
Abstract
Since the breakthrough discoveries of DNA and histone modifications, the field of RNA modifications has gained increasing interest in the scientific community. The discovery of N6-methyladenosine (m6A), a predominantly internal epigenetic modification in eukaryotes mRNA, heralded the creation of the field of epi-transcriptomics. This post-transcriptional RNA modification is dynamic and reversible, and is regulated by methylases, demethylases and proteins that preferentially recognize m6A modifications. Altered m6A levels affect RNA processing, degradation and translation, thereby disrupting gene expression and key cellular processes, ultimately resulting in tumor initiation and progression. Furthermore, inhibitors and regulators of m6A-related factors have been explored as therapeutic approaches for treating cancer. In the present review, the mechanisms of m6A RNA modification, the clinicopathological relevance of m6A alterations, the type and frequency of alterations and the multiple functions it regulates in different types of cancer are discussed.
Similar content being viewed by others
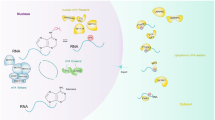
The N6-methyladenosine (m6A) RNA modification
M6A RNA modification, describes a methylation at the N6 position of adenosine, and is the most abundant internal modification in eukaryotes mRNA [1]. Since its discovery in 1974 [1], research on m6A has flourished owing to improvements in detection methods and the identification of important regulatory proteins and it recently reported that m6A modifications regulate the generation and function of transfer RNA (tRNA), ribosomal RNA (rRNA) and non-coding RNAs (ncRNAs), such as microRNA (miRNAs), long non-coding RNA (lncRNAs), and circular RNAs (circRNAs). Gene examination technology and high-throughput sequencing methods have demonstrated that the m6A modification is not randomly distributed, but is enriched near stop codons and 3′-untranslated terminal regions (UTRs) and translated near 5′-UTR or in long exons [2]. The m6A modification of RNA is dynamically and reversibly regulated by two important catalytic proteins, demethylases (writers) and methyltransferases (erasers) [3]. It is also recognized by a group of binding proteins so-called “readers” that decode m6A methylation and mediate recruitment of downstream functional complexes. A summary of known the machinery regulating m6A modifications is shown in Fig. 1.

Chemical basis and molecular composition of m6A RNA methylation
M6A modifications influence RNA maturation, transcription, localization, translation and metabolism [4]. The biological significance of m6A is demonstrated by the vital molecular functions regulated by this modification in mammals, including nervous system development, the circadian rhythm, DNA damage response, heat shock response and tumorigenesis amongst others [5]. Furthermore, m6A regulators are tightly correlated with the activation and inhibition of cancer-associated signaling pathways. In the present review, a summary of the literature and hypotheses surrounding m6A modifications is provided with a focus on the functional mechanisms of this ubiquitous RNA modification in carcinogenesis. Additionally, the mechanisms underlying therapeutic approaches which target m6A regulators for the development of anti-cancer drugs are described.
The methyltransferase complex writes the m6A modification
The m6A methyltransferase of RNA consists of “writer” proteins including: METTL3, METTL5, METTL14, METTL16 and their cofactors Wilms tumor 1associated protein (WTAP), RNA-binding motif protein 15 (RBM15/15B), Cbl proto-oncogene-like 1 (CBLL1; also known as HAKAI), zinc finger CCCH-type containing 13 (ZC3H13) and Vir-like m6A methyltransferase-associated (VIRMA; also known as KIAA1429). In 1997, METTL3 was demonstrated to serve as the primary methyltransferase critical for m6A methylation and aberrant expression of METTL3 could alter the total m6A methylation levels [6]. METTL14 serves as structural support for METTL3, and together they form the core methyltransferase complex inducing m6A modification synergistically [7]. WTAP stabilizes the core complex and promotes m6A by recruiting the complex to nuclear speckles [8]. RBM15/15B functions to assist binding of METTL3 and WTAP, directing the two proteins to their target sites [9]. VIRMA preferentially locates mRNA methylation modifications near the 3′-UTR and stop codon regions [10]. Other proteins, such as ZC3H13 and CBLL1, in concert with additional cofactors, including WTAP, control nuclear m6A methylation [11]. Recently, ZCCHC4, another CCHC zinc-finger-containing protein, was identified as a novel methyltransferase which was involved in the modification of the 28S rRNA, mediating rRNA ribosome subunit distribution and global translation [12].
METTL16 was proposed to act as an independent mRNA methyltransferase in 2017 [13]. It may regulate mRNA stability and splicing and its binding sites do not overlap with those of METTL3/METTL14 methylation complexes, suggesting that it functions independently [14]. Accordingly, METTL16 was confirmed to initiate splicing when a construct of METTL16 with a mutated catalytic domain was overexpressed [15]. Additionally, METTL16 could function alone and catalyze m6A on U6 snRNA and regulate tumorigenesis by targeting pre-mRNAs and ncRNAs [13, 16]. However, there are few studies which have shown that METTL3/16 may function as an m6A ‘reader’ [3]. Several writers, such as METTL3/16, are multifunctional enzymes with prominent non-catalytic activities. In the absence of an enzyme cofactor presence, m6A writers function as readers and bind to unmodified substrates constitutively, thus triggering non-catalytic functions [16]. Recently, METTL5 was identified as a novel methyltransferase responsible for 18S rRNA m6A modification [17, 18]. METTL5 forms a heterodimer with TRMT112 increasing its metabolic stability and modification area on precursor and mature forms of 18S rRNA. Similar to the complex of METTL3/METTL14, TRMT112 is a coactivator of METTL5. The atomic resolution structure of METTL5-TRMT112, supports the hypothesis that its RNA-binding mode differs distinctly from that of other m6A writers [17].
Additionally, 26 core interacting factors amongst hundreds of WTAP-binding proteins have been identified by co-immunoprecipitation studies, and > 100 proteins may bind to METTL3 or METTL14 [19]. Thus, there may be other components of the m6A methyltransferase complexes which remain to be discovered.
M6A methylation is removed via specific demethylases
Unlike the large multi-subunit m6A methyltransferase complex, only two m6A demethylases, FTO and AlkB homolog (ALKBH)5, have been identified. The two proteins are predominantly localized in the nucleus where the removal of m6A modification occurs. As a member of the AlkB family with a well-conserved catalytic domain, FTO was the first protein to be identified to catalyze m6A demethylation [20] and the notion of reversible m6A methylation in RNA was described. The hypothesis that FTO affects human obesity resulted in interest in examining its function [21]. ALKBH5 was the second RNA demethylase to be identified that could oxidatively reverse m6A modifications. ALKBH5 is expressed in the majority of the tissues, and its expression is particularly abundant in the testes [22]. Recently, the unique crystal structure of ALKBH5 have been resolved by several groups [22, 23]. Remarkably, FTO could mediate m6Am (N6,20-O-dimethyladenosine) demethylation as well. Unlike FTO, ALKBH5 seems to be an m6A-specific demethylase in mRNA [24]. These findings have greatly facilitated the development of inhibitors of m6A demethylases.
In addition, recent studies have shown that ALKBH3 may serve as a novel demethylase of m6A modifications [25, 26]. They identified m6A in mammalian tRNA as a novel ALKBH3 substrate and ALKBH3 preferentially modifies tRNA over mRNA or rRNA [25, 26].
M6A readers recognize m6A modification and confer specific phenotypic outcomes
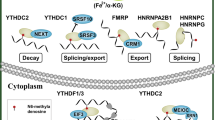
“Readers” are comprised of YTH domain-containing proteins (YTHDF1/2/3 and YTHDC1/2), heterogeneous nuclear ribonucleoproteins (including hnRNPC, hnRNPG and hnRNPA2B1) and insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs YTHDF1/2/3 and YTHDC1/2). In the cytosol, YTHDF1 interacts with initiation factors to facilitate RNA translation initiation [21]. YTHDF2 selectively binds m6A-methylated mRNA and regulates RNA degradation. YTHDF3 facilitates translation by promoting protein synthesis in synergy with YTHDF1 and affects mRNA decay mediated by YTHDF2 [27]. All three YTHDF proteins function co-operatively in fundamental biological pathways [28]. Moreover, YTHDF1 and YTHDF2 recognize circRNAs m6A marks and modify circRNAs expression [29]. YTHDC1 could increase the export of circNSUN2 to the cytoplasm [30]. In contrast to the functions of YTHDF2, IGF2BPs enhance the stability and translation of their target mRNAs by recognizing m6A modifications under normal and stressed conditions [31]. HNRNPC selectively recognizes m6A-induced splicing in mRNA secondary structures, whereas HNRNPA2B1 recognizes pri-miRNA m6A marks and interacts with DGCR8, thus stimulating miRNA processing [32].
