
Abstract
Monocytes and macrophages are targets of HIV-1 infection and play critical roles in multiple aspects of viral pathogenesis. HIV-1 can replicate in blood monocytes, although only a minor proportion of circulating monocytes harbor viral DNA. Resident macrophages in tissues can be infected and function as viral reservoirs. However, their susceptibility to infection, and their capacity to actively replicate the virus, varies greatly depending on the tissue localization and cytokine environment. The susceptibility of monocytes to HIV-1 infection in vitro depends on their differentiation status. Monocytes are refractory to infection and become permissive upon differentiation into macrophages. In addition, the capacity of monocyte-derived macrophages to sustain viral replication varies between individuals. Host determinants regulate HIV-1 replication in monocytes and macrophages, limiting several steps of the viral life-cycle, from viral entry to virus release. Some host factors responsible for HIV-1 restriction are shared with T lymphocytes, but several anti-viral mechanisms are specific to either monocytes or macrophages. Whilst a number of these mechanisms have been identified in monocytes or in monocyte-derived macrophages in vitro, some of them have also been implicated in the regulation of HIV-1 infection in vivo, in particular in the brain and the lung where macrophages are the main cell type infected by HIV-1. This review focuses on cellular factors that have been reported to interfere with HIV-1 infection in monocytes and macrophages, and examines the evidences supporting their role in vivo, highlighting unique aspects of HIV-1 restriction in these two cell types.
Similar content being viewed by others


Introduction
Bone marrow-derived monocytes (Mos) are released into the blood where they circulate for a few days (the half-life of circulating Mos in normal healthy individuals is 71 h [1]) before subsequent extravasation into the lungs, gastrointestinal tract, kidney, primary and secondary lymphoid organs and the central nervous system (CNS). In tissues, Mos undergo differentiation into tissue-specific macrophages (Mφ) and dendritic cells (DC). HIV-infected mononuclear phagocytes (bone marrow (BM) and blood Mo, tissue Mφ, microglia, and DC) can thus serve as vehicles for dissemination and reservoirs of HIV-1 infection [2]. In the macaque model, the blood Mo count increases during the first few days following SIV infection [3], and high Mo turnover during SIV infection is a predictive marker for AIDS progression [4]. Subsets of activated Mo that express CD16 and/or CD163 are expanded both in HIV-infected individuals and in SIV-infected macaques [5]. During acute infection, activated Mos migrate into different tissues, including the CNS ([3]and accompanying review by G. Gras and M. Kaul). Relatively few Mos in the blood bear HIV-1 DNA (<0.1%) [6], reviewed in [7], whereas Mφ vary greatly in their permissivity to HIV-1 infection depending on their tissue localization [8]. Viral replication in tissue Mφ is probably governed not only by the cytokine network, but also by other environmental factors. In vitro, Mφ differentiated from blood Mos (Mo-derived macrophages, MDMs) display a great heterogeneity in their capacities to replicate HIV-1, depending on the donor (up to a 3 log difference in viral production between donors) [9–11]. In contrast, HIV-1 replication kinetics were similar in MDM from pairs of identical twins [9]. These observations strongly argue in favor of the influence of the genetic background on viral replication in Mo/Mφ [12], as has also been suggested for CD4+ T cells [13]. Indeed, the CCR5Δ32 genotype has been associated with a restricted infection of MDM and CD4+ T cells by HIV-1 strains that use the CCR5 co-receptor (R5 HIV-1) [11, 14, 15]. Thus both constitutive and environmental factors appear to regulate HIV-1 replication in Mo/Mφ. Due to the difficulty of assessing HIV-1 infection in resident tissue Mφ, most studies have addressed the regulation of HIV-1 infection in Mo/Mφ in the MDM model. Methodological differences in the purification and differentiation of Mos therefore add further variability to the heterogeneity of these cells with respect to infection by the virus. Several recent reviews have addressed the influence of cytokines and other endogenous and exogenous stimuli on HIV-1 infection of Mo/Mφ [16–18](see also the accompanying review by G. Herbein and A. Varin). This review will focus on the mechanisms of HIV-1 restriction in Mo and Mφ. In vitro data will be discussed for their potential relevance in the light of our knowledge concerning the in vivo infection of these cells.
Molecular shields against HIV-1 replication in monocytes
Although infectious virus can be recovered from peripheral blood Mos taken from HIV-1-infected patients (see below), freshly isolated Mos are highly resistant to HIV-1 infection in vitro [19–21]. There are divergent reports on the level of refractivity of freshly isolated quiescent Mos, in vitro, to HIV-1 infection, varying from absolute to relative. Methodological parameters including the viral strain and infectious dose, the time of Mo infection after their isolation from blood (immediately or following some hours of culture), the Mo condition at the time of infection (fresh or thawed), and the time lapse of monitoring viral replication after infection, may explain the reported differences in refractivity to HIV-1 replication [22–26]. In addition, the markers used to evaluate Mo differentiation differ depending on the study [24, 27, 28], and may not completely reflect phenotypic changes associated with maturation. Even when cultured in the absence of human serum or exogenous cytokines such as M-CSF or GM-CSF, Mos may undergo partial differentiation that could modify their capacity to support viral replication [29, 30]. Indeed, permissiveness to HIV-1 infection in vitro increases with Mo differentiation to Mφ [19, 28, 31]. The association of Mo maturation with an enhancement of viral replication appears to be a conserved phenomenon among the lentiviruses, as it has also been described for non-primate lentiviruses such as the caprine arthritis-encephalitis virus and maedi-visna virus (MVV) [32, 33]. However, while MVV replication in monocytes appears to be restricted at transcriptional level [34, 35], distinct mechanisms of restriction contribute to render Mo resistant to HIV-1 infection, at least in vitro (Fig. 1A). The relative weight of the restrictions affecting different steps of viral replication is still subject of debate, although pre-integrative blocks appear to play a determinant role.

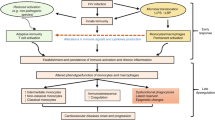
Schematic representation of host restriction factors in human Mos and Mφ. On the left, low levels of CD4 and CCR5 may limit viral entry in monocytes. Low expression of thymidine phosphorylase associated with a limited stock of dTTP reduces RT rate. APOBEC3A and 3G may interfere with HIV-1 RT in Mos. HIV-2/SIV Vpx antagonizes the restriction of HIV-1 in Mos and Mφ by counteracting an unidentified host factor. Cellular miRNAs have been proposed to target the 3'UTR of HIV-1 transcripts. miR-198 may repress CycT1 expression that contributes to Tat transactivation. On the right, the CCR5Δ32 mutation restricts viral entry of R5 HIV-1 in Mφ. LPS targets the early phases of the HIV-1 cycle in Mφ, through the down-regulation of CCR5 expression and the LTR-driven transcription by IL-10/IFN-β-induced expression of 16 kDa C/EBPβ. p21Waf1 interferes with both RT and integration and is induced by FcγR engagement. CTIP2 and TRIM22 have been implicated in the inhibition of HIV-1 transcription. Urokinase-type plasminogen activator (uPA) blocks the release of viral particles from intracellular vacuoles.
Restrictions at early steps of HIV-1 replication in monocytes
The early events of viral entry are represented by the engagement between CD4 receptors at the membrane of target cells and the viral envelope proteins gp120-gp41. The consequent conformational changes in the structure of gp120 allow the interaction with the CXCR4 or CCR5 co-receptors, the latter being the primary co-receptor used by macrophage-tropic HIV-1 strains. Increasing susceptibility of maturing Mos to R5 HIV-1 infection has been associated with an increasing expression of CCR5 at the cell surface that enhances viral entry into the cells [28, 36]. However, HIV-1 restriction in Mos does not appear to be due to limiting amounts of HIV-1 co-receptors, and has been attributed to post-entry blocks. Indeed, Mos do not support transduction with HIV-1-based vectors pseudotyped with the VSV-G or MLV-A envelopes, that mediate viral entry by pathways independent of the HIV-1 receptor and co-receptors [27, 29], indicating that the block to HIV-1 infection is independent of the route of viral entry. Furthermore, efficient entry of HIV-1 pseudoviruses has been directly demonstrated using a β-lactamase entry assay [27]. Post-entry blocks in infected Mos have been localized either prior to or at the reverse transcription (RT) step of viral replication [26, 27] or at the level of nuclear translocation of viral cDNA [29].
A recent study challenges these conclusions, claiming that the relative HIV-1 restriction in Mos, in comparison with Mφ and HeLa-P4 cells, is related to a defect in viral entry followed by a delay in the preintegrative steps [24]. In this work, the inhibition of viral entry into Mos was measured using a fusion assay and was found to be independent of HIV-1 Env, since it also affected VSV-G pseudotyped viruses. Subsequent post-entry steps, RT and integration were not totally blocked, although they proceeded with very slow kinetics (tIN50% = 7-8 days) [24]. Neither the nature of the entry block in Mos nor the potential impact of different endocytic/phagocytic capacities of Mos and Mφ with respect to entry of viral particles into cells was addressed in this study.
Using VSV-G pseudotyped HIV-1 and qPCR, Triques and Stevenson showed that reverse transcription is restricted in Mos, and they suggested that the absence of reverse transcription-favouring cellular cofactors is the limiting circumstance [27]. It has been suggested that the defect in reverse transcription observed in Mos, as well as the slow reverse transcription seen in MDMs, is due to a limited availability of nucleotide precursors in these non-dividing cells [37, 38]. In particular, Mos contain very low levels of deoxythymidine triphosphate (dTTP), associated with low levels of thymidine phosphorylase, the enzyme that converts thymine into thymidine [27]. Both dTTP and thymidine phosphorylase levels increase during maturation to Mφ. However, D-thymidine supplementation of Mo cultures increased the dTTP levels but did not relieve the reverse transcription block [27], suggesting that other factors are involved in the restriction. In addition, reverse transcriptase from lentiviruses have been shown to be able to efficiently catalyze DNA synthesis even at low dNTP concentrations, in contrast to the RT of gammaretroviruses, which are unable to replicate in non-dividing cells [39].
In contrast to the hypothesis that links Mo resistance to HIV-1 with a lack of cellular cofactors needed for viral replication, Peng et al. proposed that viral replication in Mos is restricted because of factors belonging to the APOBEC3 cytidine deaminase family [40]. The best-characterized member of this family concerning its anti-retroviral activity, including HIV-1 restriction, is APOBEC3G [41–44]. APOBEC3G is incorporated into HIV-1 virions and deaminates dC to dU in minus single-strand nascent cDNA within newly infected cells; resulting in lethal G-to-A hypermutations in the single stranded viral intermediates. This antiviral activity is counteracted by the Vif protein, that induces degradation of APOBEC3G and prevents its incorporation into virions (recently reviewed in [45]). A deaminase-independent anti-viral activity, not counteracted by Vif, has also been described that affects the accumulation of reverse transcripts in infected cells [46, 47]. Several mechanisms have been proposed for such antiviral activity, including the inhibition of viral cDNA synthesis by a block in the translocation of reverse transcriptase along the template RNA genome and the destabilization of viral core morphology and stability during virion assembly [47], reviewed in [48]. The APOBEC3G non-enzymatic activity has been proposed to account for the post-entry HIV-1 restrictions in quiescent resting CD4+ T cells [49] and in DC [50], although its role in quiescent CD4+ T-cells has been recently contested [51, 52]. The expression of APOBEC3G, and of another member of the same family APOBEC3A, has been shown to be down-regulated during Mo differentiation to Mφ [40]. siRNA-mediated silencing of each of the two genes allowed HIV-1 replication in Mos, whereas induction of APOBEC3A and 3G by IFNs was associated with the inhibition of HIV-1 replication in Mφ [40]. However, the way in which APOBEC3A and 3G interfere with HIV-1 replication in Mos remains to be determined.
Experiments of transduction of heterokaryons formed by the fusion of Mos and permissive HeLa cells with HIV-1 vectors showed that the heterokaryons were refractory to transduction, suggesting the presence of a dominant restriction factor in the parental Mos [53]. HIV-1 restriction in Mo/HeLa heterokaryons could be alleviated by providing the Vpx protein from SIV, either in trans or packaged into HIV-1 virions [53]. Vpx has been shown to be required for the replication of HIV-2 and SIV in Mφ, and it has been hypothesized that it diverts a cullin-ubiquitine ligase complex to inactivate a factor that restricts HIV-2 and SIV infection. Vpx expression also enhanced HIV-1 transduction of Mφ, pointing to a common mechanism of restriction [53]. The role of Vpx and the mechanisms underlying its activity in overcoming a retroviral restriction in myeloid cells [54] is discussed in an accompanying review (Ayinde D. et al.).
