
Abstract
Background
Mounting evidences displayed that miRNAs play crucial roles in tumor initiation and development. However, the regulation and relevant mechanism of miR-1825 in glioblastoma (GBM) remain unclear.
Methods
qRT-PCR was used to detect miR-1825 and CDK14 mRNA expression. Western blot was applied for testing protein levels (VEGF, E-cadherin, N-cadherin, vimentin, β-catenin, c-myc, p-c-Jun). MTT and transwell assays were used for detecting GBM cell progression, including cell viability, migration, and invasion.
Results
The results showed that miR-1825 was decreased in GBM tissue specimens by qRT-PCR and it was confirmed as a prognostic marker of GBM by Kaplan-Meier survival analysis. Moreover, we also found that miR-1825 upregulation suppressed GBM cell viability, tumor growth, invasion, and migration. Furthermore, CDK14 was first identified as the direct target of miR-1825 by Luciferase reporter assay. CDK14 acted as an oncogene in GBM development by immunohistochemistry. In addition, Western blot analysis demonstrated that miR-1825 regulated Wnt/β-catenin signaling pathway in GBM development.
Conclusion
In conclusion, miR-1825 upregulation suppressed GBM progression by targeting CDK14 through Wnt/β-catenin pathway.
Similar content being viewed by others


Introduction
Glioblastoma (GBM) are tumors derived from neuroepithelial tumors, accounting for 40 to 50% of brain tumors [1, 2]. They are the most common intracranial malignant tumors, which are caused by the interaction of congenital genetic high-risk factors and environmental carcinogenic factors [3, 4]. Although the current treatment for GBM patients has made great improvement, the survival rate of GBM patients was still very poor. Moreover, the high recurrence of GBM makes it very difficult to cure. Thus, it is very urgent to understand the underlying mechanism of GBM and to explore novel targets for GBM treatment.
Currently, miRNAs were determined as the potential in cancer treatment [5, 6] and it can modulate a tumor’s development and progression by regulating their target genes [7,8,9]. In GBM, many miRNAs have been proved to participate in cell progression and development. For instance, miR-365 displayed the inhibitory effect on GBM cell viability and migration via targeting PAX6 [10]. Besides, miR-210 was proved to inhibit GBM cell invasion and migration by targeting BNDF [11]. Moreover, the findings of Cui T et al displayed that miR-4516 can target PTPN14 to promote GBM cell progression and miR-4516/ PTPN14 axis provided an insight for treating GBM [12]. However, the researches of miR-1825 in GBM development and progression are very little. Xing and his colleagues showed that the expression of miR-1825 was decreased in glioma and it could suppress cell proliferation and invasion and facilitate cell apoptosis of glioma cell [13]. These findings suggested the possibility that miR-1825 might have an important role in regulating in GBM development and progression.
Cyclin-dependent kinases (CDKs), which are critical regulatory enzymes that drive cell cycle transition, are serine/threonine kinases characterized by their need for a separate subunit a cyclin—to provide the essential domains for the enzymatic activity of the CDKs [14, 15]. CDK14, also called PFTK1 (PFTAIRE protein kinase 1), is well known to regulate cell cycle and play important roles in cellular activities [16]. As previous studies displayed, CDK14 took part in multiple cancers’ development as a target of miRNAs [17]. For example, it was the target of miR-542 and involved in ovarian cancer cell proliferation, invasion, and tumorigenesis [18]. Also, CDK14 acted as the target of miR-431 in regulating pancreas cancer development [19]. Furthermore, CDK14 functioned as an oncogene in glioma and served as a target gene of miR-613 [20]. Based on the studies above, we want to verify whether CDK14 is the target of miR-1825 in regulating GBM progression, which has not been reported until now.
Epithelial-mesenchymal transition (EMT) is an important biological process for the migration and invasion of epithelial-derived malignant cells [21]. The activation of Wnt/β-catenin signaling pathway promotes tumor progression [22]. Therefore, we further investigated miR-1825 effect on EMT and Wnt/β-catenin axis. Collectively, the goal of this study was to investigate the role of miR-1825 in GBM progression and explore whether miR-1825 inhibited cell proliferation, invasion, and migration by regulating CDK14 and Wnt/β-catenin signaling pathway.