Furthermore, several novel readers of m6A have been identified. In the cytoplasm, mRNA translation is also stimulated by the direct readers, eukaryotic initiation factor 3, Fragile X mental retardation 1 (FMR1) and ATP binding cassette subfamily F member 1 [33]. Taken together, the intricate interactions between m6A modifications and RNA-binding proteins may regulate mRNA expression at multiple levels.
m6A in cancer
Numerous studies have confirmed the effects of m6A modifications and its ability to fine-tune and coordinate gene expression [5, 25, 34,35,36,37,38,39]. The alterations of m6A levels may profoundly influence cancer hallmarks, including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, reprogramming energy metabolism, genomic instability and mutation, evading immune destruction and tumor-promoting inflammation, suggesting that m6A may function as an oncogenic or suppressive role in malignant tumors [25, 35, 37, 38, 40]. Certain proteins require m6A modification to participate in the mechanisms underlying the development of cancer, but it is not clear whether they make an effect in m6A modification or not. The specific roles of m6A proteins in human cancers are summarized in Tables 1 and 2.
Hematological malignancy: acute myeloid leukemia (AML)
AML is the result of uncontrolled proliferation and defects in cell differentiation of myeloid white blood cells, with distinct genetic aberrations, for which the therapeutic options remain unsatisfactory [111]. Mechanistically, several studies have shown that MELLT3 and METTL14 serve an oncogenic role in AML by promoting the translation of MYC, MYB, BCL2, SP1 and PTEN, thus increasing the levels of phospho-AKT [41, 42]. Additionally, METTL3 has been also shown to be mis-localized in the cytoplasm and results in a concomitant increase in WTAP expression, and WTAP has been demonstrated to function as a tumor suppressor gene. However, Bansal et al identified the oncogenic role of WTAP and its target, which is involved in the mTOR signaling pathway, in AML [43]. RBM15 exhibits a well-established oncogenic role in the development of hematologic malignancies [112] and is a fusion partner of the MKL1 gene in acute megakaryoblastic leukemia, a subtype of pediatric AML [113]. Notably, FTO expression is increased in AML with t(11q23)/MLL rearrangements, t(15; 17)/PML-RARA, FLT3-ITD, and/or NPM1 mutations. Downregulation of FTO inhibits the proliferation and differentiation capacity through reducing the abundance of m6A on the transcripts of ASB2 and RARA [44]. As core readers, YTHDFs and IGF2BPs may mediate the majority of the resultant phenotypes through regulation of MYC [46]. IGF2BP1 is a novel downstream target of LIN28B and functions via miRNA let-7 in AML, thus leading to cell cycle arrest, inhibition of cell proliferation and colony formation [47].
These studies corroborate the significance of m6A in AML. The regulators of m6A are all oncogenic in AML. METTL3, METTL14 and RBM15 expression are all upregulated in AML compared with other types of cancer [112]. Intriguingly, writers and erasers both serve a synergistic role in AML, and this may be due to the FTO-targeted sites, which exhibit effects on mRNA distinct from the known reading processes [114]. In previous studies, expression of METTL3, METTL14, FTO and YTHDFs were all correlated with MYC, highlighting the importance of precise regulation of MYC, and the notable impact dysregulation of MYC has on tumorigenesis [41, 42, 44, 46, 115]. Furthermore, hematopoietic stem cells (HSCs) notably influence AML. Abnormal or blocked differentiation of HSCs is a shared feature in AML. M6A regulates symmetric division of HSCs by modulating MYC mRNA levels, which is required for rapid regeneration during tissue damage and stress [116]. Mouse HSCs with METTL14 deleted obtained from primary leukemia blasts exhibit significantly delayed AML onset when implanted in mice [42]. RBM15 directly binds to and controls the differentiation of HSCs by regulating genes, such as GATA1, RUNX1, c-MPL and TAL1, which are critical for HSC self-renewal [113]. Suppression of YTHDF2 promotes expansion of HSCs ex vivo by stabilizing Tal1 mRNAs [46]. Therefore, focusing on HSCs or inhibition of MYC may serve as potential targets for treatment of AML.
Neurological tumors: glioblastoma (GBM)
GBM is the most lethal type of primary brain tumor. Studies on the role of METTL3 in GBM have produced contradictory results. Initially, Cui et al demonstrated that METTL3 and METTL14 inhibited growth and tumorigenesis of glioblastoma stem-like cells (GSCs) by downregulating the ADAM19/EPHA3/KLF4 pathway [49]. However, the same year, another group produced contradictory results which showed that METTL3 promoted GSC growth by upregulating SOX2 expression and protected GSCs from radiation-induced cytotoxicity [48]. Other studies have proposed that FTO and ALKBH5 expression are associated with a less favorable prognosis in patients with GBM. The lncRNA antisense to FOXM1 promotes an interaction between ALKBH5 and FOXM1 and subsequently ALKBH5 demethylates FOXM1 nascent transcripts, enhancing FOXM1 expression, thus maintaining tumorigenicity of GBM [50]. FTO constrains the progression of GBM progression and significantly shortens the lifespan of GSC-grafted mice [49].
The phenotypic differences associated with METTL3 may be explained by a differing reliance on m6A-modified RNAs in different types of GBM cells and differences in genetic heterogeneity. In addition, the mechanisms by which METTL3 exerts its effects can be divided into two modes: m6A-dependent and m6A-independent. METTL3 may exert oncogenic functions independent of its catalytic activity or its downstream readers. Consequently, METTL3 itself or possibly an unknown METTL3-complex components may function as m6A reader proteins, and this may underlie the dual functions of METTL3 under specific conditions [51]. Notably, GSCs are almost ubiquitously used in studies regarding GBM. GSCs can self-renew, are resistant to conventional therapy and in in vivo models, they give rise to tumor recurrence [117]. These findings may pave avenues for developing effective therapeutic strategies for treatment of GBM [50].
Respiratory tumors: lung cancer and nasopharyngeal carcinoma (NPC)
Lung cancer is the major cause of cancer-associated mortality worldwide. METTL3 acts as an oncogene in lung cancer via different mechanisms. METTL3 enhances translation of epidermal growth factor receptor (EGFR), the Hippo pathway effector TAZ and MAPKAPK2 (MK2) [51]. MiR-33a suppresses proliferation of non-small cell lung cancer cells via reducing the expression of METTL3 [52]. Additionally, METTL3 promotes YAP translation, increasing YAP activity via miR-1914-3p to induce drug resistance and metastasis [54]. Also, METTL3 facilitates the biogenesis of miR-143-3p to promote the brain metastasis of lung cancer via regulation of VASH1 [55]. In lung squamous cell carcinoma, METTL3 interacts with eukaryotic translation initiation 3 h to accelerate tumorigenicity by promoting translation of oncogenic mRNAs, such as Bromodomain-containing protein 4 (BRD4) [53]. Sumoylation of METTL3 also enhances tumorigenesis [118]. FTO expression is associated with a less favorable poor prognosis by increasing the expression levels of myeloid zinc finger protein 1 expression and the stability of ubiquitin-specific protease mRNA [56, 57]. Among the m6A readers, YTHDF2 facilitates METTL3-induced oncogenic effects by increasing degradation of SOCS2 [58]. IGF2BP1 is associated with a less favorable prognosis by increasing serum response factor mRNA stability and promoting cancer phenotypes in lung cancer [59].
In NPC, METTL3 is negatively associated with tumor repressor ZNF750, which is part of a ZNF750-FGF14 signaling axis that inhibits NPC growth [60]. LncRNA FAM225A, where m6A levels are highly enriched, functions as a competing endogenous RNA (ceRNA) sponging miR-590-3p and miR-1275, leading to the activation of FAK/PI3K/AKT signaling to promote proliferation and invasion of NPC cells [119]. Taken together, METTL3 governs in respiratory tumors. Moreover, m6A proteins could affect the biogenesis process of miRNAs/lncRNAs eventually influencing on development of tumor.
