
Abstract
Contrast-induced nephropathy (CIN) or contrast-induced acute kidney injury (CI-AKI) is an iatrogenic acute kidney injury observed after intravascular administration of contrast media for intravascular diagnostic procedures or therapeutic angiographic intervention. High risk patients including those with chronic kidney disease (CKD), diabetes mellitus with impaired renal function, congestive heart failure, intraarterial intervention, higher volume of contrast, volume depletion, old age, multiple myeloma, hypertension, and hyperuricemia had increased prevalence of CIN. Although CIN is reversible by itself, some patients suffer this condition without renal recovery leading to CKD or even end-stage renal disease which required long term renal replacement therapy. In addition, both CIN and CKD have been associated with increasing of mortality. Three pathophysiological mechanisms have been proposed including direct tubular toxicity, intrarenal vasoconstriction, and excessive production of reactive oxygen species (ROS), all of which lead to impaired renal function. Reports from basic and clinical studies showing potential preventive strategies for CIN pathophysiology including low- or iso-osmolar contrast media are summarized and discussed. In addition, reports on pharmacological interventions to reduce ROS and attenuate CIN are summarized, highlighting potential for use in clinical practice. Understanding this contributory mechanism could pave ways to improve therapeutic strategies in combating CIN.
Similar content being viewed by others


Introduction
Contrast-induced nephropathy (CIN) or contrast-induced acute kidney injury (CI-AKI) is an iatrogenic acute kidney injury (AKI) observed after intravascular administration of contrast media (CM) for diagnostic procedures or therapeutic angiographic interventions [1,2,3,4]. Chemical hypersensitivity has also been reported as another side effect of CM [5]. According to the Kidney Disease Improving Global Outcomes (KDIGO) clinical practice guidelines for AKI, a serum creatinine (Cr) increase of at least 0.3 mg/dL (or 26.5 µmol/L) over the baseline value within 48 h after exposure to CM, or an increase greater than 1.5 times over the baseline value within 7 days after exposure to CM, or a urinary volume of less than 0.5 mL/kg/h for at least 6 h after exposure, are the definition of this condition [6]. Incidence of CIN has been reported in 1–25% of cases of hospital-acquired AKI, and is the third common cause of acute tubular necrosis in hospitalized patients leading to prolonged hospitalization [1]. In the general population, the CIN incidence is 1–2% [7]. Although CIN can be reversible, up to 15% of the patients may need temporary dialysis [8]. In patients without renal recovery, CKD can develop 4% progressing to end-stage renal disease (ESRD) [9]. The mortality rate of CIN varies from 3.8 to 64% [10, 11]. Patients with high risk of developing CIN include chronic kidney disease (CKD) and diabetes mellitus (DM) with impaired renal function. Other associated risks include congestive heart failure, volume depletion, old age, hypertension, and hyperuricemia increasing CIN prevalence by up to 25% [7].
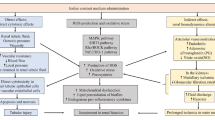
The pathophysiological mechanisms of CIN have not been completely elucidated. Currently, several mechanisms including direct effect, indirect effect, and generation of reactive oxygen species (ROS) have been proposed (Fig. 1). In direct effects, CM with high osmolality can directly cause cytotoxicity in nephrons including renal tubular epithelial cells and endothelial cells, leading to mitochondrial dysfunction, cellular apoptosis or necrosis and interstitial inflammation [12]. In indirect effects, CM can alter renal hemodynamics, leading to intrarenal vasoconstriction contributing to medullary hypoxia [12]. Regarding ROS generation, CM can either cause excessive ROS production or reduce antioxidant enzyme activity, resulting in increased oxidative stress and leading to impaired renal function [13]. In addition, medullary hypoxia also leads to enhanced ROS formation, resulting in mitochondrial oxidative stress and mitochondrial dysfunction [13]. Overall, it can be seen that mitochondrial function and oxidative stress play important roles in the pathophysiology of CIN [13]. Therefore, strategies that reduce oxidative stress as well as protecting mitochondrial dysfunction are potential targets for CIN prevention.

Pathophysiology of CIN. Pathogenesis of CIN consists of 3 mechanisms; direct effect, indirect effect, and generation of ROS. Direct effects include, direct cytotoxicity of CM to nephron leading to cellular apoptosis or necrosis and tubular injury. Indirect effects are that CM could alter renal hemodynamics, leading to intrarenal vasoconstriction, contributing to medullary hypoxia. This mechanism is mediated by the increase in vasoconstrictive mediators including renin, angiotensin II, and endothelin along with the decreasing of vasodilatory mediators including nitric oxide and PGI2. Lastly, CM can generate ROS and also reduce antioxidant enzyme activity as a result of various complex mechanisms which result in oxidative stress, leading to progression of impaired renal function. CIN, contrast-induced nephropathy; CM, contrast media; PGI2, prostaglandin I2; ROS, reactive oxygen species
KDIGO clinical practice guidelines for AKI state there is no definitive treatment available for established CIN [6]. Thus, the prevention of CIN is the best option. This review aims to comprehensively summarize the available in vitro, in vivo, and clinical reports regarding the pathophysiologic roles of mitochondria and ROS in CIN. Reports on pharmacological interventions to prevent CIN by targeting ROS and mitochondria are also presented and discussed with their potential for clinical use in in the future.
Searching methodology and selection criteria
A comprehensive search of the literature was performed using PubMed covering the period from database inception to September 2019. The search for literature included only articles written in English. An article was rejected if it was clearly a letter or case report. The search used the following keywords: contrast-induced nephropathy; oxidative stress; mitochondria; prevention; and statin either in the title, abstracts, or in the text. The relevance of the subject and eligibility of all publications detected was further evaluated, and data were then extracted from relevant papers to be included in this comprehensive review.
Pathogenesis of CIN via ROS generation: reports from in vitro studies
In vitro studies offer the unique opportunity to evaluate the activation of intracellular signaling pathways involved in cellular apoptosis or necrosis, which could pave ways for developing specific therapies to be used in in vivo and clinical studies. However, a major shortcoming of preclinical models of CIN relates to the fact that contrast administration alone does not cause AKI in animals. Multiple stressors are required to be utilized concomitantly to inflict CIN; such as inhibition of nitric oxide (NO), dehydration, and use of prostaglandin inhibitor. A summary of findings from in vitro reports is shown in Table 1.
Roles of mitogen-activated protein kinase (MAPK) pathways in CIN
ROS induced by CM could activate the MAPK signaling pathway through 4 cascades, including extracellular signal-related kinases (ERK) 1 and 2, c-JUN N-terminal kinase (JNK) 1, 2, and 3, p38-MAPK, and ERK5 [14]. These pathways contribute to the activation of caspase-9 and caspase-3, thus inducing apoptosis [15]. In HK-2 cells, CM increased cell injury and decreased cell viability, leading to severe mitochondrial vacuolar degeneration and nucleus fragmentation [16]. CM also increased ROS production via upregulation of nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2), Nox4 and p22phox, [16] and triggered apoptosis via induction of caspase 3/7 activity, MAPK pathways (including p38, JNK and ERK pathways), and B-cell lymphoma 2-associated X protein (Bax) expression [17]. Transfection of Nox4 short interfering ribonucleic acid (siRNA) caused a reduction in ROS production and apoptosis [17]. These findings indicated that both Nox and MAPK pathways are involved in the CM-induced ROS production.