Restriction of transcription and later events in HIV-1 replication in monocytes
Besides restrictions at early post-entry steps of viral replication, transcriptional restriction has also been reported to contribute to Mo resistance to HIV-1 [55]. The 5' LTR of integrated provirus contains several cis-regulatory elements necessary for the binding of cellular transcription factors (NFκB sites, C/EBP sites, Sp1 sites and a TATA cassette) and is recognized by the RNA polymerase II as a promoter. The viral Tat protein is recruited to the 5' LTR sequence, interacts with a 59-nucleotide structure called the transactivation response (TAR) element and acts as a stimulator of transcriptional elongation soon after the generation of short terminated transcripts. Tat interacts with the host cyclin T1 protein (CycT1), which recruits the cyclin-dependent kinase 9 (CDK9) to the TAR element. The complex formed by CycT1 and CDK9 is called P-TEFb (for positive transcription elongation factor b). The cooperation of Tat and P-TEFb at the TAR sequence produces a hyperphosphorylation of the C-terminus of RNA polymerase II, stimulating the elongation of viral RNA. After transfection of the HIV-1 genome or of an LTR-reporter construction, neither viral production nor Tat transactivation were detected in undifferentiated Mo [25]. Heterokaryons between Mo and 293 T cells restored the Tat transactivation function of the LTR, suggesting that Mo lack factors required for transactivation. The level of the CycT1 P-TEFb component required for Tat transactivation was below the detection threshold in Mos, in agreement with previous reports [56, 57]. The regulation of CycT1 seems to occur at a post-transcriptional level and is likely to involve proteasome-mediated proteolysis [58]. Interestingly, lack of CycT1 expression in Mos has recently been linked to a translational repression by the miR-198 microRNA [59]. It has been proposed that miR-198 contributes to HIV-1 restriction in Mos by repressing CycT1 expression, while miR-198 is down-regulated during Mo differentiation to Mφ [59]. However, transient expression of CycT1 did not rescue Tat transactivation in Mos [25], suggesting that this is not sufficient to relieve HIV-1 transcriptional restriction. Increased permissivity to HIV-1 infection during Mo differentiation to Mφ was associated with both increased expression of CycT1 [25, 57] and phosphorylation of the CycT1 P-TEFb partner, CDK9 [25]. It has therefore been suggested that the transcriptional restriction of HIV-1 in Mos may involve regulation of P-TEFb function [25].
Some recent reports have suggested the implication of cellular microRNA (miRNA) in Mo resistance to HIV-1 infection. Wang et al. showed that four miRNA, previously shown to target the 3'UTR of HIV-1 transcripts [60, 61], are down-regulated during Mo differentiation to Mφ [62]. This rather preliminary report does not go further into the analysis of miRNA effect on HIV-1 replication. miRNA might target HIV-1 directly or indirectly by side effects on the cell biology [63]. An indirect effect of an miRNA on HIV-1 replication that targets the RNA polymerase II positive transcription elongation factor P-TEFb has indeed been described (see below) [59].
When HIV-1 meets monocytes in vivo...
In spite of the resistance to HIV-1 infection exhibited by Mos in vitro, circulating peripheral blood Mos from HIV-1 infected individuals harbor HIV-1 DNA, although at a low frequency (<0.1%) [64, 65]. Replication competent virus could be recovered from circulating Mos, even those of patients receiving HAART and with a viral load below detectable levels that would indicate their role as a viral reservoir [66–68]. Compelling evidence for active replication in Mos in vivo is supplied by the detection of unintegrated circularized forms of viral DNA (2-LTR circles) and multiply spliced HIV mRNA species in freshly isolated blood Mos [64, 68, 69], and by markers of compartmentalization and viral evolution in this compartment [70–73].
How can observations pertaining to the in vitro and in vivo contexts be reconciled? It has been suggested that Mos may be infected before leaving the bone marrow (BM) at the stage of precursors, and that they then migrate to other organs, including secondary lymphoid organs, lungs and brain, where they differentiate into Mφ [74] (Fig. 2A). Viral replication will then be reactivated and probably lead to the dissemination of infection to neighboring cells [75] (Fig. 2A). A similar scenario has been hypothesized for MVV infection: infected monocytes carrying the viral genome without expressing viral proteins can enter the organs by a "Trojan Horse" mechanism, avoiding immune surveillance [76, 77]. Otherwise, Mo refractivity to HIV-1 may simply not be absolute, and Mo subsets may be permissive to infection. Mos may become permissive to infection after being activated in the BM or in the blood of HIV-1 infected patients, owing to the inflammatory environment and immune activation [78]. Considering the extraordinary plasticity of Mo/Mφ [79], it may also be hypothesized that infected Mos can transmigrate back to the blood [80] after meeting either the virus or infected cells in inflamed tissues (Fig 2A). In support of this possibility, recent evidence has been provided for Mos recirculation from tissues to the BM in a murine model (reviewed in [81]). A subset of circulating Mos that displays pro-inflammatory characteristics is actually expanded in HIV-infected individuals. One minor subset of Mos that expresses the CD16 (FcγRIII) molecule, and represents 5%-15% of circulating Mos in healthy individuals, is expanded in HIV-1 patients and may reach up to 40% of the total circulating Mo population during the progression to AIDS [82]. This Mo subset expresses the CX3CR1 receptor, and its members migrate into tissues that express CXC3CL1, produce pro-inflammatory cytokines (including TNF and IL-1), and can activate resting T-cells by producing CCR3 and CCR4 ligands [83–86]. CD16+ Mos exhibit some characteristics of tissue Mφ and display a transcriptional profile closer to Mφ and DC profiles than to that of CD16- Mos [87–89]. The CD16+ subset of circulating Mos have been shown to be preferentially infected by HIV-1 in vivo [90, 91] and in vitro [90] (Fig 2). Increased susceptibility to R5 HIV-1 was associated with a higher level of CCR5 expression in this cell subset, compared to the CD14highCD16- Mos, and to a shift in the APOBEC3G distribution towards high molecular mass forms [90]. Whether the CD16+ Mos represent a higher level of Mo differentiation and may thus reconcile the findings of Mo restriction to HIV-1 infection in vitro and the presence of a fraction of infected Mos in vivo remains to be clarified.

Schematic model of infection of monocytes and macrophages. A) Hypothetical ways to infect monocytes. Mo precursors may be infected before leaving the bone marrow (1) and then migrate to peripheral tissues where they differentiate into Mφ (2). Viral replication will then be reactivated leading to viral production and infection of neighboring cells (3). Alternatively, Mo subsets may become permissive to infection after being activated in the bone marrow or in the blood, owing to the inflammatory environment (4). Mos may be infected after encountering the virus or infected cells in inflamed tissues (5), where they then differentiate to Mφ. However, infected Mos might also transmigrate back to the blood (6). A Mo subset expressing CD16 that displays pro-inflammatory characteristics appears to be preferentially infected by HIV-1. B) Dissemination and control of HIV-1 infection in tissue macrophages. Infected Mos migrate to peripheral tissues such as brain, lungs and gastrointestinal tract where they differentiate and disseminate infection to resident microglial cells, alveolar Mφ or mucosal Mφ. The CD16+ subset has an enhanced capacity to transmigrate into tissues. Various factors that may control HIV-1 replication are present in peripheral compartments. Mφ from the mucosa of the gastrointestinal tract, where exposure to LPS is frequent, do not express CCR5 and are resistant to HIV-1 infection. An increased expression of the inhibitory C/EBPβ may suppress viral transcription in Mφ in brain and lungs, contributing to viral latency. Transcriptional silencing of the HIV-1 LTR by CTIP2 may contribute to HIV-1 latency in the CNS. uPA is also involved in the control of HIV-1 replication in the CNS and is sequestered by the soluble receptor suPAR in CNS disease.
Limits to macrophage permissivity to HIV-1 infection
Mo differentiation to Mφ is accompanied by an increased permissivity to HIV-1 infection, both in vitro and in vivo (see above). Nevertheless, a great heterogeneity in the capacity to sustain viral replication is observed in MDM from different donors, and HIV-1 infection of resident Mφ varies depending on their tissue localization. In addition, only a fraction of MDM, which varies in size depending on the blood donor, is able to replicate the virus. Some studies suggest that only Mφ which maintain their capacity to proliferate can support a productive HIV-1 infection [31, 92]. However, this fact cannot account for differences in the capacity of MDM to replicate HIV-1, since the percentage of cells capable of DNA synthesis is far lower than the percentage of HIV-1-infected cells in MDM cultures [29, 93]. Therefore, it appears that HIV-1 replication in Mφ is also regulated by host factors, at the level of both single cells and the individual. Variability in MDM permissivity to HIV-1 infection among individuals has been attributed to host genetic factors that mainly influence pre-reverse transcription steps [12]. The reverse transcription process appears to be the main limiting step of HIV-1 replication, not only in Mo (see above) but also in MDM [10, 38, 53, 94]. However, several other steps of the HIV-1 life cycle that can be restricted in MDM have been described.
Restrictions at early steps of HIV-1 replication in macrophages
CCR5 co-receptor expression levels at the cell surface are an important determinant for MDM susceptibility to HIV-1 infection. A lack of expression of the CCR5 molecule at the cell surface, linked to a homozygous CCR5Δ32 mutation, blocks the entry of R5 HIV-1 into both CD4+ T cells and MDM [15, 95, 96]. The heterozygous CCR5Δ32 genotype has also been associated with a decreased susceptibility of MDM to R5 HIV-1 infection [11, 14]. A strong and sustained down-regulation of CCR5 expression, independent of ex novo protein synthesis but rather due to an altered recycling of chemokine receptors, is induced by exposure of Mφ to lipopolysaccharide (LPS) [97]. This and other mechanisms that underlie LPS-induced restriction of HIV-1 replication in Mφ have been reviewed elsewhere and will not be described here [98]. However, it is worth mentioning that LPS, a major constituent of the cell wall of Gram-negative bacteria, is one of the main stimuli for human Mφ activation and a potent HIV-1 inhibitory factor in these cells that express a wide number of Toll-like receptors (TLRs) and the GPI-anchored CD14 receptor that is responsible for LPS binding. Exposure of Mφ to LPS in physiological conditions might limit viral replication in these cells. In fact, Mφ isolated from the mucosa of the gastrointestinal tract, where exposure to gram-negative bacteria and subsequently to LPS is enhanced, do not express CCR5 at their surface and are resistant to HIV-1 infection [99] (Fig 2B). In addition to the CD4/CCR5 mediated entry of HIV-1 into the cell by membrane fusion, an alternative route of infection has been described in Mφ that involves the uptake of the virus via macropinocytosis [100, 101]. This process requires an intact lipid raft, and notably the correct amount and distribution of cholesterol molecules. Cholesterol is a structural component of biological membranes that forms ordered lipid assemblies called lipid rafts, essential for the fluidity of membranes and for the mobility of proteins at the cell surface. Cholesterol may be considered a limiting molecule that can modulate infection by different enveloped viruses, including vaccinia virus, SV40, and herpes simplex virus [102–104]. Chemical cholesterol depletion of target cells has been shown to disrupt HIV-1 entry into primary T lymphocytes and T cell lines as well as into MDM, possibly by reducing the fusion capacity with the HIV-1 envelope and CCR5-mediated CCR5 signaling [105–107].
Mo and Mφ express receptors for the Fc portion of G immunoglobulins (IgG), called FcγR [108]. FcγR molecules form a family of integral membrane proteins that can either activate or inhibit cell functions. The activating receptors expressed on Mφ are the high affinity receptor for monomeric IgG FcγRI (CD64) and two low affinity receptors that only bind the Ag-Ab immune complexes (ICs) FcγRIIA/C (CD32) and FcγRIIIA (CD16). The aggregation of FcγR after binding of IC induces the phosphorylation of their ITAM (immunoglobulin tyrosine activating motif) intracellular activating portion and triggers major responses to pathogens (endocytosis, phagocytosis and cytokine production). FcγRIIB is the inhibitory receptor containing an ITIM (immunoglobulin tyrosine inhibitory motif) in its intracytoplasmic tail, which negatively regulates cell functions induced by the activating FcγR. The stimulation of MDM by IC immobilized on culture plates through activating FcγRs strongly inhibits HIV-1 replication independently of the use of the CXCR4 or CCR5 co-receptors [109, 110]. Using one cycle infections, we showed that HIV-1 entry and post-integration steps of the viral replication are not affected in IC-activated MDM, whereas levels of reverse transcription products and integrated proviruses are strongly decreased [110]. Remarkably, other lentiviruses, such as HIV-2, SIVmac and SIVagm, are affected by FcγR engagement, suggesting that the restriction targets either a protein conserved among these viruses or a common function. Recent work showed that the cyclin-dependent kinase inhibitor p21Cip1/Waf1 (p21) knock-down rescues the replication of HIV-1, SIVmac and HIV-2 restoring the levels of reverse transcription products and integrated proviruses in IC-activated MDM [111]. Moreover, p21 silencing also increased HIV-1 replication in unstimulated MDM by enhancing reverse transcription and integration. These results suggest that p21 whose expression is enhanced by FcγR engagement acts as an inhibitory factor of lentiviral infection in macrophages. First described as a cell cycle inhibitor, that blocks cell cycling at the G1/S interface and plays a critical role in the control of cell growth, p21 is also involved in the regulation of apoptosis and differentiation [112–114]. Controversial data have been published in the last few years concerning p21 effects on HIV-1 replication in Mφ and in other cell types. Vazquez et al. reported that p21 enhances HIV-1 infection in Mφ 12-14 days after challenge with the R5 BaL viral strain, and proposed that an increased p21 expression after HIV-1 infection was linked to an accumulation of Vpr in infected cells [30]. The reasons for the contrasting findings reported by Vazquez and by ourselves are unclear. They might underlie a dual role for p21 in HIV-1 infection depending on the time after infection: a block of preintegrative steps of HIV-1 replication in acute infection, or an activation of HIV-1 gene expression, synergistically with Vpr, in chronic infection [115]. In T lymphocytes, HIV-1 infection was associated with a loss of p21 expression [116], and 9-aminoacridine (9AA), that induces p21 expression via p53-dependent pathways, significantly inhibits HIV-1 replication in activated PBMCs [117]. p21 was described as a unique molecular barrier for HIV-1 replication in primitive hematopoietic cells that are normally resistant to HIV-1 infection [118]. p21 knockdown in bone marrow CD34+ cells resulted in a strong increase in HIV-1 infection by alleviating a nuclear block to viral genome integration [118]. Zhang et al. showed that p21 was associated with HIV-1 PIC and proposed that the antiviral activity of p21 depends on its ability to interact with HIV-1 integrase (IN). We did not detect interactions between p21 and HIV-1 proteins, including IN, in yeast two-hybrid, pull down or co-immunoprecipitation assays, suggesting that p21 may affect viral replication independently of a specific interaction with an HIV-1 component [111]. Further investigations are needed to precisely determine the interplay between p21 and HIV-1 (see also data reported in the accompanying review by Le Doucet V. et al).