Gastrointestinal tumors: hepatocellular carcinoma (HCC), colorectal cancer (CRC), pancreas cancer and gastric carcinoma (GC)
HCC is a significant public burden, and the incidence is rising worldwide [120]. As mentioned above, METTL3 and METTL14 exert an oncogenic role in HCC via YTHDF2-dependent post-transcriptional silencing of SOCS2 [51]. METTL3 and YTHDF1 act as opposing prognostic factors of overall survival of patients with HCC via regulation of Snail, a key translator of EMT [61]. Additionally, KIAA1429 facilitates migration and invasion of HCC by inhibiting ID2 [62]. GATA3-AS functions as a guide lncRNA that promotes a malignant phenotype driven by KIAA1429 [63]. WTAP promotes the proliferative capacity of HCC through a p21/p27-dependent pattern mediated by ETS proto-oncogene 1(ETSI) [64]. However, Ma et al demonstrated that METTL14 is an anti-metastatic factor, positively modulating DGCR8 binding to primary miR126 (pri-miR126) [100]. Amongst the m6A readers, YTHDF1 overexpression is associated with a poor prognosis in HCC [121] and YTHDF2 is closely associated with the malignancy of HCC through interactions with miR-145 [65]. In contrast, two groups have shown that YTHDF2 suppresses the development of HCC development through stabilization of EGFR or interleukin 11 mRNA [102]. YTHDF2 downregulation increased inflammation and abnormal vascularization, degrading the mRNA of tumor suppressor genes in HCC [101]. In cholangiocarcinoma, WTAP is correlated with HCC metastasis [122]. In hepatoblastoma, METTL3 promotes development of hepatoblastoma development through increasing the expression of CTNNB1 via regulation of the Wnt/β-catenin pathway [66].
CRC has the second highest incidence of death worldwide [123]. METTL3 exhibits dual roles in CRC. METTL3 increases the expression of lncRNA RP11, which subsequently stimulates Zeb1 expression, initiating the dissemination of CRC cells [67]. Li et al demonstrated that METTL3 facilitates tumor progression via maintenance of expression of the stem cell marker SOX2, in an IGF2BP2-dependent manner in CRC [68]. They also suggested that METTL3 may serve as a marker of cancer stem cells (CSCs) due to its role in promoting stemness. Meanwhile, METTL3-mediated m6A modification and IGF2BP1 binding directly to CBX8 mRNA both could induce aberrant overexpression of CBX8, thus maintaining the stemness and inhibiting the chemosensitivity of CRC [70]. Peng et al confirmed that METTL3 advances the maturation of pri-miR-1246, where it further reverses the inhibition of the MAPK pathway, thus promoting metastasis [69]. However, recently METTL3 and METTL14 were reported to proliferation and migration of suppress CRC through regulating the p38/ERK pathway and tumor suppressor miR-375, respectively [103, 104]. Zhang et al showed that WTAP was a novel oncogene in CRC by Wnt signaling pathway [73]. In regards to erasers, FTO promoted progression of CRC cells through degrading expression of miR-1266, or initiation of the cellular signaling molecules STAT3, cyclin D1 and MMPs [71, 72]. YTHDC2, YTHDF1 and IGF2BPs are all hypothesized to promote metastasis of CRC by upregulating HIF-1α or c-Myc expression [31, 74, 75]. Yang et a showed that the specific mechanism by which YTHDF1 functions in CRC was through inhibition of the Wnt/β-catenin pathway, thus accelerating tumorigenicity and CSC activity [76]. Most recently, Wang et al introduced that lncRNA LINRIS stabilizes IGF2BP2 and promotes progression of CRC via aerobic glycolysis pathway [77].
Pancreatic cancer is a lethal malignancy, and is one of the most aggressive types of cancer [124]. Chen et al showed that YTHDF2 performed dual cellular functions in pancreatic cancer cells [79]: Promoting proliferation and inhibiting migration via different pathways, forming a phenomenon termed the migration-proliferation dichotomy. A novel mechanism was unveiled by which ALKBH5 inhibits the motility of pancreatic cancer by demethylating lncRNA KCNK15-AS1 [105]. Cigarette smoke condensate promotes aberrant overexpression of METTL3 in smokers, significantly promoting maturation of the oncogene, primary miR-25-3p, which activates AKT-p70S6K oncogenic signaling [78]. Bioinformatics analysis drew a consistent conclusion that METTL3 and FTO may promote proliferation and invasion of pancreatic cancer [125].
Despite the decline in the death rate of patients with GC, it is still the fifth most common malignancy worldwide [126]. miR-660 reduces proliferation by regulating expression of the oncogene E2F3 via m6A modifications in GC [127]. METTL3 promotes GC angiogenesis and glycolysis by increasing the stability of HDGF mRNA and activating the AKT signaling pathway, respectively [80]. ALKBH5 promotes invasion and metastasis of GC by decreasing methylation of the lncRNA NEAT1 [81]. Bioinformatics analysis predicted that m6A suppression promotes GC development through activating the Wnt/PI3K-AKT signaling pathway, whereas increasing m6A levels reversed these phenotypical and molecular changes [128, 129].
Cumulatively, emerging studies have focused on gastrointestinal tumors in 2019. These findings highlight the interaction between miRNAs/lncRNAs with m6A proteins in gastrointestinal tumors, such as pri-miR-126, miR-145, miR-1266, miR-1246, miR-25-3p, lncRNA NEAT1, KCNK15-AS1 and GATA3-AS. m6A promotes tumorigenesis via dysregulation of miRNAs/lncRNAs to modulate metastatic progression and increasing chromosomal instability [130, 131]. For example, METTL3 promotes the maturation of miRNAs such as let-7e, miR221/222, miR-4485, miR-25, miR-93, miR-126, miR-1246 and miR-335. METTL16 is associated with various ncRNAs, lncRNAs and pre-mRNAs, including MALAT1 lncRNA. IGF2BP1 enhances an aggressive phenotype in tumor cells by impairing miRNA-directed downregulation of oncogenic factors [132]. Therefore, further identification of tumor-related miRNAs/lncRNAs and investigations of their functions may highlight other interactions where m6A modifications are involved. CSCs and oncogene MYC exert powerful effects on gastrointestinal tumors, similar to those observed in GBM. METTL14 and YTHDF2 however, exert the opposite effect to that observed in HCC, and the same is true of the METTL3 and CRC. These targets may highlight potentially effective therapeutic strategies for treatment of gastrointestinal tumors.
Urological tumors: bladder cancer (BCA), renal cell carcinoma (RCC) and prostate cancer (PCA)
In 2019, several groups explored the function of m6A in bladder cancer [82,83,84, 106]. Cheng et al showed that METTL3 promoted the progression of BCA via an AFF4/NF-κB/MYC signaling network [83]. Shortly after, other groups showed that METTL3 promoted proliferation of BCA cells by accelerating pri-miR221/222 maturation and upregulating the expression of the oncogene CDCP1 [82, 84]. Bioinformatics analysis showed that m6A RNA methylation regulators can contribute to the malignant progression of BCA [133]. Gu et al demonstrated that METTL14 inhibited the self-renewal capacity of BCA initiating cells through targeting Notch1 [106]. These recent studies provide novel insights into new avenues for BCA therapy, and determining the inter-associations between the different underlying mechanisms may facilitate this.
Relatively fewer studies have been reported on the role of m6A modifications in PCA. METTL3 silencing decreases expression of GLI1, an important apoptotic factor involved in the hedgehog pathway [86]. YTHDF2 and miR-493-3p are cited as two crucial oncogenes, involved in the progression of PCA by indirectly modulating m6A levels [85].
Among urological malignancies, RCC is the most lethal [126]. Methylenetetrahydrofolate dehydrogenase 2 overexpression enhances m6A modification of HIF-2α and forms a positive feedforward loop in RCC, resulting in malignant phenotypes [134]. Li et al demonstrated that METTL3 could suppress proliferation, migration and epithelial-to-mesenchymal transition (EMT) of RCC cells via regulation of the PI3K-AKT-mTOR pathway [107]. METTL14 inhibits P2RX6 protein translation and modulates ATP-P2RX6-Ca2+-p-ERK1/2-MMP9 signaling to prevent migration and invasion of RCC cells [135]. Additionally, WTAP promotes tumorigenesis by enhancing CDK2 expression [87] and FTO is expression is decreased in clear cell RCC, reducing tumor growth via increasing the expression of PGC-1α, a central regulator of mitochondrial function in the PPARγ co-activator family [108].