Different types of high-osmolar CM were studied in renal cortical cells isolated from male Fischer 344 rats (Table 1). CM was shown to induce renal cell injury in a dose-dependent manner regardless of type of high-osmolar CM [18]. In human embryonic kidney 293 T cells, CM activated JNK/activating transcriptional factor 2 (ATF2) signaling pathways and decreased cell viability. Transfection with ATF2 siRNA caused reduced apoptosis in those CM-treated cells [19]. These findings indicated that JNK/ATF2 pathways are involved in CM-induced ROS production (Table 1).
Roles of silent information regulator 1 (SIRT1) in CIN
SIRT1 is a histone deacetylase of nicotinamide adenine dinucleotide (NAD+), which mainly exists in the nucleus [20]. In NRK-52E rodent tubular cells, CM caused oxidative stress and decreased cell viability by downregulation of SIRT1 [21]. Transfection with SIRT1 siRNA resulted in increased apoptosis in these cells treated with CM [21]. These findings indicated that CM downregulated SIRT1, leading to increased cell apoptosis (Table 1).
Pathogenesis of CIN via ROS generation: reports from in vivo studies
Consistent with in vitro reports, data from in vivo studies demonstrated that CM increased ROS levels and apoptosis, leading to impaired renal function [19]. A summary of these in vivo reports is shown in Table 2.
Roles of MAPK and SIRT1 in CIN
In mice, CM administration activated the Nox4/Nox2 axis, resulting in increased ROS production, and involving the MAPK pathway (including p38, JNK and ERK pathways) resulting in apoptosis, leading to impaired renal function [17]. CM administration also downregulated SIRT1 and upregulated peroxisome proliferator-activated receptor gamma-assisted activating factor-1α-Forkhead-box transcription factor 1 (PGC-1α-FoxO1) signaling mediated oxidative stress and apoptosis, leading to impaired renal function (Table 2) [21].
Roles of Rho/Rho-kinase (Rho/ROCK) pathway in CIN
The Rho/ROCK pathway is an important regulator in vascular smooth muscle cell contraction, cell migration, proliferation and differentiation [22]. Administration of CM in mice increased Rho/ROCK pathway activity, contributing to increased nuclear factor-kB (NF-kB) transcriptional activity, oxidative stress, inflammation and apoptosis, finally resulting in impaired renal function (Table 2) [23].
Roles of nuclear factor erythroid 2-related factor 2/heme oxygenase 1 (Nrf-2/HO-1) pathway in CIN
The Nrf-2/HO-1 pathway is involved in many functions including mitochondrial oxidative stress, autophagy, and programmed cell death [24]. Nrf-2, when translocated into the nucleus, stimulates transcription of genes that encode detoxifying and antioxidant enzymes, contributing to cellular protection by reducing oxidative stress [25]. In CM-treated rats, the Nrf-2/HO-1 pathway was upregulated to develop adaptive cytoprotective responses to counteract tissue damage, increased oxidative stress and apoptosis caused by CM (Table 2) [26,27,28]. Fig. 2 illustrates the mechanisms involved in the pathogenesis of CIN from in vitro and in vivo reports.

Mechanism of CIN via complex pathways of ROS from in vitro and in vivo studies. Contrast media can generate ROS especially in high risk patients such as DM and CKD through 4 major pathways: (1) MAPK pathway including ERK, JNK and p38; (2) SIRT1 pathway including SIRT1, FoxO, NF-kB, PGC-1 and p53; (3) Rho/ROCK pathway including MYPT-1 and NF-kB; (4) Nrf-2/HO-1 pathway including Nrf-2, NQO1, GSH and HO-1. CIN, contrast-induced nephropathy; CKD, chronic kidney disease; DM, diabetes mellitus; ERK, extracellular signal-related kinases; FoxO, Forkhead-box transcription factor; GSH, glutathione; JNK, c-JUN N-terminal kinase; MAPK, mitogen-activated protein kinase; MYPT-1, myosin-phosphatase target unit; NF-kB, nuclear factor-kB; NQO1, nicotinamide adenine dinucleotide phosphate quinone oxidoreductase 1; Nrf-2/HO-1, nuclear factor erythroid 2-related factor 2/heme oxygenase 1; PGC-1, peroxisome proliferator-activated receptor gamma-assisted activating factor-1; ROCK, rho-kinase; ROS, reactive oxygen species; SIRT1, silent information regulator 1
Interventions targeting ROS for CIN prevention: evidence from in vitro reports
In HK-2 cells [16] and MDCK cells, [29] atorvastatin attenuated CM-induced cytotoxicity through the downregulation of Nox4 and p22phox, and activation of MAPK pathways via JNK and tumor suppressor p53 activation [16, 29]. GKT137831, a specific Nox1/4 inhibitor, also decreased Nox2 expression, leading to decreased ROS production, and reduced apoptosis via decreasing caspase 3/7 activity and Bax along with activating the phosphorylation of p38, JNK and ERK [17].
Resveratrol, a known SIRT1 activator, was shown to increase SIRT1, PGC-1α expression, and superoxide dismutase 2 (SOD2), and increased cell viability in NRK-52E rodent tubular cells [21]. These findings indicated that resveratrol attenuated CM-induced nephrotoxicity via activating SIRT1-PGC-1α-FoxO1 signaling, leading to reduced oxidative stress and apoptosis [21]. In addition, Sulforaphane, an Nrf-2 activator, decreased ROS production and increased cell viability in HK-2 cells [27]. These reports are summarized in Table 3 and Fig. 3.

Intervention to reduce ROS for the prevention of CIN: evidence from in vitro, in vivo and clinical studies. In response to the mechanisms involved in ROS production in CIN, interventions to reduce ROS via complex pathways are illustrated. The MAPK pathway was inhibited by statins, GKT137831 and probucol. The SIRT1 pathway was inhibited by resveratrol. Rho/ROCK pathway was inhibited by fasudil. The Nrf-2/HO-1 pathway was inhibited by sulforaphane and salvianolic acid B. Antioxidant agents reported to exert benefits in CIN prevention have also been shown in this figure. CIN, contrast-induced nephropathy; GLP-1, glucagon-like peptide-1; MAPK, mitogen-activated protein kinase; MESNA, sodium-2-mercaptoethane sulphonate; mTOR, mammalian target of rapamycin; Nrf-2/HO-1, nuclear factor erythroid 2-related factor 2/heme oxygenase 1; ROCK, rho-kinase; ROS, reactive oxygen species; SIRT1, silent information regulator
Interventions targeting ROS for CIN prevention: evidence from in vivo reports
Interventions targeting MAPK, SIRT1, Rho/ROCK and Nrf-2/HO-1 pathways
In mice, GKT137831 could ameliorate oxidative stress via increased SOD and decreased Nox2, reducing apoptosis through the phosphorylation of p38, JNK, ERK, thus resulting in improved renal function [17]. Resveratrol was shown to attenuate CIN in rats via increasing SIRT1, PGC-1α, and SOD2, and decreasing phosphorylation of Ser256 FoxO1 expression, leading to a reduction in oxidative stress, apoptosis, improving renal function [21].
