
Abstract
Chimeric antigen receptor (CAR) T cell (CAR-T cell) therapy based on gene editing technology represents a significant breakthrough in personalized immunotherapy for human cancer. This strategy uses genetic modification to enable T cells to target tumor-specific antigens, attack specific cancer cells, and bypass tumor cell apoptosis avoidance mechanisms to some extent. This method has been extensively used to treat hematologic diseases, but the therapeutic effect in solid tumors is not ideal. Tumor antigen escape, treatment-related toxicity, and the immunosuppressive tumor microenvironment (TME) limit their use of it. Target selection is the most critical aspect in determining the prognosis of patients receiving this treatment. This review provides a comprehensive summary of all therapeutic targets used in the clinic or shown promising potential. We summarize CAR-T cell therapies’ clinical trials, applications, research frontiers, and limitations in treating different cancers. We also explore coping strategies when encountering sub-optimal tumor-associated antigens (TAA) or TAA loss. Moreover, the importance of CAR-T cell therapy in cancer immunotherapy is emphasized.
Similar content being viewed by others
Background
CAR-T cell therapy specific to tumor antigens is a rapidly evolving concept that has shown remarkable results when applied clinically and has transformed the treatment paradigm for hematologic malignancies. In August of 2017, the Food and Drug Administration (FDA) of the United States approved the use of CAR-T cell therapy in treating patients who suffered from relapsed or refractory B-acute lymphoblastic leukemia (r/r B-ALL). Since then, this field has entered an era of fast-paced, innovative development. Many clinical trials of CAR-T cell therapy have been conducted over the years.
It is widely acknowledged that each T cell has an extremely sensitive and specific T cell receptor (TCR) that constantly checks the organism for ‘non-self’ signals and triggers a cascade of immune responses when abnormal peptides are identified as a precise killer of pathogens. In the TME, T cells are specific to the mutant proteins of cancer cells. Interestingly, these cells could be extracted from a patient’s tumor tissue. After amplification in vitro, they are injected back into the patient, producing a long-lasting antitumor response. However, the method is mainly used for solid tumor treatment and is limited by the collection method, amplification effect, etc. The production scale is small, and its application in the clinic is not satisfactory [1]. CAR-T cell therapy involves genetically engineering T cells to express antigen-specific, non-MHC-restricted receptors that could target and attack specific pathological cells to exert a therapeutic effect on patients [2, 3].
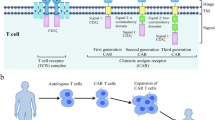
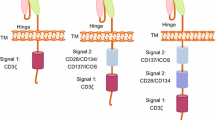
The structure of CAR has been constantly updated (Fig. 1). First-generation CAR consisted of an extracellular structural domain recognizing antigen and a single intracellular motif. Still, there were no costimulatory molecules in the structure, making it difficult for CAR-T cells to persist in patients and ineffective against tumours [4]. Second-generation CARs have added an intracellular motif consisting of the signaling domain of a costimulatory receptor to their structure [5]. Even in the absence of exogenous costimulatory molecules, second-generation CAR-T cells could continue to proliferate and release cytokines to exert anti-tumor effects and are the most widely used in clinical practice [6]. The third generation CAR contains two costimulatory molecules designed to enhance further the killing ability of CAR-T cells [7, 8]. The fourth generation CARs inserted additional molecular elements to express functional transgenic proteins, such as interleukin genes or suicide genes, enhancing the killing power and safety of CAR-T cells [9, 10]. To improve the flexibility in target recognition of the CAR, the universal CAR-T cells are designed using BBIR CAR or SUPRA CAR. The tumour-specific scFv extracellular structural domain used in previous generations of CAR-T cells is replaced in the universal CAR-T cells with an adapter-specific recognition structural domain that binds to an adapter molecule targeting a tumour-specific target. This design allows the antigen-targeting structural domain to be separated from the t-cell signalling unit, thus giving CAR-T cells the ability to recognise multiple antigens. At the same time, this CAR-T cell only functions to recognise and attack cells when the adapter is provided, thus increasing the user’s control over the CAR-T cell and facilitating its use in the body [11,12,13]. In addition, single-domain antibodies, also known as single variable domain on a heavy chain (VHH) or nano-antibodies, are also used as targeting domains for CAR-T. Nanobody-based CAR-T cells have been proved to inhibit the growth of solid tumors in immunocompetent mice [14]. Moreover, nanobodies could not aggregate on the surface of T cells because of their monomeric structure [15]. Furthermore, nanobodies do not have the limitation of affinity loss which is recognized as a possible side effect in the design of the conventional single-chain fragment variable (scFv) used as the antigen-targeting domain of CAR [16].