The genetic expression of members of the APOBEC family of cellular polynucleotide cytidine deaminases that have been involved in Mo resistance to HIV-1 infection, including APOBEC3G and APOBEC3A, is down-regulated in Mφ [40]. Besides the species-specific restriction factor TRIM5α [119], an increasing number of TRIM proteins have been found to inhibit several viral infections, including HIV-1 [120]. For instance, TRIM22 (Staf50) has been shown to inhibit HIV-1 replication in MDMs, although its mechanism of action and the step at which the block occurs remain unclear, other than that it appears to affect late steps of HIV-1 replication. Using cell lines, the block has been localized either at the step of viral transcription from the LTR or at that of viral assembly and release [121–123]. TRIM25 participates in RIG-I-mediated antiviral activity through its E3 ubiquitin ligase activity [124]. Although the relevance of antiviral effects of members of the TRIM family has not yet been documented in human Mφ, several TRIM proteins are expressed in these cells and are modulated by external stimuli [125]. Therefore it may be worthwhile to investigate the potential of TRIM proteins for antiretroviral activity. A recent systematic analysis of TRIM gene expression levels in primary human PBMCs and MDM in response to interferons and FcγR engagement may be a helpful tool for further functional studies in this direction [126].
It has been suggested that lentiviruses, unlike other retroviruses, can infect non-dividing cells such as resting T lymphocytes, DC and Mφ, due to the capacity of their cDNA to enter the nuclei through an intact nuclear membrane. A number of mechanisms underlying the interaction of the lentiviral PIC with the cell nuclear import machinery have been proposed to account for this property [127], as reviewed in [128]. In particular, it was proposed that the reduced ability of Vpr-deficient HIV-1 to replicate in MDM reflects the relevance of Vpr-dependent nuclear import in these cells [129]. However, the role of Vpr in the nuclear transport of HIV-1 and in HIV-1 replication in Mφ remains unclear (see accompanying review, Ayinde D. et al.). Several studies show redundant nucleophilic determinants in HIV-1 proteins that independently allow the nuclear localization of viral DNA and virus replication in MDM [130–132]. However, a recent study reported that the deletion of all the nuclear localization signals described in HIV-1 proteins did not abrogate HIV-1 infection of resting CD4+ T cells and Mφ [133]. The authors proposed that the limiting step that determines the capacity of HIV-1 and MLV to infect non-dividing cells is the uncoating of the entering viral particles, independently of nuclear entry. In the same vein, recent studies concerning HIV-1 and HIV-2 infection of Mφ led to the conclusion that nuclear entry may not be the limiting step for HIV-1 infection, but that the restrictions affect earlier steps before or during reverse transcription [27, 134–137].
After passing the nuclear membrane barrier, HIV-1 cDNA is oriented to chromosome targets where viral integrase (IN) catalyzes integration into the host genome [138]. A network of intermediate filament proteins, called lamins, expressed on the inner nuclear membrane ensures the close association between the nuclear envelope and chromatin. The barrier to autointegration factor (BAF), a small DNA-binding protein, is a component of the HIV-1 PIC that promotes integration of the viral cDNA into cell chromosomes and prevents intramolecular integrations. BAF interacts with LEM domain proteins of the inner nuclear membrane (lamina-associated polypeptide 2 (LAP2), emerin, manin). One of its binding partners is emerin, an integral inner-nuclear-envelope protein that participates in chromatin organization and bridges the interface between the inner nuclear envelope and chromosomes. The group of Stevenson proposed that emerin, as well as LAP2α, was required for HIV-1 infection in Mφ to assist the targeting of HIV DNA to the chromatin [139]. The binding of emerin and LAP2α to the viral genome was found to be indirect, and LEM-mediated interaction with BAF was essential to promote integration through the association of these proteins and the viral cDNA. In Mφ that lack emerin or BAF, HIV-1 cDNA entered the nuclear compartment, but was rapidly converted into non-functional episomal DNA that accumulated in the nuclear matrix. Integration into the host genome was therefore dramatically impaired. These results were shortly contradicted by the observation of HIV-1 infection of HeLa-P4 cells following potent down-regulation of emerin, BAF or LAP2α with specific siRNAs [140]. To clarify these conflicting data based on RNA interference-mediated gene knockdown, which were therefore highly dependent on the silencing efficiency, another group demonstrated that HIV-1 efficiently infects embryonic fibroblasts taken from emerin knockout, LAP2α knockout or emerin-LAP2α double knockout mice [141]. The same results were found in Mφ from wild-type and knockout mice transduced with HIV-1, indicating that emerin and LAP2α are dispensable for HIV-1 infection in mouse/human dividing/non-dividing cells. A third experimental approach based on the use of dominant negative emerin molecules presenting mutations in the LEM domain confirmed that HIV-1 infections occur even in the presence of high levels of mutant proteins [141]. Future studies of the role of BAF and its associated nuclear lamin proteins in vivo during HIV-1 infection could possibly add further clarification to the interaction of PIC with the nuclear membrane.
Transcriptional control of HIV-1 in macrophages
Transcriptional regulation has been involved in viral latency of integrated HIV-1 and the formation of viral reservoirs in Mos (see above) and in Mφ. LPS acts as a potent modulator of HIV-1 transcription, displaying opposite effects on Mos and Mφ. Early reports showed that LPS potently stimulates HIV-1 LTR expression in monocytic cell lines by induction of NFκB [142] and through the activation of PU.1 Ets proteins [143]. LPS induces the phosphorylation of PU.1, which allows its interaction with the LTR promoter [143] and with the NFκB transcription factor bound to the downstream binding site. The ability of LPS to induce or suppress transcription from the HIV LTR is linked to the maturation state of monocytic cells. In freshly isolated Mos, LTR-driven gene transcription is enhanced by LPS stimulation, whereas it is suppressed in MDM [144]. A factor contributing to this dichotomy could be the different expression of CycT1, required for Tat transactivation, that is undetectable in Mos, but is induced during the differentiation to Mφ [57] (see above). Although CycT1 is later down-regulated in differentiated MDM, its expression is enhanced by HIV-1 infection [58].
Further insight pertaining to the dual effect of LPS on HIV-1 gene expression came from the observation that LPS modulates the expression of the CCAAT enhancer binding protein β (C/EBPβ) transcription factors differently in Mos and in Mφ [145, 146]. C/EBPβ is a member of the C/EBP transcription factor family that is associated with myelomonocytic differentiation [147]. C/EBP binding sites are required for the control of viral replication in Mo/Mφ but not in T lymphocytes [148, 149]. Three C/EBP binding sites are localized upstream of the transcriptional start site within the HIV-1 LTR [150]. The C/EBPβ gene has no introns. However, two different proteins can originate from the same mRNA: a large isoform of 30-37 kDa that stimulates gene transcription, and a small isoform of 16-21 kDa that has repressive activity [151, 152]. The small inhibitory form of the protein is produced when an internal ribosome entry site is used by ribosomes to start translation [151]. The 16 kDa C/EBPβ protein can be considered to be a dominant negative transcription factor since it blocks DNA transcription even when it is expressed at relatively low levels (20%) compared to the 37 kDa activating isoform [151]. The complex regulation pattern of HIV-1 gene expression by C/EBPβ in Mos and Mφ has been addressed in a series of studies by M. Weiden et al. concerning HIV-1 replication in lung Mφ during pulmonary tuberculosis [145, 153–155]. In HIV-1 infected patients, alveolar Mφ (AM) do not show active viral replication, whereas they represent a major source of virus in pulmonary tuberculosis [156]. The inhibitory 16 kDa C/EBPβ isoform is highly expressed in resting AM of healthy individuals, and may be responsible for viral latency in these cells after HIV-1 infection, but it is strongly suppressed after M. tuberculosis infection [153]. However, in vitro infection with M. tuberculosis or stimulation of PMA-differentiated THP-1 monocytic cells and primary Mφ with LPS did not enhance HIV-1 infection, and even suppressed viral replication [145]. In contrast, M. tuberculosis and LPS enhanced HIV-1 replication in undifferentiated THP-1 monocytic cells. These opposing effects were reflected by significant changes in the C/EBPβ isoform balance upon exposure to M. tuberculosis and LPS in Mos and Mφ: a high amount of activating C/EBPβ transcription factor was induced in Mos, whereas a strong expression of the inhibitory 16 kDa form was induced in Mφ. It turned out that the production of the dominant negative C/EBPβ isoform is mediated by IFNβ in Mφ but not in Mos [145]. LPS and M. tuberculosis trigger IFNβ production in both Mo and Mφ. However, while in Mφ IFNβ induces inhibitory C/EBPβ gene expression by stimulating the nuclear translocation and the DNA binding of ISGF-3 (a heterotrimeric complex formed by the interferon regulatory factor IRF-9, STAT-1 and STAT-2), these two stimuli are not sufficient to activate ISGF-3 in Mos [145]. LPS or M. tuberculosis-derived lipoarabinomannan induction of IL-10 can also trigger the production of the inhibitory C/EBPβ in differentiated THP-1 Mφ, but not in undifferentiated Mos, through STAT-3 signaling [155]. Thus, differentiation-induced post-translational regulations govern the production of inhibitory C/EBPβ in response to either IFNβ or IL-10 in Mφ. An explanation for the apparent discrepancy between the M. tuberculosis-mediated HIV-1 suppression in Mφ in vitro and the enhancement of HIV-1 replication in AM in vivo was proposed in another study by the same group [154]. The addition of activated T lymphocytes to AM reduced inhibitory C/EBPβ and activated the NF-κB pathway, leading to activation of the HIV-1 LTR and increased viral replication. Down-regulation of inhibitory C/EBPβ expression and subsequent de-repression of the HIV-1 LTR were mediated by the interaction of T cell-expressed co-stimulatory molecules, including CD40L, VLA-4 and CD28, and the cognate macrophage-expressed ligands. The induction of NF-κB was mediated by cytokines secreted from activated T-cell, including TNFβ, IL-1β and IL-6 [154]. Erythromycin A derivatives counteract the positive effect of CD4+ T cells on HIV-1 replication in resistant Mφ by blocking MAPK activation and C/EBPβ induction [157]. Moreover, erythromycin A derivatives render tissue Mφ resistant to HIV-1 infection by inducing the inhibitory C/EBPβ isoform and by down-regulating the activity of hematopoietic cell kinase (Hck) [157]. Recently, IFNβ was shown to induce the truncated inhibitory C/EBPβ isoform and to suppress SIV replication in primary Mφ of rhesus macaques [158]. A downstream effector of class I IFNs, CUGBP1 (CUG-repeat RNA-binding protein 1), was shown to induce the expression of the inhibitory C/EBPβ form by alternative translation of its mRNA [158]. Indeed, the inhibition of SIV replication and the increase of 16 kDa C/EBPβ by IFNβ were associated with and dependent on the phosphorylation of CUGBP1 and the formation of CUGBP1-C/EBPβ mRNA complexes.