Materials and methods
GBM tissues
Fifty-five paired fresh tissue specimens were collected from GBM patients who were recruited between March 2013 and September 2017 at Jinan Zhangqiu District Hospital of TCM. All GBM patients have not received any treatment before surgery. The fresh tissues were verified by pathologists and stored at a − 80 °C refrigerator for further use. All patients have provided written consent to allow for research purposes prior to the collection of tissue samples. The histological features of all specimens were confirmed by pathologists according to the WHO criteria. The research was approved by the ethic committee of Jinan Zhangqiu District Hospital of TCM.
Cell culture
GBM cell lines U251, U87, and A172 were obtained from BeNa Culture Collection (Suzhou, China). Normal human astrocytes (NHA) were purchased from BeNa Culture Collection (Suzhou, China). All the cells were cultured as previously described [23]. Then the cells were maintained in a humidified incubator containing 5% CO2 at 37 °C.
MiR-1825 mimic and miR-1825 inhibitor
The mimic or inhibitor of miR-1825 was purchased from Shanghai GenePharma Co., Ltd. (Shanghai, China). They were used for increasing or decreasing the expression of miR-1825. A172 cells were the selected cells for further analysis. The transfection was conducted for 48 h following the instructions of Lipofectamine 2000 reagent (Invitrogen, Thermo Fisher Scientific).
Real-time PCR (RT-PCR)
The mRNA expression of miR-1825 and CDK14 was measured by RT-PCR. Total RNAs were firstly isolated from GBM tissues and cells using TRIzol reagent (Invitrogen) for both mRNA and miRNA analyses. The miScript Reverse Transcription kit (Beyotime, Haimen, Jiangsu, China) was carried out for producing cDNA. Then, quantitative PCR was conducted by a miScript SYBR-Green PCR kit (Beyotime) at an ABI 7500 Real-time PCR system (Applied Biosystems, Thermo Fisher Scientific). U6 was applied for normalizing miR-1825 relative mRNA expression and GAPDH for normalizing CDK14. 2−ΔΔCq method was used for calculating the gene expression. The primers are shown in supplemental table 1.
Western blot
The relative protein level was tested by Western blot. In brief, the total proteins were exacted from GBM cells using RIPA lysis buffer and then conducted the protein concentration by BCA kit (Beyotime). After the equal protein separated by 10% SDS-PAGE, they were transferred to the NC membranes. Then, the membranes were blocked with 5% skimmed milk powder at 37 °C for 1 h, incubated with primary antibodies (CDK14, ab167928, 1:1,000; E-cadherin, ab76055, 1:1,000; N-cadherin, ab18203, 1:1,000; Vimentin, ab92547, 1:1,000; β-catenin, ab32572, 1:1,000; c-myc, ab32072, 1:1,000; p-c-Jun, ab32385, 1:1,000; GAPDH, ab181602, 1:1,000; all from Abcam) at 4 °C overnight, and secondary antibodies at 37 °C for 1 h. Finally, an enhanced chemiluminescence (ECL) method and the ImageJ software were applied for detecting the immune complexes and quantified protein levels, respectively.
Cell proliferation analysis
Cell proliferation was evaluated by 3- (4, 5-dimethylthiazolyl-2)- 2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, A172 cells were placed in 96-well plates at a density of 2000 cells/well. When the cells were cultured for 1, 2, 3, and 4 days, MTT solution (20 μl) was added to each well and incubated for another 4 h at 37 °C. Then, the medium was removed and dimethyl sulfoxide (DMSO) was added to dissolve formazan crystals by swirling gently. Finally, the optical density was detected at a wavelength of 490 nm using a microplate reader.
Cell migration and invasion analysis
The invasiveness and metastasis of U87 cells were measured by Transwell assay. Detection of invasion and migration was similar, except for the upper chamber coated with Matrigel. Briefly, the cells were added to the upper chamber, and DMEM containing 20% FBS was added to the lower chamber. After incubation for 24 h, the cells that traversed the membrane were fixed, stained, and counted. These traversed cells were used to evaluate cell invasion and migration.
