and detachment from ECs. Enhanced permeability of the BBB and subsequent leakage of blood contents exacerbate BBB disruption, fueling a vicious cycle detrimental to the imbalance of the CNS, ultimately contributing to neuronal degeneration, and driving neurodegenerative disorders pathophysiology.
CBF reduction
ASL-MRI studies indicated region-specific decreased CBF in neurodegenerative disorders like AD, PD, and MS. Decreased CBF observed in the early stages of AD, non-demented PD patients, ALS cases before cognitive decline, and in normally appearing white matter of MS patients without cognitive impairments, implies that CBF reduction may serve as an early biomarker, preceding neurodegenerative changes. Perfusion abnormalities hinder the clearance of metabolic byproducts, compromising cerebrovascular health, and enhancing vulnerability to disease initiation. Moreover, CBF reduction directly damages the cerebrovascular system, as evidenced by the widespread degeneration, detachment, and loss of pericytes, phenomena that are consistently observed in regions experiencing reduced CBF in various neurodegenerative diseases [94].
Vascular inflammation
A ubiquitous feature among neurodegenerative diseases is vascular inflammation, triggered by advancing pathology, BBB compromise, and oxygen deficiency. The vasculature is persistently challenged by blood-borne toxins, leukocytes, and metabolic waste from both the circulatory and the brain. When the integrity of the BBB is breached, infiltrating blood components and immune cells activate perivascular glia, igniting an inflammatory cascade that accelerates neuronal damage. The disease-specific mechanisms stimulate atypical glial cell activity, releasing a cascade of inflammatory mediators that target blood vessels, further deteriorating the BBB. In later stages, heightened central nervous system inflammation fuels disease progression and exacerbates BBB dysfunction, with heightened levels of enzymes like MMP-9 exacerbating this breakdown.
The distinctive clinical manifestations and singular pathophysiological mechanisms underpinning AD, PD, ALS, and MS engender unique profiles of BBB dysfunction tailored to each disorder. In AD, the excessive accumulation of Aβ overwhelms clearance mechanisms, leading to an imbalance implicated in BBB impairment. The APOE ε4 allele, a significant genetic risk factor for sporadic AD, contributes to BBB dysfunction and exacerbates AD pathogenesis through the APOE4-CypA-MMP9 pathway. In PD, disruptions to the BBB facilitate the spread of αSyn within the brain, while αSyn aggregates reciprocally worsen BBB damage by instigating immune reactions or compromising BBB integrity, perpetuating a detrimental feedback loop that accelerates PD's pathology. The progression of ALS is driven by a complex interplay among astrocyte abnormalities, chronic neuroinflammation, and TDP-43 aggregation, forming a unique web of interconnected pathologies. Astrocyte dysfunction, beyond disrupting water and potassium homeostasis through altered AQP4 and Kir4.1 expression, amplifies neuroinflammation around degenerating motor neurons, exacerbating their deterioration. MS is marked by persistent, chronic inflammation throughout its progression, characterized by immune cell attacks on the myelin sheath. Leukocytes, T lymphocytes, B lymphocytes, and macrophages infiltrate the CNS via a compromised BBB, targeting myelin and perpetuating the cycle of inflammation and demyelination.
Therapeutic implications and future directions
Currently, the available evidence demonstrates that region-specific BBB dysfunction including reduced CBF (shortfalls and dysregulations), impaired BBB integrity (increased BBB permeability and transport system), and vascular inflammation (extravasated plasma components and peri-vascular inflammatory responses) contributes to the onset and progression of multiple neurodegenerative disorders. Undoubtedly, treatment for BBB disruption shows great potential for neurodegenerative diseases by regulating the pathological processes [24]. The subsequent sections elucidate methods for restoring the structural and functional BBB.
Normalizing barrier function
Various pharmacological agents have shown efficacy in protecting against neurodegenerative diseases by enhancing BBB integrity. Activated Protein C (APC), which cleaves protease-activated receptor 1 in brain endothelium and targets Rac1 GTPase, strengthens the BBB via the β-arrestin-2-dependent biased signaling pathway [285]. APC has also demonstrated therapeutic potential in models of AD, ALS, and multiple sclerosis by reducing BBB breakdown, inflammatory responses, and neuronal damage [286,287,288,289,290]. In murine models, cyclosporine, functioning as a CypA inhibitor, diminishes the CypA-NF-κB-MMP-9 pathway associated with BBB degradation and neurodegenerative changes. This results in the restoration of BBB integrity and the reversal of neuronal dysfunction and degeneration [291].
BBB breakdown is often accompanied by the loss of TJs. Several pharmacological agents targeting TJs have demonstrated their efficacy in enhancing BBB functions. 10-O-(N, N-dimethylaminoethyl) ginkgolide B methanesulfonate (XQ-1H) has been shown to normalize the expression of claudin-5, occludin, ZO-1, and β-catenin in ischemic stroke model mice, leading to reduced BBB permeability and protection against oxygen–glucose deprivation/reoxygenation injuries [292]. In intracerebral hemorrhage mouse models, the normalization of tight junction proteins ZO-1 and occludin by fibroblasts has been observed to enhance BBB integrity [293]. Additionally, emerging research on gut-derived microbial metabolites and induced pluripotent stem cells (iPSCs) offers new therapeutic directions. For instance, mouse models lacking gut microbiota exhibit diminished expression of occludin and claudin-5, along with increased BBB permeability [294]. Mesenchymal cell transplantation has also shown promise in vascular regeneration therapy and BBB repair [295, 296]. Importantly, methods for drug delivery involving transient disruption of TJs are emerging as potential treatment strategies.
Normalizing clearance function
The BBB serves as a crucial clearance site for various brain-originated toxic substances within the CNS, particularly for the removal of Aβ in AD and α-synuclein in PD. Low-density LRP1 on ECs facilitates the trans-endothelial clearance of these neurotoxins [297]. Enhanced Aβ clearance, achieved through the delivery of the LRP1 minigene to the BBB via viral vectors, has been shown to reduce the accumulation of Aβ in the brain parenchyma and alleviate Aβ pathology [19]. Additionally, α-synuclein, found in Lewy bodies of both Lewy body dementia and PD, can also be cleared from the brain via LRP1-mediated transcytosis [298]. Animal studies have revealed that allopregnanolone promotes the clearance of Aβ and cholesterol [299]. Inhibiting RAGE may suppress Aβ accumulation in the brain parenchyma, and small molecule RAGE inhibitors are currently advancing to clinical trials [19, 300]. Thus, enhancing the BBB's ability to eliminate pathogenic factors that accumulate in the CNS holds considerable promise for improving outcomes in neurodegenerative diseases.
Targeting vascular inflammation
A crucial approach for treating vascular inflammation involves controlling pro-inflammatory cytokines, MMPs, and infiltrating leukocytes [301]. Glucocorticoids, long regarded as the frontline therapy for inflammatory conditions, bolster BBB integrity by enhancing TJs and suppressing MMPs and inflammation [302]. Resolvin D (RvD) has demonstrated promise in reducing inflammation by decreasing leukocyte-EC interactions [303]. Furthermore, astrocytes and microglia are gaining recognition as pivotal therapeutic targets in neuroinflammation. Regulating astrocyte reactivity through metabolic pathways has proven effective. For instance, glucosylceramide synthase inhibitors can mitigate neuroinflammation by interrupting immunometabolic pathways in pathogenic astrocytes [304]. Minocycline acts by inhibiting MMP-9 and the p38 mitogen-activated protein kinase signaling pathways, thereby reducing the production of glutamate, IL-1β, and NO by microglia [305]. Despite these advances, it is imperative to recognize the limitations encountered in clinical trials of anti-inflammatory medications aimed at vascular inflammation. High doses of glucocorticoids have adverse outcomes in stroke interventions, as they impair EC barrier function [306]. Similarly, endeavors in clinical research to mend the BBB through attenuation of brain inflammation or the targeting of cerebral Aβ in AD patients have, to date, proven unsuccessful [307]. These setbacks may stem from the swift degeneration of neurons, supportive glial cells, and cerebral vasculature post-trauma, complicating the management of inflammation in injured brains [308]. Navigating the landscape of vascular inflammation therapy is fraught with obstacles, yet it concurrently harbors untapped molecular pathways brimming with potential.
