
Abstract
Alzheimer’s disease (AD) is a chronic neurodegenerative disease, with the characteristics of neurofibrillary tangle (NFT) and senile plaque (SP) formation. Although great progresses have been made in clinical trials based on relevant hypotheses, these studies are also accompanied by the emergence of toxic and side effects, and it is an urgent task to explore the underlying mechanisms for the benefits to prevent and treat AD. Herein, based on animal experiments and a few clinical trials, neuroinflammation in AD is characterized by long-term activation of pro-inflammatory microglia and the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasomes. Damaged signals from the periphery and within the brain continuously activate microglia, thus resulting in a constant source of inflammatory responses. The long-term chronic inflammatory response also exacerbates endoplasmic reticulum oxidative stress in microglia, which triggers microglia-dependent immune responses, ultimately leading to the occurrence and deterioration of AD. In this review, we systematically summarized and sorted out that exercise ameliorates AD by directly and indirectly regulating immune response of the central nervous system and promoting hippocampal neurogenesis to provide a new direction for exploring the neuroinflammation activity in AD.
Similar content being viewed by others
Background
Alzheimer's disease (AD) is one of the most common neurodegenerative diseases. Nowadays, it is estimated to be approximately 50 million AD patients all over the world, and the number will reach up to 76.14 million in 2030 [1]. Although the understanding of AD concept and pathogenesis has advanced since the first case was reported in 1907 [2], there are no effective treatment strategies. According to the amyloid cascade hypothesis, AD is caused by the excessive accumulation of amyloid-β peptide (Aβ) generated from amyloid precursor protein (APP) in the brain, especially Aβ42 and its polymers. The decreased clearance rate of Aβ is also an essential factor for Aβ deposition [3]. In addition, Aβ also can induce the hyperphosphorylation of microtubule-associated protein Tau [4], which promotes the dissociation of tubulin and the aggregation of tubulin into bundles, thereby resulting in pairs of helical filaments and neurofibrillary tangles (NFTs), and eventually causing neuronal dysfunction and even death [5]. Tau protein deposition is also considered to be another critical pathological feature of AD. During exploring the broad neuropathology of the human brain across the lifespan, the experiments in many primates tell us that the deposition of Aβ may be an aging-related by-product. The abnormal increase in hyperphosphorylation of Tau protein is likely to be the initial cause of AD pathogenesis [6, 7]. Consequently, further exploration of the pathogenesis and treatments of AD is highly desired from other perspectives.
Since neuroinflammation hypothesis was proposed in 1992, neuroinflammation caused by disturbed inflammatory neuroimmune system has become the third core pathological feature of AD [8]. Several studies have demonstrated inflammatory responses accompanied by the activation of immune cells in the brains of early clinical AD patients and postmortem pathological tissues as well as animal models [9,10,11,12]. Many antibodies that target these tissues are determined in the cerebrospinal fluid (CSF) of AD patients, indicating that AD is likely to be an inflammatory disease accompanied by autoimmune activation [13]. Recently, the Food and Drug Administration (FDA) has approved sodium oligomannate, a seaweed extract, for the treatment of AD patients in China, which can suppress the neuroinflammatory response by inhibiting the accumulation of phenylalanine and isoleucine in the blood [14]. Moreover, the drug has successfully passed the phase III clinical trial [15]. Although epidemiological studies suggest the applications of non-steroid anti-inflammatory drugs (NSAIDs) to prevent AD, most clinical trials have been unsuccessful [16]. The major reasons may be correlated with the precision targets [17], blood-brain barrier [18], adverse reactions [19], and medication timing [20]. It is worth noting that more and more studies have confirmed that scientific and reasonable exercise can effectively regulate the neuroimmune system, which has been proven in cardiovascular disease, respiratory system, and other diseases [21, 22]. Relevant animal and clinical experiments have also demonstrated that exercise can alleviate the symptoms of neurodegenerative diseases and delay the pathological progression in various ways [23], indicating that there is a close connection between exercise and the immune system [24]. In recent years, exercise has gradually received extensive attention in preventing and treating AD. Therefore, we systematically summarized the relationships among exercise, neuroinflammation and AD, which will provide relevant theoretical references for the suppression of neuroinflammation to realize the prevention and treatment of AD upon exercise interventions.
A new perspective on AD-related hypotheses
AD is an age-dependent neurodegenerative disease accompanied by cognitive impairment, memory loss, and abnormal behavior [3]. From the cholinergic hypothesis proposed in 1976 [25], to the amyloid cascade hypothesis in 1991 [26], and even to the recent hypotheses with cellular aging [27] and dysfunctional immune regulation [28], a large number of scholars have contributed academic explanation of AD pathogenesis. Over the past three decades, many experimental studies have documented that Aβ is the culprit of neurodegeneration in AD [26, 29], thereby confirming the deposition of Aβ and the accumulation of hyperphosphorylated Tau protein in neurons as the major pathogenesis of AD to lead to impaired synaptic plasticity and cognitive dysfunction, as well as the occurrence of dementia. Due to the complexity of pathological process of AD, a single hypothesis could not fully clarify the specific pathogenesis of AD, which also provides the explanation for failed clinical trials of drug candidates [30]. Many developed drugs have not shown the positive therapeutic effects on AD patients and even can cause severe adverse reactions, such as meningitis [31] and cognitive impairment [32]. Recent animal and clinical studies have reported that Tau pathology can spread in the brain and cause cognitive impairment without the accumulation of Aβ, which may be due to the speeded clearance rate of Aβ in the brain upon anti-Aβ treatment for AD [33]. In addition, some non-amyloid treatment methods, such as circadian rhythm intervention [34], have gradually gained the attention by scholars. Recently, several new treatment proposals after summarizing a large number of clinical trial data have been put forward: including developing drugs to simultaneously or continuously target Aβ and Tau protein; non-biological targeted treatment strategies; or selecting mild cognitive impairment (MCI) patients or early AD patients as clinical trial subjects [35]. When APP is subjected to the cleavage by α-secretase and β-secretase, the β-carboxyl terminus of APP is released, and the Aβ48 and Aβ49 are produced through the endo-proteolytic(ε) cleavage by γ-secretase. Then, Aβ48 and Aβ49 can be γ-cleaved in the order of every three amino acid residues to generate shorter Aβ peptides, such as Aβ40 and Aβ37 [36]. The contents of these smaller Aβ peptides can be important indicators for evaluating γ-secretase activity. Among them, Aβ40 and Aβ42, mainly accumulated in nerve cells, are the most abundant and exhibit the substantial neurotoxicity [37]. Therefore, they are often used as biomarkers to predict mild cognitive impairment and AD. However, the Aβ37/Aβ42 ratio in CSF is more accurate in diagnosing AD because the change in Aβ37 level can better reflect the functional status of γ-secretase than Aβ40 [38]. Recently, based on the linear causal relationship in the amyloid cascade hypothesis, a new probabilistic model of A(Aβ)-T(Tau)-N(Neurodegeneration) has been proposed. Compared with the traditional hypothetical model, this model can be applied to different clinical types of AD [39]. However, recent animal or clinical studies have found that APP [40] or Aβ [41] in the brain does not always seem to be harmful. Only 30-40% of people with Aβ deposition in the brain will develop AD after the age of 70 years old, while approximately 50% of the populations never experience a cognitive decline for whole life [42, 43]. In recent years, when many scholars are conducting clinical trials of Aβ antibodies, they have found that many subjects have significantly reduced Aβ deposition in the brain. However, cognitive function has not been improved [44]. Similarly, neurodegeneration in the brain is persistent after APP is knocked out in animal models [45]. After analyzing brain imaging of 2,700 ordinary people, patients with mild cognitive impairment and AD patients at different periods, the appearance of AD symptoms may be caused by the reduction of soluble Aβ42 [41].
Interestingly, it is found that dense-core plaques are formed after activated microglia phagocytosed loosely organized Aβ plaques to relieve inflammation, thereby inducing the protective effects [46]. In addition, cognitive impairment is likely due to functional and structural impairment of neurons caused by hyperphosphorylation of Tau protein rather than Aβ [47], which reminds us that there may be a greater need to explore the difference with amyloid cascade hypothesis of AD. With the in-depth exploration of AD, new concepts and perspectives on the pathogenesis of AD continue to emerge, which gradually enriches the theoretical references of AD, and will be helpful for developing effective prevention and treatment strategies for AD [48].
Inflammation and AD
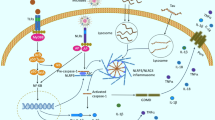
According to the epidemiology, AD is mainly divided into two categories: early-onset familial AD (EOAD) and late-onset AD (LOAD), but their pathophysiology is very similar. From a genetic point of view, the pathogenesis of EOAD is closely related to the mutation of Aβ-related genes APP, presenilin-1 and presenilin-2. However, most AD cases (> 95%) belong to LOAD, whose most vital genetic risk factor is apolipoprotein E ε4 (APOEε4) [5]. In APOEε4 heterozygotes, the risk of AD is approximately 400%, when compared with approximately 1500% in homozygotes [49]. Dementia patients typically experience four stages of health, prodromal AD, mild cognitive impairment, and AD [50]. Prodromal AD refers to the stage from the first neuropathological changes in the brain to the first symptoms of AD. Diagnosis in the preclinical stage requires the absence of clinical signs and symptoms of AD and the presence of at least one biomarker of AD, which often requires early biomarkers for diagnosis [51, 52]. Long-term clinical cohort studies have documented the importance of platelet Tau variants as early diagnosis or the prevention of neurodegenerative diseases including AD [53, 54], which is consistent with relevant results, indicating that phosphorylated Tau in plasma may be an essential biomarker in the early stage of AD [55, 56]. Mild cognitive impairment and AD are assessed according to the severity of cognitive impairment. According to the 2022 World Alzheimer's Disease Report, up to 40% of dementia are correlated with lifestyles, including work stress, growth environment, and living standards [57]. Changing these adverse factors can prevent and delay AD to a certain extent [50]. The most significant risk factor for LOAD is aging, and other factors include obesity, diabetes, and cardiovascular diseases. The common feature of these factors is systemic chronic low-level inflammatory response [58]. Multiple meta-analyses have documented the inflammatory responses to AD [59, 60], and more and more studies have validated that inflammatory diseases such as bacterial infection [61], oral infection [62], and imbalanced intestinal flora are closely related to AD [63]. With the extension of age, body function reveals the gradual decline, and the integrity and permeability of the blood-brain barrier (BBB) to protect the brain from peripheral immunity are destroyed. The damaged BBB allows peripheral hormones, bacterial metabolites, and immunoglobulins to enter the brain to activate microglia, thereby activating the central immune system [58, 64, 65]. The levels of CD45 lymphocytes, interleukin-17 (IL-17), interferon-G (IFN-G), and interleukin-6 (IL-6) in CSF and blood of patients with AD are significantly increased [66], suggesting that activated immune cells can also participate in the immune response by entering the brain from the periphery. The presence of many reactive hyperplasia of microglia and astrocytes around SPs imply that the immune system is an essential event in the pathogenesis of AD. Observational studies have found that elevated glial fibrillary acidic protein (GFAP) and triggering receptor expressed on myeloid cell 2 (TREM2) in blood and CSF can serve as biomarker events for the diagnosis of early AD [67, 68]. TREM2 levels in CSF reveal the reduction before AD and the increase in early AD, and the final decrease again in late AD [69]. Plasma GFAP can more accurately reflect the changes in Aβ burden (not Tau protein) and disease severity in pre-symptomatic AD when compared with GFAP and TREM2 in CSF [70,71,72]. At the same time, TREM2 levels in CSF are closely related to Tau protein [73]. In addition, a biomarker analysis of healthy elderly, MCI patients, and AD patients has found that MCI patients with low levels of TREM2 in CSF or high levels of plasma TREM2 are more likely to accelerate the progression of AD [71]. In AD and other neurodegenerative diseases, the function of TREM2-mediated activation of microglia depends on the stage of disease progression and the type of microglia [74]. After summarizing the recent studies, relevant scholars propose that neuroinflammation is more than an incidental phenomenon of AD pathology. Conversely, neuroinflammation may help to drive the pathogenesis of AD. Epidemiological studies have confirmed that long-term users of anti-inflammatory drugs are less likely to suffer from AD [75]. In addition, according to the neuroinflammation hypothesis, new treatments for the alleviation of AD using natural anti-inflammatory products such as curcumin are gaining attention [76,77,78]. Therefore, AD has regarded as one of the secondary neurological diseases of chronic inflammation [79] (Fig. 1).

