
Abstract
Drug resistance in cancer cells significantly diminishes treatment efficacy, leading to recurrence and metastasis. A critical factor contributing to this resistance is the epigenetic alteration of gene expression via RNA modifications, such as N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), 7-methylguanosine (m7G), pseudouridine (Ψ), and adenosine-to-inosine (A-to-I) editing. These modifications are pivotal in regulating RNA splicing, translation, transport, degradation, and stability. Governed by “writers,” “readers,” and “erasers,” RNA modifications impact numerous biological processes and cancer progression, including cell proliferation, stemness, autophagy, invasion, and apoptosis. Aberrant RNA modifications can lead to drug resistance and adverse outcomes in various cancers. Thus, targeting RNA modification regulators offers a promising strategy for overcoming drug resistance and enhancing treatment efficacy. This review consolidates recent research on the role of prevalent RNA modifications in cancer drug resistance, with a focus on m6A, m1A, m5C, m7G, Ψ, and A-to-I editing. Additionally, it examines the regulatory mechanisms of RNA modifications linked to drug resistance in cancer and underscores the existing limitations in this field.
Similar content being viewed by others

Introduction
Chemotherapy, targeted therapy, and immunotherapy serve as primary treatment strategies for cancer [1,2,3,4,5]. Nonetheless, patients with advanced cancer often show poor responses to these interventions [6,7,8,9,10]. Cancer cells circumvent these treatments’ toxic effects through intrinsic resistance, stemming from pre-existing genetic and epigenetic variations, or acquired resistance, which emerges during therapy [11,12,13,14,15,16,17]. Principal mechanisms of therapeutic resistance include increased drug efflux, altered drug metabolism, and modified expression of therapeutic targets [8, 18,19,20]. Additionally, cancer cells can activate anti-apoptotic pathways, enhance DNA repair, initiate autophagy, remodel the tumor microenvironment (TME), and induce epithelial-mesenchymal transition (EMT), all contributing significantly to cancer cell survival and drug resistance [14, 21,22,23,24].
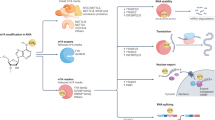
Beyond these factors, epigenetic regulation is pivotal in mediating drug resistance [25,26,27,28,29]. Epigenetic modifications, such as RNA modifications, influence gene expression without altering the DNA sequence [30,31,32,33]. With advancements in high-throughput detection methods, RNA methylation has been recognized for its critical role in global epigenetic remodeling [34,35,36,37,38]. RNA modifications occur on various types of RNAs and are essential for regulating RNA processing, including splicing, localization, transport, translation, and degradation [39,40,41,42]. These modifications alter the chemical and topological properties of the four basic nucleotides, thereby influencing the stability and function of the modified RNAs. Similar to DNA and proteins, RNAs undergo methylation and demethylation by specific enzymes known as methyltransferases (“writers”) and demethylases (“erasers”) [43,44,45,46,47]. Writers install RNA modifications by recognizing the sequence and structure of substrate RNAs, interacting with the transcriptional machinery or RNA-binding proteins (RBPs), while demethylases reverse these modifications (Fig. 1) [48,49,50,51]. Predominant RNA modifications include 7-methylguanosine (m7G), 5-methylcytosine (m5C), N1-methyladenosine (m1A), and N6-methyladenosine (m6A) [52,53,54,55,56]. Other widespread RNA modifications, such as pseudouridine (Ψ) and adenosine-to-inosine (A-to-I) editing, are detected in most RNA types [57,58,59,60].

Overview of main RNA modifications closely associated with cancer development. The most critical RNA modifications implicated in tumorigenesis include 7-methylguanosine (m7G), 5-methylcytosine (m5C), N1-methyladenosine (m1A), N6-methyladenosine (m6A), pseudouridine (Ψ), and adenosine-to-inosine (A-to-I) editing. These modifications alter the chemical structure of RNAs, directly affecting gene expression and various biological processes. RNA modifications are dynamic and often reversible. “Writer” proteins add chemical groups to RNA molecules, thereby enhancing or modifying their function. “Readers” identify and bind to these modified RNAs, influencing subsequent RNA processing steps such as splicing, localization, export, translation, stability, and degradation. Conversely, “erasers” remove RNA modifications, potentially restoring the original RNA functions. The intricate interplay among writers, readers, and erasers is essential for cellular responses and adaptations, particularly in cancer cells
Numerous studies have demonstrated the integral role of RNA modifications in diverse biological processes, including stem cell differentiation, tissue development, and responses to external stress [61,62,63]. Aberrant levels of RNA modifications and their regulatory proteins, whether higher or lower than normal, are frequently associated with cancer progression, drug resistance, and disease relapse. Normal reference levels are typically based on each type of healthy tissue or cell lines [64,65,66,67,68]. Specifically, RNA modifications may drive drug response in various cancers by modulating drug transport and metabolism, target receptors, cancer stemness, and DNA repair [69,70,71,72]. Targeting RNA modifications offers a promising strategy for enhancing traditional anti-cancer treatments by inhibiting overactive enzymes and activating those that are underactive [73,74,75,76]. However, the majority of RNA modification functions in pathological processes are intricately linked to the unique disease environment, presenting challenges in targeting RNA modifications for cancer treatment [62, 77, 78]. Understanding the role of RNA modifications in drug resistance can facilitate the development of novel strategies to overcome drug resistance and improve cancer treatment outcomes [79,80,81,82]. This review summarizes recent research on the role of RNA modifications, specifically m6A, m1A, m5C, m7G, Ψ, and A-to-I editing, in cancer therapeutic resistance. Additionally, it explores the mechanisms underlying RNA modifications associated with drug resistance and discusses strategies for targeting these modifications to reverse drug resistance and improve patient prognosis.
Overview of key RNA modifications
Adenosine methylation
N6-methyladenosine modification
m6A, a significant epigenetic modification, involves adding a methyl group to the nitrogen atom at the sixth position of adenosine. This modification is present in various RNA species, including messenger RNAs (mRNAs), long non-coding RNAs (lncRNAs), ribosomal RNAs (rRNAs), and polyadenylated RNAs [83,84,85,86,87]. Predominantly occurring in the 3’-untranslated regions (3’-UTRs) and near mRNA stop codons, m6A is regulated by specific methyltransferases (“writers”), demethylases (“erasers”), and m6A-binding proteins (“readers”) [43, 88,89,90,91]. The core methyltransferase complex comprises Methyltransferase-like 3 (METTL3) and Methyltransferase-like 14 (METTL14), with METTL3 serving as the catalytic core. Additional proteins such as Wilms tumor 1-associated protein (WTAP), VIR-like m6A methyltransferase associated (VIRMA), RNA-binding motif protein 15 (RBM15), and zinc finger CCCH-type containing 13 (ZC3H13) support this complex, facilitating the co-transcriptional deposition of m6A on nascent pre-mRNAs [73, 92,93,94,95,96,97]. Demethylation is executed by enzymes such as Fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) [98,99,100,101]. Reader proteins recognizing and interpreting m6A marks include members of the YTH domain family (YTHDF1-3, YTHDC1-2) and others like insulin-like growth factor 2 mRNA-binding proteins (IGFBPs), Musashi2 (MSI2), Proline-rich coiled-coil containing protein 2 A (PRRC2A), and Heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) [102,103,104,105,106].
N1-methyladenosine modification
m1A, an essential internal RNA modification, involves adding a methyl group to the nitrogen atom at the first position of adenosine [107,108,109,110,111]. This modification is found in various RNA types, including transfer RNAs (tRNAs), rRNAs, mRNAs, lncRNAs, and mitochondrial RNAs [112,113,114,115]. m1A is particularly enriched near the start codon and just upstream of the first splice site, playing a pivotal role in the initiation of translation [116,117,118,119]. The methyltransferase complex responsible for installing m1A in tRNAs includes tRNA methyltransferase 10 homolog A (TRMT10) and the TRM6-TRM61 complex [108, 120,121,122,123,124]. The AlkB family of demethylases, such as ALKBH1 and ALKBH3, reverse the m1A modification, with ALKBH3 being the sole enzyme known to remove m1A from mRNAs [125,126,127,128,129]. YTH domain-containing proteins, including YTHDF1-3 and YTHDC1, primarily recognize and bind to m1A-modified RNAs [130, 131].
Other RNA modifications
5-methylcytosine Modification
5–Methylcytosine (m5C) is a prevalent and extensively studied RNA modification [132,133,134,135] It involves the addition of a methyl group to the fifth carbon of cytosine, found in various RNA classes, including tRNAs, mRNAs, rRNAs, and enhancer RNAs (eRNAs) [136,137,138,139,140]. m5C is particularly enriched near stop codons and within coding sequences in mRNAs and at loop structures in tRNAs. This modification is primarily catalyzed by the NOL1/NOP2/SUN domain (NSUN) family of SAM-dependent methyltransferases and DNA methyltransferase-like 2 (DNMT2) [137, 141, 142]. In mammalian cells, the ten-eleven translocation (TET) family and ALKBH1 are responsible for reversing m5C modification [143,144,145]. Aly/REF export factor (ALYREF) and Y-box binding protein 1 (YBX1) act as reader proteins that specifically interact with m5C-modified RNAs, playing pivotal roles in regulating RNA metabolism and function [136, 146,147,148]. Fragile X mental retardation protein (FMRP) is another m5C reader that coordinates between m5C writers and erasers, facilitating transcription-coupled homologous recombination [149,150,151].
7-methylguanosine Modification
N7-methylguanosine (m7G) is a key RNA modification primarily found at the 5’ cap of eukaryotic mRNAs, and also internally in rRNAs, tRNAs, and microRNAs (miRNAs) [152,153,154,155,156]. This modification involves the addition of a methyl group to the N7 position of ribo-guanosine [157,158,159,160]. Methyltransferase-like 1 (METTL1), along with its cofactor WD repeat domain 4 (WDR4), is the primary enzyme responsible for m7G modification in mammals [161,162,163,164,165]. In human 18S rRNA, the m7G modification at position 1639 is mediated by the Williams–Beuren syndrome chromosomal region 22 (WBSCR22)–TRMT112 complex [166,167,168,169]. RNA guanine-7 methyltransferase (RNMT) and RNMT-activating miniprotein (RAM) specifically target the 5’ cap of mRNAs for m7G modification [170,171,172]. To date, no enzymes responsible for the removal or specific recognition of m7G have been identified [173, 174].
Pseudouridine modification
Pseudouridine (Ψ) is the most abundant RNA modification in various RNA classes in humans, including tRNAs, mRNAs, small nuclear RNAs (snRNAs), and other non-coding RNAs [175,176,177,178]. Ψ, an isomer of uridine, is formed by relocating the N-C glycosidic bond from the sixth to the fifth carbon position [179,180,181]. This modification is enriched within the coding region and 3’-UTR of mRNAs [182, 183]. Of the 14 known pseudouridine writers, dyskerin pseudouridine synthase 1 (DKC1) is part of a small nucleolar ribonucleoprotein complex and primarily targets rRNAs, snRNAs, small nucleolar RNAs (snoRNAs), and the non-coding RNA component of telomerase (TERC) [184,185,186,187,188]. Currently, there are no known enzymes that erase or specifically recognize pseudouridine modifications.
Adenosine-to-inosine editing
Adenosine-to-inosine (A-to-I) editing is among the most prevalent co-transcriptional and post-transcriptional RNA modifications in mammals [189,190,191,192,193]. Unlike reversible modifications such as m6A and m5C, A-to-I editing induces a permanent alteration within RNAs [194,195,196,197]. This modification is mediated by a family of enzymes known as adenosine deaminases acting on RNAs (ADARs), which preferentially edit adenines flanked by 5’-uridine and 3’-guanosine [198,199,200]. The primary targets of A-to-I editing are double-stranded RNAs (dsRNAs) derived from inverted Alu repetitive elements (Alu dsRNAs), which are abundant in the human genome [201,202,203,204]. A-to-I editing occurs at various RNA locations, leading to diverse functional outcomes crucial for cellular dynamics and genetic regulation [205, 206].
Association of aberrant RNA modifications with Drug Resistance
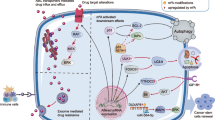
Aberrant RNA modifications have been linked to the development of various human diseases, including cancer [207,208,209,210,211]. Loss-of-function mutations in genes encoding RNA modification regulators, as well as point mutations that hinder proper RNA modifications, contribute to RNA modification abnormalities (Fig. 2) [47, 212,213,214,215]. Multiple factors lead to the deregulation of RNA modifications, consequently affecting drug resistance through mechanisms such as changes in gene expression, metabolic reprogramming, and immune evasion. These alterations influence the sensitivity of tumor cells to chemotherapy, targeted therapy, and immunotherapy (Table 1) (Fig. 3) [70, 216,217,218,219].

Mechanisms of abnormal RNA modifications. Aberrant RNA modifications can drive cancer progression through complex genetic and environmental interactions within the tumor microenvironment. These modifications often result from genetic mutations in enzymes that add or remove methyl groups, or in RNA-binding proteins, as well as from mutations in target genes that disrupt normal RNA modification processes

General Mechanisms of abnormal RNA modifications in drug resistance. Aberrant RNA modifications and their regulators are frequently linked to chemotherapy resistance in various cancers, including those of the digestive, genitourinary, and respiratory systems, as well as hematologic malignancies. These modifications can alter gene expression, affect DNA damage repair (DDR), and induce immune evasion, thereby contributing to drug resistance. Collectively, these drug resistance-related processes accelerate cancer progression, underscoring the significant role of RNA modifications in shaping the malignant behavior of cancer cells and influencing therapeutic outcomes
Aberrant RNA modifications interfere with gene expression by altering the stability and translation efficiency of mRNAs encoding proteins essential for DNA repair, cell cycle control, and apoptosis. This interference leads to increased DNA mutation frequency and genomic instability [70, 149, 217, 218, 269]. For instance, abnormal modifications in the cap, tail, and internal regions of tumor suppressor gene RNA can disrupt transcription, thereby promoting tumorigenesis and resistance to anticancer drugs [270,271,272,273]. Dysregulation of RNA modifications also affects the expression of drug-metabolizing enzymes or transporters involved in drug metabolism and efflux, such as ATP-binding cassette (ABC) transporters, enhancing drug efflux and reducing intracellular drug concentration [274,275,276,277].
Cancer cells can modify metabolic processes to support rapid, uncontrolled growth and proliferation [278,279,280,281,282]. RNA modifications critically impact metabolic pathways involved in cancer cell survival under drug-induced stress. Increasing evidence suggests that RNA modifications influence the expression and activity of key enzymes and transcription factors involved in the Warburg effect, enabling cancer cells to prefer glycolysis over oxidative phosphorylation [278, 283, 284]. This metabolic shift supports rapid cancer cell growth and contributes to drug resistance by enhancing cellular adaptation to the toxic effects of chemotherapy.
RNA modifications significantly contribute to immune evasion, indirectly promoting drug resistance [285,286,287,288]. Studies have shown that FTO enhances the self-renewal and immune evasion capabilities of cancer stem cells (CSCs) [289,290,291]. Reducing FTO expression through genetic manipulation or pharmacological intervention to lower immune checkpoint gene expression has demonstrated potent therapeutic effects against various cancer types [65, 101]. In the innate immune system, cells detect pathogens via encoded receptors, primarily RIG-I-like receptors (RLRs), endosomal toll-like receptors (TLRs), and protein kinase R (PKR) [292,293,294]. The m6A modification can disrupt RIG-I-mediated activation of innate immunity, aiding cancer cells in evading immune responses. Additionally, A-to-I RNA editing is crucial for immune system development, influencing T and B cell maturation by affecting innate immune tolerance to self-RNAs [295,296,297,298]. Antigen presentation is vital for the immune system’s recognition of cancer cells. Certain RNA modifications impede antigen presentation in cancer cells, reducing the visibility of antigens to immune cells [299,300,301]. For instance, the loss of YTHDF1 in dendritic cells (DCs) enhances the cross-presentation of tumor antigens and activation of CD8 + T cells, thereby improving the immune response to cancer [266, 302]. The intricate interplay of RNA modifications in cancer cells underscores their importance in immune evasion, highlighting potential therapeutic targets to enhance immunotherapy efficacy [303,304,305,306].
The diverse impacts of RNA modifications on gene expression, metabolic processes, and immune system interactions underscore their critical role in the development of drug resistance in cancer cells. Targeting these RNA modifications offers a promising strategy for overcoming resistance and improving the efficacy of existing cancer therapies [307, 308].
Profiles and mechanisms of aberrant RNA modifications in Chemotherapy Resistance in Cancer
Chemotherapy effectively suppresses tumor cell proliferation, but resistance frequently leads to treatment failure, promoting relapse or metastasis [309,310,311]. Common chemotherapeutic agents include alkylating agents like cyclophosphamide; antimetabolites such as 5-fluorouracil and cytarabine; DNA crosslinkers like cisplatin and carboplatin; anthracycline antibiotics including doxorubicin, idarubicin, and mitoxantrone; anti-microtubule agents such as paclitaxel and docetaxel; topoisomerase inhibitors like etoposide; nucleoside analogs such as gemcitabine; DNA methyltransferase inhibitors like 5-azacytidine; and proteasome inhibitors such as bortezomib, melphalan, and carfilzomib. These drugs exert their therapeutic effects through distinct mechanisms. The advent of immune checkpoint inhibitors targeting PD-1/PD-L1 and CTLA-4 has revolutionized cancer treatment [312,313,314]. However, resistance can arise from complex interactions between these inhibitors and the TME. Resistance to targeted therapies remains a significant challenge, affecting the prognosis of various tumors, including hepatocellular carcinoma (HCC) and non-small cell lung cancer (NSCLC) [315,316,317,318,319]. Therapeutic resistance in cancer involves a complex interplay of factors such as oncogene activation, impaired DNA repair, hypoxic TME, and metabolic alterations. CSCs significantly contribute to therapeutic resistance due to their self-renewal capabilities, aberrant differentiation, and enhanced drug efflux [320,321,322,323]. Recent studies on RNA modifications have revealed that epigenetic mechanisms play a pivotal role in developing therapeutic resistance in cancer [324, 325]. Investigating the patterns and mechanisms of aberrant RNA modifications in cancer can enhance the understanding of therapeutic resistance and provide novel avenues for developing effective therapeutic strategies (Table 2).