Gynecological oncology: breast cancer (BC), cervical squamous cell carcinoma (CSCC), epithelial ovarian cancer (EOC) and endometrial cancer (EC)
Breast cancer is the most common type of cancer in women worldwide [136]. Cai et al showed that METTL3 increases the expression of mammalian hepatitis B X-interacting protein (HBXIP), thus driving the aggressiveness of BC. HBXIP upregulates the expression of METTL3 via inhibiting the function of the tumor suppressor miRNA let-7 g, forming a positive feedback loop of METTL3/HBXIP/let-7 g/METTL3 [90]. Consistent with this, a recent study showed that METTL3 promoted BC progression by targeting BCL-2 [91]. Hypoxia induces ALKBH5 to demethylate NANOG mRNA and enhance its stability in BC stem cells (BCSCs) [92]. The group further demonstrated that ZNF217 and ALKBH5 play complementary roles in negatively regulating m6A levels, eventually increasing the number of BCSCs under hypoxic conditions [137]. Furthermore, ALKBH5 and METTL14 interact with each other and inhibit YTHDF3 activity, thus accelerating tumor angiogenesis. They stated that METTL14 and ALKBH5 constitute a positive feedback loop with HuR to regulate the target genes of cell cycle progression, EMT and angiogenesis [93]. In 2019, Jessica et al proposed that through regulating m6A methylation, far upstream binding protein 1 (FUBP1) globally affects alternative splicing to promote the activity of proteins associated with BC neoplastic transformation, including BRCA1, MAGI3 and CASP8 [138]. Niu et al showed that FTO promoted tumor development via inhibiting BNIP3, a pro-apoptotic gene of the BCL-2 family [94]. Notably, members of the BCL-2 family have repeatedly been shown to involved in the development and progression of BC, and are targeted by FTO and METTL3 [91]. The translation process of BCL-2 is also promoted by METTL3 in AML and ALKBH5 in epithelial ovarian cancer [41, 96]. Thus, m6A proteins may act to inhibit the activity of members of the BCL-2 family at various stages of the BCL-2 signaling process, thereby providing a favorable therapeutic response.
M6A also serves a role in several other types of gynecological cancer. In CSCC, FTO enhances chemoradiotherapy resistance by targeting β-catenin [88]. FTO also interacts with the transcripts of E2F1 and MYC to facilitate proliferation and migration [89]. In EC, Liu et al demonstrated that reduced levels of METTL3/METTL14 and an accumulation of FTO induced by estrogen enhanced AKT/mTOR signaling to promote tumorigenicity [109]. In EOC, METTL3 promotes tumorigenicity through regulating translation of AXL and EMT [95]. ALKBH5 acts as a candidate oncogene, inhibiting cancer autophagy through miR-7 and BCL-2, eventually activating an EGFR-PI3K-AKT-mTOR signaling pathway [96]. The PI3K/AKT/mTOR pathway has been implicated in the development of various types of cancer regulated by m6A proteins, including METTL3/WTAP in AML [41, 43], METTL3 in RCC/PDAC [78, 107], ALKBH5 in EOC [96], and FTO in melanoma [97] and EC [109]. YTHDF2 and RBM15 expression are also correlated with activation of this pathway [139]. Activation of this pathway in AML and EC may be inhibited by rapamycin, an mTOR specific inhibitor. These data suggest that preventing communication between mTOR signaling and m6A regulators may present a potential avenue for treatment of various types of cancer.
Skin neoplasm: melanoma and cutaneous squamous cell carcinoma (cSCC)
Melanoma is notorious for its high rate of mortality and its resistance to available therapies [140]. In 2019, two groups probed for the mechanisms underlying development and progression of melanoma [97, 110]. YTHDF1 suppresses ocular melanoma through modulation of mRNA translation of histidine triad nucleotide-binding protein 2, a tumor suppressor in ocular melanoma [110]. Another group showed that induction of FTO promotes tumorigenicity via mTOR signaling through m6A-mediated tuning of the PD-1 gene. Subsequently, IFN-γ downregulates FTO expression and may mediate the effect of FTO knockdown in PD-1 blockade [97]. In this study, m6A effective proteins influence the immune response by controlling signal transduction. Immune checkpoint blockade therapy has demonstrated an unprecedented anti-tumor response rate in patients with advanced cancer. Therefore, the complete mechanism of immune regulation by PD-1 blockade with m6A modifications in melanoma should be determined. Additionally, in cSCC, METTL3 upregulates ΔNp63 expression to promote tumorigenesis [98].
The landscape of alterations of m6A regulators in human cancers
The vast majority of existing studies have focused on the m6A perturbation mediated via knockdown or overexpression of m6A related protein in cell death, proliferation, impaired self-renewal capacity and developmental defects. Meanwhile, mutations may cause gain or loss of m6A sites, thus affecting cellular m6A modification and associated with human cancers [24]. So, we investigated the alterations frequency (overexpression, down-regulation and mutation) of m6A proteins in cancers based on the cBioPortal database. The overall average alterations frequency of m6A proteins ranged from 0 to 16%. M6A related proteins exhibited a relatively higher alterations frequency in EC and melanoma. Besides, among m6A proteins, ZC3H13, KIAA1429, YTHDC2and IGF2BP1 showed higher alterations frequencies while m6A erasers seldom had a genetic mutation (Fig. 2a). Moreover, m6A proteins exhibited few alterations in several cancer types like PCA and AML (Fig. 2a). All data were collected from the cBioPortal database (Additional file 1: Table S1). The overexpression of m6A proteins was more frequent than the down-expression and mutation, which indicated that m6A proteins always served as an oncogenic role in cancers. To exemplify, METT14 alteration made up about 5% in CRC and the down-expression of METT14 mainly occurred in CRC (Fig. 2b), which is consistent with the previous reports [141]. Besides, in PCA, the incidence of IGF2BP1 alteration was 4% and it is the overexpression of IGF2BP1 rather than the down-expression or mutation that mainly occurred (Fig. 2c). The type of alterations m6A proteins in other cancers were also provided (Additional file 2: Figure S1).

The alterations frequency of m6A regulators in cancers from cBioPortal data analysis. Totally 15 different TCGA projects were included (TCGA, PanCancer Atlas), and each project represents a specific cancer type. Oncoprints in cBioPortal were also used to represent the proportion and distribution of samples with altered m6A regulators. a The overall alterations frequency of m6A regulators across 15 cancer types. b The type and percentages of m6A regulators alterations in CRC. cThe type and percentages of m6A regulators alterations in PCA
Li et al systematically studied the mutation of m6A regulators across 33 cancer types and they found that the average mutation frequency of m6A regulators was low, ranging from 0.02–8.07%. M6A regulators exhibited a relatively higher mutation frequency of EC and melanoma. YTHDC1, IGF2BP1, YTHDC2, FTO and the writers showed relatively higher mutation frequencies [139]. While in AML, mutations of m6A regulatory genes were low (2.6%) and were significantly associated with poorer cytogenetic and genotypes risk, predicting poorer OS and EFS independently [142]. As well, Wu et al found genetic mutations of m6A enzymes occurred 24% of 2051 patients with BC. Nevertheless, the reduced level of the m6A members METTL3, METTL14, WTAP and FTO but not their mutation and overexpression was tightly associated with poor survival [143]. Zhang et al illustrated that mutations of METTL3, METTL14, ALKBH5, FTO, YTHDF1, YTHDF2 and YTHDF3 were rare in GC. The content and functions of m6A in GC might be impaired by specific mutations, thus predicting malignant phenotypes and augmenting Wnt/PI3K-Akt signaling in GC [144]. Liu et al found that the hotspot R298P mutation in METTL14 was more prevalent than other mutations in EC and occurred in ~ 1.5% of EC patients. The mutation eventually regulated AKT activity to promote the proliferation and tumorigenicity of EC [109]. In GBM, the genetic change (mutation or copy number variations) frequencies of the m6A RNA methylation regulators were very low (all≤1.1%) and the expression changes of these regulators were not caused by the genetic changes of the corresponding genes [145]. The mutations of m6A genes in several cancers were summarized in Table 3.
These results revealed a highly heterogeneous genetic and expression alteration landscape of m6A regulators across cancers. The landscape of alterations m6A regulators regulating in tumors laid a critical foundation for understanding the dysregulation of RNA methylation.
