
Abstract
HIV-associated neurological disorders (HAND) affect up to 50% of people living with HIV (PLWH), even in the era of combination antiretroviral therapy (cART). HIV-DNA can be detected in the cerebral spinal fluid (CSF) of approximately half of aviremic ART-suppressed PLWH and its presence is associated with poorer neurocognitive performance. HIV DNA + and HIV RNA + cells have also been observed in postmortem brain tissue of individuals with sustained cART suppression. In this review, we provide an overview of how HIV invades the brain and HIV infection of resident brain glial cells (astrocytes and microglia). We also discuss the role of resident glial cells in persistent neuroinflammation and HAND in PLWH and their potential contribution to the HIV reservoir. HIV eradication strategies that target persistently infected glia cells will likely be needed to achieve HIV cure.
Similar content being viewed by others

Background
HIV-associated neurocognitive disorders (HAND) is observed in approximately half of people living with HIV (PLWH) since the beginning of the HIV/AIDS epidemic [1,2,3]. HAND encompass a spectrum of cognitive impairments that cannot be attributed to co-infections or other causes. They are classified based on severity as asymptomatic neurocognitive impairment (ANI), mild neurocognitive disorder (MND), and HIV-associated dementia (HAD) [4]. In the pre-cART era, HIV associated dementia (HAD), the most severe form of HAND, was associated with advanced HIV infection and characterized by a progressive cognitive impairment, often accompanied by motor symptoms and behavioral changes, resulting in death within months [5,6,7]. The prevalence of HAD has dramatically declined in the cART era from ~ 15% to 2% [2, 8,9,10]. In contrast, the incidence of ANI and MND in PLWH has not declined despite widespread access to highly suppressive cART [2, 8, 10].
The persistence of HAND has been attributed to multiple factors including early CD4+ T cell depletion and neuronal damage prior to cART initiation, antiretroviral neurotoxicity, cardiovascular disease, and CNS inflammation [1, 3, 11]. Potential factors that contribute to sustained CNS inflammation in cART treated individuals include circulating microbial products and byproducts derived from the intestinal microbiome, conversion of proteasomes to immunoproteasomes in CNS cells, and the production of viral proteins such as gp120, Tat, and Vpr which promote CNS inflammation and neuronal damage, suggesting the persistence of HIV-infected cells in the CNS [3, 11,12,13,14,15,16]. In this review, we will address non-classical HIV infection of resident glia (astrocytes and microglia), their role as a reservoir for HIV and their contribution to persistent neuroinflammation, which together complicate cure strategies that must address the role of the brain as an HIV reservoir.
Maintext
HIV neuroinvasion
HIV invades the brain within approximately two weeks of infection, as demonstrated by both animal [17,18,19,20,21] and human [22, 23] studies. The consequence of this neuroinvasion is two-fold, induction of inflammatory responses that culminate in the manifestation of HIV-Associated Neurocognitive Disorders (HAND) and the establishment of the brain as a reservoir for HIV. Yet, two decades after identifying HIV as the etiologic agent of AIDS, there is still some controversy regarding which infected immune cells disseminate HIV into the brain. Earlier studies identified monocytes as the trojan horse of HIV, and especially those that are CD14 + CD16 + cells [24,25,26,27]. However, monocytes are not productively infected by HIV and only when they differentiate to macrophages can they support productive HIV replication [28,29,30]. Recent studies also demonstrated that while monocytes harbor HIV DNA they also express higher level of an HIV-repressor factor, β-catenin, but loose that expression once they differentiate into macrophages, supporting HIV replication in macrophages [31]. Further, the level of HIV replication in macrophages likely depends on their phenotype (M1/M-2-like) and tissue site. Hence, the paradigm that exists in HIV neuroinvasion is that monocytes harbor HIV DNA, migrate to the CNS, differentiate into macrophages allowing dissemination of HIV into the brain. Emerging evidence supports a role for CD4 + T cells in HIV neuroinvasion [32,33,34], especially since there is a greater appreciation that the brain is not an immune privileged site [35]. T cells home into the brain at low numbers to survey the brain and in response to neuroinflammation they home at greater numbers, although with slower kinetics. There is also a greater appreciation for a lymphatic system in the brain which was first described in the mid-1940s and re-evaluated using sophisticated studies led by the Kipnis group [36,37,38]. These observations then provide a rationale by which infected CD4 + T cells can disseminate HIV in the brain. Indeed, CD4 + T cells are detected in the brain of SIV-infected macaques [39, 40] and in human CSF [41]. Further, HIV-infected CD4+ T cells migrated into the CNS in humanized mice [17]. Despite these studies, the presence of infected CD4 + T cells in the brain parenchyma of deceased HIV + individuals are not welldocumented. On the other hand, CD8 +T cells are found. Modeling the role of lymphocytes in HIV neuroinvasion in humanized mice, a recent study found that infected CD4 + T cells can home to the brain but die fairly quickly, which can potentially explain the inability to find infected CD4 + T cells from human post mortem brains [17]. Of interest, is a recent observation that a unique population of CD8 + T cells which express CD4 on its surface, called CD4dimCD8bright T cells mediates HIV neuroinvasion [17, 42,43,44,45,46,47]. CD4dimCD8bright T cells constitute ~ 3–5% of total CD8 + T cells among healthy and HIV + chronically infected patients and up to 15% of CD8 + T cells from HIV infected long-term non-progressors [43]. Blood CD4dimCD8bright T cells are highly enriched in anti-HIV responses [43], constituting approximately 60% of the anti-HIV tetramer responses and are polyfunctional and cytolytic [43]. β-catenin, the central mediator of the Wnt/β-catenin pathway, mediates CD4 expression on mature CD8+ T cells [46]. Using NOD/SCID/IL-2rcγ-/- mice reconstituted with human PBMCs (NSG-huPBMC), CD4dimCD8bright T cells were reported to mediate HIV neuroinvasion yet due to a high level of the antiapoptotic protein, Bcl-XL, they resist HIV-mediated cytopathy [33, 48]. Further the Wnt-rich environment in the CNS induced CD8 single positive T cells to become CD4dimCD8bright T cells both in vitro and in vivo[17]. Expression of CD4dimCD8bright (not CD8 single positive) T cells was inversely associated with HIV in the brain and brain CD4dimCD8bright T cells of HIV-infected NSG-huPMBC mice exert anti-HIV cytolytic activity [17]. These findings highlight a significant role for CD4dimCD8bright T cells in HIV neuroinvasion (https://www.nimh.nih.gov/news/research-highlights/2022/t-cells-help-hiv-enter-and-persist-in-the-brain). A model of key cell types, to date, reported to mediate HIV neuroinvasion is illustrated in Fig. 1.

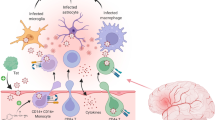
Immune cells-mediating HIV neuroinvasion. To date, four immune cell types are described to support HIV invasion into the CNS. Those include CD14 + CD16 + monocytes, perivascular macrophages, CD4 + T cells, and CD4dimCD8bright T cells. CD4dimCD8bright T cell may invade the brain as such, or the phenotype is generated in the brain as CD8 + T cells enter the brain and through the Wnt-rich environment in the CNS induce CD4 expression on their surface. These HIV-infected immune cells then released HIV in the brain to support infection of microglia and to a lesser extent, non-classical infection, of astrocytes.
Mechanisms of CD4-independent HIV infection of non-classical cells
The primary and most efficient means by which HIV enters cells is through binding of its envelope (gp120) to the host CD4 molecule (its receptor) leading to a conformational change in both gp120 and CD4 exposing the V3 region of gp120 allowing it to bind to one of the chemokine-coreceptors (CCR5, CXCR4) and for HIV gp41 to mediate fusion with the host cell membrane to release the viral core and initiate the cascade of HIV replication from uncoating, to reverse transcription, to integration, to transcription, to protein synthesis, to assembly, and finally budding [49,50,51]. However, two alternative mechanism(s) of HIV infection, albeit less efficient, are described, which are cell-to-cell transfer of HIV and endocytosis [52, 53] (Fig. 2). Both of these mechanisms have been described for HIV entry into astrocytes [54,55,56,57,58,59,60,61,62,63].