Recent advancements in research have greatly enhanced our comprehension of BBB dysfunction and furnished us with strategies aiming at restoring the structural and functional BBB in neurodegenerative diseases. Nonetheless, opportunities and challenges remain regarding the BBB in the context of diseases. First, how can we identify the degree of BBB breakdown and its intricate connection to neurodegenerative diseases within a clinical setting? The bulk of clinical evidence about BBB breakdown is derived from postmortem human brain tissue analyses, which inherently limits our ability to assess real-time dysfunction or track disease progression [27, 309]. Moreover, there is an inadequate availability of preclinical models effectively exhibiting BBB dysfunction characteristics akin to those observed in neurodegenerative conditions [12, 310]. This deficiency poses an obstacle to monitoring or discerning such dysfunction as a measurable outcome in vivo studies concerning pathology. Besides, the precise correlation between varied functional alterations in distinct brain regions affected by different neurodegenerative disorders and the corresponding modifications in brain vasculature and CBF within those areas remains elusive and not clearly defined. As for the research of inflammatory response, how significant is the role of the BBB in mediating the effects of systemic inflammation on neurological disease progression needs more evidence to demonstrate. Despite there are quantity of BBB-targeted treatment strategies based on animal models, the situation is disappointing when used in clinical trials for the limited rate of success. The specificity and effectiveness of related treatment strategies should be strengthened in the subsequent research.
Conclusion
The recognition of BBB dysfunction as a common feature in multiple neurodegenerative disorders has underscored the importance of maintaining healthy blood vessels for optimal neural function. Understanding the specific manifestations and shared mechanisms of BBB breakdown in prevalent neurodegenerative diseases opens new research and therapeutic opportunities. This review provides a comprehensive overview of the interplay between vascular stability, CBF regulation, and vascular inflammation in the context of the BBB, with a particular emphasis on the dysfunction of these mechanisms in the pathophysiology of neurodegenerative diseases such as AD, PD, ALS, and MS. It underscores the critical role these factors play in disease progression and highlights the importance of understanding their complex interactions for the development of novel therapeutic strategies aimed at preserving BBB integrity and mitigating neurodegeneration. Recent therapeutic strategies targeting BBB deterioration and related neurodegenerative conditions focus on enhancing drug delivery to the CNS and restoring BBB integrity. However, current research on the BBB and methods for repairing it are predominantly limited to animal models. Future studies are needed to elucidate the molecular and cellular aspects of human brain vasculature in health and disease. The application of single-cell RNA sequencing and advancements in neuroimaging, such as PET imaging and MRI, are invaluable for understanding the molecular and cellular underpinnings of BBB dysfunction. These approaches also aid in identifying novel vascular biomarkers and are essential in developing effective therapies for various neurological diseases.
Availability of data and materials
Not applicable.
Abbreviations
- BBB:
-
Blood–brain barrier
- CNS:
-
Central nervous system
- NVU:
-
Neurovascular unit
- ECs:
-
Endothelial cells
- CBF:
-
Cerebrovascular blood flow
- TJs:
-
Tight junctions
- AJs:
-
Adherens junctions
- JAMs:
-
Junctional adhesion molecules
- ZO:
-
Zonula occludin
- VE-cadherin:
-
Vascular endothelial cadherin
- PECAM-1:
-
Platelet endothelial cell adhesion molecule-1
- CMT:
-
Carrier-mediated transport
- RMT:
-
Receptor-mediated transport
- Mfsd2a:
-
Major facilitator superfamily domain containing 2a
- Glut1:
-
Glucose carrier 1
- LRP1:
-
Lipoprotein receptor 1
- LRP2:
-
Lipoprotein receptor 2
- RAGE:
-
Advanced glycation end products
- ABC transporters:
-
ATP-binding cassette transporters
- P-gp:
-
P-glycoprotein
- BCRP:
-
Breast cancer resistance protein
- ICAMs:
-
Intercellular adhesion molecules
- Aβ:
-
Amyloid-beta
- AQP4:
-
Aquaporin-4
- PDGF-BB:
-
Platelet-derived growth factor-BB
- PDGFRβ:
-
PDGF receptor-β
- VEGF-A:
-
Vascular endothelial growth factor-A
- VEGFR2:
-
VEGF receptor 2
- TGF-β:
-
Transforming growth factor-β
- TGFβR2:
-
TGF-β receptor 2
- Ang1:
-
Angiopoietin-1
- Ang2:
-
Angiopoietin-2
- SMCs:
-
Smooth muscle cells
- A2AR:
-
Adenosine A2A receptors
- NA:
-
Noradrenaline
- α2A:
-
α2-Adrenergic receptors
- ACh:
-
Acetylcholine
- MRs:
-
Muscarinic ACh receptors
- NO:
-
Nitric oxide
- mGluR:
-
Metabotropic glutamate receptors
- AA:
-
Arachidonic acid
- 20-HETE:
-
20-Hydroxy-eicosatetraenoic acid
- VGCCs:
-
Voltage-gated calcium channels
- ET1:
-
Endothelin 1
- IGF1:
-
Insulin-like growth factor 1
- MRs:
-
Muscarinic ACh receptors
- LPS:
-
Lipopolysaccharide
- TNF-α:
-
Tumor necrosis factor-alpha
- IL-1β:
-
Interleukin-1 beta
- MAPK:
-
Mitogen-activated protein kinase
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IFN-γ:
-
Interferon-gamma
- CCLs:
-
Chemokine ligands
- MMPs:
-
Matrix metalloproteinases
- ROCK:
-
Rho-associated protein kinase
- VCAM-1:
-
Vascular cell adhesion molecule 1
- IgG:
-
Immunoglobulin G
- GM-CSF:
-
Granulocyte–macrophage colony-stimulating factor
- C1q:
-
Component subunit 1q
- A1:
-
Reactive astrocytes
- Mac-1:
-
Macrophage-1 antigen
- APOE4:
-
Apolipoprotein E
- CypA:
-
Cyclophilin A
- MCI:
-
Mild cognitive impairment
- p-tau:
-
Phosphorylated tau
- APOJ:
-
Apolipoprotein J
- ISF:
-
Interstitial fluid
- PCSK9:
-
Proprotein convertase subtilisin/kexin type 9
- BACE1:
-
β-Site amyloid precursor protein cleaving enzyme 1
- NOXs:
-
NADPH oxidases
- CAA:
-
Cerebral amyloid angiopathy
- eNOS:
-
Endothelial nitric oxide synthase
- LRP1-ICD:
-
Intracellular domain of LRP1
- αSyn:
-
Alpha-synuclein preformed fibrils
- PARP1:
-
Poly (adenosine 5′-diphosphate-ribose) polymerase 1
- proBDNF:
-
Pro-brain-derived neurotrophic factor
- 6-OHDA:
-
6-Hydroxydopamine
- COX-2:
-
Cyclooxygenase-2
- iNOS:
-
Inducible nitric oxide synthase
- MCP-1:
-
Chemoattractant protein-1
- OLs:
-
Oligodendrocytes
- EAE:
-
Experimental autoimmune encephalomyelitis
- ROS:
-
Reactive oxygen and nitrogen species
- NRs:
-
NMDA receptors
- APC:
-
Activated Protein C
- XQ-1H:
-
10-O-(N, N-dimethylaminoethyl) ginkgolide B methanesulfonate
- iPSCs:
-
Induced pluripotent stem cells
- RvD:
-
Resolvin D
References
Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50.
Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96(1):17–42.
Vicario N, Parenti R. Connexins signatures of the neurovascular unit and their physio-pathological functions. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23179510.
Hartmann DA, Berthiaume AA, Grant RI, Harrill SA, Koski T, Tieu T, et al. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat Neurosci. 2021;24(5):633–45.
Berthiaume AA, Grant RI, McDowell KP, Underly RG, Hartmann DA, Levy M, et al. Dynamic remodeling of pericytes maintains capillary coverage in the adult mouse brain. Cell Rep. 2018;22(1):8–16.
Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22(7):1089–98.
Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18(7):419–34.
Sato Y, Falcone-Juengert J, Tominaga T, Su H, Liu J. Remodeling of the neurovascular unit following cerebral ischemia and hemorrhage. Cells. 2022. https://doi.org/10.3390/cells11182823.
Ahmad A, Patel V, Xiao J, Khan MM. The role of neurovascular system in neurodegenerative diseases. Mol Neurobiol. 2020;57(11):4373–93.
Xingi E, Koutsoudaki PN, Thanou I, Phan MS, Margariti M, Scheller A, et al. LPS-induced systemic inflammation affects the dynamic interactions of astrocytes and microglia with the vasculature of the mouse brain cortex. Cells. 2023. https://doi.org/10.3390/cells12101418.
Sturtzel C. Endothelial cells. Adv Exp Med Biol. 2017;1003:71–91.
