
Abstract
Skeletal system disease (SSD) is defined as a class of chronic disorders of skeletal system with poor prognosis and causes heavy economic burden. m6A, methylation at the N6 position of adenosine in RNA, is a reversible and dynamic modification in posttranscriptional mRNA. Evidences suggest that m6A modifications play a crucial role in regulating biological processes of all kinds of diseases, such as malignancy. Recently studies have revealed that as the most abundant epigentic modification, m6A is involved in the progression of SSD. However, the function of m6A modification in SSD is not fully illustrated. Therefore, make clear the relationship between m6A modification and SSD pathogenesis might provide novel sights for prevention and targeted treatment of SSD. This article will summarize the recent advances of m6A regulation in the biological processes of SSD, including osteoporosis, osteosarcoma, rheumatoid arthritis and osteoarthritis, and discuss the potential clinical value, research challenge and future prospect of m6A modification in SSD.
Similar content being viewed by others

Introduction
Skeletal system disease (SSD) is defined as a class of chronic disorders of skeletal system, such as bone, cartilage and joint, characterized by cartilage destruction, limitation of movement and significant disability, and commonly includes osteoporosis (OP), osteosarcoma (OS), rheumatoid arthritis (RA), osteoarthritis (OA) and so on [1,2,3,4]. As the incidence of SSD is increasing year by year, it has become one of the heaviest burdens on global health and economics [5]. In recent decades, SSD has gained increasing attention, however, the pathogenesis remains unsystematically discussed and few effective therapeutic options for patients [6]. Therefore, it is imperative to understand the molecular mechanism of SSD and more efforts are needed to develop new therapeutic strategies.
N6‐methyladenosine (m6A) modification, involved in posttranscriptional RNA regulation, is the most common epigenetic modification that perpetuate alternative gene expression and function without changing gene sequence [7, 8]. And it is widely involved in regulating RNA metabolism, such as RNA splicing, transportation, localization, translation, degradation and so on [9, 10]. As the most abundant modification of mRNA and lncRNA among mammals, m6A modification plays a critical role in understanding the pathogenesis of diseases [11, 12]. For example, it has been found that m6A modification can regulate glioblastoma stem cells growth and self-renewal [13]. Therefore, m6A modification has become a popular research topic and researchers have made great progress in exploring pathogenesis and new therapeutic strategies. Similarly, current studies have found m6A modification plays a key role in the occurrence and development of SSD [14]. Such as Pan et al. found that silencing of WTAP retarded OS progress which could be partially eliminated by knockdown of ALB [15].
Therefore, in the current work, we summarize the latest research concerning roles that m6A plays in common SSD including osteoporosis, osteosarcoma, rheumatoid arthritis and osteoarthritis. And we also discuss the novel potential of m6A modification as a therapeutic target of SSD.
Enzymes and proteins involved in modification by m6A
m6A methylation, first discovered in 1974 [16], can be understood simply that a methyl group is added to the sixth position of the nitrogen atom of adenosine on RNA [17]. It is shown that m6A methylation is a reversible and dynamic posttranscriptional modification of RNA which is distinct from other kinds of epigenetic modifications [18] (Fig. 1). There are three kinds of enzymes and proteins involved in m6A modification, including writers, erasers and, readers [19]. Firstly, m6A writers, means methyltransferases, include methyltransferase-like 3 (METTL3), METTL5, METTL14, METTL16, Wilm’s tumor 1-associated protein (WTAP), RBM15/15B, ZC3H13 and VIRMA, also called KIAA1429. Secondly, m6A erasers, means demethylases, include fat mass and obesity-associated protein (FTO) and α-ketoglutarate-dependent dioxygenase alk B homolog 5 (ALKBH5). Lastly, m6A readers are kind of RNA binding proteins (RBPs) which could recognize and bind to the m6A modification position of RNA. These RBPs consisted of YT521-B homology (YTH) domain family (YTHDF1/2/3, YTHDC1/2), heterogeneous nuclear ribonucleoproteins (hnRNP) family (HNRNPC/G/A2B1), insulin-like growth factor 2 mRNA-binding proteins family (IGF2BP1/2/3) and ELAV-like protein 1 (ELAVL1) [20,21,22,23].

Structural schematic diagram of reversible and dynamic posttranscriptional m6A modification of RNA. a. In the nucleus, methyltransferases and demethylases regulated the m6A modifications of target mRNAs. b. In the nucleus, m6A readers such as hnRNPA2B1 regulated the splicing process of target mRNAs. c. In the cytoplasm, m6A readers such as YTHDF1 regulated the translation process of target mRNAs. d. In the cytoplasm, m6A readers such as IGF2BP1 regulated the stability of target mRNAs. e. In the cytoplasm, m6A readers such as YTHDF3 mediated the degradation of target mRNAs
m6A writers
METTL3, first identified m6A methyltransferase [24], is the only catalytic subunit of the methyltransferase complex (MTC) by which writers promote m6A methylation on posttranscriptional RNA [25]. METTL14, the most important accessory subunit of MTC, can enhance the catalytic activity of METTL3 and stabilize MTC [26]. Moreover, METTL3 has no catalytic activity without the help of METTL14 and stable METTL3-METTL14 complex is the core component of MTC [27, 28]. WTAP, another accessory subunit, has been proved to regulate m6A methylation by recognizing and recruiting METTL3-METTL14 complex to target RNA [29]. RBM15/15B, RNA binding domain protein, can work with WTAP to recruit m6A complex to RNA binding sites [30]. Besides, it has been found that METTL5 and METTL16 catalyze m6A methylation independent of the MTC, METTL5 is involved in rRNA m6A [31] and METTL16 promotes depositing m6A, splicing and translation of mRNA transcripts [32]. Moreover, ZC3H13 is responsible for m6A installation and plays critical roles in the proliferation of cancer cells [33]. VIRMA, also called KIAA1429, is important for specially depositing m6A methylation to 3′UTR region of mRNA [34, 35].
m6A erasers
m6A erasers, demethylases, remove m6A modifications from target RNA and are essential to mediate the reversible and dynamic process of m6A methylation [36, 37]. FTO is the first m6A eraser which was discovered in 2011 [38]. The demethylation mechanism of FTO is to firstly oxidate m6A to N6-hydroxymethyladenosine (hm6A), secondly transform hm6A into N6-formyladenosine (f6A), finally change f6A into adenosine [39]. It is proved that FTO downregulation cooperating with YTHDF2 promotes mRNA degradation of ATG5 and ATG7 [40] and FTO enhances STAT3 mRNA stability in a demethylation-mediated m6A manner [41]. Luo et al. reported that FTO regulated ADRB1 mRNA level through mRNA alternative splicing [42]. As the second demethylase discovered in 2013 [43], ALKBH5 can catalyze m6A into adenosine directly which is different from FTO [44]. Studies have shown that ALKBH5 upregulated, HuR bound to the unmethylated 3′UTR and then promoted the expression of FOXM1 nascent transcripts [45], meanwhile, knockout of ALKBH5 inhibited he translation efficiency of FOXM1 mRNA [46]. Moreover, ALKBH5 affects mRNA stability rather than translation to regulate its targets’ expression level [47].
m6A readers
m6A readers, effectively binding to and recognizing m6A, make sure that m6A can regulate the metabolism of m6A-modified RNA [48]. YTH domain family is the most common “reader” of m6A and YTHDF2 is the first identified reader [49]. YTHDF1-3 are highly homologous in the sequence of their YTH domain but play different roles in processing mRNA [37]. YTHDF1 promotes mRNA translation whereas YTHDF2 catalyzes the degradation of mRNA, and YTHDF3 participates in the metabolism of mRNA in synergy with YTHDF1 and YTHDF2 [50]. For example, Bai et al. found that YTHDF2 promotes its degradation in an m6A-dependent manner through binds to the 3'-UTR of DAPK3 mRNA [51]. It is reported that YTHDF3 and YTHDF1 interactions with proteins associated with mRNA translation are blocked by O-GlcNAcylation, which inhibits the translation-promoting function [52]. Furthermore, YTHDC1-2 were another important m6A reader involved in RNA-related processes [53, 54]. For example, YTHDC1 works as a splicing protein alternatively splicing its targets’ mRNA [55]. And YTHDC2 was reported to recognize and bind to the m6A site "GGACA" in LIMK1 mRNA, thereby increasing LIMK1 mRNA stability and expression [56].
In addition to YTH domain family, there are other m6A readers involved in RNA splicing, translation and transport [57]. It has been reported that HNRNPC and HNRNPG regulate RNA metabolism via m6A switch, which is distinct from other m6A readers [58]. As for m6A switch, m6A-induced RNA structural alteration, was first identified in 2015 and can promote HNRNP proteins binding to mRNA for RNA processing [58, 59]. Conversely, HNRNPA2B1 binds to mRNA directly, not through m6A switch [60]. It is reported that HNRNPA2B1 binds to Specific RNA substrates and DNA motifs and then regulates RNA metabolism processes [61]. Moreover, IGF2BP1-3 cooperate with ELAVL1 to enhance the stability and translation efficiency of mRNA [62]. For example, it has been reported that IGF2BP1 coworking with ELAVL1 enhances the stability of MIR210HG [63]. IGF2BP3/ELAVL1 complex recognizes and enhances mRNA stability which prolongs half-lives of the mRNA molecules and increases target genes expression [64]. Furthermore, SR family is another kind of RNA binding proteins regulating RNA alternative splicing [65]. And it is reported that SRSF9 binds to and stabilizes DSN1 mRNA in an m6A-related manner, which is weakened by METTL3 downregulation [66].
m6A and skeletal system disease
m6A and osteoarthritis
OA, mainly characterized by progressive cartilage degeneration and synovial inflammation in pathology, is the most common chronic, degenerative joint disease in aging population and the symptom of OA is pain, stiffness, limitation of movement and joint deformity [67, 68]. The exact pathogenesis of OA is still ambiguous and the role of m6A modifications in OA has attracted great attention of researchers in recent 5 years. We show in Table 1 the function m6A regulation in osteoarthritis.