Immunohistochemistry analysis
Immunohistochemistry was applied for detecting CDK14 protein density. Firstly, the paraffin sections were obtained from GBM tissues. Then they were incubated with 3% H2O2 for 15 min. After blocking with goat serum for 2 h at room temperature, the sections were incubated with primary antibodies at 4 °C overnight and secondary antibodies at 37 °C for 1 h. Next, the sections were stained using DAB solution, followed by alcohol dehydration, xylene decolorization, and neutral resin sealing. Finally, the results were observed and photographed by a microscope.
Tumor growth analysis
Xenograft tumor formation assays were applied for measuring the tumor growth in vivo. The Animal Research Committee of Jinan Zhangqiu District Hospital of TCM approved these animal experiments. The operation meets the standards for laboratory animal care and use in Jinan Zhangqiu District Hospital of TCM. A172 cells (1 × 105) treated with miR-1825 mimic or NC were injected into right flank of nude mice subcutaneously, which were purchased from Shanghai Laboratory Animal Center (Shanghai, China). Then, the tumor size and weight was measured every 4 days for 28 days by a vernier caliper and electronic scale, respectively. All mouse experiments were carried out in accordance with institutional guidelines and regulations of the government.
Luciferase activity analysis
Bioinformatic analysis algorithm TargetScan (http://www.targetscan.org/vert_72/) was used to predict the targets of miR-1825. The binding site of miR-1825 in the 3′-untranslated region (3′-UTR) of CDK14 (wild-type or mutant) was cloned into pGL3-reporter vector (Promega, Madison, WI, USA). Then, A172 cells were co-transfected with the NC vector and miR-1825 mimic using Lipofectamine 2000 (Invitrogen). After transfection for 48 h, the luciferase activity was tested by the Dual-Luciferase Reporter Assay System (Promega), and Renilla luciferase activity was used to normalize the data.
Statistics analysis
The values were represented as mean ± SD. All experimental conditions were repeated in duplicate independently. Data was analyzed by SPSS 22.0 statistical software (SPSS, Inc.) and the statistics was performed by GraphPad Prism 6. Student’s t test was applied for comparing the difference between two groups, and Tukey’s post hoc test of one-way analysis of variance (one-way ANOVA) was carried out for comparing the differences between more than two groups. The Pearson test was applied for determining the relationship between miR-1825 and CDK14. Log-rank test was applied for analyzing the survival rate. P < 0.05 was considered statistically significant.
Results
MiR-1825 downregulation was associated with poor prognosis
Here, we detected miR-1825 expression in GBM tissue specimens. The findings displayed that expression of miR-1825 was downregulated in GBM tissues compared with that in normal tissues (Fig. 1a). Moreover, we investigated whether the differential expression of miR-1825 was related to patients’ survival rate. The results of Fig. 1b display that GBM with high expression of miR-1825 predicted better prognosis, while low expression predicted poorer prognosis. Moreover, miR-1825 was significantly associated with GBM clinicopathological features, including WHO grade (Table 1). These findings demonstrated that miR-1825 downregulation served as an indicator for poorer prognosis of GBM patients.

The association of miR-1825 differential expression with overall survival in GBM. a Decreased expression of miR-1825 in GBM tissue samples (n = 55). b Higher expression of miR-1825 in GBM patients exhibited a higher survival rate of GBM patients. **P < 0.01
MiR-1825 suppressed GBM progression
The role of miR-1825 in GBM was investigated by examining the levels of miR-1825 in GBM cell lines (U251, U87, and A172) and normal human astrocytes (NHA) by qRT-PCR. As shown in Fig. 2a, the expression level of miR-1825 was reduced in all GBM cell lines was significantly lower than that in NHA cell line. We then selected A172 cells for the following experiments. To see miR-1825 role in GBM cell viability, invasiveness, and metastasis, miR-1825 expression was increased or decreased by mimic or inhibitor. As we saw in Fig. 2b, results showed that expression levels of miR-1825 were significantly upregulated by miR-1825 mimic but reduced by miR-1825 inhibitor. Next, MTT assay was carried out for testing A172 cell viability. The findings displayed that increasing miR-1825 inhibited while decreasing miR-1825 enhanced GBM cell viability (Fig. 2c). Transwell assay revealed that A172 cell migration was reduced by miR-1825 mimic but enhanced by miR-1825 inhibitor (Fig. 2c). For invasion, upregulating of miR-1825 expression inhibited A172 cell invasion, while downregulating of miR-1825 expression enhanced A172 cell invasion (Fig. 2d). The above findings indicated that miR-1825 exhibited hindrance effect on cell proliferation, invasion, and migration.