A distinct mechanism of HIV-1 transcriptional repression was described in a human microglial cell line. A co-repressor known as the COUP-TF interacting protein 2 (CTIP2) potently inhibited Tat transactivation, and over-expression of CTIP2 disrupted Tat nuclear localization and its recruitment to CTIP2-induced nuclear structures [159]. The authors proposed that Tat inactivation occurs through subnuclear relocalization within inactive regions of the chromosomes [159]. CTIP2 inhibited Sp1- and COUP-TF-mediated activation of HIV-1 gene transcription in microglial nuclei [159]. Indeed, CTIP2 was recruited to the HIV-1 LTR promoter via its interaction with Sp1 bound to the GC-box sequences. CTIP2 co-localized with Sp1, COUP-TF and the heterochromatin-associated protein HP1α that is normally detected in transcriptionally repressed heterochromatic regions [160]. In addition, HDAC1, HDAC2 and the histone methyltransferase SUV39H1 were recruited to the chromatin by CTIP2 and promoted the association of HP1 to the HIV LTR region, thereby silencing viral gene transcription. CTIP2 thus induces HIV-1 gene silencing by forcing the transcriptionally repressed environment onto the LTR promoter [160](see also the accompanying review by Le Douce V. et al.).
Restriction of late events in HIV-1 replication in macrophages
HIV-1 assembly in infected Mφ occurs within intracellular compartments associated with the tetraspanin proteins CD63, CD81, CD9 and CD53 [161]. The nature of these vesicular structures is uncertain, some authors claiming that they belong to the system of late endosomes/multivesicular bodies (LE/MVB), others that they represent deep invaginations of the plasma membrane (reviewed in [107], see also the accompanying review by Benaroch P. et al.). Virions that have accumulated in these vesicles can be released into the extracellular fluid either directly or after fusion with the plasma membrane, depending on the hypothesis invoked. Urokinase-type plasminogen activator (uPA) signaling has been shown to inhibit a post-translational step of HIV-1 replication in MDM by promoting the sequestration of HIV-1 particles in intracellular vacuoles, possibly related to MVB, which affects the maturation and release of HIV-1 from infected cells [162, 163]. uPA is a serine protease that interacts with a specific GPI-anchored receptor, uPAR (CD87), at the cell surface [164]. uPAR is expressed by inflammatory cells including T cells, Mos and Mφ, and regulates cellular functions such as adhesion, proliferation and activation. The interest in the uPA/uPAR system in AIDS has risen from the observation that in a cohort of HIV-1 infected patients, the serum level of the suPAR soluble receptor was closely correlated to the mortality rate before anti-retroviral treatment and was an independent predictor of survival [165]. uPAR expression is up-regulated in vivo and in vitro by HIV-1 infection [166, 167]. In 2001 an HIV-1 suppressor factor was identified from the culture supernatants of an immortalized CD8+ T cell clone [163]. This factor corresponded to the amino-terminal fragment (ATF) of uPA. Urokinase can be found as two enzymatically active isoforms, a high molecular weight form (HMW-uPA) and a low molecular weight form (LMW-uPA) that lacks 135 amino acids of the N-terminus tail of the HMW-uPA. The 135 amino acid peptide, which is naturally cleaved during the processing of HMW-uPA, corresponds to ATF and is catalytically inactive. Late steps of viral replication, such as budding or viral particle assembly, are affected by ATF [163, 168]. Similarly, uPA inhibits HIV-1 replication in primary MDM, lymphocytes and monocytic cell lines. The uncleaved inactive precursor of uPA, pro-uPA, which interacts with the same membrane receptor, also inhibits the replication of HIV-1 in MDM, activated PBMCs and ex vivo cultures of lymphoid tissue that have been infected in vitro [162, 168]. The uPA-uPAR interaction interferes with late events of HIV-1 replication in MDM and U937 pro-monocytic cells, as well as in PMA- and TNFα-differentiated U1 cells, inhibiting the release of virions from cells [162, 163, 168]. The association of the receptor with other signaling competent receptors was required for this inhibitory activity. In particular, the engagement of β1 and β2 integrins, as well as Mac-1 integrin bound to fibrinogen, was identified as mediator of the uPA antiviral effect [168]. Interestingly, cross-linking of Mac-1 also inhibited viral replication. Signaling from uPA/uPAR interaction and assembly of Mac-1 are thus able to interfere with virion assembly and release in Mφ independently of uPAR [168]. The interaction of uPAR and integrins may occur at the level of lipid rafts of the plasma membrane that have been previously described to be a limiting factor for viral entry and budding (see above) suggesting an additional role of these structures in the accumulation/release of viral particles from infected cells.
Regulation of HIV-1 infection in tissue macrophages
Resident Mφ in tissues are heterogeneous in terms of phenotype, morphology and function [169]. Their characteristics probably depend on the specific tissue microenvironment, as well as on the conditions of inflammation during infections. Accordingly, Mφ from different tissues, such as lung, brain, gastrointestinal and genital tracts, while comprising many HIV reservoirs, display different susceptibilities to HIV infection (Fig. 2B).
Two reports concerning the use of cervical and vaginal explants and purified cell populations from vaginal mucosa provided evidence that subepithelial Mφ are susceptible to infection with monocytotropic R5 HIV-1 strains, and suggested that these cells may represent the main target for HIV infection in the female genital tract [8, 170]. In contrast, jejunum intestinal Mφ did not support viral replication [8]. The basis for this differential permissiveness to HIV-1 infection was related to differences in the expression of the CCR5 co-receptor. Mφ from the vaginal mucosa display a similar phenotypic profile to that of blood Mos, and express CD4 and CCR5, whereas Mφ from the jejunum intestinal mucosa express a distinct phenotype, with very low levels of CD4 and virtually no CCR5 [8, 171, 172]. Therefore the latter cells could resist HIV-1 infection by restricting viral entry due to a lack of the CCR5 co-receptor or to an inappropriate CCR5/CD4 stochiometry [28, 173] (Fig. 2B). Susceptibility of Mφ in the intestinal mucosa to HIV-1 infection may however vary depending on their localization at different sites of the intestinal tract, for example in the jejunum versus in the rectum, as well as according to the level of local inflammation. Indeed, HIV-1 and SIV infected CD68+ Mφ are found in the colon mucosa of HIV-infected patients or SIV-infected macaques respectively [174, 175].
The main cells infected by HIV-1 in the CNS are perivascular Mφ and resident microglial cells, and in the lung are AM [19, 156, 176–182] (Fig. 2B). However, although HIV-1 entry into the CNS occurs during acute infection [183, 184], viral RNA is almost undetectable during the asymptomatic phase of infection. Few AM in bronchoalveolar lavages (BAL) from HIV-1 infected patients harbor viral DNA, and low genetic variability in viral sequences argues against active viral replication [185]. However, in spite of low or undetectable HIV-1 RNA levels in AM in infected patients, viral replication could be reactivated by stimulation of AM from BAL in vitro with granulocyte/macrophage colony-stimulating factor (GM-CSF) and TNF-α, or with M. tuberculosis and its purified protein derivative [186, 187]. More importantly, reactivation of latent HIV-1 replication in AM occurs during co-infections, including those with opportunistic pathogens such as M. avium and Pneumocystis carinii [156]. Longitudinal studies showed that SIV infection is established in brain and lungs of infected macaques already in acute infection, but HIV-1 replication is then rapidly controlled, and viral RNA becomes undetectable [188, 189]. These data suggest that HIV/SIV infection of Mφ in brain and lung is mostly latent before the onset of symptomatic disease, due to the suppression of viral replication. A unifying hypothesis that accounts for the suppression of viral replication in brain and lung has been proposed by Clements et al. Acute HIV/SIV infection induces the production of IFN-β in brain and lung tissues. IFN-β in turn is proposed to induce alternative translation of C/EBPβ mRNA, switching the balance between activating and inhibitory C/EBPβ isoforms in favor of the inhibitory form that suppresses LTR-driven transcription of viral genes [[189, 190]. Several other studies support a role for C/EBPβ regulation in HIV-1 expression in the CNS and in neuropathogenesis [149]. In the lung, pulmonary tuberculosis markedly up-regulates HIV-1 replication in AM by repressing the expression of inhibitory C/EBPβ [153]. Lung infiltration of T lymphocytes leads to the loss of inhibitory C/EBPβ mediated by the interaction of cell surface co-stimulatory molecules and ligands during T cell/macrophage contact [154]. Meanwhile, production of pro-inflammatory cytokines by activated T lymphocytes boosts infection by triggering NF-κB activation [154]. It is conceivable that a similar scenario may also occur in brain and other tissues of HIV-infected individuals. Indeed, although HIV-1 infects the CNS during acute primary infection, active viral replication is observed after the loss of immune control, and this coincides with increased immune activation (reviewed in [191]). Reactivation of viral replication from latently infected microglia may occur concomitantly with trafficking of activated Mo/Mφ into the CNS [192]. Interestingly, enteropathy in chronically SIV infected macaques has also been associated with increased expression of the activating isoform of C/EBPβ localized predominantly in Mφ in the jejunum and colon mucosa [175]. Thus, increased expression of activating C/EBPβ may contribute to the maintenance of inflammation and the activation of viral replication in Mφ of different organs. However, other mechanisms of transcriptional silencing of the HIV-1 LTR that have been described in microglial cells may contribute to HIV-1 latency in the CNS [160].
The deregulation of uPA and its receptor uPAR has also been implicated in the loss of control of HIV-1 replication in the CNS [193]. uPA signaling through uPAR inhibits late steps of HIV-1 replication [162, 168] (see above). High levels of expression of uPAR were detected in HIV-infected microglial cells and reactive Mφ in the brain of patients with encephalitis and other HIV-related neurological lesions, whereas uPA was detected in few cells [193–195]. In addition, higher levels of the soluble form of uPAR (suPAR), uPA and suPAR/uPA complexes were found in the cerebrospinal fluid (CSF) in HIV-infected patients than in HIV-negative controls, and in patients with ADC or opportunistic CNS infections than in neurologically asymptomatic patients. suPAR levels correlated with CSF HIV-1 RNA [193]. It has thus been proposed that HIV-1 infection induces over-expression of uPAR and consequently overproduction of suPAR. The excess suPAR in the CSF would bind most of the extracellular uPA, preventing its binding to cell surface uPAR and its signaling-induced inhibition of HIV-replication [193].
Conclusions
The list of putative mechanisms of control of HIV-1 infection in Mo/Mφ is rapidly growing (Table 1). HIV-1 replication is restricted at different steps. More research is, of course, needed to gain further insights into the molecular mechanisms underlying each restriction event. However, importantly, evidence for the relevance of some of these mechanisms in vivo is now coming from studies concerning HIV-1 infected individuals and from the SIV/macaque model, as reviewed above for the IFNβ/C/EBPβ or uPA/uPAR pathways. The contribution of other mechanisms of HIV-1 restriction that have been identified in vitro to the control of Mo/Mφ infection in vivo is still uncertain and requires further studies. For example, whether and how the balance of the inhibitory and enhancing effects of p21 influences the replication of HIV-1 in tissue Mφ remains an open question. The answer to this question may be directly relevant to therapy: modulation of p21 expression is currently studied in anticancer therapy. The role of the restriction factors of the TRIM or the APOBEC3 family in HIV-1 infection of Mo/Mφ is unclear. In particular, APOBEC3A and 3G are strongly induced by IFNs in Mφ [40, 196], which is in favor of their antiviral role. Whether and by which mechanisms these molecules contribute to the control of HIV-1 infection in the cells remains to be elucidated [52, 196].
New high throughput screening techniques are being used to discover host molecules relevant to HIV-1 replication. Microarray analyses have revealed alterations of gene expression in Mo/Mφ that are associated to HIV-1 infection (reviewed in [197]), but they have not allowed the identification of new molecules involved in the control of HIV-1 replication. Genome-wide screenings, including small interfering RNA (siRNA) screening have provided a huge amount of information [198–201]. However, while they have a high potential to identify host cofactors required for HIV-1 replication and to drive the search for host targets for HIV therapeutics, such approaches may be less suited to unveil factors that inhibit viral replication [201]. For example APOBEC3G was not detected by RNA interference screens [198]. In addition, some factors may act differentially depending on the cell type (for example, the restriction overcome by Vpx in Mos/Mφ and DC). Only studies focused on the relevant cell type will be able to detect these factors.
Last but not least, a question about the interplay between host restriction mechanisms and viral infection is: what is really the ultimate effect of the host factors that hinder HIV-1 replication in host cells and are thought to exert an antiviral activity beneficial to the host. It may be that an effective mechanism of restriction that blocks viral replication in a cell may be subverted by the virus at the organism level, in the complex interplay between the virus and the host. For example, while inhibitory C/EBPβ is an effective anti-viral lock that suppresses HIV-1 transcription, it also allows the virus to remain latent in the brain and lung until the host immune response is declining, and the infection is then unlocked when immune activation boosts viral replication. This could be a conserved strategy of persistence in lentiviral infections: a post-transcriptional block also restricts MVV replication in macrophages in the CNS and lung macrophages unless inflammatory lesions promote viral expression [202, 203]. Therapeutic strategies based on restriction mechanisms may therefore be aimed either at enhancing some restriction mechanisms to limit infection or to thwart other events that would otherwise reactivate viral replication and drain viral reservoirs.
References
Whitelaw DM: Observations on human monocyte kinetics after pulse labeling. Cell Tissue Kinet. 1972, 5: 311-7.
Crowe S, Zhu T, Muller WA: The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J Leukoc Biol. 2003, 74: 635-41. 10.1189/jlb.0503204.