Increasing evidences have shown that METTL3, WTAP, FTO and ALKBH5 are aberrantly expressed in OA chondrocytes and involved in OA pathogenesis by regulating chondrocyte proliferation, apoptosis, extracellular matrix (ECM) degradation through related signal pathways [69, 72, 80, 84]. For example, Lin et al. have revealed that WTAP-mediated miR-92b-5p/TIMP4 axis plays crucial role in OA development. Overexpression of WTAP suppressed cell proliferation enhanced apoptosis and ECM degradation in an LPS-induced OA chondrocyte model and promoting cartilage damage in a destabilizing the medial meniscus (DMM)-induced OA mice model [69]. Furthermore, the fibroblast-like synoviocyte (FLS) senescence is tightly associated with OA progression. It has been displayed that autophagy-related 7 (ATG7) mRNA regulates FLS senescence through autophagy-GATA4 axis in an METTL3/YTHDF2 dependent manner. And targeted METTL3 inhibition has been proved to enhance autophagy and reduce senescence- associated secretory phenotype expression in senescent FLS to decelerate OA development in DMM-induced mice model [78]. Furthermore, Lv et al. found that FTO cooperated with exosomal OANCT from dysfunctional chondrocytes could affect PIK3R5 mRNA stability, and then promoted OA progression via PI3K/AKT/mTOR pathway [82].
Lange-Brokaar’s study found that immunocytes, including T cells, B cells, NK cells and so on, contributed to cartilage injury and repair [87]. Recently study revealed that m6A was proved involved in OA progression by regulating immune responses [88]. Evidence found that IGFBP1 and RBM15B were strongly correlated with infiltrating immunocytes [89], YTHDF2 was positively related with regulatory T cells, IGFBP2 was negatively associated with dendritic cells [90], and IGF2BP3 upregulation promoted macrophage M1 polarization in OA [91].
Long non-coding RNA (lncRNA), transcript lacking protein-coding ability but mediating gene expression, is believed to play an essential role in OA development [92, 93]. It has been found that METTL3-mediated upregulation of IGFBP7-OT via DNMT1/DNMT3a-IGFBP7 axis promotes OA progression [72]. Moreover, Ren et al. found that METTL3 overexpression increased the LINC0068 level in OA, which repressed chondrocyte proliferation and accelerated ECM degradation [76]. Conversely, ALKBH5-mediated upregulation of HS3ST3B1-IT1 suppresses OA progression [84]. What’s more, Yang et al. showed that FTO suppressed the expression of AC008, which promoted chondrocyte apoptosis and ECM degradation in OA [83]. The above findings suggest that the relationship between m6A and lncRNA may provide new ideas for the future therapy of OA.
Recent studies mainly focus on the regulatory role of METTL3 in OA and the molecular mechanisms of METTL3 in OA progress are revealed in detail. Therefore, METTL3 may serve as potential therapeutic targets to alleviate OA [73]. For example, Xiong et al. proved that METTL3 interacted with NEK7 to inhibit OA progress [70]. Zhou et al. reported that hucMSCs-EVs could alleviate OA through combining with METTL3 to lower the m6A level of NLRP3 mRNA [74]. And BMSC-Exos were applied to prevent OA progression via disrupting METTL3-m6A-ACSL4 axis [94]. In addition, FTO and ALKBH5 are identified as potential targets to alleviate OA. For example, Liu et al. reported that overexpression of FTO inhibited the miR-3591-5p maturation via regulating demethylation of pri-miR-3591, and then downregulated PRKAA2 to alleviate cartilage damage in OA [80]. Cai and colleagues found that upregulating FTO promoted proliferation, inhibited apoptosis and inflammation in LPS-induced C28/I2 cells through the miR-515-5p/ TLR4/MyD88/NF-κB axis [81]. Moreover, senescent mesenchymal stem cell (MSC) can be another target to alleviate OA progression. Ye et al. have proved that overexpression of ALKBH5 inhibits MSC senescence by degrading CYP1B1 mRNA via IGF2BP1 [85].
However, there still some limitations in the study of m6A and osteoarthritis. Firstly, studies about m6A in regulation immune microenvironment of OA are mostly based on bioinformatics analysis [95, 96], they need to be verified by more experiments. Secondly, although many differentially expressed m6A regulators have been identified via bioinformatic analysis, only the mechanism of METTL3, WTAP, FTO, ALKBH5, YTHDF2 and IGF2BP3 in OA have been shown in recent studies. Therefore, more experiments are needed to reveal mechanisms of m6A regulators and their targets in OA. In addition, m6A regulators can be potential therapeutic targets for OA [97], but there are lack of studies exploring potential role of m6A regulators in diagnosis, treatment and prognosis of OA.
m6A and rheumatoid arthritis
Rheumatoid arthritis (RA), characterized by persistent synovial inflammation and joint destruction, is the most common chronic autoimmune joint disorder and the main pathological feature is immune cells infiltration, proliferation of fibroblast-like synoviocytes (FLSs) and cartilage erosion [98, 99]. It has been proven that epigenetic regulation is involved in RA pathogenesis [100] and studies of m6A modifications in RA are increasingly significant in recent years.
FLSs are not only the main cells involved in joint destruction of RA, but also responsible for synovial inflammation by releasing inflammatory cytokines like interleukin-6 [101, 102]. Recently, Ye et al. using single-cell analysis identified IGFBP2 and METTL3 were key factors in regulating m6A of NPR3 and GHR in synovial fibroblasts, and then mediated the development and progression of RA [103]. It has been revealed that overexpression of METTL3 not only promoted proliferation, migration and invasion, but also increased the expression of inflammatory cytokines in RA-FLSs via the NF-κB signaling pathway [104]. Wang et al. confirmed that METTL3 overexpression inhibited releasing inflammatory cytokines of macrophages in RA through NF-κB signaling [105]. Furthermore, in Zhang’s study, METTL3 coordinated with YTHDF2 to enhance the inflammatory response in monocytes depend on suppressing the expression of PGC-1α [106]. Together that the immunopathogenesis of RA is complex that METTL3 can play different roles in regulating inflammatory response of different target cells in RA. The reasons may attribute to: Different signaling pathways and different immunocytes. Wang et al. investigated that METTL3 regulated macrophages through NF-κB signaling, meanwhile, Zhang et al. investigated that METTL3 regulated monocytes through suppressing PGC-1α. The roles of METTL3 in inflammatory response of of RA are limited, which may be the future research directions.
Except for METTL3, studies also revealed that METTL14 and ALKBH5 participated in migration, invasion, proliferation and related inflammatory response of RA-FLSs [107, 108]. For example, it has been reported that METTL14 regulates TNFAIP3 expression via m6A-related mRNA stability and involved the inflammatory response of active rheumatoid arthritis [109]. Tan et al. shown that METTL14 improved the mRNA stability of LASP1 through m6A modification and promoted FLSs activation via the LASP1/SRC/AKT axis in RA [107]. Furthermore, Xu et al. reported that ALKBH5 and YTHDF2 regulated m6A modification of MYO1C and contributed to synovial aggression and joint destruction in RA [110]. Mechanistically, ALKBH5 enhances JARID2 mRNA stability through IGF2BP3 and suppressed NLRP3 mRNA expression in cooperation with YTHDF2 are crucial for proliferation, migration, and invasion of RA FLSs [108, 111].
METTL3, METTL14 and ALKBH5 may work as therapeutic target for relieving RA due to crucial regulatory roles in RA progress. For example, Shi et al. found that METTL3 knockdown inhibited inflammatory response in human RA-FLSs and rat AIA-FLSs [104]. METTL14 silencing was proved to relieve RA progression through LASP1/SRC/AKT signaling pathway [107]. And Xiao et al. reported that ALKBH5 inhibited RA progression by suppressing NLRP3 through YTHDC2 [111]. These findings provide novel sights for developing clinical treatment strategies targeting METTL3, METTL14 and ALKBH5. In addition, through systemically analyzing the roles of m6A modifications in RA based on gene expression profiling data, novel targets were identified for RA clinical diagnosis and therapy, such as, Geng et al. found that IGF2BP3 and YTHDC2 could be used to diagnose RA accurately [112]. Besides, WTAP was involved in the m6A modification of ETS1 and regulated the macrophage polarization progression in RA [113]. Furthermore, Song et al. reported that the therapeutic benefits of infliximab can be predicted via the m6A diagnosis model, consist of 20 m6A regulators. This m6A diagnosis model classified RA patients into three clusters with distinct molecular and cellular signatures. And patient in cluster C with adaptive lymphocytes and NK-mediated cytotoxicity signatures was significantly benefited from infliximab therapy [114]. It is more believable that theoretical targets from bioinformatics analysis can be confirmed by experiments like methylated RNA immunoprecipitation, CCK8 assay, RT-qPCR, Western blot and so on. Through cross-validated work based on datasets and experiments, Lin et al. have reported that TGM2 can be a therapeutic target, regulating RA-FLS proliferation and apoptosis via activating NF-κB signaling [115].
m6A and osteoporosis
OP has become a global health problem mainly among postmenopausal women and elder people, characterized by decreasing bone mineral density, degrading bone microarchitecture and increasing bone fragile [116, 117]. And high incidence of disability and mortality due to osteoporotic fracture can be observed in OP patients. It is observed that the cumulative mortality rate is 69.38% and the 1 year mortality rate increases by 2% per year for patients with osteoporotic hip fracture from 1999 to 2015 [118]. Bone homoeostasis, maintained by osteoblasts, osteoclasts and bone marrow mesenchymal stem cells (BMSCs), is tightly associated with pathogenesis of OP [119]. And recent studies have revealed the molecular mechanisms of m6A modifications on osteoblasts, osteoclasts and BMSCs [120]. We show the functions of m6A regulators in OP in Table 2.