Effects of abnormal RNA modifications on chemotherapy resistance in gastro-intestinal cancers
5-Fluorouracil (5-FU) and the pyrimidine analog gemcitabine are widely used anti-nucleotide metabolism drugs in treating digestive system cancers, particularly pancreatic cancer (PC). Long non-coding RNAs (lncRNAs) play a critical role in maintaining cancer stemness, contributing to 5-FU resistance in PC. METTL3 enhances m6A modification in the lncRNA FOXD1-AS1 through a YTHDF1-dependent mechanism in MIA cells. Overexpressed FOXD1-AS1 then promotes self-renewal and 5-FU resistance in PC cells by acting as a competing endogenous RNA (ceRNA) that sponges miR-570-3p, resulting in increased expression of secreted phosphoprotein 1 (SPP1). Additionally, studies using patient-derived xenograft (PDX) models have highlighted FOXD1-AS1’s potential as a biomarker for predicting 5-FU treatment efficacy in PC [326]. ALKBH5 has dual roles in gemcitabine resistance in PC, either promoting or inhibiting drug resistance depending on the m6A modification of various downstream molecules. For example, ALKBH5 downregulation is linked to increased gemcitabine resistance and poor clinical outcomes. ALKBH5 demethylates WIF-1 mRNA to promote its transcription, inhibiting the Wnt signaling pathway in AsPC-1 and PANC-1 cells. This inhibition increases gemcitabine sensitivity, reducing the proliferative, colony-forming, and migratory abilities of PC cells, ultimately suppressing tumor growth and liver metastasis [220]. Conversely, ALKBH5 mediates m6A modification in the lncRNA DNA damage-inducible transcript 4 antisense transcript 1 (DDIT4-AS1), increasing its expression in PC cells. Upregulated DDIT4-AS1 disrupts DDIT4 mRNA stability and activates the mTOR pathway, enhancing cancer stemness and reducing gemcitabine sensitivity [221]. Serine/arginine-rich splicing factor 3 (SRSF3) and METTL3 induce m6A modification-related splicing of the lncRNA ANRIL in Panc1 and BXPC3 cells. This modification enables ANRIL to form a DNA homologous recombination (HR) repair complex with Ring finger protein 1B (Ring1B) and zeste homolog 2 (EZH2), enhancing DNA repair capabilities and gemcitabine resistance in PC cells [222]. RNA modifications also influence 5-FU resistance in colorectal cancer (CRC). METTL3 is crucial for aberrant m6A modifications contributing to chemoresistance in CRC. High m6A levels are observed in CRC tissues and cell lines. IGF2BP1 binds to METTL3-modified Sect. 62 mRNA, enhancing its stability and expression in CRC cells. Upregulated Sect. 62 interacts with β-catenin to activate the Wnt signaling pathway, which enhances stemness and chemoresistance in CRC [224]. Exosomal miR-181d-5p derived from cancer-associated fibroblasts (CAFs) regulates 5-FU sensitivity in CRC by targeting neurocalcin δ (NCALD), decreasing CRC cell sensitivity to 5-FU. METTL3-dependent m6A modification promotes miR-181d-5p processing by recognizing DiGeorge syndrome critical region 8 (DGCR8), highlighting the complex interplay between m6A and CAF-derived exosomes in chemoresistance [225]. Furthermore, FTO plays a significant role in promoting chemoresistance in CRC by mediating the demethylation of m6A and inducing the degradation of SIVA1 mRNA, a gene related to apoptosis, via a YTHDF2-dependent mechanism. Inhibiting FTO reduces 5-FU tolerance, accelerates tumor cell apoptosis, and inhibits tumor growth via the FTO-SIVA1 axis in both 5-FU-resistant CRC cells and tumor xenograft-bearing mouse models [226]. Overexpressed METTL1 can increase cisplatin sensitivity in CRC cells by modulating the miR-149-3p/small calcium-binding protein A4 (S100A4)/p53 axis. Specifically, METTL1 suppresses S100A4 expression and decreases cisplatin resistance in CRC cells by upregulating miR-149-3p through a p53-dependent mechanism [228].
HNRNPA2B1 is upregulated in multidrug-resistant gastric cancer (GC) cells and is linked to poor prognosis. It stabilizes the lncRNA NEAT1 via m6A modification, which subsequently activates the Wnt/β-catenin signaling pathway. This activation bestows GC cells with stemness properties and heightens their resistance to 5-FU [229]. The lncRNA ARHGAP5-AS1 recruits METTL3 to stimulate m6A modification of ARHGAP5 mRNA, maintaining its stability in the cytoplasm and preventing its autophagic degradation. This process leads to resistance against cisplatin, Adriamycin (ADR), and 5-FU in GC cells [230]. Additionally, upregulated LINC00942 stabilizes and enhances c-Myc mRNA expression by interacting with MSI2 in chemo-resistant GC cells. Targeting the LINC00942–MSI2–c-Myc axis has demonstrated potential in reversing chemoresistance, promoting apoptosis, and reducing stemness in GC cells (Fig. 4) [231].

Mechanisms of abnormal RNA modifications involved in chemotherapy resistance in digestive system cancers. Abnormal RNA methylation significantly contributes to chemotherapy resistance in digestive system cancers. In pancreatic cancer, METTL3-mediated m6A methylation of lncRNA FOXD1-AS1 enhances 5-FU resistance by promoting cell self-renewal through miR-570-3p sequestration. In gastric cancer, HNRNPA2B1 stabilizes lncRNA NEAT1, activating the Wnt/β-catenin pathway and increasing 5-FU resistance. Additionally, the dysregulation of RNA modification enzymes such as ALKBH5 affects gemcitabine sensitivity in pancreatic cancer by altering WIF-1 mRNA methylation and influencing the Wnt signaling pathway. These examples highlight the complex role of RNA methylation in modulating drug response and resistance across various gastrointestinal cancers
Effects of abnormal RNA modifications on chemotherapy resistance in genitourinary cancers
Since the 1970s, anthracyclines have been integral to chemotherapy, often used in neoadjuvant and combination treatments for breast cancer (BC) [327,328,329,330]. Recent studies indicate that m6A modification, particularly through METTL3, promotes chemoresistance in BC. In BC cell lines MCF-7 and MDA-MB-231, METTL3 enhances resistance to ADR by increasing homologous recombination (HR) efficiency and reducing ADR-induced DNA damage. This resistance mechanism involves METTL3-mediated m6A modification of epidermal growth factor (EGF) mRNA, which subsequently increases the expression of RAD51, a critical gene in HR. The RNA-binding protein YTHDC1 interacts with m6A-modified EGF mRNA to stabilize it, thereby enhancing HR and cellular resistance to ADR [235]. In ADR-resistant MCF-7 cells, upregulated METTL3 promotes the maturation of pri-miR-221-3p via m6A modification, leading to increased miR-221-3p expression. This miRNA downregulates the pro-apoptotic kinase Homeodomain-Interacting Protein Kinase 2 (HIPK2) while upregulating its target, AATF (Che-1), raising the IC50 value of ADR and reinforcing resistance [236]. Forkhead box protein O1 (FOXO1)-mediated reduction of reactive oxygen species (ROS) is another key mechanism underlying ADR resistance in BC. In ADR-resistant MDA-MB-231 and BT549 cells, ALKBH5-mediated m6A demethylation enhances FOXO1 mRNA stability. Consequently, upregulated FOXO1 increases superoxide dismutase (SOD2) levels, effectively reducing intracellular ROS and maintaining cancer stemness and ADR resistance [237]. HIF1α also enhances paclitaxel (PTX) resistance by promoting BC cell stemness [331]. A key mechanism involves the upregulation of lncRNA LINC00115 in MDA-MB-231 and BT549 cells, which activates HIF1α and subsequently upregulates ALKBH5. ALKBH5 demethylates YTHDF2-mediated m6A modification of LINC00115, stabilizing its expression. This feedback loop sustains the resistance of BC stem cells (BCSCs) to PTX and enhances their metastatic ability. Targeting this pathway with antisense oligonucleotides (ASO) against LINC00115, in combination with PTX, can effectively reduce the proliferative and invasive abilities of PTX-resistant BC cells. This strategy also significantly reduces tumor burden, prevents lung metastasis, and improves survival in animal models, representing a promising approach to overcoming PTX resistance in BC [238]. Chemotherapy resistance poses a significant challenge to improving ovarian cancer (OC) prognosis. Recent studies highlight the role of upregulated YTHDF1 in maintaining stem-like features in cisplatin-resistant OC cells. YTHDF1 enhances the translation of tripartite motif protein 29 (TRIM29) mRNA, promoting colony- and spheroid-forming abilities and invasiveness of cisplatin-resistant SKOV3/DDP and A2780/DDP cells. Upregulated TRIM29 facilitates tumorigenesis in nude mice with OC, indicating its impact on aggressiveness and chemotherapy resistance [239]. Additionally, upregulated ALKBH5 enhances epithelial cell proliferation, tumor growth, and cisplatin resistance in OC. ALKBH5 interacts with homeobox A10 (HOXA10), establishing a positive feedback loop that sustains its overexpression in cisplatin-resistant A2780-DDP and HO8910-DDP cells. This interaction prevents YTHDF2-mediated degradation of Janus kinase 2 (JAK2) mRNA, leading to increased JAK2 expression. Activated JAK2 then phosphorylates signal transducer and activator of transcription 3 (STAT3), initiating the JAK2/STAT3 signaling pathway, which drives cell proliferation and enhances cisplatin resistance [240]. In HeLa and SiHa cervical cancer (CC) cells, ZC3H13 induces hypermethylation of centromere protein K (CENPK) mRNA, increasing CENPK expression. Upregulated CENPK interacts with SOX6, enhancing Wnt signaling and suppressing p53 signaling. These changes stimulate CC stemness and increase resistance to chemotherapeutic agents like cisplatin and carboplatin, promoting tumor growth and lung metastasis [241].
Research indicates that ALKBH5 is significantly downregulated in bladder cancer (BCa) tissues compared to healthy bladder tissues, with reduced expression linked to favorable clinical outcomes. ALKBH5 diminishes the stability of casein kinase 2α (CK2α) mRNA via an m6A-dependent mechanism, thereby lowering CK2α expression. This reduction impairs glycolysis-related processes, including glucose utilization, lactate production, and intracellular ATP generation in BCa cells [295]. Elevated ALKBH5 levels increase BCa cells’ sensitivity to cisplatin by targeting CK2α-mediated glycolysis, thereby inhibiting cell proliferation and migration. Furthermore, this mechanism reduces tumor size and weight in BCa mouse models [242]. Circular RNAs (circRNAs) regulate m6A modification of mRNAs, influencing cisplatin resistance in BCa. Higher levels of circ0008399 and WTAP correlate with cisplatin resistance and poor clinical outcomes in BCa. circ0008399 interacts with WTAP to promote the formation of the WTAP/METTL3/METTL14 complex, enhancing m6A modification in BCa cells [296]. This interaction notably stabilizes and increases TNF alpha-induced protein 3 (TNFAIP3) mRNA expression in an m6A-dependent manner, reducing BCa cells’ sensitivity to cisplatin [243]. Recent studies have revealed that cisplatin treatment can induce the upregulation of the N4-acetylcytosine (ac4C) modification writer N-acetyltransferase 10 (NAT10) through NF-κB signaling activation [332]. Consequently, NAT10 stabilizes desmoyokin (AHNAK) mRNA, enhancing the DNA damage response (DDR) and promoting cisplatin resistance in BCa [244]. m6A sequencing of cancer tissues has shown higher overall m6A modification levels in castration-resistant prostate cancer (CRPC) compared to castration-sensitive prostate cancer (CSPC) [333, 334]. The AZGP1P2/UBA1/RBM15/TPM1 cascade is a critical pathway that enhances docetaxel sensitivity in CSPC. AZGP1 pseudogene 2 (AZGP1P2) knockdown increases the stemness of prostate cancer stem cells (PCSCs) and promotes tumor growth and metastasis, indicating AZGP1P2’s essential role in preventing docetaxel resistance. AZGP1P2 forms a complex with ubiquitin-like modifier activating enzyme 1 (UBA1) and RNA-binding motif protein 15 (RBM15), facilitating RBM15 ubiquitination and degradation. This process reduces the m6A-dependent decay of tropomyosin 1 (TPM1) mRNA, stabilizing TPM1 and inhibiting prostate cancer cell growth, migration, and metastasis. Targeting the AZGP1P2/UBA1/RBM15/TPM1 pathway may improve docetaxel therapeutic efficacy in CRPC (Fig. 5) [245].

Mechanisms of abnormal RNA modifications involved in chemotherapy resistance in genitourinary system cancers
Aberrant RNA modifications are frequently linked to chemotherapy resistance by altering gene expression and signaling pathways in genitourinary system cancers, including breast, ovarian, cervical, prostate, and bladder cancers. In breast cancer, METTL3 enhances ADR resistance by methylating EGF mRNA, which increases homologous recombination (HR) repair efficiency and reduces DNA damage. In ovarian cancer, elevated ALKBH5 levels increase cisplatin resistance by demethylating HOXA10, thereby promoting cell proliferation. Cervical cancer shows increased CENPK expression via ZC3H13-mediated mRNA methylation, which enhances stemness and drug resistance. Conversely, in bladder cancer, downregulation of ALKBH5 is associated with improved cisplatin sensitivity due to reduced CK2α-mediated glycolysis. Targeting RNA methylation pathways thus presents a promising strategy for overcoming drug resistance in genitourinary system cancers
Effects of abnormal RNA modifications on chemotherapy resistance in hematological malignancies
IGF2BP1 is highly expressed in various leukemia subtypes and is essential for maintaining leukemogenesis and promoting stemness. It enhances resistance to all-trans retinoic acid (ATRA), doxorubicin, cytarabine, and cyclophosphamide, preventing cell death and differentiation in 697(EU3) and K562 leukemia cells by post-transcriptionally regulating key self-renewal genes, including homeobox B4 (HOXB4), MYB, and aldehyde dehydrogenase 1 family member A1 (ALDH1A1) [246]. METTL3 facilitates homing and engraftment in the bone marrow (BM) and subsequent idarubicin (IDA) resistance in acute myeloid leukemia (AML) by increasing m6A modification and protein expression of integrin subunit alpha 4 (ITGA4) mRNA. The METTL3 inhibitor STM2457 has been shown to counteract the homing/engraftment capabilities of IDA-resistant AML cells, reversing their chemotherapeutic resistance [247]. Mitoxantrone-resistant AML cells exhibit more m7G sites, with resistance-related genes regulated through the circRNA–miRNA–mRNA co-expression network [248]. Enhanced m5C modification can influence 5-azacitidine (5-AZA) resistance in leukemia and myelodysplastic syndrome (MDS) by forming 5-AZA-sensitive/resistant chromatin structures on nascent RNAs in leukemia cells [249].
HNRNPK interacts with lineage-determining transcription factors (TFs) GATA1 and SPI1/PU.1, and with CDK9/P-TEFb, to recruit RNA polymerase II to nascent RNAs, forming 5-AZA-sensitive chromatin structures. In chronic myeloid leukemia (CML), LINC00470, a METTL3 regulator, enhances METTL3 recruitment to PTEN mRNA, reducing PTEN expression. This interaction activates the AKT signaling pathway, inhibiting autophagy and promoting ADR resistance in CML [250]. Multiple myeloma (MM) cells increase METTL7A activity through EZH2-mediated protein methylation. Upregulated METTL7A enriches LOC606724 and lncRNA SNHG1 in adipocyte-derived exosomes, protecting MM cells from chemotherapy-induced apoptosis and enhancing resistance to bortezomib, melphalan, and carfilzomib (Fig. 6) [335].

Mechanisms of abnormal RNA modifications involved in chemotherapy resistance in hematologic malignancies and respiratory system cancers. Abnormal RNA methylation plays a significant role in chemotherapy resistance across various cancers. In leukemia, IGF2BP1 enhances resistance by regulating genes such as HOXB4 and MYB, impacting responses to treatments like ATRA and doxorubicin. METTL3 further contributes to resistance through m6A methylation of ITGA4, affecting homing and engraftment. Additionally, LINC00470 decreases PTEN expression via METTL3, activating AKT signaling and increasing resistance to ADR. In multiple myeloma, elevated METTL7A levels promote chemotherapy evasion by enhancing exosomal mechanisms. In lung cancer and nasopharyngeal carcinoma, higher levels of METTL3 and METTL5 lead to chemoresistance by activating mitochondrial autophagy and enhancing the translation of oncogenic factors, respectively. These findings underscore the complex role of RNA modifications in
treatment resistance, highlighting the urgent need for targeted therapeutic strategies
Effects of abnormal RNA modifications on chemotherapy resistance in respiratory system cancers and other cancers
Recent studies have elucidated the impact of aberrant RNA modifications on chemotherapy resistance in respiratory system cancers, such as lung cancer (LC). In small-cell lung cancer (SCLC), elevated METTL3 levels induce chemoresistance by downregulating decapping protein 2 (DCP2) [313]. DCP2 suppression activates the Pink1–Parkin mitochondrial autophagy pathway, mitigating mitochondrial damage and conferring resistance to cisplatin and etoposide in H69 and H446 cells [259].
In nasopharyngeal carcinoma (NPC), increased METTL5/TRMT112 mediates m6A modification at position 1832 (m6A1832) in 18 S rRNA, essential for 80 S ribosome assembly and enhancing global mRNA translation [305]. METTL5 promotes the translation of heat shock transcription factor 4b (HSF4b), which subsequently activates heat shock protein 90 beta family member 1 (HSP90B1). HSP90B1 interacts with oncogenic mutant p53 (mutp53), inhibiting its ubiquitination, thereby promoting tumorigenesis and resistance to cisplatin and docetaxel in NPC [252]. METTL1 and its mediated m7G tRNA modification are also pivotal in NPC chemoresistance. METTL1/WDR4 upregulates the translation of mRNAs involved in the WNT/β-catenin signaling pathway, promoting epithelial–mesenchymal transition (EMT) and enhancing resistance to cisplatin and docetaxel. Aryl hydrocarbon receptor nuclear translocator (ARNT) acts as a negative regulator of METTL1, reducing its oncogenic potential in NPC [253].
In glioma, the Warburg effect stimulates exosomal circ_0072083 release, which upregulates ALKBH5 expression, enhancing NANOG mRNA demethylation and stability, thereby promoting Temozolomide (TMZ) resistance [336]. Additionally, the long non-coding RNA JPX interacts with FTO, boosting FTO-mediated demethylation of phosphoinositide-dependent kinase-1 (PDK1) mRNA. This interaction increases PDK1 expression, further promoting aerobic glycolysis and TMZ resistance in glioma [337]. In osteosarcoma, overexpressed METTL1/WDR4 enhances m7G tRNA modification and the translation of oncogenic lysyl oxidase-like 2 (LOXL2), facilitating tumor progression and ADR resistance [254]. Moreover, the interplay between DNA cytosine methylation and m5C modification affects chemotherapy-induced apoptosis in osteosarcoma. DNA methyltransferase I (DNMT1) suppresses NSUN2 expression and subsequent m5C modification on both DNA promoters and mRNAs of anti-apoptotic target genes AXL, NOTCH2, and YAP1, inducing apoptosis in osteosarcoma cells treated with cisplatin or ADR [255].