Conventional and unconventional mechanisms of HIV infection of target cells: HIV infection of target cells through gp120/CD4/chemokine receptor interactions is the most efficient means of HIV infection. Alternative mechanisms include cell-to-cell transfer of HIV between infected cell and a target cells and endocytic pathway of viral infection.
In an elegant study, human astrocytes and HIV-infected lymphocytes were co-cultured and HIV infection from lymphocyte to astrocyte was monitored by sophisticated imaging approaches, which included live-imaging, confocal and three-dimensional electron microscopy [55]. Images show HIV transmitted by cell-to-cell contact from T cells to astrocytes. The images also show two means of contact, one where the infected lymphocytes were folded into astrocytes and another where a virologic synapses was formed between the two cells allowing for the immature budding of HIV particles through filopodial extensions from lymphocytes to astrocytes.
The endocytic pathway is often hijacked by viruses to enter host cells [64]. Although there are four described mechanisms of endocytosis: phagocytosis, micropinocytosis, caveolin-mediated endocytosis, and clathrin-mediated endocytosis, described based on particle size and other markers, caveolin-mediated endocytosis is the most commonly used by viruses, including HIV [65]. Once HIV is within the endosome, its fate can be either secreted as the endosome recycles and releases its content, degraded due to the acidic pH of the endosome as it matures from an endosome to a lysosome, or the virus fuses with the vesicular membranes to release its core to the cytoplasm and undergoes its normal replicative cycle [58, 65]. Factors that can disrupt the maturation of endosome to lysosome, such as methamphetamine which increases pH of endosome or chloroquine, can allow escape of HIV from the endosome to the cytoplasm [63]. While endocytosis is not a highly efficient mechanism of HIV entry, it can promote HIV infection in non-classically (e.g. CD4 negative) target cells [65, 66].
HIV infection of astrocytes
Astrocytes, depending on their location, constitute 30–70% of the brain and perform vital functions to maintain brain homeostasis, including serving as the food source for the brain through storing glycogen which is then hydrolyzed to ATP and glucose and lactate, they regulate the level of water and other extracellular ions such as potassium and sodium, they secrete neurotrophic factors, and regulate the level of neurotransmitters such as glutamate which in excess is neurotoxic, and their end feet wrap around endothelial cells to maintain the integrity of the blood brain barrier. Astrocytes also have immune modulatory functions including release of cytokines and chemokines as a signal to alert immune cells to sites of brain inflammation/injury, promote myelination and phagocytosis of the ends of damaged neurons, and secret factors that promote neurogenesis. Due to these prolific functions, any disruption in their role can negatively impact the brain. Indeed, over the last several years, there is a greater appreciation for the role of astrocytes in health and disease, where in a number of neurodegenerative diseases, astrocytes either have a loss or gain of function [67]. In HIV, dysregulation of astrocytes is well documented [68, 69].
Astrocytes are CD4 negative [70], express CXCR-4, and depending on their developmental and activation state they may express CCR5. While HIV infection of astrocytes is independent of CD4, several alternative receptors were identified, albeit, still controversial as some are identified in astrocytoma cell lines and not primary human astrocytes. These alternative receptors for HIV entry in astrocytes include an orphan chemokine co-receptor called D6, galactocerebroside, human mannose receptor, and an unidentified molecule [62, 71,72,73]. In the case of mannose receptor, its binding to gp120 leads to HIV endocytosis with low levels of viral escape from the endocytic compartment, reverse transcription, and integration of HIV [59, 62, 63]. Further, astrocytes are infected by cell-to-cell contact, as described above. Table 1 highlights some of the studies demonstrating HIV infection of astrocytes using primary human astrocytes, astrocytoma cell lines, animal models (mice and macaque), and human brain tissue samples.
HIV infection in astrocytes is not robust and is either low-level or latent. An extensive body of literature has demonstrated that astrocytes fulfill classical criteria defining “HIV reservoirs” which include: (1) Harboring provirus: In vitro and in vivo studies indicates that astrocytes harbor integrated HIV DNA (provirus) [74,75,76,77]. In fact, 1–3% of astrocytes from HIV infected brains harbor HIV provirus [75, 78]. The size of astrocytic pool of HIV is impressive considering their vast number in the brain and in context of the size of HIV latently infected resting CD4 + T cells, which is quite small constituting approximately 105–107 of CD4 + T cell, with 62-fold higher numbers of CD4 + T cells harboring provirus that is replication competent, yet non-inducible [79, 80]. Further, these calculations do not take into consideration other cellular reservoirs and are probably a vast under estimation of the size of the HIV reservoir in the entire body. Astrocytes in essence could constitute a comparable pool of latent HIV as that defined for resting T cells since there are billions of astrocytes in the human brain. (2) The provirus can be reactivated and is infectious: HIV from infected astrocytes is induced/reactivated by inflammatory signals [81,82,83]. (3) Detection of low-level HIV transcripts without protein expression: HIV-infected astrocytes express very low levels of early and late transcripts and no detectable protein can be measured. (4) HIV promoter is under epigenetic regulation in astrocytes: class I histone deacetylases (HDACs) and a lysine-specific histone methyltransferase, SU (VAR)3–9, regulate silencing of HIV transcription in astrocytes [84]. (5) Evidence for HIV integrated DNA in post-mortem tissue described by a number of groups (Table 1) and more recently in [85].
HIV in latently infected astrocytes is responsive to latency reversing agents [84, 86], although just as was described for resting CD4 + T cells, there is integrated HIV that is not responsive to latency reversing agents and yet are replication competent [79, 87]. Further, HIV DNA and protein were detected in SIV infected macaques [88, 89]. The rate of integrated HIV DNA in astrocytes, based on in vitro [84, 90] and ex vivo studies (post-mortem tissue from HIV infected individuals) [74, 78] is approximately 1–3%. Other studies have shown that the percentage of infected astrocytes correlates with their proximity to myeloid cells, with up to 20% of astrocytes positive for HIV DNA when in close proximity to infected macrophages among donors who had dementia/HIV encephalitis, [74], however, this may be an over estimate as a result of astrocytes engulfing HIV-infected macrophages [70] or potential contamination of astrocytes with macrophages. Nonetheless, in captured astrocytes from postmortem brain of HIV-infected individuals without dementia/encephalitis, whereby the astrocytes were distant from microglia or perivascular macrophages, HIV in astrocytes was close to 1 and 6% from individuals with both dementia and advanced encephalitis [74]. This is consistent with recent data using advanced technology such as RNA and DNAscope in combination with laser capture of astrocytes from human brains [85]. Given the sheer number of astrocytes in the human brain, 1% of infected astrocytes can far exceed the resting memory T cell reservoir pool. The question remains whether astrocytes can harbor replication competent HIV in vivo. A recent study modeled this in vivo using NOD/scid-IL-2Rgc null (NSG) mice xenotransplanted with human astrocytes and reconstituted with human peripheral blood mononuclear cells (huAstro/HuPBMCs). The model targeted a specific question and by design was reductionist in nature, and it demonstrated that infected astrocytes harbor replication competent HIV and release it to periphery [85]. The authors demonstrated that astrocytes support HIV infection in vivo and egress to peripheral organs, at least in part, through trafficking of infected CD4 + T cells out of the brain [85].