Osteoclasts are responsible for bone resorption which is important for maintaining bone homoeostasis [119]. Increasing studies have shown that m6A methylation is involved in OP by regulating osteoclast differentiation [121, 133]. Deng and colleagues found that METTL14 was downregulated in postmenopausal osteoporotic women and overexpression of METTL14 can suppress osteoclast formation to ameliorate osteoporosis by stabilizing GPX4 [133]. Moreover, Wang et al. illustrated that METTL14 could alleviate OP via upregulating m6A level of SIRT1 mRNA [134]. Meanwhile, FTO and WTAP participate in alleviating OP through negatively regulation of osteoclast differentiation [120, 140]. Furthermore, Li et al. revealed that METTL3 regulated osteoclast differentiation and function through different mechanisms involving ATP6V0D2 mRNA degradation mediated by YTHDF2 and TRAF6 mRNA nuclear export [148]. And EGR1 positively promotes osteoclastogenesis in osteoporosis by increasing METTL3 and CHI3L1 levels [121]. In addition, YTHDF1 was reported that promoted inflammatory osteoclast differentiation by regulating ER stress and TNFRSF11A mRNA stability [149].
Bone homoeostasis is a dynamic process including removing old bone and promoting new bone formation. Conversed with osteoclasts for bone absorption, m6A contribute to bone homoeostasis by regulating osteoblast activity for new bone formation [150]. Wang et al. found that METTL14 protects against OP via TCF1/RUNX2 axis [135]. Interestingly METTL14, targeted by miR-103-3p, can also inhibit osteoblast activity to promote OP [139]. Likewise, METTL3 was found to promote osteoblast differentiation through piRNA-36741 overexpression [128] and activate the ferroptosis in osteoblasts via ASK1-p38 signaling pathway in diabetic bone loss [127]. Moreover, Zhang et al. reported that METTL3 promoted osteoblast differentiation via Smad signaling and MAPK signaling by stabilizing Smad7 and Smurf1 in YTHDF2-dependent manner [151]. However, METTL3 and YTHDF2 mediated osteoblast apoptosis by regulating endoplasmic reticulum stress during LPS-induced inflammation [152]. Furthermore, FTO was reported that play important function in regulating the maintenance of bone mass and protecting osteoblasts from genotoxic damage [153]. Wu and colleagues demonstrated that YTHDF1 regulated osteogenesis of MC3T3-E1 cells under hypoxia via enhancing the stability of THBS1 [154]. In addition, natural compound Ecliptae herba and wedelolactone can enhance the expression of METTL3 to upregulate the level of HIF-1α, VEGF-A, and RASSF1 and then regulating osteoblastogenesis [122].
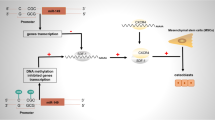
As the cellular source for bone reconstruction, BMSCs are known as the ability of self-renewal and multilineage differentiation [119]. And whether BMSCs differentiate into osteogenic cells or adipocytes is linked to the pathogenesis of OP. Recent studies have revealed several mechanisms of m6A modifications to promoted osteogenic differentiation and inhibited adipogenic differentiation of BMSCs [137, 141, 147]. Growing evidences have shown the relationship between m6A modification and BMSC differentiation. We show in Fig. 2 the schematic model of m6A related protein in regulating osteogenesis in osteoporosis. Among these studies, Liu et al. identified that WTAP-mediated m6A methylation regulated BMSCs differentiation through the miR-29b-3p/HDAC4 axis [140]. You and coworkers found that upregulation of WTAP promoted MiR-181a and miR-181c expression via YTHDC1 recognization which sequentially inhibited mRNA expression of SFRP1 to promote BMSCs osteogenic differentiation [141]. Accumulating evidences have reported that METTL3 is another key m6A regulator playing essential roles in BMSCs differentiation. Cooperating with MIR99AHG and LINC00657, METTL3 respectively increases the expression of BMPR1B by sponging miR-144-3p and targets miR-4660 to promote osteogenic differentiation of BMSCs [124, 126]. Moreover, METTL3 overexpression can protect BMSCs from OP via PTH /Pth1r, pre-miR-320/RUNX2 and the Wnt signaling pathway [125, 129, 130]. Furthermore, METTL14, FTO, YTHDF1 and ALKBH1 have been confirmed to regulate BMSCs differentiation, which will help to understand the molecular mechanisms of OP deeply and develop potential therapeutic strategies for OP [132, 142, 147, 155].

Structural schematic diagram of m6A related protein in regulating osteogenesis in osteoporosis. The role of m6A writers, easers work with readers in regulating the osteogenesis of BMSC and then affacting osteoporosis
Taken together, m6A modifications, including METTL3, METTL14, WTAP, FTO, YTHDF1 and ALKBH1, play essential roles in regulating bone homoeostasis. METTL3 and METTL14 can be novel targets for OP treatment due to diverse regulatory roles on osteoblasts, osteoclasts and BMSCs. For example, METTL14 overexpression suppressed osteoclast differentiation via enhancing GPX4 mRNA stability [133], and METTL14 overexpression increased SIRT1 mRNA expression to promoted osteoblast differentiation [134], and METTL14 overexpression was found to promote the osteogenic differentiation ability of BMSCs [137]. Although diverse regulatory roles of METTL3, METTL14 and FTO show the novel targets for OP treatment, there is lack of research to develop therapeutic strategies targeting METTL3 and METTL14. And m6A regulators identified as therapeutic targets for OP are limited, thus, more attention should be paid to validating the mechanisms of potential therapeutic targets and developing future treatment strategies for OP.
m6A and osteosarcoma
OS is the most malignant bone tumor mainly occurring in children and adolescents, and the prognosis of OS is poor due to strong aggression, early metastasis, fast growth and high mortality. The definitive diagnosis of OS needs bone biopsy, including needle biopsy and incisional biopsy, and the standard therapeutic strategies consists of neoadjuvant chemotherapy pre/postoperatively and surgical resection [156, 157]. Recently, m6A methylation has become a hot blot in understanding the molecular mechanism of OS pathogenesis to overcome the delayed diagnosis, low survival rate, metastasis and recurrence of OS [158]. We summarize the recent findings related to m6A methylation in OS in Table 3.
Proliferation, migration, invasion and metastasis are the main biological processes of OS, of which the underlying mechanisms may help to understand the pathogenesis and prognosis of OS [199]. It is reported that METTL3 plays an essential role in promoting OS progression by cooperating with noncoding RNAs, circRNAs and other targets [159,160,161]. For example, Zhang et al. found that METTL3 promoted the proliferation and migration of OS via increasing the stability of MALAT1 [159]. And inhibiting MALAT1 in a METTL3-dependant manner suppresses cell migration and invasion in Ewing's sarcoma through miR-124-3p/CDK4 axis [200]. Moreover, Zhou et al. found that silencing of another METTL3-mediated noncoding RNA DANCR can also inhibit OS cells proliferation, migration, and invasion [162]. METTL3 and METTL14 co-treatment suppressing DIRAS1 expression can reserve the inhibitory effect on malignant behaviors of HOS cells [165]. Furthermore, evidence have shown that circRNF220 and circNRIP1 are identified as oncogenic roles in OS progression via METTL3 methylation. And METTL3-induced circRNF220 and circNRIP1 promote OS proliferation, invasion by modulating miR-330-5p/survivin axis or sponging miR-199a respectively [160, 168]. In addition, targeting the METTL3/ZBTB7C axis, METTL3/USP13/ATG5 axis, METTL3/COPS5 axis, METTL3/LEF1/ Wnt/β-catenin axis, METTL3/TRAF6 axis and METTL3/HDAC5/miR-142-5p/ARMC8 axis may help to understand OS pathogenesis and develop the novel strategies for OS therapy [161, 163, 164, 166, 167, 172].
Besides METTL3, KIAA1429, YTHDF3, METTL16, WTAP, RBM15 and ALKBH5 are identified to regulate OS progression [15, 181, 185, 187, 192, 201]. It is reported that KIAA1429 knockdown decreases the activity of JAK2/STAT3 signal to decreased cell proliferation, migration, and invasion of OS, which can be rescued by JAK2/STAT3 stimulator colivelin [188]. And KIAA1429 can also act as a crucial gene to regulate Ewing sarcoma progression after CRISPR-Cas9 knockout [187]. Moreover, researchers found that aerobic glycolysis is essential to make sure OS cells obtain metabolic survival advantage compared with other cells [192]. Yang et al. have proved that circ-CTNNB1 interacted with RBM15 drives aerobic glycolysis and OS progression by elevating key aerobic glycolysis genes expression, such as PGK1 [185].
Increasing evidences have reported that m6A regulators can act as prognostic biomarkers of OS [202]. For example, high expression of METTL3, IGF2BP2, YTHDC1, KIAA1429 and HNRNPA2B1 and low expression of FTO, METTL14 and YTHDF2 have been identified to result in poor prognosis of OS [4, 203, 204]. Through a comprehensive bioinformatic analysis, Kaplan–Meier and Cox regression analyses, Li et al. concluded that low expression of FTO was prognostic biomarker for poor prognosis in OS [203]. Interestingly, upregulated FTO was proved to predict a poorer prognosis of OS via the FTO/DACT1 axis [176]and overexpression of FTO was correlated with low prognosis survival of OS patients by regulating KLF3 expression [205]. In summary, the findings between bioinformatic analysis and experimental validation may be different. Thus, more experiments are necessary to validate the prognostic biomarkers identified through comprehensive bioinformatic analysis.
