
Abstract
Cognitive decline covers a broad spectrum of disorders, not only resulting from brain diseases but also from systemic diseases, which seriously influence the quality of life and life expectancy of patients. As a highly selective anatomical and functional interface between the brain and systemic circulation, the blood-brain barrier (BBB) plays a pivotal role in maintaining brain homeostasis and normal function. The pathogenesis underlying cognitive decline may vary, nevertheless, accumulating evidences support the role of BBB disruption as the most prevalent contributing factor. This may mainly be attributed to inflammation, metabolic dysfunction, cell senescence, oxidative/nitrosative stress and excitotoxicity. However, direct evidence showing that BBB disruption causes cognitive decline is scarce, and interestingly, manipulation of the BBB opening alone may exert beneficial or detrimental neurological effects. A broad overview of the present literature shows a close relationship between BBB disruption and cognitive decline, the risk factors of BBB disruption, as well as the cellular and molecular mechanisms underlying BBB disruption. Additionally, we discussed the possible causes leading to cognitive decline by BBB disruption and potential therapeutic strategies to prevent BBB disruption or enhance BBB repair. This review aims to foster more investigations on early diagnosis, effective therapeutics, and rapid restoration against BBB disruption, which would yield better cognitive outcomes in patients with dysregulated BBB function, although their causative relationship has not yet been completely established.
Similar content being viewed by others
Introduction
The blood-brain barrier (BBB) is a semi-permeable barrier between the central nervous system (CNS) and the bloodstream, which dynamically and tightly regulates the bidirectional exchange of fluid, molecules, extracellular vesicles (EVs), and cells. Consequently, the BBB protects the CNS from harmful insults by selective substance permeation to prevent the passage of harmful substances into the brain parenchyma, and thereby provides a stable environment essential for maintaining brain homeostasis and function. Since the BBB plays a key role in the communication between the two sides during the physiological processes, its structural and functional damage may influence brain homeostasis, leading to brain dysfunction including cognitive decline.
Cellular components of the BBB
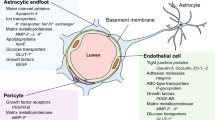
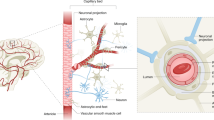
The BBB comprises a cerebrovascular network that forms a structural and chemical barrier. Histologically, it consists of non-fenestrated endothelial cells (ECs), pericytes, astrocyte endfeet, perivascular macrophages, and endothelial and parenchymal basal membranes [1]. Anatomically, the components of the BBB are defined as a part of neurovascular unit (NVU), consisting mainly of specialized ECs that establish intimate interactions with astrocytes and pericytes via basal lamina [2]. The basal lamina, a thin, dense cross-linked network of extracellular matrix proteins, is synthesized by ECs, astrocytes, and pericytes. Its components mainly include laminin, collagen IV, nidogen/entactin, and heparin sulfate proteoglycans. As a scaffold, it adheres those cellular components to provide a structural support for BBB integrity. These cellular components of the NVU can be activated in pathological conditions, which triggers BBB instability and severely damages its integrity. Figure 1 shows an intact BBB structure.

Schematic representation of the cellular and molecular structure of an intact BBB. The molecular and cellular components constitute an intact BBB which maintain brain homeostasis and function. The distribution of blood vessels and cells are shown on the left, a schematic cross-section of a blood vessel is shown in the center, and the molecular connections between endothelial cells are shown on the right. JAM: junctional adhesion molecule; PECAM: platelet endothelial cell adhesion molecule
Cerebrovascular ECs comprise around 5% of brain cells, which constitute the most important component of the BBB. Unlike that in peripheral ECs, the unique lipid composition in cerebrovascular ECs underlies the specific function of the BBB [3]. The ECs maintain BBB integrity through its interactions with astrocytes and pericytes, and regulate cerebral blood flow (CBF) respond to changes in neural activities via the release of vasoactive substances.
Astrocytes are the most abundant cells in the brain, with long processes that form endfeet. These astrocytic endfeet are intimate with the ECs and form a tightly overlapping barrier, which limits paracellular communication and diffusion [4, 5]. The astrocyte-EC complex, as the anatomical support of the BBB, controls the influx and efflux of biological substances at extremely low rates, driving transcellular vesicular transport. This process depends on the glycolytic metabolism of ECs [6]. Interestingly, a specific subset of Dmp1-expressing astrocytes can transfer mitochondria to ECs via their endfeet for maintaining BBB integrity [7]. Astrocytes are also reported to control the vascular tone, and hence CBF, through complex signaling cascades involving calcium elevation, release of arachidonic acid, and production of vasodilatory mediators such as prostaglandin E2 (PGE2), epoxyeicosatrienoic acids, and vasoconstrictive mediator 20-hydroxyeicosatetraenoic acid [8]. Therefore they appear to be essential during the early BBB development, and BBB maintenance and repair later in life.
Pericytes are located on the abluminal side of ECs and embedded in the basement membrane. Although they exist throughout the vascular network, pericytes are most abundantly present in the brain and are recruited during brain development for the stabilization of newly formed blood vessels and the integrity of the BBB. Pericyte-EC interaction is also essential during the BBB development. Initially, adhesion molecule CD146 is expressed in the ECs of immature capillaries in the absence of pericytes; however, once pericytes coverage occurs, CD146 is only detected in the pericytes while not in the ECs. Knockdown of CD146 in mouse ECs results in lower brain endothelial claudin-5 expression and BBB breakdown, whereas conditional deletion of CD146 in pericytes leads to defects in pericyte coverage and BBB integrity [9], suggesting its role in coordinating the pericyte-EC interaction. Furthermore, pericyte-EC interaction via endothelial nitric oxide (NO) synthase (eNOS)-derived NO can promote formation of functional vascular networks [10], implying that restoring pericyte-EC interaction after a stroke may improve brain functional recovery. Pericytes can regulate the BBB-specific gene expression patterns in ECs, promote oligodendrocyte progenitor cell (OPC) differentiation, and induce polarization of astrocytic endfeet [11]. Pericyte-deficient adult brains display ongoing angiogenic sprouting without concomitant cell proliferation, whereas a lack of pericytes recruitment results in increased BBB permeability and rupture of microaneurysms associated with cerebral hemorrhage [12]. Platelet-derived growth factor B (PDGFB) released from ECs is indispensable for the recruitment of pericytes expressing the PDGFB receptor beta (PDGFRβ) during angiogenesis and maintenance of pericyte longitudinal capillary coverage via PDGFB–PDGFRβ signaling. Interestingly, a recent study showed that PDGFB released from ECs in neonatal mice was converted to release from microglia in adult mice [13]. Acute deletion of microglial PDGFB greatly impairs BBB integrity in adult mice but not in newborn ones. In contrast, acute knockdown of endothelial PDGFB severely influences CNS microvasculature in neonatal mice but not the BBB in adult ones [13], suggesting a role of microglia in maintaining the BBB integrity during adulthood, although they are not regarded as a component of the BBB. PDGFB deficiency can lead to an impaired BBB, characterized by a pericyte hypoplasia and a sparse, dilated, and venous-shifted brain microvasculature, as well as the formation of microvascular calcification [14]. Similarly, PDGFRβ knockout models exhibit pericyte deficiency and progressive BBB damage secondary to hemodynamic disturbances [15].
Molecular formation of the BBB
ECs are interconnected via the integral membrane proteins tight junctions (TJs) and adherens junctions (AJs). Intercellular adaptors link these TJs (e.g., occludin and claudin) and AJ (e.g., the cadherin–catenin and nectin–afadin complexes) proteins to the cytoskeleton (such as zonula occludens) for the BBB construction at the ultrastructural level [16]. The TJs between adjacent ECs are responsible for the intercellular restrictive barrier properties, the so-called “fence function”. Using direct stochastic optical reconstruction super-resolution microscopy, Sasson et al. revealed the nanoscale architecture of TJs and found sparse occluding and clustered zonula occludens 1(ZO-1)/claudin-5 molecular architecture in the normal cortical BBB of mice, and claudin-5 levels were inversely correlated with TJ functionality [17]. The AJs contain vascular endothelial cadherin (VE-cadherin), which connects neighboring ECs through its extracellular domain during homeostasis, whereas inflammatory mediators, such as histamine, thrombin, or vascular endothelial growth factor (VEGF) can disrupt endothelial barrier by affecting the VE-cadherin adhesion and distabilizing the AJ complexes.
As a dynamic structure covering the luminal side of the microvascular ECs, the cerebral endothelial glycocalyx (CeGC), consisting of proteoglycans, glycoproteins, and glycolipids, is also a significant determinant of BBB function. The CeGC is thicker in the cerebral microvasculature, suggesting its functional specialization as the first physical barrier of the BBB. Therefore CeGC degradation may initiate BBB dysfunction, leading to BBB permeability associated with cognitive impairment [18]; and plasma CeGC thus is considered a potential diagnostic biomarker of BBB breakdown.
Several endothelial proteins are also essential for BBB integrity. A recent study indicated that deletion of endothelial autophagy related 7 (Atg7) led to astrocytic endfeet detachment from the microvasculature through downregulation of fibronectin expression, a major basement membrane component of the BBB [19]. This resulted in insufficient coverage of astrocytic endfeet along the cerebral microvasculature, suggesting a role of endothelial Atg7 in astrocyte-EC interaction to maintain the BBB integrity. Additionally, endothelial β1 integrin blockade is reported to be associated with EC disintegrity and TJ disruption [20], which may account for BBB breakdown. Similarly, bone morphogenetic protein (BMP) is reported pro-angiogenic. A conditional knockout neonatal mouse model verified that BMP affected cerebral angiogenesis through its receptor BMP type IA (Bmpr1a) [21], suggesting a role of BMP signaling in BBB formation. Furthermore, astrocytic Bmpr1a knockout significantly decreased cerebral microvasculature density and VEGF level, leading to astrocytic endfeet failure to encircle the cerebral microvasculature. However, Bmpr1a blockade at the site of BBB breakdown can rescue the inhibitory effects of remyelination and promote OPC differentiation into myelinating oligodendrocytes [22]. Therefore, the overall effects of BMP signaling on functional recovery after BBB disruption remain unclear.
As a specific endothelial marker of the BBB, major facilitator superfamily domain-containing 2a (Mfsd2a) is regulated by pericytes to facilitate BBB integrity and lysophosphatidylcholine transport across the brain. Mfsd2a is also essential for sphingosine-1-phosphate (S1P) export from ECs into the brain, whereas the S1P-rich microenvironment in the cerebrovascular ECs modulates the formation and integrity of the BBB [23] through phosphatase and tensin homolog (PTEN)/Akt/Nedd4-2/Mfsd2a signaling [24]. Animal studies have shown that although normal patterning of vascular networks and TJ proteins are preserved, genetic ablation of Mfsd2a can result in a leaky BBB from the embryonic stage through to adulthood in mice, causing neural deficits and brain edema after intracerebral hemorrhage [25]. Conversely, Mfsd2a overexpression can rescue BBB disruption [26]. Further studies have indicated that Mfsd2a maintains BBB function by inhibiting caveolae-mediated transcellular transport [3, 26]. These data suggest Mfsd2a is a potential target for modulating BBB permeability. Similarly, monocarboxylate transporter 8 (MCT8), a transmembrane transporter, is expressed at the BBB. MCT8-deficient adult mouse brains are characterized by increased BBB permeability, transcytotic flux, and decreased blood vessel density [27].
Functions of the BBB
Physical barrier
Except small, nonpolar molecules, an intact BBB prevents the influx of most blood-borne substances into the brain. First, specialized TJs prevent paracellular passage of water-soluble molecules from the circulatory system to the brain parenchyma. Second, ECs exhibit extremely low levels of vesicle trafficking, known as transcytosis. Therefore, when the BBB is disrupted, blood-borne substances can enter the brain at sufficiently high concentrations through an un-saturable mechanism to trigger harmful events. However, the BBB can also act as an obstacle to impede large molecules (such as drugs, peptides, and proteins) delivery into the brain. Different therapeutic strategies, including osmotic disruption, physical interventions such as focused ultrasound (FUS), and chemical modification of drugs such as nanoparticles, have been used to transfer chemotherapeutic agents across the BBB to treat brain disorders, i.e., neurodegenerative diseases and brain tumors. Although BBB disruption promotes drug transport across into the brain, it may also exacerbate the transport of other molecules, as described below.
Substance transport
The BBB facilitates the supply of nutrients (influx, i.e., glucose and oxygen) and disposal of metabolites (efflux, i.e., amyloid beta, Aβ) via the expression of transporters and transcytotic receptors at the surface of the ECs. Transport routes across the BBB include paracellular and transcellular diffusion, and receptor-, cell-, transporter-, and absorptive-mediated transcytosis [28, 29]. In general, steroid hormones cross the BBB via a un-saturable process, transcellular diffusion, whereas thyroid hormones and many other peptides and regulatory proteins cross the BBB using transporters, a saturable process [30]. The endogenous transporters for omega-3 fatty acids [31] and reduced folic acid [32] are also located in the BBB. For example, Mfsd2a is a lysophosphatidylcholine transporter that mediates Na+-dependent uptake of ω-3 fatty acids into the brain. MCT8 is responsive to thyroid hormone transport, and transport of the active form of T3 across the BBB is strongly diminished in Mct8-knockdown animals [33]. In contrast, the BBB is also a major pathway for the clearance of brain Aβ. This process requires specific transporter proteins such as low-density lipoprotein receptor-related protein 1 (LRP1), very low-density lipoprotein receptor, and P-glycoprotein [34]. Therefore, the activity of these polarized efflux transporters can alter the BBB permeability, which affects the relationship between Alzheimer’s disease (AD)-associated biomarkers in the brain and blood. Interestingly, in individuals with high BBB permeability, the plasma Aβ42/40 level can be better identified the Aβ42/40 level in cerebrospinal fluid (CSF) and Aβ-positron emission tomography (PET) positivity. In contrast, BBB permeability does not alter the relationship between brain and plasma phosphorylated tau (p-tau) levels [35].
Immune privilege
The BBB strictly regulates the entry of proinflammatory substances and blood-borne immune cells into the brain [36]. Damage to the BBB can facilitate the entry of peripheral macrophages into the brain and activate resident immunocytes-microglia in the brain. Prenatal damage to the BBB is an important cause of maternal inflammation-induced brain injury in the offspring [37]. A recent study demonstrated that gestational maternal immune activation induces disruption of BBB formation through the proliferation of cyclooxygenase-2 (COX2)-expressing microglia in the brain parenchyma and perivascular spaces of fetal mice, leading to persistent neuroinflammation in adult offspring. In contrast, inhibition of COX2 expression prevents microglial proliferation and BBB breakdown [38], may leading to a reduction in the incidence of neuropsychiatric disorders. These data suggest that the BBB formation is important for brain development.
Regulation of CBF
The CBF is controlled by various neurophysiological mechanisms via the cerebrovascular network, including cerebral perfusion pressure, intracranial pressure, arterial blood gases, neural activity, and metabolic demand. CBF is important for the maintenance of brain homeostasis and function, while its dysregulation has been implicated in neurodegenerative diseases. The pericytes play important role in controlling neurovascular functions through modulation of CBF [39], specifically, cerebrovascular contraction and relaxation in response to neuronal activities are used to regulate the CBF, known as neurovascular coupling. Vasoactive substances such as NO and prostaglandins may contribute to this regulation. Age-dependent chronic pericytes loss can trigger capillary rarefaction, leading to CBF reduction. A recent study revealed that a subset of microglia may be associated with pericytes lacking astroglial endfeet coverage, and if pericytes lose such microglia, capillary width is decreased and CBF regulation is affected [40], which is commonly found in AD.
Neuroendocrine function
The BBB also plays a role in neuroendocrine effects. As an endocrine tissue, BBB can secrete several hormone-like substances. For example, ECs release NO, prostaglandins, and adrenomedullin [41], which may influence brain functions by acting on neuroendocrine nuclei in the brain. NO signaling is involved in controlling neuroendocrine cells in the hypothalamus, whereas PGE2 is regarded as a mediator of fever. Peripherally released pyrogenic substances (i.e., proinflammatory cytokines) bind to their receptors on cerebral microvascular ECs, which elicits COX-2-catalyzed PGE2 synthesis to evoke fever through their binding in the preoptic hypothalamus [42]. Adrenomedullin, another hormone released by ECs, plays multiple roles during brain injuries and neurodegenerative diseases. Endothelial dysfunction significantly increases adrenomedullin expression, and adrenomedullin levels are also elevated in human AD brains and transgenic mouse models of AD.
As BBB disruption is commonly found in many brain disorders, reliable and sensitive methods to detect BBB disruption are of great clinical interest. The established methods for preclinical and clinical assessments of BBB integrity are summarized in Table 1. Recently, several plasma EC proteins associated with cerebral small vessel disease (cSVD) have been analyzed, and show promise for quantifying BBB disruption. For example, the AJ protein cadherin-5 is associated with preserved BBB integrity [43], whereas glycoprotein 1b platelet subunit beta is positively associated with a permeable BBB [44].
BBB disruption in cognitive impairments
Cognitive decline commonly refers to impairments in a single or multiple cognitive domains, including executive abilities, attention, visuospatial abilities, and memory. Since the BBB closely controls the microenvironment in the brain parenchyma, several brain disorders that cause cognitive decline have been linked to widespread BBB disruption. In turn, cerebrovascular damage can promote neurodegenerative diseases, which adversely influence cognitive functions [45]. These typical disorders can be divided into three categories: (1) primary neurodegenerative diseases such as AD, (2) primary cerebrovascular lesions such as acute stroke, and (3) secondary to systemic disorders (i.e., sepsis and diabetes). Here, we exemplify several clinically common scenarios to show a close link between BBB disruption and cognitive decline.
Neurodegenerative diseases
Neurodegenerative diseases are a wide range of neurological disorders, characterized by abnormal behavioral and cognitive changes. Human neuroimaging and biomarker studies have demonstrated that BBB breakdown is a common feature in AD, Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), multiple sclerosis, human immunodeficiency virus-1-associated dementia, and chronic traumatic encephalopathy [46].
Alzheimer’s disease
AD is the most common type of dementia, characterized by the formation of amyloid plaques and neurofibrillary tangles in the brain. Several clinical studies have demonstrated BBB disruption in AD via autopsy [47] or neuroimaging method [48]. BBB disruption is found to precede Aβ deposition, p-tau accumulation, microglial activation, and neuronal cell death. Tau protein can trigger BBB structural and functional damage driven by chronic neuroinflammation, as evidenced by low levels of inflammatory cytokines in the brain. In an amyloid precursor protein (APP) knock-in mouse model of AD, initial Aβ accumulation is sufficient to change basement membrane-associated and ribosomal protein expression at the BBB, until advanced Aβ accumulation alters protein expression in the brain parenchyma [49]. In patients with early AD, endothelial angiopoietin-2 (Ang2) is elevated in CSF, correlated strongly with the CSF/serum albumin ratio (Q-Alb), an indicator of BBB disruption [50], and BBB breakdown in the hippocampus occurs prior to the onset of brain atrophy and cognitive dysfunction [51]. Autopsy also provides direct evidence of BBB destruction, including pericytes loss, endothelial degeneration, and cell leakage in these patients [52]. Conversely, Aβ overexpression in animal AD models triggers pathogenic cerebrovascular neoangiogenesis, leading to hypervascularity, coincident TJs disruption, and BBB leakage [53]. However, when pro-angiogenic Aβ level decrease, amyloid plaque burden and cerebrovascular pathogenesis subside. While these pathological changes can be considerably relieved by an anti-cancer drug Axitinib, which effectively restores memory and cognitive performance in a preclinical model of AD. Furthermore, CSF biomarker studies have confirmed BBB breakdown in patients with AD by measuring the Q-Alb and soluble PDGFRβ (a pericyte injury marker) levels both in the CSF and plasma [54, 55]. Interestingly, BBB permeability to small molecules (i.e., water) rather than to large molecules (i.e., albumin) may have a greater impact on cognitive performance [54]. In patients with mild cognitive impairment or dementia, higher cortical BBB permeability is associated with a higher CSF Aβ42/40 ratio and lower hippocampal volume and cognitive scores in Aβ-PET positive patients; BBB breakdown is positively related to CSF total tau level but negatively related to p-tau level [56]. These evidences address the relationship among BBB permeability, AD-specific biomarkers, and cognition, although this relationship varies in the presence of Aβ accumulation. Taken together, BBB breakdown likely plays an early and important role in the pathogenesis of AD.
Parkinson’s disease
PD is characterized by abnormal accumulation of alpha-synuclein (αSyn) aggregates, loss of dopaminergic neurons, and gliosis of the substantia nigra. The risk of cognitive impairment in patients with PD increases with disease progression, the spectrum including subjective cognitive decline, mild cognitive impairment, and dementia [57]. In vivo and in vitro PD models have been reported BBB disruption. In a rotenone-induced mouse PD model, rotenone dose-dependently induced nigral dopaminergic neurodegeneration via decreasing the expression of the TJ proteins ZO-1, claudin-5, and occludin, and increasing Evans blue content, fibrinogen accumulation, and microglial activation [58]. Inhibition of microglial activation in a mouse PD model can attenuate the activation of matrix metallopeptidase (MMP)-2/9 and BBB disruption, thereby improving dopaminergic neurodegeneration and motor dysfunction [59]. Using dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), PD patients have also been found to have significantly higher BBB leakage [60]. The Q-Alb and plasma protein markers reflect BBB disruption in patients with idiopathic PD and patients with PD dementia and/or dementia with Lewy bodies, whereas greater BBB breakdown is seen in Lewy body disease with cognitive impairments [61].
Huntington’s disease
HD is an inherited neurodegenerative disease which causes involuntary movement, severe emotional disturbances, and cognitive decline. The clinical cognitive features include executive dysfunction, psychomotor symptoms, visuospatial deficits, perceptual deficits, memory loss, and learning difficulty. Experimental studies have demonstrated that BBB permeability progressively increases in multiple brain regions in HD models using sodium fluorescein staining [62], suggesting that BBB hyperpermeability is a driving factor in HD, leading to disease progression [63]. Increased MMP-9 expression may contribute to striatal BBB disruption in a HD animal model, as evidenced by high Evans blue extravasation and low expression of BBB markers: endothelial brain barrier antigen, ZO-1, and laminin [64].
Overall, BBB disruption is evident in these neurodegenerative diseases. A longitudinal study in a memory unit showed that increased BBB disruption contributed to deterioration in cognitive decline in patients with pre-existing cognitive impairment [65], further linking the BBB disruption to cognitive dysfunction. Therefore, the BBB may be a promising target for the treatment of neurodegenerative diseases.
Psychotic disorders
Patients with psychiatric disorders, such as schizophrenia (SZ), bipolar disorder (BD), and depression have more social cognitive and nonsocial cognitive impairments than healthy individuals [66]. Several studies suggest that BBB disruption contributes to the pathogenesis of these psychiatric disorders [67]. To determine whether BBB deficits are intrinsic to brain microvascular ECs or arise via the effects of peripheral inflammatory cytokines, Lizano et al. examined TJ proteins expression and BBB permeability in brain microvascular ECs (BMECs) derived from induced pluripotent stem cells from SZ, BD, and healthy control individuals, and found that BMECs from patients with psychotic disorders did not show differences in BBB integrity or permeability compared to control BMECs [68]. However, those patients with SZ and BD showed a BBB-deficit subtype with reduced claudin-5 level and barrier function, increased MMP-1 activity and barrier permeability [68]. In particular, the Q-Alb is higher in patients with SZ than in healthy controls. Furthermore, subgroup analyse showed that patients in partial symptom remission had a higher Q-Alb than those patients in full symptom remission [69]. The meta-analysis also indicates that neuroinflammation and BBB permeability are found in patients with unipolar depression [70]. These studies support the contribution of BBB hyperpermeability to the development of psychotic disorders.
Claudin-5 is downregulated in numerous depression models. Reduced endothelial claudin-5 expression and abnormal microvessel morphology in the nucleus accumbens (NAc), a brain region associated with mood regulation, are found in chronic social defeat stress models. Knockdown of claudin-5 in the NAc is sufficient to induce depression-like behaviors through promoting peripheral interleukin-6 (IL-6) across the BBB into the brain, whereas chronic antidepressant treatment can reverse the claudin-5 loss [71]. Further, low claudin-5 levels at the BBB is associated with depression in a chronic unpredictable mild stress model, whereas increased claudin-5 expression can alleviate hippocampal tumor necrosis factor alpha (TNFα) expression, and depression-like behaviors [72]. Similarly, a prolonged learned helplessness depression model presents higher levels of hippocampal glycogen synthase kinase-3 and proinflammatory factors, lower levels of TJ proteins, and BBB hyperpermeability [73]. Similarly, Peng et al. showed that chronic restraint stress-induced depression exhibited BBB disruption in the mouse dorsal striatum [74]. TNFα/nuclear factor kappa B (NF-κB) signaling and histone deacetylase 1 (HDAC1) have been identified as mediators of claudin-5 and occludin loss [75]. Furthermore, inhibitors of TNFα, MMP-1, and HDAC1 can ameliorate BBB deficits in these depression models, suggesting that prolonged exposure to peripheral inflammation may contribute to BBB disruption in psychotic disorders. The convergent evidence indicates that BBB disruption may contribute to behavioral and cognitive symptoms of psychotic disorders via affecting brain perfusion and homeostasis [76], however, whether BBB disruption causally triggers cognitive impairments in psychiatric disorders is still not clear.
Cerebrovascular diseases
The neuroinflammation and BBB damage can result in disruption of brain homeostasis, both contributing to the pathogenesis of cerebrovascular diseases [77]. Gadolinium-based imaging studies in humans have demonstrated active BBB breakdown in patients with subcortical ischemic vascular disease, vascular cognitive impairment, and chronic cortical ischemic stroke lesions, therefore loss of BBB integrity in cerebrovascular diseases may be an early biomarker of human cognitive decline [78].
Acute cerebrovascular diseases
BBB disruption develops during ischemic and hemorrhagic strokes, which increases the influx of water molecules and blood components into the brain extracellular space. This results in angiogenic edema and further hemorrhage, thus exacerbating brain homeostasis and neuronal damage [79, 80]. Therefore, BBB disruption in stroke may induce cognitive impairments.