Lochhead JJ, Yang JZ, Ronaldson PT, Davis TP. Structure, function, and regulation of the blood-brain barrier tight junction in central nervous system disorders. Front Physiol. 2020. https://doi.org/10.3389/fphys.2020.00914.
Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol. 1969;40(3):648–77.
Westergaard E, Brightman MW. Transport of proteins across normal cerebral arterioles. J Comp Neurol. 1973;152(1):17–44.
Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47–62.
Tietz S, Engelhardt B. Brain barriers: crosstalk between complex tight junctions and adherens junctions. J Cell Biol. 2015;209(4):493–506.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214(11):3151–69.
Zhao Y, Gan L, Ren L, Lin Y, Ma C, Lin X. Factors influencing the blood-brain barrier permeability. Brain Res. 2022;1788: 147937.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21–78.
Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Et Biophysica Acta Biomembr. 2008;1778(3):660–9.
Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol Cell Physiol. 2018;315(3):C343–56.
Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72.
Langen UH, Ayloo S, Gu C. Development and cell biology of the blood-brain barrier. Annu Rev Cell Dev Biol. 2019;35:591–613.
Knox EG, Aburto MR, Clarke G, Cryan JF, O’Driscoll CM. The blood-brain barrier in aging and neurodegeneration. Mol Psychiatry. 2022;27(6):2659–73.
Teixeira MI, Lopes CM, Amaral MH, Costa PC. Current insights on lipid nanocarrier-assisted drug delivery in the treatment of neurodegenerative diseases. Eur J Pharm Biopharm. 2020;149:192–217.
Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018;21(10):1318–31.
Beyond the Amyloid Hypothesis of Alzheimer’s Disease. Tau pathology takes center stage. ACS Chem Neurosci. 2018;9(11):2519.
Jones AR, Shusta EV. Blood-brain barrier transport of therapeutics via receptor-mediation. Pharm Res. 2007;24(9):1759–71.
Pardridge WM. Drug and gene targeting to the brain with molecular trojan horses. Nat Rev Drug Discov. 2002;1(2):131–9.
Miller DS. Regulation of ABC transporters blood-brain barrier: the good, the bad, and the ugly. Adv Cancer Res. 2015;125:43–70.
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–78.
Hindle SJ, Munji RN, Dolghih E, Gaskins G, Orng S, Ishimoto H, et al. Evolutionarily conserved roles for blood-brain barrier xenobiotic transporters in endogenous steroid partitioning and behavior. Cell Rep. 2017;21(5):1304–16.
Langen UH, Ayloo S, Gu C. Development and cell biology of the blood-brain barrier. Annu Rev Cell Dev Biol. 2019;35:591–613.
Aird WC. Phenotypic heterogeneity of the endothelium: II representative vascular beds. Circ Res. 2007;100(2):174–90.
Daneman R, Zhou L, Agalliu D, Cahoy JD, Kaushal A, Barres BA. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS ONE. 2010;5(10): e13741.
Achon Buil B, Tackenberg C, Rust R. Editing a gateway for cell therapy across the blood-brain barrier. Brain. 2023;146(3):823–41.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12(12):723–38.
Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274(1–2):23–6.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19(6):771–83.
Yang AC, Vest RT, Kern F, Lee DP, Agam M, Maat CA, et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature. 2022;603(7903):885–92.
Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. 2020. https://doi.org/10.1084/jem.20190062.
Winkler EA, Bell RD, Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci. 2011;14(11):1398–405.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21(2):193–215.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468(7323):562–6.
Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, et al. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932.
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013;125(1):111–20.
Zheng Z, Chopp M, Chen J. Multifaceted roles of pericytes in central nervous system homeostasis and disease. J Cereb Blood Flow Metab. 2020;40(7):1381–401.
Pandey K, Bessieres B, Sheng SL, Taranda J, Osten P, Sandovici I, et al. Neuronal activity drives IGF2 expression from pericytes to form long-term memory. Neuron. 2023. https://doi.org/10.1016/j.neuron.2023.08.030.
Diaz-Castro B, Robel S, Mishra A. Astrocyte endfeet in brain function and pathology: open questions. Annu Rev Neurosci. 2023;46:101–21.
Bardehle S, Kruger M, Buggenthin F, Schwausch J, Ninkovic J, Clevers H, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nat Neurosci. 2013;16(5):580–6.
Yao Y, Chen ZL, Norris EH, Strickland S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat Commun. 2014;5:3413.
Michinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20030571.
Heithoff BP, George KK, Phares AN, Zuidhoek IA, Munoz-Ballester C, Robel S. Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia. 2021;69(2):436–72.
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. Glial and neuronal control of brain blood flow. Nature. 2010;468(7321):232–43.
Cohen-Salmon M, Slaoui L, Mazare N, Gilbert A, Oudart M, Alvear-Perez R, et al. Astrocytes in the regulation of cerebrovascular functions. Glia. 2021;69(4):817–41.
Zhou B, Zuo YX, Jiang RT. Astrocyte morphology: diversity, plasticity, and role in neurological diseases. CNS Neurosci Ther. 2019;25(6):665–73.
Trillo-Contreras JL, Ramirez-Lorca R, Villadiego J, Echevarria M. Cellular distribution of brain aquaporins and their contribution to cerebrospinal fluid homeostasis and hydrocephalus. Biomolecules. 2022. https://doi.org/10.3390/biom12040530.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012. https://doi.org/10.1126/scitranslmed.3003748.
Aldea R, Weller RO, Wilcock DM, Carare RO, Richardson G. Cerebrovascular smooth muscle cells as the drivers of intramural periarterial drainage of the brain. Front Aging Neurosci. 2019;11:1.
Diem AK, Carare RO, Weller RO, Bressloff NW. A control mechanism for intra-mural peri-arterial drainage via astrocytes: How neuronal activity could improve waste clearance from the brain. PLoS ONE. 2018;13(10): e0205276.
Xue X, Zhang W, Zhu J, Chen X, Zhou S, Xu Z, et al. Aquaporin-4 deficiency reduces TGF-beta1 in mouse midbrains and exacerbates pathology in experimental Parkinson’s disease. J Cell Mol Med. 2019;23(4):2568–82.
Morris AW, Sharp MM, Albargothy NJ, Fernandes R, Hawkes CA, Verma A, et al. Vascular basement membranes as pathways for the passage of fluid into and out of the brain. Acta Neuropathol. 2016;131(5):725–36.
Thomsen MS, Routhe LJ, Moos T. The vascular basement membrane in the healthy and pathological brain. J Cereb Blood Flow Metab. 2017;37(10):3300–17.
Ceafalan LC, Fertig TE, Gheorghe TC, Hinescu ME, Popescu BO, Pahnke J, et al. Age-related ultrastructural changes of the basement membrane in the mouse blood-brain barrier. J Cell Mol Med. 2019;23(2):819–27.
Thomsen MS, Birkelund S, Burkhart A, Stensballe A, Moos T. Synthesis and deposition of basement membrane proteins by primary brain capillary endothelial cells in a murine model of the blood-brain barrier. J Neurochem. 2017;140(5):741–54.
Gautam J, Zhang X, Yao Y. The role of pericytic laminin in blood brain barrier integrity maintenance. Sci Rep. 2016;6:36450.
Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood-brain barrier. J Neurosci. 2014;34(46):15260–80.
Kenne E, Soehnlein O, Genove G, Rotzius P, Eriksson EE, Lindbom L. Immune cell recruitment to inflammatory loci is impaired in mice deficient in basement membrane protein laminin alpha4. J Leukoc Biol. 2010;88(3):523–8.
Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131(7):1619–28.
Nakamura K, Ikeuchi T, Nara K, Rhodes CS, Zhang P, Chiba Y, et al. Perlecan regulates pericyte dynamics in the maintenance and repair of the blood–brain barrier. J Cell Biol. 2019;218(10):3506–25.
Trout AL, Rutkai I, Biose IJ, Bix GJ. Review of alterations in perlecan-associated vascular risk factors in dementia. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21020679.
Lord MS, Chuang CY, Melrose J, Davies MJ, Iozzo RV, Whitelock JM. The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 2014;35:112–22.
Tallquist MD, French WJ, Soriano P. Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol. 2003;1(2):E52.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68(3):409–27.
Franco M, Roswall P, Cortez E, Hanahan D, Pietras K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression. Blood. 2011;118(10):2906–17.
Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125(Pt 11):2549–57.
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci U S A. 2009;106(6):1977–82.