Effective neoadjuvant chemotherapy is helpful to improve survival of OS, but chemotherapy resistance is a big challenge for OS therapy [206]. Thus, it is necessary to explore the mechanism of chemotherapy resistance. Recently, researchers have observed higher levels of METTL3, ALKBH5, METTL14 and IGF2BP1 in chemotherapy-resistant OS cells [173, 193, 207], and METTL14 and YTHDF2 are associated with multiple drug sensitivity in Ewing's sarcoma. Different from higher levels of METTL3 observed in Wang’s study [207], Zhou et al. found that lower levels of METTL3 and YTHDF2 contributed to higher TRIM7 expression which promoted chemotherapy resistance in OS through ubiquitination of BRMS1 [169]. The possible reasons of this phenomenon include but not limited to: (1) experiment methods and materials between Wang’s study and Zhou’s study are different; (2) different RNA binding proteins can lead to different functional effects of m6A methylation on downstream processes, like translation [208]. Furthermore, IGF2BP1/ERRα axis can regulate the metabolic reprogramming of Doxorubicin-resistant OS cells [193] and METTL14-IGF2BP2-MN1 panel is responsible for all-trans-retinoic acid resistance in osteosarcoma [173]. The above findings suggest that m6A modification can provide a novel sight in understanding mechanism of chemotherapy resistance in OS and develop effective chemotherapy strategies for OS. Nevertheless, more studies are still needed to enrich our limited understanding of the relationship between m6A methylation and chemotherapy resistance in OS. And possible controversial findings of chemotherapy resistance in OS should be validated by further detailed studies.
Discussion
Clinical value of m6A modifications for SSD
Given that dysregulation of m6A modifications plays a crucial role in: (1) Affecting ECM degradation, immune microenvironment and apoptosis in OA chondrocyte; (2) Regulating inflammatory response of immunocytes, and rheumatoid fibroblast-like synoviocytes proliferation, migration and invasion; (3) Maintaining bone homoeostasis of OP, including osteoblasts, osteoclasts and BMSCs; (4) Proliferation, and metastasis of OS [108, 209]. It is believable that m6A modifications can offer novel ideas for early diagnosis, clinical therapy and prognosis of skeletal system diseases [210]. For example, Luo et al. firstly revealed that the expression of ALKBH5, FTO, and YTHDF2 in peripheral blood of RA patients were significant low compared to control patients [211]. YTHDF2 was significant decreased while IL-1β expression was increased in RA patients’ peripheral blood mononuclear cells [212]. These findings make it possible to early diagnose RA, even skeletal system diseases, via detecting the level of m6A regulators in peripheral blood. Moreover, Bian and colleagues concluded that YTHDF2 was a crucial m6A regulators with high diagnostic value in OA, based on the protein and mRNA contents of YTHDF2 were significantly lower in OA patients via WB and qRT-PCR, which was consistent with the results of bioinformatic analysis [213]. Similarly, in Qiao’s study, METTL16, CBLL1, FTO, and YTHDF2 were applied to build a diagnostic model in OP. METTL16 and FTO were identified as risk factors in promoting OP progress, whereas CBLL1 and YTHDF2 were protective factors [214]. Furthermore, m6A modified noncoding RNAs were also could be act as biomarkers for disease diagnosis. Such as, Chen et al. reveled that hyper-methylated hsa_circ_0007259 activated STAT3 signaling pathway via sponge miR-21-5p and could be acted as a potential biomarker in RA [215]. Besides, it has been reported that m6A-related lncRNAs regulated the tumor immune microenvironment and predicted the overall survival of OS patients [190, 216]. Although researchers have made progress in exploring the diagnosis value of m6A methylation in SSD, further detailed studies are needed to verify the effectiveness, accuracy and feasibility of these findings in larger samples.
In addition to diagnosis, researchers have explored treating SSD via targeting m6A regulators. For example, Chen et al. found that knockdown of METTL3 could inhibit SASP expression in OA-FLS and relieve the cartilage destruction in DMM mice model [78]. In Ye’s study, overexpression of ALKBH5 could alleviated MSC senescence through inhibiting the expression of CYP1B1 and alleviating mitochondrial dysfunction [85]. Moreover, Wang and his coworkers reported that knockdown METTL3 could inhibit EGR1 expression to suppress osteoclastogenesis, and then alleviate OP [121]. These results illustrated that targeting m6A regulators expression might be novel strategy for SSD treatment. Besides modified the expression of m6A regulators in the molecular level for SSD therapy, natural compounds or small molecules were also shown significant role in regulating the expression of m6A regulators. Such as, Natural Compound Radicicol was identified as a potent FTO inhibitor and exhibited a dose-dependent inhibition of FTO demethylation activity with an IC50 value of 16.04 μM [217]. Moreover, Gao et al. have shown that acetaminophen treatment could recover m6A levels and related protein expression mainly including, downregulating METTL3 and upregulating ALKBH5, and suppress inflammatory cytokines secretion in IL-1β-treated chondrocyte cells [218]. Moreover, Chinese Ecliptae herba extract and its component wedelolactone enhances osteoblastogenesis of BMSCs by targeting METTL3-mediated m6A methylation of HIF-1α, VEGF-A, and RASSF1 [122].
And recent studied found that drug resistance of OS may attribute to m6A methylation. Evidence suggested that IGF2BP1/ERRα axis was involved in metabolic reprogramming which led to chemoresistance of OS cells [193]. What’s more, METTL3 and YTHDF2 METTL3 and YTHDF2 mediated N6-Methyladenosine modification of TRIM7 positively regulates chemoresistance in OS through ubiquitination of BRMS1 [169]. However, the understanding of m6A methylation in clinical application of SSD is still in infant stage, lacking exploring signaling pathways, m6A methylation targeted drugs and developing effective therapeutic strategies. And more studies should focus on explore the mechanisms of m6A methylation in diagnosis, clinical therapy and prognosis of SSD based on the breakthroughs of m6A methylation in SSD pathogenesis.
Challenges of m6A methylation in SSD
First, recent advances of m6A methylation in SSD mainly focus on METTL3, METTL14, WTAP, FTO, ALKBH5, other m6A writers (eg. METTL16, RBM15/15B and KIAA1429) and m6A readers (eg. YTHDF1/2/3, YTHDC1/2, HNRNPC/G/A2B1 and IGF2BP1/2/3) should obtain more research attention. Second, study of m6A methylation in immune microenvironment of SSD is limited. It is known that m6A interacted with noncoding RNA is associated with biological processes of many diseases. Thus, the role of m6A and noncoding RNA in immune microenvironment of SSD might be a hot blot for future research. Third, the dual roles of m6A methylation in SSD may make it more complex to understand SSD pathogenesis. For example, METTL3 was proved to promote osteoclastogenesis in osteoporosis [121], meanwhile, Tian et al. showed that upregulation of METTL3 could enhance osteoblastogenesis of BMSC [122]. The reasons of the dual roles of METTL3 and METTL14 in OP may attribute to: (1) Different study purposes. Tian et al. explored the molecular mechanism of Ecliptae herba on osteoblast differentiation in OP while Wang et al. investigated EGR1-mediated METTL3/m6A/CHI3L1 axis in OP. (2) Different research material and methods. Tian et al. treated BMSC isolated from BALB/c mice with MHL while Wang et al. used OVX mouse osteoporosis model and stimulated mouse BMMs with cytokines. (3) Different signaling pathways. Tian et al. found that MHL targeted METTL3 and then uoregulated METTL3 to promote osteoblastogenesis. And Wang et al. found that EGR1 upregulated METTL3 and then increased CHI3L1 level to promote osteoclastogenesis. Although the dual roles of m6A regulators are limited in current studies, further studies are needed to reveal the underlying mechanism of m6A methylation dual roles.
Conclusion
Taken together, this article summarizes the recent advances of regulatory role of m6A modifications in common SSD (OA, RA, OP and OS). Although m6A modifications have brought significant breakthroughs for underlying mechanisms of biological and pathological processes in SSD, there is a lack of detailed studies on m6A modification involved in clinical diagnosis, treatment and prognosis of SSD. Hopefully, future studies will show deeper understand of m6A regulation in SSD pathogenesis and make the clinical value of m6A modification come true.
Availability of data and materials
Not applicable.
Abbreviations
- OS:
-
Osteosarcoma
- SSD:
-
Skeletal system disease
- m6A:
-
N (6)-methyladenine
- OA:
-
Osteoarthritis
- OP:
-
Osteoporosis
- RA:
-
Rheumatoid arthritis
- mRNA:
-
Messenger RNA
- lncRNA:
-
Long non-coding RNA
- YTH:
-
RNA binding protein DC1/2
- MDR1:
-
Multidrug resistance protein 1
- ALB:
-
Albumin
- RBPs:
-
RNA binding proteins
- ELAVL1:
-
ELAV-like protein 1
- MTC:
-
Methyltransferase complex;
- hm6A:
-
N6-hydroxymethyladenosine
- f6A:
-
N6-formyladenosine
- ACL-T:
-
Anterior cruciate ligament transection
- MIA:
-
Maternal immune activation
- BMSC-Exos:
-
Bone marrow stem cell exosome
- AIA-FLS:
-
Adjuvant-induced arthritis fibroblast-like synoviocyte
- OVX:
-
Ovariectomized
- ASC:
-
Adipose stem cell
- ECM:
-
Extracellular matrix
- LPS:
-
Lipopolysaccharide
- DMM:
-
Destabilizing the medial meniscus
- FLS:
-
Fibroblast-like synoviocyte
- mTOR:
-
Rapamycin
- MSC:
-
Mesenchymal stem cell
- CCK8:
-
Cell counting kit 8
- BMSCs:
-
Bone marrow mesenchymal stem cells
- ER:
-
Endoplasmic reticulum
- circRNAs:
-
Circular RNAs
- ES:
-
Ewing’s sarcoma
- IL-1β:
-
Interleukin-1β
- SASP:
-
senescence-associate secretory phenotype
References
Zhai G, et al. Regulatory role of N6-methyladenosine (m6A) modification in osteoarthritis. Front Cell Dev Biol. 2022;10:946219.