Hindrance effect of miR-1825 on GBM progression. a Decreased expression of miR-1825 in GBM cell lines. b Decreased miR-1825 expression in miR-1825 inhibitor group and increased miR-1825 expression in miR-1825 mimic group in A172 cells. c Cell viability was suppressed by miR-1825 mimic and promoted by miR-1825 inhibitor in A172 cells. d Cell migration was suppressed by miR-1825 mimic and promoted by miR-1825 inhibitor in A172 cells. e Cell invasion was suppressed by miR-1825 mimic and facilitated by miR-1825 inhibitor in A172 cells. *P < 0.05, **P < 0.01
MiR-1825 upregulation blocked tumor growth in vivo
Then we tested miR-1825 effect on the size and weight of tumors extracted from GBM mice. As we saw in Fig. 3a, the tumor size in miR-1825 mimic group was smaller than that in NC group. Also, miR-1825 mimic made the tumor growth rate slower than the normal control (Fig. 3b). Moreover, miR-1825 mimic reduced the tumor weight in comparison with the NC group (Fig. 3c). All the results demonstrated that miR-1825 upregulation suppressed tumor growth.

Inhibition effect of miR-1825 on tumor growth. a The representative pictures of the size of the tumors in mice transfected with miR-1825 mimic or normal control (NC). b The slow growth of tumors in the miR-1825 mimic group compared to that in the NC group. c The weight-loss tumors in mice treated with miR-1825 mimic compared to NC. *P < 0.05, **P < 0.01
MiR-1825 negatively modulated CDK14 expression
As TargetScan prediction, CDK14 was the possible target of miR-1825 and Fig. 4a displayed the binding sides of miR-1825 with CDK14. Furthermore, luciferase reporter assay was applied for further confirming CDK14 was the target of miR-1825. As Fig. 4b displayed that overexpression of miR-1825 significantly suppressed firefly luciferase reporter activity of the WT-CDK14 3′-UTR; however, they did not affect the luciferase activity of MuT-CDK14 3′-UTR. Next, qRT-PCR and Western blot analyses were carried out for detecting the effect of miR-1825 on CDK14 expression in mRNA and the protein level, respectively The findings exerted that miR-1825 mimic exhibited a reduced expression of CDK14, while miR-1825 inhibitor displayed a raised CDK14 expression (Fig. 4c, d). Due to the opposite expression of miR-1825 and CDK14 in GBM, we detected the correlation between miR-1825 and CDK14. Results showed that their relationship was negative (Fig. 4e). These results indicated that CDK14 was the target of miR-1825.

Negatively regulation of CDK14 by miR-1825. a Predicted binding site between miR-1825 and 3′-UTR of CDK14. b Luciferase activity in A172 cells transfected with WT or MuT CDK14 and miR-1825 mimic or con mimic. c Decreased CDK14 level by miR-1825 mimic and increased CDK14 level by miR-1825 inhibitor. d Decreased CDK14 mRNA expression by miR-1825 mimic and increased CDK14 mRNA expression by miR-1825 inhibitor. e Negatively relationship between CDK14 and miR-1825 in GBM tissue (r = −0.7550, P < 0.0001). **P < 0.01
CDK14 upregulation was associated with poor prognosis
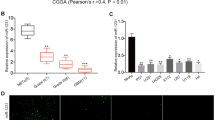
We then checked CDK14 expression in GBM tissue samples. The findings displayed that CDK14 was located in cell membrane (Fig. 5a) and its protein density was raised in GBM tissues (Fig. 5b). Moreover, we investigated whether the differential expression of CDK14 was related to patients’ survival rate. The results of Fig. 5c display that the low expression of CDK14 in GBM tissues predicted better prognosis, while high expression of CDK14 predicted worse prognosis. The findings demonstrated that CDK14 upregulation served as an indicator of poorer prognosis in GBM patients.