Hemorrhagic stroke or aneurysmal subarachnoid hemorrhage (SAH) is associated with disrupted TJ proteins levels, and increased BBB permeability and cerebral edema. The C-C motif chemokine receptor 5 (CCR5) expression increases in the acute phase after cerebral ischemia, and contributes to BBB disruption and neuroinflammation [81]. The matricellular protein tenascin-C is induced when mice are subjected to SAH, while its knockout prevents BBB disruption, brain edema, and neurological impairments following SAH [82]. These neuroprotective effects appear to be associated with the MMP-9 inhibition and ZO-1 degradation. A recent preclinical study has showed that overexpression of MMP-9 derived from reactive astrocytes after SAH plays a deleterious role in the BBB permeability. Deletion of astrocytic N-myc downstream-regulated gene 2 can greatly decrease MMP-9 production, resulting in improvement of BBB damage [83]. SAH-induced BBB disruption can promote further hematoma expansion [84], therefore, markers of BBB remodeling can be used to predict the occurrence of hematoma expansion [85]. In acute cerebrovascular diseases, hypoxia induces BBB breakdown and a subsequent inflammatory response, as evidenced by increases in hypoxia inducible factor-α (HIF-α) and cytokine levels in the stroke area [86]. Thus, to control the onset of BBB disruption in acute cerebrovascular disease may assist in reducing adverse neurological outcomes, including long-term cognitive decline.
Chronic cerebrovascular diseases
Vascular damage owing to aging, hypertension, and diabetes can also trigger cognitive impairments, known as vascular cognitive impairment. BBB damage has been found in the course of these chronic cerebrovascular diseases, as evidenced by changes in CSF biochemical indicators and neuroimaging findings [87]. This BBB destruction is caused by microvascular injury and appears to be secondary to hypoxia-associated MMPs overexpression [88]. For example, in patients with reversible cerebral vasoconstriction syndrome, dynamic changes in BBB permeability are associated with impaired cerebral microvascular compliance [89], whereas regional BBB changes are associated with a reduction in the local CBF. Through activation of signaling pathways related to excitotoxicity, oxidative/nitrosative stress, inflammation, and MMPs overexpression, downstream perivascular damage and leukocyte infiltration are exacerbated in the brain. This chronic cerebral hypoperfusion initiates a vicious cycle of BBB damage, and contributes to further damage to brain homeostasis, leading to vascular cognitive impairment [90]. A recent genome-wide transcriptomic analysis of cerebral microvessels revealed vascular damage (e.g., serpine1/plasminogen activator inhibitor-1 and hemoxygenase-1), endothelial activation (e.g., Ang2), and sphingolipid signaling (e.g., S1P receptor 2) -associated molecular features in a mouse model of stroke and human chronic stroke lesions [91]. These molecular alterations provide novel insights into the mechanisms underlying BBB disruption and the therapeutic potential for BBB protection in stroke.
Traumatic brain injury (TBI)
Numerous studies have indicated that BBB disruption is a core component of TBI pathophysiology. Excessive neuroinflammation after the acute phase of TBI triggers the MMPs overexpression, which proteolytically degrade TJ proteins and disrupt BBB integrity not only in focal lesions, but also in peri-lesional and non-lesional regions [89]. Consequently, BBB hyperpermeability in turn leads to secondary long-term neuropsychiatric symptoms after TBI. Supplementation with vitamin D3 or ethyl pyruvate can attenuate TBI-induced brain edema and neurobehavioral deficits [92, 93]. DCE-MRI can confirm that BBB permeability is remarkably elevated in association with focal traumatic lesions during a median of 23 days after hospitalization for TBI, and significant BBB disruption remained several months later [94]. Further neuroimaging study showed a subtle yet significant BBB disruption up to 1.5 years following TBI, validated by histological examination [95]; whereas microdialysis study demonstrated that BBB disruption after TBI could cause long-term neurotoxic dysregulation, including glutamate excitotoxicity, excessive free radical generation, and neuroinflammatory responses, leading to further BBB damage and secondary brain injury [96]. Although the BBB disruption is most frequently observed in the anterior and posterior brain regions in a spatial distribution, the correlation between BBB disruption and cognitive functions such as verbal learning is inconsistent [94, 97].
ECs are an important regulator of the cerebrovascular response to TBI. Conditional knockout of the EC-specific ephrin receptor A4 can protect BBB integrity from TBI, including increases in the expression of TJs and actin cytoskeletal regulators, and a decrease in Ang2 expression, which correlates with motor improvement and CBF recovery in a controlled cortical impact mouse model [98]. Intriguingly, because soluble Ang2 is also increased in individuals with acute TBI, it may be a promising serum biomarker indicating an early BBB disruption. Apoptosis signal-regulating kinase 1 (ASK1) plays a key role in the degradation of TJ proteins and disruption of BBB integrity, leading to the infiltration of peripheral immune cells into the brain parenchyma and increased number of proinflammatory microglia/macrophages following TBI [99], suggesting the importance of the BBB in the evolution of TBI. While ASK1 inhibition displayed better recognition memory, social novelty recognition ability, and spatial cognitive function 25–35 days post-TBI in TBI model mice [99], indicating ASK1 could be a therapeutic target for improvement of TBI-induced long-term cognitive deficits.
Sleep disorders
Sleep disorders, such as chronic sleep restriction and fragmentation, are becoming increasingly a common health problem. Since circadian oscillations dynamically regulates specific properties of the BBB, there is a link between sleep or circadian disturbances and neurodegenerative diseases including AD [100]. Sleep deprivation (SD) can induce deterioration in mental and cognitive performance, and BBB disruption has been found in animals and patients with SD [101, 102]. For example, SD can diminish expressions of eNOS and inducible NOS, endothelin1, and glucose transporter in cerebral microvessels, and consequently, SD mice have lower expression of TJ proteins and impairment of BBB transport [103], while exhibiting an elevated BBB catabolism [101].
Sleep restriction can promote astrocyte phagocytosis and microglial activation, which may be associated with abnormal slow-wave electroencephalographic activity during sleep [104]. This leads to altered vascular reactivity, BBB inflammation, endothelial detachment from pericytes, and thus BBB destruction [102, 103, 105]. Studies have demonstrated that loss of rapid eye movement sleep can induce disturbances in brain homeostasis secondary to BBB breakdown, whereas sleep replenishment via rebound sleep or administration of gaboxadol can rapidly and effectively normalize BBB permeability [106], suggesting this effect of SD on the BBB is reversible. Sleep disorders lead to a systemic low-level but sustained inflammatory status that promotes BBB disruption [107]. However, in prolonged SD, prostaglandin D2 is overproduced in the brain and effused to the bloodstream across the BBB to induce a cytokine storm, leading to multi-organ dysfunction and death [108]. Sleep disorders also increase caveolae formation in cerebral microvascular ECs, inducing BBB hyperpermeability. Conversely, altering BBB permeability through moody G protein-coupled receptor signaling can induce robust sleep in Drosophila [109], indicating an interesting role of the BBB in sleep regulation. Moreover, sleep disorders-induced BBB disruption appears age-related, for instance, sleep fragmentation disrupts the BBB and increase TNF-α transport in aged rather than young mice [110]. Further, deletion of a major circadian clock transcriptional activator, brain and muscle aryl hydrocarbon receptor nuclear translocator-like protein 1, can also cause an age-dependent loss of vascular pericyte coverage and BBB breakdown in the mouse brain [111]. These data suggest that an older BBB may be more susceptible to sleep disorders.
Systemic diseases
Numerous systemic diseases also contribute to BBB disruption. Here, we illustrate the link between BBB disruption and systemic disease-associated cognitive decline.
Infectious diseases
Sepsis-associated encephalopathy (SAE)
Diffuse cerebral dysfunction is often secondary to acute sepsis, commonly manifesting as delirium [112], also known as SAE. Approximately one in six patients with sepsis have long-term cognitive, motor, and mental impairments [113]. Increased cytokines production and BBB disruption are involved in the pathogenesis of SAE. MRI investigation has shown increased brain permeability in rodent sepsis models [114]. Specially, cellular components of the BBB are damaged or altered during sepsis [115,116,117]. As BBB impairment has an impact on sepsis-induced long-term cognitive function, targeting BBB damage could limit the progression of neurological deficits, including cognitive decline. Recent studies also suggest regulation of intestinal flora plays an important role in SAE. The key microbial mediators in the brain-gut axis have been found to maintain BBB integrity and prevent cognitive decline in SAE. In vitro studies have shown that enterobacterial metabolites alter the cytoskeletal arrangement of brain ECs, thus increasing TJ proteins [118, 119].
Bacterial meningitis
One defining feature of bacterial meningitis is elevated BBB permeability. This BBB breakdown is mediated by a combination of increased bacterial virulence factors (including bacterial surface structures, hemolysins, and associated enzymes) and signals from the host that regulate the BBB (including cytokines, angiogenic factors, apoptotic factors, transcription factors, metalloproteinases, and non-coding RNAs) [120]. A recent study showed that streptococcal M family protein SzM released during streptococcal meningitis forms SzM-bound membrane vesicles, and endocytosis of these vesicles by human brain microvascular ECs could lead to BBB disruption [121]. While PTEN-activated autophagic EC death contributes to SzM-induced cytotoxicity in microvascular cells. Therefore, targeting the pathological factors that contribute to BBB hyperpermeability could be an effective therapeutic complement to antimicrobial therapy against bacterial meningitis.
Prion disease
Most of cognitive domains, especially the frontal executive, language, and parietal functions are impaired in patients with prion disease. BBB disruption in a prion disease model was reported three decades ago. In an in vitro BBB model, the prion protein reduced the expression of TJ proteins and the ratio of cytoskeletal proteins F-actin/G-actin [122], suggesting a prion-induced BBB disruption. A recent in vivo study showed that in prion-infected mice, the loss of ECs and downregulation of TJ and AJ proteins, including occludin, claudin-5, and VE-cadherin, appeared to be driven by reactive astrocytes prior to disease onset [123]. However, aquaporin 4 (AQP4) expression in astrocytic processes is significantly increased while its expression in astrocytic endfeet is remarkedly decreased at the BBB, suggesting a detachment of astrocytic endfeet from ECs and a loss of BBB integrity. In vitro experiments further showed that reactive astrocytes from prion-infected mice impaired intercellular adhesion and barrier function of primary mouse brain ECs. This may be attributed to proinflammatory factor IL-6 secreted from reactive astrocytes.
COVID-19 infection
Coronavirus disease 2019 (COVID-19) has been reported to affect brain structure and function via direct or indirect immune activation [124, 125]. Because ECs, pericytes, and astrocytes highly express angiotensin converting enzyme 2 (ACE2), severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) may disturb BBB components, including cells and TJ and AJ proteins. Furthermore, viruses can cross microvascular ECs through the ACE2 receptor-mediated pathway and trigger a neuroinflammatory response. In contrast, by using a BBB-alveolus chip system, Wang et al. found that SARS-CoV-2 caused mild BBB changes, whereas conditioned medium from the infected alveolus could trigger more severe BBB damage, resulting from endothelial dysfunction, pericyte detachment, and neuroinflammation [126]. Lung infection with a low viral load can induce early cerebral microvascular damage in the transgenic mice expressing human ACE2, suggesting systemic inflammation rather than direct viral neural invasion may contribute to neuropathogenesis. SARS-CoV-2-induced cytokines release and systemic inflammation can disrupt the BBB integrity by reducing TJ proteins expression [127]. BBB disruption has been detected in patients with COVID-19, along with encephalopathy and CSF abnormalities [128]. Interestingly, sustained systemic inflammation and BBB disruption have been found in patients with long COVID, so-called “brain fog” [129]. Serum from patients with long COVID can induce increases in inflammatory markers in brain ECs, which provides an insight into the mechanism underlying long COVID. The link between BBB disruption and COVID-19-associated cognitive deficits has been discussed elsewhere [130, 131].
Vital organ dysfunction
End-stage diseases typically have CNS involvement, manifesting as cognitive dysfunction. BBB breakdown, including TJ proteins reduction and endothelial disruption, can be observed in animal models and patients with chronic hepatic encephalopathy and fulminant hepatitis, which is associated with increased MMP-9 activity [132,133,134]. In addition, common neuropathological features in acute liver failure, such as astrocytes swelling and microglial activation, can lead to BBB abnormalities [135]. In patients with chronic kidney disease, uremic toxins such as indoxyl sulfate, may play a role in the development of cognitive impairment. An increase in serum indoxyl sulfate concentrations is associated with higher BBB permeability and a stronger impairment in several cognition domains. This can be explained by the fact that most of uremic toxins are agonists of the transcription factor aryl hydrocarbon receptor (AhR) widely expressed in cerebrovascular ECs. An experimental study has shown that AhR knockout could protect against indoxyl sulfate-induced BBB disruption and cognitive impairments [136].
Postoperative neurocognitive disorders (PND)
Anesthesia/surgery can induce PND, predisposing patients to long-term cognitive impairment, especially in elderly patients and those with pre-existing cognitive disorders. Several MRI studies have indicated that neurovascular rather than neurodegenerative changes, are consistently associated with postoperative delirium (POD), characterized by disturbances in awareness, attention, and cognition [137, 138].
Intravenous anesthetic propofol is reported to trigger excessive production of inflammatory factors (IL-1β, IL-8, and monocyte chemoattractant protein-1 [MCP-1]), adhesion molecules (ICAM-1 and VCAM-1) and MMP-2 in human brain microvascular ECs, which increases permeability of an in vitro BBB model [139]. Inhalational anesthetics isoflurane and sevoflurane can also induce BBB opening, leading to cognitive decline. Plasma immunoglobulin G leakage from cortical microvessels is found to be significantly increased in rat exposed to 3% sevoflurane or isoflurane for 3 h, which is associated with brain microvascular ECs death [140]. Moreover, the BBB in immature and aged brains may be more susceptible to anesthetic-induced neurotoxicity [141, 142]. Inflammation and oxidative stress may be responsible for this anesthetic-induced BBB damage and thus memory loss [143].
Surgical trauma can elicit a systemic inflammatory response. The upregulated proinflammatory cytokines IL-1β and IL-6 levels can promote BBB damage, microglial activation, and peripheral immune cells infiltration, leading to delirium-like behavior in mice [144, 145]. In an animal model of delirium, impaired astrocytic-TJ interaction is found after orthopedic surgery [146]. BBB impairment in PND is also reported to be inflammation-mediated and age-dependent [147].
The patients undergoing coronary artery bypass grafting under cardiopulmonary bypass have a significant increase in BBB disruption, evidenced by frontal permeability and peripheral immune cells infiltration into the brain parenchyma, resulting in neuroinflammation, and cognitive impairments [147, 148]. In these patients, the postoperative global cognitive score is reduced, predominantly in executive and attention frontal functions. Furthermore, the intensity of BBB permeability is correlated with the magnitude of cognitive impairment. The biomarkers of BBB disruption can also be found in post-surgical CSF biochemical tests [149]. In a recent clinical trial, for example, in older adults undergoing non-cardiac surgery, the Q-Alb increased in both patients with and without POD, however, preoperative to postoperative change in Q-Alb was greater in patients with POD [150], suggesting that postoperative BBB hyperpermeability is independently associated with POD. Similarly, in a small sample-size clinical study, a change in cortical BBB permeability was most strongly associated with a lower composite cognitive score 6 weeks after surgery, and patients with lower cognitive scores had the BBB in a greater number of brain regions involved, including the cortex, deep gray, and cerebellum [151]. Moreover, pre-existing BBB damage can increase the incidence of POD [152].
Current data suggests a contributing role of BBB disruption in PND, therefore several investigators have claimed that POD or PND is mainly mediated by BBB dysfunction [148, 153, 154]. However, other studies indicated that early transient increases in the astroglial biomarkers S-100β and glial fibrillary acidic protein in the CSF, or Q-Alb, failed to predict long-term cognitive decline following major surgery [155]; and the BBB integrity was found similar between baseline and 1 month postoperatively [156]. Therefore, it appears too arbitrary to consider BBB disruption as the sole pathogenesis of PND.
Cancer/cancer treatment-related cognitive impairments
Cognitive complications associated with cancer and cancer treatment have become an increasing concern. Numerous studies support the notion that a certain subset of cancer survivors is vulnerable to cognitive impairments after treatment [157]. One of the mechanisms is BBB disruption associated with neuroinflammation and oxidative stress [158].
Chemotherapy-induced cognitive impairment (also known as “chemobrain”) is a common neurological complication in treatment for the control of malignancies. Cognitive impairments, including deficits in learning, memory, attention, executive function, and processing speed, have been observed in cancer survivors receiving chemotherapy. Therefore, chemobrain places a significant psychosocial burden on cancer survivors and has a considerable impact on their daily activities. Several chemotherapeutic agents, including oxaliplatin and irinotecan, can directly alter BBB permeability, while others such as adriamycin do not directly cross the BBB, but they disrupt BBB integrity may through microbiota-gut-brain axis [159] or peripheral immune cells-released cytokines and reactive oxygen species (ROS) [160]. In particular, TNF-α causes neuronal death and eventually cognitive impairment by binding to its receptors at the BBB and being translocated to the parenchyma via receptor-mediated endocytosis [160]. Chemotherapy can also promote cerebrovascular senescence. For example, paclitaxel-induced ECs senescence can drive neuroinflammation, consequently, decrease microvascular density, dysregulate neurovascular coupling and increase BBB permeability. These changes impair spatial memory performance in paclitaxel-treated mice [161]. Therefore, elimination of senescent ECs may be a promising treatment for “chemobrain”.
Radiotherapy, another therapeutic paradigm for cancer patients, can induce adverse neurological consequences. Cognitive decline after partial or whole-brain irradiation for primary brain tumors and metastases has been demonstrated in clinical trials and experimental studies [162, 163], marked by reduced verbal and spatial memory, attention, and novel problem-solving abilities. Whole-brain irradiation-induced cognitive deficits is considered associated with cerebrovascular damage [164]. Preclinical studies also demonstrate that irradiation-induced cognitive deficits is related to neuroinflammation, BBB disruption, demyelination, and a decline in neurogenesis. In a mouse model, radiation-induced decreases in BBB integrity and cerebral perfusion are attributed to the loss of pericytes secondary to decreased PDGFRβ expression [165], endothelial damage [166] and senescence [167]. Standard radiation therapy commonly elicits short-term BBB damage by disrupting supportive cells, vasculature volume, and TJ proteins such as occludin and claudin-5. A newly developed modality of irradiation can preserve cerebral microvasculature integrity by optimizing physical parameters to minimize cognitive impairment [168].
Emerging evidence has shown that tumor-secreted exosomes induce microvascular ECs damage to disrupt BBB integrity, and have an impact on the formation of brain metastases. The components of exosomes, such as non-coding RNAs, may mediate loss of TJs, whereas inhibiting these non-coding RNAs can alleviate tumor-secreted exosomes-induced BBB hyperpermeability [169]. Several studies have suggested that patients with extracranial cancers while without brain metastases experience cognitive deficits. For example, cognitive impairment in patients with advanced lung cancer is mainly characterized by impaired visuospatial/executive function and delayed recall [170, 171]. These cognitive changes may be associated with tumor-induced immune activation, chronic neuroinflammation, and BBB breakdown.
Risk factors of BBB disruption
Cognitive decline owing to BBB disruption is influenced by a variety of genetic, physiological, vascular, metabolic, and environmental factors. Therefore, early recognition of certain risk factors may be helpful in identifying those patients at a high risk of cognitive impairments. Figure 2 summarizes the risk factors related to BBB disruption.

The risk factors attributing to BBB disruption
Genetic factors
Several genotypes may be directly associated with microvascular impairment. The ε4 allele of apolipoprotein E 4 (APOE4) is a genetic risk factor for AD, independent of AD pathology. APOE4-related cognitive dysfunction is associated with BBB disruption [172]. More severe pericyte degeneration and BBB breakdown are observed in APOE4 than in APOE3 carriers [172, 173], which begins from middle age, although this difference is subtle [174]. This change in BBB permeability is associated with activation of the cyclophilin A (CypA)-MMP-9 pathway in cerebral microvessels [175]. Furthermore, activation of CypA-NFκB-MMP-9 pathway has been reported to be associated with the degradation of TJs and basement membrane proteins at the BBB. APOE4 can also exacerbate astrocyte activity, leading to BBB impairment in the aging brain, which is associated with vascular cognitive impairment and dementia [176]. Interestingly, peripheral but not cerebral APOE4 expression also compromises synaptic plasticity and cognitive function. In an amyloid mouse model, liver-expressed APOE4 exacerbated AD pathology by impairing cerebrovascular ECs function, whereas liver-expressed APOE3 reduced Aβ deposits [177]. Another genetic factor, presenilin 1 (PSEN1), encodes an aspartate protease that promotes Aβ accumulation by cleaving the APP [178]. PSEN1 mutation or knockout can lead to vascular lesions, including a reduced number of microvessels, BBB breakdown, pericytes degeneration, and Aβ deposits in small arteries [179]. Furthermore, genetic mutation of PDGFRβ is an etiology of primary familial brain calcification, presenting as a variety of neuropsychiatric disorders [180]. PDGFRβ knockout model exhibits pericyte deficiency and progressive BBB damage secondary to hemodynamic disturbances [15], causing vascular and perivascular calcium accumulation. In turn, cerebrovascular damage can promote neurodegenerative diseases, which adversely influence cognitive function [45].
Aging
Aging induces molecular, cellular, and functional changes in the brain, which increases brain vulnerability and drives cognitive decline. BBB components are also subject to changes with aging [181], single-cell RNA-sequencing analysis has shown age-related changes in brain ECs and other cellular components at the BBB [182]. Clinical neuroimaging study measuring water exchange across the BBB with arterial spin labeling (ASL) technique has shown that BBB permeability increases in the aging brain [35, 183]. Vascular senescence is considered a feature of early BBB disruption in AD [184]. Accumulation of senescent ECs and pericytes is associated with damaged BBB integrity and increased peripheral immunocytes aggregation, displaying decreases in TJs expression and transcytosis capacity, and region- and segment-specific increases in inflammatory mediators in the cerebral microvessels [185, 186]. While the astrocytes reduce their mitochondrial transfer efficiency to ECs, leading to the age-associated decrease in BBB integrity [7]. And in the aging brain ligand-specific receptor-mediated transport shifts to non-specific caveolar transcytosis. Consequently, Aβ clearance via BBB transport and/or proteolysis significantly declines [187]. Heterochronic parabiosis, a robust method to improve the function of aging tissues, regulates aging profiles in a cell-type-specific manner. Cerebral ECs appear to be especially malleable to this intervention when compared to other brain cell types [188]. The decreased pericytes coverage and increased neuronal senescence during brain aging also affect the BBB integrity by activating neuroinflammatory response [189].
Several molecular events may drive aging-related changes in the BBB integrity: (1) increases in neuronal CREB-regulated transcription co-activator 1/COX-2 expression with aging may be responsible for neuroinflammation and ECs senescence, leading to BBB breakdown; (2) decreases in connexin 43 (CX43) expression in cadherin-5+ cerebral vascular cells in the aging brain promotes BBB disruption owing to mitochondrial dysfunction through nicotinamide adenine dinucleotide (NAD+)-dependent sirtuin 3, whereas nicotinamide mononucleotide supplementation rescues NAD+ levels to alleviate aging-associated BBB leakage [190]; (3) age-related activation of vasoactive mediators, astrocytes and monocytes, upregulation of ion channels and downregulation of ECs surface carriers are also associated with age-related BBB hyperpermeability [191]; (4) injured pericytes release PDGFRβ into the CSF during aging, whereas a higher PDGFRβ concentration in the CSF is associated with poorer BBB integrity and higher neuroinflammatory biomarkers [192]. In addition, aging also alters the calcium coupling of astrocytic endfeet to the cerebrovascular system, which may directly or indirectly trigger BBB dysfunction [193].
Vascular factors
Increase in cerebrovascular risk has been shown to promote the onset and progression of cognitive dysfunction [194, 195]. Hypertension can affect cerebrovascular structure as well as CBF regulation, leading to hypoperfusion. The BBB hyperpermeability has been observed in animal models of hypertension, characterized by a decrease in TJ proteins at EC junctions, including claudin-5, ZO-1, and occludin [196, 197]. And hypertensive patients with early signs of BBB impairment show cognitive impairments [198]. Moreover, transient extremes of hypertension, hypoxemia, and hypercapnia can induce vascular injury. For example, in neurosurgical patients intraoperative hypertension or hemodynamic fluctuations can open the BBB and raise intracranial pressure; or in divers breath-holding-induced hypoxemia increases BBB permeability [199].
Spontaneous intracerebral hemorrhage is commonly attributed to cerebrovascular abnormalities, such as aneurysms, arteriovenous malformations, and cavernous hemangiomas. Intracerebral hemorrhage compromises the BBB integrity, leading to subsequent infiltration of blood-bore components and aggravation of brain injury. Therefore, targeting the BBB may improve brain edema and neurological injury [200]. In contrast, Moyamoya disease, a rare cerebrovasular disorder, can cause recurrent transient ischemic attack or stroke. Ring finger protein 213 (RNF213) is a genetic factor that increases susceptibility to Moyamoya disease. Since Rnf213 knockout causes significant decreases in the density of pericytes and TJ proteins occludin, claudin-5, and ZO-1 [201], Rnf213-deficient mice have a disrupted BBB.
cSVD is the most common chronic and progressive vascular disease, which affects the arterioles, capillaries, and small veins supplying the white matter and deep brain structures. Disruption of the BBB is one key feature of cSVD that has been linked to cognitive impairment [202]. Neuroimaging studies using DCE-MRI and ASL techniques have measured the BBB impairments in cSVD [203], providing an insight into the mechanisms underlying this cerebrovascular disorder.
Metabolic factors
Metabolic disorders, including type 1 and 2 diabetes and chronic obesity, are primary risk factors for AD. Diabetes is also a major vascular risk factor for cognitive dysfunction after stroke. Increased BBB permeability has been demonstrated in patients with type 2 diabetes and in animal models [204, 205]. Hyperglycemia significantly promotes the release of proinflammatory cytokines, mitochondrial dysfunction, and the accumulation of ROS and nitrogen species. Cerebral microvascular inflammation and oxidative/nitrosative stress activate NF-κB, MMPs, Toll-like receptors (TLRs) and receptors for advanced glycation end-products(RAGE), which promote BBB damage, including basement membrane thickening, TJs degrade, pericyte loss, and astrocyte dysfunction [206]. In vivo study has shown that high glucose level increases glycation of basal occludin and accumulation of methylglyoxal, a carbonyl stressor, leading to fewer occludin-positive microvessels in the diabetic brain [207], suggesting hyperglycemia can exacerbate brain microvascular endothelial dysfunction. However, a recent experimental study indicated that prolonged systemic hyperglycemia did not lead to BBB leakage, as evidenced by the unaffected transcriptional and morphological profiles of the BBB and its supporting cellular components [208]. Therefore, cognitive impairments found in diabetes may be caused by other mechanisms.