Fasudil, a Rho kinase inhibitor, was shown to decrease ROS and increase SOD-1, and reduced Inflammation via the reduction of NF-kB p65, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) [23]. Moreover, apoptosis was decreased via a reduction in cleaved caspase-3 and Bax, together with increased B-cell lymphoma-2 (Bcl-2) and p-Akt/total Akt ratio. These benefits on ROS and apoptosis attenuation led to improved renal function [23]. Similarly, sulforaphane was shown to exert CIN protection in rats via the Nrf-2/HO-1 pathway, resulting in reduced renal damage and improved Cr [27]. Salvianolic acid B, a component of Danshen (Salvia miltiorrhiza root), attenuated CIN in rats via decreasing malondialdehyde (MDA) and increasing Nrf-2-positive cells, p-Akt/Akt, Nrf-2/Histone H3, and HO-1/Actin, with antioxidative effects through PI3K/Akt/Nrf2 pathway, leading to improved renal function [26]. Table 4 and Fig. 3 show a summary of these reports.
Lipid-lowering agents as interventions to reduce CIN
Lipid-lowering agents including rosuvastatin, [30, 31] simvastatin, [31, 32] atorvastatin, [31, 33] xuezhikang (containing lovastatin), [33] and probucol [34] were investigated as potential pharmacological interventions in CIN animal models. These interventions appeared to effectively attenuate CIN as indicated by decreased level of kidney thiobarbiturates (TBARS), serum or renal MDA, serum protein carbonyl content (PCC), and increased serum thiol and glutathione (GSH) [29,30,31,32,33,34]. Inflammatory markers were also ameliorated as indicated by reduced IL-6, TNF-α, monocyte chemotactic protein-1 (MCP-1), myeloperoxidase (MPO), and increased NO [29,30,31,32,33,34]. The apoptotic markers were also reduced [29,30,31,32,33,34]. Furthermore, an appearance of unfavorable histological findings was decreased in an ischemic-reperfusion injury model [29,30,31,32,33,34]. These findings suggested that statins and probucol could attenuate CIN by modulation of NO, inflammatory responses, oxidative stress and apoptotic processes, leading to improved renal function [29,30,31,32,33,34]. A summary of these reports on the effects of lipid lowering agents on the protection of CIN is shown in Table 4. There are few clinical studies in this area so these statins are not recommended in the guidelines for CIN prevention.
Antioxidants as interventions to reduce CIN
Many antioxidants; such as vitamin E, [35] L-carnitine, [36] human serum albumin-thioredoxin-1 fusion protein (HSA-Trx), [37] paricalcitol, [38] N-acetylcysteine (NAC), [39, 40] recombinant manganese SOD (rMnSOD), [41] and agomelatine and melatonin; [42,43,44] were investigated for their potential effects to prevent CIN in rat models. All of the studies demonstrated the renoprotective effect by attenuating serum Cr and renal histological damage through their antioxidant activities (Table 4). Both inflammatory process and apoptosis were decreased following antioxidant treatments [35,36,37,38, 42,43,44].
Active component of herbs; such as astragaloside, [45] curcumin, [46] grape seed proanthocyanidin, [47] lycopene, [48] magnolin (major active ingredient of herb Magnolia fargesii), [49] Phyllanthus emblica extract, [50] salidroside, [40] sesame oil, [51] and silymarin, [52] were investigated in CIN in rats (Table 4). All studies demonstrated their benefits in attenuating CIN and AKI biomarkers such as cystatin C, neutrophil gelatinase-associated lipocalin (NGAL), and urine kidney injury molecule-1 (KIM-1), due to reduced oxidative stress and apoptosis.
Other agents such as cardiotrophin-1 and antithrombin III, [53, 54] exendin-4, [55] β-receptor antagonist, [56, 57] phosphodiesterase-5 inhibitor, [58,59,60,61] an mTOR inhibitor, [62] exogenous sphingosylphosphorylcholine, [63] and sodium bicarbonate; [64] have been investigated in CIN models (Table 4 and Fig. 3). They all effectively reduced oxidative stress, inflammation and apoptosis, with improved renal histopathology. These findings suggested that these pharmacological interventions prevented CIN through a reduction in oxidative stress, inflammation and apoptosis, leading to improved renal function in rats.
Pharmacological interventions to reduce CIN: evidence from clinical reports
Effects of statins on the prevention of CIN
Statins have been shown to exert renoprotective effects in CIN via inhibition of uptake of contrast into renal tubular cells, attenuation of endothelial dysfunction and oxidative stress, anti-inflammation, anti-proliferation of mesangial cells, and protection of podocytes [9]. Clinical studies of statins on the prevention of CIN are summarized in Table 5 and Fig. 3.
In a retrospective study of 29,409 patients undergoing percutaneous coronary intervention (PCI), initiating statin therapy before PCI reduced risk of CIN [65]. Many randomized-controlled trials of atorvastatin for CIN prevention were done in patients undergoing coronary angiography (CAG). In high risk patients, short-term pretreatment with high-dose atorvastatin decreased incidence of CIN [29, 66,67,68,69,70,71,72,73], reducing C-reactive protein (CRP) [68].
The largest randomized-controlled trial in rosuvastatin was done in 2,998 patients with type 2 DM and CKD who underwent coronary or peripheral angiography, receiving either pre and post-intervention rosuvastatin or standard care. The rosuvastatin-treated group had lower incidence of CIN and high-sensitivity CRP (hsCRP) [74]. In the PRATO-ACS trial, the incidence of CIN in non-ST elevated ACS patients undergoing CAG who receive rosuvastatin in statin-naïve patients was lower than in control group [75]. With simvastatin, the prospective randomized-controlled trials in patients undergoing CAG demonstrated that short-term pretreatment of high-dose simvastatin reduced the incidence of CIN [76, 77]. Simvastatin also reduced inflammation by decreasing hsCRP, P-selectin, and intracellular cell adhesion molecule 1 (ICAM-1) [77].
Despite these promising findings, inconsistent reports exist. The PROMISS trial failed to show a difference between simvastatin and placebo with respect to a primary end point based on the mean peak increase in plasma Cr within 48 h after CAG in patients with CKD [78]. Also, another randomized-controlled trial demonstrated that short-term administration of high-dose atorvastatin with oral NAC did not decrease incidence of CIN in pre-existing CKD patients, [79] and a retrospective study, statin given before non-emergent PCI increased the incidence of CIN [80]. Similarly, a prospective cohort study in patients with or without CKD undergoing CAG demonstrated that high plasma atorvastatin or rosuvastatin was associated with increased CIN risk [81]. Therefore, currently statins are not recommended in the guidelines for CIN prevention.
Effects of antioxidants on the prevention of CIN
Many clinical trials have investigated the effects of various antioxidants on the prevention of CIN. These include NAC, ascorbic acid, sodium bicarbonate, sodium-2-mercaptoethane sulphonate (MESNA), and nebivolol. A summary of these reports is shown in Table 6 and Fig. 3.
N-Acetylcysteine (NAC)
For nearly two decades, many randomized-controlled trials have investigated the roles of oral NAC for CIN prevention in patients receiving PCI or diagnostic CAG undergoing computed tomography. They demonstrated the protective effect of NAC to CIN in both low and high risk patients compared with placebo [82,83,84,85,86,87,88,89,90,91]. The prospective studies demonstrated that NAC prevented the decline in urinary NO end-products [85, 91]. However, many conflicting reports exist, with no significant evident benefits of oral NAC in CIN prevention [92,93,94,95,96,97,98,99,100,101,102,103,104,105,106]. Therefore, routine administration of NAC for the prevention of CIN or longer-term adverse events after angiographic procedures is not recommended [93].