Despite these extensive studies demonstrating low level of HIV replication and/or latency in astrocytes (Table 1), there is still a level of skepticism regarding HIV infection of astrocytes. Two studies in particular are in opposition of the majority of published studies [70, 91]. Ko et al. [91] analyzed post-mortem tissue from HIV + donors for HIV presence using DNA and/or RNA scope. Specifically, using RNA- and DNAscope with immunohistochemistry to detect HIV DNA and RNA in macrophages (defined as CD68 + or CD206 +) and astrocytes (defined as GFAP +) from post-mortem brains of individuals with undetectable plasma viral load, they concluded that astrocytes do not harbor HIV in these samples [91]. There were several technical concerns that negate the conclusion that astrocytes do not harbor HIV in vivo, which were rebutted in a letter to editor [92] and responded to [93]. Most significantly, the title and conclusion of this study is rather bold and affirmative that HIV is found in macrophages only and not astrocytes, which is problematic because this is based on limited donors, limited regions of the brain, and most critically astrocytes are heterogenous and they were defined by GFAP expression only and did not include other markers such as S100β. Indeed, a caveat that plagues the field is the heterogeneity of astrocytes which should not be defined by GFAP expression only and as the data are based on GFAP + it is a far reach to conclude the lack of infection of all astrocytes as no other markers were used. The study by Sattentau et al. employed luciferase and green fluorescent protein (GFP) reporter viruses and cell fusion and imaging assays and concluded that HIV does not infect human fetal astrocytes [70]. This assay, although elegant, has its limitation as it may not detect signal from a small fraction of cells and the assay does not measure HIV integration, which likely occurs through non-classical means, as well as culturing conditions and developmental stage of HFAs may be a factor in their ability to be infected, albeit, a restricted infection.
Overcoming restricted and/or latent HIV infection in astrocytes
Astrocytes infected with VSV-G pseudotyped virus to bypass receptor entry recruitment, demonstrated that there is no intrinsic restriction to HIV release [94], yet in comparison to virus output from VSVg-G pseudotyped infected HeLa cells, the level is much lower. This suggests that there are intrinsic host factors that suppress robust HIV replication in astrocytes. Several were identified which include low constitutive expression of an essential protein for Rev function known as Sam68 [95,96,97], enhanced double-stranded (ds) RNA-activated protein kinase (PKR) associated with a block in protein synthesis of TAR RNA binding protein (TRBP) [98,99,100,101], endogenous expression of the downstream effector of the Wnt signaling pathway, T cell factor 4 (TCF-4) that represses HIV LTR activation [102] and replication [103], and abundance of Beclin1, an autophagic protein [104]. This restricted replication can be overcome by certain signals such as cytokines by suppressing the repressor as in the case of IFNγ, GM-CSF, or even Methamphetamine which inhibit β-catenin signaling to overcome TCF-4 mediated suppression of HIV transcription [90, 103, 105,106,107] or RNA interference of Beclin [104]. As such, these conditions may explain the controversial nature of whether astrocytes are infected or not as the microenvironment, activation state, and even developmental stage of astrocytes may dictate their extent of permissiveness to HIV infection, replication, latency, and/or reactivation.
HIV infection of Microglia
Soon after neurological impairments were described in HIV/AIDS patients, analyses of brain tissue obtained post-mortem from pediatric and adult patients that succumbed to AIDS demonstrated the presence of HIV protein and nucleic acid in the brain. Further studies revealed the presence of HIV-infected macrophages/microglia in brain tissue via immunohistochemistry, in situ hybridization, and laser capture microdissection followed by PCR [75, 78, 91, 108,109,110,111,112,113,114]. Brain macrophages and microglia express similar surface markers (e.g. CD68, CD14, and CD45) and therefore, it can be difficult to differentiate microglia from macrophages. However, based on location and morphology one study estimated that microglia account for at least two-thirds of HIV-infected cells in the brain of people with HIV encephalitis [108].
Microglia express low levels of CD4 and the HIV co-receptor CCR5 [115, 116]. Viruses detected in the CNS of PLWH are predominately CCR5-tropic and many infect cells expressing low levels of CD4 (macrophage-tropic or M-tropic) [117]. Accordingly, multiple CCR5-tropic M-tropic HIV strains have been shown to infect primary microglia in vitro [115, 118,119,120,121,122]. HIV infection of microglia is not restricted despite high expression levels of the sterile alpha motif and histidine/aspartic acid domain-containing protein 1 (SAMHD1), a deoxynucleoside triphosphohydrolase (dNTPase) that can restrict HIV infection by reducing cellular dNTP pools [123]. This is attributed to microglia residing in a G1-like state which promotes upregulation of cyclin kinase 1 resulting in phosphorylation and inactivation of SAMHD1 [124].
HIV infection of microglia results in the production of viral proteins and the release proinflammatory cytokines/chemokines, reactive oxygen species (ROS), and reactive nitrogen species (RNS) creating an inflammatory environment resulting in activation of bystander microglia and astrocytes [125,126,127,128]. Viral proteins like gp120, Tat, and Vpr can directly or indirectly mediate neuronal cell injury resulting in neuronal cell damage and loss which may contribute to the development of HAND [126, 128]. HIV gp120 and Tat also stimulate microglia to release proinflammatory cytokines TFN-α and IL-1β [129, 130]. Elevated levels of TFN-α and IL-1β have been observed in the CSF of PLWH with HAD [131]. TNF-α is directly toxic to neurons in vitro and both TNF-α and IL-1β can facilitate neuronal injury by increasing the release of neurotoxic molecules (e.g. ceramide, L-cysteine) [125, 129, 130, 132, 133].
CNS inflammation persists in PLWH receiving cART [2, 8, 10], suggesting the continued presence of HIV-infected cells in the brain. Indeed, HIV DNA + cells have been detected in the CSF of aviremic ART-suppressed PLWH [134]. HIV DNA + and HIV RNA + cells have also been observed in postmortem brain samples of individuals with sustained cART suppression that died of unrelated causes [91, 109]. Microglia are thought to represent a key cellular reservoir of HIV in the brain under ART as they are long-lived, slowly self-renew, and are resistant to HIV-induced apoptosis [135,136,137]. In SIV-infected, ART suppressed rhesus macaques, latently-infected macrophages/microglia have been isolated from the brain [138, 139]. Latent HIV infection has also been demonstrated in iPSC-derived microglia and immortalized human microglia cell line models [140,141,142,143,144,145,146,147].
Whereas there is vast information regarding HIV latency in CD4 + T cells, particularly in peripheral blood CD4 + T cells, there is much less knowledge regarding key aspects of HIV latency in microglia. HIV latency is established in CD4 + T cells when activated infected CD4 + T cells transition to resting memory cells resulting in transcriptional silencing of the virus [148]. Multiple mechanisms are involved in maintaining HIV latency in CD4 + T cells including epigenetic modifications of chromatin and the levels of host transcriptional activators and repressors [148]. Microglia cannot be readily obtained from PLWH to study the mechanisms of HIV latency. In cell line models, Coup-TF interacting protein 2 (CTIP2) has been identified as an important regulator of HIV transcription in human microglia [145,146,147]. CTIP2 promotes latency by recruiting HDAC1, HDAC2, and HMT Suv39H1 to the HIV promoter resulting in epigenetic changes and the formation of heterochromatin [145,146,147]. Host transcriptional factor nuclear receptor related 1 (Nurr1) has also been shown to induce silencing of HIV transcription in an immortalized microglia cell line model. Nurr1 binds directly to the HIV LTR and recruits CoREST transcription repressor complexes (CoREST, HDAC1, G9a, and EZH2) resulting in chromatin remodeling [142]. Activation of the glucocorticoid receptor, which directly associates with the HIV LTR, has also been shown to promote HIV latency in a microglia cell line model [143]. Interestingly, co-culture with healthy GABAergic cortical and dopaminergic neurons (but not motor neurons) has also been shown to promote HIV latency in a human microglia cell line model and in iPSC-derived microglia [140]. GABAergic cortical and dopaminergic neurons produce glucocorticoids while motor neurons do not, further supporting a role for the glucocorticoid receptor in regulating HIV latency in microglia [140].