The evolution of CAR-T cell structure. There are five generations of CAR-T cells to date. scFv is currently the most commonly used targeting domain for CAR-T cells, and VHH is also emerging as a targeting domain for CAR-T cells with great potential. VH: heavy chain variable domain; VL: light chain variable domain; scFv: single-chain fragment variable; VHH: single variable domain on a heavy chain
The fact that tumor recognition is not dependent on the major histocompatibility complex (MHC) constitutes their primary benefit. Even though CAR-T cell treatment has shown promising outcomes in clinical trials, considerable challenges remain in cancer treatment using CAR-T cells, such as tumor antigen escape and treatment-related toxicity [17]. CAR-T cell therapies for solid tumors face more significant difficulties due to tumor antigen heterogeneity, difficulty transporting to and infiltrating tumor sites, and challenges with immunosuppressive TME.
The selection of optimal target antigens is the key to addressing these challenges. Typically, the target ought to be a protein, carbohydrate, or glycolipid molecule particularly common in cancer cells. The specificity of the target antigen is essential to prevent toxic effects; the ideal target should be minimally expressed in normal tissue. It is well-recognized that cancer cells could evolve through complex genomic evolutionary mechanisms to evade their destruction by immune cells gradually. Consequently, the target antigen’s stability is vital in avoiding the immunological escape of malignancies. For the security and efficacy of CAR-T cells, ideal targets should include high levels of malignant cell coverage, specificity, and stability [18]. Indeed, antigens that play a crucial role in disease pathophysiology are more suitable as targets. Researchers focus on multi-antigen targeted CAR-T cell therapy to prevent relapse following treatment directed toward a single antigen. This review summarizes and discusses CAR-T cell therapies for different targets in hematological diseases and solid tumors, ranked by disease incidence in Western countries, and highlights the importance of CAR-T cells in oncology treatment.
Haematologic cancers
Lymphoma
B-cell non-Hodgkin’s lymphoma (B-NHL)
Non-Hodgkin’s lymphoma (NHL) is the most prevalent hematologic tumor, with diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and follicular lymphoma (FL) representing the most common types. The conventional treatment includes radiation therapy, chemotherapy, etc. However, about 20–30% of patients develop tolerance to these treatments [19]. Hematopoietic stem cell (HSC) transplantation (HSCT) is effective, but many patients are not candidates for this treatment and are prone to relapse after treatment [20]. However, the anti-CD20 antibody rituximab could significantly improve the prognosis of B-NHL, the prognosis of patients who are resistant to immunochemotherapy or relapse after HSCT is extremely poor [19]. Greatly, CAR-T cell therapy could potentially enhance the prognosis of B-NHL patients.
B-NHL is a malignant tumor with high heterogeneity. CD19 is a transmembrane glycoprotein that regulates B lymphocyte activation and proliferation. Its expression in normal tissues is limited to B lymphocyte lines and could be found at high levels in most malignant B cell tumors [21]. The CD19 CAR-T cell therapy product has the highest safety and effectiveness and is the most advanced CAR-T cell therapy product (Fig. 2). Tisagenlecleucel (Kymriah), lisocabtagene maraleucel (Breyanzi), and axicabtagene ciloleucel (Yescarta) are FDA-approved drugs that target CD19 for treating relapsed or refractory DLBCL with good efficacy and manageable adverse events (NCT02445248, JULIET; NCT02631044, TRANSCEND; NCT02348216, ZUMA-1; NCT03391466, ZUMA-7) [22,23,24,25]. In a multicenter, single-arm, phase 2 study (ZUMA-12, NCT03761056), axicabtagene ciloleucel yielded highly significant treatment outcomes in 37 patients with high-risk DLBCL, with patients achieving a complete remission (CR) rate (CRR) of 78% and an objective response (OR) rate (ORR) of 89%. Eighty-six percent of patients were alive at the time of data cut-off (median follow-up of 15.9 months), while ≥ grade 3 cytokine release syndrome (CRS) and neurological events occurred in 8 and 23% of patients, respectively. Adverse events were monitored according to the National Cancer Institute’s Common Terminology Criteria for Adverse Events (CTCAE) v5.0 [26]. Compared with the previous median survival of only 6.3 months in high-risk DLBCL patients, CAR-T cell therapy has substantially improved patient survival [19]. The use of axicabtagene ciloleucel is also recommended for treating FL that has recurred or is resistant to therapy [27]. In a multicenter, single-arm, phase 2 trial (ZUMA-5, NCT03105336), 104 patients with relapsed or refractory FL and marginal zone lymphoma were treated with conditioning chemotherapy followed