Consumption of a high-fat diet can promote the proinflammatory cytokines TNFα and IL-6 release and increase BBB permeability in the hippocampus [209]. Structural and functional impairments of the BBB promote leukocytes across the BBB, which activates microglia to initiate a neuroinflammatory response. Hyperlipidemia is another risk factor that promotes BBB damage. Hyperlipidemic animal models with APOE deficiency exhibit a significant increase in BBB permeability [210]. Furthermore, hyperlipidemia can exacerbate BBB breakdown after ischemic brain injury [211]. Cellular and molecular mechanisms underlying the deterioration of BBB catabolism include ECs impairment, MMP activation, and oxidative/nitrosative stress [212,213,214]. Therefore, improving the BBB integrity may alleviate metabolic disorder-associated cognitive decline.
Environmental factors
Environmental factors, including heavy metal and chemical substance pollution, may contribute to BBB damage. Here, we focus on the effects of cadmium, lead, and fine particulate matter (PM2.5) exposure on the BBB integrity.
PM2.5 PM2.5 is defined as inhalable particles generally ≤ 2.5 µM in diameter. Owing to their special physicochemical properties, recent studies have shown that ambient PM2.5 can cause neurodegenerative diseases including AD and PD via direct crossing of the BBB or the indirect nasal olfactory pathway [215]. Chronic PM2.5 exposure leads to cognitive decline in mice, as detected using the Morris water maze test [216]. The etiologies of PM2.5 pollution-associated neurological disorders include activation of microglia, increased ROS levels, and neuronal loss in the CNS. The relationship between neuroinflammation and learning, memory, and behavioral impairments was investigated in a mouse model exposed to PM2.5. PM2.5-induced microglial activation occurred earlier in the hypothalamus and olfactory bulb than in other brain regions, characterized by the release of proinflammatory cytokines such as IL-1β and TNFα. PM2.5-induced peripheral inflammatory mediators also easily cross the BBB and enter the brain parenchyma to stimulate innate immune responses. Dependent on P-glycoprotein transport activity, PM2.5-mediated TJ proteins downregulation and MMP-2/9 upregulation in the vasculature increase BBB permeability in animal models and human exposure studies [217]. This may be related to the lectin-like oxidized low-density lipoprotein receptor 1-mediated signaling pathway, as evidenced by increased oxidative stress and proinflammatory cytokine expression of MCP-1, VCAM-1, and ICAM-1 in brain microvessels [218], which may eventually lead to stroke and AD. Generally, PM2.5 can cross the BBB into the brain to trigger neuroinflammation and neurodegeneration, leading to progressive cognitive decline.
Heavy metals Cadmium (Cd) is a highly toxic environmental pollutant, and its toxicity is associated with neurodegenerative diseases such as AD and PD, typically presenting with cognitive impairments. Cd-induced pathogenesis of neurodegenerative diseases is related to dysfunction of the BBB. In an in vitro rat brain EC line model, Cd-induced dose- and time-dependent ROS increases, coincident with reduced expression of TJ proteins ZO-1, F-actin, and vimentin [219]. Lead has been demonstrated to decrease the expressions of TJ proteins occludin and ZO-1 at the BBB, mediated by the intracellular non-receptor protein tyrosine kinase Src, whereas redistribution of glucose regulatory protein 78 contributed to lead-induced Src activation and the ensuing reduction in occludin level [220]. A human study showed that patients with high blood lead concentration had a higher serum S100β concentration [221], suggesting a BBB hyperpermeability.
Mechanisms underlying BBB disruption
Impairments of cellular and molecular components at the BBB greatly affect BBB integrity. Understanding the mechanisms underlying BBB disruption can help develop novel therapeutic modalities to prevent or repair BBB damage, accordingly, to potentially slow or improve cognitive decline. The underlying mechanisms are illustrated in Fig. 3.

The possible mechanisms underlying the BBB disruption. (a) Inflammatory factors can lead to down-regulation and degradation of TJs, mediated by VEGF and NO. MMPs, ROS and others can also induce a decrease in TJ protein expression. (b) A variety of factors including inflammation, ischemia and insulin resistance can lead to disintegration and death of ECs, leading to loss of ECs. Overexpression of endothelial adhesion molecules induces migration of leukocyte; activation of Adora2a decreases TJs expression; down-regulation of glucose transport proteins affects endothelial metabolism and inhibits LRP1 transcription, which disrupts the BBB integrity; and activation of TREM2 triggers endothelial oxidative stress. (c) Stress factors induce pericyte contraction and apoptosis, and hypoxia and inflammation trigger pericytes detachment from the basal lamina, leading to loss of pericytes. (d) Pathologic factors trigger astrocytes destruction and endfeet loss, and induce reactive astrocytes to secrete MMPs, proinflammatory cytokines, VEGF-A, ECGF1, and extracellular vesicles (EVs) containing inflammation-related proteins. (e) Activated microglia release proinflammatory factors, promote phagocytosis of astrocytic perivascular endfeet, and lower the expression of TJs. These mechanisms greatly impair molecular and cellular components and dysregulate cell-cell interaction, which underlie BBB disruption. BBB: blood brain barrier; TJs: tight junctions; ECs: endothelial cells; VEGF: vascular endothelial growth factor; ECGF1: endothelial cell growth factor 1; NO: nitrous oxide; MMPs: matrix metalloproteinases; ROS: reactive oxygen species; Adora2a: adenosine receptor 2a; LRP1: low-denisity lipoprotein receptor-related protein 1; TREM2: triggering receptor expressed on myeloid cells 2
Tight junction proteins
Destruction of TJs can directly lead to BBB breakdown. Many proinflammatory factors including IL-1β, IL-6, IL-9, IL-17, and interferon γ (IFN-γ) directly or indirectly degrade TJs or reduce their expressions [222,223,224]. While inflammatory signaling, including the NF-κB pathway, can lead to downregulation and denaturation of the most important TJ protein, claudin-5 [225, 226]. Two cytokines, TNFα and IL-1β, are reported to increase endolysosomal protein degradation and stroke-induced TJ strand disassembly through activating endothelial small GTPase Rab7a. This greatly promotes paracellular BBB permeability and exacerbates neuronal outcomes [227]. Furthermore, hypoxia-induced TJ reorganization appears to be mediated by VEGF and NO [228]. Unlike that derived from ECs, VEGF-A released from hypoxic OPCs is linked to BBB impairment by affecting OPC-EC interaction in mouse cortical areas during cerebral hypoperfusion [229]. In contrast, a compensatory increase in VEGF expression in macrophages at the BBB in response to a high-fat diet-induced decrease in glucose transporter protein isoform 1 (GLUT1) expression and brain glucose uptake, can restore cerebral glucose metabolism, limit neurodegeneration and preserve cognitive function. And myeloid cell-specific deletion of VEGF exaggerates neuroinflammation and cognitive decline in obesity [230]. Therefore, the role of VEGF in BBB integrity may depend on the cell type in a context-dependent manner.
During the neuroinflammatory response, MMPs, ROS, and aspartic proteinases (Saps), also play the role in decreasing TJ proteins expression [231,232,233], through: (1) the activity of permeability glycoprotein, one of the main efflux transporters, can be inhibited to increase MMPs secretion and ROS overproduction, mediated by HIF1α-dependent mechanism [234, 235]. Meanwhile, decreases in MMPs inhibitors - tissue inhibitors of metalloproteases (TIMPs) is also the main cause of BBB breakdown [236]; (2) ECs and recruited leukocytes can also secrete MMP-9 to proteolyze neurovascular basement membrane proteins and TJ proteins, and thus degradate basal lamina and TJs. Therefore, MMP inhibitors can prevent the TJs loss, further indicating a key role of MMPs in the BBB disruption; (3) cell-cell interactions are affected through downregulating AJs, which promotes extravasation of peripheral immune cells [237, 238]; (4) Saps, especially Sap2, secreted from Candida albicans could degrade endothelial TJ protein occludin-1, to permit fungal invasion into the brain parenchyma from the blood through the disrupted BBB [239]. Interestingly, after fungi enter the mouse brain, Saps cleave neuronally expressed APP into Aβ-like peptides that bind to TLR4 to activate microglia, promoting neuroinflammation.
Integrins are transmembrane glycoprotein receptors in the extracellular matrix that physiologically interact with the basement membrane components and regulate BBB permeability. They are also upregulated by cytokines to alter cell-matrix junctions [240]. Integrin α3 expressed in CNS-infiltrating Th17 cells can promote BBB disruption and Th17 cells transmigration into the brain, while knockout of integrin α3 enhances the retention of CD4+ T cells in the perivascular space and maintains the BBB integrity [241]. Furthermore, IL-17 A secreted from Th17 cells can induce NADPH oxidase- or xanthine oxidase-dependent ROS production, and activate the endothelial contractile machinery, leading to occludin downregulation and BBB disruption [242]. Furthermore, vascular β-site APP-cleaving enzyme 1 (BACE1) may be involved in cerebral microvessel injury. BACE1 can induce membrane accumulation of caveolin-1 and caveolin-1-mediated endocytosis through cleaving the endothelial TJ protein occludin, thus resulting in the lysosomal degradation of other TJ proteins, and finally BBB breakdown. In contrast, inhibition of BACE1 activity can ameliorate TJs loss, endothelial dysfunction, and cognitive deficits [243].
Endothelial cells
ECs are another important target of BBB damage. Proinflammatory and other toxic factors can directly cause structural disintegration and even ECs death. Apoptosis of ECs and downregulation of TJ proteins, together with the overexpression of EC adhesion molecules and leukocyte transmigration, are mediated by astrocyte-derived VEGFs, MMPs, NO, and endothelins [244]. High mobility group box 1 (HMGB1), acting as a damage-associated molecular pattern, can induce vascular inflammation by activating the Nod-like receptor protein 3 (NLRP3) inflammasome, thereby initiate the ECs pyroptosis and disrupt the BBB integrity [245]. Wei et al. found cytosolic lipopolysaccharide (LPS) sensor caspase-11 could activate endothelial pore-forming protein GSDMD, which induced pyroptotic endothelia, abnormal TJs and vasculature detachment from the basement membrane in vitro and in mice. GSDMD-neutralizing nanobody can block this inflammatory BBB disruption [246]. Further, in obesity and insulin resistance, activation of endothelial adenosine receptor 2a (Adora2a) disrupts the TJs between the microvascular ECs, whereas inhibition of Adora2a prevents BBB breakdown via decrease in vascular inflammation, thus improves synaptic plasticity and hippocampus-dependent memory [247]. Similarly, loss of ECs expressing transforming growth factor β1 type I (TGF-β1) receptor, activin receptor-like kinase (ALK)-1, and ALK-5, increases vascular permeability, whereas the administration of TGF-β1 protects BBB integrity [248].
GLUT1 is the main glucose transporter that supports ECs metabolism. Surgery-induced GLUT1 reduction in the hippocampal microvasculature, along with downregulation of TJ proteins, significantly contributes to postoperative cognitive deficits in aged mice [249]. Similarly, GLUT1 deficiency in an AD mouse model leads to cerebral microvascular degeneration, CBF reduction, BBB disruption, and Aβ pathology progression [250]. Mechanistically, GLUT1 deficiency induces transcriptional repression of LRP1 in cerebrovascular ECs, and adenovirus-mediated re-expression of GLUT1 restores LRP1 levels [250], and increases transvascular clearance of 70–85% Aβ under physiological conditions [251]. A significant reduction in LRP1 expression in the AD brain microvascular ECs has also been validated in several clinical studies [251,252,253]. Conversely, inhibition of LRP1 expression accelerates Aβ accumulation in the brain, thereby worsening microvascular damage [254]. Loss of endothelial LRP1 in the brain leads to BBB disruption, neuronal loss, and cognitive deficits in mice, all of which can be reversed by endothelial-specific LRP1 gene therapy [255]. Furthermore, LRP1 oxidation significantly reduces Aβ binding and efflux from the brain [256]. A recent study provides another mechanism underlying the role of GLUT1 in ECs. EC-derived lactate participates in EC-pericyte interactions. EC-derived lactate is transported into pericytes to support their functions. TBI can disrupt EC-pericyte crosstalk and increase BBB permeability [257]. Deletion of GLUT1 in ECs reduces lactate production, which significantly impairs pericyte metabolism including energy generation and amino acid biosynthesis. Inhibition of EC-derived lactate production in mice results in loss of pericyte coverage in the brain vasculature and BBB hyperpermeability [258], suggesting that ECs metabolic support for pericytes may be essential for maintenance of BBB integrity. However, in an earlier study, Veys et al. found that GLUT1 knockout in adult animal ECs neither disrupted BBB integrity nor altered the expression of TJ genes [259]. Moreover, during ischemic stroke, endothelial Ca2+-permeable non-selective cation channel transient receptor potential melastatin 2 (TRPM2) can be activated, whereas inhibition or deletion of TRPM2 greatly attenuates BBB hyperpermeability by inhibiting endothelial oxidative stress and Ca2+ overload [260]. Therefore, endothelial dysfunction is an early and typical pathology of neurodegenerative and cerebral microvascular diseases.
Pericytes
Pericytes play a key role in maintenance of the BBB integrity. Bohannon et al. identified two subsets of brain microvascular pericytes in rhesus macaques: type 1 in young adults and type 2 in older adults. There is a strong positive correlation between the degree of BBB disruption and the percentage of type 2 pericytes regardless of age [261]. Compared with ECs, pericytes are more susceptible to stress conditions, such as aging, diabetes, or ischemia. Hypoxia/ischemia can increase the expression of several vasoactive mediators, including PDGFRβ, adenosine, and NO in the pericytes to induce contraction. During prolonged hypoxia and LPS-induced sepsis, pericytes can detach from the basal lamina, leading to BBB disruption [262, 263]. In addition, pericytes mediate hypoxia-induced leukocyte infiltration, which exacerbates BBB destruction via the inflammatory response [264]. Concurrently, pericytes may differentiate into microglia and increase phagocytosis under hypoxia, which participates in the inflammatory response [265]. Although the role of pericytes in neurodegenerative diseases has not been completely elucidated, several studies have shown that the morphology, function, and PDGFRβ signaling of pericytes are altered in AD [266]. The loss of pericytes results in a significant increase in BBB leakage, brain Aβ levels, tau pathology, and neuronal loss in the AD mouse model [267]. The number of pericytes is also decreased in the white matter in post-stroke dementia and vascular dementia [268].
Recently, Liu et al. investigated the relationship between bone aging and neurodegenerative diseases, and found that bone preosteoclast-derived PDGF-BB in aged mice promoted hippocampal vascular impairment [269]. A high circulating PDGF-BB level is responsible for capillary reduction, pericyte loss, and BBB hyperpermeability in the hippocampus of aged mice, whereas preosteoclast-specific Pdgfb knockout reduces this BBB impairment. Exposure of brain pericytes to high concentration PDGF-BB in vitro upregulates MMP-14, which promotes PDGFRβ shedding from the pericyte surface, while MMP inhibition alleviates hippocampal pericyte loss in conditional Pdgfb transgenic mice and BBB hyperpermeability in aged mice. Surprisingly, PDGF-BB is also reported to promote the pericytes proliferation and protects pericytes against apoptosis through extracellular signal-regulated kinase signaling, whereas PDGF augments pericyte-derived inflammatory secretions through Akt pathway [270].
Astrocytes
Astrocytes interact closely with the cerebral vasculature through perivascular endfeet, where the transmembrane protein AQP4 and the gap junction protein CX43 exhibit high expressions. Deletion of CX43 in the astrocytes induces a significant reduction in AQP4 levels, followed by edema formation and BBB disruption. In contrast, deletion of AQP4 does not alter the ECs ultrastructure, TJs expression, and BBB permeability [271]. Therefore, astrocytes play a significant role in the BBB formation and maintenance [272, 273], thus astrocytes destruction is associated with BBB disruption. Furthermore, astrocytes modulate EC permeability by direct cell-cell interactions, soluble factors secretion, or mitochondrial transfer, endothelial dysfunction in turn activates astrocyte, which directly influences the astrocyte-EC interaction and BBB hyperpermeability [274]. Astrocytes modulate BBB development and function under pathophysiological conditions also by secreting a series of molecules that regulate immune responses, and release of EVs containing inflammation-related proteins and miRNAs. Triggered by inflammation, activated astrocytes disrupt the BBB structural components by secreting MMP-9 and proinflammatory cytokines IL-1β, IL-6, and TNFα [83, 273].
In a PD model, oligomeric αSyn, but not monomeric or fibrillar αSyn, results in a significant BBB breakdown [275]. This process involves the expression and release of VEGF-A in astrocytes, suggesting that astrocyte-derived VEGF-A may be an essential mediator of PD-associated BBB disruption. Another key astrocyte-derived factor endothelial cell growth factor 1 (ECGF1), co-localizing with VEGF-A in reactive astrocytes, also correlates with BBB permeability [276]. Interestingly, inactivating VEGF-A induces an increase in ECGF1 expression. The interaction between the ECGF1 product 2-deoxy-d-ribose and VEGF-A cooperatively inhibits TJ proteins expression and drives BBB permeability. Inhibiting either of them can reduce BBB disruption. For example, the anti-VEGF antibody bevacizumab can block VEGF binding to its receptors, which effectively reverses the BBB damage and vascular smooth muscle contraction. This eventually improves memory performance in a mouse AD model [277]. While antagonizing both of them enhances BBB preservation and alleviates neurological pathology [276], suggesting that astrocytic ECGF1 and VEGF-A co-operatively promote BBB disruption.
Microglia
Microglia can interact with the vascular components at the BBB. Microglia-BBB interactions are critical in microglial response to NVU damage [278]; however, depletion of microglia alone does not alter BBB permeability [279], but increases LPS-induced BBB permeability in adult mice [280]. Systemic inflammatory factors can activate both of microglial pro- and anti-inflammatory responses. Proinflammatory microglia promote further release of inflammatory cytokines which augment BBB damage by increasing paracellular permeability and activating adsorptive endocytosis. Microglial activation also promotes phagocytosis of astrocytic perivascular endfeet and lowers the expression of two TJ proteins: occludin and claudin 5. These changes aid leukocyte infiltration across the BBB, and further exacerbate neuroinflammation and neurodegeneration [281]. Therefore, increased BBB permeability following prolonged systemic inflammation may contribute to cognitive dysfunction. In contrast, anti-inflammatory microglia protect the BBB from inflammatory attacks by elevating the levels of anti-inflammatory factors, including IL-10, IL-4, IL-13, and TGF-β [282, 283]. Therefore, depending on the perivascular microenvironment, the highly dynamic switch of microglial phenotype between pro- and anti-inflammatory states relates to their impact on BBB integrity. Interestingly, during systemic inflammation induced by LPS or IFN-α, microglia migrate to the cerebral vasculature through a C-C motif chemokine ligand 5/CCR5-dependent mechanism. These microglia initially maintain BBB integrity via expressing TJ protein claudin-5 and contacting with ECs. However, sustained inflammation facilitates microglial phagocytosis of astrocytic endfeet and impairs BBB function [280], suggesting a dual role of microglia in maintaining BBB integrity.
Molecular mechanisms
Several molecular events that are included but not limited to these participate in BBB disruption: (1) plasma insulin-like growth factor-1 (IGF-1) level is associated with brain microvascular dysfunction and cognitive impairments. Endothelial loss of IGF1 receptor impairs neurovascular coupling, increases senescent ECs and BBB disruption in mice [284]. Further, knockdown of astrocytic IGF-1 receptor in cerebral ischemic mice can trigger greater BBB disruption and larger ischemic lesions. These evidences suggest IGF-1 signaling plays a crucial role in preserving the BBB integrity. (2) in the absence of ischemia, recurrent seizures induced by local potassium channel blocker 4-AP or γ-aminobutyric acid subtype A (GABAA) receptor blocker PTX can promote vascular permeability via N-methyl-D-aspartate (NMDA) receptors-mediated excessive release of glutamate in the cerebral cortex of rat. Furthermore, in a focal cerebral ischemia animal model, GABAA receptors inhibitor bicuculline enhances BBB disruption, although it increases regional CBF [285]. Similarly, exposure of cultured brain ECs to glutamate can result in NMDA receptor-mediated decreases in the TJ protein occludin levels and cellular redistribution, as well as increases in BBB permeability. Conversely, NMDA receptor antagonists reduce BBB permeability of the peri-ischemic cortical region in the stroke photothrombosis rat model [286], suggesting a role of NMDA receptor activation in BBB disruption; (3) mitochondrial protein polymerase delta-interacting protein 2 (Poldip2), a multifunctional protein, is necessary for vascular integrity and function. In a transient middle cerebral artery occlusion model, cerebral ischemia significantly upregulated Poldip2 expression; however, deletion of Poldip2 inhibited the expressions of TNFα, IL-6, MCP-1, VEGF, and MMPs, and thus BBB hyperpermeability [287]. Similarly, in an SAE model, LPS induced increases in Poldip2 expression, proinflammatory cytokines, and thus BBB permeability [288]. Further, alterations in BBB integrity were preceded by the induction of Poldip2, whereas silencing of Poldip2 inhibited LPS-mediated NF-κB subunit p65 translocation and COX-2 induction in rat brain microvascular ECs, which effectively alleviated BBB disruption.
Overall, these data suggest that under pathological conditions, the ultrastructural degradation between cells, dysregulated cell-cell interactions, as well as metabolic, angiotrophic, supportive, immune changes at the BBB contribute to barrier disruption, whereas neuroinflammation appears to play a central role in this process. However, whether changes in different BBB properties leads to specific cognitive impairment is not known.
Protection against BBB disruption
BBB disruption is commonly presumed reversible after harmful insults are removed; however, re-establishment of the BBB integrity after stroke is incomplete [289], and could influence rehabilitation, increase the risk of repeated stroke, and further develop to vascular dementia. Therefore there is an urgent need for new therapies to protect and restore BBB structure and function. Several strategies to stabilize the BBB and promote its restoration have been developed.
Anti-inflammatory agents
In clinical practice, steroidal glucocorticoids (GCs) are still the mainstream agents used to treat BBB damage. Preclinical data has shown that GCs could significantly reduce BBB permeability and successive brain edema formation in a stroke animal model [290], through limiting the inflammatory cascade and immune system over-activation at the BBB. These findings have also extended to their role in BBB repair, including partial reversal of TJ proteins downregulation and MMP-9 upregulation [291], increase in anti-inflammatory protein membrane association protein A1 (Annexin A1) expression to protect inter-endothelial junctions [292, 293], and decrease in astrocytic VEGF level to reverse reduced TJ proteins expression through elevated endothelial NOS production [294]. However, GCs are largely ineffective in patients with acute ischemic stroke [295], although they may be effective in treating acute multiple sclerosis. Preoperative administration of dexamethasone has been reported to reduce the proinflammatory response, thereby decrease the risk of early postoperative cognitive dysfunction after cardiac surgery [296]. However, the latest evidence fails to support the idea that GCs can prevent BBB disruption in patients undergoing surgery. Although preventive treatment with high-dose methylprednisolone could attenuate the systemic inflammatory response in open-heart surgery under cardiopulmonary bypass, it does not prevent BBB hyperpermeability or neuroinflammatory responses [148]. Similarly, intraoperative use of dexamethasone in pediatric cardiac surgery also does not reduce major neurological complications within postoperative 30 days [297]. These findings question the effectiveness of GCs in the BBB restoration and cognitive improvement; therefore, it is not justified to recommend the use of GCs during the perioperative period for preventing postoperative cognitive dysfunction.
In addition to GCs, there are other effective medications to reduce neuroinflammation by inhibiting the immune response in the brain, including the gas molecules hydrogen and hydrogen sulfide. Minocycline, an inhibitor of microglial/macrophage activation, can reduce BBB permeability by inhibiting the expressions of MMP-9 and inflammatory mediators [298]. Alternatively, dexmedetomidine, a sedative agent, can also exert neuroprotective effects by inhibiting the release of proinflammatory HMGB1 from astrocytes and microglia in response to a variety of stressors, including hypoxia, ischemia, and cytokines [299]. Other drugs including Tanshinone II, Berberine, and Ginsenosides that target HMGB-1, can also stabilize the BBB [300]. NF-κB/NLRP3 signaling plays an important role in the inflammatory response. Direct inhibition of NF-κB/NLRP3 pathway can modulate neuroinflammation to maintain BBB integrity in an animal study [288]. In addition, statins also have an anti-inflammatory effect, in vitro experiment has revealed that statins could inhibit Aβ-induced inflammatory and cytotoxic effects on cultured cerebral ECs and astrocytes [301], suggesting their protective role in BBB integrity.
Anti-oxidative agents have also been reported to prevent BBB injury via inhibiting peripheral inflammation. These anti-oxidative agents, including resveratrol, curcumin, and apigenin, exhibit an anti-inflammatory property and thus provide neuroprotective effects by inhibiting inflammatory pathways (i.e., TLR4/NF-κB) and BBB breakdown. Similarly, administration of antifibrinolytic drugs, including tranexamic acid, can provide acute anti-inflammatory protection and alleviate BBB damage following TBI [302, 303]. The proinflammatory cytokines IL-1β and TNF-α are considered etiologic triggers for BBB breakdown; therefore, IL-1 receptor antagonists [304], or TNF-α inhibitors [73] can reduce BBB damage and thus prevent neurobehavioral impairments in the BBB-disrupted animal models. Collectively, although anti-inflammatory agents protect the BBB integrity against neuroinflammation, it is not clear whether this BBB protection can translate into cognitive improvement.
Vascular dysfunction blockers
Several vascular leakage blockers have been developed. CU06-1004, an EC dysfunction blocker, prevents vascular leakage and enhances vascular integrity in a cerebral ischemia/reperfusion injury model [305]. Furthermore, long-term administration of CU06-1004 also alleviates age-associated cerebral microvascular senescence and BBB damage in in vitro and in vivo models, significantly improving cognitive function in aged mice [306]. Another vascular leakage blocker, Sac-1004, can also rescue TJ-related proteins, decrease BBB leakage, and inhibit inflammation, and thus effectively ameliorate ischemic damage and neurological deficits in a cerebral ischemia/reperfusion injury model [307]. Nevertheless, these vascular leakage blockers have not been validated or approved for clinical use.
Mitochondrial protectants
A novel target for BBB protection is the mitochondrion. Impaired mitochondrial energy metabolism directly affects BBB structure and function, and dynamin-related protein 1 (Drp1)-mitochondrial fission protein 1 (Fis1)-mediated mitochondrial dysfunction can lead to BBB disruption, which can be ameliorated by Drp1-Fis1 inhibitors [308]. In fact, rotenone, a mitochondrial electron transport inhibitor, impairs learning and memory in mice via microglia-mediated BBB disruption and neuronal apoptosis [309]. Methylene blue (MB), a clinically available mitochondrial protectant, is neuroprotective and potentially effective against neurodegenerative diseases. Our study revealed that the intravenous administration of 2 mg/kg MB during major surgical procedures could decrease the incidence of early PND in elderly patients [310]. A single intravenous injection of MB (1 mg/kg) 30 min after TBI was shown to significantly reduce the TBI-induced increase in S100β expression and BBB permeability [311], and thus improved cognitive and motor functions [312]. Recently, a mitochondrial-targeting fluorescent small molecule, IR-780, was shown to reduce neuroinflammation and restore TJs expression at the BBB, leading to cognitive improvement after whole-brain irradiation [313]. These studies suggest that targeting mitochondrial metabolism may promote the recovery of BBB and brain function.