Reyahi A, Nik AM, Ghiami M, Gritli-Linde A, Ponten F, Johansson BR, et al. Foxf2 is required for brain pericyte differentiation and development and maintenance of the blood-brain barrier. Dev Cell. 2015;34(1):19–32.
Darland DC, D’Amore PA. TGF beta is required for the formation of capillary-like structures in three-dimensional cocultures of 10T1/2 and endothelial cells. Angiogenesis. 2001;4(1):11–20.
Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K, et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of caveolae-mediated transcytosis. Neuron. 2017. https://doi.org/10.1016/j.neuron.2017.03.043.
Qu C, Song H, Shen J, Xu L, Li Y, Qu C, et al. Mfsd2a reverses spatial learning and memory impairment caused by chronic cerebral hypoperfusion via protection of the blood-brain barrier. Front Neurosci. 2020. https://doi.org/10.3389/fnins.2020.00461.
Dietrich JB. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J Neuroimmunol. 2002;128(1–2):58–68.
Eilam R, Segal M, Malach R, Sela M, Arnon R, Aharoni R. Astrocyte disruption of neurovascular communication is linked to cortical damage in an animal model of multiple sclerosis. Glia. 2018;66(5):1098–117.
Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A, et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. 2019;10(1):5816.
Yu X, Ji C, Shao A. Neurovascular unit dysfunction and neurodegenerative disorders. Front Neurosci. 2020;14:334.
Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab. 2001;21(10):1133–45.
Horlyck S, Cai C, Helms HCC, Lauritzen M, Brodin B. ATP induces contraction of cultured brain capillary pericytes via activation of P2Y-type purinergic receptors. Am J Physiol Heart Circ Physiol. 2021;320(2):H699–712.
Ko KR, Ngai AC, Winn HR. Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am J Physiol. 1990;259(6 Pt 2):H1703-1708.
Jackson WF. Tuning the signal: ATP-sensitive K (+) channels direct blood flow to cerebral capillaries. Sci Signal. 2022;15(727):eabo1118.
Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443(7112):700–4.
Tota S, Goel R, Pachauri SD, Rajasekar N, Najmi AK, Hanif K, et al. Effect of angiotensin II on spatial memory, cerebral blood flow, cholinergic neurotransmission, and brain derived neurotrophic factor in rats. Psychopharmacology. 2013;226(2):357–69.
Lecrux C, Hamel E. Neuronal networks and mediators of cortical neurovascular coupling responses in normal and altered brain states. Philos Trans R Soc Lond B Biol Sci. 2016. https://doi.org/10.1098/rstb.2015.0350.
Hamilton NB, Attwell D, Hall CN. Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenergetics. 2010. https://doi.org/10.3389/fnene.2010.00005.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55–60.
Kitchen P, Salman MM, Halsey AM, Clarke-Bland C, MacDonald JA, Ishida H, et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020. https://doi.org/10.1016/j.cell.2020.03.037.
Stokum JA, Kurland DB, Gerzanich V, Simard JM. Mechanisms of astrocyte-mediated cerebral edema. Neurochem Res. 2015;40(2):317–28.
Patabendige A, Singh A, Jenkins S, Sen J, Chen R. Astrocyte activation in neurovascular damage and repair following ischaemic stroke. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22084280.
Bordoni L, Thoren AE, Gutierrez-Jimenez E, Abjorsbraten KS, Bjornstad DM, Tang W, et al. Deletion of aquaporin-4 improves capillary blood flow distribution in brain edema. Glia. 2023;71(11):2559–72.
Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, et al. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science. 2019. https://doi.org/10.1126/science.aav9518.
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20(3):406–16.
Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22(7):1089–98.
Hirunpattarasilp C, Attwell D, Freitas F. The role of pericytes in brain disorders: from the periphery to the brain. J Neurochem. 2019;150(6):648–65.
Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci. 2016;19(12):1619–27.
Zhou SY, Guo ZN, Zhang DH, Qu Y, Jin H. The role of pericytes in ischemic stroke: fom cellular functions to therapeutic targets. Front Mol Neurosci. 2022;15: 866700.
Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380(6573):435–9.
Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, et al. Capillary K(+)-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci. 2017;20(5):717–26.
Szutowicz A, Bielarczyk H, Ronowska A, Gul-Hinc S, Klimaszewska-Tata J, Dys A, et al. Intracellular redistribution of acetyl-CoA, the pivotal point in differential susceptibility of cholinergic neurons and glial cells to neurodegenerative signals. Biochem Soc T. 2014;42:1101–6.
Garcia-Bonilla L, Brea D, Benakis C, Lane DA, Murphy M, Moore J, et al. Endogenous protection from ischemic brain injury by preconditioned monocytes. J Neurosci. 2018;38(30):6722–36.
Quaranta DV, Weaver RR, Baumann KK, Fujimoto T, Williams LM, Kim HC, et al. Transport of the proinflammatory chemokines C-C motif chemokine ligand 2 (MCP-1) and C-C motif chemokine ligand 5 (RANTES) across the intact mouse blood-brain barrier is inhibited by heparin and eprodisate and increased with systemic inflammation. J Pharmacol Exp Ther. 2023;384(1):205–23.
Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain Behav Immun. 2010;24(1):102–9.
Herkenham M, Lee HY, Baker RA. Temporal and spatial patterns ofc-fos mRNA induced by intravenous interleukin-1: a cascade of non-neuronal cellular activation at the blood-brain barrier. J Comp Neurol. 1998;400(2):175–96.
Quan N, He L, Lai W. Endothelial activation is an intermediate step for peripheral lipopolysaccharide induced activation of paraventricular nucleus. Brain Res Bull. 2003;59(6):447–52.
Wilhelms DB, Kirilov M, Mirrasekhian E, Eskilsson A, Kugelberg UO, Klar C, et al. Deletion of prostaglandin E2 synthesizing enzymes in brain endothelial cells attenuates inflammatory fever. J Neurosci. 2014;34(35):11684–90.
Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett. 2011;585(23):3770–80.
Labus J, Woltje K, Stolte KN, Hackel S, Kim KS, Hildmann A, et al. IL-1beta promotes transendothelial migration of PBMCs by upregulation of the FN/alpha(5)beta(1) signalling pathway in immortalised human brain microvascular endothelial cells. Exp Cell Res. 2018;373(1–2):99–111.
Tan S, Shan Y, Lin Y, Liao S, Zhang B, Zeng Q, et al. Neutralization of interleukin-9 ameliorates experimental stroke by repairing the blood-brain barrier via down-regulation of astrocyte-derived vascular endothelial growth factor-A. FASEB J. 2019;33(3):4376–87.
Labus J, Hackel S, Lucka L, Danker K. Interleukin-1beta induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Method. 2014;228:35–45.
Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci. 2017;20(12):1752–60.
Chen W, Ju XZ, Lu Y, Ding XW, Miao CH, Chen JW. Propofol improved hypoxia-impaired integrity of blood-brain barrier via modulating the expression and phosphorylation of zonula occludens-1. CNS Neurosci Ther. 2019;25(6):704–13.
Wong D, Dorovini-Zis K, Vincent SR. Cytokines, nitric oxide, and cGMP modulate the permeability of an in vitro model of the human blood-brain barrier. Exp Neurol. 2004;190(2):446–55.
Qin LH, Huang W, Mo XA, Chen YL, Wu XH. LPS induces occludin dysregulation in cerebral microvascular endothelial cells via MAPK signaling and augmenting MMP-2 levels. Oxid Med Cell Longev. 2015;2015: 120641.
Han D, Fang W, Zhang R, Wei J, Kodithuwakku ND, Sha L, et al. Clematichinenoside protects blood brain barrier against ischemic stroke superimposed on systemic inflammatory challenges through up-regulating A20. Brain Behav Immun. 2016;51:56–69.
Liu X, Sui B, Sun J. Blood-brain barrier dysfunction induced by silica NPs in vitro and in vivo: Involvement of oxidative stress and Rho-kinase/JNK signaling pathways. Biomaterials. 2017;121:64–82.
Gao M, Lu W, Shu Y, Yang Z, Sun S, Xu J, et al. Poldip2 mediates blood-brain barrier disruption and cerebral edema by inducing AQP4 polarity loss in mouse bacterial meningitis model. CNS Neurosci Ther. 2020;26(12):1288–302.
Huang X, Hussain B, Chang J. Peripheral inflammation and blood-brain barrier disruption: effects and mechanisms. CNS Neurosci Ther. 2021;27(1):36–47.
Nakamuta S, Endo H, Higashi Y, Kousaka A, Yamada H, Yano M, et al. Human immunodeficiency virus type 1 gp120-mediated disruption of tight junction proteins by induction of proteasome-mediated degradation of zonula occludens-1 and -2 in human brain microvascular endothelial cells. J Neurovirol. 2008;14(3):186–95.