Decisive factors in NAC for CIN prevention are dosage and treatment duration. NAC was commonly given only for two days prior to CM administration in those trials. It is possible that the duration of NAC treatment was too short to be effective in counteracting CIN-induced ROS production. Furthermore, since NAC has a very short plasma half-life, dosing twice daily could be insufficient to achieve consistent renal protective effects [7]. Future studies are needed to test this hypothesis. A summary of these reports on the effects of oral administration of NAC to prevent CIN is shown in Table 6.
Effects of intravenous administration of NAC on CIN protection has been investigated in patients requiring emergent CAG. In a prospective randomized-controlled study in low risk patients, short-term intravenous NAC treatment could prevent CIN [107]. However, 7% of the patients developed anaphylactoid reactions in that report. In addition, conflicting reports exist [108, 109]. Intravenous NAC reduced oxidative stress after reperfusion of myocardial infarction, however it did not provide additional clinical benefit to nephropathy [108, 109]. Also, intravenous NAC at higher doses could be associated with significant side effects (anaphylactoid reaction, hypotension, bronchospasm) [12]. Despite equipoise on its efficacy, NAC has been widely used in clinical practice in high risk patients due to its low cost, ready availability, easy administration, and limited toxicity in an oral form.
Ascorbic acid, sodium bicarbonate and sodium-2-mercaptoethane sulphonate (MESNA)
In a randomized, double-blind, placebo-controlled trial of patients with Cr ≥ 1.2 mg/dL undergoing CAG, the use of ascorbic acid was associated with a significant reduction in the rate of CIN [110]. For sodium bicarbonate, a randomized trial in patients undergoing CAG demonstrated that bicarbonate supplementation prevented CIN when it was given 6 h prior to CAG and administered continuously for another 6 h after CAG [111]. MESNA has the potential to act as a ROS scavenger. In a pilot study in CKD patients undergoing CAG, renal function improved after MESNA pre and posttreatment [112]. These findings indicated that these antioxidants could prevent CIN in patients with renal impairment. However, future studies may add weight to these limited reports.
Nebivolol
Although in vivo studies report the benefits of antioxidant effects by β-receptor antagonists leading to prevention of CIN, [56, 57] a report from a clinical study demonstrated otherwise. In a cross-sectional case–control study, the patients with risk factor for CIN that received nebivolol had no change in renal function before and after CAG, and did not prevent CIN in patients undergoing CAG [113]. These inconsistent reports could be due to a small sample size from a single center and different times of follow-up on Cr since a 1 month follow up might allow the development of a tolerance to the vasodilatory effects of the drug.
In summary, this review provides a comprehensive narrative review that summarizes findings of the pathogenesis of CIN from in vitro and in vivo studies and the novel interventions for prevention of CIN. Consistent as well as controversial reports regarding the clinical findings are also summarized for potential interventions to prevent CIN. This review provides fundamental knowledge for future basic and clinical studies to find novel interventions to prevent CIN in a clinical setting. However, since our review does not include the non-English original articles it is possible that other potential interventions to prevent CIN be missing.
Conclusions
CIN is associated with adverse outcomes. These include renal replacement therapy, prolonged hospitalization, increased mortality, and increased financial burden. For this reason, the appropriate prophylactic interventions before CM administration in high-risk patients are important in reducing CIN. Since hypoxic-toxic injury, including altered renal microcirculation, medullary hypoxia and ROS-mediated cellular injury is a fundamental pathogenesis of CIN, understanding these various complex pathways could lead to prevention. Although a variety of experimental studies and clinical trials have demonstrated potential pharmacological interventions to prevent CIN, inconclusive results exist. Future double-blinded randomized-controlled trials with large populations of oral or intravenous antioxidants as well as other novel compounds are needed to warrant their use to prevent CIN in patients exposed to CM.
Availability of data and materials
Not applicable.
Abbreviations
- AKI:
-
Acute kidney injury
- ATF2:
-
Activating transcriptional factor 2
- Bax:
-
B-cell lymphoma 2-associated X protein
- Bcl-2:
-
B-cell lymphoma-2
- CAG:
-
Coronary angiography
- CI-AKI:
-
Contrast-induced acute kidney injury
- CIN:
-
Contrast-induced nephropathy
- CKD:
-
Chronic kidney disease
- CM:
-
Contrast media
- Cr:
-
Creatinine
- ERK:
-
Extracellular signal-related kinases
- ESRD:
-
End-stage renal disease
- GSH:
-
Glutathione
- HO-1:
-
Heme oxygenase 1
- HSA-Trx:
-
Human serum albumin-thioredoxin-1 fusion protein
- hsCRP:
-
High-sensitivity C-reactive protein
- ICAM-1:
-
Intracellular cell adhesion molecule 1
- IL:
-
Interleukin
- JNK:
-
C-JUN N-terminal kinase
- KDIGO:
-
Kidney Disease Improving Global Outcomes
- KIM-1:
-
Kidney injury molecule-1
- MAPK:
-
Mitogen-activated protein kinase
- MCP-1:
-
Monocyte chemotactic protein-1
- MDA:
-
Malondialdehyde
- MESNA:
-
Sodium-2-mercaptoethane sulphonate
- MPO:
-
Myeloperoxidase
- NAC:
-
N-Acetylcysteine
- NAD:
-
Nicotinamide adenine dinucleotide
- NF-kB:
-
Nuclear factor-kB
- NGAL:
-
Neutrophil gelatinase-associated lipocalin
- NO:
-
Nitric oxide
- Nox:
-
Nicotinamide adenine dinucleotide phosphate oxidase
- Nrf-2:
-
Nuclear factor erythroid 2-related factor 2
- PCC:
-
Protein carbonyl content
- PCI:
-
Percutaneous coronary intervention
- PGC-1α-FoxO1:
-
Peroxisome proliferator-activated receptor gamma-assisted activating factor-1α-Forkhead-box transcription factor 1
- rMnSOD:
-
Recombinant manganese superoxide dismutase
- ROCK:
-
Rho-kinase
- ROS:
-
Reactive oxygen species
- siRNA:
-
Short interfering ribonucleic acid
- SIRT1:
-
Silent information regulator 1
- SOD:
-
Superoxide dismutase
- TBARS:
-
Thiobarbiturates
- TNF-α:
-
Tumor necrosis factor-α
References
Pattharanitima P, Tasanarong A. Pharmacological strategies to prevent contrast-induced acute kidney injury. Biomed Res Int. 2014;2014:236930.
Lopez-Ruiz A, Chandrashekar K, Juncos LA. Changing paradigms in contrast nephropathy. J Am Soc Nephrol. 2017;28(2):397–9.
Ehrmann S, Aronson D, Hinson JS. Contrast-associated acute kidney injury is a myth: yes. Intensive Care Med. 2018;44(1):104–6.
Haq MFU, Yip CS, Arora P. The conundrum of contrast-induced acute kidney injury. J Thorac Dis. 2020;12(4):1721–7.
Iordache AM, Docea AO, Buga AM, Mitrut R, Albulescu D, Zlatian O, et al. The incidence of skin lesions in contrast media-induced chemical hypersensitivity. Exp Ther Med. 2019;17(2):1113–24.
Work Group Membership. Kidney Int Suppl. 2012;2(1):2.