The molecular regulation of neuroinflammation for AD. At the early stage of AD, the body presents a chronic low-inflammatory state induced by aging, hypertension, type II diabetes, obesity, infection, and other risk factors, which can release a variety of danger-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) to activate immune responses of the nervous system. Resting microglia can be converted into a pro-inflammatory M1 phenotype to clear these danger signals for returning to a resting state. With the progression of the disease, under the continuous stimulation of DAMPs dominated by hyperphosphorylated Tau protein, extensive endoplasmic reticulum stress, oxidative stress, and the formation of NOD-like receptor protein 3 (NLRP3) inflammatory cells after assembly by NLRP3 in activated microglia are triggered, which promotes the entry of nuclear factor kappa-B (NF-κB) into the nucleus, thus resulting in the up-regulation of inflammatory genes (such as IL-1β and IL-18) to restore neural homeostasis. Thus, the accumulation of mis-folded proteins, M1-phenotype microglia, and inflammatory factors contribute to the neuroinflammatory microenvironment. Neuroinflammation also promotes the diffusion of hyperphosphorylated Tau protein in the brain, thereby creating a positive feedback loop to drive AD
Pathogenesis of neuroinflammation in AD
Neuroinflammation has been first proposed as the essential inducer of AD. Long-term stimulation of "damaged signals" can activate the innate immune system and trigger a series of inflammatory cascades, which is closely related to neurodegeneration in AD [80,81,82]. Relevant studies have pointed out that the importance of Tau in AD neuroinflammation may be far greater than that of Aβ [81]. Furthermore, the neuroinflammation hypothesis also seems to hold for other neurodegenerative diseases with tauopathy [83]. The body is exposed to various stimuli in daily life, including exogenous PAMPs and in vivo DAMPs, such as bacterial endotoxins and mis-folded proteins. As a defense mechanism, neuroinflammation protects the brain mainly by clearing DAMPs or PAMPs.
PAMPs and DAMPs play a protective role by binding to pattern recognition receptors (PRRs) to initiate the activation of microglia and promote the release of mature pro-inflammatory cytokines from microglia to clear DAMPs and PAMPs. However, dysregulated DAMPs and PAMPs can pathologically exacerbate the levels of chronic inflammation with the extension of age [84]. When these stimuli are chronically present, microglia remain activated and become a long-term source of inflammatory factors [85]. However, long-term inflammatory stimulation may show a wrong side [86]. Inflammatory factors can also activate protein-related kinases, such as cyclin-dependent kinase 5, to promote the formation of NFTs from hyperphosphorylated Tau protein and further aggravate the inflammatory response in the brain [87,88,89]. Blocking the formation of inflammasomes in microglia can reduce neuroinflammation and delay the pathogenesis of AD [90]. On the one hand, communication between microglia and astrocytes plays a vital role in neuroinflammation [91]. As the most numerous glial cells in the brain, astrocytes regulate blood flow, maintain the blood-brain barrier, and maintain a stable environment for synapses and neurotransmitters [92]. The switch of both glial cells between pro-inflammatory and anti-inflammatory phenotypes promotes the transformation of each other. For example, IL-1α, TNF-α, and complement secreted by activated microglia can convert astrocytes to a pro-inflammatory phenotype [93]. Interestingly, neuroinflammation due to the activation of astrocytes is responsible for the degenerative development of tauopathies [94]. Recent animal and clinical studies have shown that a subset of astrocytes can influence microglial function by releasing IL-3, thereby allowing microglia to focus on clearing phosphorylated Tau and NFTs without destroying neurons [95]. In addition, the primary source of the protective IL-3 is the brain, and its expression is reduced in the brain of 5xFAD mice [95]. On the other hand, both high-risk factors for AD (aging and APOE4) and astrocyte damage disrupt the integrity of the blood-brain barrier, thus allowing the immune privilege in the brain to be disrupted [96]. Blood-borne DAMPs and PAMPs, such as complement, monocytes, and gut microbiota metabolites, enter the compromised blood-brain barrier to promote specific immune responses and exacerbate the burden on the innate system, thereby accelerating neurodegeneration [97]. In addition to the activation of peripheral T cells, the number of CD8+ T cells in the CSF of MCI and AD patients is significantly higher than that of normal subjects [98]. The experimental results of AD model animals have also confirmed that the neuroinflammatory events of AD include not only the activation of immune cells, but also the infiltration of activated peripheral immune cells in the brain [97]. Peripheral immune cells can penetrate brain tissue and bind to glial cells, thus reducing AD pathology and cognitive impairment in AD-transgenic mice [99, 100]. However, the role of peripherally derived immune cells remains controversial. In brain tissues of AD patients, the level of CD3+ T cells in peripheral blood vessels is related to Tau protein, rather than Aβ [101], suggesting that Tau-related neurodegenerative changes drive the intervention of peripheral immune substances. Thus, the interaction between microglia and astrocytes and the disruption of brain immune privilege plays essential roles in neuroinflammation (Fig. 2).

The pathogenesis and process of neuroinflammation in AD. After damaged signals (DAMPs and PAMPs) invade the brain, microglia become pro-inflammatory cells and secrete inflammatory factors to clear these signals. Subsequently, anti-inflammatory microglia emerge and secrete anti-inflammatory factors to counteract inflammatory factors. Damage signals in AD cannot be entirely cleared by microglia, so pro-inflammatory microglia keep secreting inflammatory factors to damage neurons. Inflammatory factors continuously intensify the phosphorylation of Tau protein and the formation of NFTs, which are the major substances of damaged signals in AD, finally forming a vicious circle
Neuroinflammation as an upstream core event in AD pathogenesis
The immune system of the brain and the levels of inflammatory biomarkers in blood largely contribute to the occurrence and development of AD due to hyperphosphorylated Tau protein accumulation, neuronal damage and cognitive impairment [102]. In a study with CSF examination and brain magnetic resonance images of 300 people over the age of 60 years old, it is found that even people without dementia symptoms exhibit a trend of significantly higher inflammatory biomarkers [103, 104]. When lipopolysaccharide (LPS) induces a chronic inflammatory state in mice, AD-related pathological processes in brain tissues of mice are significantly aggravated [105]. These experimental data in animal models and humans suggest that neuroinflammation is an upstream event in AD pathogenesis. Large-scale samples also show the increased C-reactive protein (CRP) in the blood of the people at middle age, which is similar with AD patients [106]. However, some clinical studies have also reported that lower plasma CRP levels are more prone to AD [107]. It is worth noting that CRP elevation is an acute inflammation as the feedback adaptation, indicating that chronic inflammation may be an essential factor for inducing AD. In addition, neuroinflammation can induce M1-phenotype microglia to release pro-inflammatory factors, whose complements modulate neuronal and synaptic damage, thereby resulting in the spread of Tau protein from the medulla or pons to the entire cortex [108]. In animal experiments, sustained expression of interleukin-1 beta (IL-1β) in the hippocampus can activate astrocytes and microglia to trigger a robust inflammatory response, ultimately leading to memory impairment [109]. For AD patients with long-term application of NSAIDs, symptomatic or asymptomatic progression of AD is significantly slowed down [78]. Therefore, neuroinflammation may be the upstream core event with the function of triggering AD.
Critical features of neuroinflammation in AD
Microglial activation has been recognized as a critical hallmark event in the pathogenesis of AD [110]. The activation process of microglia is closely related to the release of endocannabinoids. Microglia are the primary source of endocannabinoids under physiological conditions, but the expression of cannabinoid receptors in resting microglia is low [111]. Under normal conditions, microglia play a vital role in removing the aggregation of mis-folded proteins or forming a glial barrier to prevent mis-folded proteins from accumulation and spreading [112]. Activated microglia can also express cannabinoid receptors to activate endogenous cannabinoid signaling to regulate the polarization and proliferation of microglia, ultimately reducing the inflammatory response [113, 114]. According to the microglial dysfunction hypothesis, when the damage factors exist for a long time, the chronically activated microglia will suffer from malnutrition and apoptosis, so they cannot perform their average clearance and monitoring functions, eventually accelerating the formation of neuroinflammation and neuronal degeneration [115,116,117]. Using the tracking technology of microglia, positron emission tomography (PET) data show that microglial activation is observed in the elderly and patients with MCI. Moreover, AD patients show a sequential increase in activated microglia, and this phenomenon may be positively correlated with the level of phosphorylated Tau protein in the brain [108], which is also consistent with the results from RNA-seq analysis of brain tissue samples from AD patients [118]. Moreover, chronically activated microglia-mediated inflammatory events may be also related to the maturation of precursor interleukins induced by Aβ-activated NLRP3 in microglia [119]. The initiation signals of NLRP3 include several internal and external activators, such as PAMPs and DAMPs. Mis-folded proteins are the most typical DAMPs in AD. In APP/PS1, APP/PS1/NLRP3−/−, and APP/PS1/Caspase-1−/− model mice, reduced NLRP3 and Caspase-1 activity can promote the clearance of DAMPs and restore learning and memory capacity of the mice [90]. Interestingly, in APP/PS1/NLRP3−/− mice, M2 phenotype microglia are the majority, indicating that the NLRP3-Caspase-1 axis plays a vital role in the pathological process of AD, and this axis may be a potential therapeutic target through suppressing neuroinflammation. At the same time, the increased expression of Caspase-1 in brain tissues of MCI and AD patients is determined [90], implying that the NLRP3 inflammasome is at a chronic activation state during the pathogenesis of AD. Indeed, apoptosis-associated speck-like protein containing caspase recruitment domain (CARD) (ASC) specks in the inflammasomes are released into the intercellular space. Adjacent microglia can take up these specks, thus leading to the spreading of inflammatory factors in the brain and long-term activation of the immune system. However, neurodegeneration is ameliorated after applying the ASC speck antibody [120,121,122]. High expression of NLRP3 and Caspase-1 can suppress the function of microglia and then accelerate the pathological process of AD [123, 124]. Notably, the changes in inflammatory responses appear to be triggered by Tau pathology alone. Previous experiments have shown that the inflammatory response in the AD brain is pronounced in the presence of Tau pathology only, with the essential role in the involvement of microglia [125, 126]. Furthermore, in AD, Tau protein propagates itself into the brain via the NLRP3-ASC signal pathway, so the aggregation of Tau protein appears to be more inflammation than Aβ and is associated with the progression of AD symptoms [127], indicating that Tau protein is likely to be the dominant player in neuroinflammation. In in vitro studies, microglial activation may lead to pathological aggravation of Tau proteins again, which supports this positive feedback mechanism and explains the rapid progression of cortical NFTs in AD [128]. Consequently, reducing the expression of these inflammatory factors and the activation of NLRP3 in microglia are helpful for alleviating cognitive impairment of AD mice [129]. The inhibition of NF-κB and NLRP3/Caspase-1 signal pathways in microglia is a potential therapeutic strategy for AD [130]. Therefore, chronic activation of microglia and NLRP3 are the significant features of neuroinflammation in AD.
Regulatory mechanisms of neuroinflammation in AD
Long-term endoplasmic reticulum dysfunction is closely related to cognitive impairment and memory loss in AD [131]. In the AD brain, the continuous accumulation of Aβ and hyperphosphorylated Tau leads to the continuous increase of endoplasmic reticulum stress (ERS), which sequentially activates the unfolded protein response (URP) and NF-κB, through the PKR-like ER kinase (PERK)/JAK1/STAT3 and inositol-requiring enzyme 1 (IRE1)/thioredoxin-interacting protein (TXNIP) signal pathways, thereby causing aseptic inflammatory responses [132, 133]. The significant elevation of URP markers such as immunoglobulin heavy-chain-binding protein (BiP) is detected in the hippocampus of AD patients, especially in neurons of the CA1 and CA2 regions [134]. Moreover, ERS and hyperphosphorylation of Tau protein can mutually induce and promote each other, thereby exacerbating the pathogenesis of AD [135]. In addition to participating the process of relieving ERS, URP itself can activate inflammatory pathways in immune responses, including NF-κB, mitogen-activated protein kinase (MAPK) family protein c-Jun N-terminal kinase (JNK), and p38 [136]. It has been reported that ERS induced by Aβ and NFTs in the hippocampus of 5xFAD mice could also up-regulate TXNIP, although it does not affect the expression level of its negative regulator or redox regulator thioredoxin (TRX) [137]. ERS also can lead to increased expression and secretion of IL-1β in the hippocampus via the activation of TXNIP/NLPR3 signal pathway in the brain [138], which is consistent with the previous report that ERS can induce inflammation in diabetes to modulate islet β cell death [139]. The expression of cytokines and chemokines is enhanced by the activated PERK signaling, and conditional knockout of PERK can enhance synaptic plasticity and memory function in APP/PS1 transgenic mice [140, 141].