by axicabtagene ciloleucel. Ninety-two percent of patients had an overall response, 74% had a CR. The most common adverse events greater than or equal to grade 3 were haemocytopenia (70% of patients) and infection (18% of patients), which suggests that axicabtagene ciloleucel has good efficacy in indolent non-Hodgkin lymphoma with manageable adverse effects (CTCAE v4.03) [28]. Primary central nervous system (CNS) lymphoma (PCNSL) tends to have a worse prognosis than other lymphomas, and first-line treatment often leads to neurotoxicity. There is little research into treatment options for this disease [29]. In a phase 1/2 clinical trial (NCT02445248), 12 patients with relapsed PCNSL were treated with tisagenlecleucel, of which six patients had a CR, and only one developed immune cell-associated neurotoxicity syndrome, demonstrating the safety and efficacy of tisagenlecleucel in this refractory patient group [30]. Brexucabtagene autoleucel (Tecartus), a CD19 CAR-T cell product, has been given the go-ahead for managing recurrent or refractory MCL. In a phase 2 multicentre clinical trial (ZUMA-2, NCT02601313), 74 patients were enrolled. The primary efficacy analysis showed that 93% of patients receiving brexucabtagene autoleucel had an ORR, 67% had a CR, and estimated progression-free survival (PFS) and overall survival (OS) at 12 months was 61 and 83%, respectively [31]. The everyday adverse events in grade 3 or higher were hematogenic (94%) and infection (32%), with no fatal adverse events (CTCAE v4.03) [31]. In children, Burkitt lymphoma (BL) is perhaps the most prevalent form of NHL [32]. Currently, lentiviral or retroviral technology is often used to produce CAR-T cells. Still, these approaches often hinder CAR expression, carry a high tumor risk, and are more expensive to manufacture [33,34,35,36,37]. A paper published in Nature reports that by using non-viral targeted integration, researchers have prepared CD19 CAR-T cells (AAVS1-19bbz) that effectively eradicate tumor cells in the BL cell line Raji and cell line-derived xenograft mouse models [38]. In this study, the researchers also produced CD19 CAR-T cells (PD1-19bbz) with programmed cell death 1 (PD1) knocked out by CRISPR-Cas9 technology, which showed strong eradication ability against Raji cells that were either high or low in programmed death-ligand 1 (PD-L1) expression [38]. In the phase 1 clinical trial using PD1-19bbz cells, seven out of eight relapsed/refractory B-NHL patients achieved CR and the rest PR, and no CAR-T cell-related grade 3 or higher adverse events were observed (CTCAE v5.0) [38]. This demonstrates the high efficacy and safety profile of the cells. However, there remains a greater likelihood of recurrence after CAR-T cell treatment; the leading cause of relapse is the loss of CD19 molecules. Accordingly, CAR-T cells could be harnessed to target new targets to help solve this problem.

Common CAR-T cell therapy targets in hematological malignancies. B-ALL: B-acute lymphoblastic leukemia; BAFF-R: B-cell activating factor receptor; TSLPR: thymic stromal lymphopoietin receptor; T-ALL: T-acute lymphoblastic leukemia; AML: acute myeloid leukemia; NKG2DL: natural killer group 2 member D ligand; CLL1: C-type lectin like molecule 1; FLT3: FMS-like tyrosine kinase 3; WT1: wilms tumor 1; CLL: chronic lymphocytic leukemia; ROR: receptor tyrosine kinase like orphan receptor; BCMA: B-cell maturation antigen; MM: multiple myeloma; SLAMF7: signaling lymphocytic activation molecule F7; GPRC5D: G protein-coupled receptor class-C group-5 member-D; B-NHL: B-cell non-Hodgkin’s lymphoma; T-NHL: T-cell non-Hodgkin’s lymphoma; TRBC: T cell receptor β-chain constant domains; HL: Hodgkin’s lymphoma
CD22 is a sialic acid adhesin family member which regulates B-cell activation [39] (Fig. 2). It is expressed only in B cell lineages except for plasma cells in normal tissues. It is expressed in most B lymphoma cells and has become a popular therapeutic target for the disease. In a phase 1 dose-escalation study (NCT04088890), three patients had a tumor relapse after recovering from treatment with CD19 CAR-T cells. Still, they had a CR following treatment with CD22 CAR-T cells, and no non-hematological adverse severe events were observed (CTCAE v5.0) [40].
CD20 is highly expressed in malignant cells and could regulate cell activation and proliferation. It has also emerged as an alternative CAR-T therapeutic target (Fig. 2). Clinical trials have confirmed its good efficacy, and no serious adverse effects have been observed in the trials (NCT00621452, 12 participants, CTCAE v3.0; NCT01735604, 50 participants, CTCAE v3.0) [41, 42]. A study reported that a patient with BL with no significant response to CD19 CAR-T cell therapy experienced partial remission (PR) but rapidly relapsed after CD22 CAR-T cell therapy. After CD20 CAR-T cell therapy, he achieved CR with event-free survival (EFS) of 16 months (CTCAE v4.03) [43].