Pflueger A, Abramowitz D, Calvin AD. Role of oxidative stress in contrast-induced acute kidney injury in diabetes mellitus. Med Sci Monit. 2009;15(6):125–36.
Mohammed NM, Mahfouz A, Achkar K, Rafie IM, Hajar R. Contrast-induced nephropathy. Heart Views. 2013;14(3):106–16.
McCullough PA, Choi JP, Feghali GA, Schussler JM, Stoler RM, Vallabahn RC, et al. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2016;68(13):1465–73.
Briguori C, Marenzi G. Contrast-induced nephropathy: pharmacological prophylaxis. Kidney Int Suppl. 2006;100:S30–8.
Kini AS, Mitre CA, Kamran M, Suleman J, Kim M, Duffy ME, et al. Changing trends in incidence and predictors of radiographic contrast nephropathy after percutaneous coronary intervention with use of fenoldopam. Am J Cardiol. 2002;89(8):999–1002.
Mehran R, Dangas GD, Weisbord SD. Contrast-associated acute kidney injury. N Engl J Med. 2019;380(22):2146–55.
Heyman SN, Rosen S, Khamaisi M, Idee JM, Rosenberger C. Reactive oxygen species and the pathogenesis of radiocontrast-induced nephropathy. Invest Radiol. 2010;45(4):188–95.
Pritchard AL, Hayward NK. Molecular pathways: mitogen-activated protein kinase pathway mutations and drug resistance. Clin Cancer Res. 2013;19(9):2301–9.
Pisani A, Riccio E, Andreucci M, Faga T, Ashour M, Di Nuzzi A, et al. Role of reactive oxygen species in pathogenesis of radiocontrast-induced nephropathy. Biomed Res Int. 2013;2013:868321.
Liu GL, Lei R, Duan SB, Tang MM, Luo M, Xu Q. Atorvastatin alleviates iodinated contrast media-induced cytotoxicity in human proximal renal tubular epithelial cells. Exp Ther Med. 2017;14(4):3309–13.
Jeong BY, Lee HY, Park CG, Kang J, Yu SL, Choi DR, et al. Oxidative stress caused by activation of NADPH oxidase 4 promotes contrast-induced acute kidney injury. PLoS ONE. 2018;13(1):e0191034.
Harmon RC, Duffy SP, Terneus MV, Ball JG, Valentovic MA. Characterization of a novel model for investigation of radiocontrast nephrotoxicity. Nephrol Dial Transplant. 2009;24(3):763–8.
Lee HC, Sheu SH, Yen HW, Lai WT, Chang JG. JNK/ATF2 pathway is involved in iodinated contrast media-induced apoptosis. Am J Nephrol. 2010;31(2):125–33.
Ren Z, He H, Zuo Z, Xu Z, Wei Z, Deng J. The role of different SIRT1-mediated signaling pathways in toxic injury. Cell Mol Biol Lett. 2019;24:36.
Hong YA, Bae SY, Ahn SY, Kim J, Kwon YJ, Jung WY, et al. Resveratrol ameliorates contrast induced nephropathy through the activation of SIRT1-PGC-1alpha-Foxo1 signaling in mice. Kidney Blood Press Res. 2017;42(4):641–53.
Wirth A. Rho kinase and hypertension. Biochim Biophys Acta. 2010;1802(12):1276–84.
Wang Y, Zhang H, Yang Z, Miao D, Zhang D. Rho kinase inhibitor, Fasudil, attenuates contrast-induced acute kidney injury. Basic Clin Pharmacol Toxicol. 2018;122(2):278–87.
Zhang X, Ding M, Zhu P, Huang H, Zhuang Q, Shen J, et al. New insights into the Nrf-2/HO-1 signaling axis and its application in pediatric respiratory diseases. Oxid Med Cell Longev. 2019;2019:3214196.
Mamoulakis C, Tsarouhas K, Fragkiadoulaki I, Heretis I, Wilks MF, Spandidos DA, et al. Contrast-induced nephropathy: basic concepts, pathophysiological implications and prevention strategies. Pharmacol Ther. 2017;180:99–112.
Tongqiang L, Shaopeng L, Xiaofang Y, Nana S, Xialian X, Jiachang H, et al. Salvianolic acid B prevents iodinated contrast media-induced acute renal injury in rats via the PI3K/Akt/Nrf2 pathway. Oxid Med Cell Longev. 2016;2016:7079487.
Zhao Z, Liao G, Zhou Q, Lv D, Holthfer H, Zou H. Sulforaphane attenuates contrast-induced nephropathy in rats via Nrf2/HO-1 pathway. Oxid Med Cell Longev. 2016;2016:9825623.
Goodman AI, Olszanecki R, Yang LM, Quan S, Li M, Omura S, et al. Heme oxygenase-1 protects against radiocontrast-induced acute kidney injury by regulating anti-apoptotic proteins. Kidney Int. 2007;72(8):945–53.
Quintavalle C, Fiore D, De Micco F, Visconti G, Focaccio A, Golia B, et al. Impact of a high loading dose of atorvastatin on contrast-induced acute kidney injury. Circulation. 2012;126(25):3008–16.
Deng J, Wu G, Yang C, Li Y, Jing Q, Han Y. Rosuvastatin attenuates contrast-induced nephropathy through modulation of nitric oxide, inflammatory responses, oxidative stress and apoptosis in diabetic male rats. J Transl Med. 2015;13:53.
Wang X-L, Zhang T, Hu L-H, Sun S-Q, Zhang W-F, Sun Z, et al. Comparison of effects of different statins on contrast-induced acute kidney injury in rats: histopathological and biochemical findings. Oxidative Med Cell Longevity. 2017;2017:1–10.
Al-Otaibi KE, Al Elaiwi AM, Tariq M, Al-Asmari AK. Simvastatin attenuates contrast-induced nephropathy through modulation of oxidative stress, proinflammatory myeloperoxidase, and nitric oxide. Oxid Med Cell Longev. 2012;2012:831748.
Chu S, Hu L, Wang X, Sun S, Zhang T, Sun Z, et al. Xuezhikang ameliorates contrast media-induced nephropathy in rats via suppression of oxidative stress, inflammatory responses and apoptosis. Ren Fail. 2016;38(10):1717–25.
Ma X, Jiao Z, Liu Y, Chen J, Li G, Liu T, et al. Probucol Protects against contrast-induced acute kidney injury via the extracellular signal-regulated kinases 1 and 2 (ERK1/2)/JNK-caspase 3 pathway in diabetic Rats. Med Sci Monit. 2019;25:1038–45.
Kongkham S, Sriwong S, Tasanarong A. Protective effect of alpha tocopherol on contrast-induced nephropathy in rats. Nefrologia. 2013;33(1):116–23.
Kunak CS, Ugan RA, Cadirci E, Karakus E, Polat B, Un H, et al. Nephroprotective potential of carnitine against glycerol and contrast-induced kidney injury in rats through modulation of oxidative stress, proinflammatory cytokines, and apoptosis. Br J Radiol. 2016;89(1058):20140724.
Kodama A, Watanabe H, Tanaka R, Tanaka H, Chuang VT, Miyamoto Y, et al. A human serum albumin-thioredoxin fusion protein prevents experimental contrast-induced nephropathy. Kidney Int. 2013;83(3):446–54.