Maintenance of tight junction proteins
Preventing degradation of TJ proteins and promoting TJ proteins expression after their destruction can accelerate BBB repair [294]. The TJ complex is a dynamic structure that maintains the rigid structure of the barrier. This dynamic structure is regulated by many transport proteins and signaling pathways in the BBB, which may facilitate the rapid localization of nascent TJs and promote BBB repair [314]. As the role of MMPs in degradation of microvascular basal lamina and TJs, MMPs inhibitors are assumed to protect against BBB disruption. Recent pre-clinical and clinical studies have demonstrated that several compounds with specific or non-specific MMP-9 inhibitory properties have potential as therapeutic agents for inhibiting BBB breakdown after TBI [315].
One study found that astrocytes were enriched in genes for laminin and laminin receptors, and knockdown of these genes could reduce the expressions of TJ proteins claudin 5 and ZO-1. Therefore, it is essential to maintain astrocytic function during BBB repair [316]. In vitro experiment has shown estrogen upregulates claudin-5 and has a protective effect against BBB damage, with its target site located at TJ proteins and their regulators [317]. Cortistatin is a neuropeptide that exerts beneficial effects on the cerebral ECs. Endogenous cortistatin deficiency predisposes TJs to breakdown and leads to the weakening of brain ECs, BBB hyperpermeability and inflammatory response, whereas exogenous cortistatin supplementation can reverse the TJs disruption, hyperpermeability, and immune activity in murine brain ECs, and reduce BBB leakage in LPS-treated adult mice [318].
Outgrowth ECs
The outgrowth ECs, derived from peripheral blood progenitor cells or moncytes, have a high proliferative and expansion potential in vitro, stable phenotype and ability to expand in vivo with no apparent toxicity [319]. A in vitro study revealed that treatment with outgrowth ECs effectively restored BBB integrity and function in an oxygen-glucose deprivation (OGD) human BBB model through inhibiting oxidative stress [320]. While in a triple culture model of BBB with astrocytes, pericytes and ECs, adding exogenous outgrowth ECs effectively repaired the damaged BBB induced by OGD treatment [321]. Similarly, administration of outgrowth ECs provides better BBB protection in rats subjected to middle cerebral artery occlusion [322]. Finally, factors that promote BBB integrity by restricting transcytotic flux and maintaining the ECs stability, including angiopoietin-1, S1P, activator protein C, water channel AQP4, MMP inhibitors, and omega-3 fatty acids are under preclinical examination.
Endothelial senescence results in chronic BBB disruption and microvascular rarefaction. Therefore, clearance of senescent ECs may improve BBB integrity and cognitive function. When mice subjected to whole-brain irradiation are treated with ABT263, a well-known senolytic drug, for 3 months, senescent ECs are selectively and effectively removed, BBB permeability decreases, and cortical microvascular density tends to increase [166]. Similarly, when aged rats are administered ABT263 for 6 months, this senolytic drug can preserve BBB integrity, prevent hippocampal NMDA receptor hypofunction, accordingly, rescue memory and cognitive impairments [189]. In a “chemobrain” model, genetic (ganciclovir) or pharmacological (senolytic drug ABT263) treatments effectively eliminate senescent ECs, and likewise rescue BBB integrity and capillarization, leading to improved neurovascular coupling and cognitive performance [161], suggesting a causal relationship between clearance of senescent ECs, improved BBB integrity, and better cognitive functions.
Cellular and gene therapies
Mesenchymal stromal cells (MSCs) is another therapeutic with great potential for BBB reparation. MSCs are multipotent, non-hematopoietic cells found in the perivascular area and serve as immunomodulators and regenerative agents [323]. MSCs can promote angiogenesis and barrier repair in animal stroke models. Intravenous administration of MSCs to animals which have had an induced-stroke could reduce BBB hyperpermeability, increase vascular remodeling, and improve functional deficiency through secretion of VEGF and angiopoietin-1 [324]. The immunoreactivity of the mitogenic factor PDGFRβ from MSCs can be induced by salubrinal, a small molecular activator of the integrated stress response, which can inhibit the activation of immune cells at the BBB and promote restoration of the BBB integrity [325]. Recently, MSC-derived and cerebral EC-derived exosomes have been used to treat BBB disruption-associated cognitive dysfunction by inhibiting neuroinflammation. The exosome-treated animals had a significantly shorter time to complete neurological recovery. Williams et al. found that early single-dose treatment with MSC-derived exosomes after TBI or hemorrhagic shock enhanced TJ proteins expression and improved BBB integrity in a swine model [326]. This enhanced recovery of BBB integrity may accelerate neurocognitive rehabilitation [327]. In two rat stroke models, serum-derived exosomes [328] and bone marrow MSC-derived exosomes [329] improved neurological outcomes by reducing microvascular EC apoptosis and reversing autophagy-mediated TJ proteins degradation. In addition, inducible pluripotent stem cell-derived small EVs could significantly attenuate damage to the BBB integrity via activation of the eNOS-Sirt1 axis [330]. Non-coding RNAs, including miRNAs, lncRNAs, and circRNAs encapsulated in exosomes, have been implicated in the modulation of BBB structure and function, and play a role in BBB breakdown or repair. It is essential to identify potential candidate miRNAs to establish novel therapeutics for BBB recovery [331]. For example, Campbell et al. used miRNA directed against claudin-5 loss to decrease BBB breakdown after TBI, which eventually improved focal cerebral edema and cognitive function [332].
Recent studies indicate that gene therapy plays an important role in the development of safe, efficient, and specifically targeted vectors. For example, the brain microvascular EC-specific viral vector AAV-BR1 can be successfully expressed in ECs [333]. As previously mentioned, GLUT1-mediated glycolysis plays an important role in the metabolism of ECs and pericytes [334], therefore their GLUT1 levels in AD patients may be a novel target to rescue BBB dysfunction. Using the AAV-BR1 vector to upregulate endothelial GLUT1 levels may improve metabolism in ECs and, thereby, BBB integrity. Another innovative gene therapy is the development of a nanoparticle or engineered ligand targeting those brain regions with reduced expression of BBB molecules, such as claudin-1 and β-catenin. The nanoparticles can accumulate and remain in the brain NVU, and promote claudin-1 expression [335]. Newly synthesized claudin-1 is present on the cell membrane and can be incorporated into the TJ complex with established interactions with ZO-1 [336]. Since Wnt-mediated BBB maintenance, upregulation of Wnt/β-catenin signaling can reduce BBB disruption and immune cell infiltration in the brain [337, 338], while delivery of cerebrovascular-targeted, engineered Wnt7a ligands can protect BBB integrity, reduce T-cell infiltration and microglial activation in SARS-CoV-2-infected brain, and improve cognitive deficits in SARS-CoV-2-infected mouse brain through enhancing Wnt/β-catenin activity [339]. It is expected that these gene therapies can be used as a precision and safe tool for BBB restoration.
Other preclinical strategies
To provide innovative therapeutics for BBB restoration, investigators have also identified other experimental strategies for maintenance and repair of the BBB integrity, including: (1) application of new materials, such as biomimetic oxygen-boosted hybrid membrane nanovesicles, to promote angiogenesis and inhibit TJ proteins degradation [340]; (2) use of vitamin B12 to restore BBB integrity in AD through reducing homocysteine levels [341]; (3) administration of selective TGF-β receptor inhibitors to prevent BBB opening caused by cortical spreading depolarization after mild TBI [342]; (4) RAGE-specific inhibitors such as FPS-ZM1 and azeiragon, which control Aβ accumulation in the animal brain, can inhibit sustained cellular perturbation and vascular disruption [343]. Furthermore, soluble RAGE (sRAGE) is also available as a decoy to attenuate the adverse effects of Aβ and may improve CBF, BBB integrity, and neuroinflammation in animals [344]; (5) the non-immunosuppressive CypA inhibitor Debio-025 shows significant improvement in BBB integrity and neurobehavioral deficits independent of AD pathological alterations [345]; and (6) the fibroblast and fibroblast growth factor-21 are used to improve BBB permeability through a TIMP2-dependent pathway [346] or an Nrf2-dependent pathway [347]. TIMP2 has been reported to inhibit VEGF-induced vascular hyperpermeability and hypoxic plus inflammatory insult-induced TJs degradation and translocation to reduce paracellular permeability in human brain microvascular ECs through an MMP-independent mechanism [348]. Unlike TIMP2, TIMP1 has been found protects TJs dependent on an MMP-mediated degradation [349]. Although these promising strategies for BBB restoration, they are still at the bench and lack specificity toward brain regions.
Although restoration of the BBB integrity serves as a robust hallmark for better neurocognitive outcomes in animal studies, how endogenous signals induce BBB repair, and whether recovery of the BBB integrity causally improve cognitive impairments in patients are not fully understood. Therefore clinically effective BBB protectants and strategies for restoring the BBB are lacking.
Medications undergoing clinical trials
Thalidomide is used today to effectively treat inflammatory and cancer conditions, which initially acted as a sedative and an antiemetic until the discovery of its teratogenicity. Recently, it is found to treat radiation-induced brain injury (RIBI). In a clinical phase 2 study, thalidomide could reduce cerebral edema on fluid-attenuated inversion recovery-MRI in patients with RIBI, and 62.1% patients experienced cognitive improvement based on the Montreal cognitive assessment scores [164]. Further, in a mouse model of RIBI, thalidomide increased PDGFRβ expression to rescue pericytes loss and thus restored the BBB integrity and cerebral perfusion.
Another medication is ongoing examined in clinical phase II trial is bevacizumab, an anti-VEGF antibody. It is used in adult patients with steroid-refractory RIBI after their BBB disruption. The results showed that bevacizumab significantly reduced volume of brain edema and brain necrosis induced by radiation, while improved white matter recovery and neurocognitive scores [350]. Similarly, in animal study, bevacizumab is found to alleviate the BBB dysfunction and improve cognitive function through inhibiting VEGF signaling in AD mice [277]. Natalizumab, targeting the α4 integrin on immune cells, can prevent their interaction with endothelial VCAM-1, greatly reduces BBB disruption and thus new lesion formation in MS.
Although large-size sample, randomized controlled trials are needed, these novel therapies have provided an promising insight into treatment of cognitive impairments through improvement of BBB disruption.
Perspectives
Why BBB disruption can lead to cognitive decline?
BBB disruption is critical for initiation and perpetuation of diverse diseases at the concomitance with other neuropathological changes, therefore this disruption has been demonstrated to be an early biomarker in human cognitive impairment. Some evidences suggest sepsis or chemotherapy disrupts the BBB while directly affects brain function. However, several studies have suggested that BBB disruption is sufficient but not necessary for cognitive decline. For example, in stroke patients, the massive death of neurons can directly lead to cognitive deficits, while subsequent BBB disruption only plays a role as a deteriorating factor. Another example is AD. Those AD individuals with BBB breakdown suffer from early impairments in memory, attention/executive function and language, global cognition dependent of AD-specific biomarkers Aβ and tau [351]; In contrast, other studies have indicated that the severity of BBB permeability does not parallel AD pathology, such as brain Aβ and tau levels, in cognitively normal or impaired patients [35, 192, 352], suggesting that BBB dysfunction may only represent cerebrovascular damage but is not inherent to AD and cognitive impairments. Further, a clinical study indicated that a more intact barrier was associated with more prominent neurodegeneration, as reflected by higher CSF macrophage inflammatory protein-1α and TNFα levels in patients with mild cognitive impairment or early AD [46]. Therefore it is not clear that BBB disruption is the cause or result of these neuropathological changes. However, the BBB has combined functions of several physiological properties, thus critical for neural function and protection. Several reasons why BBB disruption can cause cognitive decline directly and indirectly can be speculated.
1. The BBB disruption facilitates xenobiotics and blood-borne substances entering into the brain. The numerous molecules including toxins, drugs, and other xenobiotics can enter brain parenchyma and directly influence neural fates. Further, the immune cells migration and plasma proteins infiltration into the brain could induce neuroinflammatory response [353]. The activated immune cells, including macrophage, mast cells and T cells, release proinflammatory factors by themselves, concurrently, they recruit the innate immune cells microglia/astrocytes in the brain, which triggers their activation to further release proinflammatory factors such as IL-1β, IL-6 and TNFɑ, leading to inflammatory cascade and significant neuroinflammation. While neuroinflammation-induced cognitive decline is well known.The circulating proinflammatory factors can also cross the disrupted BBB, and trigger neuroinflammatory response. Several plasma proteins fibrinogen and fibrin can enter and deposit in the CNS after BBB disruption, which bind to CD11b and CD11c on innate immune cells, triggering an inflammatory response that is toxic to neurons [354], while fibrin depletion protects against cognitive deficits. Plasma albumin is also reported to activate microglia and astrocytes when leaking through the disrupted BBB, which induces neuronal apoptosis, neuroinflammation, and increased tau phosphorylation, eventually impairing the spatial learning and memory abilities [355].Other plasma proteins such as sclerostin across the BBB in the 22-month rather than 10-month mice, can increase Aβ production through β-catenin-BACE1 signalling, accordingly, high sclerostin levels in AD patients are associated with severe cognitive impairment [356]. Further, pathological proteins such as β-amyloid, Tau and α-synuclein can promote BBB disruption, which conversely exacerbates brain damage [357]. Since the BBB maintains the chemical composition of the neuronal “milieu”, which is required for normal neural circuits, synaptic transmission and remodeling, and neurogenesis in the brain, therefore BBB breakdown would disrupt brain homeostasis, regardless of the intracerebral or systemic causes, thereby affecting brain cognitive performance.
2. The BBB facilitates the supply of nutrients for the brain. Several endogenous transporters and receptors expressed in the ECs and pericytes at the BBB, for example, GLUT1 mediates the transport of glucose, Mfsd2a for transport of omega-3 fatty acids, LAT1 for amino acids, MCT1 for lactate while MCT8 for thyroid hormone, transferrin receptor for iron, and proton-coupled folate transporterr for folate [358] across the BBB into the brain. And latest studies confirm that endothelial FLVCR2 (also known as MFSD7C) is a BBB transorter of choline and responsible for the majority of choline uptake into the brain [359, 360]. Disruption of these nutrient transporters would affect the entry of components critical for brain function, development and repair, as well as BBB integrity [25, 361]. For example, Mfsd2a deficiency could reduce docosahexaenoic acid level in the brain, leading to microcephaly and hypomyelination; impaired cerebral folate transport is associated with various types of neurological and neurodevelopmental complications, while FLVCR2 deficiency may influence choline uptake into the brain, which could alter membrane synthesis, methylation of DNA and protein, and acetylcholine formation, a neurotransmitter that drives cognition and memory. Physiological blood-brain transport is impaired with age, these age-dependent changes could adversely influence cognitive function. However, whether changes in different BBB properties cause damage in specific cognitive domain is not known.
On the other side, BBB is one of routes for the clearance of waste metabolites out of the brain. The accumulation of waste metabolites around the vasculatures are the neuropathological findings of p-tau protein in cerebral amyloid angiopathy and aggregation of Aβ in AD. These observations clearly indicate that BBB may directly participates in the clearance of p-tau protein and Aβ protein accumulated in the brain. LRP1 is a critical transporter for eliminating Aβ protein. It is reported that BBB-mediated transport accounts for 54% of brain Aβ40 clearance or 80% Aβ protein clearance through trans-vascular transcytosis [362]. Although such findings are based on clearance of small Aβ peptide in normal physiological condition, since these waste metabolites aggregation is detrimental to neurological function, it is expected that BBB damage would impair the clearance of metabolites out of the brain, and consequently leading to abnormal accumulation of p-tau and Aβ, which eventually cause cognitive dysfunction.
3. The dysfunctional BBB themselves have cumulative effects of inflammation, oxidative stress, and metabolic dysfunction, which can produce structural and biochemical alterations to the nervous system. For example, senescence of cerebral ECs can upregulate proinflammatory cell adhesion molecules and cytokines [254]. Further, dysfunctional ECs secrete heat shock protein 90α, which blocks oligodendroglial differentiation, contributing to impaired myelination in cSVD [363].The injured pericytes at the BBB release PDGFRβ into the CSF [192], loss of vascular PDGFRβ is found significantly associated with increased vascular Aβ40 and Aβ42 burden in mild cognitive impairment (MCI) and AD human donors compared to cognitively normal controls [364]. While RAGE can stimulate TGF-β release by pericytes as well as VEGF and MMP-2 release by ECs to disrupt BBB [365]. Further, normal ECs express low levels of leukocyte adhesion molecules, instead, upregulation of leukocyte adhesion molecules on ECs surface after their damage can initiate binding of leukocytes, the first step of their entrance into the brain parenchyma. Moreover, damaged BBB can serve as a CNS entry route for certain virus, for example, SARS-Cov-2 through Neuropilin-1-mediated transport [366]. Therefore the BBB dysfunction may be the first change in development of certain brain disorders and cognitive impairments.
4. The BBB is also responsible for neurovascular development, angiogenesis and CBF regulation. ECs, pericytes and astrocyte endfeet all have a major role in the control of CBF, therefore BBB is important for the coordination of neurovascular coupling and maintenance of brain function, and its dysregulation can cause brain hypoperfusion or hyperemia, namely, loss of CBF autoregulation, which has been implicated in AD.The BBB disruption induced by stroke not only recruits immune cells into ischemic region, but also increases water exchange, leading vasogenic edema. This can greatly decrease CBF and increase neuronal damage, which potentially impairs the cognitive function. While protecting against BBB disruption can maintain microcirculation and cerebral perfusion, contributing to cognitive improvement [367]. On other hand, by stimulating vessel growth, angiogenesis may stabilize brain perfusion, thereby promoting neuronal survival, brain plasticity, and neurologic recovery after ischemic stroke [368].
Whether BBB disruption definitely cause cognitive decline?
This question may be answered from the relationship between deliberate BBB opening and cognitive function. FUS coupled with intravenous administration of microbubbles is a noninvasive technique that has been shown to open the BBB reliably and reversibly in rodents and non-human primates (NHP). This technique is promising for drug or gene delivery into the brain. The potential neural effects of long-term FUS application have been investigated. In mice, repeated BBB opening for up to 6 months does not cause motor impairment, morphological and behavioral changes [369], similarly, treatment with FUS combined with microbubbles for up to 20 months induces neither significant edema nor microhemorrhage in most of the NHP brains assessed by MRI, nor visual, cognitive, motivational, and motor dysfunctions detected by quantitative cognitive testing [370]. In several small sample-size clinical trials, FUS-mediated BBB disruption showed no significant effect on cognition evolution in patients with mild AD [371, 372], whereas in a larger clinical trial, FUS-based repeated BBB opening in multiple cortical and deep brain regions decreased amyloid plaque burden without worsening cognitive function in patients with mild AD at 1 year follow-up [373]. These data suggest that the repeated BBB opening using FUS with microbubbles is clinically safe. Further studies have shown that the safety (lack of edema or microhemorrhage) of FUS was improved in awake NHP compared to those anesthetized [374]. Interestingly, FUS-mediated transient BBB opening significantly increased the expression of brain-derived neurotrophic factor and early growth response protein 1, neurogenesis in the hippocampus, and acetylcholinesterase activity in the frontal cortex and hippocampus of adult rats, leading to spatial memory improvement [375]. Furthermore, FUS-mediated BBB opening modulates tau hyperphosphorylation and Aβ accumulation to alleviate behavioral deficits in AD animal models [376, 377].
Another suggested approach to open BBB is to inhibit endothelial S1P receptor-1. This transient BBB opening can be used for the delivery of small molecules into the CNS. However, in endothelial S1P receptor-1 knockout mice, chronic BBB leakiness can induce cognitive impairments through TJ proteins subcellular redistribution rather than neuroinflammation [378]. This may be the first evidence showing that BBB disruption alone can directly lead to cognitive decline. Several studies suggest that the cerebral effects of short-term controlled BBB opening by FUS may be related to changes in neural metabolism and activity, as well as proteomic profiles, but not the BBB opening itself [379, 380]. Similarly, scanning ultrasound for BBB opening is effective not only for restoring long-term potential but also for ameliorating spatial learning deficits in aged mice [381]. Recently, a pilot study examined the effects of low-intensity transcranial FUS on hippocampal BBB opening, regional cerebral glucose metabolic rate, and cognition in patients with AD, and found that FUS did not activate BBB opening, but could improve cerebral metabolism and cognitive function [382]. Therefore, BBB opening adversely influences cognitive function or not seems dependent on whether the structural components of BBB are disrupted.
Interestingly, severity of BBB permeability is not a key to cognitive impairment. A recent clinical study showed that BBB permeability to water rather than large molecule albumin was related to AD markers Aβ and p-tau levels in CSF, and was a predictor of cognitive function in the patients with mild cognitive impairment [54]. Further, the regional heterogeneity of the BBB permeability may also matter. Several studies have revealed that BBB permeability among brain regions differs during aging. The study showed that the water exchange rate (kw) in the frontoparietal brain regions was positively associated with cognitive performance in elder people, whereas the kw in the basal ganglia was negatively associated with cognitive performance [383]. In terms of cognitive function, the clinical relevance of BBB disruption in regional heterogeneity remains unclear. Nevertheless, given that BBB leakage in the hippocampal region is most relevant to cognitive function, age-dependent breakdown of hippocampal BBB is considered to exacerbate cognitive function [384].Therefore, dependent on the brain region where the BBB is disrupted, cognitive status does not necessarily deteriorate, although BBB permeability increases with aging.
Summary
As a physical, transport, and metabolic interface between the CNS and circulatory blood flow, BBB plays a critical role in maintaining homeostasis and function of the brain. And the role of the BBB has now been extended to the delivery of neuroprotective and antitumor drugs to improve neurological outcomes [385]. BBB disruption predisposes to edema, excitotoxicity, and infiltration of blood-borne components, and an impaired BBB is often accompanied by neuroinflammation and neurodegeneration, which greatly influences cognitive brain function. Although the neural mechanisms underlying cognitive decline are diverse and complex, BBB disruption is increasingly being seen as a key event and early biomarker in various brain and systemic disorders leading to cognitive decline. It is not clear whether such changes in the BBB are initial etiology of cognitive impairments, and if so, how much of BBB dysfunction directly accounts for cognitive decline. As severity of BBB disruption and cognitive decline commonly both tend to increase with disease progression, few direct evidence confirms that BBB disruption causally leads to cognitive decline, however, current data appears to indirectly support the notion that BBB integrity is essential for normal cognitive function. Therefore, although many questions remain unanswered [386], targeting BBB disruption has important clinical implications for cognitive improvement if it can be detected and identified early. Clarifying the cellular and molecular mechanisms underlying BBB disruption may provide multiple opportunities to obtain a desirable therapy for cognitive decline. To date, there is limited efficacy of therapy to prevent or repair BBB damage at the cerebral microvascular level. Novel strategies targeting BBB disruption and restoration are still awaiting development.Therefore, further studies should focus on clinically effective and safe interventions to protect and restore the structural and functional integrity of the BBB in patients with cognitive decline, followed by reverse homeostatic imbalance in the brain, which will finally improve their cognitive functions regardless of the pathogenesis.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ACE2:
-
Angiotensin Converting Enzyme 2
- AD:
-
Alzheimer’s Disease
- AhR:
-
Aryl hydrocarbon Receptor
- AJs:
-
Adherens Junctions
- ALK:
-
Activin receptor-Like Kinase
- Ang2:
-
Angiopoietin-2
- Annexin A1:
-
Membrane association protein A1
- APOE4:
-
ApolipoProtein E 4
- APP:
-
Amyloid Precursor Protein
- AQP4:
-
AQuaPorin 4
- ASK1:
-
Apoptosis Signal-regulating Kinase 1
- ASL:
-
Arterial Spin Labeling
- Atg7:
-
Autophagy related 7
- BACE1:
-
β-site APP-Cleaving Enzyme 1
- BBB:
-
Blood-Brain Barrier
- BD:
-
Bipolar Disorder
- BMECs:
-
Brain Microvascular Endothelial Cells
- BMP:
-
Bone Morphogenetic Protein
- Bmpr1a:
-
BMP type IA
- CBF:
-
Cerebral Blood Flow
- CCR5:
-
C-C motif chemokine Receptor 5
- Cd:
-
Cadmium
- CeGC:
-
Cerebral endothelial Glycocalyx
- CNS:
-
Central Nervous System
- COVID-19:
-
Coronavirus Disease 2019
- COX2:
-
Cyclooxygenase-2
- CSF:
-
Cerebrospinal Fluid
- CSVD:
-
Cerebral Small Vessel Disease
- CX43:
-
ConneXin 43
- CypA:
-
Cyclophilin A
- DCE-MRI:
-
Dynamic Contrast-Enhanced magnetic resonance imaging
- Drp1:
-
Dynamin-related protein 1
- ECGF1:
-
Endothelial Cell Growth Factor 1
- ECs:
-
Endothelial Cells
- eNOS:
-
endothelial Nitric Oxide Synthase
- EVs:
-
Extracellular Vesicles
- Fis1:
-
mitochondrial Fission protein 1
- FUS:
-
Focused UltraSound
- GCs:
-
GlucoCorticoids
- GABAA :
-
γ-AminoButyric Acid subtype A
- GLUT1:
-
Glucose Transporter protein isoform 1
- HDAC1:
-
Histone deacetylase 1
- HMGB1:
-
High Mobility Group Box 1
- IL:
-
InterLeukin
- IFN-γ:
-
InterFeroNγ
- Kw:
-
Water exchange rate
- LPS:
-
LipoPolySaccharide
- LRP1:
-
Low-density lipoprotein Receptor-related Protein 1
- MB:
-
Methylene Blue
- MCP-1:
-
Monocyte Chemoattractant Protein-1
- MCT8:
-
MonoCarboxylate Transporter 8
- Mfsd2a:
-
Major facilitator superfamily domain-containing 2a
- MMP:
-
Matrix MetallopePtidase
- MSCs:
-
Mesenchymal Stromal Cells
- NAc:
-
Nucleus Accumbens
- NAD:
-
Nicotinamide Adenine Dinucleotide
- NHP:
-
Non-Human Primates
- NLRP3:
-
Nod-Like Receptor Protein 3
- NMDA:
-
N-Methyl-D-Aspartate
- NO:
-
Nitric Oxide
- NVU:
-
NeuroVascular Units
- OGD:
-
Oxygen-Glucose Deprivation
- OPC:
-
Oligodendrocyte Progenitor Cell
- PDGFB:
-
Platelet-Derived Growth Factor B
- PDGFRβ:
-
Platelet-Derived Growth Factor Receptor beta
- PD:
-
Parkinson’s Disease
- PET:
-
Positron Emission Tomography
- PGE2:
-
ProstaGlandin E2
- PM2.5:
-
fine Particulate Matter
- PND:
-
Postoperative Neurocognitive Disorders
- POD:
-
PostOperative Delirium
- p-tau:
-
phosphorylated tau
- Poldip2:
-
Polymerase delta-interacting protein 2
- PSEN1:
-
PreSENilin 1
- PTEN:
-
Phosphatase and TENsin homolog
- Q-Alb:
-
cerebrospinal fluid/serum Albumin ratio
- RAGE:
-
Advanced Glycation End pRoduct
- RNF213:
-
RiNg Finger protein 213
- ROS:
-
Reactive Oxygen Species
- S1P:
-
Sphingosine-1-Phosphate
- SAE:
-
Sepsis-Associated Encephalopathy
- SAH:
-
SubarAchnoid Hemorrhage
- Saps:
-
Secreted aspartic proteinases
- SARS-CoV-2:
-
Severe Acute Respiratory Syndrome CoronaVirus 2
- SD:
-
Sleep Deprivation
- SZ:
-
SchiZophrenia
- TBI:
-
Traumatic Brain Injury
- TGF-β1:
-
Transforming Growth Factor β1 type I
- TIMPs:
-
Tissue Inhibitors of MetalloProteases
- TJs:
-
Tight Junctions
- TLRs:
-
Toll-Like Receptors
- TNFα:
-
Tumor Necrosis Factor alpha
- TRPM2:
-
Transient Receptor Potential Melastatin 2
- VE-cadherin:
-
Vascular Endothelial cadherin
- VEGF:
-
Vascular Endothelial Growth Factor
- ZO-1:
-
Zonula Occludens 1
References
Duarte-Delgado NP, Vásquez G, Ortiz-Reyes BL. Blood-brain barrier disruption and neuroinflammation as pathophysiological mechanisms of the diffuse manifestations of neuropsychiatric systemic lupus erythematosus. Autoimmun Rev. [Journal Article; Review]. 2019 2019-04-01;18(4):426–32.