Role of abnormal RNA modifications in resistance to other Anti-cancer therapies
Role of abnormal RNA modifications in resistance to targeted therapy
Sorafenib, the first FDA-approved drug for advanced hepatocellular carcinoma (HCC), faces limited efficacy due to resistance development. Hepatocyte nuclear factor 3-gamma (HNF3γ), a regulator of hepatocyte and liver cancer stem cell differentiation, is pivotal in sorafenib resistance. Recent research indicates that METTL14 reduces HNF3γ mRNA expression via m6A modification. Lower HNF3γ levels hinder the activation of organic anion-transporting polypeptides 1B1 (OATP1B1) and 1B3 (OATP1B3), which are essential for drug uptake. Consequently, reduced HNF3γ diminishes HCC cells’ sensitivity to sorafenib’s growth-inhibiting and pro-apoptotic effects, undermining its therapeutic efficacy [232]. Forkhead box O3 (FOXO3)-mediated autophagy contributes to sorafenib resistance in HCC. Under hypoxia, METTL3 depletion suppresses m6A modification of FOXO3 mRNA through a YTHDF1-dependent mechanism, leading to decreased FOXO3 expression. Reduced FOXO3 enhances angiogenesis and autophagic flux, promoting sorafenib resistance and tumor growth in HCC [233]. Lenvatinib-resistant HCC tissues exhibit increased m7G modification levels and upregulated METTL1 and WDR4, correlating with poor clinical outcomes [164, 234]. WDR4 enhances TRIM28 translation, increasing stemness and lenvatinib resistance in HCC cells. Leukemia, a severe malignancy, often involves activating receptor tyrosine kinase (RTK) mutations, making tyrosine kinase inhibitors (TKIs) like erlotinib and imatinib essential treatments. The m6A modification plays a pivotal role in TKI resistance development in leukemia. The FTO–m6A axis has emerged as a novel biomarker for TKI resistance. Overexpression of FTO and reduced m6A modification levels are associated with genetically homogeneous leukemia cells harboring BCR/ABL, KIT, or FLT3 mutations. Thus, targeting the FTO–m6A axis offers a promising strategy to overcome TKI resistance in leukemia [256]. Crizotinib, a TKI targeting c-MET/ALK/ROS1, is an FDA-approved first-line treatment for NSCLC with ALK mutations, ROS1 rearrangement, or c-MET overactivation. Recent studies suggest that the histone deacetylase inhibitor chidamide enhances crizotinib’s effectiveness in ALK mutant-negative NSCLC cell lines, particularly those with high c-MET expression. In HCC827 and H661 NSCLC cells, chidamide downregulates METTL3 and WTAP, inhibiting c-MET mRNA modification and increasing NSCLC cells’ sensitivity to crizotinib, thereby inhibiting cell growth [257]. Furthermore, METTL3-mediated autophagy counters the autophagy suppression induced by the anticancer drug β-elemene, thereby enhancing gefitinib resistance in NSCLC [258]. m5C hypermethylation and NSUN2 are linked to intrinsic gefitinib resistance and tumor recurrence in EGFR-mutant NSCLC. Overexpressed NSUN2 interacts with YBX1, boosting the translation of quiescin sulfhydryl oxidase 1 (QSOX1) mRNA, thus enhancing the proliferative and colony-forming capabilities of gefitinib-resistant NSCLC cells [260]. Another mechanism underlying TKI resistance in NSCLC involves the upregulation of the hepatocyte growth factor receptor MET. Increased A-to-I editing of miR-411-5p directly decreases MET protein expression and reduces downstream ERK signaling activity, thereby sensitizing NSCLC cells to gefitinib and Osimertinib [261]. In PC9 and A549 lung adenocarcinoma (LUAD) cells, METTL3 stabilizes the lncRNA SNHG17, leading to its upregulation. Elevated SNHG17 facilitates the epigenetic suppression of large tumor suppressor kinase 2 (LATS2) via interaction with EZH2, promoting gefitinib resistance, cell migration, invasion, and EMT [262]. METTL3, along with YTHDF2, promotes miR-146a expression while suppressing TUSC7 expression in an m6A-dependent manner, maintaining active Notch signaling and exacerbating erlotinib resistance in LUAD [263]. METTL7B increases the levels of three key ROS scavengers—glutathione peroxidase 4 (GPX4), superoxide dismutase 1 (SOD1), and heme oxygenase 1 (HMOX1)—in PC9 and HCC827 LUAD cells, thereby inducing glutathione metabolism and enhancing resistance to gefitinib and Osimertinib [264]. In A375R melanoma cells, METTL3 enhances the m6A modification of EGFR mRNA, significantly improving its translation efficiency. Upregulated EGFR activates the RAF/MEK/ERK signaling pathway, contributing to resistance against the BRAF (V600E) kinase inhibitor PLX4032 in melanoma [265].
Role of abnormal RNA modifications in resistance to immunotherapy
Despite the abundance of neoantigens in patients, tumor-associated antigens are essential for eliciting spontaneous anti-tumor immune responses. Research indicates that RNA modification regulators significantly influence the response to immune checkpoint blockade (Fig. 7). Most studies on m6A modification related to resistance to immune checkpoint inhibitors have concentrated on PD-1/PD-L1 inhibitors. In melanoma, YTHDF1 specifically recognizes m6A-modified mRNAs encoding lysosomal proteases, enhancing their translation in dendritic cells. This mechanism limits tumor antigen cross-presentation and impairs the cross-priming of CD8 + T cells’ anti-tumor responses during anti-PD-1 immunotherapy [266]. Under metabolic stress, melanoma cells elevate FTO levels via autophagy and NF-κB signaling, leading to resistance to interferon-gamma (IFN-γ) and anti-PD-1 treatment. FTO knockdown increases the RNA decay of intrinsic pro-tumorigenic genes, including PD-1, CXCR4, and SOX10, through YTHDF2 in melanoma cells [267]. In colorectal cancer (CRC), METTL3 and METTL14 negatively impact genes involved in IFN-γ and IFN-β pathways, such as STAT1 and IRF1, reducing cytotoxic CD8 + T cell infiltration and diminishing the response to anti-PD-1 immunotherapy, especially in mismatch repair-proficient or low microsatellite instability CRC cases [227]. Furthermore, glucose directly activates NSUN2, stabilizing three prime repair exonuclease 2 (TREX2) mRNA and suppressing cGAS/stimulator of interferon genes (STING) signaling. This cGAS/STING axis drives resistance in immunologically “cold” tumors to anti-PD-1 immunotherapy and promotes tumorigenesis [268]. Recent studies have underscored that A-to-I editing affects therapeutic responses in cancer by generating RNA editing events linked to drug resistance [338, 339]. In PDAC, often classified as an immunologically “cold” tumor, elevated METTL3 enhances anti-tumor immunity by blocking A-to-I editing of m6A-modified transcripts and promoting endogenous dsRNA accumulation. dsRNA stress activates RLRs, triggering signaling cascades that amplify the anti-tumor immune response [223].

Mechanisms of abnormal RNA modifications involved in resistance to targeted therapy and immunotherapy in cancer. Beyond contributing to chemotherapy resistance, abnormal RNA modifications can diminish the efficacy of targeted therapies and immunotherapies, resulting in treatment failure or cancer recurrence. Modifications such as m6A, m7G, m5C, and A-to-I editing promote resistance to tyrosine kinase inhibitors in various cancers, including hepatocellular carcinoma, leukemia, and lung cancer. For example, METTL3 reduces the therapeutic efficacy of the BRAF inhibitor PLX4032 in melanoma by activating the epidermal growth factor receptor (EGFR) pathway. Additionally, RNA modifications can disrupt gene expression and intracellular signaling pathways, leading to abnormal expression of immune checkpoints, interference with antigen presentation and recognition, suppression of cytotoxic T-cell infiltration, and the development of immunotherapy resistance
Optimizing Treatment Efficacy Through the Combination of Traditional Anti-cancer Drugs and Modulation of RNA Modifications
Recent research advancements underscore the pivotal role of RNA modifications in fostering therapeutic resistance in cancer, paving the way for innovative strategies to enhance the effectiveness of conventional anti-cancer treatments. One promising method focuses on inhibiting the enzymatic activity or expression of proteins associated with RNA modification [340]. This approach includes the deployment of small molecule inhibitors, such as the extensively studied FTO inhibitors Rhein, R-2-hydroxyglutarate (R-2HG), meclofenamic acid (MA), FB23, and FB23-2 [101, 341,342,343,344,345]. Notably, the combination of Rhein with TKIs exhibits synergistic effects, effectively targeting TKI-resistant leukemia cells, including those from patients with relapsed or refractory leukemia [256]. Additionally, Rhein significantly enhances the antiproliferative impact of the anti-PD-L1 antibody atezolizumab in 4T1 BC xenografts by augmenting the proportion of CD8 + T cells in the spleen and tumor tissues and elevating apoptotic factors in BC tissues [346]. Another direct FTO inhibitor, Saikosaponin-d (SsD), displays broad anti-proliferative effects in leukemia and improves TKI resistance mediated by FTO [347]. Moreover, R-2HG, in conjunction with first-line chemotherapeutic drugs such as ATRA, AZA, decitabine, and ADR, synergistically induces cell cycle arrest and apoptosis in leukemia and glioma cells, underscoring the efficacy of targeting RNA modification regulators in cancer treatment strategies [342]. The FTO inhibitor 18,097 has been shown to suppress cell growth and lung colonization in BC animal studies, while FB23-2 demonstrates therapeutic potential for clear cell renal cell carcinoma (ccRCC) by inhibiting tumor growth and prolonging survival in PDX model mice [348, 349].
Inhibitors targeting other RNA modification-related enzymes also exhibit therapeutic potential across various cancer types. For instance, ALK-04, a small-molecule inhibitor of ALKBH5, enhances the efficacy of anti-PD-1 immunotherapy in melanoma by modifying the TME [350]. TAS0612, targeting the m5C reader YBX1, reduces antiestrogen resistance in patients with triple-negative BC [351]. Targeting METTL3 presents considerable promise in overcoming cancer drug resistance [352, 353]. A significant development is STM2457, a highly potent and selective catalytic inhibitor of METTL3, which shows potential against AML by targeting key stem cell subpopulations, leading to diminished AML growth, improved differentiation, and increased apoptosis [354]. STM2457 effectively reverses homing/engraftment and drug resistance in IDA-resistant AML cells, contributing to decreased proliferation and increased apoptosis [247, 289, 352, 355]. In primary resistant HCC cell line MHCC97H and acquired resistant Huh7-LR cells, as well as multiple mouse HCC models, STM2457 enhances tumor response to lenvatinib and alleviates gemcitabine resistance in intrahepatic cholangiocarcinoma (ICC) by inhibiting glycolytic reprogramming [216, 281]. In SHH subgroup medulloblastoma (SHH-MB), targeting METTL3 with STM2457 represses Sonic hedgehog signaling, thereby restraining tumor progression [356]. WD6305, a PROTAC degrader of the METTL3-METTL14 complex, exhibits more effective anti-leukemic effects than its parent inhibitor by suppressing m6A modification and affecting various signaling pathways involved in AML development and proliferation [357]. Additionally, a designed stapled peptide inhibitor (RSM3) targeting the METTL3-METTL14 binding interface and inhibiting global RNA methylation significantly suppresses tumor growth in diverse cancer types, particularly in prostate cancer (PCa) cell lines [358].
Increasing evidence underscores the pivotal role of RNA modifications in cancer immunotherapy, significantly enhancing the efficacy of immune checkpoint blockade (ICB) and chimeric antigen receptor (CAR) T-cell therapy. RNA modifications achieve this by directly suppressing tumor growth, modulating immune cell activation, and promoting immune cell infiltration into the TME [359]. A Phase I clinical trial is currently evaluating the anti-tumor efficacy of the METTL3 inhibitor STC-15, both as a monotherapy and in combination with an anti-PD-1 antibody for treating solid tumors. In various pre-clinical cancer models, STC-15 activates innate immune pathways and enhances CD8 + T cell-mediated tumor cell death. It also amplifies the anti-tumor effects of anti-PD-1 therapy, generating a durable anti-tumor immune response, indicating significant potential for METTL3 inhibition in cancer treatment [360]. In non-alcoholic fatty liver disease-associated HCC (NAFLD-HCC), the combination of STM2457 and ICB therapy dramatically boosts the cytotoxic CD8 + T cell-mediated antitumor response and overcomes anti-PD-1 resistance, leading to a synergistic suppression of NAFLD-HCC. In NSCLC, STM2457 upregulates PD-L1 expression, thereby improving immunotherapy outcomes both in vivo and in vitro [361]. While small molecule inhibitors can be rapidly developed and tested in clinical trials, offering a scalable and cost-effective treatment strategy, their application is restricted by potential off-target effects, the development of resistance over time, and limited bioavailability and stability [27, 362, 363].
In addition to small molecule inhibitors targeting RNA modification regulators, advanced techniques such as CRISPR/Cas9, RNA interference (RNAi), and nanomedicine-based technology provide further methods to target and suppress these regulators [316, 364,365,366,367]. The CRISPR/Cas9 system, in particular, offers significant potential for enhancing drug sensitivity and enabling personalized therapies in cancer treatment [368,369,370]. For instance, METTL3 knockdown via CRISPR/Cas9 in head and neck squamous cell carcinoma has been shown to increase cancer cell sensitivity to cisplatin [371]. A novel approach by Liu et al. involved developing a programmable RNA m6A editing system by fusing CRISPR/Cas9 with a single-chain m6A methyltransferase, allowing site-specific methylation in different mRNA regions using guide RNA [372]. Additionally, CRISPR/Cas9 fused with ALKBH5 or FTO, m6A ‘erasers,’ can achieve site-specific RNA demethylation. The increasing versatility of CRISPR/Cas enzymes suggests the potential for creating m6A editors with smaller sizes, higher specificity, and adjustable features, enabling precise m6A modifications on RNA without altering the primary sequence. The high efficiency and precision of the CRISPR/Cas9 technique make it a valuable tool for exploring drug targets [373,374,375]. For example, genome-wide CRISPR/Cas9 knockout screening across over 800 cancer cell lines revealed a stronger dependency on METTL16 in leukemias compared to other cancer types, particularly in AML cell lines [376]. Despite these advancements, CRISPR/Cas9-based RNA modifications face limitations, including unstable genome editing efficiency and specificity, off-target effects, and the induction of immune responses [368]. While the CRISPR/Cas9 system boasts high efficiency, the safe and effective delivery of CRISPR components to target cells is crucial for achieving high-efficiency gene editing [377]. The choice and optimization of delivery systems are key factors; viral vectors offer high transfection efficiency but pose safety concerns such as oncogenicity, insertional mutagenesis, and immunogenicity. Off-target effects also reduce the specificity of CRISPR/Cas9 gene editing [378,379,380]. Furthermore, nanomedicine-based RNA modification has greatly expanded, improving drug bioavailability, tumor targeting, and intracellular release efficiency, thus enhancing therapeutic outcomes while mitigating adverse effects [381,382,383,384,385,386]. An innovative application of nanotechnology in cancer therapy involves utilizing nanoparticles engineered to deliver mRNA or small interfering RNA (siRNA) targeting RNA modification-related regulators, effectively remodeling the TME and achieving anti-cancer effects [355, 367, 387, 388]. For example, a PLGA-based nanoplatform loaded with LINC00958 siRNA significantly decreased the proliferative ability of HCC cells and reduced xenograft tumor growth by inhibiting m6A-mediated upregulation of LINC00958, demonstrating the potential of targeted nanomedicine strategies in controlling tumor progression [389]. Additionally, combining an antigen-capturing nanoplatform designed for co-delivering tumor-associated antigens (TAAs) and FTO into tumor-infiltrating dendritic cells (TIDCs) with thermal ablation significantly enhanced DC maturation and triggered tumor infiltration of effector T cells, thereby boosting the anti-tumor effects of ICB treatment in HCC [390]. Moreover, using a Toll-like receptor 9 agonist-conjugated siRNA to selectively target YTHDF2 in tumor-associated macrophages (TAMs) reprogrammed TAMs towards an anti-tumoral phenotype, increasing their antigen cross-presentation capabilities and improving CD8 + T cell-mediated antitumor immunity. This reprogramming not only restrained tumor growth but also significantly boosted the efficacy of PD-L1 antibody therapy [391]. However, challenges such as the complexity of nanoparticle design and manufacturing, long-term safety, potential toxicity, and off-target effects hinder the clinical reliability of nanotechnology applications [392,393,394].
The investigation of RNA modification mechanisms in drug resistance provides a promising route for developing innovative therapeutic strategies to combat cancer resistance. Nonetheless, the field of translational drug discovery targeting RNA modifications is nascent, with no clinical trials yet employing m6A inhibitors for cancer treatment [395,396,397]. The global effects of methylation and demethylation induced by RNA modification inhibitors or activators demand thorough investigation due to their extensive distribution across nearly all RNA species and significant biological implications. RNA modifications display cellular heterogeneity, meaning the same writer, eraser, and reader proteins may function differently across various cellular contexts. Additionally, the impact of m6A modifications varies depending on their location within different regions of the same RNA. This complex landscape necessitates precise targeting and modulation of RNA modifications to maximize therapeutic efficacy while minimizing potential adverse effects [176, 256, 335, 398, 399]. Future clinical trials must explore the synergistic anti-cancer effects of combining traditional anti-cancer drugs with RNA modification inhibitors, potentially paving new pathways for effectively treating resistant cancers.
Conclusions and outlooks
Recent advancements in epigenetic research have highlighted the pivotal role of RNA modifications in tumorigenesis and drug resistance. Modifications such as m6A, m1A, m5C, m7G, Ψ, and A-to-I editing significantly influence gene expression by affecting RNA splicing, stability, translation, and degradation, thereby impacting the function and malignancy of tumor cells. Many RNA modification regulators are overexpressed in cancer cells, contributing to enhanced proliferation, stemness, and resistance to conventional therapies, leading to poor prognosis and high recurrence rates in most cancers.
The dysregulation of RNA modification regulators and their downstream pathways is linked to various mechanisms of therapeutic resistance, including altered drug targets, disrupted drug efflux, and evasion of drug-induced apoptosis. Emerging studies reveal both oncogenic and tumor-suppressive roles of these regulators in different cancers. For example, METTL3 functions as an oncogene in multiple cancer types, enhancing drug resistance through various signaling pathways, yet it also potentially improves the therapeutic efficacy of sorafenib in HCC. These dual roles underscore the complexity of RNA modifications and necessitate further investigation into their specific functions in different tumor contexts to develop effective treatments.
Research on RNA modification inhibitors, particularly those targeting m6A, is gaining traction as a novel strategy to overcome cancer drug resistance. However, specific drugs targeting other RNA modifications, such as m1A, m5C, and m7G, remain scarce due to the intricate mechanisms involved and the limited understanding of their roles and quantification in cancer. Consequently, developing precise methods for detecting RNA modifications and identifying key modified RNAs in resistant cancers is imperative.
Despite the therapeutic potential of RNA modification inhibitors, drug development in this area is still in its infancy, with only a few candidates undergoing clinical testing. This gap highlights both a challenge and a significant opportunity for therapeutic innovation. Expanding research to include a broader array of RNA modifications could yield more therapeutic options. Combining RNA modification inhibitors with traditional anti-cancer therapies is a promising strategy for enhancing treatment efficacy and achieving synergistic anti-tumor effects. Developing targeted therapies that modulate RNA modifications without disrupting the overall epigenetic landscape could significantly advance cancer treatment. To fully harness the therapeutic potential of RNA modification inhibitors, a comprehensive investigation into the complex interplay among various RNA modifications and their interactions within the TME is essential. Large-scale clinical trials are necessary to validate the efficacy and safety of RNA modification-based anti-cancer therapies.
In conclusion, RNA modifications offer promising avenues for improving cancer treatment efficacy. Understanding how these modifications influence tumor cell responses to drugs may guide the development of novel cancer treatments and facilitate the design of individualized therapies tailored to the unique epigenetic profiles of individual cancers.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- m6A:
-
N6-methyladenosine
- m1A:
-
N1-methyladenosine
- m5C:
-
5-methylcytosine
- m7G:
-
7-methylguanosine
- Ψ:
-
Pseudouridine
- A-to-I editing:
-
Adenosine-to-inosine editing
- TME:
-
Tumor microenvironment
- EMT:
-
Epithelial–mesenchymal transition
- mRNAs:
-
messenger RNAs
- lncRNAs:
-
long non-coding RNAs
- rRNAs:
-
ribosomal RNAs
- tRNAs:
-
transfer RNAs
- miRNAs:
-
microRNAs
- dsRNAs:
-
double-stranded RNAs
- ceRNA:
-
endogenous RNA
- circRNAs:
-
circular RNAs
- DDR:
-
DNA damage repair
- PC:
-
Pancreatic cancer
- CRC:
-
Colorectal cancer
- GC:
-
Gastric cancer
- BC:
-
Breast cancer
- OC:
-
Ovarian cancer
- BCa:
-
Bladder cancer
- CRPC:
-
Castration-resistant prostate cancer
- CSPC:
-
Castration-sensitive prostate cancer
- MDS:
-
Myelodysplastic syndrome
- AML:
-
Acute myeloid leukemia
- CML:
-
Chronic myeloid leukemia
- MM:
-
Multiple myeloma
- NPC:
-
Nasopharyngeal carcinoma
- HCC:
-
Hepatocellular carcinoma
- LC:
-
Lung cancer
- NSCLC:
-
Non-small cell lung cancer
- SCLC:
-
Small-cell lung cancer
- CSCs:
-
Cancer stem cells
- 5-FU:
-
5-fluorouracil
- TKIs:
-
Tyrosine kinase inhibitors
- METTL3:
-
Methyltransferase-like 3
- WTAP:
-
Wilms tumor 1-associated protein
- VIRMA:
-
VIR-like m6A methyltransferase associated
- RBM15:
-
RNA-binding motif protein 15
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- FTO:
-
Fat mass and obesity-associated protein
- ALKBH5:
-
AlkB homolog 5
- YTHDF1:
-
YTH N6-methyladenosine RNA binding protein 1
- YTHDC1:
-
YTH domain-containing protein 1
- IGFBPs:
-
Insulin-like growth factor 1 mRNA-binding proteins
- MSI2:
-
Musashi2
- PRRC2A:
-
Proline-rich coiled-coil containing protein 2 A
- HNRNPA2B1:
-
Heterogeneous nuclear ribonucleoprotein A2/B1
- TRMT10:
-
tRNA methyltransferase 10 homolog A
- NSUN:
-
NOL1/NOP2/SUN domain
- DNMT2:
-
DNA methyltransferase-like 2
- TET:
-
Ten-eleven translocation
- ALYREF:
-
Aly/REF export factor
- YBX1:
-
Y-box binding protein 1
- FMRP:
-
Fragile X mental retardation protein
- WDR4:
-
WD repeat domain 4
- WBSCR22:
-
Williams–Beuren syndrome chromosomal region 22
- RNMT:
-
RNA guanine-7 methyltransferase
- RAM:
-
RNMT-activating miniprotein
- DKC1:
-
Dyskerin pseudouridine synthase 1
- TERC:
-
Telomerase RNA component
- ADARs:
-
Adenosine deaminases acting on RNAs
References
Yang Q, Xu J, Gu J, Shi H, Zhang J, Zhang J, et al. Extracellular vesicles in Cancer Drug Resistance: roles, mechanisms, and implications. Adv Sci (Weinh). 2022;9:e2201609. https://doi.org/10.1002/advs.202201609.