Ari E, Kedrah AE, Alahdab Y, Bulut G, Eren Z, Baytekin O, et al. Antioxidant and renoprotective effects of paricalcitol on experimental contrast-induced nephropathy model. Br J Radiol. 2012;85(1016):1038–43.
Ozturk O, Eroglu HA, Ustebay S, Kuzucu M, Adali Y. An experimental study on the preventive effects of N-acetyl cysteine and ozone treatment against contrast-induced nephropathy. Acta Cir Bras. 2018;33(6):508–17.
Xing Y, Wei RB, Tang L, Yang Y, Zheng XY, Wang ZC, et al. Protective effect of salidroside on contrast-induced nephropathy in comparison with N-acetylcysteine and its underlying mechanism. Chin J Integr Med. 2015;21(4):266–73.
Pisani A, Sabbatini M, Riccio E, Rossano R, Andreucci M, Capasso C, et al. Effect of a recombinant manganese superoxide dismutase on prevention of contrast-induced acute kidney injury. Clin Exp Nephrol. 2014;18(3):424–31.
Karaman A, Diyarbakir B, Durur-Subasi I, Kose D, Ozbek-Bilgin A, Topcu A, et al. A novel approach to contrast-induced nephrotoxicity: the melatonergic agent agomelatine. Br J Radiol. 2016;89(1061):20150716.
Gazi S, Altun A, Erdogan O. Contrast-induced nephropathy: preventive and protective effects of melatonin. J Pineal Res. 2006;41(1):53–7.
Onk D, Onk OA, Turkmen K, Erol HS, Ayazoglu TA, Keles ON, et al. Melatonin attenuates contrast-induced nephropathy in diabetic rats: the role of interleukin-33 and oxidative stress. Mediators Inflamm. 2016;2016:9050828.
Gui D, Huang J, Liu W, Guo Y, Xiao W, Wang N. Astragaloside IV prevents acute kidney injury in two rodent models by inhibiting oxidative stress and apoptosis pathways. Apoptosis. 2013;18(4):409–22.
Buyuklu M, Kandemir FM, Ozkaraca M, Set T, Bakirci EM, Topal E. Protective effect of curcumin against contrast induced nephropathy in rat kidney: what is happening to oxidative stress, inflammation, autophagy and apoptosis? Eur Rev Med Pharmacol Sci. 2014;18(4):461–70.
Ozkan G, Ulusoy S, Orem A, Ersoz S, Alkanat M, Yucesan FB, et al. Protective effect of the grape seed proanthocyanidin extract in a rat model of contrast-induced nephropathy. Kidney Blood Press Res. 2012;35(6):445–53.
Buyuklu M, Kandemir FM, Ozkaraca M, Set T, Bakirci EM, Topal E, et al. Benefical effects of lycopene against contrast medium-induced oxidative stress, inflammation, autophagy, and apoptosis in rat kidney. Hum Exp Toxicol. 2015;34(5):487–96.
Wang F, Zhang G, Zhou Y, Gui D, Li J, Xing T, et al. Magnolin protects against contrast-induced nephropathy in rats via antioxidation and antiapoptosis. Oxid Med Cell Longev. 2014;2014:203458.
Tasanarong A, Kongkham S, Itharat A. Antioxidant effect of Phyllanthus emblica extract prevents contrast-induced acute kidney injury. BMC Complement Altern Med. 2014;14:138.
Hsu DZ, Li YH, Chu PY, Periasamy S, Liu MY. Sesame oil prevents acute kidney injury induced by the synergistic action of aminoglycoside and iodinated contrast in rats. Antimicrob Agents Chemother. 2011;55(6):2532–6.
de Souza SV, Peters B, Coco LZ, Alves GM, de Assis A, Nogueira BV, et al. Silymarin protects against radiocontrast-induced nephropathy in mice. Life Sci. 2019;228:305–15.
Lu Z, Cheng D, Yin J, Wu R, Zhang G, Zhao Q, et al. Antithrombin III protects against contrast-induced nephropathy. EBioMedicine. 2017;17:101–7.
Quiros Y, Sanchez-Gonzalez PD, Lopez-Hernandez FJ, Morales AI, Lopez-Novoa JM. Cardiotrophin-1 administration prevents the renal toxicity of iodinated contrast media in rats. Toxicol Sci. 2013;132(2):493–501.
Hussien NI, Sorour SM, El-Kerdasy HI, Abdelrahman BA. The glucagon-like peptide-1 receptor agonist Exendin-4, ameliorates contrast-induced nephropathy through suppression of oxidative stress, vascular dysfunction and apoptosis independent of glycaemia. Clin Exp Pharmacol Physiol. 2018;45(8):808–18.
Toprak O, Cirit M, Tanrisev M, Yazici C, Canoz O, Sipahioglu M, et al. Preventive effect of nebivolol on contrast-induced nephropathy in rats. Nephrol Dial Transplant. 2008;23(3):853–9.
Akgullu C, Hekim T, Eryilmaz U, Boyacioglu M, Gungor H, Meteoglu I, et al. The usefulness of carvedilol and nebivolol in preventing contrast nephropathy in rats. Ren Fail. 2015;37(3):511–7.
Almeida LS, Barboza JR, Freitas FPS, Porto ML, Vasquez EC, Meyrelles SS, et al. Sildenafil prevents renal dysfunction in contrast media-induced nephropathy in Wistar rats. Hum Exp Toxicol. 2016;35(11):1194–202.
Iordache AM, Buga AM, Albulescu D, Vasile RC, Mitrut R, Georgiadis G, et al. Phosphodiesterase-5 inhibitors ameliorate structural kidney damage in a rat model of contrast-induced nephropathy. Food Chem Toxicol. 2020;143:111535.
Iordache AM, Docea AO, Buga AM, Zlatian O, Ciurea ME, Rogoveanu OC, et al. Sildenafil and tadalafil reduce the risk of contrast-induced nephropathy by modulating the oxidant/antioxidant balance in a murine model. Food Chem Toxicol. 2020;135:111038.
Georgiadis G, Zisis IE, Docea AO, Tsarouhas K, Fragkiadoulaki I, Mavridis C, et al. Current concepts on the reno-protective effects of phosphodiesterase 5 inhibitors in acute kidney injury: systematic search and review. J Clin Med. 2020;9:5.
Yang X, Yan X, Yang D, Zhou J, Song J, Yang D. Rapamycin attenuates mitochondrial injury and renal tubular cell apoptosis in experimental contrast-induced acute kidney injury in rats. Biosci Rep. 2018;38:6.
Aksu F, Aksu B, Unlu N, Karaca T, Ayvaz S, Erman H, et al. Antioxidant and renoprotective effects of sphingosylphosphorylcholine on contrast-induced nephropathy in rats. Ren Fail. 2016;38(7):1089–98.
Barlak A, Akar H, Yenicerioglu Y, Yenisey C, Meteoglu I, Yilmaz O. Effect of sodium bicarbonate in an experimental model of radiocontrast nephropathy. Ren Fail. 2010;32(8):992–9.
Khanal S, Attallah N, Smith DE, Kline-Rogers E, Share D, O’Donnell MJ, et al. Statin therapy reduces contrast-induced nephropathy: an analysis of contemporary percutaneous interventions. Am J Med. 2005;118(8):843–9.
Khosravi A, Dolatkhah M, Hashemi HS, Rostami Z. Preventive effect of atorvastatin (80 mg) on contrast-induced nephropathy after angiography in high-risk patients: double-blind randomized clinical trial. Nephrourol Mon. 2016;8(3):e29574.