Clay CC, Rodrigues DS, Ho YS, Fallert BA, Janatpour K, Reinhart TA, Esser U: Neuroinvasion of fluorescein-positive monocytes in acute simian immunodeficiency virus infection. J Virol. 2007, 81: 12040-8. 10.1128/JVI.00133-07.
Hasegawa A, Liu H, Ling B, Borda JT, Alvarez X, Sugimoto C, Vinet-Oliphant H, Kim WK, Williams KC, Ribeiro RM, Lackner AA, Veazey RS, Kuroda MJ: The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood. 2009, 114 (14): 2917-25. 10.1182/blood-2009-02-204263.
Fischer-Smith T, Tedaldi EM, Rappaport J: CD163/CD16 coexpression by circulating monocytes/macrophages in HIV: potential biomarkers for HIV infection and AIDS progression. AIDS Res Hum Retroviruses. 2008, 24: 417-21. 10.1089/aid.2007.0193.
Lewin SR, Kirihara J, Sonza S, Irving L, Mills J, Crowe SM: HIV-1 DNA and mRNA concentrations are similar in peripheral blood monocytes and alveolar macrophages in HIV-1-infected individuals. Aids. 1998, 12: 719-27. 10.1097/00002030-199807000-00008.
Crowe SM, Sonza S: HIV-1 can be recovered from a variety of cells including peripheral blood monocytes of patients receiving highly active antiretroviral therapy: a further obstacle to eradication. J Leukoc Biol. 2000, 68: 345-50.
Shen R, Richter HE, Clements RH, Novak L, Huff K, Bimczok D, Sankaran-Walters S, Dandekar S, Clapham PR, Smythies LE, Smith PD: Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J Virol. 2009, 83: 3258-67. 10.1128/JVI.01796-08.
Chang J, Naif HM, Li S, Sullivan JS, Randle CM, Cunningham AL: Twin studies demonstrate a host cell genetic effect on productive human immunodeficiency virus infection of human monocytes and macrophages in vitro. J Virol. 1996, 70: 7792-803.
Eisert V, Kreutz M, Becker K, Königs C, Alex U, Rübsamen-Waigmann H, Andreesen R, von Briesen H: Analysis of cellular factors influencing the replication of human immunodeficiency virus type I in human macrophages derived from blood of different healthy donors. Virology. 2001, 286: 31-44. 10.1006/viro.2001.0940.
Bol SM, van Remmerden Y, Sietzema JG, Kootstra NA, Schuitemaker H, van't Wout AB: Donor variation in in vitro HIV-1 susceptibility of monocyte-derived macrophages. Virology. 2009, 390: 205-11. 10.1016/j.virol.2009.05.027.
Naif HM, Li S, Alali M, Chang J, Mayne C, Sullivan J, Cunningham AL: Definition of the stage of host cell genetic restriction of replication of human immunodeficiency virus type 1 in monocytes and monocyte-derived macrophages by using twins. J Virol. 1999, 73: 4866-81.
Bleiber G, May M, Martinez R, Meylan P, Ott J, Beckmann JS, Telenti A: Use of a combined ex vivo/in vivo population approach for screening of human genes involved in the human immunodeficiency virus type 1 life cycle for variants influencing disease progression. J Virol. 2005, 79: 12674-80. 10.1128/JVI.79.20.12674-12680.2005.
Ometto L, Zanchetta M, Cabrelle A, Esposito G, Mainardi M, Chieco-Bianchi L, De Rossi A: Restriction of HIV type 1 infection in macrophages heterozygous for a deletion in the CC-chemokine receptor 5 gene. AIDS Res Hum Retroviruses. 1999, 15: 1441-52. 10.1089/088922299309955.
Connor R, Paxton WA, Sheridan KE, Koup RA: Macrophages and CD4+ T lymphocytes from Two multiply exposed uninfected individuals resist infection with primary non-syncitium-inducind isolates of human immunodeficiency virus type 1. J Virol. 1996, 70: 8758-8764.
Kedzierska K, Crowe SM, Turville S, Cunningham AL: The influence of cytokines, chemokines and their receptors on HIV-1 replication in monocytes and macrophages. Rev Med Virol. 2003, 13: 39-56. 10.1002/rmv.369.
Schmidtmayerova H, Ancuta P, Pancino G: Monocyte and macrophage susceptibility to HIV infection: pathophysiological implications. HIV and the Macrophage. Edited by: Herbein G. 2007, Kerala, India: Transworld Research Network, 1-34.
M Alfano, A Crotti, E Vicenzi, G Poli: New players in cytokine control of HIV infection. Curr HIV/AIDS Rep. 2008, 5: 27-32. 10.1007/s11904-008-0005-5.
Rich EA, Chen IS, Zack JA, Leonard ML, O'Brien WA: Increased susceptibility of differentiated mononuclear phagocytes to productive infection with human immunodeficiency virus-1 (HIV-1). J Clin Invest. 1992, 89: 176-83. 10.1172/JCI115559.
Coleman CM, Wu L: HIV interactions with monocytes and dendritic cells: viral latency and reservoirs. Retrovirology. 2009, 6: 51-10.1186/1742-4690-6-51.
Sonza S, Maerz A, Uren S, Violo A, Hunter S, Boyle W, Crowe S: Susceptibility of human monocytes to HIV type 1 infection in vitro is not dependent on their level of CD4 expression. AIDS Res Hum Retroviruses. 1995, 11: 769-76. 10.1089/aid.1995.11.769.
Kazazi F, Mathijs JM, Foley P, Cunningham AL: Variations in CD4 expression by human monocytes and macrophages and their relationships to infection with the human immunodeficiency virus. J Gen Virol. 1989, 70: 2661-72. 10.1099/0022-1317-70-10-2661.
Valentin A, Von Gegerfelt A, Matsuda S, Nilsson K, Asjo B: In vitro maturation of mononuclear phagocytes and susceptibility to HIV-1 infection. J Acquir Immune Defic Syndr. 1991, 4: 751-9.
Arfi V, Riviere L, Jarrosson-Wuilleme L, Goujon C, Rigal D, Darlix JL, Cimarelli A: Characterization of the early steps of infection of primary blood monocytes by human immunodeficiency virus type 1. J Virol. 2008, 82: 6557-65. 10.1128/JVI.02321-07.
Dong C, Kwas C, Wu L: Transcriptional restriction of human immunodeficiency virus type 1 gene expression in undifferentiated primary monocytes. J Virol. 2009, 83: 3518-27. 10.1128/JVI.02665-08.
Sonza S, Maerz A, Deacon N, Meanger J, Mills J, Crowe S: Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J Virol. 1996, 70: 3863-9.
Triques K, Stevenson M: Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J Virol. 2004, 78: 5523-7. 10.1128/JVI.78.10.5523-5527.2004.
Naif HM, Li S, Alali M, Sloane A, Wu L, Kelly M, Lynch G, Lloyd A, Cunningham AL: CCR5 expression correlates with susceptibility of maturing monocytes to human immunodeficiency virus type 1 infection. J Virol. 1998, 72: 830-6.
Neil S, Martin F, Ikeda Y, Collins M: Postentry restriction to human immunodeficiency virus-based vector transduction in human monocytes. J Virol. 2001, 75: 5448-56. 10.1128/JVI.75.12.5448-5456.2001.
Vazquez N, Greenwell-Wild T, Marinos NJ, Swaim WD, Nares S, Ott DE, Schubert U, Henklein P, Orenstein JM, Sporn MB, Wahl SM: Human immunodeficiency virus type 1-induced macrophage gene expression includes the p21 gene, a target for viral regulation. J Virol. 2005, 79: 4479-91. 10.1128/JVI.79.7.4479-4491.2005.
Schuitemaker H, Kootstra NA, Koppelman MH, Bruisten SM, Huisman HG, Tersmette M, Miedema F: Proliferation-dependent HIV-1 infection of monocytes occurs during differentiation into macrophages. J Clin Invest. 1992, 89: 1154-60. 10.1172/JCI115697.
Narayan O, Kennedy-Stoskopf S, Sheffer D, Griffin DE, Clements JE: Activation of caprine arthritis-encephalitis virus expression during maturation of monocytes to macrophages. Infect Immun. 1983, 41: 67-73.
Gendelman HE, Narayan O, Kennedy-Stoskopf S, Kennedy PG, Ghotbi Z, Clements JE, Stanley J, Pezeshkpour G: Tropism of sheep lentiviruses for monocytes: susceptibility to infection and virus gene expression increase during maturation of monocytes to macrophages. J Virol. 1986, 58: 67-74.
Haase AT, Stowring L, Narayan P, Griffin D, Price D: Slow persistent infection caused by visna virus: role of host restriction. Science. 1977, 195: 175-7. 10.1126/science.188133.
JClements JE, Zink MC, Narayan O, Gabuzda DH: Lentivirus infection of macrophages. Immunol Ser. 1994, 60: 589-600.
Tuttle DL, Harrison JK, Anders C, Sleasman JW, Goodenow MM: Expression of CCR5 increases during monocyte differentiation and directly mediates macrophage susceptibility to infection by human immunodeficiency virus type 1. J Virol. 1998, 72: 4962-9.
Terai C, Carson DA: Pyrimidine nucleotide and nucleic acid synthesis in human monocytes and macrophages. Exp Cell Res. 1991, 193: 375-81. 10.1016/0014-4827(91)90110-G.
O'Brien WA, Namazi A, Kalhor H, Mao SH, Zack JA, Chen IS: Kinetics of human immunodeficiency virus type 1 reverse transcription in blood mononuclear phagocytes are slowed by limitations of nucleotide precursors. J Virol. 1994, 68: 1258-63.
Diamond TL, Roshal M, Jamburuthugoda VK, Reynolds HM, Merriam AR, Lee KY, Balakrishnan M, Bambara RA, Planelles V, Dewhurst S, Kim B: Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J Biol Chem. 2004, 279: 51545-53. 10.1074/jbc.M408573200.
Peng G, Greenwell-Wild T, Nares S, Jin W, Lei KJ, Rangel ZG, Munson PJ, Wahl SM: Myeloid differentiation and susceptibility to HIV-1 are linked to APOBEC3 expression. Blood. 2007, 110: 393-400. 10.1182/blood-2006-10-051763.
Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D: Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003, 424: 99-103. 10.1038/nature01709.
Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt In, Neuberger MS, Malim MH: DNA deamination mediates innate immunity to retroviral infection. Cell. 2003, 113: 803-9. 10.1016/S0092-8674(03)00423-9.
Sheehy AM, Gaddis NC, Choi JD, Malim MH: Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002, 418: 646-50. 10.1038/nature00939.
Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L: The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003, 424: 94-8. 10.1038/nature01707.
Goila-Gaur R, Strebel K: HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008, 5: 51-10.1186/1742-4690-5-51.
Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM: Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005, 15: 166-70. 10.1016/j.cub.2004.12.068.
Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH: APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4: e1000231-10.1371/journal.ppat.1000231.
Henriet S, Mercenne G, Bernacchi S, Paillart JC, Marquet R: Tumultuous relationship between the human immunodeficiency virus type 1 viral infectivity factor (Vif) and the human APOBEC-3G and APOBEC-3F restriction factors. Microbiol Mol Biol Rev. 2009, 73: 211-32. 10.1128/MMBR.00040-08.
Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC: Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005, 435: 108-14. 10.1038/nature03493.
Pion M, Granelli-Piperno A, Mangeat B, Stalder R, Correa R, Steinman RM, Piguet V: APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med. 2006, 203: 2887-93. 10.1084/jem.20061519.
Kamata M, Nagaoka Y, Chen IS: Reassessing the role of APOBEC3G in human immunodeficiency virus type 1 infection of quiescent CD4+ T-cells. PLoS Pathog. 2009, 5: e1000342-10.1371/journal.ppat.1000342.
Santoni de Sio FR, Trono D: APOBEC3G-depleted resting CD4+ T cells remain refractory to HIV1 infection. PLoS One. 2009, 4: e6571-10.1371/journal.pone.0006571.
Kaushik R, Zhu X, Stranska R, Wu Y, Stevenson M: A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe. 2009, 6: 68-80. 10.1016/j.chom.2009.05.022.
Goujon C, Riviere L, Jarrosson-Wuilleme L, Bernaud J, Rigal D, Darlix JL, Cimarelli A: SIVSM/HIV-2 Vpx proteins promote retroviral escape from a proteasome-dependent restriction pathway present in human dendritic cells. Retrovirology. 2007, 4: 2-10.1186/1742-4690-4-2.
Kilareski EM, Shah S, Nonnemacher MR, Wigdahl B: Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology. 2009, 6: 118-10.1186/1742-4690-6-118.
Yu W, Wang Y, Shaw CA, Qin XF, Rice AP: Induction of the HIV-1 Tat co-factor cyclin T1 during monocyte differentiation is required for the regulated expression of a large portion of cellular mRNAs. Retrovirology. 2006, 3: 32-10.1186/1742-4690-3-32.
Liou LY, Herrmann CH, Rice AP: Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J Virol. 2002, 76: 10579-87. 10.1128/JVI.76.21.10579-10587.2002.