The association of CDK14 differential expression with overall survival. a Location of CDK14 in the cell of GBM tissues. b Increased protein density of CDK14 in GBM tissues. c Low expression of CDK14 in GBM patients exhibited high survival rate of GBM patients. *P < 0.05
MiR-1825 upregulation blocked EMT and Wnt/β-catenin signaling pathway
To further examine the mechanism of miR-1825 in modulating GBM progression, the expression of the downstream genes of EMT and Wnt/β-catenin pathway was tested by Western blot. The findings showed that miR-1825 upregulation enhanced E-cadherin expression, but inhibited N-cadherin and Vimentin expression. However, miR-1825 downregulation displayed the opposite effect (Fig. 6a). As shown in Fig. 6b, increasing miR-1825 reduced β-catenin, c-myc, and p-c-Jun expression, while decreasing miR-1825 exhibited the opposite effect on these levels. The results demonstrated that miR-1825 upregulation blocked Wnt/β-catenin signaling pathway in GBM cells.

The activation of EMT and Wnt/β-catenin signaling pathway by miR-1825 downregulation. a Decreased expression of E-cadherin expression, but increased N-cadherin and vimentin expression by miR-1825 mimic. Increased E-cadherin expression, but decreased N-cadherin and vimentin expression by miR-1825 inhibitor. b Reduced β-catenin, c-myc, and p-c-Jun expression by miR-1825 mimic and increased expression of β-catenin, c-myc, and p-c-Jun expression by miR-1825 inhibitor
Discussion
Glioblastoma is the most common type of malignant primary intracranial tumor and is characterized by the hallmarks of cellular heterogeneity, rapid proliferation, angiogenesis, extensive invasion, hypoxia, and necrosis [24, 25]. It is an urgent need to gain deeper understanding of molecular mechanisms implicated in glioblastoma progression and development [26]. Many studies have revealed that miRNAs affect GBM development, including miR-146b, miR-148a, and miR-34a [27,28,29]. These outcomes have attracted in-depth research into miRNAs in GBM.
Previous study has shown that miR-1825 may play an important role in the development of human glioma including apoptosis, cell proliferation, and invasion by microRNA-Gene Ontology network [13]. Here, in our study, we revealed that miR-1825 expression was declined in GBM tissue specimens and its downregulation predicted poor prognosis. However, we also found that CDK14 expression was raised in GBM tissues and its upregulation predicted poor prognosis. Moreover, increasing miR-1825 impeded tumor growth, GBM cell proliferation, invasion, and migration. MiR-1825 negatively regulated the CDK14 expression and Wnt/β-catenin axis.
Researches on miR-1825 in tumors revealed that it served as a potential biomarker in multiple cancers. For example, miR-1825 was highly expressed in prostate cancer and it acted as a prostate cancer biomarker [30]. Also, miR-1825 was upregulated in larynx cancer and involved in tumor progression [31]. However, miR-1825 was downregulated in glioma and associated with tumorigenesis [13]. In this study, we revealed that miR-1825 expression was declined in GBM and used as a predictor for prognosis. Moreover, re-expression of miR-1825 impeded GBM cell viability, invasiveness, and metastasis, which is in line with the research that miR-1825 played an important role in glioma cell proliferation, apoptosis, and invasion [13].