Future studies in SIV/NHP and HIV/humanized mouse models may provide further insight into the establishment of HIV latency in microglia and the maintenance of latently infected cells. Sequence analysis of near-full-length genome proviruses isolated from T cells of ART-treated PLWH suggest that clonal proliferation of CD4 + TH1 cells drives stabilization of the HIV reservoir in T cells [149]. However, the role of clonal expansion in maintaining the HIV reservoir in microglia in vivo is not known. It will also be important to determine the relative contribution of microglia to the HIV reservoir and to virus rebound during therapy interruption.
HIV compartmentalization within resident brain cells
There is clear evidence for genetic evolution of HIV compartmentalization in the brain that is different from that of lymphoid tissue [23, 117, 150,151,152]. HIV in CSF had a unique half-life compared to blood among a subset of patients who have evolved M-tropic HIV in the CNS [153]. Characterization of HIV quasispecies in CSF revealed that compartmentalization of HIV in the CNS is associated with HAND, implicating a role for HIV genetics in the development of neuropathology [154]. Early studies investigating CNS strains in CSF and brain parenchyma found distinct envelope, LTR, and other genes [155], however, new virologic techniques developed to unambiguously and physiologically determine HIV genotypes and phenotypes have not been applied to CNS HIV. Limited studies evaluated the extent of HIV compartmentalization within resident brain cells (astrocytes vs. macrophages/microglia). To date, a small study based on brain tissue from two HIV-E patients found evidence for HIV compartmentalization in astrocytes that is distinct from macrophages/microglia [156]. There is a need for more detailed studies, especially those evaluating HIV genetic evolution in era of cART and in relation to various clinical stages of HAND.
Conclusions
Elimination of the persistent reservoir of latently-infected cells is the greatest challenge to HIV eradication. Latently-infected CD4 + T cells are considered the most important HIV reservoir and obstacle to an HIV cure [157, 158]. However, prior research has also shown that resident glial cells in the brain are infected by HIV and may establish latency [81,82,83, 140, 143, 145,146,147]. Therefore, elimination of the T cell reservoir alone may not eradicate HIV and HIV cure approaches may be needed that penetrate the brain and target alternative cell types like glial cells. Regardless of whether HIV establishes true latency in glial cells, HIV-infection persist in the brain of cART-suppressed PLWH and contributes to neuroinflammation and dysfunction. Therefore, future studies are needed to better understand the mechanisms of HIV persistence in glial cells in vivo and their relative contribution to the HIV reservoir of virus rebound.
Availability of data and materials
Not applicable.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- ANI:
-
Asymptomatic neurocognitive impairment
- ATP:
-
Adenosine triphosphate
- cART:
-
Combination antiretroviral therapy
- CNS:
-
Central nervous system
- CoREST:
-
REST co-repressor 1
- CSF:
-
Cerebral spinal fluid
- CTIP2:
-
Coup-TF interacting protein 2
- DNA:
-
Deoxyribonucleic acid
- dNTPase:
-
Deoxynucleoside triphosphohydrolase
- DS:
-
Double stranded
- GFAP:
-
Glial fibrillary acidic protein
- HAD:
-
HIV-associated dementia
- HAND:
-
HIV-associated neurocognitive disorders
- HDAC:
-
Histone deacetylase
- HIV:
-
Human immunodeficiency virus
- LTR:
-
Long terminal repeat
- MND:
-
Mild neurocognitive disorder
- NSG:
-
NOD/scid-IL-2Rgc null
- Nurr1:
-
Nuclear receptor related 1
- PCR:
-
Polymerase chain reaction
- PKR:
-
RNA activated protein kinase
- PLWH:
-
People living with HIV
- ROS:
-
Reactive oxygen species
- RNA:
-
Ribonucleic acid
- RNS:
-
Reactive nitrogen species
- PBMC:
-
Peripheral blood mononuclear cell
- SAMHD1:
-
Sterile alpha motif and histidine/aspartic acid domain containing protein 1
- SIV:
-
Simian immunodeficiency virus
- TCF-4:
-
T cell factor 4
- TRBP:
-
TAR DNA binding protein
- VSV:
-
Vesicular stomatitis virus
References
Clifford DB, Ances BM. HIV-associated neurocognitive disorder. Lancet Infect Dis. 2013;13(11):976–86.
Heaton RK, Clifford DB, Franklin DR Jr, Woods SP, Ake C, Vaida F, et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: charter Study. Neurology. 2010;75(23):2087–96.
Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, et al. HIV-associated neurocognitive disorder—pathogenesis and prospects for treatment. Nat Rev Neurol. 2016;12(5):309.
Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69(18):1789–99.
Price RW, Brew B, Sidtis J, Rosenblum M, Scheck AC, Cleary P. The brain in AIDS: central nervous system HIV-1 infection and AIDS dementia complex. Science. 1988;239(4840):586–92.
Navia BA, Jordan BD, Price RW. The AIDS dementia complex: I. Clinical Featur Ann Neurol. 1986;19(6):517–24.
Navia BA, Cho ES, Petito CK, Price RW. The AIDS dementia complex: II. Neuropathol Ann Neurol. 1986;19(6):525–35.
Heaton RK, Franklin DR, Ellis RJ, McCutchan JA, Letendre SL, Leblanc S, et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16.
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, et al. Dementia in AIDS patients: incidence and risk factors. Multicent AIDS Cohort Study Neurol. 1993;43(11):2245–52.
Sacktor N, Skolasky RL, Seaberg E, Munro C, Becker JT, Martin E, et al. Prevalence of HIV-associated neurocognitive disorders in the multicenter AIDS Cohort Study. Neurology. 2016;86(4):334–40.
Alford K, Vera JH. Cognitive Impairment in people living with HIV in the ART era: a review. Br Med Bull. 2018;127(1):55–68.
Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, et al. HIV-1 tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci. 2003;24(1):224–37.
Chompre G, Cruz E, Maldonado L, Rivera-Amill V, Porter JT, Noel RJ Jr. Astrocytic expression of HIV-1 Nef impairs spatial and recognition memory. Neurobiol Dis. 2013;49:128–36.
van Marle G, Henry S, Todoruk T, Sullivan A, Silva C, Rourke SB, et al. Human immunodeficiency virus type 1 Nef protein mediates neural cell death: a neurotoxic role for IP-10. Virology. 2004;329(2):302–18.
Nguyen TP, Soukup VM, Gelman BB. Persistent hijacking of brain proteasomes in HIV-associated dementia. Am J Pathol. 2010;176(2):893–902.
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE. 2008;3(6):e2516.
Richards MH, Narasipura SD, Seaton MS, Lutgen V, Al-Harthi L. Migration of CD8+ T Cells into the central nervous system gives rise to highly potent anti-HIV CD4dimCD8bright T cells in a Wnt signaling-dependent manner. J Immunol. 2016;196(1):317–27.
Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206(2):275–82.
Lackner AA. Pathology of simian immunodeficiency virus induced disease. Curr Top Microbiol Immunol. 1994;188:35–64.
Chakrabarti L, Hurtrel M, Maire MA, Vazeux R, Dormont D, Montagnier L, et al. Early viral replication in the brain of SIV-infected rhesus monkeys. Am J Pathol. 1991;139(6):1273–80.
Honeycutt JB, Liao B, Nixon CC, Cleary RA, Thayer WO, Birath SL, et al. T cells establish and maintain CNS viral infection in HIV-infected humanized mice. J Clin Invest. 2018;128(7):2862–76.
Gega A, Kozal MJ, Chiarella J, Lee E, Peterson J, Hecht FM, et al. Deep sequencing of HIV-1 variants from paired plasma and cerebrospinal fluid during primary HIV infection. J Virus Erad. 2015;1(4):264–8.
Schnell G, Price RW, Swanstrom R, Spudich S. Compartmentalization and clonal amplification of HIV-1 variants in the cerebrospinal fluid during primary infection. J Virol. 2010;84(5):2395–407.