De Las Rivas J, Brozovic A, Izraely S, Casas-Pais A, Witz IP, Figueroa A. Cancer drug resistance induced by EMT: novel therapeutic strategies. Arch Toxicol. 2021;95:2279–97. https://doi.org/10.1007/s00204-021-03063-7.
Ward RA, Fawell S, Floc’h N, Flemington V, McKerrecher D, Smith PD. Challenges and opportunities in Cancer Drug Resistance. Chem Rev. 2021;121:3297–351. https://doi.org/10.1021/acs.chemrev.0c00383.
Song H, Liu D, Dong S, Zeng L, Wu Z, Zhao P, et al. Epitranscriptomics and epiproteomics in cancer drug resistance: therapeutic implications. Signal Transduct Target Ther. 2020;5:193. https://doi.org/10.1038/s41392-020-00300-w.
Di Donato M, Medici N, Migliaccio A, Castoria G, Giovannelli P. Exosomes: emerging modulators of pancreatic Cancer Drug Resistance. Cancers (Basel). 2023;15. https://doi.org/10.3390/cancers15194714.
Gonzalez-Fierro A, Dueñas-González A. Drug repurposing for cancer therapy, easier said than done. Semin Cancer Biol. 2021;68:123–31. https://doi.org/10.1016/j.semcancer.2019.12.012.
Xue C, Yao Q, Gu X, Shi Q, Yuan X, Chu Q, et al. Evolving cognition of the JAK-STAT signaling pathway: autoimmune disorders and cancer. Signal Transduct Target Ther. 2023;8:204. https://doi.org/10.1038/s41392-023-01468-7.
Chen S, Zhao Y, Liu S, Zhang J, Assaraf YG, Cui W, et al. Epigenetic enzyme mutations as mediators of anti-cancer drug resistance. Drug Resist Updat. 2022;61:100821. https://doi.org/10.1016/j.drup.2022.100821.
Jo H, Shim K, Jeoung D. Targeting HDAC6 to Overcome Autophagy-promoted Anti-cancer Drug Resistance. Int J Mol Sci. 2022;23. https://doi.org/10.3390/ijms23179592.
Chan YT, Lu Y, Wu J, Zhang C, Tan HY, Bian ZX, et al. CRISPR-Cas9 library screening approach for anti-cancer drug discovery: overview and perspectives. Theranostics. 2022;12:3329–44. https://doi.org/10.7150/thno.71144.
Gao L, Wu ZX, Assaraf YG, Chen ZS, Wang L. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist Updat. 2021;57:100770. https://doi.org/10.1016/j.drup.2021.100770.
Namee NM, O’Driscoll L. Extracellular vesicles and anti-cancer drug resistance. Biochim Biophys Acta Rev Cancer. 2018;1870:123–36. https://doi.org/10.1016/j.bbcan.2018.07.003.
Zhao Z, Mei Y, Wang Z, He W. The effect of oxidative phosphorylation on Cancer Drug Resistance. Cancers (Basel). 2022;15. https://doi.org/10.3390/cancers15010062.
Qin Y, Ashrafizadeh M, Mongiardini V, Grimaldi B, Crea F, Rietdorf K, et al. Autophagy and cancer drug resistance in dialogue: pre-clinical and clinical evidence. Cancer Lett. 2023;570:216307. https://doi.org/10.1016/j.canlet.2023.216307.
Pi M, Kuang H, Yue C, Yang Q, Wu A, Li Y, et al. Targeting metabolism to overcome cancer drug resistance: a promising therapeutic strategy for diffuse large B cell lymphoma. Drug Resist Updat. 2022;61:100822. https://doi.org/10.1016/j.drup.2022.100822.
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. https://doi.org/10.1038/nrd1984.
Gonçalves AC, Richiardone E, Jorge J, Polónia B, Xavier CPR, Salaroglio IC, et al. Impact of cancer metabolism on therapy resistance - clinical implications. Drug Resist Updat. 2021;59:100797. https://doi.org/10.1016/j.drup.2021.100797.
Wang J, Tian T, Li X, Zhang Y. Noncoding RNAs emerging as drugs or drug targets: their Chemical Modification, Bio-conjugation and Intracellular Regulation. Molecules. 2022;27. https://doi.org/10.3390/molecules27196717.
Zahedipour F, Jamialahmadi K, Karimi G. The role of noncoding RNAs and sirtuins in cancer drug resistance. Eur J Pharmacol. 2020;877:173094. https://doi.org/10.1016/j.ejphar.2020.173094.
Cui Q, Wang JQ, Assaraf YG, Ren L, Gupta P, Wei L, et al. Modulating ROS to overcome multidrug resistance in cancer. Drug Resist Updat. 2018;41:1–25. https://doi.org/10.1016/j.drup.2018.11.001.
Kong M, Yu X, Guo W, Guo R. The bidirectional interplay between ncRNAs and methylation modifications in gastrointestinal tumors. Int J Biol Sci. 2023;19:4834–48. https://doi.org/10.7150/ijbs.87028.
Zhang Z, Zhang C, Luo Y, Zhang G, Wu P, Sun N, et al. RNA N(6) -methyladenosine modification in the lethal teamwork of cancer stem cells and the tumor immune microenvironment: current landscape and therapeutic potential. Clin Transl Med. 2021;11:e525. https://doi.org/10.1002/ctm2.525.
Piperi C, Markouli M, Gargalionis AN, Papavassiliou KA, Papavassiliou AG. Deciphering glioma epitranscriptome: focus on RNA modifications. Oncogene. 2023;42:2197–206. https://doi.org/10.1038/s41388-023-02746-y.
Icard P, Shulman S, Farhat D, Steyaert JM, Alifano M, Lincet H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist Updat. 2018;38:1–11. https://doi.org/10.1016/j.drup.2018.03.001.
Wang N, Ma T, Yu B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct Target Ther. 2023;8:69. https://doi.org/10.1038/s41392-023-01341-7.
Li B, Jiang J, Assaraf YG, Xiao H, Chen ZS, Huang C. Surmounting cancer drug resistance: new insights from the perspective of N(6)-methyladenosine RNA modification. Drug Resist Updat. 2020;53:100720. https://doi.org/10.1016/j.drup.2020.100720.
Liu Z, Zou H, Dang Q, Xu H, Liu L, Zhang Y, et al. Biological and pharmacological roles of m(6)a modifications in cancer drug resistance. Mol Cancer. 2022;21:220. https://doi.org/10.1186/s12943-022-01680-z.
Li F, Zheng Z, Chen W, Li D, Zhang H, Zhu Y, et al. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resist Updat. 2023;68:100938. https://doi.org/10.1016/j.drup.2023.100938.
Du B, Shim JS. Targeting epithelial-mesenchymal transition (EMT) to Overcome Drug Resistance in Cancer. Molecules. 2016;21. https://doi.org/10.3390/molecules21070965.
Xie W, Sun H, Li X, Lin F, Wang Z, Wang X. Ovarian cancer: epigenetics, drug resistance, and progression. Cancer Cell Int. 2021;21:434. https://doi.org/10.1186/s12935-021-02136-y.
Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13:877–919. https://doi.org/10.1007/s13238-021-00846-7.
Zhao LY, Song J, Liu Y, Song CX, Yi C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell. 2020;11:792–808. https://doi.org/10.1007/s13238-020-00733-7.
Orsolic I, Carrier A, Esteller M. Genetic and epigenetic defects of the RNA modification machinery in cancer. Trends Genet. 2023;39:74–88. https://doi.org/10.1016/j.tig.2022.10.004.
Zhang Y, Lu L, Li X. Detection technologies for RNA modifications. Exp Mol Med. 2022;54:1601–16. https://doi.org/10.1038/s12276-022-00821-0.
Sağlam B, Akgül B. An overview of current detection methods for RNA methylation. Int J Mol Sci. 2024;25. https://doi.org/10.3390/ijms25063098.
Leger A, Amaral PP, Pandolfini L, Capitanchik C, Capraro F, Miano V, et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat Commun. 2021;12:7198. https://doi.org/10.1038/s41467-021-27393-3.
Ofusa K, Chijimatsu R, Ishii H. Detection techniques for epitranscriptomic marks. Am J Physiol Cell Physiol. 2022;322:C787–93. https://doi.org/10.1152/ajpcell.00460.2021.
Zhang Y, Jiang J, Ma J, Wei Z, Wang Y, Song B, et al. DirectRMDB: a database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Res. 2023;51:D106–16. https://doi.org/10.1093/nar/gkac1061.
Lin S, Kuang M. RNA modification-mediated mRNA translation regulation in liver cancer: mechanisms and clinical perspectives. Nat Rev Gastroenterol Hepatol. 2024;21:267–81. https://doi.org/10.1038/s41575-023-00884-y.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in Gene expression regulation. Cell. 2017;169:1187–200. https://doi.org/10.1016/j.cell.2017.05.045.
Delaunay S, Helm M, Frye M. RNA modifications in physiology and disease: towards clinical applications. Nat Rev Genet. 2024;25:104–22. https://doi.org/10.1038/s41576-023-00645-2.
Li G, Zhu Y, Gu J, Zhang T, Wang F, Huang K, et al. RNA modification patterns based on major RNA modifications define tumor microenvironment characteristics in glioblastoma. Sci Rep. 2022;12:10278. https://doi.org/10.1038/s41598-022-14539-6.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24. https://doi.org/10.1038/s41580-019-0168-5.
Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–26. https://doi.org/10.1038/nrm3785.
Ju G, Lei J, Cai S, Liu S, Yin X, Peng C. The Emerging, Multifaceted Role of WTAP in Cancer and Cancer therapeutics. Cancers (Basel). 2023;15. https://doi.org/10.3390/cancers15113053.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m⁶A RNA methylation. Nat Rev Genet. 2014;15:293–306. https://doi.org/10.1038/nrg3724.
Zhou W, Wang X, Chang J, Cheng C, Miao C. The molecular structure and biological functions of RNA methylation, with special emphasis on the roles of RNA methylation in autoimmune diseases. Crit Rev Clin Lab Sci. 2022;59:203–18. https://doi.org/10.1080/10408363.2021.2002256.
Das AS, Alfonzo JD, Accornero F. The importance of RNA modifications: from cells to muscle physiology. Wiley Interdiscip Rev RNA. 2022;13:e1700. https://doi.org/10.1002/wrna.1700.
Jiapaer Z, Su D, Hua L, Lehmann HI, Gokulnath P, Vulugundam G, et al. Regulation and roles of RNA modifications in aging-related diseases. Aging Cell. 2022;21:e13657. https://doi.org/10.1111/acel.13657.
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12:121. https://doi.org/10.1186/s13045-019-0805-7.
Li B, Qu L, Yang J, RNA-Guided RNA, Modifications. Biogenesis, functions, and applications. Acc Chem Res. 2023;56:3198–210. https://doi.org/10.1021/acs.accounts.3c00474.
Tomikawa C. 7-Methylguanosine modifications in transfer RNA (tRNA). Int J Mol Sci. 2018;19. https://doi.org/10.3390/ijms19124080.
Kumari R, Ranjan P, Suleiman ZG, Goswami SK, Li J, Prasad R, et al. mRNA modifications in cardiovascular biology and disease: with a focus on m6A modification. Cardiovasc Res. 2022;118:1680–92. https://doi.org/10.1093/cvr/cvab160.
Wen T, Li T, Xu Y, Zhang Y, Pan H, Wang Y. The role of m6A epigenetic modifications in tumor coding and non-coding RNA processing. Cell Commun Signal. 2023;21:355. https://doi.org/10.1186/s12964-023-01385-w.
Song J, Yi C. Chemical modifications to RNA: a New Layer of Gene expression regulation. ACS Chem Biol. 2017;12:316–25. https://doi.org/10.1021/acschembio.6b00960.
He Y, Sun MM, Zhang GG, Yang J, Chen KS, Xu WW, et al. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct Target Ther. 2021;6:425. https://doi.org/10.1038/s41392-021-00828-5.
Frezza V, Chellini L, Del Verme A, Paronetto MP. RNA editing in Cancer Progression. Cancers (Basel). 2023;15. https://doi.org/10.3390/cancers15215277.
Jin Y, Fan Z. New insights into the interaction between m6A modification and lncRNA in cancer drug resistance. Cell Prolif. 2024;57:e13578. https://doi.org/10.1111/cpr.13578.
Pomaville MM, He C. Advances in targeting RNA modifications for anticancer therapy. Trends Cancer. 2023;9:528–42. https://doi.org/10.1016/j.trecan.2023.04.003.
Cayir A. RNA modifications as emerging therapeutic targets. Wiley Interdiscip Rev RNA. 2022;13:e1702. https://doi.org/10.1002/wrna.1702.
Mao Z, Wang B, Zhang T, Cui B. The roles of m6A methylation in cervical cancer: functions, molecular mechanisms, and clinical applications. Cell Death Dis. 2023;14:734. https://doi.org/10.1038/s41419-023-06265-2.
Delaunay S, Frye M. RNA modifications regulating cell fate in cancer. Nat Cell Biol. 2019;21:552–9. https://doi.org/10.1038/s41556-019-0319-0.
Gatsiou A, Stellos K. RNA modifications in cardiovascular health and disease. Nat Rev Cardiol. 2023;20:325–46. https://doi.org/10.1038/s41569-022-00804-8.
Han SH, Choe J. Diverse molecular functions of m(6)a mRNA modification in cancer. Exp Mol Med. 2020;52:738–49. https://doi.org/10.1038/s12276-020-0432-y.
Li Y, Su R, Deng X, Chen Y, Chen J. FTO in cancer: functions, molecular mechanisms, and therapeutic implications. Trends Cancer. 2022;8:598–614. https://doi.org/10.1016/j.trecan.2022.02.010.
Lin H, Wang Y, Wang P, Long F, Wang T. Mutual regulation between N6-methyladenosine (m6A) modification and circular RNAs in cancer: impacts on therapeutic resistance. Mol Cancer. 2022;21:148. https://doi.org/10.1186/s12943-022-01620-x.
Jo H, Shim K, Jeoung D. Roles of RNA methylations in Cancer Progression, Autophagy, and Anticancer Drug Resistance. Int J Mol Sci. 2023;24. https://doi.org/10.3390/ijms24044225.
Wang L, Li X, Zhang W, Yang Y, Meng Q, Wang C, et al. miR24-2 promotes malignant progression of Human Liver Cancer Stem cells by enhancing tyrosine kinase Src Epigenetically. Mol Ther. 2020;28:572–86. https://doi.org/10.1016/j.ymthe.2019.10.015.
Zhuang H, Yu B, Tao D, Xu X, Xu Y, Wang J, et al. The role of m6A methylation in therapy resistance in cancer. Mol Cancer. 2023;22:91. https://doi.org/10.1186/s12943-023-01782-2.
Bai Y, Zhao H, Liu H, Wang W, Dong H, Zhao C. RNA methylation, homologous recombination repair and therapeutic resistance. Biomed Pharmacother. 2023;166:115409. https://doi.org/10.1016/j.biopha.2023.115409.
Xu Z, Peng B, Cai Y, Wu G, Huang J, Gao M, et al. N6-methyladenosine RNA modification in cancer therapeutic resistance: current status and perspectives. Biochem Pharmacol. 2020;182:114258. https://doi.org/10.1016/j.bcp.2020.114258.
Luo P, Li S, Long X. N6-methyladenosine RNA modification in PD-1/PD-L1: novel implications for immunotherapy. Biochim Biophys Acta Rev Cancer. 2023;1878:188873. https://doi.org/10.1016/j.bbcan.2023.188873.
Qi YN, Liu Z, Hong LL, Li P, Ling ZQ. Methyltransferase-like proteins in cancer biology and potential therapeutic targeting. J Hematol Oncol. 2023;16:89. https://doi.org/10.1186/s13045-023-01477-7.
Lan Q, Liu PY, Bell JL, Wang JY, Hüttelmaier S, Zhang XD, et al. The emerging roles of RNA m(6)a methylation and demethylation as critical regulators of Tumorigenesis, Drug Sensitivity, and resistance. Cancer Res. 2021;81:3431–40. https://doi.org/10.1158/0008-5472.Can-20-4107.
Vembuli H, Gor R, Ramalingam S, Perales S, Rajasingh J. RNA binding proteins in cancer chemotherapeutic drug resistance. Front Cell Dev Biol. 2024;12:1308102. https://doi.org/10.3389/fcell.2024.1308102.
Chen Z, Hu Y, Jin L, Yang F, Ding H, Zhang L, et al. The emerging role of N6-Methyladenosine RNA methylation as regulators in Cancer Therapy and Drug Resistance. Front Pharmacol. 2022;13:873030. https://doi.org/10.3389/fphar.2022.873030.
Huang G, Ding Q, Xie D, Cai Z, Zhao Z. Technical challenges in defining RNA modifications. Semin Cell Dev Biol. 2022;127:155–65. https://doi.org/10.1016/j.semcdb.2021.11.009.
Ye L, Yao X, Xu B, Chen W, Lou H, Tong X, et al. RNA epigenetic modifications in ovarian cancer: the changes, chances, and challenges. Wiley Interdiscip Rev RNA. 2023;14:e1784. https://doi.org/10.1002/wrna.1784.
Luo M, Yang X, Chen HN, Nice EC, Huang C. Drug resistance in colorectal cancer: an epigenetic overview. Biochim Biophys Acta Rev Cancer. 2021;1876:188623. https://doi.org/10.1016/j.bbcan.2021.188623.
Wang L, Tang Y. N6-methyladenosine (m6A) in cancer stem cell: from molecular mechanisms to therapeutic implications. Biomed Pharmacother. 2023;163:114846. https://doi.org/10.1016/j.biopha.2023.114846.
Liu L, Liang L, Li H, Shao W, Yang C, Lin F, et al. The role of m6A-mediated PD-1/PD-L1 in antitumor immunity. Biochem Pharmacol. 2023;210:115460. https://doi.org/10.1016/j.bcp.2023.115460.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. https://doi.org/10.1016/j.cell.2012.06.013.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74. https://doi.org/10.1038/s41392-020-00450-x.