On the other hand, oxidative stress is a proximal event in AD pathogenesis prior to AD symptoms [142]. In SAMP8 mice with significant oxidative stress, the cognitive function of the mice can be alleviated or even reversed after the application of antioxidants, Aβ antibodies, and APP antibodies, respectively [143, 144]. Similarly, enhancing antioxidant capacity can suppress the expression of Aβ and APP and ultimately restore the impaired memory capacity of 3xTG transgenic mice. At the same time, the reduction of superoxidase dismutase (SOD) activity in the cytoplasm can lead to the formation of more Aβ oligomers [145, 146]. Oxidative damage can disrupt oxidative homeostasis, thus resulting in the production of reactive oxygen species (ROS) [147]. In the cerebral cortex of APP/PS1 transgenic mice at the age of 7, 12 and 20 months, the ratio of NAD+/NADH is decreased when compared with that of wild-type mice, and the inflammatory signal pathway in the hippocampal tissues is decreased upon the treatment with NAD+ supplementation nicotinamide riboside (NR), as shown in reducing neuroinflammation and alleviating cellular senescence through the cGAS-STING signal pathway [148]. Interestingly, TXNIP is not only implicated in ERS, but also acts as an endogenous inhibitor of the antioxidant TRX [149]. TRX is a major intracellular thiol-reducing and ROS-scavenging protein, and the binding of TXNIP to TRX can inhibit TRX activation and trigger oxidative stress. In AD, the antioxidant effect can reveal a decreasing trend with the decrease of nuclear factor-related factor 2 (Nrf2) level [150], as confirmed that Dl-3-n-butylphthalide (NBP) inhibits NLRP3 inflammasomes and delays the pathological process of AD through the Nrf2-TXNIP-TRX signal pathway [151, 152]. Therefore, ERS and oxidative stress, at the early stage of AD, can execute the protective mechanism and the suppression of neuroinflammation in AD.
Exercise and inflammation
Today, it is widely accepted that physical activity is essential for maintaining and promoting health. In 2020, the World Health Organization (WHO) proposed that all adults should have 150–300 min of moderate-intensity physical activity or 75–150 min of vigorous-intensity physical activity per week [153]. Exercise has a wide range of effects on the immune system. Exercise can increase the inflammatory state in the body by promoting the hypothalamus-pituitary-adrenal axis, improving cell survival environment and anti-apoptosis, optimizing the functional status of autophagy, and regulating endocrine. Exercise can also trigger an inflammatory response, thus releasing ROS and reactive nitrogen species (RNS) by damaging muscle, suppressing immune systems, activating inflammation, and depleting glycogen [23, 154]. According to the “open-window” hypothesis, a compromised immune system after strenuous exercise increases the risk of contracting an upper respiratory tract infection [155]. However, strenuous exercise also increases immune activity by redistributing immune cells to desired tissues, thereby reducing the chance of infection [156]. The effect of exercise on inflammation is related to the type, intensity, duration of exercise training, and individual or tissue differences [157]. For example, regular moderate-intensity physical activity can promote an anti-inflammatory state, but high-intensity physical activity or competition has been shown to activate the inflammatory response [158]. An acute exercise results in peak inflammation in muscle tissue within the first few hours [159], and this initial pro-inflammatory response is quickly counteracted by anti-inflammatory effects after regular exercise [160]. In addition, studies have shown that exercise intensity can be adjusted by assessing the level of neopterin, an endogenous immune activation marker, to determine the level of inflammation in the body during or after exercise [157]. In an animal model of LPS-induced inflammation, 3-week moderate-intensity treadmill exercise reduces neopterin levels and suppresses immune-inflammatory responses [161]. Regular moderate-intensity physical activity is thought to have immunomodulatory effects, enhancing defenses against infection and reducing the incidence of chronic diseases [155]. Therefore, the anti-inflammatory effects of exercise are more likely to be triggered by long-term moderate-intensity physical activity.
Increasing animal and clinical studies show that scientific and reasonable exercise can stimulate the body to produce an anti-inflammatory phenotype state [162]. In addition to enhancing memory and cognitive capacity, regular aerobic exercise can also reduce the levels of CRP, IL-6, tumor necrosis factor alpha (TNF-α), soluble tumor necrosis factor receptor-1 (sTNFR1), and soluble tumor necrosis factor receptor-2 (sTNFR2) and promote the production of anti-inflammatory factors such as interleukin 10 (IL-10), IL-1 receptor antagonist (IL-1RA), interleukin 4 (IL-4) and transforming growth factor beta-1 (TGF-β1) [163, 164]. For example, regular aerobic exercise of patients with metabolic syndromes can reduce IL-6 by 30%, TNF-α by 15% and leukocyte counts by 15% in blood [165]. Not only that, regular physical activity also can exert an anti-inflammatory effect on chronic and inflammatory diseases. Similarly, weekly moderate-intensity aerobic exercise can be helpful to reduce peripheral inflammation levels of the people with type 2 diabetes [166]. Regular physical activity can promote the tropism of neutrophils and natural killer (NK) cells to optimize their functional status [167]. Similarly, the elderly at the average age of 71 years old reveal a threefold decrease in the number of pro-inflammatory monocytes CD14 and CD16 in their blood after strength training of legs and chest [168]. In addition, regular exercise (aerobic and resistance exercise) can also reduce the secretion of pro-inflammatory cytokines in young people and induce skeletal muscle to release anti-inflammatory mediators such as IL-6 [169]. Exercise-induced increase of circulating IL-6 and increased plasma levels of anti-inflammatory factors, such as IL-1RA and IL-10. IL-1RA can inhibit IL-1β signaling, while IL-10 can inhibit the production of inflammatory factors, including TNF-α [170]. Notably, the expression of toll-like receptors on the monocyte membrane is decreased after an acute prolonged exercise, thereby affecting the secretion of pro-inflammatory factors [164]. In addition, prolonged exercise can also affect the number of different T cells including regulatory T cells for influencing the immune system [171]. Therefore, exercise may be one of the crucial ways to regulate immune and inflammatory responses.
Exercise and AD
Since current drugs for AD have not achieved good clinical efficacy, people have focused their attention on changing lifestyles for the prevention and treatment of AD. The 2022 report on AD lists high-risk factors for AD at all stages. For example, one of early AD risk factors includes poor education; late risk factors are smoking, physical inactivity, depression, social isolation, diabetes, and air pollution. In addition, 40% of AD patients can be prevented or delayed by modulating these controllable risk factors [172]. Several decades observational studies have confirmed this fact [173, 174]. Therefore, the study recommends the prevention of dementia based on the entire life cycle, such as regular physical activity in middle and old age [175]. It is well known that a sedentary lifestyle is associated with impaired cognitive function in AD populations, so one possible approach to ameliorating AD is regular physical activity [176]. A recent retrospective analysis including 160,000 participants has found that participants with regular and active exercise have a lower risk of developing AD by 45% [177], suggesting that physical activity has a more positive role in preventing AD, and similar results are also reported in other literature [178]. Comparable results are also achieved in a prospective study of 716 elderly subjects [179]. In addition, exercise positively affects high-risk factors for AD, including hypertension, type II diabetes, obesity, and hyperlipidemia [180]. On the other hand, regular physical activity has been reported to have multiple benefits in relieving AD symptoms in both human and animal experiments [181]. For example, physical activity has improved learning and memory capacity by increasing long-term potentiation (LTP) and neurogenesis [182], suggesting that physical activity may also be associated with structural and functional changes in the brain. The 5-month voluntary wheel running can reduce Aβ40 and Aβ42 levels in the brains of 3xTG transgenic mice [183]. Similarly, 3-month-old APP/PS1 transgenic mice reveal the significantly reduced Aβ accumulation in the brain after five months of treadmill running [184]. In addition to reducing the expression level of β-secretase in transgenic mice, exercise can also change the activity of γ-secretase [185] and promote the activity of α-secretase in a Sirt1-dependent manner [186]. Furthermore, exercise also can promote lactate secretion in skeletal muscle in a Sirt1-dependent manner to increase the level of brain-derived neurotrophic factor (BDNF) in the hippocampus for enhancing neuronal function and ultimately restoring the memory capacity of mice. At the same time, BDNF can reduce β-site amyloid precursor protein cleaving enzyme 1 (BACE1) activity [187, 188]. The capability of the brain to synthesize BDNF reveals an increase by approximately 2–3 folds during prolonged exercise [189]. Therefore, exercise may reduce the content of Aβ in AD model mice by regulating the activities of α-, β-, and γ-secretase. Accumulating evidence suggests that microglia play essential roles in many aspects including the regulation of Aβ, hyperphosphorylated Tau, neuronal function, and synaptic plasticity [190]. For example, microglia can promote the phagocytosis capacity of Aβ [191]. Astrocytes can promote the positional change of the aquaporin 4 (AQP4) to increase the clearance rate of Aβ. Similarly, a transient and prominent microglial activation state in the hippocampus of 3xTG transgenic mice after 3 weeks of voluntary wheel running is also observed [192]. Reducing the deposition of Aβ in the hippocampus can increase learning and memory capacity of mice. Therefore, the effects of exercise on preventing and delaying AD are multifaceted. After 6-month resistance exercise in 100 MCI patients aged 55–85 years old, their memory, attention, and executive skills are improved significantly during and 12 months after exercise [193]. In terms of different AD patients, MCI patients, or healthy people, exercise is effective against high-risk factors for AD. It also can enhance the resilience against AD, thereby improving cognitive reserve function and brain tissue plasticity [194]. However, some human experiments have shown that 16-week aerobic exercise does not reduce the content of Aβ in the brain of AD patients [195]. This result may suggest that the anti- and pro-inflammatory effects of exercise may depend on various factors such as exercise intensity and duration.
Exercise suppresses neuroinflammation for ameliorating AD
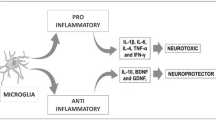
AD has been recognized as a neurodegenerative disease caused by a chronic inflammatory response [196]. The aging of organisms, the accumulation of progressive damage, and the loss of the reserve function of each organ may be related to the inflammatory response [197]. Long-term high-frequency physical exercise can alleviate the degeneration of the body system for delaying aging and improving physical fitness [198]. In studies on naturally aging animals and healthy people with different age, it is found that lifelong exercise can effectively alleviate the systemic inflammatory response in mice by inhibiting the levels of pro-inflammatory factors and increasing the levels of anti-inflammatory factors, respectively [199, 200]. A recent review proposes that the anti-inflammatory properties of exercise can suppress the inflammatory state of AD and ameliorate the pathophysiological characteristics of AD [201]. Numerous studies have shown the decreased immune responses and ameliorated cognitive impairment in elderly, and MCI and AD patients after a period of physical activity [202]. The studies on exercise-mediated neuroinflammation in AD models are listed in Table 1. Non-work-related exercise has a specific protective effect on AD. For example, exercise can improve the judgment and problem-solving capacity of AD patients and modulate serum inflammatory markers in AD patients [203]. Similarly, human and animal data show that exercise can increase TREM2 levels in CSF of AD patients, maintain plasma TREM2 levels in APP/PS1 mice, and reduce plasma GFAP levels in multiple sclerosis patients [204,205,206]. The plasma of long-term exerciser has been shown to suppress inflammatory responses in the hippocampus by inhibiting complement-related signal pathways and preventing neuroinflammation [207]. Although these causal mechanisms are still under debate, the anti-inflammatory effects of exercise are meaningful and feasible as a therapeutic strategy for aging-related neurodegenerative diseases. Therefore, the inhibition of inflammatory responses may be a potential target for ameliorating AD (Fig. 3).

Exercise modulates neuroinflammatory responses to ameliorate AD. First, exercise suppresses chronic inflammation in the body through reducing circulating levels of pro-inflammatory factors and immune cells. Second, exercise restores the permeability and integrity of the BBB by repairing damaged endothelial cells and tight junctions, ultimately preventing inflammatory factors and immune cells from entering the brain. Third, exercise inhibits the pro-inflammatory M1 phenotype and stimulates the anti-inflammatory M2 phenotype to increase the levels of anti-inflammatory factors in the brain, thereby restoring homeostasis. Finally, exercise triggers adult hippocampal neurogenesis (AHN) by inducing the expression of BDNF in the brain and muscle, thus leading to the continuous formation of new neurons, astrocytes, and oligodendrocytes. These new cells can replace the corresponding senescent and damaged cells, thus remodeling the high-loading state caused by neuroinflammation. Therefore, exercise inhibits the neuroinflammatory response through four effects and ultimately delays the pathological process of AD and alleviates symptoms
Neuroimmune-modifying effects upon exercise interventions
The innate immune system is the primary defense against exogenous pathogens and endogenous infections in the body. PAMPs or DAMPs bind to pattern recognition receptors (PPRs) on the membrane of glial cells to release cytokines, nitric oxide (NO), and other factors for defense [208]. In many animal models and humans with AD, astrocytes and microglia are significantly activated in areas with the initiation of AD pathology (frontal cortex and hippocampus) [209]. Activated microglia and astrocytes transform from a rest state to a pro-inflammatory state and play a protective role in clearing these damaged insults. However, under the long-term stimulation of injury factors, glial cells are continuously activated to stimulate the release of pro-inflammatory factors for restoring the cellular homeostasis in the body. As mentioned above, the activation state of microglia and corresponding phenotypic transformation play a key role in inflammatory responses in AD. After 10-day treadmill running of 4-month-old and 18-month-old C57BL/6 mice, exercise can promote the phagocytic capacity of mouse microglia, thereby reducing the content of IL-1β and reversing the decline of hippocampal neurons and memory capacity [210]. In multiple studies, the activation of microglia is suppressed in the hippocampus of APP/PS1 mice after 4 or 5 months of aerobic exercise [211, 212]. In addition, the treadmill running for 12 weeks promotes the transformation of microglia from the M1 phenotype to the M2 phenotype, thereby reducing inflammation and oxidative damage in hippocampal tissues, and finally resulting in the improvement of cognitive performance of mice [213].