Wu S, et al. N(6) -methyladenosine and rheumatoid arthritis: a comprehensive review. Front Immunol. 2021;12:731842.
Bai Q, et al. Comprehensive analysis of the m6A-related molecular patterns and diagnostic biomarkers in osteoporosis. Front Endocrinol. 2022;13:957742.
Zhang W, et al. m6A regulators are associated with osteosarcoma metastasis and have prognostic significance: a study based on public databases. Medicine. 2021;100(20):e25952.
Liu H, Zheng YL, Wang XQ. The emerging roles of N(6)-methyladenosine in osteoarthritis. Front Mol Neurosci. 2022;15:1040699.
Han J, et al. Novel insights into the interaction between N6-methyladenosine methylation and noncoding RNAs in musculoskeletal disorders. Cell Prolif. 2022;55(10):e13294.
Zhang M, et al. N(6)-methyladenosine RNA modification regulates photosynthesis during photodamage in plants. Nat Commun. 2022;13(1):7441.
Yang C, et al. The crucial mechanism and therapeutic implication of RNA methylation in bone pathophysiology. Ageing Res Rev. 2022;79:101641.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21(1):14.
Zhang Q, Xu K. The role of regulators of RNA m(6)A methylation in lung cancer. Genes Dis. 2023;10(2):495–504.
Hui L, et al. Epigenetic regulations in autoimmunity and cancer: from basic science to translational medicine. Eur J Immunol. 2023;53(4):e2048980.
Allegri L, et al. Role of m6A RNA methylation in thyroid cancer cell lines. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms231911516.
Cui Q, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Tian C, et al. Mettl3 regulates osteogenic differentiation and alternative splicing of vegfa in bone marrow mesenchymal stem cells. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20030551.
Pan Q, et al. WTAP contributes to the tumorigenesis of osteosarcoma via modulating ALB in an m6A-dependent manner. Genomics. 2023. https://doi.org/10.1016/j.ygeno.2023.110566.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A, 1974;71(10):3971–5.
Zhang Z, et al. RNA m(6)A reader YTHDF2 facilitates precursor miR-126 maturation to promote acute myeloid leukemia progression. Genes Dis. 2024;11(1):382–96.
Chen Y, et al. The functions and mechanisms of post-translational modification in protein regulators of RNA methylation: Current status and future perspectives. Int J Biol Macromol. 2023;253(Pt 2):126773.
Li Y, et al. Rna M(6) a methylation regulates glycolysis of beige fat and contributes to systemic metabolic homeostasis. Adv Sci. 2023;10(25):e2300436.
Zhang J, et al. A lncRNA from the FTO locus acts as a suppressor of the m(6)A writer complex and p53 tumor suppression signaling. Mol Cell. 2023;83(15):2692–708.
Yu Y, et al. RNA N6-methyladenosine methylation and skin diseases. Autoimmunity. 2023;56(1):2167983.
Xu Y, et al. Novel insights into the METTL3-METTL14 complex in musculoskeletal diseases. Cell Death Discov. 2023;9(1):170.
Fang Z, et al. Role of m6A writers, erasers and readers in cancer. Exp Hematol Oncol. 2022;11(1):45.
Bujnicki JM, et al. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT-A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol. 2002;55(4):431–44.
Qi YN, et al. Methyltransferase-like proteins in cancer biology and potential therapeutic targeting. J Hematol Oncol. 2023;16(1):89.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74(4):640–50.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17.
Wang X, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–8.
Tao X, et al. PGE(2) -EP3 axis promotes brown adipose tissue formation through stabilization of WTAP RNA methyltransferase. EMBO J. 2022;41(16):e110439.
Yuan J, et al. RBM15-mediating MDR1 mRNA m(6)A methylation regulated by the TGF-beta signaling pathway in paclitaxel-resistant ovarian cancer. Int J Oncol. 2023. https://doi.org/10.3892/ijo.2023.5560.
Wang L, Peng JL. METTL5 serves as a diagnostic and prognostic biomarker in hepatocellular carcinoma by influencing the immune microenvironment. Sci Rep. 2023;13(1):10755.
Su R, et al. METTL16 exerts an m(6)A-independent function to facilitate translation and tumorigenesis. Nat Cell Biol. 2022;24(2):205–16.
Xie R, et al. Overexpressed ZC3H13 suppresses papillary thyroid carcinoma growth through m6A modification-mediated IQGAP1 degradation. J Formos Med Assoc. 2023;122(8):738–46.
Li N, et al. KIAA1429/VIRMA promotes breast cancer progression by m(6) A-dependent cytosolic HAS2 stabilization. EMBO Rep. 2023;24(10):e55506.
Lee Q, et al. Overexpression of VIRMA confers vulnerability to breast cancers via the m(6)A-dependent regulation of unfolded protein response. Cell Mol Life Sci. 2023;80(6):157.
Dai Z, Asgari S. ALKBH8 as a potential N(6) -methyladenosine (m(6) A) eraser in insects. Insect Mol Biol. 2023;32(5):461–8.
Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505(7481):117–20.
Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Wang T, et al. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88.
Wang X, et al. m(6)A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy. 2020;16(7):1221–35.
Sun Z, et al. FTO promotes proliferation and migration of bladder cancer via enhancing stability of STAT3 mRNA in an m6A-dependent manner. Epigenetics. 2023;18(1):2242688.
Luo G, et al. FTO promoted adipocyte differentiation by regulating ADRB1 gene through m(6)A modification in Hycole rabbits. Anim Biotechnol. 2023;34(7):2565–70.
Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29.
Luo J, Xu T, Sun K. N6-methyladenosine RNA modification in inflammation: roles, mechanisms, and applications. Front Cell Dev Biol. 2021;9:670711.
Zhang S, et al. m(6)A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606.
Chao Y, Shang J, Ji W. ALKBH5-m(6)A-FOXM1 signaling axis promotes proliferation and invasion of lung adenocarcinoma cells under intermittent hypoxia. Biochem Biophys Res Commun. 2020;521(2):499–506.
Shen C, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self-renewal in acute myeloid leukemia. Cell Stem Cell. 2020;27(1):64–80.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–42.
Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Shi H, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–28.
Bai X, et al. YTHDF2 promotes gallbladder cancer progression and gemcitabine resistance via m6A-dependent DAPK3 degradation. Cancer Sci. 2023. https://doi.org/10.1111/cas.15953.
Chen Y, et al. O-GlcNAcylation determines the translational regulation and phase separation of YTHDF proteins. Nat Cell Biol. 2023;25(11):1676–90.
Jin Z, Liu Y. The m6A reader YTHDC1-mediated lncRNA CTBP1-AS2 m6A modification accelerates cholangiocarcinoma progression. Heliyon. 2023;9(9):e19816.
Tanabe A, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1alpha mRNA is translated. Cancer Lett. 2016;376(1):34–42.
Luxton HJ, et al. The oncogene metadherin interacts with the known splicing proteins YTHDC1, Sam68 and T-STAR and plays a novel role in alternative mRNA splicing. Cancers. 2019. https://doi.org/10.3390/cancers11091233.
Chen L, et al. LIMK1 m(6)A-RNA methylation recognized by YTHDC2 induces 5-FU chemoresistance in colorectal cancer via endoplasmic reticulum stress and stress granule formation. Cancer Lett. 2023;576:216420.
Zhao Y, et al. m(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13(1):35.
Liu N, et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Liu N, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Alarcon CR, et al. HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell. 2015;162(6):1299–308.
Zhou Z, et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol Cancer. 2020;19(1):104.
Huang H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Shi W, et al. MIR210HG promotes breast cancer progression by IGF2BP1 mediated m6A modification. Cell Biosci. 2022;12(1):38.
Li K, et al. Stabilization of oncogenic transcripts by the IGF2BP3/ELAVL1 complex promotes tumorigenicity in colorectal cancer. Am J Cancer Res. 2020;10(8):2480–94.
Wang H, et al. The cisplatin-induced lncRNA PANDAR dictates the chemoresistance of ovarian cancer via regulating SFRS2-mediated p53 phosphorylation. Cell Death Dis. 2018;9(11):1103.
Wang X, et al. SRSF9 promotes colorectal cancer progression via stabilizing DSN1 mRNA in an m6A-related manner. J Transl Med. 2022;20(1):198.
Jang S, Lee K, Ju JH. Recent updates of diagnosis, pathophysiology, and treatment on osteoarthritis of the knee. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22052619.
Liu Y, et al. N(6) -methyladenosine-modified circRNA RERE modulates osteoarthritis by regulating beta-catenin ubiquitination and degradation. Cell Prolif. 2023;56(1):e13297.