A phase 1 clinical trial (54 participants) of κ light chain CAR-T cell therapy treating B-NHL confirmed that the κ light chain is a prospective target and that this type of CAR-T cell has definite anti-lymphoma activity while ensuring feasibility and safety (NCT00881920, CTCAE v4.03) [44] (Fig. 2).
The most common B-NHL subtype associated with λ light chain expression is MCL, which has an λ:κ expression ratio of approximately 2:1. The efficacy of λ CAR-T has been demonstrated in Igλ + lymphoma cell lines (Maver-1, SP53) and xenograft Igλ + lymphoma mouse models [45].
In addition, chemokine receptor (CXCR) 5 CAR-T cells could target both B-NHL cells and follicular T helper cells, effectively inhibiting lymphoma growth in a mouse xenograft model [46].
Bispecific CAR-T cells are gradually being used in the treatment of B-NHL. Recently, CAR-T cell treatments targeting CD19/CD22 and CD19/CD20 have proven to be highly successful in clinical studies (NCT03233854, NCT03196830, ChiCTR1800015575, NCT03097770, NCT03019055) [47,48,49,50,51]. Such CAR-T cells are particularly helpful in addressing the problem of disease relapse due to antigen loss and deserve the attention of clinicians.
T-cell non-Hodgkin’s lymphoma (T-NHL)
Few effective treatments are available for T-NHL, and patients generally have poor prognoses. Moreover, the relapse rate of this disease group is high. Despite CAR-T cell therapy’s relatively good results in treating B-cell malignancies, its application to T-NHL still faces many difficulties. Firstly, manufacturing autologous CAR-T products is difficult because the malignant cells are presented with normal T cells when immune cell extraction is performed on the patient [52]. Secondly, the major CAR-T targeting antigens (e.g., CD5 and CD7) are also expressed in normal T cells [53,54,55]. The use of CAR-T cells results in the clearance of normal T cells referred to as T cell dysplasia [56]. In addition, target antigens expressed in CAR-T cells themselves could cause CAR-T cells to attack each other, i.e., fratricide [57]. These factors limit the use of CAR-T cell therapy in this disease.
CD5 is a characteristic surface marker of malignant T cells that is extremely important for cell survival and is only expressed in a subset of immune system cells in normal tissues [58, 59] (Fig. 2). The investigators created a CD5 CAR-T cell that could secrete IL-15 with the enhanced anti-tumor response, which rapidly and potently improved the condition of a T-NHL patient with CNS involvement in a phase 1 clinical trial with 20 participants enrolled (NCT04594135) and only grade 1 CRS was observed (CTCAE v4.0) [60]. CD5 is rapidly internalized upon binding to ligands, leading to a reduction in its availability on the cell surface and a consequent decrease in CAR-CD5 interactions [61]. CAR-T cells targeting CD5 downregulate their CD5 expression to counteract self-mutilation and ensure their ability to continue to function [60]. In addition, IL-15 is thought to promote T cell proliferation, which could reduce the impact of self-mutilation on T cell numbers [62]. This minimizes the impact effect of fratricide in this CAR-T cell type. This trial suggests that CD5 CAR-T cells may be an excellent way to treat T-NHL, but further large-scale trials are needed to validate this.
CD7 is expressed in T-NHL malignant cells, normal T cells, and natural killer cells (NK cells). CAR-T cell therapy targeting CD7 is reportedly effective in T-NHL in preclinical studies [53] (Fig. 2).
The mutually exclusive expression of T cell receptor β-chain constant domains (TRBC) 1 and 2 enables immunotherapy to completely eradicate malignant T cells while maintaining a sufficient number of normal T cells to sustain cell-mediated immunity [63]. This could be extremely important for the application of CAR-T therapeutic approaches (Fig. 2).
CD30 CAR-T cells are based on the novel costimulatory combination CD28.OX40 showed vigorous anti-lymphoma activity in anaplastic large cell lymphoma xenograft immunodeficient mouse model, which has an excellent prospect in clinical application [64] (Fig. 2). The costimulatory combination plays a vital role in this.
CD4 is expressed in most T-cell lymphoma subtypes. Patients with peripheral T-cell lymphoma, not otherwise specified, most commonly have a CD4+/CD8- phenotype, with only a few being CD4/CD8 +/+ or −/−, demonstrating the potential of anti-CD4 CAR-T cells [65].