Moreover, exercise can also inhibit the excessive activation of hippocampal microglia in aged mice, maintain the homeostasis of the nervous system, and effectively prevent neuronal damage in aged mice [214]. Furthermore, in streptozotocin (STZ)-induced diabetic model rats with AD-like symptoms, treadmill running increases the proportion of M2 phenotype microglia and attenuates oxidative stress-induced injury in the brain [215]. Therefore, exercise protects brain by suppressing the immune-inflammatory response by increasing the number and phagocytic capacity of M2 phenotype microglia, whose mechanism may be related to PPRs expressed on the microglial membrane. When the AIM2-like receptor 2 (ALR2) function is lost, it can induce the transformation of microglia from M1 phenotype to M2 phenotype and reduce the inflammatory response [216]. Exercise can up-regulate the expression of triggering receptor expressed on myeloid cell 2 (TREM2) and scavenger receptor A (SR-A) to enhance the neuroprotective function of microglia [205]. Moreover, a recent study has pointed out that four weeks of treadmill running can reduce NLRP3 content and Caspase-1 activity in the hippocampus of the mice after lateral ventricle injection of Aβ40 [217]. Numerous animal studies have also confirmed that inhibiting NLRP3 in microglia is a potential target for the treatment of AD in the future [218, 219], indicating that the regulation of PPRs in microglia by exercise is a potential immunotherapeutic direction. However, single-cell transcriptomics have revealed that microglia might have multiple phenotypes, including disease-associated microglia (DAM), and microglia with the coexistence of different phenotypes during the progression of AD [220, 221]. It is warranted in the future to investigate whether exercise has differential effects on these subdivided microglial phenotypes. Furthermore, whether exercise has a similar effect on astrocytes also needs to be explored [222].
On the other hand, exercise can reduce the impact of peripheral inflammatory factors on the central system. The BBB comprises endothelial cells, a basement membrane containing pericytes and astrocytes. Previous studies have confirmed that BBB is closely related to the occurrence and development of AD and is also a key point in preventing and treating AD [223]. Loss of BBB integrity allows cytokines and immune cells to enter the central nervous system (CNS), thereby activating glial cells and leading to changes in the extracellular milieu. Exercise can reduce TNF-α level in blood and enhance BBB function in patients with type II diabetes. Regular exercise training down-regulates pro-inflammatory cytokines, such as IL-6, and TNF-α, associated with low-grade systemic inflammation [224]. In AD, the accumulation of ROS activates metalloproteinases, thus leading to the disruption of BBB integrity [225]. BBB dysfunction during the progression of AD affects Aβ clearance and endothelial trafficking, impairs endothelial and pericyte function, disrupts tight junction (TJ) integrity, activates glial cells, and promotes leukocyte recruitment in the brain [226]. Physical activity inhibits neuroinflammation by up-regulating Aβ transporter activity to clear Aβ [227]. Aerobic exercise has been shown to alleviate the reduction of aging-induced cerebral blood flow (CBF) and cognitive performance in healthy individuals [228]. In addition, exercise also promotes the return of tight junction proteins in the BBB to their original levels, thereby restoring the permeability of the BBB. Long-term aerobic exercise from the midlife to old age prevents aging-related neurovascular decline, reduces the entry of inflammatory substances into the brain, and increases synaptic plasticity and overall behavioral capacity in aged mice [229]. Relevant studies have found that high-intensity continuous training (high-intensity interval training and high-intensity circuit training) has better immunomodulatory effects than moderate-intensity training [230]. Moreover, high-intensity circuit training is more effective in suppressing the proliferation of T cells and macrophages. In contrast, high-intensity interval training is better at inducing the anti-inflammatory phenotype polarization of immune cells [231]. Therefore, exercise suppresses neuroinflammation by altering the levels of peripheral inflammatory mediators and modulating microglia in AD models.
Exercise promotes hippocampal neurogenesis through suppressing neuroinflammation
During adulthood, neural stem cells in the hippocampus of humans can continuously proliferate and differentiate to generate new neurons, termed adult hippocampal neurogenesis (AHN), which is closely related to the learning and memory capacity [232]. AHN exists only in the subventricular zone (SVZ) and subgranular zone (SGZ) in hippocampal tissues of the mammalian brain. AHN can promote brain plasticity in adulthood, and the newly generated neurons can re-establish the connections between damaged neurons [233]. Emerging evidence suggests that AHN is impaired prior to onset in classical mouse models of AD [234]. During the intraperitoneal injection of LPS in the brains of 3xTG mice, the number of immature neurons in the hippocampal tissues of 3xTG mice reveals the significant reduction when compared with wild-type mice, even leading to hippocampal-dependent memory loss [235]. The aggregation of Aβ leads to impaired function of neuronal stem cells in the adult hippocampus. Restoring stem cell function and reducing neuroinflammation are considered as the key therapeutic strategies for AD [236]. In addition, the immune system is an essential regulator of AHN. During the progression of AD, chronic inflammatory responses down-regulate AHN through anti-neurogenic effects. A study indicates that activated microglia, particularly the M1 phenotype, can promote inflammatory responses by reducing the survival of neural precursor cells and play an essential role in suppressing AHN [236]. It has been reported that intra-cerebroventricular injection of STZ in rats induces persistent neuroinflammatory responses in the hippocampal SVZ and SGZ, and inhibits the proliferation, differentiation, and maturation of neural precursor cells, thus leading to memory loss [237]. Extensive evidence confirms that exercise can promote the generation of new neurons in the lateral ventricle and the differentiation of immature neurons in young, middle-aged, and elderly subjects [238, 239]. Experiments show that 4 months of voluntary wheeling running promotes hippocampal neurogenesis in AD model mice [240]. Furthermore, exercise stimulates the secretion of selenoprotein for promoting the proliferation, differentiation and migration of hippocampal precursor cells [241]. However, drug-induced AHN does not affect cognitive function in 5xFAD mice, and only exercise-induced AHN can ameliorate cognitive impairment in mice [242]. According to previous reports, exercise can awaken dormant neural stem cells and clear senescent neural stem cells to enhance AHN and reverse Aβ-induced cognitive impairment, which may be related to BDNF [243,244,245]. Chronic inflammation impairs neural stem cell function, and the administration of NSAID inhibits LPS-induced systemic inflammatory responses in mice, elevates IL-6 level, and enhances neurogenesis [246]. The activation of inflammatory response-mediated glial cells and chemokines in AD can inhibit AHN in the brain and promote the pathological process of AD, especially at the later stage of AD. However, reducing chronic inflammation in AD-transgenic mice can increase the proliferation of hippocampal stem cells and delay the occurrence and progression of AD [236]. In addition, exercise can restore the damaged nerve regeneration mediated by neuroinflammation through PGC-1α/FNDC5/BNDF signal pathway, thereby inducing sympathetic activation and the generation of uric acid [247, 248]. The newly generated neurons can restore standard memory-storing neural circuits, increase the number of neuronal dendritic spines, and restore regular expression of some neuronal genes [249]. However, recent animal studies have found that the differentiation tendency of neural stem cells is related to the conditions of exercise [250]. For example, for a long-term runner, neural stem cells are more inclined to differentiate into astrocytes, and the self-proliferation capacity of microglia is also enhanced. Glial cells have a particular regeneration capacity and can be induced to reprogram or convert into neurons, thereby extensively replenishing damaged neurons [251]. Therefore, exercise suppresses inflammation and increases the number of neurons and glial cells in the hippocampus to repair the irreversible damage caused by inflammation, which may be essential for early prevention and improvement of AD.
Exercise, neuroinflammation and neurodegenerative diseases
Numerous studies have shown that neuroinflammation is a common feature of neurodegenerative diseases [83]. A pathological hallmark of age-related degenerative diseases is the accumulation of excessive mis-folded proteins in neurons. These diseases cover tauopathy dominated by AD and synucleinopathies represented by Parkinson's disease (PD) and dementia with Lewy bodies [252]. These abnormal proteins appear to share the features such as the formation and insolubility of amyloid fibril structures that make them less susceptible to clearance by defense mechanisms in the body and induce the conversion of normal proteins to irregular forms in a prion-like manner [253,254,255,256]. The constantly emerging and accumulating erroneous proteins are likely the results of glial-neuron interactions in neuroinflammation. In addition, the studies on serial pathological section observations of postmortem brain tissue have found that the accumulation of these abnormal proteins is closely related to clinical symptoms and spreading in the brain in a specific way [257,258,259]. In various neurodegenerative disease models, exercise increases the number of anti-inflammatory phenotypes of microglia, thereby reducing the formation of faulty proteins, ultimately delaying disease progression and mitigating symptoms of neurodegenerative diseases, which also may be also related to the down-regulation of pattern recognition receptors in microglia and neuronal apoptosis [213, 260,261,262,263]. Likewise, the propagation of pathologically faulty proteins across various brain regions can cause the loss of differentiated mature neurons and impair the regeneration capacity of new neurons [264]. An experiment with autopsy specimens has demonstrated that increased susceptibility to AHN in different neurodegenerative diseases, suggesting that the functional decline of hippocampal neural stem cells may underlie cognitive impairment during pathological aging in humans [265]. In addition, abnormal glial function is associated with fragile AHN in aging and neurodegenerative diseases [265]. Neuroinflammation can effectively inhibit AHN, and exercise can accelerate the formation of new neurons, thereby resisting the damage caused by the inflammatory response and ultimately improving cognitive capacity [266]. Therefore, the role of exercise in immunomodulation and the repairing of damaged neurons appear to be informative in studying other neurodegenerative diseases. However, the causal relationship between exercise and neuroinflammation in neurodegenerative diseases has been less studied.
Limitations and future directions
Possibly due to ethical restriction and the difficulties in obtaining human brain tissue samples, there are only a few human studies on the relationship between exercise and inflammation in AD, mainly based on animal experiments. Moreover, animal models used to explore the underlying mechanisms for neuroinflammation of AD vary widely. Even exercise has many negative results for AD and inflammation [267,268,269], because studies on exercise and inflammation in AD are still sparse, our current understanding of the effects of exercise on regulating inflammation and the role of inflammatory responses in AD is limited. Moreover, the exercise conditions are not the same in animal or human experiments. Therefore, in terms of the current research results, we raise some thought-provoking questions and views: which chronic neuroinflammation level can induce AD? Is Aβ as a bystander in AD neuroimmune responses? Whether the restoration of inflammation can reduce the prevalence of AD or improve the prognosis of patients with MCI and AD is unclear. What kind of exercise has the best anti-inflammatory effect for AD patients is not defined? Does exercise have immunomodulatory effects on non-polarized state, or non-M1/M2 phenotypes of microglia? Whether microglia and their intracellular NLRP3 are potential therapeutic targets for immunology-based drugs or biomarker development still needs to be clarified. The roles of astrocytes in neuroinflammation still need to be explored. Which "exerkines" are involved in the anti-inflammatory effects of exercise? Are the anti-inflammatory effects of exercise sustained throughout the lifespan? Does physical exercise affect plasma GFAP or TREM2 levels in subjects with MCI or AD? Is the anti-inflammatory effect of exercise on other neurodegenerative diseases? All above unsolved questions should be the future directions for the prevention and treatments of AD through exercise interventions, which are also highly needed for the resolutions based on animal or cell experiments or the identification of potential biomarkers for clinical trials or practice.