Syed MH, Khandelwal PN, Thawani VR, Katare SS. Efficacy of atorvastatin in prevention of contrast-induced nephropathy in high-risk patients undergoing angiography: a double-blind randomized controlled trial. J Pharmacol Pharmacother. 2017;8(2):50–3.
Patti G, Ricottini E, Nusca A, Colonna G, Pasceri V, D’Ambrosio A, et al. Short-term, high-dose Atorvastatin pretreatment to prevent contrast-induced nephropathy in patients with acute coronary syndromes undergoing percutaneous coronary intervention (from the ARMYDA-CIN [atorvastatin for reduction of myocardial damage during angioplasty–contrast-induced nephropathy] trial. Am J Cardiol. 2011;108(1):1–7.
Li W, Fu X, Wang Y, Li X, Yang Z, Wang X, et al. Beneficial effects of high-dose atorvastatin pretreatment on renal function in patients with acute ST-segment elevation myocardial infarction undergoing emergency percutaneous coronary intervention. Cardiology. 2012;122(3):195–202.
Ozhan H, Erden I, Ordu S, Aydin M, Caglar O, Basar C, et al. Efficacy of short-term high-dose atorvastatin for prevention of contrast-induced nephropathy in patients undergoing coronary angiography. Angiology. 2010;61(7):711–4.
Sanei H, Hajian-Nejad A, Sajjadieh-Kajouei A, Nazemzadeh N, Alizadeh N, Bidram P, et al. Short term high dose atorvastatin for the prevention of contrast-induced nephropathy in patients undergoing computed tomography angiography. ARYA Atheroscler. 2014;10(5):252–8.
Zhou X, Jin YZ, Wang Q, Min R, Zhang XY. Efficacy of high dose atorvastatin on preventing contrast induced nephropathy in patients underwent coronary angiography. Zhonghua Xin Xue Guan Bing Za Zhi. 2009;37(5):394–6.
Acikel S, Muderrisoglu H, Yildirir A, Aydinalp A, Sade E, Bayraktar N, et al. Prevention of contrast-induced impairment of renal function by short-term or long-term statin therapy in patients undergoing elective coronary angiography. Blood Coagul Fibrinolysis. 2010;21(8):750–7.
Han Y, Zhu G, Han L, Hou F, Huang W, Liu H, et al. Short-term rosuvastatin therapy for prevention of contrast-induced acute kidney injury in patients with diabetes and chronic kidney disease. J Am Coll Cardiol. 2014;63(1):62–70.
Leoncini M, Toso A, Maioli M, Tropeano F, Villani S, Bellandi F. Early high-dose rosuvastatin for contrast-induced nephropathy prevention in acute coronary syndrome: results from the PRATO-ACS Study (Protective Effect of Rosuvastatin and Antiplatelet Therapy On contrast-induced acute kidney injury and myocardial damage in patients with Acute Coronary Syndrome). J Am Coll Cardiol. 2014;63(1):71–9.
Sanadgol H, Abdani S, Tabatabaiee P, Mohammadi M. Protective effect of high dose short term statin therapy with normal saline in prevention of contrast-induced nephropathy among iodixanol-receiving patients. J Renal Inj Prev. 2012;1(1):43–5.
Xinwei J, Xianghua F, Jing Z, Xinshun G, Ling X, Weize F, et al. Comparison of usefulness of simvastatin 20 mg versus 80 mg in preventing contrast-induced nephropathy in patients with acute coronary syndrome undergoing percutaneous coronary intervention. Am J Cardiol. 2009;104(4):519–24.
Jo SH, Koo BK, Park JS, Kang HJ, Cho YS, Kim YJ, et al. Prevention of radiocontrast medium-induced nephropathy using short-term high-dose simvastatin in patients with renal insufficiency undergoing coronary angiography (PROMISS) trial–a randomized controlled study. Am Heart J. 2008;155(3):499.
Toso A, Maioli M, Leoncini M, Gallopin M, Tedeschi D, Micheletti C, et al. Usefulness of atorvastatin (80 mg) in prevention of contrast-induced nephropathy in patients with chronic renal disease. Am J Cardiol. 2010;105(3):288–92.
Kandula P, Shah R, Singh N, Markwell SJ, Bhensdadia N, Navaneethan SD. Statins for prevention of contrast-induced nephropathy in patients undergoing non-emergent percutaneous coronary intervention. Nephrology (Carlton). 2010;15(2):165–70.
Cai L, Bai X, Lei H, Wu H, Liu Y, Zhu Q, et al. High plasma exposure of statins associated with increased risk of contrast-induced acute kidney injury in chinese patients with coronary artery disease. Front Pharmacol. 2018;9:427.
Tepel M, van der Giet M, Schwarzfeld C, Laufer U, Liermann D, Zidek W. Prevention of radiographic-contrast-agent-induced reductions in renal function by acetylcysteine. N Engl J Med. 2000;343(3):180–4.
Briguori C, Manganelli F, Scarpato P, Elia PP, Golia B, Riviezzo G, et al. Acetylcysteine and contrast agent-associated nephrotoxicity. J Am Coll Cardiol. 2002;40(2):298–303.
Diaz-Sandoval LJ, Kosowsky BD, Losordo DW. Acetylcysteine to prevent angiography-related renal tissue injury (the APART trial). Am J Cardiol. 2002;89(3):356–8.
Efrati S, Dishy V, Averbukh M, Blatt A, Krakover R, Weisgarten J, et al. The effect of N-acetylcysteine on renal function, nitric oxide, and oxidative stress after angiography. Kidney Int. 2003;64(6):2182–7.
Kay J, Chow WH, Chan TM, Lo SK, Kwok OH, Yip A, et al. Acetylcysteine for prevention of acute deterioration of renal function following elective coronary angiography and intervention: a randomized controlled trial. JAMA. 2003;289(5):553–8.
MacNeill BD, Harding SA, Bazari H, Patton KK, Colon-Hernadez P, DeJoseph D, et al. Prophylaxis of contrast-induced nephropathy in patients undergoing coronary angiography. Catheter Cardiovasc Interv. 2003;60(4):458–61.
Ochoa A, Pellizzon G, Addala S, Grines C, Isayenko Y, Boura J, et al. Abbreviated dosing of N-acetylcysteine prevents contrast-induced nephropathy after elective and urgent coronary angiography and intervention. J Interv Cardiol. 2004;17(3):159–65.
Shyu KG, Cheng JJ, Kuan P. Acetylcysteine protects against acute renal damage in patients with abnormal renal function undergoing a coronary procedure. J Am Coll Cardiol. 2002;40(8):1383–8.
Baskurt M, Okcun B, Abaci O, Dogan GM, Kilickesmez K, Ozkan AA, et al. N-acetylcysteine versus N-acetylcysteine + theophylline for the prevention of contrast nephropathy. Eur J Clin Invest. 2009;39(9):793–9.
Drager LF, Andrade L, Toledo JF, Laurindo FR, MachadoCesar LA, Seguro AC. Renal effects of N-acetylcysteine in patients at risk for contrast nephropathy: decrease in oxidant stress-mediated renal tubular injury. Nephrol Dial Transplant. 2004;19(7):1803–7.