CDK14 was used as an oncogene in several cancers. For instance, its expression was raised in hepatocellular carcinoma as a target of miR-1202 [32]. Besides, CDK14 expression was higher in glioma than normal and took part in glioma progression regulated by miR-613 [20]. All these results supported our above research. Moreover, we first time revealed that CDK14 was the direct target of miR-1825. We also revealed that CDK14 high expression was associated with the poor survival time of GBM patients. Wnt/β-catenin pathway played important roles in tumorigenesis, including GBM. Lots of studies displayed that miRNAs regulated GBM development by modulating Wnt/β-catenin signaling pathway, such as miR-34a [33], miR-328 [34], and miR-21 [35]. Here, we displayed that miR-1825 repressed GBM progression by inhibiting the activation of Wnt/β-catenin signaling pathway. Furthermore, certain reports have suggested that CDK14can regulates a number of pathways, including the Wnt/β-catenin signaling pathway and phosphoinositide 3 kinase (PI3K)/Akt signaling pathway, and cellular mechanisms to act as an oncogene [36, 37]. In the absence of Wnt signaling, the mitosis-specific CDK14-Cyclin Y kinase complex phosphorylates Ser-1490 of LRP5/6 are co-receptors for Wnt ligands at the G2/M stage, thereby triggering the receptor for Wnt-induced phosphorylation [38, 39]. The above studies showed that there is a connection between the regulation of CDK14 and Wnt signaling. We will show the effect of CDK14 on GBM progression and Wnt/β-catenin pathway in our further study. In conclusion, miR-1825 was underexpressed in GBM and suppressed GBM progression, whereas CDK14 was overexpressed in GBM. Moreover, CDK14 was shown as the direct target of miR-1825 in GBM. MiR-1825 upregulation blocked the EMT and Wnt/β-catenin signaling pathway.
Conclusions
In conclusion, the identification of miR-1825 as a tumor suppressive miRNA in human GBM that acts by targeting CDK14 provides additional evidence of a pivotal role for miRNAs in GBM progression. Given that miR-1825 is downregulated in GBM, the introduction of this mature miRNA into the tumor tissue could serve as a therapeutic strategy by regulating the expression of target genes. MiRNA-based therapeutic patterns are still in their infancy; however, our findings are encouraging and suggest that this miRNA could be targeted for the development of a treatment for patients with GBM in the future.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
References
Hofer S, Roelcke U, Herrmann R. New aspects of interdisciplinary therapy for malignant gliomas in adults. Schweiz Med Wochenschr. 1999;129:1332–41.
Shah U, Morrison T. A review of the symptomatic management of malignant gliomas in adults. J Natl Compr Cancer Netw. 2013;11:424–9.
Wen PY, Kesari S. Malignant gliomas in adults. N Engl J Med. 2008;359:492–507.
Niyazi M, Siefert A, Schwarz SB, Ganswindt U, Kreth FW, Tonn JC, Belka C. Therapeutic options for recurrent malignant glioma. Radiother Oncol. 2011;98:1–14.
Pardini B, Calin GA. MicroRNAs and long non-coding RNAs and their hormone-like activities in cancer. Cancers (Basel). 2019;11.
Gajos-Michniewicz A, Czyz M. Role of miRNAs in melanoma metastasis. Cancers (Basel). 2019;11.
Ma M, Dai J, Tang H, Xu T, Yu S, Si L, Cui C, Sheng X, Chi Z, Mao L, et al. MicroRNA-23a-3p inhibits mucosal melanoma growth and progression through targeting adenylate cyclase 1 and attenuating cAMP and MAPK pathways. Theranostics. 2019;9:945–60.
Gao R, Zhang N, Yang J, Zhu Y, Zhang Z, Wang J, Xu X, Li Z, Liu X, Li Z, et al. Long non-coding RNA ZEB1-AS1 regulates miR-200b/FSCN1 signaling and enhances migration and invasion induced by TGF-beta1 in bladder cancer cells. J Exp Clin Cancer Res. 2019;38:111.
Amodio N, Gallo Cantafio ME, Botta C, Agosti V, Federico C, Caracciolo D, Ronchetti D, Rossi M, Driessen C, Neri A, et al. Replacement of miR-155 elicits tumor suppressive activity and antagonizes bortezomib resistance in multiple myeloma. Cancers (Basel). 2019;11.