Covelo-Molares H, Bartosovic M, Vanacova S. RNA methylation in nuclear pre-mRNA processing. Wiley Interdiscip Rev RNA. 2018;9:e1489. https://doi.org/10.1002/wrna.1489.
Xie S, Chen W, Chen K, Chang Y, Yang F, Lin A, et al. Emerging roles of RNA methylation in gastrointestinal cancers. Cancer Cell Int. 2020;20:585. https://doi.org/10.1186/s12935-020-01679-w.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88. https://doi.org/10.1186/s12943-020-01204-7.
Alarcón CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519:482–5. https://doi.org/10.1038/nature14281.
Zhang H, Shi X, Huang T, Zhao X, Chen W, Gu N, et al. Dynamic landscape and evolution of m6A methylation in human. Nucleic Acids Res. 2020;48:6251–64. https://doi.org/10.1093/nar/gkaa347.
Meyer KD, Jaffrey SR. Rethinking m(6)a readers, writers, and Erasers. Annu Rev Cell Dev Biol. 2017;33:319–42. https://doi.org/10.1146/annurev-cellbio-100616-060758.
Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)a mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–4. https://doi.org/10.1038/nature15377.
He PC, He C. M(6) a RNA methylation: from mechanisms to therapeutic potential. Embo j. 2021;40:e105977. https://doi.org/10.15252/embj.2020105977.
Zhang SY, Zhang SW, Liu L, Meng J, Huang Y. m6A-Driver: identifying context-specific mRNA m6A methylation-driven Gene Interaction Networks. PLoS Comput Biol. 2016;12:e1005287. https://doi.org/10.1371/journal.pcbi.1005287.
Shao N, Ye T, Xuan W, Zhang M, Chen Q, Liu J, et al. The effects of N(6)-methyladenosine RNA methylation on the nervous system. Mol Cell Biochem. 2023;478:2657–69. https://doi.org/10.1007/s11010-023-04691-6.
Huang L, Zhu J, Kong W, Li P, Zhu S. Expression and prognostic characteristics of m6A RNA methylation regulators in Colon cancer. Int J Mol Sci. 2021;22. https://doi.org/10.3390/ijms22042134.
Sun HL, Zhu AC, Gao Y, Terajima H, Fei Q, Liu S, et al. Stabilization of ERK-Phosphorylated METTL3 by USP5 increases m(6)a methylation. Mol Cell. 2020;80:633–47. https://doi.org/10.1016/j.molcel.2020.10.026. .e7.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5. https://doi.org/10.1038/nchembio.1432.
Wu Y, Xie L, Wang M, Xiong Q, Guo Y, Liang Y, et al. Mettl3-mediated m(6)a RNA methylation regulates the fate of bone marrow mesenchymal stem cells and osteoporosis. Nat Commun. 2018;9:4772. https://doi.org/10.1038/s41467-018-06898-4.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7. https://doi.org/10.1038/nchembio.687.
Song T, Yang Y, Wei H, Xie X, Lu J, Zeng Q, et al. Zfp217 mediates m6A mRNA methylation to orchestrate transcriptional and post-transcriptional regulation to promote adipogenic differentiation. Nucleic Acids Res. 2019;47:6130–44. https://doi.org/10.1093/nar/gkz312.
Zhou C, She X, Gu C, Hu Y, Ma M, Qiu Q, et al. FTO fuels diabetes-induced vascular endothelial dysfunction associated with inflammation by erasing m6A methylation of TNIP1. J Clin Invest. 2023;133. https://doi.org/10.1172/jci160517.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in Acute myeloid leukemia. Cancer Cell. 2019;35:677–91. https://doi.org/10.1016/j.ccell.2019.03.006. .e10.
Liao S, Sun H, Xu CYTH, Domain. A family of N(6)-methyladenosine (m(6)A) readers. Genomics Proteom Bioinf. 2018;16:99–107. https://doi.org/10.1016/j.gpb.2018.04.002.
Zaccara S, Jaffrey SR. A unified model for the function of YTHDF proteins in regulating m(6)A-Modified mRNA. Cell. 2020;181:1582–95. https://doi.org/10.1016/j.cell.2020.05.012. .e18.
Alarcón CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-Dependent Nuclear RNA Processing events. Cell. 2015;162:1299–308. https://doi.org/10.1016/j.cell.2015.08.011.
Hao W, Chen Z, Tang J, Yang R, Gao WQ, Xu H. hnRNPA2B1 promotes the occurrence and progression of hepatocellular carcinoma by downregulating PCK1 mRNA via a m6A RNA methylation manner. J Transl Med. 2023;21:861. https://doi.org/10.1186/s12967-023-04704-4.
Jiang L, Lin W, Zhang C, Ash PEA, Verma M, Kwan J, et al. Interaction of tau with HNRNPA2B1 and N(6)-methyladenosine RNA mediates the progression of tauopathy. Mol Cell. 2021;81:4209–27. .e12.
Oerum S, Dégut C, Barraud P, Tisné C. m1A post-transcriptional modification in tRNAs. Biomolecules. 2017;7. https://doi.org/10.3390/biom7010020.
Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, et al. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551:251–5. https://doi.org/10.1038/nature24456.
Li J, Zhang H, Wang H. N(1)-methyladenosine modification in cancer biology: current status and future perspectives. Comput Struct Biotechnol J. 2022;20:6578–85. https://doi.org/10.1016/j.csbj.2022.11.045.
Wang Y, Wang J, Li X, Xiong X, Wang J, Zhou Z, et al. N(1)-methyladenosine methylation in tRNA drives liver tumourigenesis by regulating cholesterol metabolism. Nat Commun. 2021;12:6314. https://doi.org/10.1038/s41467-021-26718-6.
Chen C, Ye L. The m1A modification of tRNAs: a translational accelerator of T-cell activation. Cell Mol Immunol. 2022;19:1328–9. https://doi.org/10.1038/s41423-022-00942-6.
Jiang H. Granulation of m1A-modified mRNAs protects their functionality through cellular stress. J Mol Cell Biol. 2021;12:821–2. https://doi.org/10.1093/jmcb/mjaa073.
Wu Y, Chen Z, Xie G, Zhang H, Wang Z, Zhou J, et al. RNA m(1)a methylation regulates glycolysis of cancer cells through modulating ATP5D. Proc Natl Acad Sci U S A. 2022;119:e2119038119. https://doi.org/10.1073/pnas.2119038119.
Li X, Xiong X, Zhang M, Wang K, Chen Y, Zhou J, et al. Base-resolution mapping reveals distinct m(1)a methylome in Nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68:993–e10059. https://doi.org/10.1016/j.molcel.2017.10.019.
Xiong X, Li X, Yi C. N(1)-methyladenosine methylome in messenger RNA and non-coding RNA. Curr Opin Chem Biol. 2018;45:179–86. https://doi.org/10.1016/j.cbpa.2018.06.017.
Qi Z, Zhang C, Jian H, Hou M, Lou Y, Kang Y, et al. N(1)-Methyladenosine modification of mRNA regulates neuronal gene expression and oxygen glucose deprivation/reoxygenation induction. Cell Death Discov. 2023;9:159. https://doi.org/10.1038/s41420-023-01458-2.
Chen Y, Yang S, Peng S, Li W, Wu F, Yao Q, et al. N1-Methyladenosine detection with CRISPR-Cas13a/C2c2. Chem Sci. 2019;10:2975–9. https://doi.org/10.1039/c8sc03408g.
Liu Y, Zhang S, Gao X, Ru Y, Gu X, Hu X. Research progress of N1-methyladenosine RNA modification in cancer. Cell Commun Signal. 2024;22:79. https://doi.org/10.1186/s12964-023-01401-z.
Xie G, Lu Y, He J, Yang X, Zhou J, Yi C, et al. Small molecule-inducible and Photoactivatable Cellular RNA N1-Methyladenosine editing. Angew Chem Int Ed Engl. 2024;63:e202320029. https://doi.org/10.1002/anie.202320029.
Su Z, Monshaugen I, Wilson B, Wang F, Klungland A, Ougland R, et al. TRMT6/61A-dependent base methylation of tRNA-derived fragments regulates gene-silencing activity and the unfolded protein response in bladder cancer. Nat Commun. 2022;13:2165. https://doi.org/10.1038/s41467-022-29790-8.
Ontiveros RJ, Shen H, Stoute J, Yanas A, Cui Y, Zhang Y, et al. Coordination of mRNA and tRNA methylations by TRMT10A. Proc Natl Acad Sci U S A. 2020;117:7782–91. https://doi.org/10.1073/pnas.1913448117.
Vilardo E, Amman F, Toth U, Kotter A, Helm M, Rossmanith W. Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res. 2020;48:6157–69. https://doi.org/10.1093/nar/gkaa353.
Wang M, Zhu Y, Wang C, Fan X, Jiang X, Ebrahimi M, et al. Crystal structure of the two-subunit tRNA m(1)A58 methyltransferase TRM6-TRM61 from Saccharomyces cerevisiae. Sci Rep. 2016;6:32562. https://doi.org/10.1038/srep32562.
Macari F, El-Houfi Y, Boldina G, Xu H, Khoury-Hanna S, Ollier J, et al. TRM6/61 connects PKCα with translational control through tRNAi(Met) stabilization: impact on tumorigenesis. Oncogene. 2016;35:1785–96. https://doi.org/10.1038/onc.2015.244.
Zhang L, Duan HC, Paduch M, Hu J, Zhang C, Mu Y, et al. The molecular basis of human ALKBH3 mediated RNA N(1) -methyladenosine (m(1) A) demethylation. Angew Chem Int Ed Engl. 2024;63:e202313900. https://doi.org/10.1002/anie.202313900.
You XJ, Zhang S, Chen JJ, Tang F, He J, Wang J, et al. Formation and removal of 1,N6-dimethyladenosine in mammalian transfer RNA. Nucleic Acids Res. 2022;50:9858–72. https://doi.org/10.1093/nar/gkac770.
Zhang C, Jia G, Reversible RNA, Modification. N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genomics Proteom Bioinf. 2018;16:155–61. https://doi.org/10.1016/j.gpb.2018.03.003.
Kuang W, Jin H, Yang F, Chen X, Liu J, Li T, et al. ALKBH3-dependent m(1)a demethylation of Aurora A mRNA inhibits ciliogenesis. Cell Discov. 2022;8:25. https://doi.org/10.1038/s41421-022-00385-3.
Gu X, Zhuang A, Yu J, Yang L, Ge S, Ruan J, et al. Histone lactylation-boosted ALKBH3 potentiates tumor progression and diminished promyelocytic leukemia protein nuclear condensates by m1A demethylation of SP100A. Nucleic Acids Res. 2024;52:2273–89. https://doi.org/10.1093/nar/gkad1193.
Wu Y, Jiang D, Zhang H, Yin F, Guo P, Zhang X, et al. N1-Methyladenosine (m1A) regulation Associated with the pathogenesis of abdominal aortic aneurysm through YTHDF3 modulating macrophage polarization. Front Cardiovasc Med. 2022;9:883155. https://doi.org/10.3389/fcvm.2022.883155.
Dai X, Wang T, Gonzalez G, Wang Y. Identification of YTH Domain-containing proteins as the readers for N1-Methyladenosine in RNA. Anal Chem. 2018;90:6380–4. https://doi.org/10.1021/acs.analchem.8b01703.
Li M, Tao Z, Zhao Y, Li L, Zheng J, Li Z, et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J Transl Med. 2022;20:214. https://doi.org/10.1186/s12967-022-03427-2.
Bohnsack KE, Höbartner C, Bohnsack MT. Eukaryotic 5-methylcytosine (m⁵C) RNA methyltransferases: mechanisms, Cellular functions, and links to Disease. Genes (Basel). 2019;10. https://doi.org/10.3390/genes10020102.
Song H, Zhang J, Liu B, Xu J, Cai B, Yang H, et al. Biological roles of RNA m(5)C modification and its implications in Cancer immunotherapy. Biomark Res. 2022;10:15. https://doi.org/10.1186/s40364-022-00362-8.
Chen YS, Yang WL, Zhao YL, Yang YG. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip Rev RNA. 2021;12:e1639. https://doi.org/10.1002/wrna.1639.
Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol. 2019;21:978–90. https://doi.org/10.1038/s41556-019-0361-y.
Zhang T, Zhao F, Li J, Sun X, Zhang X, Wang H, et al. Programmable RNA 5-methylcytosine (m5C) modification of cellular RNAs by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2024;52:2776–91. https://doi.org/10.1093/nar/gkae110.
Ma J, Song B, Wei Z, Huang D, Zhang Y, Su J, et al. m5C-Atlas: a comprehensive database for decoding and annotating the 5-methylcytosine (m5C) epitranscriptome. Nucleic Acids Res. 2022;50:D196–203. https://doi.org/10.1093/nar/gkab1075.
Fang L, Huang H, Lv J, Chen Z, Lu C, Jiang T, et al. m5C-methylated lncRNA NR_033928 promotes gastric cancer proliferation by stabilizing GLS mRNA to promote glutamine metabolism reprogramming. Cell Death Dis. 2023;14:520. https://doi.org/10.1038/s41419-023-06049-8.
Selmi T, Hussain S, Dietmann S, Heiß M, Borland K, Flad S, et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 2021;49:1006–22. https://doi.org/10.1093/nar/gkaa1193.
Zhang H, Zhai X, Liu Y, Xia Z, Xia T, Du G, et al. NOP2-mediated m5C modification of c-Myc in an EIF3A-Dependent manner to reprogram glucose metabolism and promote Hepatocellular Carcinoma Progression. Res (Wash D C). 2023;6:0184. https://doi.org/10.34133/research.0184.
Feng J, Xu T, He M, Li J, Yao P, Ma C, et al. NSUN2-mediated m5C modification of HBV RNA positively regulates HBV replication. PLoS Pathog. 2023;19:e1011808. https://doi.org/10.1371/journal.ppat.1011808.
Shen H, Ontiveros RJ, Owens MC, Liu MY, Ghanty U, Kohli RM, et al. TET-mediated 5-methylcytosine oxidation in tRNA promotes translation. J Biol Chem. 2021;296:100087. https://doi.org/10.1074/jbc.RA120.014226.
López-Moyado IF, Ko M, Hogan PG, Rao A. TET enzymes in the Immune System: from DNA demethylation to Immunotherapy, inflammation, and Cancer. Annu Rev Immunol. 2024;42:455–88. https://doi.org/10.1146/annurev-immunol-080223-044610.
Liu D, Li G, Zuo Y. Function determinants of TET proteins: the arrangements of sequence motifs with specific codes. Brief Bioinform. 2019;20:1826–35. https://doi.org/10.1093/bib/bby053.
Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27:606–25. https://doi.org/10.1038/cr.2017.55.
Zhao Y, Xing C, Peng H. ALYREF (Aly/REF export factor): a potential biomarker for predicting cancer occurrence and therapeutic efficacy. Life Sci. 2024;338:122372. https://doi.org/10.1016/j.lfs.2023.122372.
Li YJ, Guo Q, Ye MS, Cai G, Xiao WF, Deng S, et al. YBX1 promotes type H vessel-dependent bone formation in an m5C-dependent manner. JCI Insight. 2024;9. https://doi.org/10.1172/jci.insight.172345.
Yang H, Wang Y, Xiang Y, Yadav T, Ouyang J, Phoon L, et al. FMRP promotes transcription-coupled homologous recombination via facilitating TET1-mediated m5C RNA modification demethylation. Proc Natl Acad Sci U S A. 2022;119:e2116251119. https://doi.org/10.1073/pnas.2116251119.
Zeng Q, Saghafinia S, Chryplewicz A, Fournier N, Christe L, Xie YQ, et al. Aberrant hyperexpression of the RNA binding protein FMRP in tumors mediates immune evasion. Science. 2022;378:eabl7207. https://doi.org/10.1126/science.abl7207.
Zou Z, Wei J, Chen Y, Kang Y, Shi H, Yang F, et al. FMRP phosphorylation modulates neuronal translation through YTHDF1. Mol Cell. 2023;83:4304–17. https://doi.org/10.1016/j.molcel.2023.10.028. .e8.
Chen Y, Lin H, Miao L, He J. Role of N7-methylguanosine (m(7)G) in cancer. Trends Cell Biol. 2022;32:819–24. https://doi.org/10.1016/j.tcb.2022.07.001.
Han J, Liu Q, Zhou Y, Li D, Wang R. Landscape of internal N7-methylguanosine of long non-coding RNA modifications in resistant acute myeloid leukemia. BMC Genomics. 2023;24:425. https://doi.org/10.1186/s12864-023-09526-8.
Xia X, Wang Y, Zheng JC. Internal m7G methylation: a novel epitranscriptomic contributor in brain development and diseases. Mol Ther Nucleic Acids. 2023;31:295–308. https://doi.org/10.1016/j.omtn.2023.01.003.
Wang H, Chen RB, Zhang SN, Zhang RF. N7-methylguanosine modification of lncRNAs in a rat model of hypoxic pulmonary hypertension: a comprehensive analysis. BMC Genomics. 2022;23:33. https://doi.org/10.1186/s12864-021-08188-8.
Luo Y, Yao Y, Wu P, Zi X, Sun N, He J. The potential role of N(7)-methylguanosine (m7G) in cancer. J Hematol Oncol. 2022;15:63. https://doi.org/10.1186/s13045-022-01285-5.
Chen J, Li K, Chen J, Wang X, Ling R, Cheng M, et al. Aberrant translation regulated by METTL1/WDR4-mediated tRNA N7-methylguanosine modification drives head and neck squamous cell carcinoma progression. Cancer Commun (Lond). 2022;42:223–44. https://doi.org/10.1002/cac2.12273.
García-Vílchez R, Añazco-Guenkova AM, López J, Dietmann S, Tomé M, Jimeno S, et al. N7-methylguanosine methylation of tRNAs regulates survival to stress in cancer. Oncogene. 2023;42:3169–81. https://doi.org/10.1038/s41388-023-02825-0.
Tang Q, Li L, Wang Y, Wu P, Hou X, Ouyang J, et al. RNA modifications in cancer. Br J Cancer. 2023;129:204–21. https://doi.org/10.1038/s41416-023-02275-1.
Pandolfini L, Barbieri I, Bannister AJ, Hendrick A, Andrews B, Webster N, et al. METTL1 promotes let-7 MicroRNA Processing via m7G methylation. Mol Cell. 2019;74:1278–90. .e9.
Liao J, Yi Y, Yue X, Wu X, Zhu M, Chen Y, et al. Methyltransferase 1 is required for nonhomologous end-joining repair and renders hepatocellular carcinoma resistant to radiotherapy. Hepatology. 2023;77:1896–910. https://doi.org/10.1002/hep.32615.
Jin X, Guan Z, Hu N, He C, Yin P, Gong Z, et al. Structural insight into how WDR4 promotes the tRNA N7-methylguanosine methyltransferase activity of METTL1. Cell Discov. 2023;9:65. https://doi.org/10.1038/s41421-023-00562-y.
Cheng W, Gao A, Lin H, Zhang W. Novel roles of METTL1/WDR4 in tumor via m(7)G methylation. Mol Ther Oncolytics. 2022;26:27–34. https://doi.org/10.1016/j.omto.2022.05.009.
Huang M, Long J, Yao Z, Zhao Y, Zhao Y, Liao J, et al. METTL1-Mediated m7G tRNA modification promotes Lenvatinib Resistance in Hepatocellular Carcinoma. Cancer Res. 2023;83:89–102. https://doi.org/10.1158/0008-5472.Can-22-0963.
Zhang M, Kan D, Zhang B, Chen X, Wang C, Chen S, et al. P300/SP1 complex mediating elevated METTL1 regulates CDK14 mRNA stability via internal m7G modification in CRPC. J Exp Clin Cancer Res. 2023;42:215. https://doi.org/10.1186/s13046-023-02777-z.