Lane JH, Sasseville VG, Smith MO, Vogel P, Pauley DR, Heyes MP, et al. Neuroinvasion by simian immunodeficiency virus coincides with increased numbers of perivascular macrophages/microglia and intrathecal immune activation. J Neurovirol. 1996;2(6):423–32.
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J. Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008;14(4):318–26.
León-Rivera R, Veenstra M, Donoso M, Tell E, Eugenin EA, Morgello S, et al. Central nervous system (CNS) viral seeding by mature monocytes and potential therapies to reduce CNS viral reservoirs in the cART era. mBio. 2021. https://doi.org/10.1128/mBio.03633-20.
Williams DW, Eugenin EA, Calderon TM, Berman JW. Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J Leukoc Biol. 2012;91(3):401–15.
Bergamaschi A, Pancino G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology. 2010;7:31.
Triques K, Stevenson M. Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J Virol. 2004;78(10):5523–7.
Dong C, Kwas C, Wu L. Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J Virol. 2009;83(8):3518–27.
Aljawai Y, Richards MH, Seaton MS, Narasipura SD, Al-Harthi L. beta-Catenin/TCF-4 signaling regulates susceptibility of macrophages and resistance of monocytes to HIV-1 productive infection. Curr HIV Res. 2014;12(3):164–73.
Bertin J, Jalaguier P, Barat C, Roy MA, Tremblay MJ. Exposure of human astrocytes to leukotriene C4 promotes a CX3CL1/fractalkine-mediated transmigration of HIV-1-infected CD4+ T cells across an in vitro blood-brain barrier model. Virology. 2014;454–455:128–38.
Albalawi YA, Narasipura SD, Olivares LJ, Al-Harthi L. CD4(dim) CD8(bright) T cells home to the brain and mediate HIV neuroinvasion. J Virol. 2022;96(15):e0080422.
Subra C, Trautmann L. Role of T lymphocytes in HIV neuropathogenesis. Curr HIV/AIDS Rep. 2019;16(3):236–43.
Negi N, Das BK. CNS: not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int Rev Immunol. 2018;37(1):57–68.
Sandrone S, Moreno-Zambrano D, Kipnis J, van Gijn J. A (delayed) history of the brain lymphatic system. Nat Med. 2019;25(4):538–40.
Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. 2018;21(10):1380–91.
Papadopoulos Z, Herz J, Kipnis J. Meningeal lymphatics: from anatomy to central nervous system immune surveillance. J Immunol. 2020;204(2):286–93.
Marcondes MC, Burudi EM, Huitron-Resendiz S, Sanchez-Alavez M, Watry D, Zandonatti M, et al. Highly activated CD8(+) T cells in the brain correlate with early central nervous system dysfunction in simian immunodeficiency virus infection. J Immunol. 2001;167(9):5429–38.
Marcondes MC, Sopper S, Sauermann U, Burdo TH, Watry D, Zandonatti M, et al. CD4 deficits and disease course acceleration can be driven by a collapse of the CD8 response in rhesus macaques infected with simian immunodeficiency virus. AIDS. 2008;22(12):1441–52.
Sinclair E, Ronquillo R, Lollo N, Deeks SG, Hunt P, Yiannoutsos CT, et al. Antiretroviral treatment effect on immune activation reduces cerebrospinal fluid HIV-1 infection. J Acquir Immune Defic Syndr. 2008;47(5):544–52.
Al-Harthi L. Comment on “CD4+ CD8+ T cells represent a significant portion of the anti-HIV T cell response to acute HIV infection.” J Immunol. 2012;188(12):5809.
Zloza A, Schenkel JM, Tenorio AR, Martinson JA, Jeziorczak PM, Al-Harthi L. Potent HIV-specific responses are enriched in a unique subset of CD8+ T cells that coexpresses CD4 on its surface. Blood. 2009;114(18):3841–53.
Zloza A, Sullivan YB, Connick E, Landay AL, Al-Harthi L. CD8+ T cells that express CD4 on their surface (CD4dimCD8bright T cells) recognize an antigen-specific target, are detected in vivo, and can be productively infected by T-tropic HIV. Blood. 2003;102(6):2156–64.
Zloza A, Al-Harthi L. Multiple populations of T lymphocytes are distinguished by the level of CD4 and CD8 coexpression and require individual consideration. J Leukoc Biol. 2006;79(1):4–6.
Schenkel JM, Zloza A, Li W, Narasipura SD, Al-Harthi L. Beta-catenin signaling mediates CD4 expression on mature CD8+ T cells. J Immunol. 2010;185(4):2013–9.
Sullivan YB, Landay AL, Zack JA, Kitchen SG, Al-Harthi L. Upregulation of CD4 on CD8+ T cells: CD4dimCD8bright T cells constitute an activated phenotype of CD8+ T cells. Immunology. 2001;103(3):270–80.
Albalawi YA, Narasipura SD, Al-Harthi L. Wnt/β-catenin protects lymphocytes from HIV-mediated apoptosis via induction of Bcl-xL. Viruses. 2022;14(7):1469.
Gomez C, Hope TJ. The ins and outs of HIV replication. Cell Microbiol. 2005;7(5):621–6.
Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8):a006866.
Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13(8):484–96.
Lehmann M, Nikolic DS, Piguet V. How HIV-1 takes advantage of the cytoskeleton during replication and cell-to-cell transmission. Viruses. 2011;3(9):1757–76.
Estes JD, Kityo C, Ssali F, Swainson L, Makamdop KN, Del Prete GQ, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. 2017;23(11):1271–6.
Nath A, Hartloper V, Furer M, Fowke KR. Infection of human fetal astrocytes with HIV-1: viral tropism and the role of cell to cell contact in viral transmission. J Neuropathol Exp Neurol. 1995;54(3):320–30.
Li GH, Anderson C, Jaeger L, Do T, Major EO, Nath A. Cell-to-cell contact facilitates HIV transmission from lymphocytes to astrocytes via CXCR4. AIDS. 2015;29(7):755–66.
Li GH, Maric D, Major EO, Nath A. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. AIDS. 2020;34(7):963–78.
Luo X, He JJ. Cell-cell contact viral transfer contributes to HIV infection and persistence in astrocytes. J Neurovirol. 2015;21(1):66–80.
Chauhan A, Khandkar M. Endocytosis of human immunodeficiency virus 1 (HIV-1) in astrocytes: a fiery path to its destination. Microb Pathog. 2015;78:1–6.
Chauhan A, Mehla R, Vijayakumar TS, Handy I. Endocytosis-mediated HIV-1 entry and its significance in the elusive behavior of the virus in astrocytes. Virology. 2014;456–457:1–19.
Chauhan A, Tikoo A, Patel J, Abdullah AM. HIV-1 endocytosis in astrocytes: a kiss of death or survival of the fittest? Neurosci Res. 2014;88:16–22.
Hao HN, Lyman WD. HIV infection of fetal human astrocytes: the potential role of a receptor-mediated endocytic pathway. Brain Res. 1999;823(1–2):24–32.
Liu Y, Liu H, Kim BO, Gattone VH, Li J, Nath A, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78(8):4120–33.
Vijaykumar TS, Nath A, Chauhan A. Chloroquine mediated molecular tuning of astrocytes for enhanced permissiveness to HIV infection. Virology. 2008;381(1):1–5.
Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44.
Permanyer M, Ballana E, Esté JA. Endocytosis of HIV: anything goes. Trends Microbiol. 2010;18(12):543–51.
Miyauchi K, Kim Y, Latinovic O, Morozov V, Melikyan GB. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell. 2009;137(3):433–44.
Giovannoni F, Quintana FJ. The role of astrocytes in CNS inflammation. Trends Immunol. 2020;41(9):805–19.
Pandey HS, Seth P. Friends turn foe-astrocytes contribute to neuronal damage in neuroAIDS. J Mol Neurosci. 2019;69(2):286–97.
Minagar A, Shapshak P, Fujimura R, Ownby R, Heyes M, Eisdorfer C. The role of macrophage/microglia and astrocytes in the pathogenesis of three neurologic disorders: HIV-associated dementia, Alzheimer disease, and multiple sclerosis. J Neurol Sci. 2002;202(1–2):13–23.