Yue H, Xie K, Ji X, Xu B, Wang C, Shi P. Vascularized neural constructs for ex-vivo reconstitution of blood-brain barrier function. Biomaterials. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-07-01;245:119980.
Andreone BJ, Chow BW, Tata A, Lacoste B, Ben-Zvi A, Bullock K et al. Blood-brain barrier permeability is regulated by lipid transport-dependent suppression of Caveolae-Mediated Transcytosis. Neuron [Journal Article]. 2017 2017-05-03;94(3):581–94.
Grubb S. Ultrastructure of precapillary sphincters and the neurovascular unit. Vasc Biol [Journal Article]. 2023 2023-01-01;5(1).
Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia. [Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-07-01;58(9):1094–103.
Kim ES, Kim KS, Lee CH, Jeon MT, Lee SB, Lee JH et al. Brain endothelial cells utilize glycolysis for the maintenance of the Transcellular permeability. Mol Neurobiol. [Journal Article]. 2022 2022-07-01;59(7):4315–33.
Liu D, Liao P, Li H, Tong S, Wang B, Lu Y, Gao Y, Huang Y, Zhou H, Shi L, Papadimitriou J, Zong Y, Yuan J, Chen P, Chen Z, Ding P, Zheng Y, Zhang C, Zheng M, Gao J. Regulation of blood-brain barrier integrity by Dmp1-expressing astrocytes through mitochondrial transfer. Sci Adv. 2024;10(26):eadk2913. https://doi.org/10.1126/sciadv.adk2913.
Lecrux C, Bourourou M, Hamel E. How reliable is cerebral blood flow to map changes in neuronal activity? Auton Neurosci-Basic. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2019 2019-03-01;217:71 – 9.
Chen J, Luo Y, Hui H, Cai T, Huang H, Yang F et al. CD146 coordinates brain endothelial cell-pericyte communication for blood-brain barrier development. P Natl Acad Sci USA. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-09-05;114(36):E7622–31.
Guo L, Yang Q, Wei R, Zhang W, Yin N, Chen Y et al. Enhanced pericyte-endothelial interactions through NO-boosted extracellular vesicles drive revascularization in a mouse model of ischemic injury. Nat Commun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-11-13;14(1):7334.
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C et al. Pericytes regulate the blood-brain barrier. Nature. [Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-11-25;468(7323):557–61.
Mäe MA, He L, Nordling S, Vazquez-Liebanas E, Nahar K, Jung B et al. Single-cell analysis of blood-brain barrier response to Pericyte loss. CIRC RES. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-02-19;128(4):e46–62.
Weng Y, Chen N, Zhang R, He J, Ding X, Cheng G et al. An integral blood-brain barrier in adulthood relies on microglia-derived PDGFB. Brain Behav Immun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2024 2024-01-01;115:705 – 17.
Vazquez-Liebanas E, Nahar K, Bertuzzi G, Keller A, Betsholtz C, Mäe MA. Adult-induced genetic ablation distinguishes PDGFB roles in blood-brain barrier maintenance and development. J Cerebr Blood F Met. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-02-01;42(2):264–79.
Nikolakopoulou AM, Zhao Z, Montagne A, Zlokovic BV. Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet-derived growth factor receptor-β signaling. Plos One [Journal Article]. 2017 2017-01-20;12(4):e176225.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to Disease and back. Physiol Rev. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2019 2019-01-01;99(1):21–78.
Sasson E, Anzi S, Bell B, Yakovian O, Zorsky M, Deutsch U et al. Nano-scale architecture of blood-brain barrier tight-junctions. Elife. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-12-24;10.
Stoddart P, Satchell SC, Ramnath R. Cerebral microvascular endothelial glycocalyx damage, its implications on the blood-brain barrier and a possible contributor to cognitive impairment. Brain Res [Journal Article; Review]. 2022 2022-04-01;1780:147804.
Liu H, Wei JY, Li Y, Ban M, Sun Q, Wang HJ et al. Endothelial depletion of Atg7 triggers astrocyte-microvascular disassociation at blood-brain barrier. J Cell Biol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-05-01;222(5).
Halder SK, Delorme-Walker VD, Milner R. β1 integrin is essential for blood-brain barrier integrity under stable and vascular remodelling conditions; effects differ with age. Fluids Barriers Cns. [Journal Article]. 2023 2023-07-03;20(1):52.
Araya R, Kudo M, Kawano M, Ishii K, Hashikawa T, Iwasato T et al. BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol Cell Neurosci. [Comparative Study; Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2008 2008-07-01;38(3):417–30.
Petersen MA, Tognatta R, Meyer-Franke A, Bushong EA, Mendiola AS, Yan Z et al. BMP receptor blockade overcomes extrinsic inhibition of remyelination and restores neurovascular homeostasis. Brain. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-09-04;144(8):2291–301.
Wang Z, Zheng Y, Wang F, Zhong J, Zhao T, Xie Q et al. Mfsd2a and Spns2 are essential for sphingosine-1-phosphate transport in the formation and maintenance of the blood-brain barrier. Sci Adv. [Journal Article; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2020 2020-05-01;6(22):y8627.
Cui Y, Wang Y, Song X, Ning H, Zhang Y, Teng Y et al. Brain endothelial PTEN/AKT/NEDD4-2/MFSD2A axis regulates blood-brain barrier permeability. Cell Rep. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-07-06;36(1):109327.
Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2014 2014-05-22;509(7501):507–11.
Zhao C, Ma J, Wang Z, Li H, Shen H, Li X et al. Mfsd2a attenuates blood-brain barrier disruption after sub-arachnoid hemorrhage by inhibiting Caveolae-Mediated Transcellular Transport in rats. Transl Stroke Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-10-01;11(5):1012–27.
Guillén-Yunta M, Valcárcel-Hernández V, García-Aldea Á, Soria G, García-Verdugo JM, Montero-Pedrazuela A et al. Neurovascular unit disruption and blood-brain barrier leakage in MCT8 deficiency. Fluids Barriers Cns. [Journal Article]. 2023 2023-11-03;20(1):79.
Keaney J, Campbell M. The dynamic blood-brain barrier. Febs J [Journal Article; Review]. 2015 2015-11-01;282(21):4067–79.
Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2018 2018-03-01;135(3):311–36.
Banks WA. Brain meets body: the blood-brain barrier as an endocrine interface. Endocrinology. [Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.; Review]. 2012 2012-09-01;153(9):4111-9.
Nguyen LN, Ma D, Shui G, Wong P, Cazenave-Gassiot A, Zhang X et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature. [Journal Article; Research Support, Non-U.S. Gov’t]. 2014 2014-05-22;509(7501):503–6.
Sangha V, Hoque MT, Henderson JT, Bendayan R. Novel localization of folate transport systems in the murine central nervous system. Fluids Barriers CNS. [Journal Article]. 2022 2022-11-23;19(1):92.
Mayerl S, Müller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest. 2014;124(5):1987–99. 2014-05-01.
Pflanzner T, Kuhlmann CR, Pietrzik CU. Blood-brain-barrier models for the investigation of transporter- and receptor-mediated amyloid-β clearance in Alzheimer’s disease. Curr Alzheimer Res. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2010 2010-11-01;7(7):578 – 90.
Bellaver B, Puig-Pijoan A, Ferrari-Souza JP, Leffa DT, Lussier FZ, Ferreira P et al. Blood-brain barrier integrity impacts the use of plasma amyloid-β as a proxy of brain amyloid-β pathology. Alzheimers Dement. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-09-01;19(9):3815–25.
Pachter JS, de Vries HE, Fabry Z. The blood-brain barrier and its role in immune privilege in the central nervous system. J Neuropath Exp Neur. [Journal Article; Review]. 2003 2003-06-01;62(6):593–604.
Huang L, Zhan D, Xing Y, Yan Y, Li Q, Zhang J et al. FGL2 deficiency alleviates maternal inflammation-induced blood-brain barrier damage by blocking PI3K/NF-κB mediated endothelial oxidative stress. Front Immunol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-01-20;14:1157027.
Zhao Q, Dai W, Chen HY, Jacobs RE, Zlokovic BV, Lund BT et al. Prenatal disruption of blood-brain barrier formation via cyclooxygenase activation leads to lifelong brain inflammation. P Natl Acad Sci USA. [Journal Article]. 2022 2022-04-12;119(15):e2113310119.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. [Journal Article; Research Support, Non-U.S. Gov’t]. 2014 2014-04-03;508(7494):55–60.
Morris GP, Foster CG, Courtney JM, Collins JM, Cashion JM, Brown LS et al. Microglia directly associate with pericytes in the central nervous system. GLIA. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-08-01;71(8):1847–69.
Banks WA. The blood-brain barrier as an endocrine tissue. Nat Rev Endocrinol [Journal Article; Review]. 2019 2019-08-01;15(8):444–55.
Blomqvist A, Engblom D. Neural Mechanisms of Inflammation-Induced Fever. Neuroscientist. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2018 2018-08-01;24(4):381–99.
Li W, Chen Z, Chin I, Chen Z, Dai H. The role of VE-cadherin in blood-brain Barrier Integrity under Central Nervous System pathological conditions. Curr Neuropharmacol. [Journal Article; Review]. 2018 2018-01-20;16(9):1375–84.
Cordon J, Duggan MR, Gomez GT, Pucha KA, Peng Z, Dark HE et al. Identification of clinically relevant Brain endothelial cell biomarkers in plasma. Stroke [Journal Article]. 2023 2023-11-01;54(11):2853–63.
Pelisch N, Hosomi N, Mori H, Masaki T, Nishiyama A. RAS inhibition attenuates cognitive impairment by reducing blood- brain barrier permeability in hypertensive subjects. Curr Hypertens Rev. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2013 2013-05-01;9(2):93–8.
Bruno M, Bonomi CG, Ricci F, Di Donna MG, Mercuri NB, Koch G et al. Blood-brain barrier permeability is associated with different neuroinflammatory profiles in Alzheimer’s disease. Eur J Neurol [Journal Article]. 2024 2024-01-01;31(1):e16095.
McAleese KE, Graham S, Dey M, Walker L, Erskine D, Johnson M et al. Extravascular fibrinogen in the white matter of Alzheimer’s disease and normal aged brains: implications for fibrinogen as a biomarker for Alzheimer’s disease. Brain Pathol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2019 2019-05-01;29(3):414–24.
van de Haar HJ, Jansen J, van Osch M, van Buchem MA, Muller M, Wong SM et al. Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging. [Journal Article; Research Support, Non-U.S. Gov’t]. 2016 2016-09-01;45:190–6.
Ito S, Yagi R, Ogata S, Masuda T, Saito T, Saido T et al. Proteomic alterations in the brain and blood-brain barrier during brain Aβaccumulation in an APP knock-in mouse model of Alzheimer’s disease. Fluids Barriers CNS. [Journal Article]. 2023 2023-09-14;20(1):66.
Van Hulle C, Ince S, Okonkwo OC, Bendlin BB, Johnson SC, Carlsson CM, Asthana S, Love S, Blennow K, Zetterberg H. Scott Miners J.Elevated CSF angiopoietin-2 correlates with blood-brain barrier leakiness and markers of neuronal injury in early Alzheimer’s disease. Transl Psychiatry. 2024;14(1):3. https://doi.org/10.1038/s41398-023-02706-w.
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-01-21;85(2):296–302.
Zenaro E, Pietronigro E, Della BV, Piacentino G, Marongiu L, Budui S et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-08-01;21(8):880–6.
Singh C, Choi KB, Munro L, Wang HY, Pfeifer CG, Jefferies WA. Reversing pathology in a preclinical model of Alzheimer’s disease by hacking cerebrovascular neoangiogenesis with advanced cancer therapeutics. Ebiomedicine [Journal Article]. 2021 2021-09-01;71:103503.
Lin Z, Sur S, Liu P, Li Y, Jiang D, Hou X et al. Blood-brain barrier breakdown in relationship to Alzheimer and Vascular Disease. Ann Neurol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-08-01;90(2):227–38.
Miners JS, Kehoe PG, Love S, Zetterberg H, Blennow K. CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther. [Journal Article]. 2019 2019-09-14;11(1):81.
Moon Y, Jeon HJ, Han SH, Min-Young N, Kim HJ, Kwon KJ et al. Blood-brain barrier breakdown is linked to tau pathology and neuronal injury in a differential manner according to amyloid deposition. J Cerebr Blood F Met. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-11-01;43(11):1813–25.
Aarsland D, Batzu L, Halliday GM, Geurtsen GJ, Ballard C, Ray CK et al. Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2021 2021-07-01;7(1):47.
Pediaditakis I, Kodella KR, Manatakis DV, Le CY, Hinojosa CD, Tien-Street W et al. Modeling alpha-synuclein pathology in a human brain-chip to assess blood-brain barrier disruption. Nat Commun. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-10-08;12(1):5907.
Ruan Z, Zhang D, Huang R, Sun W, Hou L, Zhao J et al. Microglial Activation Damages Dopaminergic Neurons through MMP-2/-9-Mediated Increase of Blood-Brain Barrier Permeability in a Parkinson’s Disease Mouse Model. Int J Mol Sci [Journal Article]. 2022 2022-03-03;23(5).
Al-Bachari S, Naish JH, Parker G, Emsley H, Parkes LM. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front Physiol [Journal Article]. 2020 2020-01-20;11:593026.
Wong YY, Wu CY, Yu D, Kim E, Wong M, Elez R et al. Biofluid markers of blood-brain barrier disruption and neurodegeneration in Lewy body spectrum diseases: a systematic review and meta-analysis. Parkinsonism Relat D. [Journal Article; Meta-Analysis; Research Support, Non-U.S. Gov’t; Review; Systematic Review]. 2022 2022-08-01;101:119 – 28.
Kumar V, Lee JD, Coulson EJ, Woodruff TM. A validated quantitative method for the assessment of neuroprotective barrier impairment in neurodegenerative disease models. J Neurochem. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-08-01;158(3):807–17.
Pan Y, Nicolazzo JA. Altered blood-brain barrier and blood-spinal cord barrier dynamics in amyotrophic lateral sclerosis: impact on medication efficacy and safety. Brit J Pharmacol [Journal Article; Review]. 2022 2022-06-01;179(11):2577–88.
Duran-Vilaregut J, Del VJ, Manich G, Camins A, Pallàs M, Vilaplana J et al. Role of matrix metalloproteinase-9 (MMP-9) in striatal blood-brain barrier disruption in a 3-nitropropionic acid model of Huntington’s disease. Neuropath Appl Neuro. [Journal Article; Research Support, Non-U.S. Gov’t]. 2011 2011-08-01;37(5):525–37.
Puig-Pijoan A, Jimenez-Balado J, Fernández-Lebrero A, García-Escobar G, Navalpotro-Gómez I, Contador J et al. Risk of cognitive decline progression is associated to increased blood-brain-barrier permeability: a longitudinal study in a memory unit clinical cohort. Alzheimers Dement. [Journal Article; Observational Study]. 2024 2024-01-01;20(1):538–48.
Jabben N, Arts B, van Os J, Krabbendam L. Neurocognitive functioning as intermediary phenotype and predictor of psychosocial functioning across the psychosis continuum: studies in schizophrenia and bipolar disorder. J Clin Psychiat. [Comparative study; Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-06-01;71(6):764–74.
Zhao NO, Topolski N, Tusconi M, Salarda EM, Busby CW, Lima C et al. Blood-brain barrier dysfunction in bipolar disorder: molecular mechanisms and clinical implications. Brain Behav Immun Health [Journal Article; Review]. 2022 2022-05-01;21:100441.
Lizano P, Pong S, Santarriaga S, Bannai D, Karmacharya R. Brain microvascular endothelial cells and blood-brain barrier dysfunction in psychotic disorders. Mol Psychiatr [Journal Article]. 2023 2023-09-01;28(9):3698–708.
Melkersson K, Bensing S. Signs of impaired blood-brain barrier function and lower IgG synthesis within the central nervous system in patients with schizophrenia or related psychosis, compared to that in controls. Neuroendocrinol Lett. [Journal Article]. 2018 2018-03-01;39(1):33–42.
Mousten IV, Sørensen NV, Christensen R, Benros ME. Cerebrospinal fluid biomarkers in patients with Unipolar Depression compared with Healthy Control individuals: a systematic review and Meta-analysis. Jama Psychiat. [Journal Article; Meta-Analysis; Research Support, Non-U.S. Gov’t; Systematic Review]. 2022 2022-06-01;79(6):571–81.
Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci [Journal Article]. 2017 2017-12-01;20(12):1752–60.
Sun ZW, Wang X, Zhao Y, Sun ZX, Wu YH, Hu H et al. Blood-brain barrier dysfunction mediated by the EZH2-Claudin-5 axis drives stress-induced TNF-α infiltration and depression-like behaviors. Brain Behav Immun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2024 2024-01-01;115:143 – 56.
Cheng Y, Desse S, Martinez A, Worthen RJ, Jope RS, Beurel E. TNFα disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav Immun. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2018 2018-03-01;69:556 – 67.
Peng Z, Peng S, Lin K, Zhao B, Wei L, Tuo Q et al. Chronic stress-induced depression requires the recruitment of peripheral Th17 cells into the brain. J Neuroinflamm [Journal Article]. 2022 2022-07-14;19(1):186.
Dudek KA, Dion-Albert L, Lebel M, LeClair K, Labrecque S, Tuck E et al. Molecular adaptations of the blood-brain barrier promote stress resilience vs. depression. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2020 2020-02-11;117(6):3326–36.
Najjar S, Pahlajani S, De Sanctis V, Stern J, Najjar A, Chong D. Neurovascular unit dysfunction and blood-brain barrier hyperpermeability contribute to Schizophrenia Neurobiology: a theoretical integration of clinical and experimental evidence. Front Psychiatry. [Journal Article; Review]. 2017 2017-01-20;8:83.
Thurgur H, Pinteaux E. Microglia in the neurovascular unit: blood-brain barrier-microglia interactions after Central Nervous System disorders. Neuroscience. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2019 2019-05-01;405:55–67.
Barisano G, Montagne A, Kisler K, Schneider JA, Wardlaw JM, Zlokovic BV. Blood-brain barrier link to human cognitive impairment and Alzheimer’s Disease. Nat Cardiovasc Res. [Journal Article]. 2022 2022-02-01;1(2):108–15.
Alluri H, Wiggins-Dohlvik K, Davis ML, Huang JH, Tharakan B. Blood-brain barrier dysfunction following traumatic brain injury. Metab Brain Dis. [Journal Article; Review]. 2015 2015-10-01;30(5):1093–104.
Balami JS, Chen RL, Grunwald IQ, Buchan AM. Neurological complications of acute ischaemic stroke. LAncet Neurol. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2011 2011-04-01;10(4):357–71.
Jing K, Chen F, Shi X, Guo J, Liu X. Dual effect of C-C motif chemokine receptor 5 on ischemic stroke: more harm than benefit? Eur J Pharmacol. [Journal Article; Review]. 2023 2023-08-15;953:175857.
Fujimoto M, Shiba M, Kawakita F, Liu L, Shimojo N, Imanaka-Yoshida K et al. Deficiency of tenascin-C and attenuation of blood-brain barrier disruption following experimental subarachnoid hemorrhage in mice. J Neurosurg [Journal Article]. 2016 2016-06-01;124(6):1693–702.
Feng D, Zhou J, Liu H, Wu X, Li F, Zhao J et al. Astrocytic NDRG2-PPM1A interaction exacerbates blood-brain barrier disruption after subarachnoid hemorrhage. Sci Adv. [Journal Article]. 2022 2022-09-30;8(39):q2423.
Zheng Y, Hu Q, Manaenko A, Zhang Y, Peng Y, Xu L et al. 17β-Estradiol attenuates hematoma expansion through estrogen receptor α/silent information regulator 1/nuclear factor-kappa b pathway in hyperglycemic intracerebral hemorrhage mice. Stroke. [Journal Article; Research Support, N.I.H., Extramural]. 2015 2015-02-01;46(2):485–91.
Chu H, Gao Z, Huang C, Dong J, Tang Y, Dong Q. Relationship between Hematoma Expansion Induced by Hypertension and Hyperglycemia and blood-brain barrier disruption in mice and its possible mechanism: role of Aquaporin-4 and Connexin43. Neurosci Bull. [Journal Article]. 2020 2020-11-01;36(11):1369–80.
Yang Y, Rosenberg GA. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2011 2011-11-01;42(11):3323-8.
Taheri S, Gasparovic C, Huisa BN, Adair JC, Edmonds E, Prestopnik J et al. Blood-brain barrier permeability abnormalities in vascular cognitive impairment. Stroke. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2011 2011-08-01;42(8):2158–63.
Rosenberg GA, Sullivan N, Esiri MM. White matter damage is associated with matrix metalloproteinases in vascular dementia. Stroke. [Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 2001 2001-05-01;32(5):1162–8.
Thapa K, Khan H, Singh TG, Kaur A. Traumatic Brain Injury: mechanistic insight on pathophysiology and potential therapeutic targets. J Mol Neurosci. [Journal Article; Review]. 2021 2021-09-01;71(9):1725–42.
Rajeev V, Fann DY, Dinh QN, Kim HA, De Silva TM, Lai M et al. Pathophysiology of blood brain barrier dysfunction during chronic cerebral hypoperfusion in vascular cognitive impairment. Theranostics. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2022 2022-01-20;12(4):1639–58.
Callegari K, Dash S, Uchida H, Shingai Y, Liu C, Khodarkovskaya A et al. Molecular profiling of the stroke-induced alterations in the cerebral microvasculature reveals promising therapeutic candidates. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-04-18;120(16):e2089181176.
Shi H, Wang HL, Pu HJ, Shi YJ, Zhang J, Zhang WT et al. Ethyl pyruvate protects against blood-brain barrier damage and improves long-term neurological outcomes in a rat model of traumatic brain injury. Cns Neurosci Ther. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-04-01;21(4):374–84.
Yang J, Wang K, Hu T, Wang G, Wang W, Zhang J. Vitamin D3 supplement attenuates blood-brain barrier disruption and cognitive impairments in a rat model of traumatic Brain Injury. Neuromol Med. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-12-01;23(4):491–9.
Ware JB, Sinha S, Morrison J, Walter AE, Gugger JJ, Schneider A et al. Dynamic contrast enhanced MRI for characterization of blood-brain-barrier dysfunction after traumatic brain injury. Neuroimage-Clin [Journal Article]. 2022 2022-01-20;36:103236.
Liraz ZS, Sharabi S, Guez D, Daniels D, Cooper I, Shemesh C et al. Application of delayed contrast Extravasation magnetic resonance imaging for depicting subtle blood-brain barrier disruption in a traumatic brain Injury Model. J Neurotraum. [Journal Article; Research Support, Non-U.S. Gov’t]. 2024 2024-02-01;41(3–4):430–46.
Boyko M, Gruenbaum BF, Frank D, Natanel D, Negev S, Azab AN et al. The Integrity of the blood-brain barrier as a critical factor for regulating glutamate levels in traumatic brain Injury. Int J Mol Sci [Journal Article]. 2023 2023-03-20;24(6).
Yoo RE, Choi SH, Oh BM, Do SS, Lee EJ, Shin DJ et al. Quantitative dynamic contrast-enhanced MR imaging shows widespread blood-brain barrier disruption in mild traumatic brain injury patients with post-concussion syndrome. Eur Radiol [Journal Article]. 2019 2019-03-01;29(3):1308–17.
Cash A, de Jager C, Brickler T, Soliman E, Ladner L, Kaloss AM et al. Endothelial deletion of EPH receptor A4 alters single-cell profile and Tie2/Akap12 signaling to preserve blood-brain barrier integrity. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural]. 2023 2023-10-10;120(41):e2090267176.
Meng S, Cao H, Huang Y, Shi Z, Li J, Wang Y et al. ASK1-K716R reduces neuroinflammation and white matter injury via preserving blood-brain barrier integrity after traumatic brain injury. J Neuroinflamm [Journal Article]. 2023 2023-10-24;20(1):244.
Cordeira J, Kolluru SS, Rosenblatt H, Kry J, Strecker RE, McCarley RW. Learning and memory are impaired in the object recognition task during metestrus/diestrus and after sleep deprivation. Behav Brain Res. [Journal Article]. 2018 2018-02-26;339:124–9.
Sharma A, Muresanu DF, Lafuente JV, Patnaik R, Tian ZR, Buzoianu AD et al. Sleep Deprivation-Induced blood-brain barrier breakdown and brain dysfunction are exacerbated by size-related exposure to Ag and Cu nanoparticles. Neuroprotective effects of a 5-HT3 receptor antagonist Ondansetron. Mol Neurobiol. [Journal Article; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2015 2015-10-01;52(2):867–81.