Liou LY, Herrmann CH, Rice AP: Human immunodeficiency virus type 1 infection induces cyclin T1 expression in macrophages. J Virol. 2004, 78: 8114-9. 10.1128/JVI.78.15.8114-8119.2004.
Sung TL, Rice AP: miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 2009, 5: e1000263-10.1371/journal.ppat.1000263.
Hariharan M, Scaria V, Pillai B, Brahmachari SK: Targets for human encoded microRNAs in HIV genes. Biochem Biophys Res Commun. 2005, 337: 1214-8. 10.1016/j.bbrc.2005.09.183.
Huang J, Wang F, Argyris E, Chen K, Liang Z, Tian H, Huang W, Squires K, Verlinghieri G, Zhang H: Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat Med. 2007, 13: 1241-7. 10.1038/nm1639.
Wang X, Ye L, Hou W, Zhou Y, Wang YJ, Metzger DS, Ho WZ: Cellular microRNA expression correlates with susceptibility of monocytes/macrophages to HIV-1 infection. Blood. 2009, 113: 671-4. 10.1182/blood-2008-09-175000.
Swaminathan S, Zaunders J, Wilkinson J, Suzuki K, Kelleher AD: Does the presence of anti-HIV miRNAs in monocytes explain their resistance to HIV-1 infection?. Blood. 2009, 113: 5029-30. 10.1182/blood-2009-01-196741. author reply 5030-1
Sonza S, Kiernan RE, Maerz AL, Deacon NJ, McPhee DA, Crowe SM: Accumulation of unintegrated circular viral DNA in monocytes and growth-arrested T cells following infection with HIV-1. J Leukoc Biol. 1994, 56: 289-93.
McElrath MJ, Steinman RM, Cohn ZA: Latent HIV-1 infection in enriched populations of blood monocytes and T cells from seropositive patients. J Clin Invest. 1991, 87: 27-30. 10.1172/JCI114981.
Mikovits JA, Lohrey NC, Schulof R, Courtless J, Ruscetti FW: Activation of infectious virus from latent human immunodeficiency virus infection of monocytes in vivo. J Clin Invest. 1992, 90: 1486-91. 10.1172/JCI116016.
Lambotte O, Taoufik Y, de Goer MG, Wallon C, Goujard C, Delfraissy JF: Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. Journal of Acquired Immune Deficiency Syndromes. 2000, 23: 114-119.
Sonza S, Mutimer HP, Oelrichs R, Jardine D, Harvey K, Dunne A, Purcell DF, Birch C, Crowe SM: Monocytes harbour replication-competent, non-latent HIV-1 in patients on highly active antiretroviral therapy. Aids. 2001, 15: 17-22. 10.1097/00002030-200101050-00005.
Sharkey M, Triques K, Kuritzkes DR, Stevenson M: In vivo evidence for instability of episomal human immunodeficiency virus type 1 cDNA. J Virol. 2005, 79: 5203-10. 10.1128/JVI.79.8.5203-5210.2005.
Zhu T, Muthui D, Holte S, Nickle D, Feng F, Brodie S, Hwangbo Y, Mullins JI, Corey L: Evidence for human immunodeficiency virus type 1 replication in vivo in CD14(+) monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J Virol. 2002, 76: 707-16. 10.1128/JVI.76.2.707-716.2002.
Llewellyn N, Zioni R, Zhu H, Andrus T, Xu Y, Corey L, Zhu T: Continued evolution of HIV-1 circulating in blood monocytes with antiretroviral therapy: genetic analysis of HIV-1 in monocytes and CD4+ T cells of patients with discontinued therapy. J Leukoc Biol. 2006, 80: 1118-26. 10.1189/jlb.0306144.
Delobel P, Sandres-Saune K, Cazabat M, L'Faqihi FE, Aquilina C, Obadia M, Pasquier C, Marchou B, Massip P, Izopet J: Persistence of distinct HIV-1 populations in blood monocytes and naive and memory CD4 T cells during prolonged suppressive HAART. Aids. 2005, 19: 1739-50. 10.1097/01.aids.0000183125.93958.26.
Xu Y, Zhu H, Wilcox CK, van't Wout A, Andrus T, Llewellyn N, Stamatatos L, Mullins JI, Corey L, Zhu T: Blood monocytes harbor HIV type 1 strains with diversified phenotypes including macrophage-specific CCR5 virus. J Infect Dis. 2008, 197: 309-18. 10.1086/524847.
Alexaki A, Wigdahl B: HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog. 2008, 4: e1000215-10.1371/journal.ppat.1000215.
Sharova N, Swingler C, Sharkey M, Stevenson M: Macrophages archive HIV-1 virions for dissemination in trans. Embo J. 2005, 24: 2481-9. 10.1038/sj.emboj.7600707.
Peluso R, Haase A, Stowring L, Edwards M, Ventura P: A Trojan Horse mechanism for the spread of visna virus in monocytes. Virology. 1985, 147: 231-6. 10.1016/0042-6822(85)90246-6.
Narayan O, Wolinsky JS, Clements JE, Strandberg JD, Griffin DE, Cork LC: Slow virus replication: the role of macrophages in the persistence and expression of visna viruses of sheep and goats. J Gen Virol. 1982, 59: 345-56. 10.1099/0022-1317-59-2-345.
Ancuta P, Kamat A, Kunstman KJ, Kim EY, Autissier P, Wurcel A, Zaman T, Stone D, Mefford M, Morgello S, Singer EJ, Wolinsky SM, Gabuzda D: Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One. 2008, 3: e2516-10.1371/journal.pone.0002516.
Porcheray F, Samah B, Leone C, Dereuddre-Bosquet N, Gras G: Macrophage activation and human immunodeficiency virus infection: HIV replication directs macrophages towards a pro-inflammatory phenotype while previous activation modulates macrophage susceptibility to infection and viral production. Virology. 2006, 349: 112-20. 10.1016/j.virol.2006.02.031.
Maslin CL, Kedzierska K, Webster NL, Muller WA, Crowe SM: Transendothelial migration of monocytes: the underlying molecular mechanisms and consequences of HIV-1 infection. Curr HIV Res. 2005, 3: 303-17. 10.2174/157016205774370401.
Varol C, Yona S, Jung S: Origins and tissue-context-dependent fates of blood monocytes. Immunol Cell Biol. 2009, 87: 30-8. 10.1038/icb.2008.90.
Thieblemont N, Weiss L, Sadeghi HM, Estcourt C, Haeffner-Cavaillon N: CD14lowCD16high: a cytokine-producing monocyte subset which expands during human immunodeficiency virus infection. Eur J Immunol. 1995, 25: 3418-24. 10.1002/eji.1830251232.
Pulliam L, Sun B, Rempel H: Invasive chronic inflammatory monocyte phenotype in subjects with high HIV-1 viral load. J Neuroimmunol. 2004, 157: 93-8. 10.1016/j.jneuroim.2004.08.039.
Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D: Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003, 197: 1701-7. 10.1084/jem.20022156.
Ancuta P, Wang J, Gabuzda D: CD16+ monocytes produce IL-6, CCL2, and matrix metalloproteinase-9 upon interaction with CX3CL1-expressing endothelial cells. J Leukoc Biol. 2006, 80: 1156-64. 10.1189/jlb.0206125.
Belge KU, Dayyani F, Horelt A, Siedlar M, Frankenberger M, Frankenberger B, Espevik T, Ziegler-Heitbrock L: The proinflammatory CD14+CD16+DR++ monocytes are a major source of TNF. J Immunol. 2002, 168: 3536-42.
Ziegler-Heitbrock HW, Fingerle G, Strobel M, Schraut W, Stelter F, Schutt C, Passlick B, Pforte A: The novel subset of CD14+/CD16+ blood monocytes exhibits features of tissue macrophages. Eur J Immunol. 1993, 23: 2053-8. 10.1002/eji.1830230902.
Fischer-Smith T, Croul S, Sverstiuk AE, Capini C, L'Heureux D, Regulier EG, Richardson MW, Amini S, Morgello S, Khalili K, Rappaport J: CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J Neurovirol. 2001, 7: 528-41. 10.1080/135502801753248114.
Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, Gabuzda D: Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16-monocyte subsets. BMC Genomics. 2009, 10: 403-10.1186/1471-2164-10-403.
Ellery PJ, Tippett E, Chiu YL, Paukovics G, Cameron PU, Solomon A, Lewin SR, Gorry PR, Jaworowski A, Greene WC, Sonza S, Crowe SM: The CD16+ monocyte subset is more permissive to infection and preferentially harbors HIV-1 in vivo. J Immunol. 2007, 178: 6581-9.
Jaworowski A, Kamwendo DD, Ellery P, Sonza S, Mwapasa V, Tadesse E, Molyneux ME, Rogerson SJ, Meshnick SR, Crowe SM: CD16+ monocyte subset preferentially harbors HIV-1 and is expanded in pregnant Malawian women with Plasmodium falciparum malaria and HIV-1 infection. J Infect Dis. 2007, 196: 38-42. 10.1086/518443.
Kootstra NA, Schuitemaker H: Proliferation-dependent replication in primary macrophages of macrophage-tropic HIV type 1 variants. AIDS Res Hum Retroviruses. 1998, 14: 339-45. 10.1089/aid.1998.14.339.
Schmidtmayerova H, Nuovo GJ, Bukrinsky M: Cell proliferation is not required for productive HIV-1 infection of macrophages. Virology. 1997, 232: 379-84. 10.1006/viro.1997.8584.
Collin M, Gordon S: The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology. 1994, 200: 114-20. 10.1006/viro.1994.1169.
Paxton WA, Martin SR, Tse D, O'Brien TR, Skurnick J, VanDevanter NL, Padian N, Braun JF, Kotler DP, Wolinsky SM, Koup RA: Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nat Med. 1996, 2: 412-7. 10.1038/nm0496-412.
Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996, 86: 367-77. 10.1016/S0092-8674(00)80110-5.
Franchin G, Zybarth G, Dai WW, Dubrovsky L, Reiling N, Schmidtmayerova H, Bukrinsky M, Sherry B: Lipopolysaccharide inhibits HIV-1 infection of monocyte-derived macrophages through direct and sustained down-regulation of CC chemokine receptor 5. J Immunol. 2000, 164: 2592-601.
Herbein G, Coaquette A, Perez-Bercoff D, Pancino G: Macrophage activation and HIV infection: can the Trojan horse turn into a fortress?. Curr Mol Med. 2002, 2: 723-38. 10.2174/1566524023361844.
Smith PD, Meng G, Shaw GM, Li L: Infection of gastrointestinal tract macrophages by HIV-1. J Leukoc Biol. 1997, 62: 72-7.
Schneider B, Schueller C, Utermoehlen O, Haas A: Lipid microdomain-dependent macropinocytosis determines compartmentation of Afipia felis. Traffic. 2007, 8: 226-40. 10.1111/j.1600-0854.2006.00525.x.
Watarai M, Makino S, Fujii Y, Okamoto K, Shirahata T: Modulation of Brucella-induced macropinocytosis by lipid rafts mediates intracellular replication. Cell Microbiol. 2002, 4: 341-55. 10.1046/j.1462-5822.2002.00195.x.
Chung CS, Huang CY, Chang W: Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J Virol. 2005, 79: 1623-34. 10.1128/JVI.79.3.1623-1634.2005.
Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH: Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J Virol. 2003, 77: 9542-52. 10.1128/JVI.77.17.9542-9552.2003.
Wilflingseder D, Stoiber H: Float on: lipid rafts in the lifecycle of HIV. Front Biosci. 2007, 12: 2124-35. 10.2741/2216.
Kozak SL, Heard JM, Kabat D: Segregation of CD4 and CXCR4 into distinct lipid microdomains in T lymphocytes suggests a mechanism for membrane destabilization by human immunodeficiency virus. J Virol. 2002, 76: 1802-15. 10.1128/JVI.76.4.1802-1815.2002.
Popik W, Alce TM, Au WC: Human immunodeficiency virus type 1 uses lipid raft-colocalized CD4 and chemokine receptors for productive entry into CD4(+) T cells. J Virol. 2002, 76: 4709-22. 10.1128/JVI.76.10.4709-4722.2002.
Carter GC, Bernstone L, Sangani D, Bee JW, Harder T, James W: HIV entry in macrophages is dependent on intact lipid rafts. Virology. 2009, 386: 192-202. 10.1016/j.virol.2008.12.031.
Jungi TW, Hafner S: Quantitative assessment of Fc receptor expression and function during in vitro differentiation of human monocytes to macrophages. Immunology. 1986, 58: 131-7.
Perez-Bercoff D, David A, Sudry H, Barre-Sinoussi F, Pancino G: Fcgamma receptor-mediated suppression of human immunodeficiency virus type 1 replication in primary human macrophages. J Virol. 2003, 77: 4081-94. 10.1128/JVI.77.7.4081-4094.2003.