Bispecific CAR-T cells are currently being explored for use in T-NHL. Researchers made CD5/CD7 CAR-T cells that showed anti-tumor substantial effects in malignant T cell lines such as Jurkat, CCRF-CEM, MOLT, and in xenogeneic mouse models established using CCRF-CEM-ffLuc cell injections [66]. The following clinical trials are urgently needed to validate their clinical efficacy and safety.
Hodgkin’s lymphoma (HL)
HL is a common type of B-cell lymphoma. First-line therapy is highly effective against these B-cell malignancies. However, more than 10% of patients experience disease progression after initial treatment, with higher relapse rates and limited treatment options for relapsed or refractory HL [67].
It is widely acknowledged that HL malignant cells express CD30 in abundance, and HL, after relapse, still has high CD30 expression. Interestingly, CD30 CAR-T cells could attack tumor cells with low CD30 expression and kill tumor cells that have lost sensitivity to vibutuximab (Fig. 2). In two parallel phases, 1/2 studies conducted at two independent centers (NCT02690545, NCT02917083), the ORR for 32 patients receiving a lymphatic clearance regimen followed by infusion of CD30 CAR-T cells was 72%, with 59% of patients having CR. Grade 3 or higher hematological adverse events were the most common toxicity, and no CRS or neurotoxicity beyond grade 1 (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.0) [68]. The trial also found that 27 r/r HL patients who received CD30 CAR-T cells after a lymphatic clearance had a median PFS of 352 days (NCT02690545) [69]. Despite the high clinical response rate, more extensive clinical trials are needed to verify its clinical effectiveness. Humanized scFv-based CD30 CAR-T cell has low immunogenicity, low risk of cytokine-mediated toxicity, and high persistence. It destroyed CD30+ tumor cell lines (L428 and L540) in vitro and cleared lymphoma in lymphoma-bearing mice, showing promising efficacy for the next phase of clinical trials [70]. The application of the co-stimulation combination CD28.OX40 is of great value in improving the effectiveness of CD30 CAR-T cells and is expected to be used in the following clinical trials [64].
CD19 could also be used as a therapeutic target (Fig. 2). According to research (ChiCTR2000028922), a patient treated with CD19 and CD30 CAR-T cells showed a protracted PFS and no severe adverse effects (CTCAE v4.0) [71].
Leukemia
B-acute lymphoblastic leukemia (B-ALL)
B-ALL is caused by malignant precursor B lymphocytes that affect the production of normal blood cells in the bone marrow. It is much more prevalent in adults than T-acute lymphoblastic leukemia (T-ALL) [72]. Chemotherapy is the current first-line treatment for B-ALL. However, some patients develop relapsed or refractory acute B-cell lymphoblastic leukemia (r/r B-ALL) after conventional chemotherapy and have a poor prognosis. Current evidence suggests that CAR-T cell therapy plays a significant role in treating r/r B-ALL.
CD19 is now the most commonly used and researched CAR-T target for B-ALL treatment (Fig. 2). The first CAR-T cell treatment approved by the FDA to treat r/r B-ALL is tisagenlecleucel targeting CD19. In a phase 2 study conducted in 25 centers (ELIANA, NCT02435849), 75 patients received tisagenlecleucel infusions with an ORR of 81% at 3 months, EFS rate and the rate at 12 months were 50 and 76%, respectively, and a 73% incidence of grade 3 or 4 adverse events possibly related to treatment [73]. In another phase 2 study (ENSIGN, NCT02228096), 20 of 29 patients achieved OR. Eleven had grade 3 or 4 CRS, and one had grade 3 neurological symptoms [74]. Follow-up studies on this group of patients have shown a significant improvement in their quality of life, better than conventional therapy [75]. These two trials have important implications for the commercialization of tisagenlecleucel. The other CAR-T cell product targeting CD19, brexucabtagene autoleucel, is also approved for r/r B-ALL treatment. In a phase 2 clinical trial (ZUMA-3, NCT02614066), 56% of the 55 patients receiving brexucabtagene autoleucel achieved CR, with a median of 18.2 months, and anemia (49%) was the most common adverse event at grade 3 or higher (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.03) [76]. Intravenous immunoglobulin could partially limit the side effects of the attack on normal B cells. Although CD19 CAR-T cell therapy induces very high CRR in B-ALL patients, the recurrence of the disease remains an important issue. The absence or mutation of antigens and the limited duration of CAR-T cell function in vivo may account for relapse after treatment.
In response to disease relapse, researchers have begun constructing CAR-T cells that target different targets and respond by adjusting the manufacturing process. CD22 is expressed in 90% of juvenile and 50–100% of adult patients, suggesting it is an excellent target for relapsed B-ALL treatment [77, 78] (Fig. 2). In a study with CD22 CAR-T cells for r/r B-ALL (ChiCTR-OIC-17013523), 24 of 30 patients achieved CR within 1 month, and the 12-month leukemia-free survival rate for patients was 71.6%, with most patients experiencing only minor adverse effects. No CD22 antigen loss or mutation was found in the limited number of patients who relapsed (CTCAE v4.03) [79].