Semyachkina-Glushkovskaya O, Mamedova A, Vinnik V, Klimova M, Saranceva E, Ageev V et al. Brain mechanisms of COVID-19-Sleep disorders. Int J Mol Sci [Journal Article; Review]. 2021 2021-06-28;22(13).
He J, Hsuchou H, He Y, Kastin AJ, Wang Y, Pan W. Sleep restriction impairs blood-brain barrier function. J Neurosci. 2014;34(44):14697–706. 2014-10-29.
Semyachkina-Glushkovskaya O, Postnov D, Penzel T, Kurths J. Sleep as a Novel Biomarker and a Promising Therapeutic Target for Cerebral Small Vessel Disease: A Review Focusing on Alzheimer’s Disease and the Blood-Brain Barrier. Int J Mol Sci [Journal Article; Review]. 2020 2020-08-31;21(17).
Medina-Flores F, Hurtado-Alvarado G, Contis-Montes DOA, López-Cervantes SP, Konigsberg M, Deli MA et al. Sleep loss disrupts pericyte-brain endothelial cell interactions impairing blood-brain barrier function. Brain Behav Immun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-10-01;89:118 – 32.
Gómez-González B, Hurtado-Alvarado G, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JÁ, Velázquez-Moctezuma J. REM sleep loss and recovery regulates blood-brain barrier function. Curr Neurovasc Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2013 2013-08-01;10(3):197–207.
Puech C, Badran M, Runion AR, Barrow MB, Cataldo K, Gozal D. Cognitive impairments, neuroinflammation and blood-brain barrier permeability in mice exposed to chronic sleep fragmentation during the Daylight Period. Int J Mol Sci [Journal Article]. 2023 2023-06-08;24(12).
Sang D, Lin K, Yang Y, Ran G, Li B, Chen C et al. Prolonged sleep deprivation induces a cytokine-storm-like syndrome in mammals. CELL. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-12-07;186(25):5500–16.
Axelrod S, Li X, Sun Y, Lincoln S, Terceros A, O’Neil J et al. The Drosophila blood-brain barrier regulates sleep via Moody G protein-coupled receptor signaling. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural]. 2023 2023-10-17;120(42):e1985636176.
Opp MR, George A, Ringgold KM, Hansen KM, Bullock KM, Banks WA. Sleep fragmentation and sepsis differentially impact blood-brain barrier integrity and transport of tumor necrosis factor-α in aging. Brain Behav Immun. [Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.]. 2015 2015-11-01;50:259 – 65.
Nakazato R, Kawabe K, Yamada D, Ikeno S, Mieda M, Shimba S et al. Disruption of Bmal1 impairs blood-brain Barrier Integrity via Pericyte Dysfunction. J Neurosci. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-10-18;37(42):10052–62.
Mazeraud A, Pascal Q, Verdonk F, Heming N, Chrétien F, Sharshar T. Neuroanatomy and Physiology of Brain Dysfunction in Sepsis. Clin Chest Med. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2016 2016-06-01;37(2):333–45.
Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. Jama-J Am Med Assoc. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2010 2010-10-27;304(16):1787-94.
Towner RA, Saunders D, Smith N, Towler W, Cruz M, Do S et al. Assessing long-term neuroinflammatory responses to encephalopathy using MRI approaches in a rat endotoxemia model. Geroscience. [Comparative study; Journal Article; Research Support, N.I.H., Extramural]. 2018 2018-02-01;40(1):49–60.
Catalão C, Santos-Júnior NN, Da CL, Souza AO, Alberici LC, Rocha M. Brain oxidative stress during experimental Sepsis is attenuated by Simvastatin Administration. Mol Neurobiol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-11-01;54(9):7008–18.
van der Poll T, van de Veerdonk FL, Scicluna BP, Netea MG. The immunopathology of sepsis and potential therapeutic targets. Nat Rev Immunol. [Journal Article; Review]. 2017 2017-07-01;17(7):407–20.
Zrzavy T, Höftberger R, Berger T, Rauschka H, Butovsky O, Weiner H et al. Pro-inflammatory activation of microglia in the brain of patients with sepsis. Neuropath Appl Neuro. [Journal Article; Research Support, Non-U.S. Gov’t]. 2019 2019-04-01;45(3):278–90.
Barlow B, Ponnaluri S, Barlow A, Roth W. Targeting the gut microbiome in the management of sepsis-associated encephalopathy. Front Neurol. [Journal Article; Review]. 2022 2022-01-20;13:999035.
Knox EG, Aburto MR, Tessier C, Nagpal J, Clarke G, O’Driscoll CM et al. Microbial-derived metabolites induce actin cytoskeletal rearrangement and protect blood-brain barrier function. Iscience [Journal Article]. 2022 2022-12-22;25(12):105648.
Yang R, Wang J, Wang F, Zhang H, Tan C, Chen H et al. Blood-Brain Barrier Integrity damage in bacterial meningitis: the underlying link, mechanisms, and therapeutic targets. Int J Mol Sci [Journal Article; Review]. 2023 2023-02-02;24(3).
Pan F, Zhu M, Liang Y, Yuan C, Zhang Y, Wang Y et al. Membrane vesicle delivery of a streptococcal M protein disrupts the blood-brain barrier by inducing autophagic endothelial cell death. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-06-13;120(24):e2075532176.
Song K, Han HJ, Kim S, Kwon J. Thymosin beta 4 attenuates PrP(106–126)-induced human brain endothelial cells dysfunction. Eur J Pharmacol [Journal Article]. 2020 2020-02-15;869:172891.
Kushwaha R, Li Y, Makarava N, Pandit NP, Molesworth K, Birukov KG et al. Reactive astrocytes associated with prion disease impair the blood brain barrier. Neurobiol Dis. [Journal Article; Research Support, N.I.H., Extramural]. 2023 2023-09-01;185:106264.
Mao L, Jin H, Wang M, Hu Y, Chen S, He Q et al. Neurologic manifestations of hospitalized patients with Coronavirus Disease 2019 in Wuhan, China. Jama Neurol. [Journal Article; Multicenter Study; Observational Study]. 2020 2020-06-01;77(6):683–90.
Satarker S, Nampoothiri M. Involvement of the nervous system in COVID-19: the bell should toll in the brain. Life Sci. [Journal Article; Review]. 2020 2020-12-01;262:118568.
Wang P, Jin L, Zhang M, Wu Y, Duan Z, Guo Y et al. Blood-brain barrier injury and neuroinflammation induced by SARS-CoV-2 in a lung-brain microphysiological system. Nat Biomed Eng [Journal Article]. 2023 2023-06-22.
Klein RS, Garber C, Funk KE, Salimi H, Soung A, Kanmogne M et al. Neuroinflammation during RNA viral infections. Annu Rev Immunol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.; Review]. 2019 2019-04-26;37:73–95.
Edén A. Author response: CSF biomarkers in patients with COVID-19 and neurologic symptoms: a Case Series. Neurology. [Comment; Journal Article]. 2021 2021-09-07;97(10):510.
Greene C, Connolly R, Brennan D, Laffan A, O’Keeffe E, Zaporojan L et al. Blood-brain barrier disruption and sustained systemic inflammation in individuals with long COVID-associated cognitive impairment. Nat Neurosci [Journal Article]. 2024 2024-03-01;27(3):421–32.
Jamil AM, Desa M. A review of the mechanisms of blood-brain barrier disruption during COVID-19 infection. J Neurosci Res [Journal Article; Review]. 2023 2023-11-01;101(11):1687–98.
Kempuraj D, Aenlle KK, Cohen J, Mathew A, Isler D, Pangeni RP et al. COVID-19 and long COVID: disruption of the neurovascular unit, blood-brain barrier, and tight junctions. Neuroscientist. [Journal Article; Review]. 2023 2023-09-11:2055057711.
Bleau C, Filliol A, Samson M, Lamontagne L. Brain Invasion by Mouse Hepatitis Virus depends on impairment of tight junctions and Beta Interferon Production in Brain Microvascular endothelial cells. J Virol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-10-01;89(19):9896–908.
Dhanda S, Sandhir R. Blood-brain barrier permeability is exacerbated in experimental model of hepatic Encephalopathy via MMP-9 activation and downregulation of tight Junction proteins. Mol Neurobiol. [Journal Article]. 2018 2018-05-01;55(5):3642–59.
Lv S, Song HL, Zhou Y, Li LX, Cui W, Wang W et al. Tumour necrosis factor-alpha affects blood-brain barrier permeability and tight junction-associated occludin in acute liver failure. Liver Int. [Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-09-01;30(8):1198–210.
Thumburu KK, Taneja S, Vasishta RK, Dhiman RK. Neuropathology of acute liver failure. Neurochem Int [Journal Article; Review]. 2012 2012-06-01;60(7):672–5.
Bobot M, Thomas L, Moyon A, Fernandez S, McKay N, Balasse L et al. Uremic toxic blood-brain barrier disruption mediated by AhR activation leads to cognitive impairment during experimental renal dysfunction. J Am Soc Nephrol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-07-01;31(7):1509–21.
Alam A, Hana Z, Jin Z, Suen KC, Ma D. Surgery, neuroinflammation and cognitive impairment. Ebiomedicine [Journal Article; Review]. 2018 2018-11-01;37:547 – 56.
Amado LA, Perrie H, Scribante J, Ben-Israel KA. Preoperative cognitive dysfunction in older elective noncardiac surgical patients in South Africa. Brit J Anaesth [Journal Article]. 2020 2020-09-01;125(3):275–81.
Huang Z, Huang B, Wei Q, Su X, Li X, Qin S et al. The Protective effects of Benzbromarone against Propofol-Induced inflammation and Injury in Human Brain Microvascular endothelial cells (HBMVECs). Neurotox Res. [Journal Article]. 2021 2021-10-01;39(5):1449–58.
Acharya NK, Goldwaser EL, Forsberg MM, Godsey GA, Johnson CA, Sarkar A et al. Sevoflurane and isoflurane induce structural changes in brain vascular endothelial cells and increase blood-brain barrier permeability: possible link to postoperative delirium and cognitive decline. Brain Res [Journal Article]. 2015 2015-09-16;1620:29–41.
Sun Z, Satomoto M, Adachi YU, Makita K. Blood-brain barrier disruption caused by neonatal sevoflurane-induced depends on exposure time and is reversible in mice. Korean J Anesthesiol. [Letter]. 2019 2019-08-01;72(4):389–91.
Wu C, Deng Q, Zhu L, Liu TC, Duan R, Yang L. Methylene Blue Pretreatment Protects against Repeated Neonatal Isoflurane exposure-Induced Brain Injury and Memory loss. Mol Neurobiol. [Journal Article]. 2024 2024-01-17.
Yang T, Velagapudi R, Kong C, Ko U, Kumar V, Brown P et al. Protective effects of omega-3 fatty acids in a blood-brain barrier-on-chip model and on postoperative delirium-like behaviour in mice. Brit J Anaesth [Comment; J Article]. 2023 2023-02-01;130(2):e370–80.
Hu J, Feng X, Valdearcos M, Lutrin D, Uchida Y, Koliwad SK et al. Interleukin-6 is both necessary and sufficient to produce perioperative neurocognitive disorder in mice. Brit J Anaesth [Journal Article]. 2018 2018-03-01;120(3):537–45.
Velagapudi R, Subramaniyan S, Xiong C, Porkka F, Rodriguiz RM, Wetsel WC et al. Orthopedic surgery triggers attention deficits in a Delirium-Like Mouse Model. Front Immunol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2019 2019-01-20;10:2675.
Yang S, Gu C, Mandeville ET, Dong Y, Esposito E, Zhang Y et al. Anesthesia and surgery impair blood-brain barrier and cognitive function in mice. Front Immunol. [Journal Article]. 2017 2017-01-20;8:902.
Abrahamov D, Levran O, Naparstek S, Refaeli Y, Kaptson S, Abu SM et al. Blood-brain barrier disruption after cardiopulmonary bypass: diagnosis and correlation to Cognition. Ann Thorac Surg. [Journal Article]. 2017 2017-07-01;104(1):161–9.
Danielson M, Reinsfelt B, Westerlind A, Zetterberg H, Blennow K, Ricksten SE. Effects of methylprednisolone on blood-brain barrier and cerebral inflammation in cardiac surgery-a randomized trial. J Neuroinflamm. [Journal Article; Randomized Controlled Trial]. 2018 2018-09-27;15(1):283.
Reinsfelt B, Ricksten SE, Zetterberg H, Blennow K, Fredén-Lindqvist J, Westerlind A. Cerebrospinal fluid markers of brain injury, inflammation, and blood-brain barrier dysfunction in cardiac surgery. Ann Thorac Surg. [Journal Article; Research Support, Non-U.S. Gov’t]. 2012 2012-08-01;94(2):549–55.
Devinney MJ, Wong MK, Wright MC, Marcantonio ER, Terrando N, Browndyke JN et al. Role of blood-brain barrier dysfunction in Delirium following non-cardiac surgery in older adults. Ann Neurol. [Journal Article]. 2023 2023-12-01;94(6):1024–35.
Lascola CD, Cotter SF, Klinger RY, Bisanar T, Cooter WM, Berger M et al. Blood-brain barrier permeability and cognitive dysfunction after surgery - A pilot study. J Clin Anesth. [Letter; Research Support, N.I.H., Extramural]. 2023 2023-06-01;86:111059.
Wu T, Wang X, Zhang R, Jiao Y, Yu W, Su D et al. Mice with pre-existing tumors are vulnerable to postoperative cognitive dysfunction. Brain Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-04-01;1732:146650.
Taylor J, Parker M, Casey CP, Tanabe S, Kunkel D, Rivera C et al. Postoperative delirium and changes in the blood-brain barrier, neuroinflammation, and cerebrospinal fluid lactate: a prospective cohort study. Brit J Anaesth [Journal Article]. 2022 2022-08-01;129(2):219–30.
Wang B, Li S, Cao X, Dou X, Li J, Wang L et al. Blood-brain barrier disruption leads to postoperative cognitive dysfunction. Curr Neurovasc Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-01-20;14(4):359–67.
Danielson M, Wiklund A, Granath F, Blennow K, Mkrtchian S, Nellgård B et al. Neuroinflammatory markers associate with cognitive decline after major surgery: findings of an explorative study. Ann Neurol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-03-01;87(3):370–82.
Vasunilashorn SM, Ngo LH, Dillon ST, Fong TG, Carlyle BC, Kivisäkk P et al. Plasma and cerebrospinal fluid inflammation and the blood-brain barrier in older surgical patients: the role of inflammation after surgery for elders (RISE) study. J Neuroinflamm. [Journal Article]. 2021 2021-04-30;18(1):103.
Ahles TA, Root JC, Ryan EL. Cancer- and cancer treatment-associated cognitive change: an update on the state of the science. J Clin Oncol. [Journal Article; Research Support, N.I.H., Extramural; Review]. 2012 2012-10-20;30(30):3675–86.
Cruzado JA, López-Santiago S, Martínez-Marín V, José-Moreno G, Custodio AB, Feliu J. Longitudinal study of cognitive dysfunctions induced by adjuvant chemotherapy in colon cancer patients. Support Care Cancer [Journal Article]. 2014 2014-07-01;22(7):1815–23.
Subramaniam CB, Bowen JM, Gladman MA, Lustberg MB, Mayo SJ, Wardill HR. The microbiota-gut-brain axis: An emerging therapeutic target in chemotherapy-induced cognitive impairment. Neurosci Biobehav R. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2020 2020-09-01;116:470–9.
Rummel NG, Chaiswing L, Bondada S, St CD, Butterfield DA. Chemotherapy-induced cognitive impairment: focus on the intersection of oxidative stress and TNFα. Cell Mol Life Sci. [Journal Article; Review]. 2021 2021-10-01;78(19–20):6533–40.
Ahire C, Nyul-Toth A, DelFavero J, Gulej R, Faakye JA, Tarantini S et al. Accelerated cerebromicrovascular senescence contributes to cognitive decline in a mouse model of paclitaxel (Taxol)-induced chemobrain. Aging Cell. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-07-01;22(7):e13832.
Redmond KJ, Milano MT, Kim MM, Trifiletti DM, Soltys SG, Hattangadi-Gluth JA. Reducing Radiation-Induced Cognitive toxicity: sparing the Hippocampus and Beyond. Int J Radiat Oncol. [Comment; Journal Article; Review]. 2021 2021-04-01;109(5):1131–6.
Yamada MK. A link between vascular damage and cognitive deficits after whole-brain radiation therapy for cancer: a clue to other types of dementia? Drug Discov Ther. [Journal Article; Review]. 2016 2016-01-20;10(2):79–81.
Cheng J, Jiang J, He B, Lin WJ, Li Y, Duan J et al. A phase 2 study of thalidomide for the treatment of radiation-induced blood-brain barrier injury. Sci Transl Med. [Clinical trial, phase II; Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-02-22;15(684):m6543.
Kiss T, Ungvari A, Gulej R, Nyúl-Tóth Á, Tarantini S, Benyo Z et al. Whole brain irradiation-induced endothelial dysfunction in the mouse brain. Geroscience [Journal Article]. 2024 2024-02-01;46(1):531–41.
Gulej R, Nyúl-Tóth Á, Ahire C, DelFavero J, Balasubramanian P, Kiss T et al. Elimination of senescent cells by treatment with Navitoclax/ABT263 reverses whole brain irradiation-induced blood-brain barrier disruption in the mouse brain. Geroscience. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-10-01;45(5):2983–3002.
Allen BD, Acharya MM, Montay-Gruel P, Jorge PG, Bailat C, Petit B et al. Maintenance of tight Junction Integrity in the absence of vascular dilation in the brain of mice exposed to Ultra-high-dose-rate FLASH Irradiation. Radiat Res. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2020 2020-12-01;194(6):625–35.
Curtaz CJ, Reifschläger L, Strähle L, Feldheim J, Feldheim JJ, Schmitt C et al. Analysis of microRNAs in exosomes of breast Cancer patients in search of molecular prognostic factors in Brain metastases. Int J Mol Sci [Journal Article]. 2022 2022-03-27;23(7).
Wu D, Deng S, Li L, Liu T, Zhang T, Li J et al. TGF-β1-mediated exosomal lnc-MMP2-2 increases blood-brain barrier permeability via the miRNA-1207-5p/EPB41L5 axis to promote non-small cell lung cancer brain metastasis. Cell Death Dis. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-07-20;12(8):721.
Chen D, Mackenzie L, Hossain SZ, Wang JX, Jiang PL, Wang Y et al. Cognitive impairment experienced by Chinese breast cancer survivors. Sci Rep-Uk. [Journal Article]. 2023 2023-12-14;13(1):22245.
Zhang DF, Li ZH, Zhang ZP, He YF, Shang BL, Xu XF et al. Cognitive changes are associated with increased blood-brain barrier leakage in non-brain metastases lung cancer patients. Brain Imaging Behav. [Journal Article]. 2023 2023-02-01;17(1):90–9.
Montagne A, Nation DA, Sagare AP, Barisano G, Sweeney MD, Chakhoyan A et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nat [Journal Article]. 2020 2020-05-01;581(7806):71–6.
Halliday MR, Pomara N, Sagare AP, Mack WJ, Frangione B, Zlokovic BV. Relationship between cyclophilin a levels and matrix metalloproteinase 9 activity in cerebrospinal fluid of cognitively normal apolipoprotein e4 carriers and blood-brain barrier breakdown. Jama Neurol. [Journal Article; Research Support, N.I.H., Extramural]. 2013 2013-09-01;70(9):1198–200.
Alruwais NM, Rusted JM, Tabet N, Dowell NG. Evidence of emerging BBB changes in mid-age apolipoprotein E epsilon-4 carriers. Brain Behav. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-12-01;12(12):e2806.
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. [Journal Article; Research Support, N.I.H., Extramural]. 2012 2012-05-16;485(7399):512–6.
Oue H, Yamazaki Y, Qiao W, Yuanxin C, Ren Y, Kurti A et al. LRP1 in vascular mural cells modulates cerebrovascular integrity and function in the presence of APOE4. JCI Insight. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-04-10;8(7).
Liu CC, Zhao J, Fu Y, Inoue Y, Ren Y, Chen Y et al. Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2022 2022-08-01;25(8):1020–33.
Tanzi RE. The genetics of Alzheimer disease. Csh Perspect Med. [Journal Article; review]. 2012 2012-10-01;2(10).
Armstrong RA. Spatial correlations between beta-amyloid (Abeta) deposits and blood vessels in familial Alzheimer’s disease. Folia Neuropathol [Journal Article]. 2008 2008-01-20;46(4):241–8.
Taglia I, Mignarri A, Olgiati S, Menci E, Petrocelli PL, Breedveld GJ et al. Primary familial brain calcification: genetic analysis and clinical spectrum. Movement Disord. [Journal Article; Research Support, Non-U.S. Gov’t]. 2014 2014-11-01;29(13):1691–5.
Wang Y, Du W, Sun Y, Zhang J, Ma C, Jin X. CRTC1 is a potential target to delay aging-induced cognitive deficit by protecting the integrity of the blood-brain barrier via inhibiting inflammation. J Cerebr Blood F Met. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2023 2023-07-01;43(7):1042–59.
Bieri G, Schroer AB, Villeda SA. Blood-to-brain communication in aging and rejuvenation. Nat Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2023 2023-03-01;26(3):379–93.
Mahroo A, Konstandin S, Günther M. Blood-brain barrier permeability to water measured using multiple Echo Time arterial spin labeling MRI in the Aging Human Brain. J Magn Reson Imaging. [Journal Article]. 2024 2024-04-01;59(4):1269–82.
Ting KK, Coleman P, Kim HJ, Zhao Y, Mulangala J, Cheng NC et al. Vascular senescence and leak are features of the early breakdown of the blood-brain barrier in Alzheimer’s disease models. Geroscience [Journal Article]. 2023 2023-12-01;45(6):3307–31.
Iwao T, Takata F, Matsumoto J, Goto Y, Aridome H, Yasunaga M et al. Senescence in brain pericytes attenuates blood-brain barrier function in vitro: a comparison of serially passaged and isolated pericytes from aged rat brains. Biochem Bioph Res Co. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-02-19;645:154 – 63.
Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG et al. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2016 2016-04-01;47(4):1068–77.
Elbert DL, Patterson BW, Lucey BP, Benzinger T, Bateman RJ. Importance of CSF-based Aβ clearance with age in humans increases with declining efficacy of blood-brain barrier/proteolytic pathways. Commun Biol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2022 2022-01-27;5(1):98.
Ximerakis M, Holton KM, Giadone RM, Ozek C, Saxena M, Santiago S et al. Heterochronic parabiosis reprograms the mouse brain transcriptome by shifting aging signatures in multiple cell types. Nat Aging [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-03-01;3(3):327–45.
Budamagunta V, Kumar A, Rani A, Bean L, Manohar-Sindhu S, Yang Y et al. Effect of peripheral cellular senescence on brain aging and cognitive decline. Aging Cell. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-05-01;22(5):e13817.
Zhan R, Meng X, Tian D, Xu J, Cui H, Yang J et al. NAD(+) rescues aging-induced blood-brain barrier damage via the CX43-PARP1 axis. Neuron. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-11-15;111(22):3634–49.
Kurz C, Walker L, Rauchmann BS, Perneczky R. Dysfunction of the blood-brain barrier in Alzheimer’s disease: Evidence from human studies. Neuropath Appl Neuro. [Journal Article; Review; Systematic Review]. 2022 2022-04-01;48(3):e12782.
Cicognola C, Mattsson-Carlgren N, van Westen D, Zetterberg H, Blennow K, Palmqvist S et al. Associations of CSF PDGFRβ with aging, blood-brain barrier damage, Neuroinflammation, and Alzheimer Disease pathologic changes. Neurology. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-07-04;101(1):e30–9.
Zarate SM, Huntington TE, Bagher P, Srinivasan R. Aging reduces calreticulin expression and alters spontaneous calcium signals in astrocytic endfeet of the mouse dorsolateral striatum. NPJ Aging [Journal Article]. 2023 2023-03-31;9(1):5.
Debette S, Seshadri S, Beiser A, Au R, Himali JJ, Palumbo C et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2011 2011-08-02;77(5):461–8.
Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2010 2010-12-01;68 Suppl 2(Suppl 2):S74-87.
Mohammadi MT, Dehghani GA. Acute hypertension induces brain injury and blood-brain barrier disruption through reduction of claudins mRNA expression in rat. Pathol Res Pract. [Journal Article; Research Support, Non-U.S. Gov’t]. 2014 2014-12-01;210(12):985–90.
Fan Y, Yang X, Tao Y, Lan L, Zheng L, Sun J. Tight junction disruption of blood-brain barrier in white matter lesions in chronic hypertensive rats. Neuroreport. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-12-02;26(17):1039–43.
Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2013 2013-11-01;62(5):810–7.
Bailey DM, Bain AR, Hoiland RL, Barak OF, Drvis I, Hirtz C et al. Hypoxemia increases blood-brain barrier permeability during extreme apnea in humans. J Cerebr Blood F MET. [Journal Article; Research Support, U.S. Gov’t, Non-P.H.S.]. 2022 2022-06-01;42(6):1120–35.
Chen S, Li L, Peng C, Bian C, Ocak PE, Zhang JH et al. Targeting oxidative stress and inflammatory response for blood-brain Barrier Protection in Intracerebral Hemorrhage. Antioxid Redox Sign. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2022 2022-07-01;37(1–3):115–34.
Li W, Niu X, Dai Y, Wu X, Li J, Sheng W. Rnf-213 knockout induces Pericyte reduction and blood-brain barrier impairment in mouse. Mol Neurobiol. [Journal Article]. 2023 2023-11-01;60(11):6188–200.
Teng Z, Dong Y, Zhang D, An J, Lv P. Cerebral small vessel disease and post-stroke cognitive impairment. Int J Neurosci [Journal Article; Review]. 2017 2017-09-01;127(9):824–30.
Voorter P, van Dinther M, Jansen WJ, Postma AA, Staals J, Jansen J et al. Blood-brain barrier disruption and Perivascular spaces in Small Vessel Disease and neurodegenerative diseases: a review on MRI methods and insights. J Magn Reson Imaging. [Journal Article; Review]. 2024 2024-02-01;59(2):397–411.
Fujihara R, Chiba Y, Nakagawa T, Nishi N, Murakami R, Matsumoto K et al. Albumin microvascular leakage in brains with diabetes mellitus. Microsc Res Techniq. [Journal Article]. 2016 2016-09-01;79(9):833–7.
Starr JM, Wardlaw J, Ferguson K, MacLullich A, Deary IJ, Marshall I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosur Ps. [Journal Article; Research Support, Non-U.S. Gov’t]. 2003 2003-01-01;74(1):70–6.