Conclusion
Although exercise has been considered as an essential strategy for the prevention and treatment of AD, the specific mechanisms for ameliorating AD upon exercise interventions are still not fully uncovered, which is not conducive to further in-depth studies. Recent evidence highlights a more significant role of neuroinflammatory responses in AD pathogenesis, even prior to Aβ deposition, unlike previous amyloid cascade hypotheses. Therefore, this article explores whether exercise can prevent and treat AD by suppressing inflammatory response, summarizes the key features and possible mechanisms of inflammatory response in AD, and the relationship between the immune regulation of exercise and the role of promoting AHN and neuroinflammation.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- AHN:
-
Adult hippocampal neurogenesis
- ALR2:
-
AIM2-like receptor 2
- APOEε4:
-
Apolipoprotein E ε4
- APP:
-
Amyloid precursor protein
- AQP4:
-
Aquaporin 4
- ASC:
-
Apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)
- Aβ:
-
Amyloid-β peptide
- BACE1:
-
β-Site amyloid precursor protein cleaving enzyme 1
- BBB:
-
Blood–brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- BiP:
-
Immunoglobulin heavy-chain-binding protein
- CBF:
-
Cerebral blood flow
- CNS:
-
Central nervous system
- CRP:
-
C-reactive protein
- CSF:
-
Cerebrospinal fluid
- DAM:
-
Disease-associated microglia
- DAMP:
-
Danger-associated molecular pattern
- EOAD:
-
Early-onset family AD
- ERS:
-
Endoplasmic reticulum stress
- GFAP:
-
Glial fibrillary acidic protein
- IFN-G:
-
Interferon-G
- IL-10:
-
Interleukin 10
- IL-17:
-
Interleukin-17
- IL-1RA:
-
IL-1 Receptor antagonist
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IRE1:
-
Inositol-requiring enzyme 1
- JNK:
-
C-Jun N-terminal kinase
- LOAD:
-
Late-onset AD
- LPS:
-
Lipopolysaccharide
- MAPK:
-
Mitogen-activated protein kinase
- MCI:
-
Mild cognitive impairment
- NBP:
-
Dl-3-n-butylphthalide
- NFT:
-
Neurofibrillary tangle
- NF-κB:
-
Nuclear factor kappa-B
- NLRP3:
-
NOD-like receptor protein 3
- NO:
-
Nitric oxide
- NR:
-
Nicotinamide riboside
- Nrf2:
-
Nuclear factor-related factor 2
- NSAID:
-
Non-steroid anti-inflammatory drug
- PAMP:
-
Pathogen-associated molecular pattern
- PERK:
-
PKR-like ER kinase
- PET:
-
Positron emission tomography
- PPR:
-
Pattern recognition receptor
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SGZ:
-
Subgranular zone
- SOD:
-
Superoxidase dismutase
- SP:
-
Senile plaque
- SR-A:
-
Scavenger receptor A
- sTNFR1:
-
Soluble tumor necrosis factor receptor-1
- sTNFR2:
-
Soluble tumor necrosis factor receptor-2
- STZ:
-
Streptozotocin
- SVZ:
-
Subventricular zone
- TGF-β1:
-
Transforming growth factor beta-1
- TJ:
-
Tight junction
- TNF-α:
-
Tumor necrosis factor alpha
- TREM2:
-
Triggering receptor expressed on myeloid cell 2
- TRX:
-
Thioredoxin
- TXNIP:
-
Thioredoxin-interacting protein
- URP:
-
Unfolded protein response
- WHO:
-
World Health Organization
References
Dos Santos Picanco LC, Ozela PF, de Fatima de Brito Brito M, Pinheiro AA, Padilha EC, Braga FS, et al. Alzheimer’s disease: a review from the pathophysiology to diagnosis, new perspectives for pharmacological treatment. Curr Med Chem. 2018;25:3141–59.
Morris RG, Salmon DP. The centennial of Alzheimer’s disease and the publication of “Uber eine eigenartige Erkankung der Hirnrinde” by Alöis Alzheimer. Cortex. 2007;43:821–5.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–5.
van der Kant R, Goldstein LSB, Ossenkoppele R. Amyloid-β-independent regulators of Tau pathology in Alzheimer disease. Nat Rev Neurosci. 2020;21:21–35.
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chételat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90.
Arnsten AFT, Datta D, Del Tredici K, Braak H. Hypothesis: tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021;17:115–24.
Gómez-Isla T, Frosch MP. Lesions without symptoms: understanding resilience to Alzheimer disease neuropathological changes. Nat Rev Neurol. 2022;18:323–32.
McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology. 1992;42:447–9.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–6.
Czirr E, Castello NA, Mosher KI, Castellano JM, Hinkson IV, Lucin KM, et al. Microglial complement receptor 3 regulates brain Aβ levels through secreted proteolytic activity. J Exp Med. 2017;214:1081–92.
Severini C, Barbato C, Di Certo MG, Gabanella F, Petrella C, Di Stadio A, et al. Alzheimer’s disease: new concepts on the role of autoimmunity and NLRP3 inflammasome in the pathogenesis of the disease. Curr Neuropharmacol. 2021;19:498–512.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–72.
Lim B, Prassas I, Diamandis EP. Alzheimer disease pathogenesis: the role of autoimmunity. J Appl Lab Med. 2021;6:756–64.
Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29:787–803.
Xiao S, Chan P, Wang T, Hong Z, Wang S, Kuang W, et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimers Res Ther. 2021;13:62.
Ozben T, Ozben S. Neuro-inflammation and anti-inflammatory treatment options for Alzheimer’s disease. Clin Biochem. 2019;72:87–9.
Hoozemans JJ, van Haastert ES, Veerhuis R, Arendt T, Scheper W, Eikelenboom P, et al. Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer’s disease. J Neuroinflammation. 2005;2:27.
Sánchez-López E, Ettcheto M, Egea MA, Espina M, Calpena AC, Folch J, et al. New potential strategies for Alzheimer’s disease prevention: pegylated biodegradable dexibuprofen nanospheres administration to APPswe/PS1dE9. Nanomedicine. 2017;13:1171–82.
Sharifzadeh M, Naghdi N, Khosrovani S, Ostad SN, Sharifzadeh K, Roghani A. post-training intrahippocampal infusion of the COX-2 inhibitor celecoxib impaired spatial memory retention in rats. Eur J Pharmacol. 2005;511:159–66.
Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County study. Neurology. 2002;59:880–6.
Pedersen BK, Saltin B. Exercise as medicine—evidence for prescribing exercise as therapy in 26 different chronic diseases. Scand J Med Sci Sports. 2015;25(Suppl 3):1–72.
Booth FW, Roberts CK, Thyfault JP, Ruegsegger GN, Toedebusch RG. Role of inactivity in chronic diseases: evolutionary insight and pathophysiological mechanisms. Physiol Rev. 2017;97:1351–402.
Mahalakshmi B, Maurya N, Lee SD, Bharath Kumar V. Possible neuroprotective mechanisms of physical exercise in neurodegeneration. Int J Mol Sci. 2020;21:5895.
Walsh NP, Gleeson M, Shephard RJ, Gleeson M, Woods JA, Bishop NC, et al. Position statement. Part one: immune function and exercise. Exerc Immunol Rev. 2011;17:6–63.
Davies P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403.
Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–8.
Baker DJ, Petersen RC. Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Invest. 2018;128:1208–16.
Maccioni RB, Navarrete LP, González A, González-Canacer A, Guzmán-Martínez L, Cortés N. Inflammation: a major target for compounds to control Alzheimer’s disease. J Alzheimers Dis. 2020;76:1199–213.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Makin S. The amyloid hypothesis on trial. Nature. 2018;559:S4-S7.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341–50.
Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov. 2022;21:306–18.
Nassan M, Videnovic A. Circadian rhythms in neurodegenerative disorders. Nat Rev Neurol. 2022;18:7–24.
Shi J, Sabbagh MN, Vellas B. Alzheimer’s disease beyond amyloid: strategies for future therapeutic interventions. BMJ. 2020;371: m3684.
Kakuda N, Shoji M, Arai H, Furukawa K, Ikeuchi T, Akazawa K, et al. Altered γ-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO Mol Med. 2012;4:344–52.
Yan Y, Wang C. Abeta42 is more rigid than Abeta40 at the C terminus: implications for Abeta aggregation and toxicity. J Mol Biol. 2006;364:853–62.
Liu L, Lauro BM, He A, Lee H, Bhattarai S, Wolfe MS, et al. Identification of the Aβ37/42 peptide ratio in CSF as an improved Aβ biomarker for Alzheimer’s disease. Alzheimer's Dement. 2023;19:79−96.
Frisoni GB, Altomare D, Thal DR, Ribaldi F, van der Kant R, Ossenkoppele R, et al. The probabilistic model of Alzheimer disease: the amyloid hypothesis revised. Nat Rev Neurosci. 2022;23:53–66.
Steubler V, Erdinger S, Back MK, Ludewig S, Fässler D, Richter M, et al. Loss of all three APP family members during development impairs synaptic function and plasticity, disrupts learning, and causes an autism-like phenotype. Embo J. 2021;40: e107471.
Sturchio A, Dwivedi AK, Young CB, Malm T, Marsili L, Sharma JS, et al. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine. 2021;38: 100988.
Crystal H, Dickson D, Fuld P, Masur D, Scott R, Mehler M, et al. Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology. 1988;38:1682–7.
Polvikoski T, Sulkava R, Myllykangas L, Notkola IL, Niinistö L, Verkkoniemi A, et al. Prevalence of Alzheimer’s disease in very elderly people: a prospective neuropathological study. Neurology. 2001;56:1690–6.
Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid-β immunotherapy for Alzheimer disease: is it now a long shot? Ann Neurol. 2019;85:303–15.
Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of Tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020;140:417–47.
Huang Y, Happonen KE, Burrola PG, O’Connor C, Hah N, Huang L, et al. Microglia use TAM receptors to detect and engulf amyloid β plaques. Nat Immunol. 2021;22:586–94.
Maccioni RB, Tapia JP, Guzman-Martinez L. Pathway to Tau modifications and the origins of Alzheimer’s disease. Arch Med Res. 2018;49:130–1.
Pan RY, He L, Zhang J, Liu X, Liao Y, Gao J, et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34:634−48.
Zlokovic BV. Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 2013;70:440–4.
Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, et al. Dementia prevention, intervention, and care. Lancet. 2017;390:2673–734.
Dubois B, Hampel H, Feldman HH, Scheltens P, Aisen P, Andrieu S, et al. Preclinical Alzheimer’s disease: definition, natural history, and diagnostic criteria. Alzheimers Dement. 2016;12:292–323.
Guzman-Martinez L, Maccioni RB, Farías GA, Fuentes P, Navarrete LP. Biomarkers for Alzheimer’s disease. Curr Alzheimer Res. 2019;16:518–28.
González A, Guzmán-Martínez L, Maccioni RB. Plasma Tau variants detected by a novel anti-Tau monoclonal antibody: a potential biomarker for Alzheimer’s disease. J Alzheimers Dis. 2020;77:877–83.
Farías G, Pérez P, Slachevsky A, Maccioni RB. Platelet tau pattern correlates with cognitive status in Alzheimer’s disease. J Alzheimers Dis. 2012;31:65–9.
Ashton NJ, Pascoal TA, Karikari TK, Benedet AL, Lantero-Rodriguez J, Brinkmalm G, et al. Plasma p-tau231: a new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021;141:709–24.
Palmqvist S, Tideman P, Cullen N, Zetterberg H, Blennow K, Dage JL, et al. Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med. 2021;27:1034–42.
Gauthier SWC, Servaes S, Morais JA, Rosa-Neto P. World Alzheimer Report 2022: life after diagnosis: navigating treatment, care and support Alzheimer’s disease international 2022. London: Alzheimer’s Disease International; 2022.
Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer’s disease. J Neuroinflammation. 2018;15:276.
Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, Tau, immunity and lipid processing. Nat Genet. 2019;51:414–30.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8.
Abbott A. Are infections seeding some cases of Alzheimer’s disease? Nature. 2020;587:22–5.
Olsen I, Singhrao SK. Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol. 2015;7:29143.
Qian XH, Song XX, Liu XL, Chen SD, Tang HD. Inflammatory pathways in Alzheimer’s disease mediated by gut microbiota. Ageing Res Rev. 2021;68: 101317.
Ryu JK, McLarnon JG. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med. 2009;13:2911–25.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38.
Monson NL, Ireland SJ, Ligocki AJ, Chen D, Rounds WH, Li M, et al. Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab. 2014;34:30–3.
Benedet AL, Milà-Alomà M, Vrillon A, Ashton NJ, Pascoal TA, Lussier F, et al. Differences between plasma and cerebrospinal fluid glial fibrillary acidic protein levels across the Alzheimer disease continuum. JAMA Neurol. 2021;78:1471–83.
Molinuevo JL, Ayton S, Batrla R, Bednar MM, Bittner T, Cummings J, et al. Current state of Alzheimer’s fluid biomarkers. Acta Neuropathol. 2018;136:821–53.
Brown GC, St G-H. Does soluble TREM2 protect against Alzheimer’s disease? Front Aging Neurosci. 2021;13: 834697.
Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, et al. APPlasma GF is an early marker of amyloid-β but not Tau pathology in Alzheimer’s disease. Brain. 2021;144:3505–16.
Zhao A, Jiao Y, Ye G, Kang W, Tan L, Li Y, et al. Soluble TREM2 levels associate with conversion from mild cognitive impairment to Alzheimer’s disease. J Clin Invest. 2022;132: e158708.
Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, et al. Blood-based biomarkers for Alzheimer’s disease: towards clinical implementation. Lancet Neurol. 2022;21:66–77.
Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Schlepckow K, Araque Caballero M, Franzmeier N, et al. Early increase of CSF sTREM2 in Alzheimer’s disease is associated with Tau related-neurodegeneration but not with amyloid-β pathology. Mol Neurodegener. 2019;14:1.
Carmona S, Zahs K, Wu E, Dakin K, Bras J, Guerreiro R. The role of TREM2 in Alzheimer’s disease and other neurodegenerative disorders. Lancet Neurol. 2018;17:721–30.
Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. 2018;10:83.
Morales I, Cerda-Troncoso C, Andrade V, Maccioni RB. The natural product curcumin as a potential coadjuvant in Alzheimer’s treatment. J Alzheimers Dis. 2017;60:451–60.
Maccioni RB, Calfío C, González A, Lüttges V. Novel nutraceutical compounds in Alzheimer prevention. Biomolecules. 2022;12:249.
Guzman-Martinez L, Calfío C, Farias GA, Vilches C, Prieto R, Maccioni RB. New frontiers in the prevention, diagnosis, and treatment of Alzheimer’s disease. J Alzheimers Dis. 2021;82:S51-S63.
Webers A, Heneka MT, Gleeson PA. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol. 2020;98:28–41.
Maccioni RB, Rojo LE, Fernández JA, Kuljis RO. The role of neuroimmunomodulation in Alzheimer’s disease. Ann NY Acad Sci. 2009;1153:240–6.
Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB. Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer’s disease. Arch Med Res. 2008;39:1–16.
Fernández JA, Rojo L, Kuljis RO, Maccioni RB. The damage signals hypothesis of Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2008;14:329–33.
Guzman-Martinez L, Maccioni RB, Andrade V, Navarrete LP, Pastor MG, Ramos-Escobar N. Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol. 2019;10:1008.
Chiarini A, Armato U, Hu P, Dal Prà I. Danger-sensing/patten recognition receptors and neuroinflammation in Alzheimer’s disease. Int J Mol Sci. 2020;21:9036.
Bielski BH, Cabelli DE. Highlights of current research involving superoxide and perhydroxyl radicals in aqueous solutions. Int J Radiat Biol. 1991;59:291–319.
Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease–a double-edged sword. Neuron. 2002;35:419–32.
Cortés N, Andrade V, Guzmán-Martínez L, Estrella M, Maccioni RB. Neuroimmune Tau mechanisms: their role in the progression of neuronal degeneration. Int J Mol Sci. 2018;19:956.
Cortés N, Guzmán-Martínez L, Andrade V, González A, Maccioni RB. CDK5: a unique CDK and its multiple roles in the nervous system. J Alzheimers Dis. 2019;68:843–55.
Morales I, Jiménez JM, Mancilla M, Maccioni RB. Tau oligomers and fibrils induce activation of microglial cells. J Alzheimers Dis. 2013;37:849–56.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–8.
Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease a rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Lee HJ, Kim C, Lee SJ. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev. 2010;3:283–7.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020;9:42.
Mann CN, Devi SS, Kersting CT, Bleem AV, Karch CM, Holtzman DM, et al. Astrocytic α2-Na(+)/K(+) ATPase inhibition suppresses astrocyte reactivity and reduces neurodegeneration in a tauopathy mouse model. Sci Transl Med. 2022;14:eabm4107.
McAlpine CS, Park J, Griciuc A, Kim E, Choi SH, Iwamoto Y, et al. Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature. 2021;595:701–6.
Cao W, Zheng H. Peripheral immune system in aging and Alzheimer’s disease. Mol Neurodegener. 2018;13:51.
Bettcher BM, Tansey MG, Dorothée G, Heneka MT. Peripheral and central immune system crosstalk in Alzheimer disease—a research prospectus. Nat Rev Neurol. 2021;17:689–701.
Gate D, Saligrama N, Leventhal O, Yang AC, Unger MS, Middeldorp J, et al. Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature. 2020;577:399–404.
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502.
Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–7.
Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T Cells in brains of Alzheimer disease patients correlate with Tau but not with amyloid pathology: an immunohistochemical study. Neurodegener Dis. 2018;18:49–56.
Uddin MS, Kabir MT, Mamun AA, Barreto GE, Rashid M, Perveen A, et al. Pharmacological approaches to mitigate neuroinflammation in Alzheimer’s disease. Int Immunopharmacol. 2020;84: 106479.
Brosseron F, Maass A, Kleineidam L, Ravichandran KA, González PG, McManus RM, et al. Soluble TAM receptors sAXL and sTyro3 predict structural and functional protection in Alzheimer’s disease. Neuron. 2022;110:1009-22.e4.
Grimaldi A, Pediconi N, Oieni F, Pizzarelli R, Rosito M, Giubettini M, et al. Neuroinflammatory processes, A1 astrocyte activation and protein aggregation in the retina of Alzheimer’s disease patients, possible biomarkers for early diagnosis. Front Neurosci. 2019;13:925.
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37.
Tao Q, Ang TFA, DeCarli C, Auerbach SH, Devine S, Stein TD, et al. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1: e183597.
Hegazy SH, Thomassen JQ, Rasmussen IJ, Nordestgaard BG, Tybjaerg-Hansen A, Frikke-Schmidt R. C-reactive protein levels and risk of dementia-Observational and genetic studies of 111,242 individuals from the general population. Alzheimers Dement. 2022;18:2262−71.
Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, et al. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021;27:1592–9.
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595–604.
Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88:594–604.
Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95:437–45.
Nichols MR, St-Pierre MK, Wendeln AC, Makoni NJ, Gouwens LK, Garrad EC, et al. Inflammatory mechanisms in neurodegeneration. J Neurochem. 2019;149:562–81.
Mecha M, Carrillo-Salinas FJ, Feliú A, Mestre L, Guaza C. Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol Ther. 2016;166:40–55.
Mecha M, Feliú A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutiérrez S, de Sola RG, et al. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. 2015;49:233–45.
Rapaka D, Bitra VR, Challa SR, Adiukwu PC. Potentiation of microglial endocannabinoid signaling alleviates neuroinflammation in Alzheimer’s disease. Neuropeptides. 2021;90: 102196.
Streit WJ. Microglia and Alzheimer’s disease pathogenesis. J Neurosci Res. 2004;77:1–8.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
Mathys H, Davila-Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature. 2019;570:332–7.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65.
Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature. 2017;552:355–61.
Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, et al. The adaptor ASC has extracellular and “prionoid” activities that propagate inflammation. Nat Immunol. 2014;15:727–37.
Baroja-Mazo A, Martín-Sánchez F, Gomez AI, Martínez CM, Amores-Iniesta J, Compan V, et al. The NLRP3 inflammasome is released as a particulate danger signal that amplifies the inflammatory response. Nat Immunol. 2014;15:738–48.
Furman D, Chang J, Lartigue L, Bolen CR, Haddad F, Gaudilliere B, et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat Med. 2017;23:174–84.
Lučiūnaitė A, McManus RM, Jankunec M, Rácz I, Dansokho C, Dalgėdienė I, et al. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem. 2020;155:650–61.
Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12:50.
Xue QS, Streit WJ. Microglial pathology in Down syndrome. Acta Neuropathol. 2011;122:455–66.
Stancu IC, Cremers N, Vanrusselt H, Couturier J, Vanoosthuyse A, Kessels S, et al. Aggregated Tau activates NLRP3-ASC inflammasome exacerbating exogenously seeded and non-exogenously seeded Tau pathology in vivo. Acta Neuropathol. 2019;137:599–617.
Lee M, McGeer E, McGeer PL. Activated human microglia stimulate neuroblastoma cells to upregulate production of beta amyloid protein and tau: implications for Alzheimer’s disease pathogenesis. Neurobiol Aging. 2015;36:42–52.
Cantarella G, Di Benedetto G, Puzzo D, Privitera L, Loreto C, Saccone S, et al. Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer’s disease. Brain. 2015;138:203–16.
Thawkar BS, Kaur G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J Neuroimmunol. 2019;326:62–74.
Duran-Aniotz C, Martínez G, Hetz C. Memory loss in Alzheimer’s disease: are the alterations in the UPR network involved in the cognitive impairment? Front Aging Neurosci. 2014;6:8.
Gerakis Y, Hetz C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018;285:995–1011.
Keestra-Gounder AM, Byndloss MX, Seyffert N, Young BM, Chávez-Arroyo A, Tsai AY, et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature. 2016;532:394–7.
Murray HC, Dieriks BV, Swanson MEV, Anekal PV, Turner C, Faull RLM, et al. The unfolded protein response is activated in the olfactory system in Alzheimer’s disease. Acta Neuropathol Commun. 2020;8:109.
Ho YS, Yang X, Lau JC, Hung CH, Wuwongse S, Zhang Q, et al. Endoplasmic reticulum stress induces Tau pathology and forms a vicious cycle: implication in Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2012;28:839–54.
Sprenkle NT, Sims SG, Sánchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. 2017;12:42.
Feng L, Zhang L. Resveratrol suppresses Aβ-induced microglial activation through the TXNIP/TRX/NLRP3 signaling pathway. DNA Cell Biol. 2019;38:874–9.
Li L, Ismael S, Nasoohi S, Sakata K, Liao FF, McDonald MP, et al. Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer’s disease brain. J Alzheimers Dis. 2019;68:255–65.
Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab. 2012;16:265–73.
Guthrie LN, Abiraman K, Plyler ES, Sprenkle NT, Gibson SA, McFarland BC, et al. Attenuation of PKR-like ER Kinase (PERK) signaling selectively controls endoplasmic reticulum stress-induced inflammation without compromising immunological responses. J Biol Chem. 2016;291:15830–40.
Costa-Mattioli M, Sossin WS, Klann E, Sonenberg N. Translational control of long-lasting synaptic plasticity and memory. Neuron. 2009;61:10–26.
Ali AK, Banks WA, Kumar VB, Shah GN, Lynch JL, Farr SA, et al. Nitric oxide activity and isoenzyme expression in the senescence-accelerated mouse p8 model of Alzheimer’s disease: effects of anti-amyloid antibody and antisense treatments. J Gerontol A Biol Sci Med Sci. 2009;64:1025–30.
Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, et al. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–83.
Kumar VB, Farr SA, Flood JF, Kamlesh V, Franko M, Banks WA, et al. Site-directed antisense oligonucleotide decreases the expression of amyloid precursor protein and reverses deficits in learning and memory in aged SAMP8 mice. Peptides. 2000;21:1769–75.
Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer’s disease. Faseb j. 2009;23:2459–66.
Murakami K, Murata N, Noda Y, Tahara S, Kaneko T, Kinoshita N, et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. J Biol Chem. 2011;286:44557–68.
Chen Z, Zhong C. Oxidative stress in Alzheimer’s disease. Neurosci Bull. 2014;30:271–81.
Hou Y, Wei Y, Lautrup S, Yang B, Wang Y, Cordonnier S, et al. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci USA. 2021;118:e2011226118.
Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–40.
Kanninen K, Heikkinen R, Malm T, Rolova T, Kuhmonen S, Leinonen H, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:16505–10.
Wang CY, Wang ZY, Xie JW, Wang T, Wang X, Xu Y, et al. Dl-3-n-butylphthalide-induced upregulation of antioxidant defense is involved in the enhancement of cross talk between CREB and Nrf2 in an Alzheimer’s disease mouse model. Neurobiol Aging. 2016;38:32–46.
Wang CY, Xu Y, Wang X, Guo C, Wang T, Wang ZY. Dl-3-n-Butylphthalide inhibits NLRP3 inflammasome and mitigates Alzheimer’s-like pathology via Nrf2-TXNIP-TrX axis. Antioxid Redox Signal. 2019;30:1411–31.
Bull FC, Al-Ansari SS, Biddle S, Borodulin K, Buman MP, Cardon G, et al. World Health Organization 2020 guidelines on physical activity and sedentary behaviour. Br J Sports Med. 2020;54:1451–62.
Cheng AJ, Jude B, Lanner JT. Intramuscular mechanisms of overtraining. Redox Biol. 2020;35: 101480.
Nieman DC, Wentz LM. The compelling link between physical activity and the body’s defense system. J Sport Health Sci. 2019;8:201–17.
Campbell JP, Turner JE. Debunking the myth of exercise-induced immune suppression: redefining the impact of exercise on immunological health across the lifespan. Front Immunol. 2018;9:648.
Scheffer DDL, Latini A. Exercise-induced immune system response: anti-inflammatory status on peripheral and central organs. Biochim Biophys Acta Mol Basis Dis. 2020;1866: 165823.