Yu CC, Chen HL, Chen MH, Lu CH, Tsai NW, Huang CC, et al. Vascular inflammation is a risk factor associated with brain atrophy and disease severity in Parkinson’s disease: a case-control study. Oxid Med Cell Longev. 2020;2020:2591248.
Yousef H, Czupalla CJ, Lee D, Chen MB, Burke AN, Zera KA, et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat Med. 2019;25(6):988–1000.
Souza PS, Goncalves ED, Pedroso GS, Farias HR, Junqueira SC, Marcon R, et al. Physical exercise attenuates experimental autoimmune encephalomyelitis by inhibiting peripheral immune response and blood-brain barrier disruption. Mol Neurobiol. 2017;54(6):4723–37.
Mendiola AS, Yan Z, Dixit K, Johnson JR, Bouhaddou M, Meyer-Franke A, et al. Defining blood-induced microglia functions in neurodegeneration through multiomic profiling. Nat Immunol. 2023;24(7):1173–87.
Blecharz-Lang KG, Wagner J, Fries A, Nieminen-Kelhä M, Rösner J, Schneider UC, et al. Interleukin 6-mediated endothelial barrier disturbances can be attenuated by blockade of the il6 receptor expressed in brain microvascular endothelial cells. Transl Stroke Res. 2018;9(6):631–42.
Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67.
Kant R, Halder SK, Fernandez JA, Griffin JH, Milner R. Activated protein C attenuates experimental autoimmune encephalomyelitis progression by enhancing vascular integrity and suppressing microglial activation. Front Neurosci. 2020;14:333.
Jolivel V, Bicker F, Biname F, Ploen R, Keller S, Gollan R, et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015;129(2):279–95.
van Horssen J, Vos CM, Admiraal L, van Haastert ES, Montagne L, van der Valk P, et al. Matrix metalloproteinase-19 is highly expressed in active multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2006;32(6):585–93.
Pieper C, Marek JJ, Unterberg M, Schwerdtle T, Galla HJ. Brain capillary pericytes contribute to the immune defense in response to cytokines or LPS in vitro. Brain Res. 2014;1550:1–8.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–6.
Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Perez JM, Evans AC. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun. 2016. https://doi.org/10.1038/ncomms11934.
Cullen KM, Kocsi Z, Stone J. Pericapillary haem-rich deposits: evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab. 2005;25(12):1656–67.
Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002;32(5):360–71.
Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. 2015;21(8):880–6.
Cortes-Canteli M, Paul J, Norris EH, Bronstein R, Ahn HJ, Zamolodchikov D, et al. Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: a possible contributing factor to Alzheimer’s disease. Neuron. 2010;66(5):695–709.
Hultman K, Strickland S, Norris EH. The APOE varepsilon4/varepsilon4 genotype potentiates vascular fibrin (ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab. 2013;33(8):1251–8.
Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, et al. Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging. 2007;28(7):977–86.
Inoue Y, Ueda M, Masuda T, Misumi Y, Yamashita T, Ando Y. Memantine, a noncompetitive N-methyl-D-aspartate receptor antagonist, attenuates cerebral amyloid angiopathy by increasing insulin-degrading enzyme expression. Mol Neurobiol. 2019;56(12):8573–88.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302.
van de Haar HJ, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Wong SM, et al. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging. 2016;45:190–6.
van de Haar HJ, Burgmans S, Jansen JF, van Osch MJ, van Buchem MA, Muller M, et al. Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology. 2016;281(2):527–35.
van de Haar HJ, Jansen JFA, Jeukens C, Burgmans S, van Buchem MA, Muller M, et al. Subtle blood-brain barrier leakage rate and spatial extent: considerations for dynamic contrast-enhanced MRI. Med Phys. 2017;44(8):4112–25.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–6.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, et al. Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36(1):216–27.
Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D, et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin a independently of amyloid-beta. Nat Aging. 2021;1(6):506–20.
Salloway S, Gur T, Berzin T, Tavares R, Zipser B, Correia S, et al. Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J Neurol Sci. 2002;203–204:183–7.
Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013;108:21–43.
Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol. 1997;42(1):85–94.
An Y, Varma VR, Varma S, Casanova R, Dammer E, Pletnikova O, et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimers Dement. 2018;14(3):318–29.
Chen Y, Wolk DA, Reddin JS, Korczykowski M, Martinez PM, Musiek ES, et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology. 2011;77(22):1977–85.
Gil-Iturbe E, Solas M, Cuadrado-Tejedo M, Garcia-Osta A, Escote X, Ramirez MJ, et al. GLUT12 expression in brain of mouse models of Alzheimer’s disease. Mol Neurobiol. 2020;57(2):798–805.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):280–92.
Martin-Macintosh EL, Broski SM, Johnson GB, Hunt CH, Cullen EL, Peller PJ. Multimodality imaging of neurodegenerative processes: part 1, the basics and common dementias. Am J Roentgenol. 2016;207(4):871–82.
Bailly M, Destrieux C, Hommet C, Mondon K, Cottier JP, Beaufils E, et al. Precuneus and cingulate cortex atrophy and hypometabolism in patients with Alzheimer’s disease and mild cognitive impairment: MRI and (18)F-FDG PET quantitative analysis using freesurfer. Biomed Res Int. 2015;2015: 583931.
Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol Aging. 2011;32(7):1207–18.
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D, et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. 2015;18(4):521–30.
Taccola C, Barneoud P, Cartot-Cotton S, Valente D, Schussler N, Saubamea B, et al. Modifications of physical and functional integrity of the blood-brain barrier in an inducible mouse model of neurodegeneration. Neuropharmacology. 2021;191: 108588.
Majerova P, Michalicova A, Cente M, Hanes J, Vegh J, Kittel A, et al. Trafficking of immune cells across the blood-brain barrier is modulated by neurofibrillary pathology in tauopathies. PLoS ONE. 2019;14(5): e0217216.
Storck SE, Pietrzik CU. Endothelial LRP1—a potential target for the treatment of Alzheimer’s disease : theme: drug discovery, development and delivery in Alzheimer’s disease guest editor: davide brambilla. Pharm Res. 2017;34(12):2637–51.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab. 2007;27(5):909–18.
Chai AB, Hartz AMS, Gao X, Yang A, Callaghan R, Gelissen IC. New evidence for P-gp-mediated export of amyloid-β peptides in molecular, blood-brain barrier and neuronal models. Int J Mol Sci. 2020;22(1):246.
Chiu C, Miller MC, Monahan R, Osgood DP, Stopa EG, Silverberg GD. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: preliminary observations. Neurobiol Aging. 2015;36(9):2475–82.
Park R, Kook SY, Park JC, Mook-Jung I. Abeta1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-kappaB signaling. Cell Death Dis. 2014;5(6): e1299.
Hartz AM, Zhong Y, Wolf A, LeVine H 3rd, Miller DS, Bauer B. Abeta40 reduces P-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J Neurosci. 2016;36(6):1930–41.
Cordone S, Annarumma L, Rossini PM, De Gennaro L. Sleep and beta-amyloid deposition in Alzheimer disease: insights on mechanisms and possible innovative treatments. Front Pharmacol. 2019;10:695.
Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LM, Nilsson LN, et al. Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J Alzheimers Dis. 2011;27(4):711–22.
Mestre H, Hablitz LM, Xavier AL, Feng W, Zou W, Pu T, et al. Aquaporin-4-dependent glymphatic solute transport in the rodent brain. Elife. 2018;7:e40070.
Mazura AD, Ohler A, Storck SE, Kurtyka M, Scharfenberg F, Weggen S, et al. PCSK9 acts as a key regulator of abeta clearance across the blood-brain barrier. Cell Mol Life Sci. 2022;79(4):212.
Cole SL, Vassar R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol Neurodegener. 2007;2:22.
Montagne A, Nation DA, Pa J, Sweeney MD, Toga AW, Zlokovic BV. Brain imaging of neurovascular dysfunction in Alzheimer’s disease. Acta Neuropathol. 2016;131(5):687–707.
Wierenga CE, Dev SI, Shin DD, Clark LR, Bangen KJ, Jak AJ, et al. Effect of mild cognitive impairment and APOE genotype on resting cerebral blood flow and its association with cognition. J Cereb Blood Flow Metab. 2012;32(8):1589–99.