Russell RA, Chojnacki J, Jones DM, Johnson E, Do T, Eggeling C, et al. Astrocytes resist HIV-1 fusion but engulf infected macrophage material. Cell Rep. 2017;18(6):1473–83.
Neil SJ, Aasa-Chapman MM, Clapham PR, Nibbs RJ, McKnight A, Weiss RA. The promiscuous CC chemokine receptor D6 is a functional coreceptor for primary isolates of human immunodeficiency virus type 1 (HIV-1) and HIV-2 on astrocytes. J Virol. 2005;79(15):9618–24.
Bhat S, Spitalnik SL, Gonzalez-Scarano F, Silberberg DH. Galactosyl ceramide or a derivative is an essential component of the neural receptor for human immunodeficiency virus type 1 envelope glycoprotein gp120. Proc Natl Acad Sci. 1991;88(16):7131–4.
Hao H-N, Chiu F-C, Losev L, Weidenheim KM, Rashbaum WK, Lyman WD. HIV infection of human fetal neural cells is mediated by gp120 binding to a cell membrane-associated molecule that is not CD4 nor galactocerebroside. Brain Res. 1997;764(1):149–57.
Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66(2):253–8.
Trillo-Pazos G, Diamanturos A, Rislove L, Menza T, Chao W, Belem P, et al. Detection of HIV-1 DNA in microglia/macrophages, astrocytes and neurons isolated from brain tissue with HIV-1 encephalitis by laser capture microdissection. Brain Pathol. 2003;13(2):144–54.
Dewhurst S, Sakai K, Bresser J, Stevenson M, Evinger-Hodges MJ, Volsky DJ. Persistent productive infection of human glial cells by human immunodeficiency virus (HIV) and by infectious molecular clones of HIV. J Virol. 1987;61(12):3774–82.
Dewhurst S, Bresser J, Stevenson M, Sakai K, Evinger-Hodges MJ, Volsky DJ. Susceptibility of human glial cells to infection with human immunodeficiency virus (HIV). FEBS Lett. 1987;213(1):138–43.
Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DF, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol. 2006;12(2):146–52.
Ho Y-C, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DIS, et al. Replication-competent non-induced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155(3):540–51.
Siliciano JD, Siliciano RF. Nonsuppressible HIV-1 viremia: a reflection of how the reservoir persists. J Clin Invest. 2020;130(11):5665–7.
Atwood WJ, Tornatore CS, Traub R, Conant K, Drew PD, Major EO. Stimulation of HIV type 1 gene expression and induction of NF-kappa B (p50/p65)-binding activity in tumor necrosis factor alpha-treated human fetal glial cells. AIDS Res Hum Retroviruses. 1994;10(10):1207–11.
Tornatore C, Nath A, Amemiya K, Major EO. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J Virol. 1991;65(11):6094–100.
Carroll-Anzinger D, Al-Harthi L. Gamma interferon primes productive human immunodeficiency virus infection in astrocytes. J Virol. 2006;80(1):541–4.
Narasipura SD, Kim S, Al-Harthi L. Epigenetic regulation of HIV-1 latency in astrocytes. J Virol. 2014;88(5):3031–8.
Lutgen V, Narasipura SD, Barbian HJ, Richards M, Wallace J, Razmpour R, et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020;16(6):e1008381.
Proust A, Barat C, Leboeuf M, Drouin J, Tremblay MJ. Contrasting effect of the latency-reversing agents bryostatin-1 and JQ1 on astrocyte-mediated neuroinflammation and brain neutrophil invasion. J Neuroinflamm. 2017;14(1):242.
Barat C, Proust A, Deshiere A, Leboeuf M, Drouin J, Tremblay MJ. Astrocytes sustain long-term productive HIV-1 infection without establishment of reactivable viral latency. Glia. 2018;66(7):1363–81.
Overholser ED, Coleman GD, Bennett JL, Casaday RJ, Zink MC, Barber SA, et al. Expression of simian immunodeficiency virus (SIV) nef in astrocytes during acute and terminal infection and requirement of nef for optimal replication of neurovirulent SIV in vitro. J Virol. 2003;77(12):6855–66.
Overholser ED, Babas T, Zink MC, Barber SA, Clements JE. CD4-independent entry and replication of simian immunodeficiency virus in primary rhesus macaque astrocytes are regulated by the transmembrane protein. J Virol. 2005;79(8):4944–51.
Li W, Henderson LJ, Major EO, Al-Harthi L. IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol. 2011;186(12):6771–8.
Ko A, Kang G, Hattler JB, Galadima HI, Zhang J, Li Q, et al. Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J Neuroimmune Pharmacol. 2019;14(1):110–9.
Al-Harthi L, Nath A. Letter to Editor. J Neuroimmune Pharmacol. 2019;14(1):6.
Li Q, Kim WK. Reply to Letter to the Editor. J Neuroimmune Pharmacol. 2019;14(1):7–8.
Canki M, Thai JN, Chao W, Ghorpade A, Potash MJ, Volsky DJ. Highly productive infection with pseudotyped human immunodeficiency virus type 1 (HIV-1) indicates no intracellular restrictions to HIV-1 replication in primary human astrocytes. J Virol. 2001;75(17):7925–33.
Li J, Liu Y, Kim BO, He JJ. Direct participation of Sam68, the 68-kilodalton Src-associated protein in mitosis, in the CRM1-mediated Rev nuclear export pathway. J Virol. 2002;76(16):8374–82.
Li J, Liu Y, Park IW, He JJ. Expression of exogenous Sam68, the 68-kilodalton SRC-associated protein in mitosis, is able to alleviate impaired Rev function in astrocytes. J Virol. 2002;76(9):4526–35.
Zhang J, Liu Y, Henao J, Rugeles MT, Li J, Chen T, et al. Requirement of an additional Sam68 domain for inhibition of human immunodeficiency virus type 1 replication by Sam68 dominant negative mutants lacking the nuclear localization signal. Gene. 2005;363:67–76.
Daher A, Longuet M, Dorin D, Bois F, Segeral E, Bannwarth S, et al. Two dimerization domains in the trans-activation response RNA-binding protein (TRBP) individually reverse the protein kinase R inhibition of HIV-1 long terminal repeat expression. J Biol Chem. 2001;276(36):33899–905.
Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3(1):61–71.
Dorin D, Bonnet MC, Bannwarth S, Gatignol A, Meurs EF, Vaquero C. The TAR RNA-binding protein, TRBP, stimulates the expression of TAR-containing RNAs in vitro and in vivo independently of its ability to inhibit the dsRNA-dependent kinase PKR. J Biol Chem. 2003;278(7):4440–8.
Ong CL, Thorpe JC, Gorry PR, Bannwarth S, Jaworowski A, Howard JL, et al. Low TRBP levels support an innate human immunodeficiency virus type 1 resistance in astrocytes by enhancing the PKR antiviral response. J Virol. 2005;79(20):12763–72.
Wortman B, Darbinian N, Sawaya BE, Khalili K, Amini S. Evidence for regulation of long terminal repeat transcription by Wnt transcription factor TCF-4 in human astrocytic cells. J Virol. 2002;76(21):11159–65.
Carroll-Anzinger D, Kumar A, Adarichev V, Kashanchi F, Al-Harthi L. Human immunodeficiency virus-restricted replication in astrocytes and the ability of gamma interferon to modulate this restriction are regulated by a downstream effector of the Wnt signaling pathway. J Virol. 2007;81(11):5864–71.
Rodriguez M, Soler Y, Muthu Karuppan MK, Zhao Y, Batrakova EV, El-Hage N. Targeting beclin1 as an adjunctive therapy against HIV using mannosylated polyethylenimine nanoparticles. Pharmaceutics. 2021;13(2):223.