The clinicopathological relevance to m6A alterations in tumors
The clinicopathological features resulted from m6A alterations are quite vital and could further provide us the development of drugs against m6A related proteins for cancer treatment. Most included studies illustrated that m6A related proteins may influence the prognosis of cancer patients. Overall, the high level of m6A methylation would lead to poor prognosis. However, only a few reports are available where other clinicopathological features like metastasis and invasion were provided. For example, in lung cancer, patients with high expression of METTL3 were prone to occur lymph node metastasis and distant metastasis [54]. Ma et al indicated that aberrant expression of METTL14 mRNA was correlated not only with tumor differentiation and tumor stage but also with tumor encapsulation and microvascular invasion, possibly playing a suppressive role in HCC metastasis [100]. Reduced m6A modification demonstrated adverse clinical outcomes in GC [144]. In bladder cancer, patients with high expression of METTL3 had worse prognosis and shorter survival time, compared with those with low expression of METTL3 [82]. The FTO expression was markedly declined in cancer counterpart and lost in the later stage. The reduced expression of FTO was correlated with worse OS and DFS [108]. These observed clinical changes might provide alternative, promising therapeutic targets for the treatment of m6A relevant cancers. The clinicopathological information regarding m6A proteins alterations was summarized in Table 4.
m6A-related factors in cancer treatment
The balance between methylation and demethylation of m6A at specific RNA transcripts may influence the development of numerous diseases. Thus, regulators or inhibitors of m6A proteins may serve as potential therapeutics for treatment of these diseases (Table 5). M6A inhibitors have been developed for advancing traditional and regenerative medicine, particularly inhibitors of FTO including rhein, R-2HG, IOX3, FB23, MO-I-500, meclofenamic acid and so on [36, 146]. FTO belongs to the family of Fe2+ and 2-oxoglutarate (2OG) dependent AlkB dioxygenases. Meclofenamic acid (MA) was identified as a highly selective inhibitor of FTO [147]. Treatment of GSCs with the ethyl ester form of meclofenamic acid, MA2, could suppress tumorigenesis and prolong the lifespan of GSC-engrafted mice [49]. Afterwards, FB23 and its derivative (FB23–2) display a high selectivity toward FTO. FB23–2 promotes apoptosis and suppresses proliferation of AML cells [148]. Among nonselective inhibitors of FTO, rhein was identified as the first potent FTO inhibitor [149]. As a natural product, rhein competitively binds to the FTO active site and exhibits good inhibitory activity on m6A demethylation [149]. Besides, rhein and MO-I-500 both could decrease tumorigenesis of BC cells [94, 150]. R-2HG could decrease the expression of MYC and alleviates AML and GBM [45]. In addition, knockdown of FTO could enhance the response to AML cells to all-trans retinoic acid (ATRA) treatment and promote ATRA-induced differentiation [44]. In melanoma, the combination of FTO inhibition and anti-PD-1 blockers may reduce resistance to immunotherapy [97]. These collective results indicate that FTO selective or nonselective inhibitors alone or in combination with standard therapeutic agents hold the immense therapeutic potential to cancers, especially those with high FTO expression [151].
Previous studies mostly focused on the inhibitors of FTO but other m6A proteins may also be the advantageous target for m6A related cancers. METTL3-depleted cells show a higher sensitivity to anticancer reagents such as gemcitabine, 5-fluorouracil, cisplatin and irradiation in pancreatic cancer [152]. In osteosarcoma, alteration of m6A methylation is associated with acquired chemoresistance [153]. Of mention, 3-deazaadenosine (DAA), a S-adenosylhomocysteine (SAH) hydrolysis inhibitor, has been proven to inhibit METTL3/METTL14 with broad spectrum of effectiveness [154]. Also, Simona et al discovered that small-molecule compounds activate m6A methylation with exceptionally high binding efficiencies to METTL3–14-WTAP. The compounds are experimentally characterized as METTL3–14-WTAP activators that could affect m6A methylation level in HEK293 cells [151]. Rajiv et al proposed a route for further development into potent inhibitors of METTL3. Two series of adenine derivatives were identified and showed good ligand efficiency [155]. Although the pharmacological efficacy of these small-molecule activators or inhibitors of METTL3 has not been reported before, the discovery may open up a new avenue in m6A-targeted pharmacotherapeutics.
Remarkably, several regulators that are upstream of m6A proteins could alter the m6A level via regulating m6A proteins, shedding light on the development of powerful probes and new therapies for cancers. SPI1, a hematopoietic transcription factor, inhibits the development of malignant hematopoietic cells via targeting METTL14 [42]. CA4, a member of the carbonic anhydrases, could interact WTAP and induce WTAP protein degradation, thus suppressing CRC processing through the inhibition of the Wnt signalling pathway [73].
Given that m6A modification has broad physiological functions, its impairment may be a potential novel therapeutic target for the treatment of a wide range of cancers. Specific m6A regulators suitable for clinical trials are thus required. However, it remains a major challenge to identify novel biomarkers and molecular targets to guide therapies in cancers. More selective and efficacious drugs targeting m6A-related factors should be developed and explored.
Conclusion
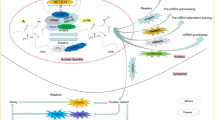
As a dominant player in gene expression, m6A is the target of numerous regulatory pathways. The disruption of these mechanisms may result in disease, sometimes with catastrophic consequences. An overview of the mechanical pathways modulated by m6A modification and their implications in human cancers are presented in Fig. 3.

The momentous mechanical pathways of m6A involved in human cancers
The cross-talk between pathways and co-operation of m6A regulation of gene expression requires considerable study, and at the present, our knowledge is limited. m6A writers, erasers and readers frequently interact with each other, particularly with writers [139]. Sorc et al proposed that METTL3 may regulate WTAP protein homeostasis, and upregulation of WTAP has an oncogenic effect only in the presence of functional METTL3 [156]. The combined outcomes of METTL14, ALKBH5 and YTHDF3 function elevate m6A expression and activity beyond the threshold required to regulate gene expression and activity of critical genes in BC [93]. Additionally, it is a commonly observed phenomena that an m6A-associated protein which participates in different types of cancer are regulated by multiple m6A proteins. For example, there have been contrasting observations suggesting that all the m6A- associated enzymes serve oncogenic roles in AML. Furthermore, an m6A-associated protein in the same type of cancer may regulate different proteins in different individuals. Thus, an extensive amount of effort is required to fully understand the m6A interactome.
Additionally, it remains possible that not all the m6A writers, erasers and readers have been identified. In 2018, Huang et al found that FMR1 and HNRNPC may serve as novel m6A binding proteins [17]. Recently, METTL5 and ZCCHC4 were confirmed to function as exclusive m6A writers of rRNA. Therefore, developing novel m6A detection methods, such as nanopore technology, will assist in the identification of m6A modifiers. For m6A erasers, only two proteins have been identified to date; whereas numerous writers have been discovered. Erasers exhibit diverse biological functions, which may result from their differing tissue distributions and localization. For example, FTO is enriched in the brain and muscle, whereas ALKBH5 is upregulated in the testes [20]. Aberrant expression of FTO or ALKBH5 only results in minor changes in the overall levels of m6A, suggesting that there may be demethylases yet to be discovered. Therefore, identifying novel m6A enzymes may result in the identification of novel regulatory mechanisms.
Importantly, m6A proteins usually serve an oncogenic role in cancer, and the oncogenic role of m6A may be attributed to either promotion of oncogene translation, or initiating the decay of tumor suppressor gene transcripts. However, it is not clear how m6A writers and erasers selectively exert their differing effects, but often still result in the same or similar outcomes, namely the progression of cancer. As an instance, METTL3 may serve dual roles in both GBM and CRC [48, 49, 68, 103]. We proposed the following hypothesis to explain this conflicting phenomena: 1) m6A proteins could function independently of its m6A catalytic activity; 2) Since the fate of m6A-modified mRNAs is also determined by the readers, the difference in the abundance, RNA affinity and cumulative binding of m6A readers may lead to divergent results; 3) The location of the m6A modifications on different regions of the same mRNA transcript may underlie the differing effects; 4) The cross-talk between pathways and co-operation of m6A regulation of gene expression requires considerable study; 5) The conflicting outcomes may also lie in differences in the cancer heterogeneity, cellular context and target specificity of the m6A proteins [37]. However, several questions remain to be answered. How does the methyltransferase family recognize their specific sites and modify them? Does the ncRNA guide the sequence selection?
Remarkably, m6A seldom acts as a tumor-suppressor, excluding METTL14. METTL14 is critical for EBV-associated tumorigenesis through interactions with viral-encoded latent oncoprotein EBNA3C, but in the majority of cases, it serves as a tumor suppressor in several types of cancer, including GBM, HCC, CRC, BCA and EC [99, 104, 139]. These results highlight the impact of m6A modification on the fate of the embedded RNA, and mediation of the RNA function following modification. These key functionally important RNA targets include miRNA, lncRNA and circRNA, amongst others, and are involved in regulating m6A, and may partially explain the mechanism of site selection of m6A. The study of m6A modification of circRNA has recently rose. CircE7 possesses m6A modifications in the cytoplasm, and is translated to produce E7, an oncoprotein, yielding novel insights into how HPV regulates infection and tumorigenesis [157]. M6A modification of circNSUN2 increases export of this circRNA to the cytoplasm, and the export is mediated through the recruitment of YTHDC1, thus enhancing the stability of HMGA2 mRNA to promote progression and metastasis of CRC [30]. Zhang et al demonstrated that m6A modification of the YAP 3′-UTR induces an interaction with miR-382-5p which resulted in the inhibition of YAP, thus impairing the tumorigenic capacity of circRNA_104075 in HCC [158]. Chen et al showed that m6A modifications on human circRNAs inhibit innate immunity through abrogation of immune gene activation, and YTHDF2 is indispensable for suppression of innate immunity [159]. The dual and opposing regulation of m6A modifications and circRNA indicates that interference with the pathway between m6A and immunogenicity of circRNA may be exploited therapeutically.