Venkat P, Chopp M, Chen J. Blood-brain barrier disruption, vascular impairment, and Ischemia/Reperfusion damage in Diabetic Stroke. J Am Heart Assoc. [Journal Article; Research Support, N.I.H., Extramural; Review]. 2017 2017-06-01;6(6).
Li W, Maloney RE, Aw TY. High glucose, glucose fluctuation and carbonyl stress enhance brain microvascular endothelial barrier dysfunction: implications for diabetic cerebral microvasculature. Redox Biol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-08-01;5:80–90.
Mäe MA, Li T, Bertuzzi G, Raschperger E, Vanlandewijck M, He L et al. Prolonged systemic hyperglycemia does not cause pericyte loss and permeability at the mouse blood-brain barrier. Sci Rep-Uk. [Journal Article; Research Support, Non-U.S. Gov’t]. 2018 2018-11-29;8(1):17462.
de Paula GC, Brunetta HS, Engel DF, Gaspar JM, Velloso LA, Engblom D et al. Hippocampal function is impaired by a short-term High-Fat Diet in mice: increased blood-brain barrier permeability and Neuroinflammation as triggering events. Front Neurosci-Switz. [Journal Article]. 2021 2021-01-20;15:734158.
Methia N, André P, Hafezi-Moghadam A, Economopoulos M, Thomas KL, Wagner DD. ApoE deficiency compromises the blood brain barrier especially after injury. Mol Med. [Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 2001 2001-12-01;7(12):810–5.
Ayata C, Shin HK, Dileköz E, Atochin DN, Kashiwagi S, Eikermann-Haerter K et al. Hyperlipidemia disrupts cerebrovascular reflexes and worsens ischemic perfusion defect. J Cerebr Blood F Met. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2013 2013-06-01;33(6):954–62.
Cao XL, Du J, Zhang Y, Yan JT, Hu XM. Hyperlipidemia exacerbates cerebral injury through oxidative stress, inflammation and neuronal apoptosis in MCAO/reperfusion rats. Exp Brain Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-10-01;233(10):2753–65.
Deng J, Zhang J, Feng C, Xiong L, Zuo Z. Critical role of matrix metalloprotease-9 in chronic high fat diet-induced cerebral vascular remodelling and increase of ischaemic brain injury in mice. Cardiovasc Res. [Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.]. 2014 2014-09-01;103(4):473–84.
Fang W, Sha L, Kodithuwakku ND, Wei J, Zhang R, Han D et al. Attenuated blood-brain barrier dysfunction by XQ-1H following ischemic stroke in hyperlipidemic rats. Mol Neurobiol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-08-01;52(1):162–75.
Shou Y, Huang Y, Zhu X, Liu C, Hu Y, Wang H. A review of the possible associations between ambient PM2.5 exposures and the development of Alzheimer’s disease. Ecotox Environ Safe. [Journal Article]. 2019 2019-06-15;174:344 – 52.
Shou Y, Zhu X, Zhu D, Yin H, Shi Y, Chen M et al. Ambient PM(2.5) chronic exposure leads to cognitive decline in mice: from pulmonary to neuronal inflammation. Toxicol Lett. [Journal Article]. 2020 2020-10-01;331:208 – 17.
Wang Y, Xiong L, Tang M. Toxicity of inhaled particulate matter on the central nervous system: neuroinflammation, neuropsychological effects and neurodegenerative disease. J Appl Toxicol. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2017 2017-06-01;37(6):644–67.
Lucero J, Suwannasual U, Herbert LM, McDonald JD, Lund AK. The role of the lectin-like oxLDL receptor (LOX-1) in traffic-generated air pollution exposure-mediated alteration of the brain microvasculature in Apolipoprotein (Apo) E knockout mice. Inhal Toxicol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2017 2017-05-01;29(6):266 – 81.
Branca J, Maresca M, Morucci G, Mello T, Becatti M, Pazzagli L et al. Effects of Cadmium on ZO-1 tight Junction Integrity of the blood brain barrier. Int J Mol Sci [Journal Article]. 2019 2019-11-29;20(23).
Song H, Zheng G, Shen XF, Liu XQ, Luo WJ, Chen JY. Reduction of brain barrier tight junctional proteins by lead exposure: role of activation of nonreceptor tyrosine kinase src via chaperon GRP78. Toxicol Sci. [Journal Article; Research Support, Non-U.S. Gov’t]. 2014 2014-04-01;138(2):393–402.
Paknejad B, Shirkhanloo H, Aliomrani M. Is there any relevance between serum Heavy Metal Concentration and BBB Leakage in multiple sclerosis patients? Biol Trace Elem Res. [Journal Article]. 2019 2019-08-01;190(2):289–94.
Abdullahi W, Tripathi D, Ronaldson PT. Blood-brain barrier dysfunction in ischemic stroke: targeting tight junctions and transporters for vascular protection. Am J Physiol-Cell PH. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2018 2018-09-01;315(3):C343-56.
Coureuil M, Mikaty G, Miller F, Lécuyer H, Bernard C, Bourdoulous S et al. Meningococcal type IV pili recruit the polarity complex to cross the brain endothelium. Science. [Journal Article; Research Support, Non-U.S. Gov’t]. 2009 2009-07-03;325(5936):83–7.
Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? Febs Lett. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2011 2011-12-01;585(23):3770–80.
Nitta T, Hata M, Gotoh S, Seo Y, Sasaki H, Hashimoto N et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2003 2003-05-12;161(3):653–60.
Wang X, Xue GX, Liu WC, Shu H, Wang M, Sun Y et al. Melatonin alleviates lipopolysaccharide-compromised integrity of blood-brain barrier through activating AMP-activated protein kinase in old mice. Aging Cell. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-04-01;16(2):414–21.
Cottarelli A, Shahriar S, Arac A, Glendinning M, Tuohy MC, Prochilo G et al. Rab7a activation promotes degradation of select tight junction proteins at the blood-brain barrier after ischemic stroke. bioRxiv. [Preprint]. 2023 2023-08-30.
Hawkins BT, Davis TP, Gov’t US. P.H.S.; Review]. 2005 2005-06-01;57(2):173–85.
Manukjan N, Majcher D, Leenders P, Caiment F, van Herwijnen M, Smeets HJ et al. Hypoxic oligodendrocyte precursor cell-derived VEGFA is associated with blood-brain barrier impairment. Acta Neuropathol Com. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-08-07;11(1):128.
Jais A, Solas M, Backes H, Chaurasia B, Kleinridders A, Theurich S et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. CELL. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2016 2016-05-05;165(4):882–95.
Gao M, Lu W, Shu Y, Yang Z, Sun S, Xu J et al. Poldip2 mediates blood-brain barrier disruption and cerebral edema by inducing AQP4 polarity loss in mouse bacterial meningitis model. Cns Neurosci Ther. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-12-01;26(12):1288–302.
Han D, Fang W, Zhang R, Wei J, Kodithuwakku ND, Sha L et al. Clematichinenoside protects blood brain barrier against ischemic stroke superimposed on systemic inflammatory challenges through up-regulating A20. Brain Behav Immun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2016 2016-01-01;51:56–69.
Liu X, Sui B, Sun J. Blood-brain barrier dysfunction induced by silica NPs in vitro and in vivo: involvement of oxidative stress and Rho-kinase/JNK signaling pathways. Biomaterials. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-03-01;121:64–82.
Bernhart E, Kogelnik N, Prasch J, Gottschalk B, Goeritzer M, Depaoli MR et al. 2-Chlorohexadecanoic acid induces ER stress and mitochondrial dysfunction in brain microvascular endothelial cells. Redox Biol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2018 2018-05-01;15:441 – 51.
Wan W, Cao L, Liu L, Zhang C, Kalionis B, Tai X et al. Aβ(1–42) oligomer-induced leakage in an in vitro blood-brain barrier model is associated with up-regulation of RAGE and metalloproteinases, and down-regulation of tight junction scaffold proteins. J Neurochem. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-07-01;134(2):382–93.
Sheikh MH, Errede M, D’Amati A, Khan NQ, Fanti S, Loiola RA et al. Impact of metabolic disorders on the structural, functional, and immunological integrity of the blood-brain barrier: therapeutic avenues. Faseb J. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-01-01;36(1):e22107.
McCaffrey G, Willis CL, Staatz WD, Nametz N, Quigley CA, Hom S et al. Occludin oligomeric assemblies at tight junctions of the blood-brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem. [Journal Article; Research Support, N.I.H., Extramural]. 2009 2009-07-01;110(1):58–71.
Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Csh Perspect Biol. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2009 2009-12-01;1(6):a2899.
Wu Y, Du S, Bimler LH, Mauk KE, Lortal L, Kichik N et al. Toll-like receptor 4 and CD11b expressed on microglia coordinate eradication of Candida albicans cerebral mycosis. Cell Rep. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2023 2023-10-31;42(10):113240.
Labus J, Wöltje K, Stolte KN, Häckel S, Kim KS, Hildmann A et al. IL-1β promotes transendothelial migration of PBMCs by upregulation of the FN/α(5)β(1) signalling pathway in immortalised human brain microvascular endothelial cells. Exp Cell Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2018 2018-12-15;373(1–2):99–111.
Park E, Barclay WE, Barrera A, Liao TC, Salzler HR, Reddy TE et al. Integrin α3 promotes T(H)17 cell polarization and extravasation during autoimmune neuroinflammation. Sci Immunol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-10-20;8(88):g7597.
Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. Faseb J. [Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-04-01;24(4):1023–34.
Zhou H, Gao F, Yang X, Lin T, Li Z, Wang Q et al. Endothelial BACE1 impairs cerebral small vessels via tight junctions and eNOS. Circ Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-04-29;130(9):1321–41.
Michinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int J Mol Sci [Journal Article; Review]. 2019 2019-01-29;20(3).
Liu L, Wang N, Kalionis B, Xia S, He Q. HMGB1 plays an important role in pyroptosis induced blood brain barrier breakdown in diabetes-associated cognitive decline. J Neuroimmunol. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2022 2022-01-15;362:577763.
Wei C, Jiang W, Wang R, Zhong H, He H, Gao X et al. Brain endothelial GSDMD activation mediates inflammatory BBB breakdown. Nat [Journal Article]. 2024 2024-05-01;629(8013):893–900.
Yamamoto M, Guo DH, Hernandez CM, Stranahan AM. Endothelial Adora2a activation promotes blood-brain barrier breakdown and cognitive impairment in mice with Diet-Induced insulin resistance. J Neurosci. [Journal Article; Research Support, N.I.H., Extramural]. 2019 2019-05-22;39(21):4179–92.
Wen H, Tan J, Tian M, Wang Y, Gao Y, Gong Y. TGF-β1 ameliorates BBB injury and improves long-term outcomes in mice after ICH. Biochem Bioph Res Co. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-04-30;654:136 – 44.
Chen Y, Joo J, Chu JM, Chang RC, Wong GT. Downregulation of the glucose transporter GLUT 1 in the cerebral microvasculature contributes to postoperative neurocognitive disorders in aged mice. J Neuroinflamm [Journal Article]. 2023 2023-10-19;20(1):237.
Winkler EA, Nishida Y, Sagare AP, Rege SV, Bell RD, Perlmutter D et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci. [Journal Article; Research Support, N.I.H., Extramural]. 2015 2015-04-01;18(4):521–30.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. [Comparative study; Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 2004 2004-08-05;43(3):333–44.
Donahue JE, Flaherty SL, Johanson CE, Duncan JR, Silverberg GD, Miller MC et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2006 2006-10-01;112(4):405–15.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. [Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 2000 2000-12-01;106(12):1489–99.
Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K et al. Central role for PICALM in amyloid-β blood-brain barrier transcytosis and clearance. Nat Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-07-01;18(7):978–87.
Nikolakopoulou AM, Wang Y, Ma Q, Sagare AP, Montagne A, Huuskonen MT et al. Endothelial LRP1 protects against neurodegeneration by blocking cyclophilin A. J Exp Med. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-04-05;218(4).
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. [Journal Article; Research Support, N.I.H., Extramural]. 2007 2007-09-01;13(9):1029–31.
Bhowmick S, D’Mello V, Caruso D, Wallerstein A, Abdul-Muneer PM. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp Neurol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2019 2019-07-01;317:260 – 70.
Lee HW, Xu Y, Zhu X, Jang C, Choi W, Bae H et al. Endothelium-derived lactate is required for pericyte function and blood-brain barrier maintenance. Embo J [Journal Article; Research Support, N.I.H., Extramural]. 2022 2022-05-02;41(9):e109890.
Veys K, Fan Z, Ghobrial M, Bouché A, García-Caballero M, Vriens K et al. Role of the GLUT1 glucose transporter in postnatal CNS angiogenesis and blood-brain Barrier Integrity. Circ Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-07-31;127(4):466–82.
Zong P, Feng J, Li CX, Jellison ER, Yue Z, Miller B et al. Activation of endothelial TRPM2 exacerbates blood-brain barrier degradation in ischemic stroke. Cardiovasc Res. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2024 2024-03-13;120(2):188–202.
Bohannon DG, Okhravi HR, Kim J, Kuroda MJ, Didier ES, Kim WK. A subtype of cerebrovascular pericytes is associated with blood-brain barrier disruption that develops during normal aging and simian immunodeficiency virus infection. Neurobiol Aging. [Journal Article; Research Support, N.I.H., Extramural]. 2020 2020-12-01;96:128 – 36.
Cai W, Liu H, Zhao J, Chen LY, Chen J, Lu Z et al. Pericytes in Brain Injury and Repair after ischemic stroke. Transl Stroke Res. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2017 2017-04-01;8(2):107–21.
Duz B, Oztas E, Erginay T, Erdogan E, Gonul E. The effect of moderate hypothermia in acute ischemic stroke on pericyte migration: an ultrastructural study. Cryobiology [Journal Article]. 2007 2007-12-01;55(3):279–84.
Seifert HA, Pennypacker KR. Molecular and cellular immune responses to ischemic brain injury. Transl Stroke Res. [Journal Article; Research Support, N.I.H., Extramural; Review]. 2014 2014-10-01;5(5):543 – 53.
Sakuma R, Kawahara M, Nakano-Doi A, Takahashi A, Tanaka Y, Narita A et al. Brain pericytes serve as microglia-generating multipotent vascular stem cells following ischemic stroke. J Neuroinflamm. [Journal Article; Research Support, Non-U.S. Gov’t]. 2016 2016-03-07;13(1):57.
Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. [Journal Article; Review]. 2001 2001-08-01;64(6):575–611.
Ding R, Hase Y, Burke M, Foster V, Stevenson W, Polvikoski T et al. Loss with ageing but preservation of frontal cortical capillary pericytes in post-stroke dementia, vascular dementia and Alzheimer’s disease. Acta Neuropathol Com. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-08-02;9(1):130.
Ding R, Hase Y, Ameen-Ali KE, Ndung’U M, Stevenson W, Barsby J et al. Loss of capillary pericytes and the blood-brain barrier in white matter in poststroke and vascular dementias and Alzheimer’s disease. Brain Pathol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-11-01;30(6):1087–101.
Liu G, Wang J, Wei Z, Fang CL, Shen K, Qian C et al. Elevated PDGF-BB from bone impairs hippocampal vasculature by inducing PDGFRβ shedding from Pericytes. Adv Sci. [Journal Article; Research Support, N.I.H., Extramural]. 2023 2023-07-01;10(20):e2206938.
Smyth L, Highet B, Jansson D, Wu J, Rustenhoven J, Aalderink M et al. Characterisation of PDGF-BB:PDGFRβ signalling pathways in human brain pericytes: evidence of disruption in Alzheimer’s disease. Commun Biol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-03-17;5(1):235.
Eilert-Olsen M, Haj-Yasein NN, Vindedal GF, Enger R, Gundersen GA, Hoddevik EH et al. Deletion of aquaporin-4 changes the perivascular glial protein scaffold without disrupting the brain endothelial barrier. Glia. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2012 2012-03-01;60(3):432–40.
Abbott NJ, Rönnbäck L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2006 2006-01-01;7(1):41–53.
Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2015 2015-05-01;16(5):249 – 63.
Siqueira M, Stipursky J. Blood brain barrier as an interface for alcohol induced neurotoxicity during development. Neurotoxicology [Journal Article; Review]. 2022 2022-05-01;90:145 – 57.
Lan G, Wang P, Chan RB, Liu Z, Yu Z, Liu X et al. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-02-01;70(2):337–53.
Chapouly C, Tadesse AA, Horng S, Castro K, Zhang J, Asp L et al. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-06-01;138(Pt 6):1548–67.
Zhang M, Zhang Z, Li H, Xia Y, Xing M, Xiao C et al. Blockage of VEGF function by bevacizumab alleviates early-stage cerebrovascular dysfunction and improves cognitive function in a mouse model of Alzheimer’s disease. Transl Neurodegener [Journal Article]. 2024 2024-01-03;13(1):1.
Da FA, Matias D, Garcia C, Amaral R, Geraldo LH, Freitas C et al. The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci. [Journal Article; review]. 2014 2014-01-20;8:362.
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2013 2013-12-19;155(7):1596–609.
Haruwaka K, Ikegami A, Tachibana Y, Ohno N, Konishi H, Hashimoto A et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun. [Journal Article; Research Support, Non-U.S. Gov’t]. 2019 2019-12-20;10(1):5816.
Pan W, Stone KP, Hsuchou H, Manda VK, Zhang Y, Kastin AJ. Cytokine signaling modulates blood-brain barrier function. Curr Pharm Design. [Journal Article; Research Support, N.I.H., Extramural; Review]. 2011 2011-11-01;17(33):3729–40.
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, Microglial and macrophage polarization—new prospects for brain repair. Nat Rev Neurol. [Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.;, Review] et al. 2015 2015-01-01;11(1):56–64.
Zheng ZV, Lyu H, Lam S, Lam PK, Poon WS, Wong G. The dynamics of Microglial polarization reveal the Resident neuroinflammatory responses after subarachnoid hemorrhage. Transl Stroke Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-06-01;11(3):433–49.
Gulej R, Csik B, Faakye J, Tarantini S, Shanmugarama S, Chandragiri SS et al. Endothelial deficiency of insulin-like growth factor-1 receptor leads to blood-brain barrier disruption and accelerated endothelial senescence in mice, mimicking aspects of the brain aging phenotype. Microcirculation [Journal Article]. 2024 2024-02-01;31(2):e12840.
Chi OZ, Hunter C, Liu X, Chi Y, Weiss HR. Effects of GABA(A) receptor blockade on regional cerebral blood flow and blood-brain barrier disruption in focal cerebral ischemia. J Neurol Sci [Journal Article]. 2011;2011–02–15(1–2):66–70.
Vazana U, Veksler R, Pell GS, Prager O, Fassler M, Chassidim Y et al. Glutamate-mediated blood-brain barrier opening: implications for Neuroprotection and Drug Delivery. J Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2016 2016-07-20;36(29):7727–39.
Hernandes MS, Lassègue B, Hilenski LL, Adams J, Gao N, Kuan CY et al. Polymerase delta-interacting protein 2 deficiency protects against blood-brain barrier permeability in the ischemic brain. J Neuroinflamm [Journal Article]. 2018 2018-02-17;15(1):45.
Kikuchi DS, Campos A, Qu H, Forrester SJ, Pagano RL, Lassègue B et al. Poldip2 mediates blood-brain barrier disruption in a model of sepsis-associated encephalopathy. J Neuroinflamm [Journal Article]. 2019 2019-11-28;16(1):241.
Okada T, Suzuki H, Travis ZD, Zhang JH. The Stroke-Induced blood-brain barrier disruption: current progress of inspection technique, mechanism, and therapeutic target. Curr Neuropharmacol. [Journal Article; Review]. 2020 2020-01-20;18(12):1187–212.
Betz AL, Coester HC. Effect of steroids on edema and sodium uptake of the brain during focal ischemia in rats. Stroke. [Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 1990 1990-08-01;21(8):1199–204.
Blecharz KG, Haghikia A, Stasiolek M, Kruse N, Drenckhahn D, Gold R et al. Glucocorticoid effects on endothelial barrier function in the murine brain endothelial cell line cEND incubated with sera from patients with multiple sclerosis. Mult Scler J [Journal Article]. 2010 2010-03-01;16(3):293–302.
Cristante E, McArthur S, Mauro C, Maggioli E, Romero IA, Wylezinska-Arridge M et al. Identification of an essential endogenous regulator of blood-brain barrier integrity, and its pathological and therapeutic implications. P Natl Acad Sci Usa. [Journal Article; Research Support, Non-U.S. Gov’t]. 2013 2013-01-15;110(3):832–41.
Go KG, Zuiderveen F, De Ley L, Ter Haar JG, Parente L, Solito E et al. Effect of steroids on brain lipocortin immunoreactivity. Acta Neurochir Suppl (Wien). [Comparative Study; Journal Article]. 1994 1994-01-19;60:101–3.
Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. [Journal Article; Research Support, American Recovery and Reinvestment Act; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2012 2012-07-01;122(7):2454–68.
Norris JW, Hachinski VC. High dose steroid treatment in cerebral infarction. Br Med J (Clin Res Ed). [Clinical Trial; Journal Article; Randomized Controlled Trial; Research Support, Non-U.S. Gov’t]. 1986 1986-01-04;292(6512):21–3.
Glumac S, Kardum G, Sodic L, Supe-Domic D, Karanovic N. Effects of dexamethasone on early cognitive decline after cardiac surgery: a randomised controlled trial. Eur J Anaesth. [Journal Article; Randomized Controlled Trial; Research Support, N.I.H., Extramural]. 2017 2017-11-01;34(11):776–84.
Lomivorotov V, Kornilov I, Boboshko V, Shmyrev V, Bondarenko I, Soynov I et al. Effect of intraoperative dexamethasone on Major complications and Mortality among infants undergoing cardiac surgery: the DECISION Randomized Clinical Trial. Jama-J Am Med Assoc. [Comparative study; Journal Article; Multicenter Study; Randomized Controlled Trial]. 2020 2020-06-23;323(24):2485–92.
Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. [Journal Article; Research Support, N.I.H., Extramural]. 2006 2006-04-01;37(4):1087–93.
Festoff BW, Sajja RK, van Dreden P, Cucullo L. HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer’s disease. J Neuroinflamm. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2016 2016-08-24;13(1):194.
Nishibori M, Wang D, Ousaka D, Wake H. High mobility Group Box-1 and blood-brain barrier disruption. Cells-Basel. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2020 2020-12-10;9(12).
Griffin JM, Kho D, Graham ES, Nicholson LF, O’Carroll SJ. Statins inhibit fibrillary β-Amyloid Induced inflammation in a model of the human blood brain barrier. Plos One. [Journal Article]. 2016 2016-01-20;11(6):e157483.
Culkin MC, Bele P, Georges AP, Lopez AJ, Niziolek G, Jacovides CL et al. Early posttraumatic brain injury tranexamic acid prevents blood-brain barrier hyperpermeability and improves surrogates of neuroclinical recovery. J Trauma Acute Care. [Journal Article]. 2023 2023-07-01;95(1):47–54.
Wallen TE, Singer KE, Baucom MR, England LG, Schuster RM, Pritts TA et al. Effects of antifibrinolytics on systemic and cerebral inflammation after traumatic brain injury. J Trauma Acute Care. [Journal Article; Research Support, N.I.H., Extramural]. 2022 2022-07-01;93(1):30–7.
Marchi N, Fan Q, Ghosh C, Fazio V, Bertolini F, Betto G et al. Antagonism of peripheral inflammation reduces the severity of status epilepticus. Neurobiol Dis. [Journal Article; Research Support, N.I.H., Extramural]. 2009 2009-02-01;33(2):171–81.
Kim A, García-García E, Straccia M, Comella-Bolla A, Miguez A, Masana M et al. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front Cell Neurosci. [Journal Article]. 2020 2020-01-20;14:163.
Kim H, Noh M, Zhang H, Kim Y, Park S, Park J et al. Long-term administration of CU06-1004 ameliorates cerebrovascular aging and BBB injury in aging mouse model. Fluids Barriers Cns. [Journal Article]. 2023 2023-02-01;20(1):9.
Zhang H, Park JH, Maharjan S, Park JA, Choi KS, Park H et al. Sac-1004, a vascular leakage blocker, reduces cerebral ischemia-reperfusion injury by suppressing blood-brain barrier disruption and inflammation. J Neuroinflamm [Journal Article]. 2017 2017-06-23;14(1):122.
Haileselassie B, Joshi AU, Minhas PS, Mukherjee R, Andreasson KI, Mochly-Rosen D. Mitochondrial dysfunction mediated through dynamin-related protein 1 (Drp1) propagates impairment in blood brain barrier in septic encephalopathy. J Neuroinflamm [Journal Article]. 2020 2020-01-27;17(1):36.
Guo Z, Ruan Z, Zhang D, Liu X, Hou L, Wang Q. Rotenone impairs learning and memory in mice through microglia-mediated blood brain barrier disruption and neuronal apoptosis. Chemosphere [Journal Article]. 2022 2022-03-01;291(Pt 2):132982.
Deng Y, Wang R, Li S, Zhu X, Wang T, Wu J et al. Methylene blue reduces incidence of early postoperative cognitive disorders in elderly patients undergoing major non-cardiac surgery: an open-label randomized controlled clinical trial. J Clin Anesth. [Journal Article; Randomized Controlled Trial]. 2021 2021-02-01;68:110108.
Genrikhs EE, Stelmashook EV, Voronkov DN, Novikova SV, Alexandrova OP, Fedorov AV et al. The single intravenous administration of methylene blue after traumatic brain injury diminishes neurological deficit, blood-brain barrier disruption and decrease in the expression of S100 protein in rats. Brain Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-08-01;1740:146854.
Shen J, Xin W, Li Q, Gao Y, Yuan L, Zhang J. Methylene Blue Reduces Neuronal Apoptosis and improves blood-brain barrier Integrity after Traumatic Brain Injury. Front Neurol. [Journal Article]. 2019 2019-01-20;10:1133.
Zhang C, Zheng J, Chen W, Yang W, Tan X, Fan X et al. Mitochondrial-targeting fluorescent small molecule IR-780 alleviates radiation-induced brain injury. Brain Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-04-15;1805:148285.
Stamatovic SM, Johnson AM, Sladojevic N, Keep RF, Andjelkovic AV. Endocytosis of tight junction proteins and the regulation of degradation and recycling. Ann Ny Acad Sci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t; Review]. 2017 2017-06-01;1397(1):54–65.
Sunny A, James RR, Menon SR, Rayaroth S, Daniel A, Thompson NA et al. Matrix Metalloproteinase-9 inhibitors as therapeutic drugs for traumatic brain injury. Neurochem Int [Journal Article; Review]. 2024 2024-01-01;172:105642.
Chen Z, Kelly JR, Morales JE, Sun RC, De A, Burkin DJ et al. The alpha7 integrin subunit in astrocytes promotes endothelial blood-brain barrier integrity. Development. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-03-15;150(6).