Lin Z, et al. N6-methyladenosine (m6A) methyltransferase WTAP-mediated miR-92b-5p accelerates osteoarthritis progression. Cell Commun Signal. 2023;21(1):199.
Xiong X, et al. METTL3 regulates the m(6)A modification of NEK7 to inhibit the formation of osteoarthritis. Cartilage. 2023. https://doi.org/10.1177/19476035231200336.
Yang S, et al. Epigenetic regulatory mechanism of ADAMTS12 expression in osteoarthritis. Mol Med. 2023;29(1):86.
Tang Y, et al. METTL3-mediated m(6)A modification of IGFBP7-OT promotes osteoarthritis progression by regulating the DNMT1/DNMT3a-IGFBP7 axis. Cell Rep. 2023;42(6):112589.
Shi L, et al. RPL38 knockdown inhibits the inflammation and apoptosis in chondrocytes through regulating METTL3-mediated SOCS2 m6A modification in osteoarthritis. Inflamm Res. 2022;71(7–8):977–89.
Zhou H, et al. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells alleviate osteoarthritis of the knee in mice model by interacting with METTL3 to reduce m6A of NLRP3 in macrophage. Stem Cell Res Ther. 2022;13(1):322.
Zhang H, et al. CREB ameliorates osteoarthritis progression through regulating chondrocytes autophagy via the miR-373/METTL3/TFEB Axis. Front Cell Dev Biol. 2021;9:778941.
Ren J, et al. N(6)-methyladenosine (m(6)A) methyltransferase METTL3-mediated LINC00680 accelerates osteoarthritis through m(6)A/SIRT1 manner. Cell Death Discov. 2022;8(1):240.
Xiao L, et al. N(6)-methyladenosine RNA methyltransferase like 3 inhibits extracellular matrix synthesis of endplate chondrocytes by downregulating sex-determining region Y-box transcription factor 9 expression under tension. Osteoarthr Cartil. 2022;30(4):613–25.
Chen X, et al. METTL3-mediated m(6)A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann Rheum Dis. 2022;81(1):87–99.
He Y, et al. Mettl3 inhibits the apoptosis and autophagy of chondrocytes in inflammation through mediating Bcl2 stability via Ythdf1-mediated m(6)A modification. Bone. 2022;154:116182.
Liu W, et al. FTO-mediated m6A demethylation of pri-miR-3591 alleviates osteoarthritis progression. Arthritis Res Ther. 2023;25(1):53.
Cai D, et al. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-κB axis. Int Immunopharmacol. 2023;114:109524.
Lv G, et al. Exosomes from dysfunctional chondrocytes affect osteoarthritis in Sprague-Dawley rats through FTO-dependent regulation of PIK3R5 mRNA stability. Bone Joint Res. 2022;11(9):652–68.
Yang J, et al. m(6)A-mediated upregulation of AC008 promotes osteoarthritis progression through the miR-328-3p-AQP1/ANKH axis. Exp Mol Med. 2021;53(11):1723–34.
Tang Y, et al. ALKBH5-mediated m(6)A demethylation of HS3ST3B1-IT1 prevents osteoarthritis progression. iScience. 2023;26(10):107838.
Ye G, et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp Mol Med. 2023;55(8):1743–56.
Yu B, et al. MiR-654-3p, reduced by the excessive ALKBH5, alleviated the inflammation in OA by targeting TNFRSF9, the trigger of the NF-κB pathway. Biochem Biophys Res Commun. 2022;634:30–9.
de Lange-Brokaar BJ, et al. Synovial inflammation, immune cells and their cytokines in osteoarthritis: a review. Osteoarthr Cartil. 2012;20(12):1484–99.
Gu Y, et al. N6-methyladenine regulator-mediated RNA methylation modification patterns in immune microenvironment regulation of osteoarthritis. Front Genet. 2023;14:1113515.
Ouyang Y, et al. Characterization of immune microenvironment infiltration and m(6)A regulator-mediated RNA methylation modification patterns in osteoarthritis. Front Immunol. 2022;13:1018701.
Liu Z, et al. Comprehensive analysis of m6A RNA methylation modification patterns and the immune microenvironment in osteoarthritis. Front Immunol. 2023;14:1128459.
Lu Y, et al. Expression pattern analysis of m6A regulators reveals IGF2BP3 as a key modulator in osteoarthritis synovial macrophages. J Transl Med. 2023;21(1):339.
Ransohoff JD, Wei Y, Khavari PA. The functions and unique features of long intergenic non-coding RNA. Nat Rev Mol Cell Biol. 2018;19(3):143–57.
Xie F, et al. Role of MicroRNA, LncRNA, and exosomes in the progression of osteoarthritis: a review of recent literature. Orthop Surg. 2020;12(3):708–16.
Cheng S, et al. Chondroprotective effects of bone marrow mesenchymal stem cell-derived exosomes in osteoarthritis. J Bioenerg Biomembr. 2023. https://doi.org/10.1007/s10863-023-09991-6.
Xie X, et al. Significance of m(6)A regulatory factor in gene expression and immune function of osteoarthritis. Front Physiol. 2022;13:918270.
Duan Y, et al. m6A regulator-mediated RNA methylation modification patterns regulate the immune microenvironment in osteoarthritis. Front Genet. 2022;13:921256.
Sang W, et al. METTL3 involves the progression of osteoarthritis probably by affecting ECM degradation and regulating the inflammatory response. Life Sci. 2021;278:119528.
Lin YJ, Anzaghe M, Schulke S. Update on the pathomechanism, diagnosis, and treatment options for rheumatoid arthritis. Cells. 2020. https://doi.org/10.3390/cells9040880.
Smolen JS, et al. Rheumatoid arthritis. Nat Rev Dis Primers. 2018;4:18001.
Ham S, et al. Epigenetic analysis in rheumatoid arthritis synoviocytes. Exp Mol Med. 2019;51(2):1–13.
Li XF, et al. Functional role of PPAR-gamma on the proliferation and migration of fibroblast-like synoviocytes in rheumatoid arthritis. Sci Rep. 2017;7(1):12671.
Zheng W, et al. Dulaglutide mitigates inflammatory response in fibroblast-like synoviocytes. Int Immunopharmacol. 2019;74:105649.
Xiao J, et al. Identification of synovial fibroblast-associated neuropeptide genes and m6A factors in rheumatoid arthritis using single-cell analysis and machine learning. Dis Markers. 2022;2022:5114697.
Shi W, et al. METTL3 promotes activation and inflammation of FLSs through the NF-kappaB signaling pathway in rheumatoid arthritis. Front Med. 2021;8:607585.
Wang J, et al. METTL3 attenuates LPS-induced inflammatory response in macrophages via NF-kappaB signaling pathway. Mediat Inflamm. 2019;2019:3120391.
Zhang X, et al. The m(6)A methyltransferase METTL3 modifies PGC-1alpha mRNA promoting mitochondrial dysfunction and oxLDL-induced inflammation in monocytes. J Biol Chem. 2021;297(3):101058.
Li X, et al. METTL14 promotes fibroblast-like synoviocytes activation via the LASP1/SRC/AKT axis in rheumatoid arthritis. Am J Physiol Cell Physiol. 2023;324(5):C1089–100.
Kuang Y, et al. ALKBH5-mediated RNA m(6) A methylation regulates the migration, invasion and proliferation of rheumatoid fibroblast-like synoviocytes. Arthr Rheumatol. 2023. https://doi.org/10.1002/art.42676.
Tang J, et al. METTL14-mediated m6A modification of TNFAIP3 involved in inflammation in patients with active rheumatoid arthritis. Arthr Rheumatol. 2023. https://doi.org/10.1002/art.42629.
Liu D, et al. SMOC2 promotes aggressive behavior of fibroblast-like synoviocytes in rheumatoid arthritis through transcriptional and post-transcriptional regulating MYO1C. Cell Death Dis. 2022;13(12):1035.
Xiao J, et al. ALKBH5-YTHDF2 m6A modification axis inhibits rheumatoid arthritis progression by suppressing NLRP3. Biochem Biophys Res Commun. 2023;668:70–6.
Geng Q, et al. Diagnostic gene signatures and aberrant pathway activation based on m6A methylation regulators in rheumatoid arthritis. Front Immunol. 2022;13:1041284.
Wan L, et al. Role of m6A modification and novel circ_0066715/ miR-486-5p/ ETS1 axis in rheumatoid arthritis macrophage polarization progression. Aging. 2022;14(24):10009–26.
Song S, et al. Predictive value of drug efficacy by M6A modification patterns in rheumatoid arthritis patients. Front Immunol. 2022;13:940918.
Lin X, et al. N6-methyladenosine modification of TGM2 mRNA contributes to the inhibitory activity of sarsasapogenin in rheumatoid arthritis fibroblast-like synoviocytes. Phytomedicine. 2022;95:153871.
Arceo-Mendoza RM, Camacho PM. Postmenopausal osteoporosis: latest guidelines. Endocrinol Metab Clin North Am. 2021;50(2):167–78.
Johnston CB, Dagar M. Osteoporosis in older adults. Med Clin North Am. 2020;104(5):873–84.
Guzon-Illescas O, et al. Mortality after osteoporotic hip fracture: incidence, trends, and associated factors. J Orthop Surg Res. 2019;14(1):203.
Sharma G, et al. Epigenetic regulation of bone remodeling and bone metastasis. Semin Cell Dev Biol. 2024;154(Pt C):275–85.
Shen X, et al. Suppression of TLR4 prevents diabetic bone loss by regulating FTO-mediated m(6)A modification. Int Immunopharmacol. 2023;122:110510.
Wang C, et al. EGR1 mediates METTL3/m(6)A/CHI3L1 to promote osteoclastogenesis in osteoporosis. Genomics. 2023;115(5):110696.