Methylation of DNA and histone has been the focus of cancer research for several decades, and the DNA methyltransferase inhibitors, azacytidine and decitabine have been approved for cancer therapy in the clinic [160]. As an important RNA epigenetic modification, it remains to be determined how m6A interacts with DNA and histone epigenetics to regulate gene expression, and whether there are potential connections between m6A modifications and other types of RNA modifications. At present, research, and our collective understanding of m6A modifications is still in its infancy.
Availability of data and materials
Not applicable.
Abbreviations
- MYB:
-
Myeloblastosis oncogene
- MYC:
-
Myelocytomatosis oncogene
- HSPC:
-
Hematopoietic stem and progenitor cells
- NANOG:
-
Nanog homeobox
- PTEN:
-
Phosphatase and tensin homolog
- RARA:
-
Retinoic acid receptor alpha
- SOX2:
-
Sex determining region Y box 2
- STAT3:
-
Activator of transcription 3
- R-2HG:
-
R-2-hydroxyglutarate
- SOCS2:
-
Suppressor of cytokine signaling 2
- CA4:
-
Carbonic anhydrase IV
- eIF:
-
eukaryotic initiation factor
- ADAM19:
-
A disintegrin and metallopeptidase domain 19
- ASB2:
-
Ankyrin repeat and SOCS box containing 2
- BCL2:
-
B cell leukaemia 2
- FOXM1:
-
Forkhead box M1
- KLF4:
-
Kruppel like factor 4
- MALAT1:
-
Metastasis-associated lung adenocarcinoma transcript 1
- AFF4:
-
AF4/FMR2 family member 4
- PPAR:
-
Peroxisome proliferator-activator
References
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–5.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–8.
Roignant JY, Soller M. m (6) a in mRNA: An ancient mechanism for fine-tuning gene expression. Trends Genet. 2017;33:380–90.
Pan Y, Ma P, Liu Y, Li W, Shu Y. Multiple functions of m (6) a RNA methylation in cancer. J Hematol Oncol. 2018;11:48.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. 2016;63:306–17.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villaseñor R, Hess D, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m (6) a machinery component Wtap/Fl (2) d. Genes Dev. 2018;32:415–29.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. VIRMA mediates preferential m (6) a mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, et al. Zc3h13 Regulates Nuclear RNA m (6) A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell. 2018;69:1028–38 e1026.
Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, Chen K, Lu Z, Chen H, Shi YG, et al. N (6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol. 2019;15:88–94.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C, Sloan KE, Bohnsack MT. Human METTL16 is a N (6)-methyladenosine (m (6) a) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004–14.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, Igarashi K. S-Adenosylmethionine synthesis is regulated by selective N (6)-adenosine methylation and mRNA degradation involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–63.
Thomas JM, Batista PJ, Meier JL. Metabolic regulation of the Epitranscriptome. ACS Chem Biol. 2019;14:316–24.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK. The U6 snRNA m (6) a methyltransferase METTL16 regulates SAM Synthetase intron retention. Cell. 2017;169:824–35 e814.
van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, Bohnsack KE, Bohnsack MT, Jaffrey SR, Graille M, Lafontaine DLJ. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47:7719–33.
Richard EM, Polla DL, Assir MZ, Contreras M, Shahzad M, Khan AA, Razzaq A, Akram J, Tarar MN, Blanpied TA, et al. Bi-allelic variants in METTL5 cause autosomal-recessive intellectual disability and microcephaly. Am J Hum Genet. 2019;105:869–78.
Scholler E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, Feederle R, Bruckmann A, Meister G. Interactions, localization, and phosphorylation of the m (6) a generating METTL3-METTL14-WTAP complex. RNA. 2018;24:499–512.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N (6)-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99.
Aik W, Scotti JS, Choi H, Gong L, Demetriades M, Schofield CJ, McDonough MA. Structure of human RNA N (6)-methyladenine demethylase ALKBH5 provides insights into its mechanisms of nucleic acid recognition and demethylation. Nucleic Acids Res. 2014;42:4741–54.
Chen W, Zhang L, Zheng G, Fu Y, Ji Q, Liu F, Chen H, He C. Crystal structure of the RNA demethylase ALKBH5 from zebrafish. FEBS Lett. 2014;588:892–8.
Huang H, Weng H, Chen J. m (6) a modification in coding and non-coding RNAs: roles and therapeutic implications in Cancer. Cancer Cell. 2020;37:270–88.
Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother. 2019;112:108613.
Ueda Y, Ooshio I, Fusamae Y, Kitae K, Kawaguchi M, Jingushi K, Hase H, Harada K, Hirata K, Tsujikawa K. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7:42271.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C. YTHDF3 facilitates translation and decay of N (6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Zhou C, Molinie B, Daneshvar K, Pondick JV, Wang J, Van Wittenberghe N, Xing Y, Giallourakis CC, Mullen AC. Genome-wide maps of m6A circRNAs identify widespread and cell-type-specific methylation patterns that are distinct from mRNAs. Cell Rep. 2017;20:2262–76.
Chen RX, Chen X, Xia LP, Zhang JX, Pan ZZ, Ma XD, Han K, Chen JW, Judde JG, Deas O, et al. N (6)-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat Commun. 2019;10:4695.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. Recognition of RNA N (6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m (6) a promotes cap-independent translation. Cell. 2015;163:999–1010.
Chandola U, Das R, Panda B. Role of the N6-methyladenosine RNA mark in gene regulation and its implications on development and disease. Brief Funct Genomics. 2015;14:169–79.
Chen XY, Zhang J, Zhu JS. The role of m (6) a RNA methylation in human cancer. Mol Cancer. 2019;18:103.
Deng X, Su R, Feng X, Wei M, Chen J. Role of N (6)-methyladenosine modification in cancer. Curr Opin Genet Dev. 2018;48:1–7.
Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The critical role of RNA m (6) a methylation in Cancer. Cancer Res. 2019;79:1285–92.
Vu LP, Cheng Y, Kharas MG. The biology of m (6) a RNA methylation in Normal and malignant hematopoiesis. Cancer Discov. 2019;9:25–33.
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, Li H, Qiu M, Xu Y, Xia W, et al. Roles of RNA methylation by means of N (6)-methyladenosine (m (6) a) in human cancers. Cancer Lett. 2017;408:112–20.
Zhao W, Qi X, Liu L, Liu Z, Ma S, Wu J. Epigenetic regulation of m (6) a modifications in human Cancer. Mol Ther Nucleic Acids. 2019;19:405–12.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. The N (6)-methyladenosine (m (6) a)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–76.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes Leukemogenesis via mRNA m (6) a modification. Cell Stem Cell. 2018;22:191–205 e199.
Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Proia D, Penalva LO, Uren PJ, Suresh U, Carew JS, et al. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014;28:1171–4.
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C, Huang H, Nachtergaele S, Dong L, Hu C, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N (6)-Methyladenosine RNA Demethylase. Cancer Cell. 2017;31:127–41.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. R-2HG exhibits anti-tumor activity by targeting FTO/mA/MYC/CEBPA signaling. Cell. 2018;172.
Li Z, Qian P, Shao W, Shi H, He XC, Gogol M, Yu Z, Wang Y, Qi M, Zhu Y, et al. Suppression of m (6) a reader Ythdf2 promotes hematopoietic stem cell expansion. Cell Res. 2018;28:904–17.
Zhou J, Bi C, Ching YQ, Chooi JY, Lu X, Quah JY, Toh SH, Chan ZL, Tan TZ, Chong PS, Chng WJ. Inhibition of LIN28B impairs leukemia cell growth and metabolism in acute myeloid leukemia. J Hematol Oncol. 2017;10:138.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, Somasundaram K. Essential role of METTL3-mediated m (6) a modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Sun G, Lu Z, Huang Y, Yang CG, et al. m (6) a RNA methylation regulates the self-renewal and Tumorigenesis of Glioblastoma stem cells. Cell Rep. 2017;18:2622–34.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O, et al. m (6) a Demethylase ALKBH5 maintains Tumorigenicity of Glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606 e596.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m (6) a methyltransferase METTL3 promotes translation in human Cancer cells. Mol Cell. 2016;62:335–45.
Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, Wang D, Huang J, Gao S, Gao Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017;482:582–9.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60.
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, Di W, Hu B, An J, Kong L, et al. m (6) a mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12:135.
Wang H, Deng Q, Lv Z, Ling Y, Hou X, Chen Z, Dinglin X, Ma S, Li D, Wu Y, et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer. 2019;18:181.
Liu J, Ren D, Du Z, Wang H, Zhang H, Jin Y. m (6) a demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem Biophys Res Commun. 2018;502:456–64.
Li J, Han Y, Zhang H, Qian Z, Jia W, Gao Y, Zheng H, Li B. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem Biophys Res Commun. 2019;512:479–85.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–70.
Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, Lederer M, Dahl A, Huang H, Chen J, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375–90.
Zhang P, He Q, Lei Y, Li Y, Wen X, Hong M, Zhang J, Ren X, Wang Y, Yang X, et al. m (6) A-mediated ZNF750 repression facilitates nasopharyngeal carcinoma progression. Cell Death Dis. 2018;9:1169.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, Luo G, Tauler J, Du J, Lin S, et al. RNA mA methylation regulates the epithelial mesenchymal transition of cancer cells and translation of snail. Nat Commun. 2019;10:2065.
Cheng X, Li M, Rao X, Zhang W, Li X, Wang L, Huang G. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 2019;12:3421–8.
Lan T, Li H, Zhang D, Xu L, Liu H, Hao X, Yan X, Liao H, Chen X, Xie K, et al. KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol Cancer. 2019;18:186.
Chen Y, Peng C, Chen J, Chen D, Yang B, He B, Hu W, Zhang Y, Liu H, Dai L, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18:127.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, Liu Y, Ye L, Li Y, Zhang X. MicroRNA-145 modulates N (6)-Methyladenosine levels by targeting the 3′-Untranslated mRNA region of the N (6)-Methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017;292:3614–23.
Liu L, Wang J, Sun G, Wu Q, Ma J, Zhang X, Huang N, Bian Z, Gu S, Xu M, et al. m (6) a mRNA methylation regulates CTNNB1 to promote the proliferation of hepatoblastoma. Mol Cancer. 2019;18:188.
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, Li J, An P, Lu L, Luo N, et al. m (6) A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18:87.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, et al. METTL3 facilitates tumor progression via an m (6) A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112.
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, Ji D, Wang Q, Zhang Z, Tang J, Sun Y. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38:393.
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, Shimamoto F, Tang B. m (6) a modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18:185.
Shen XP, Ling X, Lu H, Zhou CX, Zhang JK, Yu Q. Low expression of microRNA-1266 promotes colorectal cancer progression via targeting FTO. Eur Rev Med Pharmacol Sci. 2018;22:8220–6.
Roslan NH, Makpol S, Mohd Yusof YA. A review on dietary intervention in obesity associated Colon Cancer. Asian Pac J Cancer Prev. 2019;20:1309–19.
Zhang J, Tsoi H, Li X, Wang H, Gao J, Wang K, Go MY, Ng SC, Chan FK, Sung JJ, Yu J. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut. 2016;65:1482–93.
Tanabe A, Tanikawa K, Tsunetomi M, Takai K, Ikeda H, Konno J, Torigoe T, Maeda H, Kutomi G, Okita K, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016;376:34–42.
Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai D, et al. Oncogene c-Myc promotes epitranscriptome m (6) a reader YTHDF1 expression in colorectal cancer. Oncotarget. 2018;9:7476–86.
Bai Y, Yang C, Wu R, Huang L, Song S, Li W, Yan P, Lin C, Li D, Zhang Y. YTHDF1 regulates Tumorigenicity and Cancer stem cell-like activity in human colorectal carcinoma. Front Oncol. 2019;9:332.
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, Liu J, Luo XJ, Meng Q, Pu HY, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18:174.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, Li S, Tan L, Mai D, Li G, et al. Excessive miR-25-3p maturation via N (6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10:1858.
Chen J, Sun Y, Xu X, Wang D, He J, Zhou H, Lu Y, Zeng J, Du F, Gong A, Xu M. YTH domain family 2 orchestrates epithelial-mesenchymal transition/proliferation dichotomy in pancreatic cancer cells. Cell Cycle. 2017;16:2259–71.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, Jiang Z, Zhang Y, Xu G, Zhang J, et al. METTL3-mediated mA modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2019.
Zhang J, Guo S, Piao HY, Wang Y, Wu Y, Meng XY, Yang D, Zheng ZC, Zhao Y. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J Physiol Biochem. 2019;75:379–89.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, Liang Y, Zhu F, Zhang Y, Zhang X, et al. The m (6) a methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38:3667–80.
Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, Zhou Z, Yuan Z, Su J, Wu B, et al. Dynamic mA mRNA methylation reveals the role of METTL3-mA-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38:4755–72.
Li J, Meng S, Xu M, Wang S, He L, Xu X, Wang X, Xie L. Downregulation of N (6)-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N (6)-methyladenosine levels. Oncotarget. 2018;9:3752–64.
Cai J, Yang F, Zhan H, Situ J, Li W, Mao Y, Luo Y. RNA m (6) a methyltransferase METTL3 promotes the growth of prostate Cancer by regulating hedgehog pathway. Onco Targets Ther. 2019;12:9143–52.
Tang J, Wang F, Cheng G, Si S, Sun X, Han J, Yu H, Zhang W, Lv Q, Wei JF, Yang H. Wilms' tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J Exp Clin Cancer Res. 2018;37:40.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, Zhe H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Zou D, Dong L, Li C, Yin Z, Rao S, Zhou Q. The m (6) a eraser FTO facilitates proliferation and migration of human cervical cancer cells. Cancer Cell Int. 2019;19:321.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, Liu Y, Zhang X, Zhang W, Ye L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m (6) A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56.
Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, Abdelfattah N, Onyeagucha BC, Cui X, Lai Z, et al. Cross-talk among writers, readers, and erasers of m (6) A regulates cancer growth and progression. Sci Adv. 2018;4:eaar8263.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, Wang Y, Li X, Xiong XF, Wei B, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, Duan P. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151:356–65.
Zhu H, Gan X, Jiang X, Diao S, Wu H, Hu J. ALKBH5 inhibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J Exp Clin Cancer Res. 2019;38:163.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, Aplin AE, Lu Z, Hwang S, He C, He YY. m (6) a mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Zhou R, Gao Y, Lv D, Wang C, Wang D, Li Q. METTL3 mediated m (6) a modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating ΔNp63. Biochem Biophys Res Commun. 2019;515:310–7.
Lang F, Singh RK, Pei Y, Zhang S, Sun K, Robertson ES. EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 2019;15:e1007796.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, Wang TT, Xu QG, Zhou WP, Sun SH. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N (6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–43.
Hou J, Zhang H, Liu J, Zhao Z, Wang J, Lu Z, Hu B, Zhou J, Zhao Z, Feng M, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer. 2019;18:163.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, Ma H, Kang T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, Liu H, Deng Q, Wu X, Lan P, Deng Y. m (6) a methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco Targets Ther. 2019;12:4391–402.
Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L, Pan B, He B, Pan Y, Sun H, et al. METTL14 suppresses CRC progression via regulating N6-Methyladenosine-dependent primary miR-375 processing. Mol Ther. 2020;28:599–612.
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, Liu D, Tian L, Yin J, Jiang K, Miao Y. ALKBH5 inhibits pancreatic Cancer motility by decreasing Long non-coding RNA KCNK15-AS1 methylation. Cell Physiol Biochem. 2018;48:838–46.
Gu C, Wang Z, Zhou N, Li G, Kou Y, Luo Y, Wang Y, Yang J, Tian F. Mettl14 inhibits bladder TIC self-renewal and bladder tumorigenesis through N (6)-methyladenosine of Notch1. Mol Cancer. 2019;18:168.
Li X, Tang J, Huang W, Wang F, Li P, Qin C, Qin Z, Zou Q, Wei J, Hua L, et al. The M6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103–16.
Zhuang C, Zhuang C, Luo X, Huang X, Yao L, Li J, Li Y, Xiong T, Ye J, Zhang F, Gui Y. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1alpha signalling axis. J Cell Mol Med. 2019;23:2163–73.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al. m (6) a mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074–83.