Pinho RA, Silva LA, Pinho CA, Scheffer DL, Souza CT, Benetti M, et al. Oxidative stress and inflammatory parameters after an Ironman race. Clin J Sport Med. 2010;20:306–11.
Vella L, Markworth JF, Farnfield MM, Maddipati KR, Russell AP, Cameron-Smith D. Intramuscular inflammatory and resolving lipid profile responses to an acute bout of resistance exercise in men. Physiol Rep. 2019;7: e14108.
Pedersen BK, Toft AD. Effects of exercise on lymphocytes and cytokines. Br J Sports Med. 2000;34:246–51.
Scheffer DDL, Ghisoni K, Aguiar AS Jr, Latini A. Moderate running exercise prevents excessive immune system activation. Physiol Behav. 2019;204:248–55.
Metsios GS, Moe RH, Kitas GD. Exercise and inflammation. Best Pract Res Clin Rheumatol. 2020;34: 101504.
Hamer M, Sabia S, Batty GD, Shipley MJ, Tabák AG, Singh-Manoux A, et al. Physical activity and inflammatory markers over 10 years: follow-up in men and women from the Whitehall II cohort study. Circulation. 2012;126:928–33.
Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol. 2011;11:607–15.
Pitsavos C, Panagiotakos DB, Chrysohoou C, Kavouras S, Stefanadis C. The associations between physical activity, inflammation, and coagulation markers, in people with metabolic syndrome: the ATTICA study. Eur J Cardiovasc Prev Rehabil. 2005;12:151–8.
Kadoglou NP, Perrea D, Iliadis F, Angelopoulou N, Liapis C, Alevizos M. Exercise reduces resistin and inflammatory cytokines in patients with type 2 diabetes. Diabetes Care. 2007;30:719–21.
Benatti FB, Pedersen BK. Exercise as an anti-inflammatory therapy for rheumatic diseases-myokine regulation. Nat Rev Rheumatol. 2015;11:86–97.
Timmerman KL, Flynn MG, Coen PM, Markofski MM, Pence BD. Exercise training-induced lowering of inflammatory (CD14+CD16+) monocytes: a role in the anti-inflammatory influence of exercise? J Leukoc Biol. 2008;84:1271–8.
Della Gatta PA, Garnham AP, Peake JM, Cameron-Smith D. Effect of exercise training on skeletal muscle cytokine expression in the elderly. Brain Behav Immun. 2014;39:80–6.
Chow LS, Gerszten RE, Taylor JM, Pedersen BK, van Praag H, Trappe S, et al. Exerkines in health, resilience and disease. Nat Rev Endocrinol. 2022;18:273–89.
Weinhold M, Shimabukuro-Vornhagen A, Franke A, Theurich S, Wahl P, Hallek M, et al. Physical exercise modulates the homeostasis of human regulatory T cells. J Allergy Clin Immunol. 2016;137:1607-10.e8.
Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet commission. Lancet. 2020;396:413–46.
Zotcheva E, Bergh S, Selbæk G, Krokstad S, Håberg AK, Strand BH, et al. Midlife physical activity, psychological distress, and dementia risk: the HUNT Study. J Alzheimers Dis. 2018;66:825–33.
Hörder H, Johansson L, Guo X, Grimby G, Kern S, Östling S, et al. Midlife cardiovascular fitness and dementia: a 44-year longitudinal population study in women. Neurology. 2018;90:e1298–305.
De la Rosa A, Olaso-Gonzalez G, Arc-Chagnaud C, Millan F, Salvador-Pascual A, García-Lucerga C, et al. Physical exercise in the prevention and treatment of Alzheimer’s disease. J Sport Health Sci. 2020;9:394–404.
Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13:788–94.
Hamer M, Chida Y. Physical activity and risk of neurodegenerative disease: a systematic review of prospective evidence. Psychol Med. 2009;39:3–11.
Yu JT, Xu W, Tan CC, Andrieu S, Suckling J, Evangelou E, et al. Evidence-based prevention of Alzheimer’s disease: systematic review and meta-analysis of 243 observational prospective studies and 153 randomised controlled trials. J Neurol Neurosurg Psychiatry. 2020;91:1201–9.
Buchman AS, Boyle PA, Yu L, Shah RC, Wilson RS, Bennett DA. Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology. 2012;78:1323–9.
Silva MVF, Loures CMG, Alves LCV, de Souza LC, Borges KBG, Carvalho MDG. Alzheimer’s disease: risk factors and potentially protective measures. J Biomed Sci. 2019;26:33.
Brown BM, Peiffer JJ, Martins RN. Multiple effects of physical activity on molecular and cognitive signs of brain aging: can exercise slow neurodegeneration and delay Alzheimer’s disease? Mol Psychiatry. 2013;18:864–74.
Yu Q, Li X, Wang J, Li Y. Effect of exercise training on long-term potentiation and NMDA receptor channels in rats with cerebral infarction. Exp Ther Med. 2013;6:1431–6.
Adlard PA, Perreau VM, Pop V, Cotman CW. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J Neurosci. 2005;25:4217–21.
Liu HL, Zhao G, Zhang H, Shi LD. Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res. 2013;256:261–72.
Kang EB, Kwon IS, Koo JH, Kim EJ, Kim CH, Lee J, et al. Treadmill exercise represses neuronal cell death and inflammation during Aβ-induced ER stress by regulating unfolded protein response in aged presenilin 2 mutant mice. Apoptosis. 2013;18:1332–47.
Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–32.
El Hayek L, Khalifeh M, Zibara V, Abi Assaad R, Emmanuel N, Karnib N, et al. Lactate mediates the effects of exercise on learning and memory through SIRT1-dependent activation of hippocampal brain-derived neurotrophic factor (BDNF). J Neurosci. 2019;39:2369–82.
Baranowski BJ, Hayward GC, Marko DM, MacPherson REK. Examination of BDNF treatment on BACE1 activity and acute exercise on brain BDNF signaling. Front Cell Neurosci. 2021;15: 665867.
Rasmussen P, Brassard P, Adser H, Pedersen MV, Leick L, Hart E, et al. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol. 2009;94:1062–9.
Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217:459–72.
Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176.
Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, et al. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24.
Fiatarone Singh MA, Gates N, Saigal N, Wilson GC, Meiklejohn J, Brodaty H, et al. The study of mental and resistance training (SMART) study—resistance training and/or cognitive training in mild cognitive impairment: a randomized, double-blind, double-sham controlled trial. J Am Med Dir Assoc. 2014;15:873–80.
Huuha AM, Norevik CS, Moreira JBN, Kobro-Flatmoen A, Scrimgeour N, Kivipelto M, et al. Can exercise training teach us how to treat Alzheimer’s disease? Ageing Res Rev. 2022;75: 101559.
Tarumi T, Rossetti H, Thomas BP, Harris T, Tseng BY, Turner M, et al. Exercise training in amnestic mild cognitive impairment: a one-year Randomized Controlled Trial. J Alzheimers Dis. 2019;71:421–33.
Pereira CF, Santos AE, Moreira PI, Pereira AC, Sousa FJ, Cardoso SM, et al. Is Alzheimer’s disease an inflammasomopathy? Ageing Res Rev. 2019;56: 100966.
Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S, et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. 2020;64: 101156.
Valenzuela PL, Maffiuletti NA, Joyner MJ, Lucia A, Lepers R. Lifelong endurance exercise as a countermeasure against age-related [formula: see text] decline: physiological overview and insights from masters athletes. Sports Med. 2020;50:703–16.
Lavin KM, Perkins RK, Jemiolo B, Raue U, Trappe SW, Trappe TA. Effects of aging and lifelong aerobic exercise on basal and exercise-induced inflammation. J Appl Physiol. 1985;2020(128):87–99.
Nilsson MI, Bourgeois JM, Nederveen JP, Leite MR, Hettinga BP, Bujak AL, et al. Lifelong aerobic exercise protects against inflammaging and cancer. PLoS ONE. 2019;14: e0210863.
Valenzuela PL, Castillo-García A, Morales JS, de la Villa P, Hampel H, Emanuele E, et al. Exercise benefits on Alzheimer’s disease: state-of-the-science. Ageing Res Rev. 2020;62: 101108.
Sellami M, Gasmi M, Denham J, Hayes LD, Stratton D, Padulo J, et al. Effects of acute and chronic exercise on immunological parameters in the elderly aged: can physical activity counteract the effects of aging? Front Immunol. 2018;9:2187.
de Farias JM, Dos Santos TN, Pereira EV, de Moraes GL, Furtado BG, Tietbohl LTW, et al. Physical exercise training improves judgment and problem-solving and modulates serum biomarkers in patients with Alzheimer’s disease. Mol Neurobiol. 2021;58:4217–25.
Zhang SS, Zhu L, Peng Y, Zhang L, Chao FL, Jiang L, et al. Long-term running exercise improves cognitive function and promotes microglial glucose metabolism and morphological plasticity in the hippocampus of APP/PS1 mice. J Neuroinflammation. 2022;19:34.
Jensen CS, Bahl JM, Østergaard LB, Høgh P, Wermuth L, Heslegrave A, et al. Exercise as a potential modulator of inflammation in patients with Alzheimer’s disease measured in cerebrospinal fluid and plasma. Exp Gerontol. 2019;121:91–8.
Ercan Z, Bilek F, Demir CF. The effect of aerobic exercise on neurofilament light chain and glial fibrillary acidic protein level in patients with relapsing remitting type multiple sclerosis. Mult Scler Relat Disord. 2021;55: 103219.
De Miguel Z, Khoury N, Betley MJ, Lehallier B, Willoughby D, Olsson N, et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature. 2021;600:494–9.
Frank-Cannon TC, Alto LT, McAlpine FE, Tansey MG. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol Neurodegener. 2009;4:47.
Kamphuis W, Mamber C, Moeton M, Kooijman L, Sluijs JA, Jansen AH, et al. GFAP isoforms in adult mouse brain with a focus on neurogenic astrocytes and reactive astrogliosis in mouse models of Alzheimer disease. PLoS ONE. 2012;7: e42823.
Mela V, Mota BC, Milner M, McGinley A, Mills KHG, Kelly ÁM, et al. Exercise-induced re-programming of age-related metabolic changes in microglia is accompanied by a reduction in senescent cells. Brain Behav Immun. 2020;87:413–28.
Ke HC, Huang HJ, Liang KC, Hsieh-Li HM. Selective improvement of cognitive function in adult and aged APP/PS1 transgenic mice by continuous non-shock treadmill exercise. Brain Res. 2011;1403:1–11.
Xiong JY, Li SC, Sun YX, Zhang XS, Dong ZZ, Zhong P, et al. Long-term treadmill exercise improves spatial memory of male APPswe/PS1dE9 mice by regulation of BDNF expression and microglia activation. Biol Sport. 2015;32:295–300.
Zhang X, He Q, Huang T, Zhao N, Liang F, Xu B, et al. Treadmill exercise decreases Aβ deposition and counteracts cognitive decline in APP/PS1 mice, possibly via hippocampal microglia modifications. Front Aging Neurosci. 2019;11:78.
Giorgetti E, Panesar M, Zhang Y, Joller S, Ronco M, Obrecht M, et al. Modulation of microglia by voluntary exercise or CSF1R inhibition prevents age-related loss of functional motor units. Cell Rep. 2019;29:1539-54.e7.
Lu Y, Dong Y, Tucker D, Wang R, Ahmed ME, Brann D, et al. Treadmill exercise exerts neuroprotection and regulates microglial polarization and oxidative stress in a streptozotocin-induced rat model of sporadic Alzheimer’s disease. J Alzheimers Dis. 2017;56:1469–84.
Ma K, Guo J, Wang G, Ni Q, Liu X. Toll-Like receptor 2-mediated autophagy promotes microglial cell death by modulating the microglial M1/M2 phenotype. Inflammation. 2020;43:701–11.
Rosa JM, Camargo A, Wolin IAV, Kaster MP, Rodrigues ALS. Physical exercise prevents amyloid β(1–40)-induced disturbances in NLRP3 inflammasome pathway in the hippocampus of mice. Metab Brain Dis. 2021;36:351–9.
Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, et al. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2020;117:32145–54.
Zhang Y, Dong Z, Song W. NLRP3 inflammasome as a novel therapeutic target for Alzheimer’s disease. Signal Transduct Target Ther. 2020;5:37.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276-90.e17.
Sousa C, Golebiewska A, Poovathingal SK, Kaoma T, Pires-Afonso Y, Martina S, et al. Single-cell transcriptomics reveals distinct inflammation-induced microglia signatures. EMBO Rep. 2018;19:e46171.
Belaya I, Ivanova M, Sorvari A, Ilicic M, Loppi S, Koivisto H, et al. Astrocyte remodeling in the beneficial effects of long-term voluntary exercise in Alzheimer’s disease. J Neuroinflammation. 2020;17:271.
Montagne A, Zhao Z. Zlokovic BV Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151–69.