Kimmel M, Butscheid M, Brenner S, Kuhlmann U, Klotz U, Alscher DM. Improved estimation of glomerular filtration rate by serum cystatin C in preventing contrast induced nephropathy by N-acetylcysteine or zinc–preliminary results. Nephrol Dial Transplant. 2008;23(4):1241–5.
Weisbord SD, Gallagher M, Jneid H, Garcia S, Cass A, Thwin SS, et al. Outcomes after angiography with sodium bicarbonate and acetylcysteine. N Engl J Med. 2018;378(7):603–14.
Investigators ACT. Acetylcysteine for prevention of renal outcomes in patients undergoing coronary and peripheral vascular angiography: main results from the randomized Acetylcysteine for Contrast-induced nephropathy Trial (ACT). Circulation. 2011;124(11):1250–9.
Allaqaband S, Tumuluri R, Malik AM, Gupta A, Volkert P, Shalev Y, et al. Prospective randomized study of N-acetylcysteine, fenoldopam, and saline for prevention of radiocontrast-induced nephropathy. Catheter Cardiovasc Interv. 2002;57(3):279–83.
Amini M, Salarifar M, Amirbaigloo A, Masoudkabir F, Esfahani F. N-acetylcysteine does not prevent contrast-induced nephropathy after cardiac catheterization in patients with diabetes mellitus and chronic kidney disease: a randomized clinical trial. Trials. 2009;10:45.
Boccalandro F, Amhad M, Smalling RW, Sdringola S. Oral acetylcysteine does not protect renal function from moderate to high doses of intravenous radiographic contrast. Catheter Cardiovasc Interv. 2003;58(3):336–41.
Durham JD, Caputo C, Dokko J, Zaharakis T, Pahlavan M, Keltz J, et al. A randomized controlled trial of N-acetylcysteine to prevent contrast nephropathy in cardiac angiography. Kidney Int. 2002;62(6):2202–7.
Ferrario F, Barone MT, Landoni G, Genderini A, Heidemperger M, Trezzi M, et al. Acetylcysteine and non-ionic isosmolar contrast-induced nephropathy–a randomized controlled study. Nephrol Dial Transplant. 2009;24(10):3103–7.
Fung JW, Szeto CC, Chan WW, Kum LC, Chan AK, Wong JT, et al. Effect of N-acetylcysteine for prevention of contrast nephropathy in patients with moderate to severe renal insufficiency: a randomized trial. Am J Kidney Dis. 2004;43(5):801–8.
Gomes VO, Figueredo CE, Caramori P, Lasevitch R, Bodanese LC, Araujo A, et al. N-acetylcysteine does not prevent contrast induced nephropathy after cardiac catheterisation with an ionic low osmolality contrast medium: a multicentre clinical trial. Heart. 2005;91(6):774–8.
Goldenberg I, Shechter M, Matetzky S, Jonas M, Adam M, Pres H, et al. Oral acetylcysteine as an adjunct to saline hydration for the prevention of contrast-induced nephropathy following coronary angiography. A randomized controlled trial and review of the current literature. Eur Heart J. 2004;25(3):212–8.
Oldemeyer JB, Biddle WP, Wurdeman RL, Mooss AN, Cichowski E, Hilleman DE. Acetylcysteine in the prevention of contrast-induced nephropathy after coronary angiography. Am Heart J. 2003;146(6):E23.
Yang K, Liu W, Ren W, Lv S. Different interventions in preventing contrast-induced nephropathy after percutaneous coronary intervention. Int Urol Nephrol. 2014;46(9):1801–7.
Ozcan EE, Guneri S, Akdeniz B, Akyildiz IZ, Senaslan O, Baris N, et al. Sodium bicarbonate, N-acetylcysteine, and saline for prevention of radiocontrast-induced nephropathy. A comparison of 3 regimens for protecting contrast-induced nephropathy in patients undergoing coronary procedures. A single-center prospective controlled trial. Am Heart J. 2007;154(3):539–44.
Saitoh T, Satoh H, Nobuhara M, Machii M, Tanaka T, Ohtani H, et al. Intravenous glutathione prevents renal oxidative stress after coronary angiography more effectively than oral N-acetylcysteine. Heart Vessels. 2011;26(5):465–72.
Baker CS, Wragg A, Kumar S, De Palma R, Baker LR, Knight CJ. A rapid protocol for the prevention of contrast-induced renal dysfunction: the RAPPID study. J Am Coll Cardiol. 2003;41(12):2114–8.
Thiele H, Hildebrand L, Schirdewahn C, Eitel I, Adams V, Fuernau G, et al. Impact of high-dose N-acetylcysteine versus placebo on contrast-induced nephropathy and myocardial reperfusion injury in unselected patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. The LIPSIA-N-ACC (Prospective, Single-Blind, Placebo-Controlled, Randomized Leipzig Immediate PercutaneouS Coronary Intervention Acute Myocardial Infarction N-ACC) Trial. J Am Coll Cardiol. 2010;55(20):2201–9.
Kefer JM, Hanet CE, Boitte S, Wilmotte L, De Kock M. Acetylcysteine, coronary procedure and prevention of contrast-induced worsening of renal function: which benefit for which patient? Acta Cardiol. 2003;58(6):555–60.
Spargias K, Alexopoulos E, Kyrzopoulos S, Iokovis P, Greenwood DC, Manginas A, et al. Ascorbic acid prevents contrast-mediated nephropathy in patients with renal dysfunction undergoing coronary angiography or intervention. Circulation. 2004;110(18):2837–42.
Abouzeid S, Mosbah O. Evaluation of different sodium bicarbonate regimens for the prevention of contrast medium-induced nephropathy. Electron Physician. 2016;8(2):1973–7.
Haeussler U, Riedel M, Keller F. Free reactive oxygen species and nephrotoxicity of contrast agents. Kidney Blood Press Res. 2004;27(3):167–71.
Altunoren O, Balli M, Eren N, Tasolar H, Arpaci A, Caglayan CE, et al. Is nebivolol really effective in preventing contrast induced nephropathy? Kidney Blood Press Res. 2015;40(5):533–41.
Kaya A, Kurt M, Tanboga IH, Isik T, Ekinci M, Aksakal E, et al. Rosuvastatin versus atorvastatin to prevent contrast induced nephropathy in patients undergoing primary percutaneous coronary intervention (ROSA-cIN trial). Acta Cardiol. 2013;68(5):489–94.
Ma H, Liu Y, Xie H, Zhang G, Zhan H, Liu Z, et al. The renoprotective effects of simvastatin and atorvastatin in patients with acute coronary syndrome undergoing percutaneous coronary intervention: an observational study. Medicine (Baltimore). 2017;96(32):e7351.
Acknowledgements
Not applicable.
Funding
This work was supported by the NSTDA Research Chair grant from the National Science and Technology Development Agency Thailand (NC); the Senior Research Scholar grant from the National Research Council of Thailand (SCC), and the Chiang Mai University Center of Excellence Award (NC).
Author information
Authors and Affiliations
Contributions
PK: Data curation, formal analysis, methodology, writing original draft, review and editing; SCC: formal analysis, funding acquisition, investigation, writing-review and editing; NC: Conceptualization, formal analysis, funding acquisition, methodology, supervision, writing-review and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kusirisin, P., Chattipakorn, S.C. & Chattipakorn, N. Contrast-induced nephropathy and oxidative stress: mechanistic insights for better interventional approaches. J Transl Med 18, 400 (2020). https://doi.org/10.1186/s12967-020-02574-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02574-8