Tian S, et al. Chinese Ecliptae herba (Eclipta prostrata (L.) L.) extract and its component wedelolactone enhances osteoblastogenesis of bone marrow mesenchymal stem cells via targeting METTL3-mediated m6A RNA methylation. J Ethnopharmacol. 2023;312:116433.
Luo D, et al. Methyltransferase-like 3 modulates osteogenic differentiation of adipose-derived stem cells in osteoporotic rats. J Gene Med. 2023;25(5):e3481.
Li L, et al. METTL3-mediated long non-coding RNA MIR99AHG methylation targets miR-4660 to promote bone marrow mesenchymal stem cell osteogenic differentiation. Cell Cycle. 2023;22(4):476–93.
Wu T, et al. METTL3-m(6) A methylase regulates the osteogenic potential of bone marrow mesenchymal stem cells in osteoporotic rats via the Wnt signalling pathway. Cell Prolif. 2022;55(5):e13234.
Peng J, Zhan Y, Zong Y. METTL3-mediated LINC00657 promotes osteogenic differentiation of mesenchymal stem cells via miR-144-3p/BMPR1B axis. Cell Tissue Res. 2022;388(2):301–12.
Lin Y, et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. 2022;36(3):e22147.
Liu J, et al. piRNA-36741 regulates BMP2-mediated osteoblast differentiation via METTL3 controlled m6A modification. Aging (Albany NY). 2021;13(19):23361–75.
Yan G, et al. m(6)A Methylation of precursor-miR-320/RUNX2 controls osteogenic potential of bone marrow-derived mesenchymal stem cells. Mol Ther Nucleic Acids. 2020;19:421–36.
Wu Y, et al. Mettl3-mediated m(6)A RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat Commun. 2018;9(1):4772.
Yang JG, et al. Exosome-targeted delivery of METTL14 regulates NFATc1 m6A methylation levels to correct osteoclast-induced bone resorption. Cell Death Dis. 2023;14(11):738.
Dong X, et al. METTL14 mediates m(6)a modification on osteogenic proliferation and differentiation of bone marrow mesenchymal stem cells by regulating the processing of pri-miR-873. Mol Med Rep. 2023. https://doi.org/10.3892/mmr.2023.13053.
Deng M, et al. METTL14 represses osteoclast formation to ameliorate osteoporosis via enhancing GPX4 mRNA stability. Environ Toxicol. 2023;38(9):2057–68.
Wang C, et al. METTL14 alleviates the development of osteoporosis in ovariectomized mice by upregulating m(6)A level of SIRT1 mRNA. Bone. 2023;168:116652.
Wang X, et al. METTL14 upregulates TCF1 through m6A mRNA methylation to stimulate osteogenic activity in osteoporosis. Hum Cell. 2023;36(1):178–94.
Huang C, Wang Y. Downregulation of METTL14 improves postmenopausal osteoporosis via IGF2BP1 dependent posttranscriptional silencing of SMAD1. Cell Death Dis. 2022;13(11):919.
He M, et al. METTL14 regulates osteogenesis of bone marrow mesenchymal stem cells via inducing autophagy through m6A/IGF2BPs/Beclin-1 signal axis. Stem Cells Transl Med. 2022;11(9):987–1001.
Feng MG, et al. Osteogenic capacity and Mettl14 and Notch1 expression of adipose-derived stem cells from osteoporotic rats. Sichuan Da Xue Xue Bao Yi Xue Ban. 2021;52(3):423–9.
Sun Z, et al. MiR-103-3p targets the m(6) A methyltransferase METTL14 to inhibit osteoblastic bone formation. Aging Cell. 2021;20(2):e13298.
Liu J, et al. WTAP-mediated m6A RNA methylation regulates the differentiation of bone marrow mesenchymal stem cells via the miR-29b-3p/HDAC4 axis. Stem Cells Transl Med. 2023;12(5):307–21.
You Y, et al. WTAP-mediated m(6)A modification modulates bone marrow mesenchymal stem cells differentiation potential and osteoporosis. Cell Death Dis. 2023;14(1):33.
Chen LS, et al. The m(6)A demethylase FTO promotes the osteogenesis of mesenchymal stem cells by downregulating PPARG. Acta Pharmacol Sin. 2022;43(5):1311–23.
Zhuang J, et al. Downregulated fat mass and obesity-associated protein inhibits bone resorption and osteoclastogenesis by nuclear factor-kappa B inactivation. Cell Signal. 2021;87:110137.
Wang J, et al. RNA N6-methyladenosine demethylase FTO promotes osteoporosis through demethylating Runx2 mRNA and inhibiting osteogenic differentiation. Aging. 2021;13(17):21134–41.
Zhang X, et al. Extracellular vesicle-encapsulated miR-22-3p from bone marrow mesenchymal stem cell promotes osteogenic differentiation via FTO inhibition. Stem Cell Res Ther. 2020;11(1):227.
Li Y, et al. miR-149-3p regulates the switch between adipogenic and osteogenic differentiation of BMSCs by targeting FTO. Mol Ther Nucleic Acids. 2019;17:590–600.
Cai GP, et al. Alkbh1-mediated DNA N6-methyladenine modification regulates bone marrow mesenchymal stem cell fate during skeletal aging. Cell Prolif. 2022;55(2):e13178.
Li D, et al. METTL3 modulates osteoclast differentiation and function by controlling RNA stability and nuclear export. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21051660.
He M, et al. YTHDF1 regulates endoplasmic reticulum stress, NF-kappaB, MAPK and PI3K-AKT signaling pathways in inflammatory osteoclastogenesis. Arch Biochem Biophys. 2022;732:109464.
Anam AK, Insogna K. Update on osteoporosis screening and management. Med Clin North Am. 2021;105(6):1117–34.
Zhang Y, et al. METTL3 regulates osteoblast differentiation and inflammatory response via smad signaling and MAPK signaling. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms21010199.
Kong Y, et al. METTL3 mediates osteoblast apoptosis by regulating endoplasmic reticulum stress during LPS-induced inflammation. Cell Signal. 2022;95:110335.
Zhang Q, et al. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc Natl Acad Sci USA. 2019;116(36):17980–9.
Shi D, et al. Yth m(6)A RNA-binding protein 1 regulates osteogenesis of MC3T3-E1 cells under hypoxia via translational control of thrombospondin-1. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms24021741.
Liu T, et al. The m(6)A “reader” YTHDF1 promotes osteogenesis of bone marrow mesenchymal stem cells through translational control of ZNF839. Cell Death Dis. 2021;12(11):1078.
Simpson E, Brown HL. Understanding osteosarcomas. JAAPA. 2018;31(8):15–9.
de Azevedo JWV, et al. Biology and pathogenesis of human osteosarcoma. Oncol Lett. 2020;19(2):1099–116.
Wu Y, et al. The role of m6A methylation in osteosarcoma biological processes and its potential clinical value. Hum Genomics. 2022;16(1):12.
Zhang Y, et al. METTL3 mediated MALAT1 m6A modification promotes proliferation and metastasis in osteosarcoma cells. Mol Biotechnol. 2023. https://doi.org/10.1007/s12033-023-00953-2.
Liu F, et al. METTL3-mediated m6A modification of circRNF220 modulates miR-330–5p/survivin axis to promote osteosarcoma progression. J Cancer Res Clin Oncol. 2023. https://doi.org/10.1007/s00432-023-05455-x.
An X, et al. ZBTB7C m6A modification incurred by METTL3 aberration promotes osteosarcoma progression. Transl Res. 2023;259:62–71.
Zhou X, et al. METTL3 contributes to osteosarcoma progression by increasing DANCR mRNA stability via m6A modification. Front Cell Dev Biol. 2021;9:784719.
Wang C, et al. Deubiquitinase USP13 regulates glycolytic reprogramming and progression in osteosarcoma by stabilizing METTL3/m(6)A/ATG5 axis. Int J Biol Sci. 2023;19(7):2289–303.
Zhang C, et al. METTL3 upregulates COPS5 expression in osteosarcoma in an m(6)A-related manner to promote osteosarcoma progression. Exp Cell Res. 2022;420(2):113353.
Liu H, et al. Analysis of the function and mechanism of DIRAS1 in osteosarcoma. Tissue Cell. 2022;76:101794.
Jiang R, et al. METTL3 stabilizes HDAC5 mRNA in an m(6)A-dependent manner to facilitate malignant proliferation of osteosarcoma cells. Cell Death Discov. 2022;8(1):179.
Wang J, et al. m6A-dependent upregulation of TRAF6 by METTL3 is associated with metastatic osteosarcoma. J Bone Oncol. 2022;32:100411.
Meng Y, et al. Circular RNA circNRIP1 plays oncogenic roles in the progression of osteosarcoma. Mamm Genome. 2021;32(6):448–56.
Zhou C, et al. N6-Methyladenosine modification of the TRIM7 positively regulates tumorigenesis and chemoresistance in osteosarcoma through ubiquitination of BRMS1. EBioMedicine. 2020;59:102955.
Zhou L, et al. Silencing METTL3 inhibits the proliferation and invasion of osteosarcoma by regulating ATAD2. Biomed Pharmacother. 2020;125:109964.
Ling Z, Chen L, Zhao J. m6A-dependent up-regulation of DRG1 by METTL3 and ELAVL1 promotes growth, migration, and colony formation in osteosarcoma. 2020. Biosci Rep. https://doi.org/10.1042/BSR20200282.
Miao W, et al. The m6A methyltransferase METTL3 promotes osteosarcoma progression by regulating the m6A level of LEF1. Biochem Biophys Res Commun. 2019;516(3):719–25.