Trøseid M, Lappegård KT, Claudi T, Damås JK, Mørkrid L, Brendberg R, et al. Exercise reduces plasma levels of the chemokines MCP-1 and IL-8 in subjects with the metabolic syndrome. Eur Heart J. 2004;25:349–55.
Gamba P, Testa G, Gargiulo S, Staurenghi E, Poli G. Leonarduzzi G Oxidized cholesterol as the driving force behind the development of Alzheimer’s disease. Front Aging Neurosci. 2015;7:119.
Lepelletier FX, Mann DM, Robinson AC, Pinteaux E, Boutin H. Early changes in extracellular matrix in Alzheimer’s disease. Neuropathol Appl Neurobiol. 2017;43:167–82.
He XF, Liu DX, Zhang Q, Liang FY, Dai GY, Zeng JS, et al. Voluntary exercise promotes glymphatic clearance of amyloid Beta and reduces the activation of astrocytes and microglia in aged mice. Front Mol Neurosci. 2017;10:144.
Ainslie PN, Cotter JD, George KP, Lucas S, Murrell C, Shave R, et al. Elevation in cerebral blood flow velocity with aerobic fitness throughout healthy human ageing. J Physiol. 2008;586:4005–10.
Soto I, Graham LC, Richter HJ, Simeone SN, Radell JE, Grabowska W, et al. APOE stabilization by exercise prevents aging neurovascular dysfunction and complement induction. PLoS Biol. 2015;13: e1002279.
Fainstein N, Tyk R, Touloumi O, Lagoudaki R, Goldberg Y, Agranyoni O, et al. Exercise intensity-dependent immunomodulatory effects on encephalomyelitis. Ann Clin Transl Neurol. 2019;6:1647–58.
Goldberg Y, Fainstein N, Zaychik Y, Hamdi L, Segal S, Nabat H, et al. Continuous and interval training attenuate encephalomyelitis by separate immunomodulatory mechanisms. Ann Clin Transl Neurol. 2021;8:190–200.
Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, et al. Neurogenesis in the adult human hippocampus. Nat Med. 1998;4:1313–7.
Levone BR, Cryan JF, O’Leary OF. Role of adult hippocampal neurogenesis in stress resilience. Neurobiol Stress. 2015;1:147–55.
Mu Y, Gage FH. Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener. 2011;6:85.
Valero J, Mastrella G, Neiva I, Sánchez S, Malva JO. Long-term effects of an acute and systemic administration of LPS on adult neurogenesis and spatial memory. Front Neurosci. 2014;8:83.
Sung PS, Lin PY, Liu CH, Su HC, Tsai KJ. Neuroinflammation and neurogenesis in Alzheimer’s disease and potential therapeutic approaches. Int J Mol Sci. 2020;21:701.
Bassani TB, Bonato JM, Machado MMF, Cóppola-Segovia V, Moura ELR, Zanata SM, et al. Decrease in adult neurogenesis and neuroinflammation are involved in spatial memory impairment in the streptozotocin-induced model of sporadic Alzheimer’s disease in rats. Mol Neurobiol. 2018;55:4280–96.
Saraulli D, Costanzi M, Mastrorilli V, Farioli-Vecchioli S. The long run: neuroprotective effects of physical exercise on adult neurogenesis from youth to old age. Curr Neuropharmacol. 2017;15:519–33.
Lei X, Wu Y, Xu M, Jones OD, Ma J, Xu X. Physical exercise: bulking up neurogenesis in human adults. Cell Biosci. 2019;9:74.
Gerberding AL, Zampar S, Stazi M, Liebetanz D, Wirths O. Physical activity ameliorates impaired hippocampal neurogenesis in the Tg4-42 mouse model of Alzheimer’s disease. ASN Neuro. 2019;11:1759091419892692.
Leiter O, Zhuo Z, Rust R, Wasielewska JM, Grönnert L, Kowal S, et al. Selenium mediates exercise-induced adult neurogenesis and reverses learning deficits induced by hippocampal injury and aging. Cell Metab. 2022;34:408-23.e8.
Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, et al. Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science. 2018;361:eaan8821.
Horowitz AM, Fan X, Bieri G, Smith LK, Sanchez-Diaz CI, Schroer AB, et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science. 2020;369:167–73.
Fatt MP, Tran LM, Vetere G, Storer MA, Simonetta JV, Miller FD, et al. Restoration of hippocampal neural precursor function by ablation of senescent cells in the aging stem cell niche. Stem Cell Rep. 2022;17:259–75.
Abshenas R, Artimani T, Shahidi S, Ranjbar A, Komaki A, Salehi I, et al. Treadmill exercise enhances the promoting effects of preconditioned stem cells on memory and neurogenesis in Aβ-induced neurotoxicity in the rats. Life Sci. 2020;249: 117482.
Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–5.
Wu CW, Chen YC, Yu L, Chen HI, Jen CJ, Huang AM, et al. Treadmill exercise counteracts the suppressive effects of peripheral lipopolysaccharide on hippocampal neurogenesis and learning and memory. J Neurochem. 2007;103:2471–81.
Pedersen BK. Physical activity and muscle-brain crosstalk. Nat Rev Endocrinol. 2019;15:383–92.
Mishra R, Phan T, Kumar P, Morrissey Z, Gupta M, Hollands C, et al. Augmenting neurogenesis rescues memory impairments in Alzheimer’s disease by restoring the memory-storing neurons. J Exp Med. 2022;219: e20220391.
Huang YQ, Wu C, He XF, Wu D, He X, Liang FY, et al. Effects of voluntary wheel-running types on hippocampal neurogenesis and spatial cognition in middle-aged mice. Front Cell Neurosci. 2018;12:177.
Rao Y, Du S, Yang B, Wang Y, Li Y, Li R, et al. NeuroD1 induces microglial apoptosis and cannot induce microglia-to-neuron cross-lineage reprogramming. Neuron. 2021;109:4094-108.e5.
Tarutani A, Adachi T, Akatsu H, Hashizume Y, Hasegawa K, Saito Y, et al. Ultrastructural and biochemical classification of pathogenic Tau, α-synuclein and TDP-43. Acta Neuropathol. 2022;143:613–40.
Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, et al. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–84.
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, et al. Brain homogenates from human tauopathies induce Tau inclusions in mouse brain. Proc Natl Acad Sci USA. 2013;110:9535–40.
Hasegawa M, Arai T, Nonaka T, Kametani F, Yoshida M, Hashizume Y, et al. Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann Neurol. 2008;64:60–70.
Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4:124–34.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59.
Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211.
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38.
Koo JH, Jang YC, Hwang DJ, Um HS, Lee NH, Jung JH, et al. Treadmill exercise produces neuroprotective effects in a murine model of Parkinson’s disease by regulating the TLR2/MyD88/NF-κB signaling pathway. Neuroscience. 2017;356:102–13.
Jang Y, Koo JH, Kwon I, Kang EB, Um HS, Soya H, et al. Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson’s disease mice. Brain Res. 2017;1655:186–93.
Małkiewicz MA, Szarmach A, Sabisz A, Cubała WJ, Szurowska E, Winklewski PJ. Blood-brain barrier permeability and physical exercise. J Neuroinflammation. 2019;16:15.
Lee B, Shin M, Park Y, Won SY, Cho KS. Physical exercise-induced myokines in neurodegenerative diseases. Int J Mol Sci. 2021;22:5795.
Kohman RA, Rhodes JS. Neurogenesis, inflammation and behavior. Brain Behav Immun. 2013;27:22–32.
Terreros-Roncal J, Moreno-Jiménez EP, Flor-García M, Rodríguez-Moreno CB, Trinchero MF, Cafini F, et al. Impact of neurodegenerative diseases on human adult hippocampal neurogenesis. Science. 2021;374:1106–13.
Ryan SM, Nolan YM. Neuroinflammation negatively affects adult hippocampal neurogenesis and cognition: can exercise compensate? Neurosci Biobehav Rev. 2016;61:121–31.
Svensson M, Andersson E, Manouchehrian O, Yang Y, Deierborg T. Voluntary running does not reduce neuroinflammation or improve non-cognitive behavior in the 5xFAD mouse model of Alzheimer’s disease. Sci Rep. 2020;10:1346.
Miki Stein A, Munive V, Fernandez AM, Nuñez A, Torres AI. Acute exercise does not modify brain activity and memory performance in APP/PS1 mice. PLoS ONE. 2017;12: e0178247.
Tsai CL, Pai MC, Ukropec J, Ukropcová B. Distinctive effects of aerobic and resistance exercise modes on neurocognitive and biochemical changes in individuals with mild cognitive impairment. Curr Alzheimer Res. 2019;16:316–32.
Medhat E, Rashed L, Abdelgwad M, Aboulhoda BE, Khalifa MM, El-Din SS. Exercise enhances the effectiveness of vitamin D therapy in rats with Alzheimer’s disease: emphasis on oxidative stress and inflammation. Metab Brain Dis. 2020;35:111–20.
Wu C, Yang L, Tucker D, Dong Y, Zhu L, Duan R, et al. Beneficial effects of exercise pretreatment in a sporadic Alzheimer’s rat model. Med Sci Sports Exerc. 2018;50:945–56.
Nichol KE, Poon WW, Parachikova AI, Cribbs DH, Glabe CG, Cotman CW. Exercise alters the immune profile in Tg2576 Alzheimer mice toward a response coincident with improved cognitive performance and decreased amyloid. J Neuroinflammation. 2008;5:13.
Kohman RA, Bhattacharya TK, Wojcik E, Rhodes JS. Exercise reduces activation of microglia isolated from hippocampus and brain of aged mice. J Neuroinflammation. 2013;10:114.
Liu Y, Chu JMT, Yan T, Zhang Y, Chen Y, Chang RCC, et al. Short-term resistance exercise inhibits neuroinflammation and attenuates neuropathological changes in 3xTg Alzheimer’s disease mice. J Neuroinflammation. 2020;17:4.
Hashiguchi D, Campos HC, Wuo-Silva R, Faber J, Gomes da Silva S, Coppi AA, et al. Resistance exercise decreases amyloid load and modulates inflammatory responses in the APP/PS1 mouse model for Alzheimer’s disease. J Alzheimers Dis. 2020;73:1525–39.
Gomes da Silva S, Simões PS, Mortara RA, Scorza FA, Cavalheiro EA, da Graça Naffah-Mazzacoratti M, et al. Exercise-induced hippocampal anti-inflammatory response in aged rats. J Neuroinflammation. 2013;10:61.
Leem YH, Lee YI, Son HJ, Lee SH. Chronic exercise ameliorates the neuroinflammation in mice carrying NSE/htau23. Biochem Biophys Res Commun. 2011;406:359–65.
Bartlett DB, Fox O, McNulty CL, Greenwood HL, Murphy L, Sapey E, et al. Habitual physical activity is associated with the maintenance of neutrophil migratory dynamics in healthy older adults. Brain Behav Immun. 2016;56:12–20.
Beavers KM, Hsu FC, Isom S, Kritchevsky SB, Church T, Goodpaster B, et al. Long-term physical activity and inflammatory biomarkers in older adults. Med Sci Sports Exerc. 2010;42:2189–96.
Martins RA, Veríssimo MT, Coelho e Silva MJ, Cumming SP, Teixeira AM. Effects of aerobic and strength-based training on metabolic health indicators in older adults. Lipids Health Dis. 2010;9:76.
Li Z, Chen Q, Liu J, Du Y. Physical exercise ameliorates the cognitive function and attenuates the neuroinflammation of Alzheimer’s disease via miR-129-5p. Dement Geriatr Cogn Disord. 2020;49:163–9.
Abd El-Kader SM, Al-Jiffri OH. Aerobic exercise improves quality of life, psychological well-being and systemic inflammation in subjects with Alzheimer’s disease. Afr Health Sci. 2016;16:1045–55.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 31771318); the Key Special Project of Disciplinary Development, Hubei Superior Discipline Group of Physical Education and Health Promotion; the 14th Five-Year-Plan Advantageous and Characteristic Disciplines (Groups) of Colleges and Universities in Hubei Province for Exercise and Brain Science from Hubei Provincial Department of Education; as well as the Chutian Scholar Program and Innovative Start-Up Foundation from Wuhan Sports University to NC.
Funding
This work was financially supported by the National Natural Science Foundation of China (No. 31771318).
Author information
Authors and Affiliations
Contributions
NC and MW contributed the conception and design of the project; MW and HZ wrote the manuscript draft; HZ, JL and JH contributed the design and drawing of the figures; NC completed final editing and revision of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors have declared no conflict of interest for this manuscript.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, M., Zhang, H., Liang, J. et al. Exercise suppresses neuroinflammation for alleviating Alzheimer’s disease. J Neuroinflammation 20, 76 (2023). https://doi.org/10.1186/s12974-023-02753-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-023-02753-6