Burek M, Arias-Loza PA, Roewer N, Förster CY. Claudin-5 as a novel estrogen target in vascular endothelium. Arterioscl Throm Vas. [Journal Article; Research Support, Non-U.S. Gov’t]. 2010 2010-02-01;30(2):298–304.
Castillo-González J, Ruiz JL, Serrano-Martínez I, Forte-Lago I, Ubago-Rodriguez A, Caro M et al. Cortistatin deficiency reveals a dysfunctional brain endothelium with impaired gene pathways, exacerbated immune activation, and disrupted barrier integrity. J Neuroinflamm [Journal Article]. 2023 2023-10-04;20(1):226.
Martin-Ramirez J, Hofman M, van den Biggelaar M, Hebbel RP, Voorberg J. Establishment of outgrowth endothelial cells from peripheral blood. Nat Protoc. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2012 2012-09-01;7(9):1709–15.
Alwjwaj M, Kadir R, Bayraktutan U. Outgrowth endothelial progenitor cells restore cerebral barrier function following ischaemic damage: the impact of NOX2 inhibition. Eur J Neurosci. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-03-01;55(6):1658–70.
Abdulkadir RR, Alwjwaj M, Othman OA, Rakkar K, Bayraktutan U. Outgrowth endothelial cells form a functional cerebral barrier and restore its integrity after damage. Neural Regen Res. [Journal Article]. 2020 2020-06-01;15(6):1071–8.
Kadir R, Alwjwaj M, Bayraktutan U. Treatment with outgrowth endothelial cells protects cerebral barrier against ischemic injury. Cytotherapy. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-05-01;24(5):489–99.
Auletta JJ, Bartholomew AM, Maziarz RT, Deans RJ, Miller RH, Lazarus HM et al. The potential of mesenchymal stromal cells as a novel cellular therapy for multiple sclerosis. Immunotherapy-UK. [Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.; Review]. 2012 2012-05-01;4(5):529–47.
Zacharek A, Chen J, Cui X, Li A, Li Y, Roberts C et al. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J Cerebr Blood F Met. [Journal Article; Research Support, N.I.H., Extramural]. 2007 2007-10-01;27(10):1684–91.
Barreda-Manso MA, Yanguas-Casás N, Nieto-Sampedro M, Romero-Ramírez L. Neuroprotection and blood-brain barrier restoration by Salubrinal after a cortical stab Injury. J Cell Physiol [Journal Article]. 2017 2017-06-01;232(6):1501–10.
Williams AM, Bhatti UF, Brown JF, Biesterveld BE, Kathawate RG, Graham NJ et al. Early single-dose treatment with exosomes provides neuroprotection and improves blood-brain barrier integrity in swine model of traumatic brain injury and hemorrhagic shock. J Trauma Acute Care [Journal Article]. 2020 2020-02-01;88(2):207–18.
Williams AM, Dennahy IS, Bhatti UF, Halaweish I, Xiong Y, Chang P et al. Mesenchymal stem cell-derived exosomes provide neuroprotection and improve long-term neurologic outcomes in a Swine Model of Traumatic Brain Injury and hemorrhagic shock. J Neurotraum. [Journal Article; Research Support, U.S. Gov’t, Non-P.H.S.]. 2019 2019-01-01;36(1):54–60.
Huang LY, Song JX, Cai H, Wang PP, Yin QL, Zhang YD et al. Healthy serum-derived exosomes improve neurological outcomes and protect blood-brain barrier by inhibiting endothelial cell apoptosis and reversing autophagy-mediated tight Junction protein reduction in Rat Stroke Model. Front Cell Neurosci. [Journal Article]. 2022 2022-01-20;16:841544.
Venkat P, Zacharek A, Landschoot-Ward J, Wang F, Culmone L, Chen Z et al. Exosomes derived from bone marrow mesenchymal stem cells harvested from type two diabetes rats promotes neurorestorative effects after stroke in type two diabetes rats. Exp Neurol. [Journal Article]. 2020 2020-12-01;334:113456.
Li Q, Niu X, Yi Y, Chen Y, Yuan J, Zhang J et al. Inducible pluripotent stem cell-derived small extracellular vesicles rejuvenate senescent blood-brain barrier to protect against ischemic stroke in aged mice. Acs Nano. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-01-10;17(1):775–89.
Toyama K, Spin JM, Tsao PS. Role of microRNAs on blood brain barrier dysfunction in vascular cognitive impairment. Curr Drug Deliv. [Journal Article; Review]. 2017 2017-09-06;14(6):744–57.
Campbell M, Hanrahan F, Gobbo OL, Kelly ME, Kiang AS, Humphries MM et al. Targeted suppression of claudin-5 decreases cerebral oedema and improves cognitive outcome following traumatic brain injury. Nat Commun. [Journal Article; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2012 2012-05-22;3:849.
Körbelin J, Dogbevia G, Michelfelder S, Ridder DA, Hunger A, Wenzel J et al. A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. Embo Mol Med. [Journal Article; Research Support, Non-U.S. Gov’t]. 2016 2016-06-01;8(6):609–25.
Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC et al. Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2019 2019-01-01;15(1):158–67.
Bony BA, Tarudji AW, Miller HA, Gowrikumar S, Roy S, Curtis ET, Research Support NIH et al. Extramural; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2021 2021-11-23;15(11):18520–31.
Sladojevic N, Stamatovic SM, Johnson AM, Choi J, Hu A, Dithmer S et al. Claudin-1-Dependent destabilization of the blood-brain barrier in chronic stroke. J Neurosci. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2019 2019-01-23;39(4):743–57.
Lengfeld JE, Lutz SE, Smith JR, Diaconu C, Scott C, Kofman SB et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2017 2017-02-14;114(7):E1168–77.
Ji YB, Gao Q, Tan XX, Huang XW, Ma YZ, Fang C et al. Lithium alleviates blood-brain barrier breakdown after cerebral ischemia and reperfusion by upregulating endothelial Wnt/β-catenin signaling in mice. Neuropharmacology. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-03-15;186:108474.
Trevino TN, Fogel AB, Otkiran G, Niladhuri SB, Sanborn MA, Class J et al. Engineered Wnt7a ligands rescue blood-brain barrier and cognitive deficits in a COVID-19 mouse model. Brain. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2024 2024-05-03;147(5):1636–43.
Liu X, Geng X, Shi Y, Liang J, Zhao L. Biomimetic oxygen-boosted hybrid membrane nanovesicles as the treatment strategy for ischemic stroke with the concept of the neurovascular unit. Biomater Adv [Journal Article]. 2023 2023-05-01;148:213379.
El-Mezayen NS, Abd EMR, El-Rewini SH. Vitamin B12 as a cholinergic system modulator and blood brain barrier integrity restorer in Alzheimer’s disease. Eur J Pharm Sci [Journal Article]. 2022 2022-07-01;174:106201.
Parker E, Aboghazleh R, Mumby G, Veksler R, Ofer J, Newton J et al. Concussion susceptibility is mediated by spreading depolarization-induced neurovascular dysfunction. Brain. [Journal Article; Research Support, Non-U.S. Gov’t; Research Support, U.S. Gov’t, Non-P.H.S.]. 2022 2022-06-30;145(6):2049–63.
Deane R, Singh I, Sagare AP, Bell RD, Ross NT, LaRue B et al. A multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2012 2012-04-01;122(4):1377–92.
Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. [Journal Article; Research Support, U.S. Gov’t, P.H.S.]. 2003 2003-07-01;9(7):907–13.
Montagne A, Nikolakopoulou AM, Huuskonen MT, Sagare AP, Lawson EJ, Lazic D et al. APOE4 accelerates advanced-stage vascular and neurodegenerative disorder in old Alzheimer’s mice via cyclophilin A independently of amyloid-β. Nat Aging. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2021 2021-06-01;1(6):506–20.
Xu L, Nirwane A, Xu T, Kang M, Devasani K, Yao Y. Fibroblasts repair blood-brain barrier damage and hemorrhagic brain injury via TIMP2. Cell Rep. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2022 2022-11-22;41(8):111709.
Yu Z, Lin L, Jiang Y, Chin I, Wang X, Li X et al. Recombinant FGF21 protects against blood-brain barrier leakage through Nrf2 upregulation in type 2 diabetes mice. Mol Neurobiol. [Journal Article]. 2019 2019-04-01;56(4):2314–27.
Tang J, Kang Y, Zhou Y, Shang N, Li X, Wang H et al. TIMP2 ameliorates blood-brain barrier disruption in traumatic brain injury by inhibiting src-dependent VE-cadherin internalization. J Clin Invest [Journal Article]. 2023 2023-11-28;134(3).
Ahmadighadykolaei H, Lambert JA, Raeeszadeh-Sarmazdeh M. TIMP-1 protects tight junctions of Brain endothelial cells from MMP-Mediated degradation. Pharm Res-Dordr. [Journal Article]. 2023 2023-09-01;40(9):2121–31.
Dashti SR, Kadner RJ, Folley BS, Sheehan JP, Han DY, Kryscio RJ et al. Single low-dose targeted bevacizumab infusion in adult patients with steroid-refractory radiation necrosis of the brain: a phase II open-label prospective clinical trial. J Neurosurg. [Clinical Trial, Phase II; Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-12-01;137(6):1676–86.
Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2019 2019-02-01;25(2):270–6.
Skillbäck T, Delsing L, Synnergren J, Mattsson N, Janelidze S, Nägga K et al. CSF/serum albumin ratio in dementias: a cross-sectional study on 1861 patients. Neurobiol Aging. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-11-01;59:1–9.
Liu P, Gao Q, Guan L, Hu Y, Jiang J, Gao T et al. Atorvastatin attenuates surgery-induced BBB disruption and cognitive impairment partly by suppressing NF-κB pathway and NLRP3 inflammasome activation in aged mice. Acta Bioch Bioph Sin. [Journal Article]. 2021 2021-04-15;53(5):528–37.
Krasemann S, Haferkamp U, Pfefferle S, Woo MS, Heinrich F, Schweizer M et al. The blood-brain barrier is dysregulated in COVID-19 and serves as a CNS entry route for SARS-CoV-2. Stem Cell Rep. [Journal Article; Research Support, Non-U.S. Gov’t]. 2022 2022-02-08;17(2):307–20.
Petersen MA, Ryu JK, Akassoglou K, Review]. 2018 2018-05-01;19(5):283–301.
Hou SJ, Huang YR, Zhu J, Jia YB, Niu XY, Yang JJ et al. Mouse serum albumin induces neuronal apoptosis and tauopathies. Acta Neuropathol Com. [Journal Article; Research Support, Non-U.S. Gov’t]. 2024 2024-04-23;12(1):66.
Shi T, Shen S, Shi Y, Wang Q, Zhang G, Lin J et al. Osteocyte-derived sclerostin impairs cognitive function during ageing and Alzheimer’s disease progression. Nat Metab [Journal Article]. 2024 2024-03-01;6(3):531–49.
Wu YC, Bogale TA, Koistinaho J, Pizzi M, Rolova T, Bellucci A. The contribution of β-amyloid, tau and α-synuclein to blood-brain barrier damage in neurodegenerative disorders. Acta Neuropathol. [Journal Article; Review]. 2024 2024-02-12;147(1):39.
Cater RJ, Mukherjee D, Gil-Iturbe E, Erramilli SK, Chen T, Koo K, Santander N, Reckers A, Kloss B, Gawda T, Choy BC, Zhang Z, Katewa A, Larpthaveesarp A, Huang EJ, Mooney SWJ, Clarke OB, Yee SW, Giacomini KM, Kossiakoff AA, Quick M, Arnold T, Mancia F. Structural and molecular basis of choline uptake into the brain by FLVCR2.Nature. 2024;629(8012):704–9. https://doi.org/10.1038/s41586-024-07326-y
Nguyen XTA, Le TNU, Nguyen TQ, Thi Thuy Ha H, Artati A, Leong NCP, Nguyen DT, Lim PY, Susanto AV, Huang Q, Fam L, Leong LN, Bonne I, Lee A, Granadillo JL, Gooch C, Yu D, Huang H, Soong TW, Chang MW, Wenk MR, Adamski J, Cazenave-Gassiot A, Nguyen LN. MFSD7c functions as a transporter of choline at the blood-brain barrier. Cell Res. 2024;34(3):245–57. https://doi.org/10.1038/s41422-023-00923-y.
Guillén-Yunta M, Valcárcel-Hernández V, García-Aldea Á, Soria G, García-Verdugo JM, Montero-Pedrazuela A, Guadaño-Ferraz A. Neurovascular unit disruption and blood-brain barrier leakage in MCT8 deficiency.Fluids barriers CNS. 2023;20(1):79. https://doi.org/10.1186/s12987-023-00481-w.]
Alam C, Kondo M, O’Connor DL, Bendayan R. Clinical Implications of Folate Transport in the Central Nervous System. Trends Pharmacol Sci. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2020 2020-05-01;41(5):349–61.
Kawauchi S, Mizoguchi T, Horibe S, Tanaka T, Sasaki N, Ikeda K et al. Gliovascular interface abnormality in mice with endothelial cell senescence. Glia. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-02-01;71(2):467–79.
Rajani RM, Quick S, Ruigrok SR, Graham D, Harris SE, Verhaaren B et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci Transl Med. [Journal Article; Research Support, Non-U.S. Gov’t]. 2018 2018-07-04;10(448).
Shi H, Koronyo Y, Rentsendorj A, Regis GC, Sheyn J, Fuchs DT et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2020 2020-05-01;139(5):813–36.
Shimizu F, Sano Y, Tominaga O, Maeda T, Abe MA, Kanda T. Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-βby pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol Aging. [Journal Article; Research Support, Non-U.S. Gov’t]. 2013 2013-07-01;34(7):1902–12.
Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol-Heart C. [Journal Article; Review]. 2017 2017-01-01;312(1):H1–20.
Yang Y, Torbey MT. Angiogenesis and blood-brain barrier permeability in vascular remodeling after stroke. Curr Neuropharmacol. [Journal Article]. 2020 2020-01-20;18(12):1250–65.
Olumolade OO, Wang S, Samiotaki G, Konofagou EE. Longitudinal Motor and behavioral Assessment of blood-brain barrier opening with Transcranial Focused Ultrasound. Ultrasound Med Biol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2016 2016-09-01;42(9):2270–82.
Downs ME, Buch A, Sierra C, Karakatsani ME, Teichert T, Chen S et al. Long-term safety of repeated blood-brain barrier opening via focused Ultrasound with Microbubbles in Non-human Primates performing a cognitive Task. Plos One. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2015 2015-01-20;10(5):e125911.
Meng Y, Goubran M, Rabin JS, McSweeney M, Ottoy J, Pople CB et al. Blood-brain barrier opening of the default mode network in Alzheimer’s disease with magnetic resonance-guided focused ultrasound. Brain. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2023 2023-03-01;146(3):865–72.
Epelbaum S, Burgos N, Canney M, Matthews D, Houot M, Santin MD et al. Pilot study of repeated blood-brain barrier disruption in patients with mild Alzheimer’s disease with an implantable ultrasound device. Alzheimers Res Ther. [Clinical Trial; Journal Article; Research Support, N.I.H., Extramural; Research Support, U.S. Gov’t, Non-P.H.S.]. 2022 2022-03-08;14(1):40.
Rezai AR, Ranjan M, Haut MW, Carpenter J, D’Haese PF, Mehta RI et al. Focused ultrasound-mediated blood-brain barrier opening in Alzheimer’s disease: long-term safety, imaging, and cognitive outcomes. J Neurosurg. [Journal Article; Research Support, N.I.H., Extramural]. 2023 2023-07-01;139(1):275–83.
Downs ME, Buch A, Karakatsani ME, Konofagou EE, Ferrera VP. Blood-brain barrier opening in behaving non-human Primates via focused ultrasound with systemically administered Microbubbles. SCI REP-UK. [Journal Article; Research Support, N.I.H., Extramural]. 2015 2015-10-26;5:15076.
Shin J, Kong C, Lee J, Choi BY, Sim J, Koh CS et al. Focused ultrasound-induced blood-brain barrier opening improves adult hippocampal neurogenesis and cognitive function in a cholinergic degeneration dementia rat model. Alzheimers Res Ther. [Journal Article; Research Support, Non-U.S. Gov’t]. 2019 2019-12-27;11(1):110.
Burgess A, Dubey S, Yeung S, Hough O, Eterman N, Aubert I et al. Alzheimer disease in a mouse model: MR imaging-guided focused ultrasound targeted to the hippocampus opens the blood-brain barrier and improves pathologic abnormalities and behavior. Radiology. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2014 2014-12-01;273(3):736–45.
Leinenga G, Götz J. Scanning ultrasound removes amyloid-βand restores memory in an Alzheimer’s disease mouse model. Sci Transl Med. [Journal Article; Research Support, Non-U.S. Gov’t]. 2015 2015-03-11;7(278):r233–78.
Yanagida K, Liu CH, Faraco G, Galvani S, Smith HK, Burg N et al. Size-selective opening of the blood-brain barrier by targeting endothelial sphingosine 1-phosphate receptor 1. P Natl Acad Sci Usa. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2017 2017-04-25;114(17):4531–6.
Chen S, Nazeri A, Baek H, Ye D, Yang Y, Yuan J et al. A review of bioeffects induced by focused ultrasound combined with microbubbles on the neurovascular unit. J Cerebr Blood F Met. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2022 2022-01-01;42(1):3–26.
Shen Y, Hua L, Yeh CK, Shen L, Ying M, Zhang Z et al. Ultrasound with microbubbles improves memory, ameliorates pathology and modulates hippocampal proteomic changes in a triple transgenic mouse model of Alzheimer’s disease. Theranostics. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-01-20;10(25):11794–819.
Blackmore DG, Turpin F, Palliyaguru T, Evans HT, Chicoteau A, Lee W et al. Low-intensity ultrasound restores long-term potentiation and memory in senescent mice through pleiotropic mechanisms including NMDAR signaling. Mol Psychiatr. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-11-01;26(11):6975–91.
Jeong H, Im JJ, Park JS, Na SH, Lee W, Yoo SS et al. A pilot clinical study of low-intensity transcranial focused ultrasound in Alzheimer’s disease. Ultrasonography [Journal Article]. 2021 2021-10-01;40(4):512–9.
Zachariou V, Pappas C, Bauer CE, Shao X, Liu P, Lu H et al. Regional differences in the link between water exchange rate across the blood-brain barrier and cognitive performance in normal aging. Geroscience [Journal Article]. 2024 2024-02-01;46(1):265–82.
Bakhtiari A, Vestergaard MB, Benedek K, Fagerlund B, Mortensen EL, Osler M et al. Changes in hippocampal volume during a preceding 10-year period do not correlate with cognitive performance and hippocampal blood–brain barrier permeability in cognitively normal late-middle-aged men. Geroscience. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-04-01;45(2):1161–75.
Li J, Zheng M, Shimoni O, Banks WA, Bush AI, Gamble JR et al. Development of Novel therapeutics targeting the blood-brain barrier: from barrier to Carrier. Adv Sci. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2021 2021-08-01;8(16):e2101090.
Profaci CP, Munji RN, Pulido RS, Daneman R. The blood-brain barrier in health and disease: important unanswered questions. J Exp Med. [Journal Article; Review]. 2020 2020-04-06;217(4):e20190062.
Nicaise C, Mitrecic D, Demetter P, De Decker R, Authelet M, Boom A et al. Impaired blood-brain and blood-spinal cord barriers in mutant SOD1-linked ALS rat. Brain Res. [Journal Article; Research Support, Non-U.S. Gov’t]. 2009 2009-12-08;1301:152 – 62.
Tibbling G, Link H, Ohman S. Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand J Clin Lab Inv. [Clinical trial; comparative study; Journal Article]. 1977 1977-09-01;37(5):385–90.
Marchi N, Cavaglia M, Fazio V, Bhudia S, Hallene K, Janigro D, Research Support US, Gov’t PHS, Review]. 2004 2004-04-01;342(1–2):1–12.
Ghaffarpour S, Ghazanfari T, Kabudanian AS, Pourfarzam S, Fallahi F, Shams J et al. Correlation between MMP-9 and MMP-9/ TIMPs complex with pulmonary function in Sulfur Mustard exposed civilians: Sardasht-Iran Cohort Study. Arch Iran Med. [Journal Article]. 2017 2017-02-01;20(2):74–82.
Blyth BJ, Farahvar A, He H, Nayak A, Yang C, Shaw G et al. Elevated serum ubiquitin carboxy-terminal hydrolase L1 is associated with abnormal blood-brain barrier function after traumatic brain injury. J Neurotraum [Journal Article]. 2011 2011-12-01;28(12):2453–62.
Padden M, Leech S, Craig B, Kirk J, Brankin B, McQuaid S. Differences in expression of junctional adhesion molecule-A and beta-catenin in multiple sclerosis brain tissue: increasing evidence for the role of tight junction pathology. Acta Neuropathol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2007 2007-02-01;113(2):177–86.
Zhang Y, Nozohouri S, Abbruscato TJ. In Vivo Evaluation of BBB Integrity in the post-stroke brain. Methods Mol Biol [Journal Article]. 2023 2023-01-20;2616:191–203.
Sun P, Hamblin MH, Yin KJ. Non-coding RNAs in the regulation of blood-brain barrier functions in central nervous system disorders. Fluids Barriers Cns. [Journal Article; Review]. 2022 2022-03-26;19(1):27.
Kala D, Šulc V, Olšerová A, Svoboda J, Prysiazhniuk Y, Pošusta A et al. Evaluation of blood-brain barrier integrity by the analysis of dynamic contrast-enhanced MRI - a comparison of quantitative and semi-quantitative methods. Physiol Res [Journal Article]. 2022 2022-12-31;71(S2):S259–75.
Montagne A, Barnes SR, Nation DA, Kisler K, Toga AW, Zlokovic BV. Imaging subtle leaks in the blood-brain barrier in the aging human brain: potential pitfalls, challenges, and possible solutions. Geroscience. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2022 2022-06-01;44(3):1339-51.
Li Y, Sadiq A, Wang Z. Arterial spin labelling-based blood-brain Barrier Assessment and its applications. Investig Magn Reson Imaging [Journal Article]. 2022 2022-12-01;26(4):229–36.
Zhang Y, Wang Y, Li Z, Wang Z, Cheng J, Bai X et al. Vascular-water-exchange MRI (VEXI) enables the detection of subtle AXR alterations in Alzheimer’s disease without MRI contrast agent, which may relate to BBB integrity. Neuroimage. [Journal Article; Research Support, Non-U.S. Gov’t]. 2023 2023-04-15;270:119951.
Gilad R, Lampl Y, Eilam A, Boaz M, Loyberboim M. SPECT-DTPA as a tool for evaluating the blood-brain barrier in post-stroke seizures. J Neurol [Journal Article]. 2012 2012-10-01;259(10):2041–4.
Srinivasan B, Kolli AR, Esch MB, Abaci HE, Shuler ML, Hickman JJ. TEER measurement techniques for in vitro barrier model systems. J Lab Autom. [Journal Article; Research Support, N.I.H., Extramural; Review]. 2015 2015-04-01;20(2):107–26.
Natarajan R, Northrop N, Yamamoto B. Fluorescein Isothiocyanate (FITC)-Dextran Extravasation as a measure of blood-brain barrier permeability. Curr Protoc Neurosci [Journal Article]. 2017 2017-04-10;79:9–58.
Kincses A, Vigh JP, Petrovszki D, Valkai S, Kocsis AE, Walter FR et al. The use of sensors in blood-brain barrier-on-a-Chip Devices: current practice and future directions. Biosensors-Basel. [Journal Article; Review]. 2023 2023-03-08;13(3).
Ahishali B, Kaya M. Evaluation of blood-brain Barrier Integrity using vascular permeability markers: Evans Blue, Sodium Fluorescein, Albumin-Alexa Fluor conjugates, and Horseradish Peroxidase. Methods Mol Biol. [Journal Article; Research Support, Non-U.S. Gov’t]. 2021 2021-01-20;2367:87–103.
Ek CJ, Dziegielewska KM, Stolp H, Saunders NR. Functional effectiveness of the blood-brain barrier to small water-soluble molecules in developing and adult opossum (Monodelphis domestica). J Comp Neurol. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2006 2006-05-01;496(1):13–26.
Miah MK, Chowdhury EA, Bickel U, Mehvar R. Evaluation of [(14)C] and [(13)C]sucrose as blood-brain barrier permeability markers. J PHARM SCI-US. [Journal Article; Research Support, Non-U.S. Gov’t]. 2017 2017-06-01;106(6):1659–69.
Noorani B, Chowdhury EA, Alqahtani F, Ahn Y, Patel D, Al-Ahmad A et al. LC-MS/MS-based in vitro and in vivo investigation of blood-brain barrier integrity by simultaneous quantitation of mannitol and sucrose. Fluids Barriers Cns. [Journal Article]. 2020 2020-10-14;17(1):61.
Wang Y, Zhang R, Chen Q, Guo H, Liang X, Li T et al. Visualization of blood-brain barrier disruption with dual-wavelength high-resolution photoacoustic microscopy. Biomed Opt Express. [Journal Article]. 2022 2022-03-01;13(3):1537–50.
Sweeney MD, Sagare AP, Pachicano M, Harrington MG, Joe E, Chui HC et al. A novel sensitive assay for detection of a biomarker of pericyte injury in cerebrospinal fluid. Alzheimers Dement. [Journal Article; Research Support, Non-U.S. Gov’t]. 2020 2020-06-01;16(6):821–30.
Huang SH, Wang L, Chi F, Wu CH, Cao H, Zhang A et al. Circulating brain microvascular endothelial cells (cBMECs) as potential biomarkers of the blood-brain barrier disorders caused by microbial and non-microbial factors. Plos One. [Journal Article; Research Support, N.I.H., Extramural; Research Support, Non-U.S. Gov’t]. 2013 2013-01-20;8(4):e62164.
Kassner A, Merali Z. Assessment of blood-brain barrier disruption in stroke. Stroke. [Journal Article; Research Support, Non-U.S. Gov’t; Review]. 2015 2015-11-01;46(11):3310–5.
Acknowledgements
The figures were constructed using BioRender (https://biorender.com/) and Figdraw (https://www.figdraw.com/). This work is supported by the Natural Science Foundation of China (No.81971002 and No.82171261, to Jun Zhang).
Funding
National Natural Science Foundation of China (No. 81971002 and No. 82171261)
Ethics declarations
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
All authors agree to publish this manuscript in this journal.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Che, J., Sun, Y., Deng, Y. et al. Blood-brain barrier disruption: a culprit of cognitive decline?. Fluids Barriers CNS 21, 63 (2024). https://doi.org/10.1186/s12987-024-00563-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-024-00563-3