David A, Saez-Cirion A, Versmisse P, Malbec O, Iannascoli B, Herschke F, Lucas M, Barre-Sinoussi F, Mouscadet JF, Daeron M, Pancino G: The engagement of activating FcgammaRs inhibits primate lentivirus replication in human macrophages. J Immunol. 2006, 177: 6291-300.
Bergamaschi A, David A, Rouzic EL, Nisole S, Barré-Sinoussi F, Pancino G: The CDK inhibitor p21Cip1/WAF1 is induced by FcγR activation and restricts the replication of HIV-1 and related primate lentiviruses in human macrophages. J Virol. 2009, 83 (23): 12253-12265. 10.1128/JVI.01395-09.
Steinman RA, Johnson DE: p21WAF1 prevents down-modulation of the apoptotic inhibitor protein c-IAP1 and inhibits leukemic apoptosis. Mol Med. 2000, 6: 736-49.
Coqueret O: New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment?. Trends Cell Biol. 2003, 13: 65-70. 10.1016/S0962-8924(02)00043-0.
Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S: Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. Embo J. 1999, 18: 1223-34. 10.1093/emboj/18.5.1223.
Cui J, Tungaturthi PK, Ayyavoo V, Ghafouri M, Ariga H, Khalili K, Srinivasan A, Amini S, Sawaya BE: The role of Vpr in the regulation of HIV-1 gene expression. Cell Cycle. 2006, 5: 2626-38.
Clark E, Santiago F, Deng L, Chong S, de La Fuente C, Wang L, Fu P, Stein D, Denny T, Lanka V, Mozafari F, Okamoto T, Kashanchi F: Loss of G(1)/S checkpoint in human immunodeficiency virus type 1-infected cells is associated with a lack of cyclin-dependent kinase inhibitor p21/Waf1. J Virol. 2000, 74: 5040-52. 10.1128/JVI.74.11.5040-5052.2000.
Wu W, Kehn-Hall K, Pedati C, Zweier L, Castro I, Klase Z, Dowd CS, Dubrovsky L, Bukrinsky M, Kashanchi F: Drug 9AA reactivates p21/Waf1 and Inhibits HIV-1 progeny formation. Virol J. 2008, 5: 41-10.1186/1743-422X-5-41.
Zhang J, Scadden DT, Crumpacker CS: Primitive hematopoietic cells resist HIV-1 infection via p21. J Clin Invest. 2007, 117: 473-81. 10.1172/JCI28971.
Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J: The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature. 2004, 427: 848-53. 10.1038/nature02343.
Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W: TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog. 2008, 4: e16-10.1371/journal.ppat.0040016.
Bouazzaoui A, Kreutz M, Eisert V, Dinauer N, Heinzelmann A, Hallenberger S, Strayle J, Walker R, Rubsamen-Waigmann H, Andreesen R, von Briesen H: Stimulated trans-acting factor of 50 kDa (Staf50) inhibits HIV-1 replication in human monocyte-derived macrophages. Virology. 2006, 356: 79-94. 10.1016/j.virol.2006.07.025.
Barr SD, Smiley JR, Bushman FD: The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008, 4: e1000007-10.1371/journal.ppat.1000007.
Tissot C, Mechti N: Molecular cloning of a new interferon-induced factor that represses human immunodeficiency virus type 1 long terminal repeat expression. J Biol Chem. 1995, 270: 14891-8. 10.1074/jbc.270.25.14891.
Gack MU, Shin YC, Joo CH, Urano T, Liang C, Sun L, Takeuchi O, Akira S, Chen Z, Inoue S, Jung JU: TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature. 2007, 446: 916-920. 10.1038/nature05732.
Nisole S, Stoye JP, Saib A: TRIM family proteins: retroviral restriction and antiviral defence. Nat Rev Microbiol. 2005, 3: 799-808. 10.1038/nrmicro1248.
Carthagena L, Bergamaschi A, Luna JM, David A, Uchil PD, Margottin-Goguet F, Mothes W, Hazan U, Transy C, Pancino G, Nisole S: Human TRIM gene expression in response to interferons. PLoS One. 2009, 4: e4894-10.1371/journal.pone.0004894.
Christ F, Thys W, De Rijck J, Gijsbers R, Albanese A, Arosio D, Emiliani S, Rain JC, Benarous R, Cereseto A, Debyser Z: Transportin-SR2 imports HIV into the nucleus. Curr Biol. 2008, 18: 1192-202. 10.1016/j.cub.2008.07.079.
Suzuki Y, Craigie R: The road to chromatin - nuclear entry of retroviruses. Nat Rev Microbiol. 2007, 5: 187-96. 10.1038/nrmicro1579.
Connor RI, Chen BK, Choe S, Landau NR: Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology. 1995, 206: 935-44. 10.1006/viro.1995.1016.
von Schwedler U, Kornbluth RS, Trono D: The nuclear localization signal of the matrix protein of human immunodeficiency virus type 1 allows the establishment of infection in macrophages and quiescent T lymphocytes. Proc Natl Acad Sci USA. 1994, 91: 6992-6. 10.1073/pnas.91.15.6992.
Gallay P, Hope T, Chin D, Trono D: HIV-1 infection of nondividing cells through the recognition of integrase by the importin/karyopherin pathway. Proc Natl Acad Sci USA. 1997, 94: 9825-30. 10.1073/pnas.94.18.9825.
Zaitseva L, Cherepanov P, Leyens L, Wilson SJ, Rasaiyaah J, Fassati A: HIV-1 exploits importin 7 to maximize nuclear import of its DNA genome. Retrovirology. 2009, 6: 11-10.1186/1742-4690-6-11.
Yamashita M, Emerman M: The cell cycle independence of HIV infections is not determined by known karyophilic viral elements. PLoS Pathog. 2005, 1: e18-10.1371/journal.ppat.0010018.
Fujita M, Otsuka M, Miyoshi M, Khamsri B, Nomaguchi M, Adachi A: Vpx is critical for reverse transcription of the human immunodeficiency virus type 2 genome in macrophages. J Virol. 2008, 82: 7752-6. 10.1128/JVI.01003-07.
Sharova N, Wu Y, Zhu X, Stranska R, Kaushik R, Sharkey M, Stevenson M: Primate lentiviral Vpx commandeers DDB1 to counteract a macrophage restriction. PLoS Pathog. 2008, 4: e1000057-10.1371/journal.ppat.1000057.
Srivastava S, Swanson SK, Manel N, Florens L, Washburn MP, Skowronski J: Lentiviral Vpx accessory factor targets VprBP/DCAF1 substrate adaptor for cullin 4 E3 ubiquitin ligase to enable macrophage infection. PLoS Pathog. 2008, 4: e1000059-10.1371/journal.ppat.1000059.
Bergamaschi A, Ayinde D, David A, Le Rouzic E, Morel M, Collin G, Descamps D, Damond F, Brun-Vezinet F, Nisole S, Margottin-Goguet F, Pancino G, Transy C: The human immunodeficiency virus type 2 Vpx protein usurps the CUL4A-DDB1 DCAF1 ubiquitin ligase to overcome a postentry block in macrophage infection. J Virol. 2009, 83: 4854-60. 10.1128/JVI.00187-09.
Delelis O, Carayon K, Saib A, Deprez E, Mouscadet JF: Integrase and integration: biochemical activities of HIV-1 integrase. Retrovirology. 2008, 5: 114-10.1186/1742-4690-5-114.
Jacque JM, Stevenson M: The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature. 2006, 441: 641-5. 10.1038/nature04682.
Shun MC, Daigle JE, Vandegraaff N, Engelman A: Wild-type levels of human immunodeficiency virus type 1 infectivity in the absence of cellular emerin protein. J Virol. 2007, 81: 166-72. 10.1128/JVI.01953-06.
Mulky A, Cohen TV, Kozlov SV, Korbei B, Foisner R, Stewart CL, KewalRamani VN: The LEM domain proteins emerin and LAP2alpha are dispensable for human immunodeficiency virus type 1 and murine leukemia virus infections. J Virol. 2008, 82: 5860-8. 10.1128/JVI.00076-08.
Pomerantz RJ, Feinberg MB, Trono D, Baltimore D: Lipopolysaccharide is a potent monocyte/macrophage-specific stimulator of human immunodeficiency virus type 1 expression. J Exp Med. 1990, 172: 253-61. 10.1084/jem.172.1.253.
Lodie TA, Reiner M, Coniglio S, Viglianti G, Fenton MJ: Both PU.1 and nuclear factor-kappa B mediate lipopolysaccharide-induced HIV-1 long terminal repeat transcription in macrophages. J Immunol. 1998, 161: 268-76.
Bernstein MS, Tong-Starksen SE, Locksley RM: Activation of human monocyte--derived macrophages with lipopolysaccharide decreases human immunodeficiency virus replication in vitro at the level of gene expression. J Clin Invest. 1991, 88: 540-5. 10.1172/JCI115337.
Weiden M, Tanaka N, Qiao Y, Zhao BY, Honda Y, Nakata K, Canova A, Levy DE, Rom WN, Pine R: Differentiation of monocytes to macrophages switches the Mycobacterium tuberculosis effect on HIV-1 replication from stimulation to inhibition: modulation of interferon response and CCAAT/enhancer binding protein beta expression. J Immunol. 2000, 165: 2028-39.
Simard S, Maurais E, Gilbert C, Tremblay MJ: LPS reduces HIV-1 replication in primary human macrophages partly through an endogenous production of type I interferons. Clin Immunol. 2008, 127: 198-205. 10.1016/j.clim.2008.01.007.
Scott LM, Civin CI, Rorth P, Friedman AD: A novel temporal expression pattern of three C/EBP family members in differentiating myelomonocytic cells. Blood. 1992, 80: 1725-35.
Henderson AJ, Calame KL: CCAAT/enhancer binding protein (C/EBP) sites are required for HIV-1 replication in primary macrophages but not CD4(+) T cells. Proc Natl Acad Sci USA. 1997, 94: 8714-9. 10.1073/pnas.94.16.8714.
Hogan TH, Krebs FC, Wigdahl B: Regulation of human immunodeficiency virus type 1 gene expression and pathogenesis by CCAAT/enhancer binding proteins in cells of the monocyte/macrophage lineage. J Neurovirol. 2002, 8 (Suppl 2): 21-6. 10.1080/13550280290167911.
Tesmer VM, Rajadhyaksha A, Babin J, Bina M: NF-IL6-mediated transcriptional activation of the long terminal repeat of the human immunodeficiency virus type 1. Proc Natl Acad Sci USA. 1993, 90: 7298-302. 10.1073/pnas.90.15.7298.
Descombes P, Schibler U: A liver-enriched transcriptional activator protein, LAP, and a transcriptional inhibitory protein, LIP, are translated from the same mRNA. Cell. 1991, 67: 569-79. 10.1016/0092-8674(91)90531-3.
Ossipow V, Descombes P, Schibler U: CCAAT/enhancer-binding protein mRNA is translated into multiple proteins with different transcription activation potentials. Proc Natl Acad Sci USA. 1993, 90: 8219-23. 10.1073/pnas.90.17.8219.
Honda Y, Rogers L, Nakata K, Zhao BY, Pine R, Nakai Y, Kurosu K, Rom WN, Weiden M: Type I interferon induces inhibitory 16-kD CCAAT/enhancer binding protein (C/EBP)beta, repressing the HIV-1 long terminal repeat in macrophages: pulmonary tuberculosis alters C/EBP expression, enhancing HIV-1 replication. J Exp Med. 1998, 188: 1255-65. 10.1084/jem.188.7.1255.
Hoshino Y, Nakata K, Hoshino S, Honda Y, Tse DB, Shioda T, Rom WN, Weiden M: Maximal HIV-1 replication in alveolar macrophages during tuberculosis requires both lymphocyte contact and cytokines. J Exp Med. 2002, 195: 495-505. 10.1084/jem.20011614.
Tanaka N, Hoshino Y, Gold J, Hoshino S, Martiniuk F, Kurata T, Pine R, Levy D, Rom WN, Weiden M: Interleukin-10 induces inhibitory C/EBPbeta through STAT-3 and represses HIV-1 transcription in macrophages. Am J Respir Cell Mol Biol. 2005, 33: 406-11. 10.1165/rcmb.2005-0140OC.
Orenstein JM, Fox C, Wahl SM: Macrophages as a source of HIV during opportunistic infections. Science. 1997, 276: 1857-61. 10.1126/science.276.5320.1857.
Komuro I, Sunazuka T, Akagawa KS, Yokota Y, Iwamoto A, Omura S: Erythromycin derivatives inhibit HIV-1 replication in macrophages through modulation of MAPK activity to induce small isoforms of C/EBPbeta. Proc Natl Acad Sci USA. 2008, 105: 12509-14. 10.1073/pnas.0805504105.
Dudaronek JM, Barber SA, Clements JE: CUGBP1 is required for IFNbeta-mediated induction of dominant-negative CEBPbeta and suppression of SIV replication in macrophages. J Immunol. 2007, 179: 7262-9.