Li HB, et al. METTL14-mediated epitranscriptome modification of MN1 mRNA promote tumorigenicity and all-trans-retinoic acid resistance in osteosarcoma. EBioMedicine. 2022;82:104142.
Liu Z, et al. METTL14 overexpression promotes osteosarcoma cell apoptosis and slows tumor progression via caspase 3 activation. Cancer Manag Res. 2020;12:12759–67.
Shan HJ, et al. Fat mass and obesity associated (FTO)-mediated N6-methyladenosine modification of Krüppel-like factor 3 (KLF3) promotes osteosarcoma progression. Bioengineered. 2022;13(4):8038–50.
Lv D, et al. M(6)A demethylase FTO-mediated downregulation of DACT1 mRNA stability promotes Wnt signaling to facilitate osteosarcoma progression. Oncogene. 2022;41(12):1727–41.
Pan Q, et al. WTAP contributes to the tumorigenesis of osteosarcoma via modulating ALB in an m6A-dependent manner. Environ Toxicol. 2023;38(6):1455–65.
Ge J, et al. SNHG10/miR-141-3p/WTAP axis promotes osteosarcoma proliferation and migration. J Biochem Mol Toxicol. 2022;36(6):e23031.
Ren Z, et al. N(6)-methyladenosine methyltransferase WTAP-stabilized FOXD2-AS1 promotes the osteosarcoma progression through m(6)A/FOXM1 axis. Bioengineered. 2022;13(4):7963–73.
Chen S, et al. WTAP promotes osteosarcoma tumorigenesis by repressing HMBOX1 expression in an m(6)A-dependent manner. Cell Death Dis. 2020;11(8):659.
Yang Z, et al. ALKBH5 regulates STAT3 activity to affect the proliferation and tumorigenicity of osteosarcoma via an m6A-YTHDF2-dependent manner. EBioMedicine. 2022;80:104019.
Yadav P, et al. M6A RNA methylation regulates histone ubiquitination to support cancer growth and progression. Cancer Res. 2022;82(10):1872–89.
Yuan Y, et al. ALKBH5 suppresses tumor progression via an m(6)A-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma. Cell Death Dis. 2021;12(1):60.
Chen S, Zhou L, Wang Y. ALKBH5-mediated m(6)A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020;20:34.
Yang F, et al. Circ-CTNNB1 drives aerobic glycolysis and osteosarcoma progression via m6A modification through interacting with RBM15. Cell Prolif. 2023;56(1):e13344.
Huang H, et al. Analysis and identification of m(6)A RNA methylation regulators in metastatic osteosarcoma. Mol Ther Nucleic Acids. 2022;27:577–92.
Tan K, et al. CRISPR-Cas9 knockout screening identifies KIAA1429 as an essential gene in Ewing sarcoma. J Exp Clin Cancer Res. 2023;42(1):250.
Luo J, et al. The role and mechanism of JAK2/STAT3 signaling pathway regulated by m6A methyltransferase KIAA1429 in osteosarcoma. J Bone Oncol. 2023;39:100471.
Wei K, et al. Methylation recognition protein YTH N6-methyladenosine RNA binding protein 1 (YTHDF1) regulates the proliferation, migration and invasion of osteosarcoma by regulating m6A level of CCR4-NOT transcription complex subunit 7 (CNOT7). Bioengineered. 2022;13(3):5236–50.
Zheng D, et al. N6-methyladenosine-related lncRNAs Are potential prognostic biomarkers and correlated with tumor immune microenvironment in osteosarcoma. Front Genet. 2021;12:805607.
Yang J, et al. Circular RNA circ_0001105 inhibits progression and metastasis of osteosarcoma by sponging miR-766 and activating YTHDF2 expression. Onco Targets Ther. 2020;13:1723–36.
Liu D, et al. N(6)-methyladenosine reader YTHDF3 contributes to the aerobic glycolysis of osteosarcoma through stabilizing PGK1 stability. J Cancer Res Clin Oncol. 2023;149(8):4601–10.
He Q, et al. IGF2BP1-regulated expression of ERRalpha is involved in metabolic reprogramming of chemotherapy resistant osteosarcoma cells. J Transl Med. 2022;20(1):348.
Xu Z, et al. Mesenchymal stem cell-derived exosomes carrying microRNA-150 suppresses the proliferation and migration of osteosarcoma cells via targeting IGF2BP1. Transl Cancer Res. 2020;9(9):5323–35.
Wang L, et al. Expression of microRNA-150 and its target gene IGF2BP1 in human osteosarcoma and their clinical implications. Pathol Oncol Res. 2019;25(2):527–33.
Chen W, et al. Long non-coding RNA THOR promotes human osteosarcoma cell growth in vitro and in vivo. Biochem Biophys Res Commun. 2018;499(4):913–9.
Gu J, et al. lncRNA HCG11 suppresses human osteosarcoma growth through upregulating p27 Kip1. Aging. 2021;13(17):21743–57.
Zhang H, et al. DDX11-AS1 contributes to osteosarcoma progression via stabilizing DDX11. Life Sci. 2020;254:117392.
Gianferante DM, Mirabello L, Savage SA. Germline and somatic genetics of osteosarcoma—connecting aetiology, biology and therapy. Nat Rev Endocrinol. 2017;13(8):480–91.
He F, Ren T, Tang X. METTL3-modified lncRNA-MALAT1 regulates the molecular axis of miR-124–3p/CDK4 involved in Ewing’s sarcoma. Cell Mol Biol. 2023;69(6):193–7.
Cheng J, et al. METTL16 promotes osteosarcoma progression by downregulating VPS33B in an m(6) A-dependent manner. J Cell Physiol. 2023. https://doi.org/10.1002/jcp.31068.
Liu S, et al. Construction and validation of a potent epigenetic modification-related prognostic signature for osteosarcoma patients. J Oncol. 2021;2021:2719172.
Li J, et al. Dysregulated m6A-related regulators are associated with tumor metastasis and poor prognosis in osteosarcoma. Front Oncol. 2020;10:769.
Liu H, et al. Potential role of m6A RNA methylation regulators in osteosarcoma and its clinical prognostic value. J Orthop Surg Res. 2021;16(1):294.
Shan HJ, et al. Fat mass and obesity associated (FTO)-mediated N6-methyladenosine modification of Kruppel-like factor 3 (KLF3) promotes osteosarcoma progression. Bioengineered. 2022;13(4):8038–50.
Lindsey BA, Markel JE, Kleinerman ES. Osteosarcoma overview. Rheumatol Ther. 2017;4(1):25–43.
Wang Y, et al. Integrated analysis of transcriptome-wide m(6)A methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics. 2019;11(15):1693–715.
Zhang Z, et al. Genetic analyses support the contribution of mRNA N(6)-methyladenosine (m(6)A) modification to human disease heritability. Nat Genet. 2020;52(9):939–49.
Gu Y, et al. The essential roles of m(6)A modification in osteogenesis and common bone diseases. Genes Dis. 2024;11(1):335–45.
Zhao Z, et al. CLP1 is a prognosis-related biomarker and correlates with immune infiltrates in rheumatoid arthritis. Front Pharmacol. 2022;13:827215.
Luo Q, et al. Decreased ALKBH5, FTO, and YTHDF2 in peripheral blood are as risk factors for rheumatoid arthritis. Biomed Res Int. 2020;2020:5735279.
Yao F, et al. Expression and clinical significance of the m6A reader YTHDF2 in peripheral blood mononuclear cells from rheumatoid arthritis patients. J Immunotoxicol. 2022;19(1):53–60.
Bian A, et al. Diagnostic value and immune infiltration characterization of YTHDF2 as a critical m6A regulator in osteoarthritic synovitis. J Orthop Surg Res. 2023;18(1):535.
Qiao Y, et al. Identification and experimental validation of key m6A modification regulators as potential biomarkers of osteoporosis. Front Genet. 2022;13:1072948.
Luo H, et al. Exploring CircRNA N6-methyladenosine in human rheumatoid arthritis: hyper-methylated hsa_circ_0007259 as a potential biomarker and its involvement in the hsa_circ_0007259/hsa_miR-21-5p/STAT3 axis. Int Immunopharmacol. 2023;124(Pt A):110938.
Bi Y, et al. m6A-related lncRNAs predict overall survival of patients and regulate the tumor immune microenvironment in osteosarcoma. Comput Intell Neurosci. 2022;2022:9315283.
Wang R, et al. Identification of natural compound radicicol as a potent FTO inhibitor. Mol Pharm. 2018;15(9):4092–8.
Gao J, et al. Acetaminophen changes the RNA m(6)A levels and m(6)A-related proteins expression in IL-1beta-treated chondrocyte cells. BMC Mol Cell Biol. 2022;23(1):45.
Funding
This research was supported by the Natural Science Foundation of China (No. 82003126). Shenzhen Science and Technology Projects (No. JCYJ20210324103604013). Sichuan Science and Technology Program (No.2022YFS0609), Scientific Research Foundation of Southwest Medical University (No.2021ZKMS009). Shenzhen High-level Hospital Construction Fund (No. 1801024).
Author information
Authors and Affiliations
Contributions
YL, JL, ZY performed the literature search. Jianhui Liang and WS prepared the first draft of the manuscript. QY and WS wrote and edited the manuscript. WS draw the figures. WS supervised and QY polished the manuscript. All of the authors have read and agreed to published version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liang, J., Yi, Q., Liu, Y. et al. Recent advances of m6A methylation in skeletal system disease. J Transl Med 22, 153 (2024). https://doi.org/10.1186/s12967-024-04944-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-04944-y