Yuan F, Liu J, Pang H, Tian Y, Yuan K, Li Y, Wang J, Bian S, Zheng Y, Dong D, et al. MicroRNA-365 suppressed cell proliferation and migration via targeting PAX6 in glioblastoma. Am J Transl Res. 2019;11:361–9.
Liu S, Jiang T, Zhong Y, Yu Y. miR-210 inhibits cell migration and invasion by targeting the brain-derived neurotrophic factor in glioblastoma. J Cell Biochem. 2019.
Cui T, Bell EH, McElroy J, Becker AP, Gulati PM, Geurts M, Mladkova N, Gray A, Liu K, Yang L, et al. miR-4516 predicts poor prognosis and functions as a novel oncogene via targeting PTPN14 in human glioblastoma. Oncogene. 2018.
Xing W, Zeng C. A novel serum microRNA-based identification and classification biomarker of human glioma. Tumour Biol. 2017;39:1010428317705339.
Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93–115.
Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105.
Imawari Y, Mimoto R, Hirooka S, Morikawa T, Takeyama H, Yoshida K. Downregulation of dual-specificity tyrosine-regulated kinase 2 promotes tumor cell proliferation and invasion by enhancing cyclin-dependent kinase 14 expression in breast cancer. Cancer Sci. 2018;109:363–72.
Ji Q, Xu X, Li L, Goodman SB, Bi W, Xu M, Xu Y, Fan Z, Maloney WJ, Ye Q, Wang Y. miR-216a inhibits osteosarcoma cell proliferation, invasion and metastasis by targeting CDK14. Cell Death Dis. 2017;8:e3103.
Li J, Shao W, Feng H. MiR-542-3p, a microRNA targeting CDK14, suppresses cell proliferation, invasiveness, and tumorigenesis of epithelial ovarian cancer. Biomed Pharmacother. 2019;110:850–6.
Yang J, Zhu H, Jin Y, Song Y. MiR-431 inhibits cell proliferation and induces cell apoptosis by targeting CDK14 in pancreatic cancer. Eur Rev Med Pharmacol Sci. 2018;22:4493–9.
Li Q, Zhou L, Wang M, Wang N, Li C, Wang J, Qi L. MicroRNA-613 impedes the proliferation and invasion of glioma cells by targeting cyclin-dependent kinase 14. Biomed Pharmacother. 2018;98:636–42.
Weng YS, Tseng HY, Chen YA, Shen PC, Al Haq AT, Chen LM, Tung YC, Hsu HL. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. 2019;18:42.
Rajakulendran N, Rowland KJ, Selvadurai HJ, Ahmadi M, Park NI, Naumenko S, Dolma S, Ward RJ, So M, Lee L, et al. Wnt and Notch signaling govern self-renewal and differentiation in a subset of human glioblastoma stem cells. Genes Dev. 2019.
Sun S, Wang Q, Giang A, Cheng C, Soo C, Wang CY, Liau LM, Chiu R. Knockdown of CypA inhibits interleukin-8 (IL-8) and IL-8-mediated proliferation and tumor growth of glioblastoma cells through down-regulated NF-kappaB. J Neuro-Oncol. 2011;101:1–14.
Lv S, Sun B, Dai C, Shi R, Zhou X, Lv W, Zhong X, Wang R, Ma W. The Downregulation of MicroRNA-146a Modulates TGF-beta Signaling Pathways Activity in Glioblastoma. Mol Neurobiol. 2015;52:1257–62.
Zhang R, Luo H, Wang S, Chen W, Chen Z, Wang HW, Chen Y, Yang J, Zhang X, Wu W, et al. MicroRNA-377 inhibited proliferation and invasion of human glioblastoma cells by directly targeting specificity protein 1. Neuro-Oncology. 2014;16:1510–22.
Tanase C. miRNA interactions as a novel molecular panel for clinical outcome of glioblastoma. Biomark Med. 2013;7:201.
Khwaja SS, Cai C, Badiyan SN, Wang X, Huang J. The immune-related microRNA miR-146b is upregulated in glioblastoma recurrence. Oncotarget. 2018;9:29036–46.