Henderson LJ, Narasipura SD, Adarichev V, Kashanchi F, Al-Harthi L. Identification of novel T cell factor 4 (TCF-4) binding sites on the HIV long terminal repeat which associate with TCF-4, β-catenin, and SMAR1 to repress HIV transcription. J Virol. 2012;86(17):9495–503.
Narasipura SD, Henderson LJ, Fu SW, Chen L, Kashanchi F, Al-Harthi L. Role of β-catenin and TCF/LEF family members in transcriptional activity of HIV in astrocytes. J Virol. 2012;86(4):1911–21.
Sharma A, Hu XT, Napier TC, Al-Harthi L. Methamphetamine and HIV-1 Tat down regulate β-catenin signaling: implications for methampetamine abuse and HIV-1 co-morbidity. J Neuroimmune Pharmacol. 2011;6(4):597–607.
Cosenza MA, Zhao ML, Si Q, Lee SC. Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002;12(4):442–55.
Wallet C, De Rovere M, Van Assche J, Daouad F, De Wit S, Gautier V, et al. Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol. 2019;9:362.
Thompson KA, Cherry CL, Bell JE, McLean CA. Brain cell reservoirs of latent virus in presymptomatic HIV-infected individuals. Am J Pathol. 2011;179(4):1623–9.
Tso FY, Kang G, Kwon EH, Julius P, Li Q, West JT, et al. Brain is a potential sanctuary for subtype C HIV-1 irrespective of ART treatment outcome. PLoS ONE. 2018;13(7):e0201325.
Koenig S, Gendelman HE, Orenstein JM, Dal Canto MC, Pezeshkpour GH, Yungbluth M, et al. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233(4768):1089–93.
Kure K, Weidenheim KM, Lyman WD, Dickson DW. Morphology and distribution of HIV-1 gp41-positive microglia in subacute AIDS encephalitis pattern of involvement resembling a multisystem degeneration. Acta Neuropathol. 1990;80(4):393–400.
Vazeux R, Brousse N, Jarry A, Henin D, Marche C, Vedrenne C, et al. AIDS subacute encephalitis Identification of HIV-infected cells. Am J Pathol. 1987;126(3):403–10.
Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O’Connor MJ, et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73(1):205–13.
Jordan CA, Watkins BA, Kufta C, Dubois-Dalcq M. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J Virol. 1991;65(2):736–42.
Schnell G, Joseph S, Spudich S, Price RW, Swanstrom R. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog. 2011;7(10):e1002286.
Albright AV, Shieh JT, O’Connor MJ, Gonzalez-Scarano F. Characterization of cultured microglia that can be infected by HIV-1. J Neurovirol. 2000;6(Suppl 1):S53-60.
Ghorpade A, Nukuna A, Che M, Haggerty S, Persidsky Y, Carter E, et al. Human immunodeficiency virus neurotropism: an analysis of viral replication and cytopathicity for divergent strains in monocytes and microglia. J Virol. 1998;72(4):3340–50.
Lee SC, Hatch WC, Liu W, Kress Y, Lyman WD, Dickson DW. Productive infection of human fetal microglia by HIV-1. Am J Pathol. 1993;143(4):1032–9.
Schuenke K, Gelman BB. Human microglial cell isolation from adult autopsy brain: brain pH, regional variation, and infection with human immunodeficiency virus type 1. J Neurovirol. 2003;9(3):346–57.
Strizki JM, Albright AV, Sheng H, O’Connor M, Perrin L, Gonzalez-Scarano F. Infection of primary human microglia and monocyte-derived macrophages with human immunodeficiency virus type 1 isolates: evidence of differential tropism. J Virol. 1996;70(11):7654–62.
Rodrigues V, Ruffin N, San-Roman M, Benaroch P. Myeloid cell interaction with HIV: a complex relationship. Front Immunol. 2017;8:1698.
Mlcochova P, Sutherland KA, Watters SA, Bertoli C, de Bruin RA, Rehwinkel J, et al. A G1-like state allows HIV-1 to bypass SAMHD1 restriction in macrophages. EMBO J. 2017;36(5):604–16.
Brabers NA, Nottet HS. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006;36(7):447–58.
Borrajo A, Spuch C, Penedo MA, Olivares JM, Agis-Balboa RC. Important role of microglia in HIV-1 associated neurocognitive disorders and the molecular pathways implicated in its pathogenesis. Ann Med. 2021;53(1):43–69.
Uzasci L, Nath A, Cotter R. Oxidative stress and the HIV-infected brain proteome. J Neuroimmune Pharmacol. 2013;8(5):1167–80.
Gonzalez-Scarano F, Martin-Garcia J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5(1):69–81.
Yeung MC, Pulliam L, Lau AS. The HIV envelope protein gp120 is toxic to human brain-cell cultures through the induction of interleukin-6 and tumor necrosis factor-alpha. AIDS. 1995;9(2):137–43.
Nicolini A, Ajmone-Cat MA, Bernardo A, Levi G, Minghetti L. Human immunodeficiency virus type-1 Tat protein induces nuclear factor (NF)-kappaB activation and oxidative stress in microglial cultures by independent mechanisms. J Neurochem. 2001;79(3):713–6.
Tyor WR, Glass JD, Griffin JW, Becker PS, McArthur JC, Bezman L, et al. Cytokine expression in the brain during the acquired immunodeficiency syndrome. Ann Neurol. 1992;31(4):349–60.
Gelbard HA, Dzenko KA, DiLoreto D, del Cerro C, del Cerro M, Epstein LG. Neurotoxic effects of tumor necrosis factor alpha in primary human neuronal cultures are mediated by activation of the glutamate AMPA receptor subtype: implications for AIDS neuropathogenesis. Dev Neurosci. 1993;15(6):417–22.
Yeh MW, Kaul M, Zheng J, Nottet HS, Thylin M, Gendelman HE, et al. Cytokine-stimulated, but not HIV-infected, human monocyte-derived macrophages produce neurotoxic levels of l -cysteine. J Immunol. 2000;164(8):4265–70.
Spudich S, Robertson KR, Bosch RJ, Gandhi RT, Cyktor JC, Mar H, et al. Persistent HIV-infected cells in cerebrospinal fluid are associated with poorer neurocognitive performance. J Clin Invest. 2019;129(8):3339–46.
Castellano P, Prevedel L, Eugenin EA. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci Rep. 2017;7(1):12866.
Cosenza MA, Zhao ML, Lee SC. HIV-1 expression protects macrophages and microglia from apoptotic death. Neuropathol Appl Neurobiol. 2004;30(5):478–90.
Reu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, et al. The lifespan and turnover of microglia in the human brain. Cell Rep. 2017;20(4):779–84.
Abreu CM, Veenhuis RT, Avalos CR, Graham S, Parrilla DR, Ferreira EA, et al. Myeloid and CD4 T cells comprise the latent reservoir in antiretroviral therapy-suppressed SIVmac251-infected macaques. mBio. 2019. https://doi.org/10.1128/mBio.01659-19.
Avalos CR, Abreu CM, Queen SE, Li M, Price S, Shirk EN, et al. Brain macrophages in simian immunodeficiency virus-infected, antiretroviral-suppressed macaques: a functional latent reservoir. mBio. 2017. https://doi.org/10.1128/mBio.01186-17.
Alvarez-Carbonell D, Ye F, Ramanath N, Garcia-Mesa Y, Knapp PE, Hauser KF, et al. Cross-talk between microglia and neurons regulates HIV latency. PLoS Pathog. 2019;15(12):e1008249.
Alvarez-Carbonell D, Garcia-Mesa Y, Milne S, Das B, Dobrowolski C, Rojas R, et al. Toll-like receptor 3 activation selectively reverses HIV latency in microglial cells. Retrovirology. 2017;14(1):9.
Ye F, Alvarez-Carbonell D, Nguyen K, Leskov K, Garcia-Mesa Y, Sreeram S, et al. Recruitment of the CoREST transcription repressor complexes by nerve growth factor IB-like receptor (Nurr1/NR4A2) mediates silencing of HIV in microglial cells. PLoS Pathog. 2022;18(7):e1010110.