Haag S, Kretschmer J, Bohnsack MT. WBSCR22/Merm1 is required for late nuclear pre-ribosomal RNA processing and mediates N7-methylation of G1639 in human 18S rRNA. RNA. 2015;21:180–7. https://doi.org/10.1261/rna.047910.114.
Li J, Wang L, Hahn Q, Nowak RP, Viennet T, Orellana EA, et al. Structural basis of regulated m(7)G tRNA modification by METTL1-WDR4. Nature. 2023;613:391–7. https://doi.org/10.1038/s41586-022-05566-4.
Zhang X, Zhu WY, Shen SY, Shen JH, Chen XD. Biological roles of RNA m7G modification and its implications in cancer. Biol Direct. 2023;18:58. https://doi.org/10.1186/s13062-023-00414-5.
Han M, Huang Q, Li X, Chen X, Zhu H, Pan Y, et al. M7G-related tumor immunity: novel insights of RNA modification and potential therapeutic targets. Int J Biol Sci. 2024;20:1238–55. https://doi.org/10.7150/ijbs.90382.
Galloway A, Kaskar A, Ditsova D, Atrih A, Yoshikawa H, Gomez-Moreira C, et al. Upregulation of RNA cap methyltransferase RNMT drives ribosome biogenesis during T cell activation. Nucleic Acids Res. 2021;49:6722–38. https://doi.org/10.1093/nar/gkab465.
Osborne MJ, Volpon L, Memarpoor-Yazdi M, Pillay S, Thambipillai A, Czarnota S, et al. Identification and characterization of the Interaction between the Methyl-7-Guanosine Cap Maturation enzyme RNMT and the Cap-binding protein eIF4E. J Mol Biol. 2022;434:167451. https://doi.org/10.1016/j.jmb.2022.167451.
Culjkovic-Kraljacic B, Skrabanek L, Revuelta MV, Gasiorek J, Cowling VH, Cerchietti L, et al. The eukaryotic translation initiation factor eIF4E elevates steady-state m(7)G capping of coding and noncoding transcripts. Proc Natl Acad Sci U S A. 2020;117:26773–83. https://doi.org/10.1073/pnas.2002360117.
Enroth C, Poulsen LD, Iversen S, Kirpekar F, Albrechtsen A, Vinther J. Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 2019;47:e126. https://doi.org/10.1093/nar/gkz736.
Chen W, Feng P, Song X, Lv H, Lin H. iRNA-m7G: identifying N(7)-methylguanosine sites by Fusing multiple features. Mol Ther Nucleic Acids. 2019;18:269–74. https://doi.org/10.1016/j.omtn.2019.08.022.
Wiener D, Schwartz S. The epitranscriptome beyond m(6)a. Nat Rev Genet. 2021;22:119–31. https://doi.org/10.1038/s41576-020-00295-8.
Cerneckis J, Cui Q, He C, Yi C, Shi Y. Decoding pseudouridine: an emerging target for therapeutic development. Trends Pharmacol Sci. 2022;43:522–35. https://doi.org/10.1016/j.tips.2022.03.008.
Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, León-Ricardo BX, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–62. https://doi.org/10.1016/j.cell.2014.08.028.
Sun M, Fang X, Lin B, Mo J, Wang F, Zhou X, et al. Locus-specific detection of pseudouridine with CRISPR-Cas13a. Chem Commun (Camb). 2024;60:4088–91. https://doi.org/10.1039/d4cc00179f.
Zhang M, Jiang Z, Ma Y, Liu W, Zhuang Y, Lu B, et al. Quantitative profiling of pseudouridylation landscape in the human transcriptome. Nat Chem Biol. 2023;19:1185–95. https://doi.org/10.1038/s41589-023-01304-7.
Dai Q, Zhang LS, Sun HL, Pajdzik K, Yang L, Ye C, et al. Quantitative sequencing using BID-seq uncovers abundant pseudouridines in mammalian mRNA at base resolution. Nat Biotechnol. 2023;41:344–54. https://doi.org/10.1038/s41587-022-01505-w.
Guzzi N, Cieśla M, Ngoc PCT, Lang S, Arora S, Dimitriou M, et al. Pseudouridylation of tRNA-Derived fragments steers Translational Control in Stem cells. Cell. 2018;173:1204–16. .e26.
Burrows CJ, Fleming AM. Bisulfite and Nanopore Sequencing for pseudouridine in RNA. Acc Chem Res. 2023;56:2740–51. https://doi.org/10.1021/acs.accounts.3c00458.
Zhang LS, Ye C, Ju CW, Gao B, Feng X, Sun HL, et al. BID-seq for transcriptome-wide quantitative sequencing of mRNA pseudouridine at base resolution. Nat Protoc. 2024;19:517–38. https://doi.org/10.1038/s41596-023-00917-5.
Ko E, Kim JS, Ju S, Seo HW, Chang Y, Kang JA, et al. Oxidatively modified protein-disulfide isomerase-Associated 3 promotes dyskerin pseudouridine synthase 1-Mediated malignancy and survival of Hepatocellular Carcinoma cells. Hepatology. 2018;68:1851–64. https://doi.org/10.1002/hep.30039.
Penzo M, Rocchi L, Brugiere S, Carnicelli D, Onofrillo C, Couté Y, et al. Human ribosomes from cells with reduced dyskerin levels are intrinsically altered in translation. Faseb j. 2015;29:3472–82. https://doi.org/10.1096/fj.15-270991.
Elsharawy KA, Mohammed OJ, Aleskandarany MA, Hyder A, El-Gammal HL, Abou-Dobara MI, et al. The nucleolar-related protein dyskerin pseudouridine synthase 1 (DKC1) predicts poor prognosis in breast cancer. Br J Cancer. 2020;123:1543–52. https://doi.org/10.1038/s41416-020-01045-7.
Mochizuki Y, He J, Kulkarni S, Bessler M, Mason PJ. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc Natl Acad Sci U S A. 2004;101:10756–61. https://doi.org/10.1073/pnas.0402560101.
Kan G, Wang Z, Sheng C, Chen G, Yao C, Mao Y, et al. Dual inhibition of DKC1 and MEK1/2 synergistically restrains the growth of Colorectal Cancer cells. Adv Sci (Weinh). 2021;8:2004344. https://doi.org/10.1002/advs.202004344.
Nishikura K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol. 2016;17:83–96. https://doi.org/10.1038/nrm.2015.4.
Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, et al. RNA editing with CRISPR-Cas13. Science. 2017;358:1019–27. https://doi.org/10.1126/science.aaq0180.
Vlachogiannis NI, Tual-Chalot S, Zormpas E, Bonini F, Ntouros PA, Pappa M, et al. Adenosine-to-inosine RNA editing contributes to type I interferon responses in systemic sclerosis. J Autoimmun. 2021;125:102755. https://doi.org/10.1016/j.jaut.2021.102755.
Xu X, Wang Y, Liang H. The role of A-to-I RNA editing in cancer development. Curr Opin Genet Dev. 2018;48:51–6. https://doi.org/10.1016/j.gde.2017.10.009.
Silvestris DA, Scopa C, Hanchi S, Locatelli F, Gallo A. De Novo A-to-I RNA editing Discovery in lncRNA. Cancers (Basel). 2020;12. https://doi.org/10.3390/cancers12102959.
Marceca GP, Tomasello L, Distefano R, Acunzo M, Croce CM, Nigita G. Detecting and characterizing A-To-I microRNA editing in Cancer. Cancers (Basel). 2021;13. https://doi.org/10.3390/cancers13071699.
Yuan J, Xu L, Bao HJ, Wang JL, Zhao Y, Chen S. Biological roles of A-to-I editing: implications in innate immunity, cell death, and cancer immunotherapy. J Exp Clin Cancer Res. 2023;42:149. https://doi.org/10.1186/s13046-023-02727-9.
Walkley CR, Li JB. Rewriting the transcriptome: adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017;18:205. https://doi.org/10.1186/s13059-017-1347-3.
Slotkin W, Nishikura K. Adenosine-to-inosine RNA editing and human disease. Genome Med. 2013;5:105. https://doi.org/10.1186/gm508.
de Reuver R, Verdonck S, Dierick E, Nemegeer J, Hessmann E, Ahmad S, et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature. 2022;607:784–9. https://doi.org/10.1038/s41586-022-04974-w.
Datta R, Adamska JZ, Bhate A, Li JB, A-to-I. RNA editing by ADAR and its therapeutic applications: from viral infections to cancer immunotherapy. Wiley Interdiscip Rev RNA. 2023;e1817. https://doi.org/10.1002/wrna.1817.
Yablonovitch AL, Deng P, Jacobson D, Li JB. The evolution and adaptation of A-to-I RNA editing. PLoS Genet. 2017;13:e1007064. https://doi.org/10.1371/journal.pgen.1007064.
Ganem NS, Ben-Asher N, Lamm AT. In cancer, A-to-I RNA editing can be the driver, the passenger, or the mechanic. Drug Resist Updat. 2017;32:16–22. https://doi.org/10.1016/j.drup.2017.09.001.
Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349:1115–20. https://doi.org/10.1126/science.aac7049.
Kim DD, Kim TT, Walsh T, Kobayashi Y, Matise TC, Buyske S, et al. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–25. https://doi.org/10.1101/gr.2855504.
Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. https://doi.org/10.1371/journal.pbio.0020391.
Crews LA, Ma W, Ladel L, Pham J, Balaian L, Steel SK, et al. Reversal of malignant ADAR1 splice isoform switching with Rebecsinib. Cell Stem Cell. 2023;30:250–63. https://doi.org/10.1016/j.stem.2023.01.008. .e6.
Liao Y, Jung SH, Kim T, A-to-I. RNA editing as a tuner of noncoding RNAs in cancer. Cancer Lett. 2020;494:88–93. https://doi.org/10.1016/j.canlet.2020.08.004.
Cui L, Ma R, Cai J, Guo C, Chen Z, Yao L, et al. RNA modifications: importance in immune cell biology and related diseases. Signal Transduct Target Ther. 2022;7:334. https://doi.org/10.1038/s41392-022-01175-9.
Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–22. https://doi.org/10.1038/s41568-020-0253-2.
Teng PC, Liang Y, Yarmishyn AA, Hsiao YJ, Lin TY, Lin TW, et al. RNA modifications and epigenetics in modulation of Lung Cancer and Pulmonary diseases. Int J Mol Sci. 2021;22. https://doi.org/10.3390/ijms221910592.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68. https://doi.org/10.1038/nbt.1685.
Chen XY, Zhang J, Zhu JS. The role of m(6)a RNA methylation in human cancer. Mol Cancer. 2019;18:103. https://doi.org/10.1186/s12943-019-1033-z.
Wang Y, Wang Y, Patel H, Chen J, Wang J, Chen ZS, et al. Epigenetic modification of m(6)a regulator proteins in cancer. Mol Cancer. 2023;22:102. https://doi.org/10.1186/s12943-023-01810-1.
Qing Y, Su R, Chen J. RNA modifications in hematopoietic malignancies: a new research frontier. Blood. 2021;138:637–48. https://doi.org/10.1182/blood.2019004263.
Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020;474:127–37. https://doi.org/10.1016/j.canlet.2020.01.021.
Wu Y, Zhan S, Xu Y, Gao X. RNA modifications in cardiovascular diseases, the potential therapeutic targets. Life Sci. 2021;278:119565. https://doi.org/10.1016/j.lfs.2021.119565.
Wang L, Yang Q, Zhou Q, Fang F, Lei K, Liu Z, et al. METTL3-m(6)A-EGFR-axis drives lenvatinib resistance in hepatocellular carcinoma. Cancer Lett. 2023;559:216122. https://doi.org/10.1016/j.canlet.2023.216122.
Genois MM, Paquet ER, Laffitte MC, Maity R, Rodrigue A, Ouellette M, et al. DNA repair pathways in trypanosomatids: from DNA repair to drug resistance. Microbiol Mol Biol Rev. 2014;78:40–73. https://doi.org/10.1128/mmbr.00045-13.
Sethy C, Kundu CN. 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: implication of DNA repair inhibition. Biomed Pharmacother. 2021;137:111285. https://doi.org/10.1016/j.biopha.2021.111285.
Yang B, Wang JQ, Tan Y, Yuan R, Chen ZS, Zou C. RNA methylation and cancer treatment. Pharmacol Res. 2021;174:105937. https://doi.org/10.1016/j.phrs.2021.105937.
Tang B, Yang Y, Kang M, Wang Y, Wang Y, Bi Y, et al. M(6)a demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating wnt signaling. Mol Cancer. 2020;19:3. https://doi.org/10.1186/s12943-019-1128-6.
Zhang Y, Liu X, Wang Y, Lai S, Wang Z, Yang Y, et al. The m(6)a demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Mol Cancer. 2022;21:174. https://doi.org/10.1186/s12943-022-01647-0.
Wang ZW, Pan JJ, Hu JF, Zhang JQ, Huang L, Huang Y, et al. SRSF3-mediated regulation of N6-methyladenosine modification-related lncRNA ANRIL splicing promotes resistance of pancreatic cancer to gemcitabine. Cell Rep. 2022;39:110813. https://doi.org/10.1016/j.celrep.2022.110813.
Zhu L, Li B, Li R, Hu L, Zhang Y, Zhang Z, et al. METTL3 suppresses pancreatic ductal adenocarcinoma progression through activating endogenous dsRNA-induced anti-tumor immunity. Cell Oncol (Dordr). 2023;46:1529–41. https://doi.org/10.1007/s13402-023-00829-2.
Liu X, Su K, Sun X, Jiang Y, Wang L, Hu C, et al. Section 62 promotes stemness and chemoresistance of human colorectal cancer through activating Wnt/β-catenin pathway. J Exp Clin Cancer Res. 2021;40:132. https://doi.org/10.1186/s13046-021-01934-6.
Pan S, Deng Y, Fu J, Zhang Y, Zhang Z, Qin X. N6–methyladenosine upregulates miR–181d–5p in exosomes derived from cancer–associated fibroblasts to inhibit 5–FU sensitivity by targeting NCALD in colorectal cancer. Int J Oncol. 2022;60. https://doi.org/10.3892/ijo.2022.5304.
Lin Z, Wan AH, Sun L, Liang H, Niu Y, Deng Y, et al. N6-methyladenosine demethylase FTO enhances chemo-resistance in colorectal cancer through SIVA1-mediated apoptosis. Mol Ther. 2023;31:517–34. https://doi.org/10.1016/j.ymthe.2022.10.012.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. M(6) a RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. Embo j. 2020;39:e104514. https://doi.org/10.15252/embj.2020104514.
Liu Y, Yang C, Zhao Y, Chi Q, Wang Z, Sun B. Overexpressed methyltransferase-like 1 (METTL1) increased chemosensitivity of colon cancer cells to cisplatin by regulating miR-149-3p/S100A4/p53 axis. Aging. 2019;11:12328–44. https://doi.org/10.18632/aging.102575.
Wang J, Zhang J, Liu H, Meng L, Gao X, Zhao Y, et al. N6-methyladenosine reader hnRNPA2B1 recognizes and stabilizes NEAT1 to confer chemoresistance in gastric cancer. Cancer Commun (Lond). 2024;44:469–90. https://doi.org/10.1002/cac2.12534.
Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019;10:383. https://doi.org/10.1038/s41419-019-1585-2.
Zhu Y, Zhou B, Hu X, Ying S, Zhou Q, Xu W, et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin Transl Med. 2022;12:e703. https://doi.org/10.1002/ctm2.703.
Zhou T, Li S, Xiang D, Liu J, Sun W, Cui X, et al. m6A RNA methylation-mediated HNF3γ reduction renders hepatocellular carcinoma dedifferentiation and sorafenib resistance. Signal Transduct Target Ther. 2020;5:296. https://doi.org/10.1038/s41392-020-00299-0.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m(6) a methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. Embo j. 2020;39:e103181. https://doi.org/10.15252/embj.2019103181.
Han WY, Wang J, Zhao J, Zheng YM, Chai XQ, Gao C, et al. WDR4/TRIM28 is a novel molecular target linked to lenvatinib resistance that helps retain the stem characteristics in hepatocellular carcinomas. Cancer Lett. 2023;568:216259. https://doi.org/10.1016/j.canlet.2023.216259.
Li E, Xia M, Du Y, Long K, Ji F, Pan F, et al. METTL3 promotes homologous recombination repair and modulates chemotherapeutic response in breast cancer by regulating the EGF/RAD51 axis. Elife. 2022;11. https://doi.org/10.7554/eLife.75231.
Pan X, Hong X, Li S, Meng P, Xiao F. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp Mol Med. 2021;53:91–102. https://doi.org/10.1038/s12276-020-00510-w.
Liu X, Li P, Huang Y, Li H, Liu X, Du Y, et al. M(6)a demethylase ALKBH5 regulates FOXO1 mRNA stability and chemoresistance in triple-negative breast cancer. Redox Biol. 2024;69:102993. https://doi.org/10.1016/j.redox.2023.102993.
Luo F, Zhang M, Sun B, Xu C, Yang Y, Zhang Y, et al. LINC00115 promotes chemoresistant breast cancer stem-like cell stemness and metastasis through SETDB1/PLK3/HIF1α signaling. Mol Cancer. 2024;23:60. https://doi.org/10.1186/s12943-024-01975-3.
Hao L, Wang JM, Liu BQ, Yan J, Li C, Jiang JY, et al. m6A-YTHDF1-mediated TRIM29 upregulation facilitates the stem cell-like phenotype of cisplatin-resistant ovarian cancer cells. Biochim Biophys Acta Mol Cell Res. 2021;1868:118878. https://doi.org/10.1016/j.bbamcr.2020.118878.
Nie S, Zhang L, Liu J, Wan Y, Jiang Y, Yang J, et al. ALKBH5-HOXA10 loop-mediated JAK2 m6A demethylation and cisplatin resistance in epithelial ovarian cancer. J Exp Clin Cancer Res. 2021;40:284. https://doi.org/10.1186/s13046-021-02088-1.
Lin X, Wang F, Chen J, Liu J, Lin YB, Li L, et al. N(6)-methyladenosine modification of CENPK mRNA by ZC3H13 promotes cervical cancer stemness and chemoresistance. Mil Med Res. 2022;9:19. https://doi.org/10.1186/s40779-022-00378-z.
Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J, et al. ALKBH5 inhibited cell proliferation and sensitized bladder Cancer cells to cisplatin by m6A-CK2α-Mediated glycolysis. Mol Ther Nucleic Acids. 2021;23:27–41. https://doi.org/10.1016/j.omtn.2020.10.031.
Wei W, Sun J, Zhang H, Xiao X, Huang C, Wang L, et al. Circ0008399 Interaction with WTAP Promotes Assembly and Activity of the m(6)a methyltransferase complex and promotes cisplatin resistance in bladder Cancer. Cancer Res. 2021;81:6142–56. https://doi.org/10.1158/0008-5472.Can-21-1518.
Xie R, Cheng L, Huang M, Huang L, Chen Z, Zhang Q, et al. NAT10 drives cisplatin chemoresistance by enhancing ac4C-Associated DNA repair in bladder Cancer. Cancer Res. 2023;83:1666–83. https://doi.org/10.1158/0008-5472.Can-22-2233.
Wang H, Liu J, Zhu X, Yang B, He Z, Yao X. AZGP1P2/UBA1/RBM15 Cascade mediates the fate determinations of prostate Cancer stem cells and promotes therapeutic effect of Docetaxel in Castration-resistant prostate Cancer via TPM1 m6A modification. Res (Wash D C). 2023;6:0252. https://doi.org/10.34133/research.0252.
Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia. 2020;34:1354–63. https://doi.org/10.1038/s41375-019-0656-9.