Marban C, Redel L, Suzanne S, Van Lint C, Lecestre D, Chasserot-Golaz S, Leid M, Aunis D, Schaeffer E, Rohr O: COUP-TF interacting protein 2 represses the initial phase of HIV-1 gene transcription in human microglial cells. Nucleic Acids Res. 2005, 33: 2318-31. 10.1093/nar/gki529.
Marban C, Suzanne S, Dequiedt F, de Walque S, Redel L, Van Lint C, Aunis D, Rohr O: Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. Embo J. 2007, 26: 412-23. 10.1038/sj.emboj.7601516.
Deneka M, Pelchen-Matthews A, Byland R, Ruiz-Mateos E, Marsh M: In macrophages, HIV-1 assembles into an intracellular plasma membrane domain containing the tetraspanins CD81, CD9, and CD53. J Cell Biol. 2007, 177: 329-41. 10.1083/jcb.200609050.
Alfano M, Sidenius N, Panzeri B, Blasi F, Poli G: Urokinase-urokinase receptor interaction mediates an inhibitory signal for HIV-1 replication. Proc Natl Acad Sci USA. 2002, 99: 8862-7. 10.1073/pnas.142078099.
Wada M, Wada NA, Shirono H, Taniguchi K, Tsuchie H, Koga J: Amino-terminal fragment of urokinase-type plasminogen activator inhibits HIV-1 replication. Biochem Biophys Res Commun. 2001, 284: 346-51. 10.1006/bbrc.2001.4965.
Blasi F, Vassalli JD, Dano K: Urokinase-type plasminogen activator: proenzyme, receptor, and inhibitors. J Cell Biol. 1987, 104: 801-4. 10.1083/jcb.104.4.801.
Sidenius N, Sier CF, Ullum H, Pedersen BK, Lepri AC, Blasi F, Eugen-Olsen J: Serum level of soluble urokinase-type plasminogen activator receptor is a strong and independent predictor of survival in human immunodeficiency virus infection. Blood. 2000, 96: 4091-5.
Speth C, Pichler I, Stockl G, Mair M, Dierich MP: Urokinase plasminogen activator receptor (uPAR; CD87) expression on monocytic cells and T cells is modulated by HIV-1 infection. Immunobiology. 1998, 199: 152-62.
Alfano M, Sidenius N, Blasi F, Poli G: The role of urokinase-type plasminogen activator (uPA)/uPA receptor in HIV-1 infection. J Leukoc Biol. 2003, 74: 750-6. 10.1189/jlb.0403176.
Alfano M, Mariani SA, Elia C, Pardi R, Blasi F, Poli G: Ligand-engaged urokinase-type plasminogen activator receptor and activation of the CD11b/CD18 integrin inhibit late events of HIV expression in monocytic cells. Blood. 2009, 113: 1699-709. 10.1182/blood-2008-02-138412.
Gordon S, Taylor PR: Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005, 5: 953-64. 10.1038/nri1733.
Greenhead P, Hayes P, Watts PS, Laing KG, Griffin GE, Shattock RJ: Parameters of human immunodeficiency virus infection of human cervical tissue and inhibition by vaginal virucides. J Virol. 2000, 74: 5577-86. 10.1128/JVI.74.12.5577-5586.2000.
Li L, Meng G, Graham MF, Shaw GM, Smith PD: Intestinal macrophages display reduced permissiveness to human immunodeficiency virus 1 and decreased surface CCR5. Gastroenterology. 1999, 116: 1043-53. 10.1016/S0016-5085(99)70007-7.
Meng G, Sellers MT, Mosteller-Barnum M, Rogers TS, Shaw GM, Smith PD: Lamina propria lymphocytes, not macrophages, express CCR5 and CXCR4 and are the likely target cell for human immunodeficiency virus type 1 in the intestinal mucosa. J Infect Dis. 2000, 182: 785-91. 10.1086/315790.
Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, Kabat D: Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol. 1998, 72: 2855-64.
Lewin-Smith M, Wahl SM, Orenstein JM: Human immunodeficiency virus-rich multinucleated giant cells in the colon: a case report with transmission electron microscopy, immunohistochemistry, and in situ hybridization. Mod Pathol. 1999, 12: 75-81.
Mohan M, Aye PP, Borda JT, Alvarez X, Lackner AA: CCAAT/enhancer binding protein beta is a major mediator of inflammation and viral replication in the gastrointestinal tract of simian immunodeficiency virus-infected rhesus macaques. Am J Pathol. 2008, 173: 106-18. 10.2353/ajpath.2008.080108.
Wiley CA, Schrier RD, Nelson JA, Lampert PW, Oldstone MB: Cellular localization of human immunodeficiency virus infection within the brains of acquired immune deficiency syndrome patients. Proc Natl Acad Sci USA. 1986, 83: 7089-93. 10.1073/pnas.83.18.7089.
Fischer-Smith T, Croul S, Adeniyi A, Rybicka K, Morgello S, Khalili K, Rappaport J: Macrophage/microglial accumulation and proliferating cell nuclear antigen expression in the central nervous system in human immunodeficiency virus encephalopathy. Am J Pathol. 2004, 164: 2089-99.
Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA: Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med. 2001, 193: 905-15. 10.1084/jem.193.8.905.
Cosenza MA, Zhao ML, Si Q, Lee SC: Human brain parenchymal microglia express CD14 and CD45 and are productively infected by HIV-1 in HIV-1 encephalitis. Brain Pathol. 2002, 12: 442-55.
Salahuddin SZ, Rose RM, Groopman JE, Markham PD, Gallo RC: Human T lymphotropic virus type III infection of human alveolar macrophages. Blood. 1986, 68: 281-4.
Plata F, Autran B, Martins LP, Wain-Hobson S, Raphael M, Mayaud C, Denis M, Guillon JM, Debre P: AIDS virus-specific cytotoxic T lymphocytes in lung disorders. Nature. 1987, 328: 348-51. 10.1038/328348a0.
Meltzer MS, Kornbluth RS, Hansen B, Dhawan S, Gendelman HE: HIV infection of the lung. Role of virus-infected macrophages in the pathophysiology of pulmonary disease. Chest. 1993, 103: 103S-108S.
Davis LE, Hjelle BL, Miller VE, Palmer DL, Llewellyn AL, Merlin TL, Young SA, Mills RG, Wachsman W, Wiley CA: Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology. 1992, 42: 1736-9.
An SF, Groves M, Gray F, Scaravilli F: Early entry and widespread cellular involvement of HIV-1 DNA in brains of HIV-1 positive asymptomatic individuals. J Neuropathol Exp Neurol. 1999, 58: 1156-62. 10.1097/00005072-199911000-00005.
Nakata K, Weiden M, Harkin T, Ho D, Rom WN: Low copy number and limited variability of proviral DNA in alveolar macrophages from HIV-1-infected patients: evidence for genetic differences in HIV-1 between lung and blood macrophage populations. Mol Med. 1995, 1: 744-57.
Lebargy F, Branellec A, Deforges L, Bignon J, Bernaudin JF: HIV-1 in human alveolar macrophages from infected patients is latent in vivo but replicates after in vitro stimulation. Am J Respir Cell Mol Biol. 1994, 10: 72-8.
Toossi Z, Nicolacakis K, Xia L, Ferrari NA, Rich EA: Activation of latent HIV-1 by Mycobacterium tuberculosis and its purified protein derivative in alveolar macrophages from HIV-infected individuals in vitro. J Acquir Immune Defic Syndr Hum Retrovirol. 1997, 15: 325-31.
JClements JE, Babas T, Mankowski JL, Suryanarayana K, Piatak M, Tarwater PM, Lifson JD, Zink MC: The central nervous system as a reservoir for simian immunodeficiency virus (SIV): steady-state levels of SIV DNA in brain from acute through asymptomatic infection. J Infect Dis. 2002, 186: 905-13. 10.1086/343768.
Barber SA, Gama L, Li M, Voelker T, Anderson JE, Zink MC, Tarwater PM, Carruth LM, Clement JEs: Longitudinal analysis of simian immunodeficiency virus (SIV) replication in the lungs: compartmentalized regulation of SIV. J Infect Dis. 2006, 194: 931-8. 10.1086/507429.
Barber SA, Gama L, Dudaronek JM, Voelker T, Tarwater PM, Clements JE: Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-macaque model. J Infect Dis. 2006, 193: 963-70. 10.1086/500983.
Gonzalez-Scarano F, Martin-Garcia J: The neuropathogenesis of AIDS. Nat Rev Immunol. 2005, 5: 69-81. 10.1038/nri1527.
Fischer-Smith T, Bell C, Croul S, Lewis M, Rappaport J: Monocyte/macrophage trafficking in acquired immunodeficiency syndrome encephalitis: lessons from human and nonhuman primate studies. J Neurovirol. 2008, 14: 318-26. 10.1080/13550280802132857.
Sidenius N, Nebuloni M, Sala S, Zerbi P, Price RW, Gisslen M, Hagberg L, Vago L, Lazzarin A, Blasi F, Cinque P: Expression of the urokinase plasminogen activator and its receptor in HIV-1-associated central nervous system disease. J Neuroimmunol. 2004, 157: 133-9. 10.1016/j.jneuroim.2004.08.038.
Cinque P, Nebuloni M, Santovito ML, Price RW, Gisslen M, Hagberg L, Bestetti A, Vago G, Lazzarin A, Blasi F, Sidenius N: The urokinase receptor is overexpressed in the AIDS dementia complex and other neurological manifestations. Ann Neurol. 2004, 55: 687-94. 10.1002/ana.20076.
Nebuloni M, Cinque P, Sidenius N, Ferri A, Lauri E, Omodeo-Zorini E, Zerbi P, Vago L: Expression of the urokinase plasminogen activator receptor (uPAR) and its ligand (uPA) in brain tissues of human immunodeficiency virus patients with opportunistic cerebral diseases. J Neurovirol. 2009, 15: 99-107. 10.1080/13550280802400692.
Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH: Defining APOBEC3 Expression Patterns in Human Tissues and Hematopoietic Cell Subsets. J Virol. 2009, 83: 9474-85. 10.1128/JVI.01089-09.
Giri M, Montaner L: HIV-1 mediated monocyte/macrophage gene modulation. HIV and the Macrophage. Edited by: H G. 2007, Kerala, India: Transworld Research Network, 115-140.
Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ: Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008, 319: 921-6. 10.1126/science.1152725.
Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, Seidel S, Opaluch AM, Caldwell JS, Weitzman MD, Kuhen KL, Bandyopadhyay S, Ideker T, Orth AP, Miraglia LJ, Bushman FD, Young JA, Chanda SK: Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008, 135: 49-60. 10.1016/j.cell.2008.07.032.
Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, Espeseth AS: Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008, 4: 495-504. 10.1016/j.chom.2008.10.004.
Bushman FD, Malani N, Fernandes J, D'Orso I, Cagney G, Diamond TL, Zhou H, Hazuda DJ, Espeseth AS, Konig R, Bandyopadhyay S, Ideker T, Goff SP, Krogan NJ, Frankel AD, Young JA, Chanda SK: Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathog. 2009, 5: e1000437-10.1371/journal.ppat.1000437.
Gendelman HE, Narayan O, Molineaux S, Clements JE, Ghotbi Z: Slow, persistent replication of lentiviruses: role of tissue macrophages and macrophage precursors in bone marrow. Proc Natl Acad Sci USA. 1985, 82: 7086-90. 10.1073/pnas.82.20.7086.
Brodie SJ, Pearson LD, Zink MC, Bickle HM, Anderson BC, Marcom KA, DeMartini JC: Ovine lentivirus expression and disease. Virus replication, but not entry, is restricted to macrophages of specific tissues. Am J Pathol. 1995, 146: 250-63.
Skolnik PR, Rabbi MF, Mathys JM, Greenberg AS: Stimulation of peroxisome proliferator-activated receptors alpha and gamma blocks HIV-1 replication and TNFalpha production in acutely infected primary blood cells, chronically infected U1 cells, and alveolar macrophages from HIV-infected subjects. J Acquir Immune Defic Syndr. 2002, 31: 1-10.
Hayes MM, Lane BR, King SR, Markovitz DM, Coffey MJ: Peroxisome proliferator-activated receptor gamma agonists inhibit HIV-1 replication in macrophages by transcriptional and post-transcriptional effects. J Biol Chem. 2002, 277: 16913-9. 10.1074/jbc.M200875200.
Acknowledgements
This review is issued from a reflection conducted by the Association for Macrophage and Infection Research (AMIR) on the role of the cells of the mononuclear phagocyte lineage in HIV infection. This work was supported by the "Agence Nationale de Recherche sur le Sida et les Hépatites Virales" (ANRS) and Sidaction. France. A.B. is supported by a Sidaction fellowship. We thank P. Ancuta, Université de Montréal, and INSERM Unit 743, Montréal, Canada for critical reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
AB and GP were responsible for drafting and revising the manuscript, as well as organizing the content and editing the manuscript. GP conceptualized the figures. AB created Figures 1 and 2 and their legends. Both authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bergamaschi, A., Pancino, G. Host hindrance to HIV-1 replication in monocytes and macrophages. Retrovirology 7, 31 (2010). https://doi.org/10.1186/1742-4690-7-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4690-7-31