Jia R, Chai P, Wang S, Sun B, Xu Y, Yang Y, Ge S, Jia R, Yang YG, Fan X. mA modification suppresses ocular melanoma through modulating HINT2 mRNA translation. Mol Cancer. 2019;18:161.
Chen J, Odenike O, Rowley JD. Leukaemogenesis: more than mutant genes. Nat Rev Cancer. 2010;10:23–36.
Jaffrey SR, Kharas MG. Emerging links between m (6) a and misregulated mRNA methylation in cancer. Genome Med. 2017;9:2.
Ma Z, Morris SW, Valentine V, Li M, Herbrick JA, Cui X, Bouman D, Li Y, Mehta PK, Nizetic D, et al. Fusion of two novel genes, RBM15 and MKL1, in the t (1,22)(p13;q13) of acute megakaryoblastic leukemia. Nat Genet. 2001;28:220–1.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N (6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Weng H, Huang H, Chen J. RNA N (6)-Methyladenosine modification in Normal and malignant hematopoiesis. Adv Exp Med Biol. 2019;1143:75–93.
Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Brüning JC, Vu LP, et al. m (6) A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Rep. 2019;28:1703–16 e1706.
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195–208.
Zheng ZQ, Li ZX, Zhou GQ, Lin L, Zhang LL, Lv JW, Huang XD, Liu RQ, Chen F, He XJ, et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma Tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and Upregulate ITGB3. Cancer Res. 2019;79:4612–26.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, Chen J, Xu W, Zhong L, Sun X. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark. 2018;21:859–68.
Jo HJ, Shim HE, Han ME, Kim HJ, Kim KS, Baek S, Choi KU, Hur GY, Oh SO. WTAP regulates migration and invasion of cholangiocarcinoma cells. J Gastroenterol. 2013;48:1271–82.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang J, Lu Z, Wu P, Cai B, Miao Y, Jiang K. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathol Res Pract. 2019;215:152666.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018(68):394–424.
He X, Shu Y. RNA N6-methyladenosine modification participates in miR-660/E2F3 axis-mediated inhibition of cell proliferation in gastric cancer. Pathol Res Pract. 2019;215:152393.
Xu D, Shao W, Jiang Y, Wang X, Liu Y, Liu X. FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol Rep. 2017;38:2285–92.
Su Y, Huang J, Hu J. mA RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gastric Cancer. Front Oncol. 2019;9:1038.
Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6.
Kopp F, Mendell JT. Functional classification and experimental dissection of Long noncoding RNAs. Cell. 2018;172:393–407.
Muller S, Bley N, Glass M, Busch B, Rousseau V, Misiak D, Fuchs T, Lederer M, Huttelmaier S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018;46:6285–303.
Chen M, Nie ZY, Wen XH, Gao YH, Cao H, Zhang SF. m6A RNA methylation regulators can contribute to malignant progression and impact the prognosis of bladder cancer. Biosci Rep. 2019;39.
Green NH, Galvan DL, Badal SS, Chang BH, LeBleu VS, Long J, Jonasch E, Danesh FR. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene. 2019;38:6211–25.
Gong D, Zhang J, Chen Y, Xu Y, Ma J, Hu G, Huang Y, Zheng J, Zhai W, Xue W. The m (6) A-suppressed P2RX6 activation promotes renal cancer cells migration and invasion through ATP-induced Ca (2+) influx modulating ERK1/2 phosphorylation and MMP9 signaling pathway. J Exp Clin Cancer Res. 2019;38:233.
DeSantis CE, Ma J, Gaudet MM, Newman LA, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Breast cancer statistics, 2019. CA Cancer J Clin. 2019;69:438–51.
Zhang C, Zhi WI, Lu H, Samanta D, Chen I, Gabrielson E, Semenza GL. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016;7:64527–42.
Elman JS, Ni TK, Mengwasser KE, Jin D, Wronski A, Elledge SJ, Kuperwasser C. Identification of FUBP1 as a Long tail Cancer driver and widespread regulator of tumor suppressor and oncogene alternative splicing. Cell Rep. 2019;28:3435–49 e3435.
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, Wang Q, Li X, Zhang Y, Xu J. Molecular characterization and clinical relevance of m (6) a regulators across 33 cancer types. Mol Cancer. 2019;18:137.
Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer. 2019;19:377–91.
Yang X, Zhang S, He C, Xue P, Zhang L, He Z, Zang L, Feng B, Sun J, Zheng M. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19:46.
Kwok CT, Marshall AD, Rasko JE, Wong JJ. Genetic alterations of m (6) a regulators predict poorer survival in acute myeloid leukemia. J Hematol Oncol. 2017;10:39.
Wu L, Wu D, Ning J, Liu W, Zhang D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer. 2019;19:326.
Zhang C, Zhang M, Ge S, Huang W, Lin X, Gao J, Gong J, Shen L. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 2019;8:4766–81.
Chai R-C, Wu F, Wang Q-X, Zhang S, Zhang K-N, Liu Y-Q, Zhao Z, Jiang T, Wang Y-Z, Kang C-S. mA RNA methylation regulators contribute to malignant progression and have clinical prognostic impact in gliomas. Aging. 2019;11:1204–25.
Niu Y, Wan A, Lin Z, Lu X, Wan G. N (6)-Methyladenosine modification: a novel pharmacological target for anti-cancer drug development. Acta Pharm Sin B. 2018;8:833–43.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C, Yang CG. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, et al. Small-molecule targeting of oncogenic FTO Demethylase in acute myeloid leukemia. Cancer Cell. 2019;35.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, Peng S, Chen K, Wang M, Gong S, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of rare Panresistant triple-negative inflammatory breast Cancer cells facing a severe metabolic challenge. PLoS One. 2016;11:e0159072.
Selberg S, Blokhina D, Aatonen M, Koivisto P, Siltanen A, Mervaala E, Kankuri E, Karelson M. Discovery of small molecules that activate RNA methylation through cooperative binding to the METTL3-14-WTAP complex active site. Cell Rep. 2019;26:3762–71 e3765.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, Doki Y, Mori M, Ishii H, Ogawa K. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621–9.
Wang Y, Zeng L, Liang C, Zan R, Ji W, Zhang Z, Wei Y, Tu S, Dong Y. Integrated analysis of transcriptome-wide mA methylome of osteosarcoma stem cells enriched by chemotherapy. Epigenomics. 2019;11:1693–715.
Fustin J-M, Doi M, Yamaguchi Y, Hida H, Nishimura S, Yoshida M, Isagawa T, Morioka MS, Kakeya H, Manabe I, Okamura H. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Śledź P, Caflisch A. Small-molecule inhibitors of METTL3, the major human Epitranscriptomic writer. ChemMedChem. 2020.
Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, Marchioni M, Fazi F, Fatica A. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018;9:796.
Zhao J, Lee EE, Kim J, Yang R, Chamseddin B, Ni C, Gusho E, Xie Y, Chiang CM, Buszczak M, et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun. 2019;10:2300.
Zhang X, Xu Y, Qian Z, Zheng W, Wu Q, Chen Y, Zhu G, Liu Y, Bian Z, Xu W, et al. circRNA_104075 stimulates YAP-dependent tumorigenesis through the regulation of HNF4a and may serve as a diagnostic marker in hepatocellular carcinoma. Cell Death Dis. 2018;9:1091.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al. N6-Methyladenosine modification controls circular RNA immunity. Mol Cell. 2019;76:96–109 e109.
Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against Cancer. Annu Rev Med. 2016;67:73–89.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (grants No. 81772711 and 81602235), the Priority Academic Program Development of Jiangsu Higher Education Institutions (grant No. JX10231801), the Provincial Initiative Program for Excellency Disciplines of Jiangsu Province (grant No. BE2016791), the “333” project of Jiangsu Province (LGY2016002 and 2018055), and Jiangsu Province’s Key Provincial Talents Program (ZDRCA2016006).
Author information
Authors and Affiliations
Contributions
ZZZ, JCL and HY drafted the manuscript; JH and XY completed the `ures and tables; BRY, QKW and DXF collected the references; QL and HWY managed the article design, reviewed the manuscript and provided funding support. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All subjects have written informed consent.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
The alterations frequency of m6A regulators across 15 cancer types.
Additional file 2: Figure S1.
The type and percentages of each m6A protein alterations in tumors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Z., Lv, J., Yu, H. et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol Cancer 19, 104 (2020). https://doi.org/10.1186/s12943-020-01216-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01216-3