Li M, Ye J, Xia Y, Li M, Li G, Hu X, et al. METTL3 mediates chemoresistance by enhancing AML homing and engraftment via ITGA4. Leukemia. 2022;36:2586–95. https://doi.org/10.1038/s41375-022-01696-w.
Fu J, Si L, Zhou Y, Li D, Wang R. Distinct N7-methylguanosine profiles of circular RNAs in drug-resistant acute myeloid leukemia. Sci Rep. 2023;13:14704. https://doi.org/10.1038/s41598-023-41974-w.
Cheng JX, Chen L, Li Y, Cloe A, Yue M, Wei J, et al. RNA cytosine methylation and methyltransferases mediate chromatin organization and 5-azacytidine response and resistance in leukaemia. Nat Commun. 2018;9:1163. https://doi.org/10.1038/s41467-018-03513-4.
Lai X, Wei J, Gu XZ, Yao XM, Zhang DS, Li F, et al. Dysregulation of LINC00470 and METTL3 promotes chemoresistance and suppresses autophagy of chronic myelocytic leukaemia cells. J Cell Mol Med. 2021;25:4248–59. https://doi.org/10.1111/jcmm.16478.
Wang Z, He J, Bach DH, Huang YH, Li Z, Liu H, et al. Induction of m(6)a methylation in adipocyte exosomal LncRNAs mediates myeloma drug resistance. J Exp Clin Cancer Res. 2022;41:4. https://doi.org/10.1186/s13046-021-02209-w.
Chen B, Huang Y, He S, Yu P, Wu L, Peng H. N(6)-methyladenosine modification in 18S rRNA promotes tumorigenesis and chemoresistance via HSF4b/HSP90B1/mutant p53 axis. Cell Chem Biol. 2023;30:144–58. https://doi.org/10.1016/j.chembiol.2023.01.006. .e10.
Chen B, Jiang W, Huang Y, Zhang J, Yu P, Wu L, et al. N(7)-methylguanosine tRNA modification promotes tumorigenesis and chemoresistance through WNT/β-catenin pathway in nasopharyngeal carcinoma. Oncogene. 2022;41:2239–53. https://doi.org/10.1038/s41388-022-02250-9.
Wang Z, Yu P, Zou Y, Ma J, Han H, Wei W, et al. METTL1/WDR4-mediated tRNA m(7)G modification and mRNA translation control promote oncogenesis and doxorubicin resistance. Oncogene. 2023;42:1900–12. https://doi.org/10.1038/s41388-023-02695-6.
Shao D, Liu C, Wang Y, Lin J, Cheng X, Han P, et al. DNMT1 determines osteosarcoma cell resistance to apoptosis by associatively modulating DNA and mRNA cytosine-5 methylation. Faseb j. 2023;37:e23284. https://doi.org/10.1096/fj.202301306R.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28:1062–76. https://doi.org/10.1038/s41422-018-0097-4.
Ding N, You A, Tian W, Gu L, Deng D. Chidamide increases the sensitivity of non-small cell Lung Cancer to Crizotinib by decreasing c-MET mRNA methylation. Int J Biol Sci. 2020;16:2595–611. https://doi.org/10.7150/ijbs.45886.
Liu S, Li Q, Li G, Zhang Q, Zhuo L, Han X, et al. The mechanism of m(6)a methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by β-elemene. Cell Death Dis. 2020;11:969. https://doi.org/10.1038/s41419-020-03148-8.
Sun Y, Shen W, Hu S, Lyu Q, Wang Q, Wei T, et al. METTL3 promotes chemoresistance in small cell lung cancer by inducing mitophagy. J Exp Clin Cancer Res. 2023;42:65. https://doi.org/10.1186/s13046-023-02638-9.
Wang Y, Wei J, Feng L, Li O, Huang L, Zhou S, et al. Aberrant m5C hypermethylation mediates intrinsic resistance to gefitinib through NSUN2/YBX1/QSOX1 axis in EGFR-mutant non-small-cell lung cancer. Mol Cancer. 2023;22:81. https://doi.org/10.1186/s12943-023-01780-4.
Romano G, Le P, Nigita G, Saviana M, Micalo L, Lovat F, et al. A-to-I edited mir-411-5p targets MET and promotes TKI response in NSCLC-resistant cells. Oncogene. 2023;42:1597–606. https://doi.org/10.1038/s41388-023-02673-y.
Zhang H, Wang SQ, Wang L, Lin H, Zhu JB, Chen R, et al. m6A methyltransferase METTL3-induced lncRNA SNHG17 promotes lung adenocarcinoma gefitinib resistance by epigenetically repressing LATS2 expression. Cell Death Dis. 2022;13:657. https://doi.org/10.1038/s41419-022-05050-x.
Song H, Liu D, Wang L, Liu K, Chen C, Wang L, et al. Methyltransferase like 7B is a potential therapeutic target for reversing EGFR-TKIs resistance in lung adenocarcinoma. Mol Cancer. 2022;21:43. https://doi.org/10.1186/s12943-022-01519-7.
Li K, Peng ZY, Gao S, Wang QS, Wang R, Li X, et al. M6A associated TSUC7 inhibition contributed to Erlotinib resistance in lung adenocarcinoma through a notch signaling activation dependent way. J Exp Clin Cancer Res. 2021;40:325. https://doi.org/10.1186/s13046-021-02137-9.
Bhattarai PY, Kim G, Poudel M, Lim SC, Choi HS. METTL3 induces PLX4032 resistance in melanoma by promoting m(6)A-dependent EGFR translation. Cancer Lett. 2021;522:44–56. https://doi.org/10.1016/j.canlet.2021.09.015.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, et al. Anti-tumour immunity controlled through mRNA m(6)a methylation and YTHDF1 in dendritic cells. Nature. 2019;566:270–4. https://doi.org/10.1038/s41586-019-0916-x.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, et al. M(6)a mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782. https://doi.org/10.1038/s41467-019-10669-0.
Chen T, Xu ZG, Luo J, Manne RK, Wang Z, Hsu CC, et al. NSUN2 is a glucose sensor suppressing cGAS/STING to maintain tumorigenesis and immunotherapy resistance. Cell Metab. 2023;35:1782–98. .e8.
Fox M, Roberts JJ. Drug resistance and DNA repair. Cancer Metastasis Rev. 1987;6:261–81. https://doi.org/10.1007/bf00144267.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, et al. N(6)-methyladenosine modulates Messenger RNA translation efficiency. Cell. 2015;161:1388–99. https://doi.org/10.1016/j.cell.2015.05.014.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31–42. https://doi.org/10.1038/nrm.2016.132.
Mendel M, Delaney K, Pandey RR, Chen KM, Wenda JM, Vågbø CB, et al. Splice site m(6)a methylation prevents binding of U2AF35 to inhibit RNA splicing. Cell. 2021;184:3125–e4225. https://doi.org/10.1016/j.cell.2021.03.062.
Sciarrillo R, Wojtuszkiewicz A, Assaraf YG, Jansen G, Kaspers GJL, Giovannetti E, et al. The role of alternative splicing in cancer: from oncogenesis to drug resistance. Drug Resist Updat. 2020;53:100728. https://doi.org/10.1016/j.drup.2020.100728.
Feyzizadeh M, Barfar A, Nouri Z, Sarfraz M, Zakeri-Milani P, Valizadeh H. Overcoming multidrug resistance through targeting ABC transporters: lessons for drug discovery. Expert Opin Drug Discov. 2022;17:1013–27. https://doi.org/10.1080/17460441.2022.2112666.
Xu K, Zhang Q, Chen M, Li B, Wang N, Li C, et al. N(6)-methyladenosine modification regulates imatinib resistance of gastrointestinal stromal tumor by enhancing the expression of multidrug transporter MRP1. Cancer Lett. 2022;530:85–99. https://doi.org/10.1016/j.canlet.2022.01.008.
Li Z, Liu X, Wang L, Zhao H, Wang S, Yu G, et al. Integrated analysis of single-cell RNA-seq and bulk RNA-seq reveals RNA N6-methyladenosine modification associated with prognosis and drug resistance in acute myeloid leukemia. Front Immunol. 2023;14:1281687. https://doi.org/10.3389/fimmu.2023.1281687.
Guo C, Liu J, Zhou Q, Song J, Zhang Z, Li Z, et al. Exosomal noncoding RNAs and Tumor Drug Resistance. Cancer Res. 2020;80:4307–13. https://doi.org/10.1158/0008-5472.Can-20-0032.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14. https://doi.org/10.1186/s12943-022-01500-4.
Lin Z, Li J, Zhang J, Feng W, Lu J, Ma X, et al. Metabolic reprogramming driven by IGF2BP3 promotes Acquired Resistance to EGFR inhibitors in Non-small Cell Lung Cancer. Cancer Res. 2023;83:2187–207. https://doi.org/10.1158/0008-5472.Can-22-3059.
Han X, Wang L, Han Q. Advances in the role of m(6)a RNA modification in cancer metabolic reprogramming. Cell Biosci. 2020;10:117. https://doi.org/10.1186/s13578-020-00479-z.
Gao J, Fang Y, Chen J, Tang Z, Tian M, Jiang X, et al. Methyltransferase like 3 inhibition limits intrahepatic cholangiocarcinoma metabolic reprogramming and potentiates the efficacy of chemotherapy. Oncogene. 2023;42:2507–20. https://doi.org/10.1038/s41388-023-02760-0.
Becker LM, O’Connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of Cancer-Associated fibroblasts deregulates glucose metabolism and facilitates progression of breast Cancer. Cell Rep. 2020;31:107701. https://doi.org/10.1016/j.celrep.2020.107701.
Tan YT, Lin JF, Li T, Li JJ, Xu RH, Ju HQ. LncRNA-mediated posttranslational modifications and reprogramming of energy metabolism in cancer. Cancer Commun (Lond). 2021;41:109–20. https://doi.org/10.1002/cac2.12108.
Gu J, Cao H, Chen X, Zhang XD, Thorne RF, Liu X. RNA m6A modifications regulate crosstalk between tumor metabolism and immunity. Wiley Interdiscip Rev RNA. 2024;15:e1829. https://doi.org/10.1002/wrna.1829.
Chen H, Zhang X, Su H, Zeng J, Chan H, Li Q, et al. Immune dysregulation and RNA N6-methyladenosine modification in sepsis. Wiley Interdiscip Rev RNA. 2023;14:e1764. https://doi.org/10.1002/wrna.1764.
Lou X, Wang JJ, Wei YQ, Sun JJ. Emerging role of RNA modification N6-methyladenosine in immune evasion. Cell Death Dis. 2021;12:300. https://doi.org/10.1038/s41419-021-03585-z.
Li T, Tan YT, Chen YX, Zheng XJ, Wang W, Liao K, et al. Methionine deficiency facilitates antitumour immunity by altering m(6)a methylation of immune checkpoint transcripts. Gut. 2023;72:501–11. https://doi.org/10.1136/gutjnl-2022-326928.
Lu D, Lu J, Liu Q, Zhang Q. Emerging role of the RNA-editing enzyme ADAR1 in stem cell fate and function. Biomark Res. 2023;11:61. https://doi.org/10.1186/s40364-023-00503-7.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses Cancer Stem Cell maintenance and Immune Evasion. Cancer Cell. 2020;38:79–e9611. https://doi.org/10.1016/j.ccell.2020.04.017.
Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM. M(6)A-RNA demethylase FTO inhibitors impair Self-Renewal in Glioblastoma Stem cells. ACS Chem Biol. 2021;16:324–33. https://doi.org/10.1021/acschembio.0c00841.
Zhan L, Zhang J, Zhang JH, Liu XJ, Guo B, Chen JH, et al. METTL3 facilitates immunosurveillance by inhibiting YTHDF2-mediated NLRC5 mRNA degradation in endometrial cancer. Biomark Res. 2023;11:43. https://doi.org/10.1186/s40364-023-00479-4.
Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, et al. 5’-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc Natl Acad Sci U S A. 2009;106:12067–72. https://doi.org/10.1073/pnas.0900971106.
Anchisi S, Guerra J, Garcin D. RIG-I ATPase activity and discrimination of self-RNA versus non-self-RNA. mBio. 2015;6:e02349. https://doi.org/10.1128/mBio.02349-14.
Manivannan P, Siddiqui MA, Malathi K. RNase L amplifies Interferon Signaling by inducing protein kinase R-Mediated antiviral stress granules. J Virol. 2020;94. https://doi.org/10.1128/jvi.00205-20.
Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, et al. Human ADAR1 prevents endogenous RNA from triggering Translational Shutdown. Cell. 2018;172:811–24. https://doi.org/10.1016/j.cell.2017.12.038. .e14.
de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, et al. ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep. 2021;36:109500. https://doi.org/10.1016/j.celrep.2021.109500.
Liddicoat BJ, Chalk AM, Walkley CR. ADAR1, inosine and the immune sensing system: distinguishing self from non-self. Wiley Interdiscip Rev RNA. 2016;7:157–72. https://doi.org/10.1002/wrna.1322.
Nakahama T, Kato Y, Kim JI, Vongpipatana T, Suzuki Y, Walkley CR, et al. ADAR1-mediated RNA editing is required for thymic self-tolerance and inhibition of autoimmunity. EMBO Rep. 2018;19. https://doi.org/10.15252/embr.201846303.
Jhunjhunwala S, Hammer C, Delamarre L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nat Rev Cancer. 2021;21:298–312. https://doi.org/10.1038/s41568-021-00339-z.
Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer. 2021;20:105. https://doi.org/10.1186/s12943-021-01398-4.
Lu M, Zhang Z, Xue M, Zhao BS, Harder O, Li A, et al. N(6)-methyladenosine modification enables viral RNA to escape recognition by RNA sensor RIG-I. Nat Microbiol. 2020;5:584–98. https://doi.org/10.1038/s41564-019-0653-9.
Wang Y, Jin P, Wang X. N(6)-methyladenosine regulator YTHDF1 represses the CD8 + T cell-mediated antitumor immunity and ferroptosis in prostate cancer via m(6)A/PD-L1 manner. Apoptosis. 2024;29:142–53. https://doi.org/10.1007/s10495-023-01885-7.
Xu Y, Gao Z, Hu R, Wang Y, Wang Y, Su Z, et al. PD-L2 glycosylation promotes immune evasion and predicts anti-EGFR efficacy. J Immunother Cancer. 2021;9. https://doi.org/10.1136/jitc-2021-002699.
Ni Z, Sun P, Zheng J, Wu M, Yang C, Cheng M, et al. JNK Signaling promotes bladder Cancer Immune escape by regulating METTL3-Mediated m6A modification of PD-L1 mRNA. Cancer Res. 2022;82:1789–802. https://doi.org/10.1158/0008-5472.Can-21-1323.
Qiu X, Yang S, Wang S, Wu J, Zheng B, Wang K, et al. M(6)a demethylase ALKBH5 regulates PD-L1 expression and Tumor Immunoenvironment in Intrahepatic Cholangiocarcinoma. Cancer Res. 2021;81:4778–93. https://doi.org/10.1158/0008-5472.Can-21-0468.
Wang H, Tang A, Cui Y, Gong H, Li H. LRPPRC facilitates tumor progression and immune evasion through upregulation of m(6)a modification of PD-L1 mRNA in hepatocellular carcinoma. Front Immunol. 2023;14:1144774. https://doi.org/10.3389/fimmu.2023.1144774.
Ouyang D, Hong T, Fu M, Li Y, Zeng L, Chen Q, et al. METTL3 depletion contributes to tumour progression and drug resistance via N6 methyladenosine-dependent mechanism in HR + HER2-breast cancer. Breast Cancer Res. 2023;25:19. https://doi.org/10.1186/s13058-022-01598-w.
Sun K, Chen L, Li Y, Huang B, Yan Q, Wu C, et al. METTL14-dependent maturation of pri-miR-17 regulates mitochondrial homeostasis and induces chemoresistance in colorectal cancer. Cell Death Dis. 2023;14:148. https://doi.org/10.1038/s41419-023-05670-x.
Ippolito MR, Martis V, Martin S, Tijhuis AE, Hong C, Wardenaar R, et al. Gene copy-number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev Cell. 2021;56:2440–54. .e6.
Johnson ML, Patel JD. Chemotherapy and targeted therapeutics as maintenance of response in advanced non-small cell lung cancer. Semin Oncol. 2014;41:93–100. https://doi.org/10.1053/j.seminoncol.2013.12.007.
Chen NN, Ma XD, Miao Z, Zhang XM, Han BY, Almaamari AA, et al. Doxorubicin resistance in breast cancer is mediated via the activation of FABP5/PPARγ and CaMKII signaling pathway. Front Pharmacol. 2023;14:1150861. https://doi.org/10.3389/fphar.2023.1150861.
Rotte A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J Exp Clin Cancer Res. 2019;38:255. https://doi.org/10.1186/s13046-019-1259-z.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18:155. https://doi.org/10.1186/s12943-019-1091-2.
Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci U S A. 2010;107:4275–80. https://doi.org/10.1073/pnas.0915174107.
Zafar A, Wang W, Liu G, Wang X, Xian W, McKeon F, et al. Molecular targeting therapies for neuroblastoma: Progress and challenges. Med Res Rev. 2021;41:961–1021. https://doi.org/10.1002/med.21750.
Fang Y, Wang S, Han S, Zhao Y, Yu C, Liu H, et al. Targeted protein degrader development for cancer: advances, challenges, and opportunities. Trends Pharmacol Sci. 2023;44:303–17. https://doi.org/10.1016/j.tips.2023.03.003.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ Jr., Wu YL, et al. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311. https://doi.org/10.1016/s0140-6736(16)30958-8.
Shariati M, Meric-Bernstam F. Targeting AKT for cancer therapy. Expert Opin Investig Drugs. 2019;28:977–88. https://doi.org/10.1080/13543784.2019.1676726.
Wang Y, Deng B. Hepatocellular carcinoma: molecular mechanism, targeted therapy, and biomarkers. Cancer Metastasis Rev. 2023;42:629–52. https://doi.org/10.1007/s10555-023-10084-4.
Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, et al. Cancer Stem cells-origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol. 2020;11:1280. https://doi.org/10.3389/fimmu.2020.01280.
Zeng X, Liu C, Yao J, Wan H, Wan G, Li Y, et al. Breast cancer stem cells, heterogeneity, targeting therapies and therapeutic implications. Pharmacol Res. 2021;163:105320. https://doi.org/10.1016/j.phrs.2020.105320.
Frank MH, Wilson BJ, Gold JS, Frank NY. Clinical implications of Colorectal Cancer Stem cells in the age of single-cell omics and targeted therapies. Gastroenterology. 2021;160:1947–60. https://doi.org/10.1053/j.gastro.2020.12.080.
Olivares-Urbano MA, Griñán-Lisón C, Marchal JA, Núñez MI. CSC Radioresistance: a therapeutic challenge to improve Radiotherapy Effectiveness in Cancer. Cells. 2020;9. https://doi.org/10.3390/cells9071651.
Yu H, Liu J, Bu X, Ma Z, Yao Y, Li J, et al. Targeting METTL3 reprograms the tumor microenvironment to improve cancer immunotherapy. Cell Chem Biol. 2024;31:776–91. https://doi.org/10.1016/j.chembiol.2023.09.001. .e7.
Sun Z, Mai H, Xue C, Fan Z, Li J, Chen H, et al. Hsa-LINC02418/mmu-4930573I07Rik regulated by METTL3 dictates anti-PD-L1 immunotherapeutic efficacy via enhancement of Trim21-mediated PD-L1 ubiquitination. J Immunother Cancer. 2023;11. https://doi.org/10.1136/jitc-2023-007415.
Ouyang L, Sun MM, Zhou PS, Ren YW, Liu XY, Wei WY, et al. LncRNA FOXD1-AS1 regulates pancreatic cancer stem cell properties and 5-FU resistance by regulating the miR-570-3p/SPP1 axis as a ceRNA. Cancer Cell Int. 2024;24:4. https://doi.org/10.1186/s12935-023-03181-5.