Wirth M, Pichet Binette A, Brunecker P, Kobe T, Witte AV, Floel A. Divergent regional patterns of cerebral hypoperfusion and gray matter atrophy in mild cognitive impairment patients. J Cereb Blood Flow Metab. 2017;37(3):814–24.
Schultz N, Brannstrom K, Byman E, Moussaud S, Nielsen HM, Netherlands Brain B, et al. Amyloid-beta 1–40 is associated with alterations in NG2+ pericyte population ex vivo and in vitro. Aging Cell. 2018;17(3): e12728.
Abubaker AA, Vara D, Visconte C, Eggleston I, Torti M, Canobbio I, et al. Amyloid peptide beta1-42 induces integrin alphaiibbeta3 activation, platelet adhesion, and thrombus formation in a NADPH oxidase-dependent manner. Oxid Med Cell Longev. 2019;2019:1050476.
Park L, Zhou J, Koizumi K, Wang G, Anfray A, Ahn SJ, et al. tPA deficiency underlies neurovascular coupling dysfunction by amyloid-beta. J Neurosci. 2020;40(42):8160–73.
Albrecht D, Isenberg AL, Stradford J, Monreal T, Sagare A, Pachicano M, et al. Associations between vascular function and tau PET are associated with global cognition and amyloid. J Neurosci. 2020;40(44):8573–86.
Weller RO, Boche D, Nicoll JA. Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol. 2009;118(1):87–102.
Saito S, Ihara M. Interaction between cerebrovascular disease and Alzheimer pathology. Curr Opin Psychiatry. 2016;29(2):168–73.
Basun H, Bogdanovic N, Ingelsson M, Almkvist O, Naslund J, Axelman K, et al. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch Neurol. 2008;65(4):499–505.
Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM. TGF-beta 1 is an organizer of responses to neurodegeneration. J Cell Biochem. 1993;53(4):314–22.
Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. 2011;34(1):3–11.
Kato T, Sekine Y, Nozaki H, Uemura M, Ando S, Hirokawa S, et al. Excessive production of transforming growth factor beta1 causes mural cell depletion from cerebral small vessels. Front Aging Neurosci. 2020;12:151.
Boles Ponto LL, Magnotta VA, Moser DJ, Duff KM, Schultz SK. Global cerebral blood flow in relation to cognitive performance and reserve in subjects with mild memory deficits. Mol Imag Biol. 2006;8(6):363–72.
den Abeelen AS, Lagro J, van Beek AH, Claassen JA. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr Alzheimer Res. 2014;11(1):11–7.
Yezhuvath US, Uh J, Cheng Y, Martin-Cook K, Weiner M, Diaz-Arrastia R, et al. Forebrain-dominant deficit in cerebrovascular reactivity in Alzheimer’s disease. Neurobiol Aging. 2012;33(1):75–82.
Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, et al. Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cereb Cortex. 2014;24(4):978–88.
Rombouts SARB, Barkhof F, Veltman DJ, Machielsen WCM, Witter MP, Bierlaagh MA, et al. Functional MR imaging in Alzheimer’s disease during memory encoding. Am J Neuroradiol. 2000;21(10):1869–75.
Small SA, Perera GM, DeLaPaz R, Mayeux R, Stern Y. Differential regional dysfunction of the hippocampal formation among elderly with memory decline and Alzheimer’s disease. Ann Neurol. 1999;45(4):466–72.
Hussong SA, Banh AQ, Van Skike CE, Dorigatti AO, Hernandez SF, Hart MJ, et al. Soluble pathogenic tau enters brain vascular endothelial cells and drives cellular senescence and brain microvascular dysfunction in a mouse model of tauopathy. Nat Commun. 2023;14(1):2367.
Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, et al. Massive gliosis induced by interleukin-6 suppresses abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–59.
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117(6):1595–604.
Krabbe G, Halle A, Matyash V, Rinnenthal JL, Eom GD, Bernhardt U, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE. 2013;8(4): e60921.
Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210(1–2):3–12.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212(3):287–95.
Saresella M, Marventano I, Calabrese E, Piancone F, Rainone V, Gatti A, et al. A complex proinflammatory role for peripheral monocytes in Alzheimer’s disease. J Alzheimers Dis. 2014;38(2):403–13.
Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. 2005;25(5):593–606.
Haddick PC, Larson JL, Rathore N, Bhangale TR, Phung QT, Srinivasan K, et al. A common variant of IL-6R is associated with elevated IL-6 pathway activity in Alzheimer’s disease brains. J Alzheimers Dis. 2017;56(3):1037–54.
Decourt B, Lahiri DK, Sabbagh MN. Targeting tumor necrosis factor alpha for Alzheimer’s disease. Curr Alzheimer Res. 2017;14(4):412–25.
Forster C, Burek M, Romero IA, Weksler B, Couraud PO, Drenckhahn D. Differential effects of hydrocortisone and TNFalpha on tight junction proteins in an in vitro model of the human blood-brain barrier. J Physiol. 2008;586(7):1937–49.
Hoshi Y, Uchida Y, Tachikawa M, Ohtsuki S, Terasaki T. Actin filament-associated protein 1 (AFAP-1) is a key mediator in inflammatory signaling-induced rapid attenuation of intrinsic P-gp function in human brain capillary endothelial cells. J Neurochem. 2017;141(2):247–62.
Hartz AM, Bauer B, Fricker G, Miller DS. Rapid modulation of P-glycoprotein-mediated transport at the blood-brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol Pharmacol. 2006;69(2):462–70.
Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13(9A):2911–25.
Paul J, Strickland S, Melchor JP. Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease. J Exp Med. 2007;204(8):1999–2008.
Cortes-Canteli M, Zamolodchikov D, Ahn HJ, Strickland S, Norris EH. Fibrinogen and altered hemostasis in Alzheimer’s disease. J Alzheimers Dis. 2012;32(3):599–608.
Zamolodchikov D, Strickland S. A possible new role for Aβ in vascular and inflammatory dysfunction in Alzheimer’s disease. Thromb Res. 2016;141:S59–61.
Qi J, Goralnick S, Kreutzer DL. Fibrin regulation of interleukin-8 gene expression in human vascular endothelial cells. Blood. 1997;90(9):3595–602.
Yin X, Wright J, Wall T, Grammas P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am J Pathol. 2010;176(4):1600–6.
Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation. 2011;8:26.
Petersen MA, Ryu JK, Akassoglou K. Fibrinogen in neurological diseases: mechanisms, imaging and therapeutics. Nat Rev Neurosci. 2018;19(5):283–301.
Verstraeten A, Theuns J, Van Broeckhoven C. Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trend Genet. 2015;31(3):140–9.
Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood-brain barrier leakage is increased in Parkinson’s disease. Front Physiol. 2020;11: 593026.
Al-Bachari S, Naish JH, Parker GJM, Emsley HCA, Parkes LM. Blood-brain barrier leakage is increased in Parkinson’s disease. Front Physiol. 2020;11: 593026.
Pajares M, Ana IR, Manda G, Bosca L, Cuadrado A. Inflammation in Parkinson’s disease: mechanisms and therapeutic implications. Cells. 2020. https://doi.org/10.3390/cells9071687.
Chen X, Lan X, Roche I, Liu R, Geiger JD. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008;107(4):1147–57.
Wang W, Bodles-Brakhop AM, Barger SW. A role for P-glycoprotein in clearance of Alzheimer amyloid beta-peptide from the brain. Curr Alzheimer Res. 2016;13(6):615–20.
Cirrito JR, Deane R, Fagan AM, Spinner ML, Parsadanian M, Finn MB, et al. P-glycoprotein deficiency at the blood-brain barrier increases amyloid-beta deposition in an Alzheimer disease mouse model. J Clin Invest. 2005;115(11):3285–90.
Huang R, Gao Y, Duan Q, Zhang Q, He P, Chen J, et al. Endothelial LRP1-ICD accelerates cognition-associated alpha-synuclein pathology and neurodegeneration through PARP1 activation in a mouse model of Parkinson’s disease. Mol Neurobiol. 2023;60(2):979–1003.
Wang Y, An R, Umanah GK, Park H, Nambiar K, Eacker SM, et al. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) polymerase-1. Science. 2016. https://doi.org/10.1126/science.aad6872.
Riffault B, Kourdougli N, Dumon C, Ferrand N, Buhler E, Schaller F, et al. Pro-brain-derived neurotrophic factor (proBDNF)-mediated p75NTR activation promotes depolarizing actions of GABA and increases susceptibility to epileptic seizures. Cereb Cortex. 2018;28(2):510–27.