CD38 has been documented in r/r B-ALL malignant cells, and CD38 CAR-T cells were used to treat an r/r B-ALL patient who failed to respond to bispecific CD19/CD22 CAR-T cell therapy (Fig. 2). However, the patient developed severe complications and abandoned the treatment after 20 days of cell infusion [80].
In addition to the targets mentioned above that have proven their effectiveness in clinical trials, many promising targets are being explored. B-ALL malignant, dendritic, and HSCs express CD123 [81]. CD123 CAR-T cell therapy is an ideal solution for relapse after CD19 CAR-T treatment since it is expressed in most CD19- relapsed or innately CD19- resistant subpopulations (Fig. 2). In animal models, CD123 CAR-T cells have demonstrated high efficacy against CD19- B-ALL cells [82]. However, CD123 is expressed on normal HSCs, so CD123 CAR-T cells could potentially harm the bone marrow.
The B-cell activating factor receptor (BAFF-R) may be retained in recurrent cancer malignant cells (Fig. 2). In numerous xenogeneic animal models, including CD19 antigen deletion models, BAFF-R CAR-T cells could efficiently and accurately remove B-ALL malignant cells [83].
In addition, CRLF2 gene rearrangements produced an r/r B-ALL phenotype insensitive to standard chemotherapy regiments with poor prognosis [84]. Studies have found that thymic stromal lymphopoietin receptor (TSLPR) CAR-T cells soundly affect the subtype of diseases mentioned above [85] (Fig. 2).
The combination of CD19 and CD22 CAR-T cells has attracted significant interest recently (Fig. 2). In a trial of r/r B-ALL patients (ChiCTR-ONC-17013648), serial infusions of CD19 and CD22 CAR-T cells were given to 21 patients who relapsed after HSCT. Twenty patients achieved CR 1 month after the second infusion, including those who relapsed after the first infusion of CD19 CAR-T cells. No grade 3 or higher CRS or neurotoxicity was observed (CRS, Penn grading scale; other adverse events, CTCAE v5.0). The 12-month EFS and rates of patients were 67.5 and 88.5%, respectively. In contrast, 50–57% of patients in the group given only a single CD19 or CD22 CAR-T cells treatment relapsed within 6–8 months [86]. In another trial (NCT03185494), infusion of bispecific CD19/CD22 CAR-T cells to patients with r/r B-ALL resulted in CR in all six patients without severe adverse events (Neurological toxicities, Lee 2014 criteria; CRS, American Society for Transplantation and Cellular Therapy criteria) [87]. In a phase 1 trial conducted in Beijing (ChiCTR-OPN-16008526), 23 r/r B-ALL patients were treated with dual-targeted CD19/CD22 CAR-T cells. All 22 patients are willing to be evaluated for achieved CR, with estimated 12-month PFS rates and rates of 59.2 and 67.4%, respectively. Adverse reactions in patients greater than or equal to grade 3 included haemocytopenia, fever, and CRS. The rest of the adverse reactions were mild (CRS, American Society for Transplantation and Cellular Therapy criteria; other adverse events, CTCAE v4.03) [88]. In addition, one study found that CD19 CAR-T was an independent risk factor associated with severe CRS (ChiCTR1800015575) [89]. Bispecific CD19/CD22 CAR-T cells may lower the risk of CRS [89]. Recently, researchers have created CAR-T cells that target CD19/20/22 by co-expressing a CAR-T cell molecule on T cells using a tricistronic transgene. CD19/20/22 CAR-T cells showed superior cytotoxicity to CD19 CAR-T cells in in vitro assays against Daoy cells and primary B-ALL malignant cells. In the NSG xenograft model, CD19/20/22 CAR-T cells showed more potent inhibition of CD19(−) leukemia cells in patients who failed CD19 CAR-T cell therapy, which was challenging to inhibit CD19 CAR-T cells [90]. Dual/multi-targeted CAR-T cells could improve CRR and even reduce adverse reactions and are promising in clinical applications.
Besides, CD72 was revealed as a target for in vitro-evolved nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALL [91].
T-acute lymphoblastic leukemia
Although CAR-T cell therapy has improved the prognosis of r/r B-ALL patients, it has had less impact on T-ALL patients. Similar to what CAR-T cells face in T-NHL treatment, normal and malignant T cells often co-express target antigens, causing CAR-T cells to target normal T cells and causing severe T cell immunodeficiency. As a result, the development of CAR-T cell treatment for T-ALL remains difficult.