Lopez-Bertoni H, Kozielski KL, Rui Y, Lal B, Vaughan H, Wilson DR, Mihelson N, Eberhart CG, Laterra J, Green JJ. Bioreducible polymeric nanoparticles containing multiplexed cancer stem cell regulating miRNAs Inhibit glioblastoma growth and prolong survival. Nano Lett. 2018;18:4086–94.
Di Bari M, Bevilacqua V, De Jaco A, Laneve P, Piovesana R, Trobiani L, Talora C, Caffarelli E, Tata AM. Mir-34a-5p mediates cross-talk between M2 muscarinic receptors and Notch-1/EGFR pathways in U87MG glioblastoma cells: implication in cell proliferation. Int J Mol Sci. 2018;19.
Guo X, Han T, Hu P, Guo X, Zhu C, Wang Y, Chang S. Five microRNAs in serum as potential biomarkers for prostate cancer risk assessment and therapeutic intervention. Int Urol Nephrol. 2018;50:2193–200.
Karatas OF, Suer I, Yuceturk B, Yilmaz M, Oz B, Guven G, Cansiz H, Creighton CJ, Ittmann M, Ozen M. Identification of microRNA profile specific to cancer stem-like cells directly isolated from human larynx cancer specimens. BMC Cancer. 2016;16:853.
Du B, Zhang P, Tan Z, Xu J. MiR-1202 suppresses hepatocellular carcinoma cells migration and invasion by targeting cyclin dependent kinase 14. Biomed Pharmacother. 2017;96:1246–52.
Rathod SS, Rani SB, Khan M, Muzumdar D, Shiras A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways. FEBS Open Bio. 2014;4:485–95.
Delic S, Lottmann N, Stelzl A, Liesenberg F, Wolter M, Gotze S, Zapatka M, Shiio Y, Sabel MC, Felsberg J, et al. MiR-328 promotes glioma cell invasion via SFRP1-dependent Wnt-signaling activation. Neuro-Oncology. 2014;16:179–90.
Zhang KL, Han L, Chen LY, Shi ZD, Yang M, Ren Y, Chen LC, Zhang JX, Pu PY, Kang CS. Blockage of a miR-21/EGFR regulatory feedback loop augments anti-EGFR therapy in glioblastomas. Cancer Lett. 2014;342:139–49.
Yang L, Zhu J, Huang H, Yang Q, Cai J, Wang Q, Zhu J, Shao M, Xiao J, Cao J, et al. PFTK1 promotes gastric cancer progression by regulating proliferation, migration and invasion. PLoS One. 2015;10:e0140451.
Wang B, Zou A, Ma L, Chen X, Wang L, Zeng X, Tan T. miR-455 inhibits breast cancer cell proliferation through targeting CDK14. Eur J Pharmacol. 2017;807:138–43.
Davidson G, Niehrs C. Emerging links between CDK cell cycle regulators and Wnt signaling. Trends Cell Biol. 2010;20:453–60.
Wang X, Jia Y, Fei C, Song X, Li L. Activation/proliferation-associated Protein 2 (Caprin-2) positively regulates CDK14/Cyclin Y-mediated Lipoprotein Receptor-related Protein 5 and 6 (LRP5/6) constitutive phosphorylation. J Biol Chem. 2016;291:26427–34.
Acknowledgements
Not applicable.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
FL and CL contributed to the conception of the study. YS and TJ contributed significantly to the data analysis and study preparation. NL performed the data analyses and wrote the study. HL helped perform the analysis with constructive discussions. All authors have read and approved the final study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Approval for the study was obtained from the Ethics Committee of Jinan Zhangqiu District Hospital of TCM (Jinan, China). Informed consent was obtained from all the patients whose tissues were used in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Primer sequences for RT-PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, F., Li, C., Sun, Y. et al. Upregulation of miR-1825 inhibits the progression of glioblastoma by suppressing CDK14 though Wnt/β-catenin signaling pathway. World J Surg Onc 18, 147 (2020). https://doi.org/10.1186/s12957-020-01927-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-020-01927-3