Chan S, Friedrichs K, Noel D, Pintér T, Van Belle S, Vorobiof D, et al. Prospective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J Clin Oncol. 1999;17:2341–54. https://doi.org/10.1200/jco.1999.17.8.2341.
Alba E, Martín M, Ramos M, Adrover E, Balil A, Jara C, et al. Multicenter randomized trial comparing sequential with concomitant administration of doxorubicin and docetaxel as first-line treatment of metastatic breast cancer: a Spanish Breast Cancer Research Group (GEICAM-9903) phase III study. J Clin Oncol. 2004;22:2587–93. https://doi.org/10.1200/jco.2004.08.125.
Fisher B, Bryant J, Wolmark N, Mamounas E, Brown A, Fisher ER, et al. Effect of preoperative chemotherapy on the outcome of women with operable breast cancer. J Clin Oncol. 1998;16:2672–85. https://doi.org/10.1200/jco.1998.16.8.2672.
Martin M, Villar A, Sole-Calvo A, Gonzalez R, Massuti B, Lizon J, et al. Doxorubicin in combination with fluorouracil and cyclophosphamide (i.v. FAC regimen, day 1, 21) versus methotrexate in combination with fluorouracil and cyclophosphamide (i.v. CMF regimen, day 1, 21) as adjuvant chemotherapy for operable breast cancer: a study by the GEICAM group. Ann Oncol. 2003;14:833–42. https://doi.org/10.1093/annonc/mdg260.
Sukocheva OA, Lukina E, Friedemann M, Menschikowski M, Hagelgans A, Aliev G. The crucial role of epigenetic regulation in breast cancer anti-estrogen resistance: current findings and future perspectives. Semin Cancer Biol. 2022;82:35–59. https://doi.org/10.1016/j.semcancer.2020.12.004.
Arango D, Sturgill D, Yang R, Kanai T, Bauer P, Roy J, et al. Direct epitranscriptomic regulation of mammalian translation initiation through N4-acetylcytidine. Mol Cell. 2022;82:2797–814. https://doi.org/10.1016/j.molcel.2022.05.016. .e11.
Yuan S, He SH, Li LY, Xi S, Weng H, Zhang JH, et al. A potassium-chloride co-transporter promotes tumor progression and castration resistance of prostate cancer through m(6)a reader YTHDC1. Cell Death Dis. 2023;14:7. https://doi.org/10.1038/s41419-022-05544-8.
Li Y, Zhu S, Chen Y, Ma Q, Kan D, Yu W, et al. Post-transcriptional modification of m(6)a methylase METTL3 regulates ERK-induced androgen-deprived treatment resistance prostate cancer. Cell Death Dis. 2023;14:289. https://doi.org/10.1038/s41419-023-05773-5.
Zhou Y, Wang Q, Deng H, Xu B, Zhou Y, Liu J, et al. N6-methyladenosine demethylase FTO promotes growth and metastasis of gastric cancer via m(6)a modification of caveolin-1 and metabolic regulation of mitochondrial dynamics. Cell Death Dis. 2022;13:72. https://doi.org/10.1038/s41419-022-04503-7.
Ding C, Yi X, Chen X, Wu Z, You H, Chen X, et al. Warburg effect-promoted exosomal circ_0072083 releasing up-regulates NANGO expression through multiple pathways and enhances temozolomide resistance in glioma. J Exp Clin Cancer Res. 2021;40:164. https://doi.org/10.1186/s13046-021-01942-6.
Li XD, Wang MJ, Zheng JL, Wu YH, Wang X, Jiang XB. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021;112:4543–52. https://doi.org/10.1111/cas.15072.
Zhou X, Mitra R, Hou F, Zhou S, Wang L, Jiang W. Genomic Landscape and potential regulation of RNA editing in Drug Resistance. Adv Sci (Weinh). 2023;10:e2207357. https://doi.org/10.1002/advs.202207357.
Nakano M, Nakajima M. Significance of A-to-I RNA editing of transcripts modulating pharmacokinetics and pharmacodynamics. Pharmacol Ther. 2018;181:13–21. https://doi.org/10.1016/j.pharmthera.2017.07.003.
Huang Y, Xia W, Dong Z, Yang CG. Chemical inhibitors targeting the oncogenic m(6)a modifying proteins. Acc Chem Res. 2023;56:3010–22. https://doi.org/10.1021/acs.accounts.3c00451.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71. https://doi.org/10.1021/ja3064149.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90–e10523. https://doi.org/10.1016/j.cell.2017.11.031.
Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. 2021;81:922–39. https://doi.org/10.1016/j.molcel.2020.12.026e9.
Qiao Y, Su M, Zhao H, Liu H, Wang C, Dai X, et al. Targeting FTO induces colorectal cancer ferroptotic cell death by decreasing SLC7A11/GPX4 expression. J Exp Clin Cancer Res. 2024;43:108. https://doi.org/10.1186/s13046-024-03032-9.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84. https://doi.org/10.1093/nar/gku1276.
Shen Z, Zhu B, Li J, Qin L. Rhein augments Antiproliferative effects of Atezolizumab based on breast Cancer (4T1) regression. Planta Med. 2019;85:1143–9. https://doi.org/10.1055/a-1012-7034.
Sun K, Du Y, Hou Y, Zhao M, Li J, Du Y, et al. Saikosaponin D exhibits anti-leukemic activity by targeting FTO/m(6)a signaling. Theranostics. 2021;11:5831–46. https://doi.org/10.7150/thno.55574.
Xie G, Wu XN, Ling Y, Rui Y, Wu D, Zhou J, et al. A novel inhibitor of N (6)-methyladenosine demethylase FTO induces mRNA methylation and shows anti-cancer activities. Acta Pharm Sin B. 2022;12:853–66. https://doi.org/10.1016/j.apsb.2021.08.028.
Xu Y, Zhou J, Li L, Yang W, Zhang Z, Zhang K, et al. FTO-mediated autophagy promotes progression of clear cell renal cell carcinoma via regulating SIK2 mRNA stability. Int J Biol Sci. 2022;18:5943–62. https://doi.org/10.7150/ijbs.77774.
Li N, Kang Y, Wang L, Huff S, Tang R, Hui H, et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc Natl Acad Sci U S A. 2020;117:20159–70. https://doi.org/10.1073/pnas.1918986117.
Shibata T, Watari K, Kawahara A, Sudo T, Hattori S, Murakami Y, et al. Targeting phosphorylation of Y-Box-binding protein YBX1 by TAS0612 and Everolimus in overcoming Antiestrogen Resistance. Mol Cancer Ther. 2020;19:882–94. https://doi.org/10.1158/1535-7163.Mct-19-0690.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601. https://doi.org/10.1038/s41586-021-03536-w.
Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13:117. https://doi.org/10.1186/s13045-020-00951-w.
Li J, Gregory RI. Mining for METTL3 inhibitors to suppress cancer. Nat Struct Mol Biol. 2021;28:460–2. https://doi.org/10.1038/s41594-021-00606-5.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)a methylation promotes dendritic cell activation. Nat Commun. 2019;10:1898. https://doi.org/10.1038/s41467-019-09903-6.
Zhang ZW, Teng X, Zhao F, Ma C, Zhang J, Xiao LF, et al. METTL3 regulates m(6)a methylation of PTCH1 and GLI2 in sonic hedgehog signaling to promote tumor progression in SHH-medulloblastoma. Cell Rep. 2022;41:111530. https://doi.org/10.1016/j.celrep.2022.111530.
Du W, Huang Y, Chen X, Deng Y, Sun Y, Yang H, et al. Discovery of a PROTAC degrader for METTL3-METTL14 complex. Cell Chem Biol. 2024;31:177–83. https://doi.org/10.1016/j.chembiol.2023.12.009. .e17.
Li Z, Feng Y, Han H, Jiang X, Chen W, Ma X, et al. A stapled peptide inhibitor targeting the binding interface of N6-Adenosine-methyltransferase subunits METTL3 and METTL14 for Cancer Therapy. Angew Chem Int Ed Engl. 2024;63:e202402611. https://doi.org/10.1002/anie.202402611.
Li X, Ma S, Deng Y, Yi P, Yu J. Targeting the RNA m(6)a modification for cancer immunotherapy. Mol Cancer. 2022;21:76. https://doi.org/10.1186/s12943-022-01558-0.
Rosenfeld YO, Rausch O, McMahon, et al. STC-15, an oral small molecule inhibitor of the RNA methyltransferase METTL3, inhibits tumour growth through activation of anti-cancer immune responses associated with increased interferon signalling, and synergises with T cell checkpoint blockade. Eur J Cancer. 2022;174:S123. https://doi.org/10.1136/iitc-2022-SITC2022.1373.
Xiao H, Zhao R, Meng W, Liao Y. Effects and translatomics characteristics of a small-molecule inhibitor of METTL3 against non-small cell lung cancer. J Pharm Anal. 2023;13:625–39. https://doi.org/10.1016/j.jpha.2023.04.009.
Deng X, Qing Y, Horne D, Huang H, Chen J. The roles and implications of RNA m(6)a modification in cancer. Nat Rev Clin Oncol. 2023;20:507–26. https://doi.org/10.1038/s41571-023-00774-x.
Li G, Yao Q, Liu P, Zhang H, Liu Y, Li S, et al. Critical roles and clinical perspectives of RNA methylation in cancer. MedComm (2020). 2024;5:e559. https://doi.org/10.1002/mco2.559.
Dale B, Cheng M, Park KS, Kaniskan H, Xiong Y, Jin J. Advancing targeted protein degradation for cancer therapy. Nat Rev Cancer. 2021;21:638–54. https://doi.org/10.1038/s41568-021-00365-x.
Vojta A, Dobrinić P, Tadić V, Bočkor L, Korać P, Julg B, et al. Repurposing the CRISPR-Cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016;44:5615–28. https://doi.org/10.1093/nar/gkw159.
Zhang D, Zhang Z, Unver T, Zhang B, CRISPR/Cas. A powerful tool for gene function study and crop improvement. J Adv Res. 2021;29:207–21. https://doi.org/10.1016/j.jare.2020.10.003.
Tong J, Wang X, Liu Y, Ren X, Wang A, Chen Z, et al. Pooled CRISPR screening identifies m(6)a as a positive regulator of macrophage activation. Sci Adv. 2021;7. https://doi.org/10.1126/sciadv.abd4742.
Filippova J, Matveeva A, Zhuravlev E, Stepanov G. Guide RNA modification as a way to improve CRISPR/Cas9-based genome-editing systems. Biochimie. 2019;167:49–60. https://doi.org/10.1016/j.biochi.2019.09.003.
Wang SW, Gao C, Zheng YM, Yi L, Lu JC, Huang XY, et al. Current applications and future perspective of CRISPR/Cas9 gene editing in cancer. Mol Cancer. 2022;21:57. https://doi.org/10.1186/s12943-022-01518-8.
Nidhi S, Anand U, Oleksak P, Tripathi P, Lal JA, Thomas G, et al. Novel CRISPR-Cas systems: an updated review of the current achievements, applications, and Future Research Perspectives. Int J Mol Sci. 2021;22. https://doi.org/10.3390/ijms22073327.
Ostrowska K, Rawłuszko-Wieczorek AA, Ostapowicz J, Suchorska WM, Golusiński W. The two-faced role of RNA methyltransferase METTL3 on cellular response to cisplatin in head and neck squamous cell carcinoma in vitro model. Front Oncol. 2024;14:1402126. https://doi.org/10.3389/fonc.2024.1402126.
Liu XM, Zhou J, Mao Y, Ji Q, Qian SB. Programmable RNA N(6)-methyladenosine editing by CRISPR-Cas9 conjugates. Nat Chem Biol. 2019;15:865–71. https://doi.org/10.1038/s41589-019-0327-1.
Liu X, Xiao M, Zhang L, Li L, Zhu G, Shen E, et al. The m6A methyltransferase METTL14 inhibits the proliferation, migration, and invasion of gastric cancer by regulating the PI3K/AKT/mTOR signaling pathway. J Clin Lab Anal. 2021;35:e23655. https://doi.org/10.1002/jcla.23655.
Wilson C, Chen PJ, Miao Z, Liu DR. Programmable m(6)a modification of cellular RNAs with a Cas13-directed methyltransferase. Nat Biotechnol. 2020;38:1431–40. https://doi.org/10.1038/s41587-020-0572-6.
Ying X, Jiang X, Zhang H, Liu B, Huang Y, Zhu X, et al. Programmable N6-methyladenosine modification of CDCP1 mRNA by RCas9-methyltransferase like 3 conjugates promotes bladder cancer development. Mol Cancer. 2020;19:169. https://doi.org/10.1186/s12943-020-01289-0.
Han L, Dong L, Leung K, Zhao Z, Li Y, Gao L, et al. METTL16 drives leukemogenesis and leukemia stem cell self-renewal by reprogramming BCAA metabolism. Cell Stem Cell. 2023;30:52–e6813. https://doi.org/10.1016/j.stem.2022.12.006.
Zhang S, Shen J, Li D, Cheng Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics. 2021;11:614–48. https://doi.org/10.7150/thno.47007.
Liu Z, Shi M, Ren Y, Xu H, Weng S, Ning W, et al. Recent advances and applications of CRISPR-Cas9 in cancer immunotherapy. Mol Cancer. 2023;22:35. https://doi.org/10.1186/s12943-023-01738-6.
Tadić V, Josipović G, Zoldoš V, Vojta A. CRISPR/Cas9-based epigenome editing: an overview of dCas9-based tools with special emphasis on off-target activity. Methods. 2019. https://doi.org/10.1016/j.ymeth.2019.05.003. 164–165:109 – 19.
Naeem M, Majeed S, Hoque MZ, Ahmad I. Latest developed strategies to minimize the Off-Target effects in CRISPR-Cas-mediated genome editing. Cells. 2020;9. https://doi.org/10.3390/cells9071608.
Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65:1866–79. https://doi.org/10.1016/j.addr.2013.09.019.
Yadav P, Ambudkar SV, Rajendra Prasad N. Emerging nanotechnology-based therapeutics to combat multidrug-resistant cancer. J Nanobiotechnol. 2022;20:423. https://doi.org/10.1186/s12951-022-01626-z.
Guo S, Lv L, Shen Y, Hu Z, He Q, Chen X. A nanoparticulate pre-chemosensitizer for efficacious chemotherapy of multidrug resistant breast cancer. Sci Rep. 2016;6:21459. https://doi.org/10.1038/srep21459.
Xu Q, Lan X, Lin H, Xi Q, Wang M, Quan X, et al. Tumor microenvironment-regulating nanomedicine design to fight multi-drug resistant tumors. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2023;15:e1842. https://doi.org/10.1002/wnan.1842.
Tang Z, Tian W, Long H, Jiang S, Zhao J, Zhou J, et al. Subcellular-targeted Near-Infrared-Responsive Nanomedicine with Synergistic Chemo-Photothermal Therapy against Multidrug Resistant Cancer. Mol Pharm. 2022;19:4538–51. https://doi.org/10.1021/acs.molpharmaceut.1c00998.
Zhang R, Gao S, Wang Z, Han D, Liu L, Ma Q, et al. Multifunctional molecular Beacon Micelles for Intracellular mRNA imaging and synergistic therapy in Multidrug-Resistant Cancer cells. Adv Funct Mater. 2017;27. https://doi.org/10.1002/adfm.201701027.
Huang R, Kang T, Chen S. The role of tumor-associated macrophages in tumor immune evasion. J Cancer Res Clin Oncol. 2024;150:238. https://doi.org/10.1007/s00432-024-05777-4.
Wang H, Ning X, Wang X, Ding F, Wang Y. A versatile modular preparation strategy for targeted drug delivery systems against multidrug-resistant cancer cells. Nanotechnology. 2021;33. https://doi.org/10.1088/1361-6528/ac317c.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13:5. https://doi.org/10.1186/s13045-019-0839-x.
Xiao Z, Li T, Zheng X, Lin L, Wang X, Li B, et al. Nanodrug enhances post-ablation immunotherapy of hepatocellular carcinoma via promoting dendritic cell maturation and antigen presentation. Bioact Mater. 2023;21:57–68. https://doi.org/10.1016/j.bioactmat.2022.07.027.
Ma S, Sun B, Duan S, Han J, Barr T, Zhang J, et al. YTHDF2 orchestrates tumor-associated macrophage reprogramming and controls antitumor immunity through CD8(+) T cells. Nat Immunol. 2023;24:255–66. https://doi.org/10.1038/s41590-022-01398-6.
Zhou H, Chen DS, Hu CJ, Hong X, Shi J, Xiao Y. Stimuli-Responsive Nanotechnology for RNA delivery. Adv Sci (Weinh). 2023;10:e2303597. https://doi.org/10.1002/advs.202303597.
Sharma P, Hoorn D, Aitha A, Breier D, Peer D. The immunostimulatory nature of mRNA lipid nanoparticles. Adv Drug Deliv Rev. 2024;205:115175. https://doi.org/10.1016/j.addr.2023.115175.
Uchida S, Perche F, Pichon C, Cabral H. Nanomedicine-based approaches for mRNA delivery. Mol Pharm. 2020;17:3654–84. https://doi.org/10.1021/acs.molpharmaceut.0c00618.
Du D, He J, Ju C, Wang C, Li H, He F, et al. When N(7)-methyladenosine modification meets cancer: emerging frontiers and promising therapeutic opportunities. Cancer Lett. 2023;562:216165. https://doi.org/10.1016/j.canlet.2023.216165.
Boriack-Sjodin PA, Ribich S, Copeland RA. RNA-modifying proteins as anticancer drug targets. Nat Rev Drug Discov. 2018;17:435–53. https://doi.org/10.1038/nrd.2018.71.
Cerneckis J, Ming GL, Song H, He C, Shi Y. The rise of epitranscriptomics: recent developments and future directions. Trends Pharmacol Sci. 2024;45:24–38. https://doi.org/10.1016/j.tips.2023.11.002.
Liu WW, Zheng SQ, Li T, Fei YF, Wang C, Zhang S, et al. RNA modifications in cellular metabolism: implications for metabolism-targeted therapy and immunotherapy. Signal Transduct Target Ther. 2024;9:70. https://doi.org/10.1038/s41392-024-01777-5.
Sasso JM, Ambrose BJB, Tenchov R, Datta RS, Basel MT, DeLong RK, et al. The Progress and Promise of RNA MedicineAn Arsenal of targeted treatments. J Med Chem. 2022;65:6975–7015. https://doi.org/10.1021/acs.jmedchem.2c00024.
Acknowledgements
Not applicable.
Funding
This work was supported by the China Postdoctoral Science Foundation (2023M743200 and 2024T170826), the Postdoctoral Fellowship Program of CPSF (GZC20232425), the Science and Technology Research Program of Henan Province (242102311163), and the Henan Medical Science and Technology Joint Building Program (LHGJ20230160 and LHGJ20230239).
Author information
Authors and Affiliations
Contributions
Chen Xue, Shaohua Li, Dongming Yan and Xuqiang Zhu designed the structure of the review. Di Chen, Xinyu Gu and Yeltai Nurzat, and Lixia Xu wrote the article. Xueyuan Li, Lixin Wu, and Henan Jiao draw the figures. Di Chen, Henan Jiao and Peng Gao completed the tables. Chen Xue, Shaohua Li, Dongming Yan, and Xuqiang Zhu helped with the final revision of the review. All authors reviewed the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The corresponding author has obtained consent for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, D., Gu, X., Nurzat, Y. et al. Writers, readers, and erasers RNA modifications and drug resistance in cancer. Mol Cancer 23, 178 (2024). https://doi.org/10.1186/s12943-024-02089-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-02089-6