CD7 is expressed in 95% of T-ALL malignant cells. Eighteen of 20 T-ALL patients treated with allogeneic CD7 redirected CAR-T cells achieved CR in a single-center phase 1 clinical trial (Fig. 2). In comparison (NCT04689659), grade 3–4 haemocytopenia occurred in all patients, and grade 3–4 CRS occurred in 2 patients (haemocytopenia, CTCAE v5.0; CRS, American Society for Transplantation and Cellular Therapy criteria) [92]. In two trials (ChiCTR190002531, ISRCTN19144142), one of the two patients receiving CD7 CAR-T cells was in sustained remission for more than 1 year after treatment patients experienced grade 3 CRS (American Society for Transplantation and Cellular Therapy criteria) [93]. Despite the excellent effectiveness, the safety of the treatment needs further improvement. An important area of current research is cytosine base editors (CBEs). Unlike the induced DNA double-strand breaks (DSBs) technique used in the manufacture of most allogeneic CAR-T cells, CBEs create point mutations in T cells that silence gene expression without DSBs with an efficiency of 90 to 99%, significantly reducing the incidence of unexpected target editing results [94,95,96]. Allogeneic CD7 CAR-T cells developed based on CBEs are highly effective against T-ALL cells in a CD7+ T-ALL cell line CCRF-CEM, a model constructed by transplanting CCRF-GFP-Luc cells in NSG mice, and a mouse model created from patient-derived xenografts [96]. In addition, the removal of CD7 expression on the surface of T cells by gene editing technology could significantly inhibit the fratricide of CAR-T cells and reduce the risk of side effects. CD7 and TCR alpha chain-deficient CD7 CAR-T cells (UCART7) manufactured by CRISPR/Cas9 gene editing technology were used in the CD7+ T-ALL cell lines MOLT-3 (ACC 84), MOLT-4 (ACC 362), HSB-2 (ACC 435) and CCRF-CEM (ACC 240), the CCRF-CEM xenograft models and patient-derived xenograft models all showed better anti-tumor effects and significantly reduced fratricide [57].
CD5 is expressed in 80% of T-ALL cells. In vitro, CAR-T cells targeting CD5 successfully kill malignant T cell lines (CCRF-CEM, MOLT-4, and KARPAS-299) and primary T-ALL parent cells (Fig. 2). It also significantly slowed disease progression in a T-ALL xenograft mice model [97]. T-ALL cell lines and primary T-ALL malignant cells have been found to express natural killer group 2 member D ligand (NKG2DL). In healthy cells, it is rarely expressed. NKG2DL CAR-T cells have been shown to have remarkable in vitro activity against T-ALL cell lines (Jurkat, HPB-ALL, KOPT-K1, DND-41) [98].
Acute myeloid leukemia (AML)
The incidence of AML is high among adults, and it is the second most prevalent form of pediatric leukemia. In these patients, HSC proliferates uncontrollably and overproduces immature and functionally abnormal white blood cells [99]. Chemotherapy is a commonly used treatment strategy nowadays but often leads to poor outcomes due to the limitations of the approach, such as toxic effects on healthy tissue. HSCT becomes another option. However, the five-year survival rate for patients with relapsed AML is only about 27% [100, 101]. Given that CAR-T cells may specifically target antigens on leukemic stem cells and progenitor cells, they have enormous application potential. However, since many myeloid antigens are also expressed on healthy HSC, the critical challenge currently limiting the adoption of CAR-T cell therapies in this field is appropriate to target selection.
As genealogy-limiting antigens, CD33 and CD123 are currently the most studied CAR-T cell therapeutic targets (Fig. 2). CD33 and CD123 are expressed in approximately 99 and 78% of AML malignant cells, respectively, and CAR-T cell therapies that target them are effective in preclinical trials [102,103,104,105,106]. Many relevant clinical trials are underway. However, CD123, widely expressed in adult AML, may be less represented in children [104]. In addition, myeloid and hematopoietic progenitor cells express CD33 and CD123, which may hinder their practical application [107, 108].
AML-initiating cells express CD38, while normal human HSC does not. In a phase 1/2 clinical trial (NCT04351022), four out of six AML patients treated with CD38 CAR-T cells achieved CR, the median of 7.9 months, with no severe adverse events (CTCAE v4.0) [109] (Fig. 2).
Since it is significantly expressed in AML cells but not in healthy HSC or non-hematological cells, C-type lectin-like molecule 1 (CLL1) represents a promising target for CAR-T cells (Fig. 2). In a phase 1/2 clinical trial of CLL1 CAR-T cells for AML, 3/4 of patients achieved CR, with no high-level adverse events observed (CTCAE v5.0) [110]. Two AML patients who did not recover after multiple lines of salvage therapy, including CD38 CAR-T cell therapy, achieved molecular CR treated with CLL1 CAR-T cell therapy (NCT04884984). And again, there were no high-grade adverse events in patients (CTCAE v5.0) [111]. These trials suggest the great potential of CLL1 CAR-T cells.