Dohgu S, Takata F, Matsumoto J, Kimura I, Yamauchi A, Kataoka Y. Monomeric alpha-synuclein induces blood-brain barrier dysfunction through activated brain pericytes releasing inflammatory mediators in vitro. Microvasc Res. 2019;124:61–6.
Lan G, Wang P, Chan RB, Liu Z, Yu Z, Liu X, et al. Astrocytic VEGFA: an essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia. 2022;70(2):337–53.
Pierzchlińska A, Kwaśniak-Butowska M, Sławek J, Droździk M, Białecka M. Arterial blood pressure variability and other vascular factors contribution to the cognitive decline in Parkinson’s disease. Molecules. 2021. https://doi.org/10.3390/molecules26061523.
Rite I, Machado A, Cano J, Venero JL. Blood-brain barrier disruption induces in vivo degeneration of nigral dopaminergic neurons. J Neurochem. 2007;101(6):1567–82.
Barcia C, Emborg ME, Hirsch EC, Herrero MT. Blood vessels and parkinsonism. Front Biosci. 2004;9:277–82.
Guan J, Pavlovic D, Dalkie N, Waldvogel HJ, O’Carroll SJ, Green CR, et al. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013;23(2):154–64.
Nguyen B, Bix G, Yao Y. Basal lamina changes in neurodegenerative disorders. Mol Neurodegener. 2021;16(1):81.
Ham JH, Yi H, Sunwoo MK, Hong JY, Sohn YH, Lee PH. Cerebral microbleeds in patients with Parkinson’s disease. J Neurol. 2014;261(8):1628–35.
Biju KC, Shen Q, Hernandez ET, Mader MJ, Clark RA. Reduced cerebral blood flow in an α-synuclein transgenic mouse model of Parkinson’s disease. J Cerebr Blood F Met. 2020;40(12):2441–53.
Paul G, Elabi OF. Microvascular changes in Parkinson’s disease- focus on the neurovascular unit. Front Aging Neurosci. 2022. https://doi.org/10.3389/fnagi.2022.853372.
Shimoji M, Pagan F, Healton EB, Mocchetti I. CXCR4 and CXCL12 expression is increased in the nigro-striatal system of Parkinson’s disease. Neurotox Res. 2009;16(3):318–28.
Ding X, Zhang M, Gu R, Xu G, Wu H. Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefes Arch Clin Exp Ophthalmol. 2017;255(4):777–88.
Wu F, Liu L, Zhou H. Endothelial cell activation in central nervous system inflammation. J Leukoc Biol. 2017;101(5):1119–32.
Steinruecke M, Lonergan RM, Selvaraj BT, Chandran S, Diaz-Castro B, Stavrou M. Blood-CNS barrier dysfunction in amyotrophic lateral sclerosis: proposed mechanisms and clinical implications. J Cereb Blood Flow Metab. 2023;43(5):642–54.
Garbuzova-Davis S, Hernandez-Ontiveros DG, Rodrigues MC, Haller E, Frisina-Deyo A, Mirtyl S, et al. Impaired blood-brain/spinal cord barrier in ALS patients. Brain Res. 2012;1469:114–28.
Garbuzova-Davis S, Saporta S, Haller E, Kolomey I, Bennett SP, Potter H, et al. Evidence of compromised blood-spinal cord barrier in early and late symptomatic SOD1 mice modeling ALS. PLoS ONE. 2007;2(11): e1205.
Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, et al. Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 tesla MRI and pathology. PLoS ONE. 2012. https://doi.org/10.1371/journal.pone.0035241.
Garbuzova-Davis S, Kurien C, Haller E, Eve DJ, Navarro S, Steiner G, et al. Human bone marrow endothelial progenitor cell transplantation into symptomatic ALS mice delays disease progression and increases motor neuron survival by repairing blood-spinal cord barrier. Sci Rep. 2019;9(1):5280.
Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A. 2014;111(11):E1035-1042.
van Vliet EA, Iyer AM, Mesarosova L, Colakoglu H, Anink JJ, van Tellingen O, et al. Expression and cellular distribution of P-glycoprotein and breast cancer resistance protein in amyotrophic lateral sclerosis patients. J Neuropathol Exp Neurol. 2020;79(3):266–76.
Bataveljic D, Nikolic L, Milosevic M, Todorovic N, Andjus PR. Changes in the astrocytic aquaporin-4 and inwardly rectifying potassium channel expression in the brain of the amyotrophic lateral sclerosis SOD1(G93A) rat model. Glia. 2012;60(12):1991–2003.
Wang L, Gutmann DH, Roos RP. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum Mol Genet. 2011;20(2):286–93.
Jo M, Lee S, Jeon YM, Kim S, Kwon Y, Kim HJ. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med. 2020;52(10):1652–62.
Lee S, Kim S, Kang HY, Lim HR, Kwon Y, Jo M, et al. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory response. J Neuroinflammation. 2020;17(1):299.
Moreau C, Devos D, Brunaud-Danel V, Defebvre L, Perez T, Destee A, et al. Elevated IL-6 and TNF-alpha levels in patients with ALS: inflammation or hypoxia? Neurology. 2005;65(12):1958–60.
Zamudio F, Loon AR, Smeltzer S, Benyamine K, Navalpur Shanmugam NK, Stewart NJF, et al. TDP-43 mediated blood-brain barrier permeability and leukocyte infiltration promote neurodegeneration in a low-grade systemic inflammation mouse model. J Neuroinflammation. 2020;17(1):283.
Jara JH, Gautam M, Kocak N, Xie EF, Mao Q, Bigio EH, et al. MCP1-CCR2 and neuroinflammation in the ALS motor cortex with TDP-43 pathology. J Neuroinflammation. 2019;16(1):196.
Garbuzova-Davis S, Ehrhart J, Sanberg PR, Borlongan CV. Potential role of humoral IL-6 cytokine in mediating pro-inflammatory endothelial cell response in amyotrophic lateral sclerosis. Int J Mol Sci. 2018. https://doi.org/10.3390/ijms19020423.
Yu W, He J, Cai X, Yu Z, Zou Z, Fan D. Neuroimmune crosstalk between the peripheral and the central immune system in amyotrophic lateral sclerosis. Front Aging Neurosci. 2022. https://doi.org/10.3389/fnagi.2022.890958.
Pieper C, Pieloch P, Galla HJ. Pericytes support neutrophil transmigration via interleukin-8 across a porcine co-culture model of the blood-brain barrier. Brain Res. 2013;1524:1–11.
Ouali Alami N, Schurr C, Olde Heuvel F, Tang L, Li Q, Tasdogan A, et al. NF-kappaB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018. https://doi.org/10.15252/embj.201798697.
Jones MK, Nair A, Gupta M. Mast cells in neurodegenerative disease. Front Cell Neurosci. 2019;13:171.
Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nat Rev Immunol. 2015;15(9):545–58.
McDonald WI, Compston A, Edan G, Goodkin D, Hartung HP, Lublin FD, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the international panel on the diagnosis of multiple sclerosis. Ann Neurol. 2001;50(1):121–7.
Alvarez JI, Cayrol R, Prat A. Disruption of central nervous system barriers in multiple sclerosis. Bba-Mol Basis Dis. 2011;1812(2):252–64.
Gerwien H, Hermann S, Zhang XL, Korpos E, Song J, Kopka K, et al. Imaging matrix metalloproteinase activity in multiple sclerosis as a specific marker of leukocyte penetration of the blood-brain barrier. Sci Transl Med. 2016. https://doi.org/10.1126/scitranslmed.aaf8020.
Barreiro O, Sanchez-Madrid F. Molecular basis of leukocyte-endothelium interactions during the inflammatory response. Rev Esp Cardiol. 2009;62(5):552–62.
Hernandez-Pedro NY, Espinosa-Ramirez G, de la Cruz VP, Pineda B, Sotelo J. Initial immunopathogenesis of multiple sclerosis: innate immune response. Clin Dev Immunol. 2013;2013: 413465.
Prajeeth CK, Dittrich-Breiholz O, Talbot SR, Robert PA, Huehn J, Stangel M. IFN-gamma producing Th1 cells induce different transcriptional profiles in microglia and astrocytes. Front Cell Neurosci. 2018;12:352.
Kunkl M, Frascolla S, Amormino C, Volpe E, Tuosto L. T helper cells: the modulators of inflammation in multiple sclerosis. Cells. 2020. https://doi.org/10.3390/cells9020482.
Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116(4):508–21.
Balasa R, Barcutean L, Mosora O, Manu D. Reviewing the significance of blood-brain barrier disruption in multiple sclerosis pathology and treatment. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22168370.