Alvarez-Carbonell D, Ye F, Ramanath N, Dobrowolski C, Karn J. The glucocorticoid receptor is a critical regulator of HIV latency in human microglial cells. J Neuroimmune Pharmacol. 2019;14(1):94–109.
Garcia-Mesa Y, Jay TR, Checkley MA, Luttge B, Dobrowolski C, Valadkhan S, et al. Immortalization of primary microglia: a new platform to study HIV regulation in the central nervous system. J Neurovirol. 2017;23(1):47–66.
Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, et al. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007;26(2):412–23.
Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, et al. COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 2005;33(7):2318–31.
Rohr O, Lecestre D, Chasserot-Golaz S, Marban C, Avram D, Aunis D, et al. Recruitment of Tat to heterochromatin protein HP1 via interaction with CTIP2 inhibits human immunodeficiency virus type 1 replication in microglial cells. J Virol. 2003;77(9):5415–27.
Margolis DM, Archin NM, Cohen MS, Eron JJ, Ferrari G, Garcia JV, et al. Curing HIV: seeking to target and clear persistent infection. Cell. 2020;181(1):189–206.
Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Invest. 2017;127(7):2689–96.
Holman AG, Mefford ME, O’Connor N, Gabuzda D. HIVBrainSeqDB: a database of annotated HIV envelope sequences from brain and other anatomical sites. AIDS Res Ther. 2010;7:43.
Ellis RJ, Gamst AC, Capparelli E, Spector SA, Hsia K, Wolfson T, et al. Cerebrospinal fluid HIV RNA originates from both local CNS and systemic sources. Neurology. 2000;54(4):927–36.
Harrington PR, Schnell G, Letendre SL, Ritola K, Robertson K, Hall C, et al. Cross-sectional characterization of HIV-1 env compartmentalization in cerebrospinal fluid over the full disease course. AIDS. 2009;23(8):907–15.
Schnell G, Spudich S, Harrington P, Price RW, Swanstrom R. Compartmentalized human immunodeficiency virus type 1 originates from long-lived cells in some subjects with HIV-1-associated dementia. PLoS Pathog. 2009;5(4):e1000395.
Ritola K, Robertson K, Fiscus SA, Hall C, Swanstrom R. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J Virol. 2005;79(16):10830–4.
van Marle G, Power C. Human immunodeficiency virus type 1 genetic diversity in the nervous system: evolutionary epiphenomenon or disease determinant? J Neurovirol. 2005;11(2):107–28.
Thompson KA, Churchill MJ, Gorry PR, Sterjovski J, Oelrichs RB, Wesselingh SL, et al. Astrocyte specific viral strains in HIV dementia. Ann Neurol. 2004;56(6):873–7.
Kuo HH, Lichterfeld M. Recent progress in understanding HIV reservoirs. Curr Opin HIV AIDS. 2018;13(2):137–42.
Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. Latency reversal and viral clearance to cure HIV-1. Science. 2016;353(6297):aaf6517.
Ward JM, O’Leary TJ, Baskin GB, Benveniste R, Harris CA, Nara PL, et al. Immunohistochemical localization of human and simian immunodeficiency viral antigens in fixed tissue sections. Am J Pathol. 1987;127(2):199–205.
Gyorkey F, Melnick JL, Gyorkey P. Human immunodeficiency virus in brain biopsies of patients with AIDS and progressive encephalopathy. J Infect Dis. 1987;155(5):870–6.
Tornatore C, Chandra R, Berger JR, Major EO. HIV-1 infection of subcortical astrocytes in the pediatric central nervous system. Neurology. 1994;44(3 Pt 1):481–7.
Ranki A, Nyberg M, Ovod V, Haltia M, Elovaara I, Raininko R, et al. Abundant expression of HIV Nef and Rev proteins in brain astrocytes in vivo is associated with dementia. AIDS. 1995;9(9):1001–8.
Fiala M, Rhodes RH, Shapshak P, Nagano I, Martinez-Maza O, Diagne A, et al. Regulation of HIV-1 infection in astrocytes: expression of Nef, TNF-alpha and IL-6 is enhanced in coculture of astrocytes with macrophages. J Neurovirol. 1996;2(3):158–66.
Guillemin G, Croitoru J, Boussin FD, Le Grand R, Franck-Duchenne M, Dormont D. Astrocytes and lentivirus infection in an experimental models of macaque infected with SIVmac251. C R Seances Soc Biol Fil. 1998;192(1):179–86.
Guillemin G, Croitoru J, Le Grand RL, Franck-Duchenne M, Dormont D, Boussin FD. Simian immunodeficiency virus mac251 infection of astrocytes. J Neurovirol. 2000;6(3):173–86.
Schweighardt B, Atwood WJ. HIV type 1 infection of human astrocytes is restricted by inefficient viral entry. AIDS Res Hum Retroviruses. 2001;17(12):1133–42.
Churchill MJ, Gorry PR, Cowley D, Lal L, Sonza S, Purcell DFJ, et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol. 2006;12(2):146–52.
Eugenin EA, Clements JE, Zink MC, Berman JW. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J Neurosci. 2011;31(26):9456–65.
Gray LR, Turville SG, HItchen TL, Cheng W-J, Ellett AM, Salimi H, et al. HIV-1 entry and trans-infection of astrocytes involves CD81 vesicles. PLOS ONE. 2014;9(2):e90620.
Richards MH, Narasipura SD, Kim S, Seaton MS, Lutgen V, Al-Harthi L. Dynamic interaction between astrocytes and infiltrating PBMCs in context of neuroAIDS. Glia. 2015;63(3):441–51.
Chauhan A. Enigma of HIV-1 latent infection in astrocytes: an in-vitro study using protein kinase C agonist as a latency reversing agent. Microbes Infect. 2015;17(9):651–9.
Kunze C, Börner K, Kienle E, Orschmann T, Rusha E, Schneider M, et al. Synthetic AAV/CRISPR vectors for blocking HIV-1 expression in persistently infected astrocytes. Glia. 2018;66(2):413–27.
Rodriguez M, Lapierre J, Ojha CR, Pawitwar S, Karuppan MKM, Kashanchi F, et al. Morphine counteracts the antiviral effect of antiretroviral drugs and causes upregulation of p62/SQSTM1 and histone-modifying enzymes in HIV-infected astrocytes. J Neurovirol. 2019;25(2):263–74.
Edara VV, Ghorpade A, Borgmann K. Insights into the gene expression profiles of active and restricted red/green-HIV(+) human astrocytes: implications for shock or lock therapies in the brain. J Virol. 2020. https://doi.org/10.1128/JVI.01563-19.
Valdebenito S, Castellano P, Ajasin D, Eugenin EA. Astrocytes are HIV reservoirs in the brain: a cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J Neurochem. 2021;158(2):429–43.
Malik S, Valdebenito S, D’Amico D, Prideaux B, Eugenin EA. HIV infection of astrocytes compromises inter-organelle interactions and inositol phosphate metabolism: a potential mechanism of bystander damage and viral reservoir survival. Prog Neurobiol. 2021;206:102157.
Bauer A, Brack-Werner R. Modeling HIV latency in astrocytes with the human neural progenitor cell line HNSC. 100. Methods Mol Biol. 2022;2407:103–14.
Acknowledgements
A special thank you to Amber K. Virdi and Tanner L. Shull for assistance with graphical design of figures shown.
Funding
This work was supported in part by National Institutes of Health (NIH) Grants R01MH131441 (AW), R01MH113425 (LA), R01NS108796 (LA), R01MH100628 (LA). The funders had no role in the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AW and AL-H contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wahl, A., Al-Harthi, L. HIV infection of non-classical cells in the brain. Retrovirology 20, 1 (2023). https://doi.org/10.1186/s12977-023-00616-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-023-00616-9