Schreiner TG, Romanescu C, Popescu BO. The blood-brain barrier-a key player in multiple sclerosis disease mechanisms. Biomolecules. 2022. https://doi.org/10.3390/biom12040538.
Correale J, Gaitan MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140(3):527–46.
D’Haeseleer M, Cambron M, Vanopdenbosch L, De Keyser J. Vascular aspects of multiple sclerosis. Lancet Neurol. 2011;10(7):657–66.
Monti L, Morbidelli L, Rossi A. Impaired cerebral perfusion in multiple sclerosis: relevance of endothelial factors. Biomark Insights. 2018. https://doi.org/10.1177/1177271918774800.
De Keyser J, Wilczak N, Leta R, Streetland C. Astrocytes in multiple sclerosis lack beta-2 adrenergic receptors. Neurology. 1999;53(8):1628–33.
D’haeseleer M, Hostenbach S, Peeters I, El Sankari S, Nagels G, De Keyser J, et al. Cerebral hypoperfusion: a new pathophysiologic concept in multiple sclerosis? J Cerebr Blood F Met. 2015;35(9):1406–10.
Ouellette J, Lacoste B. From neurodevelopmental to neurodegenerative disorders: the vascular continuum. Front Aging Neurosci. 2021. https://doi.org/10.3389/fnagi.2021.749026.
Marshall O, Lu HZ, Brisset JC, Xu F, Liu PY, Herbert J, et al. Impaired cerebrovascular reactivity in multiple sclerosis. Jama Neurol. 2014;71(10):1275–81.
Wakefield AJ, More LJ, Difford J, Mclaughlin JE. Immunohistochemical study of vascular injury in acute multiple-sclerosis. J Clin Pathol. 1994;47(2):129–33.
Lyons SA, Kettenmann H. Oligodendrocytes and microglia are selectively vulnerable to combined hypoxia and hypoglycemia injury in vitro. J Cereb Blood Flow Metab. 1998;18(5):521–30.
Juhler M, Laursen H, Barry DI. The distribution of immunoglobulins and albumin in the central nervous system in acute experimental allergic encephalomyelitis. Acta Neurol Scand. 2009;73(2):119–24.
Vega-Zelaya L, Ortega GJ, Sola RG, Pastor J. Plasma albumin induces cytosolic calcium oscilations and DNA synthesis in human cultured astrocytes. Biomed Res Int. 2014;2014:1–10.
van Vliet EA, Aronica E, Gorter JA. Blood-brain barrier dysfunction, seizures and epilepsy. Semin Cell Dev Biol. 2015;38:26–34.
Tabernero A, Granda B, Medina A, Sanchez-Abarca LI, Lavado E, Medina JM. Albumin promotes neuronal survival by increasing the synthesis and release of glutamate. J Neurochem. 2002;81(4):881–91.
Andravizou A, Dardiotis E, Artemiadis A, Sokratous M, Siokas V, Tsouris Z, et al. Brain atrophy in multiple sclerosis: mechanisms, clinical relevance and treatment options. Auto Immun Highlights. 2019;10(1):7.
Choi BY, Jung JW, Suh SW. The emerging role of zinc in the pathogenesis of multiple sclerosis. Int J Mol Sci. 2017. https://doi.org/10.3390/ijms18102070.
Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C: biased for translation. Blood. 2015;125(19):2898–907.
Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, et al. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12(11):1278–85.
Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451(7182):1076–81.
Lazic D, Sagare AP, Nikolakopoulou AM, Griffin JH, Vassar R, Zlokovic BV. 3K3A-activated protein C blocks amyloidogenic BACE1 pathway and improves functional outcome in mice. J Exp Med. 2019;216(2):279–93.
Zhong Z, Ilieva H, Hallagan L, Bell R, Singh I, Paquette N, et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009;119(11):3437–49.
Griffin JH, Zlokovic BV, Mosnier LO. Activated protein C, protease activated receptor 1, and neuroprotection. Blood. 2018;132(2):159–69.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–6.
Fei Yx, Zhu Jp, Zhao B, Yin Qy, Fang Wr, Li Ym. XQ-1H regulates Wnt/GSK3β/β-catenin pathway and ameliorates the integrity of blood brain barrier in mice with acute ischemic stroke. Brain Res Bull. 2020;164:269–88.
Xu L, Nirwane A, Xu T, Kang M, Devasani K, Yao Y. Fibroblasts repair blood-brain barrier damage and hemorrhagic brain injury via TIMP2. Cell Rep. 2022;41(8): 111709.
Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Toth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6(263): 263ra158.
Geng J, Wang L, Qu M, Song Y, Lin X, Chen Y, et al. Endothelial progenitor cells transplantation attenuated blood-brain barrier damage after ischemia in diabetic mice via HIF-1alpha. Stem Cell Res Ther. 2017;8(1):163.
Leeper NJ, Hunter AL, Cooke JP. Stem cell therapy for vascular regeneration: adult, embryonic, and induced pluripotent stem cells. Circulation. 2010;122(5):517–26.
Nikolakopoulou AM, Wang Y, Ma Q, Sagare AP, Montagne A, Huuskonen MT, et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J Exp Med. 2021. https://doi.org/10.1084/jem.20202207.
Sui YT, Bullock KM, Erickson MA, Zhang J, Banks WA. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides. 2014;62:197–202.
Brinton RD, Swanson HM, Irwin RW. P4–026: allopregnanolone promotes cholesterol and amyloid-beta clearance mechanisms: assessment of a regenerative therapeutic for Alzheimer’s disease. Alzheimer Dement. 2016;12:1024–5.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9(7):907–13.
Huang Y, Chen S, Luo Y, Han Z. Crosstalk between inflammation and the BBB in stroke. Curr Neuropharmacol. 2020;18(12):1227–36.
Blecharz KG, Haghikia A, Stasiolek M, Kruse N, Drenckhahn D, Gold R, et al. Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler. 2010;16(3):293–302.
Dong X, Gao J, Zhang CY, Hayworth C, Frank M, Wang Z. Neutrophil membrane-derived nanovesicles alleviate inflammation to protect mouse brain injury from ischemic stroke. ACS Nano. 2019;13(2):1272–83.
Chao C-C, Gutiérrez-Vázquez C, Rothhammer V, Mayo L, Wheeler MA, Tjon EC, et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 2019;179(7):1483-1498.e1422.
Koistinaho M, Malm TM, Kettunen MI, Goldsteins G, Starckx S, Kauppinen RA, et al. Minocycline protects against permanent cerebral ischemia in wild type but not in matrix metalloprotease-9-deficient mice. J Cereb Blood Flow Metab. 2005;25(4):460–7.
Salvador E, Shityakov S, Förster C. Glucocorticoids and endothelial cell barrier function. Cell Tissue Res. 2014;355(3):597–605.
Li J, Zheng M, Shimoni O, Banks WA, Bush AI, Gamble JR, et al. Development of novel therapeutics targeting the blood-brain barrier: from barrier to carrier. Adv Sci. 2021. https://doi.org/10.1002/advs.202101090.
Jarrahi A, Braun M, Ahluwalia M, Gupta RV, Wilson M, Munie S, et al. Revisiting traumatic brain injury: from molecular mechanisms to therapeutic interventions. Biomedicines. 2020. https://doi.org/10.3390/biomedicines8100389.
Carrano A, Hoozemans JJM, van der Vies SM, van Horssen J, de Vries HE, Rozemuller AJM. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener Dis. 2012;10(1–4):329–31.
Nehra G, Bauer B, Hartz AMS. Blood-brain barrier leakage in Alzheimer’s disease: from discovery to clinical relevance. Pharmacol Therapeut. 2022. https://doi.org/10.1016/j.pharmthera.2022.108119.
Acknowledgements
Not applicable
Funding
This work was supported by grants: Zhejiang basic public welfare research program (Grant Numbers LGD21H250001 and LGD22H250001), Scientific Research Foundation of Zhejiang University City College (No.X-202102).
Author information
Authors and Affiliations
Contributions
WYL contributed to the conceptualization and overall structure of the manuscript. TLC, YD, and CHH did the literature search, drafted the content of the manuscript, devised all the figures, and edited the manuscript. ZHL and SZW refined and improved the manuscript during the revision stage. JY, LHZ and SSL substantially revised and edited the manuscript, ensuring its quality and coherence. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, T., Dai, Y., Hu, C. et al. Cellular and molecular mechanisms of the blood–brain barrier dysfunction in neurodegenerative diseases. Fluids Barriers CNS 21, 60 (2024). https://doi.org/10.1186/s12987-024-00557-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-024-00557-1