LewisY is less expressed in healthy tissues and may also be a good target for AML therapy, given its expression on malignant cells (Fig. 2). In a phase 1 clinical trial (CTX 08–0002), five patients with relapsed AML were enrolled. It was established that the use of LewisY CAR-T cells for the management of AML is feasible and secure [112]. Grade 3 or 4 toxicity was not observed (CTCAE v3.0). One patient achieved cytogenetic remission, one had a reduction in peripheral blood blasts, and one showed prolonged remission.
There are also many promising targets whose effects are not yet supported by clinical trial results. For example, CD7 is expressed by leukemic cells such as AML, which accounts for 30% of all cases, but not normal bone marrow cells (Fig. 2). As a consequence of this, it has the potential to be an intriguing candidate for the selective destruction of cancer cells without affecting the health of normal cells. A study found that CD7 CAR-T cells could effectively eradicate CD7+ AML cell lines (GDM-1 and Kasumi-3), primary CD7+ AML, and colony-forming cells in a xenograft mice model but did not affect the normal cells in bone marrow [113]. Besides, nanobody-based fratricide-resistant CD7-CAR T cells demonstrated a favorable and durable antitumor response in r/r T-ALL/LBL with tolerable toxicity, warranting further studies in highly aggressive CD7-positive malignancies [114]. However, since T cells express CD7, T cell self-mutilation is an issue to be considered for CD7 CAR-T cell therapy.
With standard treatment, the prognosis and clinical outcomes of AML patients with FMS-like tyrosine kinase 3 (FLT3) are poor. Targeted therapy is thus highly anticipated. In a mouse model of AML, FLT3 CAR-T cells allowed for bone marrow recovery without affecting leukemic remission [115] (Fig. 2).
Overwhelming evidence substantiates that CAR-T cells targeting CD117, Siglec-6, CD70, myeloproliferative leukemia protein (MPL), leukocyte immunoglobulin-like receptor-B4 (LILRB4), T cell immunoglobulin and mucin structural domain 3 (TIM-3), membrane-associated folate receptor β (FRβ) and CD44v6 CAR-T cells induce complete remission in immunodeficient mouse xenograft AML models [116,117,118,119,120,121,122,123] (Fig. 2).
Wilms tumor 1 (WT1) overexpression on tumor cells is linked to a poor prognosis in AML patients. In an in vitro assay, WT1 CAR-T cells identified and lyzed WT1+/HLA-A*02:01+ tumor cell lines (AML, AML-14; CML, BV173; ovarian cancer, OVCAR3) [124] (Fig. 2).
CAR-T cells targeting PR1 exhibit a significant affinity in vitro for PR1+ target cells, and they targeted human primary AML cells with a preference [125].
In addition, mesothelin (MSLN) is a possible target of CAR-T cells [126].
Chronic lymphocytic leukemia (CLL)
In Western nations, CLL is the most frequent form of adult leukemia, and its onset is associated with advancing age. However, treatment options are limited, the most effective treatment option is HSCT, but it is rarely used due to the high risks. CAR-T cell therapy might also be helpful for individuals with high-risk CLL who have not seen improvement from standard treatment.
CD19 is the main target of CAR-T cells treating CLL patients (Fig. 2). However, CD19 CAR-T is not as effective in CLL as in ALL. Only 45% of 22 CLL patients treated with CD19 CAR-T cells achieved CR in a phase 1 multicenter clinical trial in 2022 (NCT03331198). 74% of patients had CRS (9% grade 3) and 39% had neurological symptoms (22% grade 3 or 4) (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.03) [127]. In a previous study (NCT02640209), CD19 CAR-T treated 14 patients who had a CRR of only 28%. Six patients had grade 3 or higher CRS, one had grade 4 neurological symptoms lasting 2 days, and B cells were undetectable in all CR patients (CRS, American Society for Transplantation and Cellular Therapy criteria; other adverse events, CTCAE v4.03) [128]. Antigen-negative tumor escape also has a high probability of causing recurrence [129, 130]. These findings emphasize the need for new therapeutic targets and improved technologies.
B-cell maturation antigen (BCMA) is found on plasma cells and advanced B lymphocytes. It has been found to have more significant potential for immunotherapy in CLL patients [131] (Fig. 2). Because soluble BCMA levels are negatively linked with time to treatment failure and OS, but not with the CLL International Prognostic Index, therapeutic methods targeting BCMA may improve the prognosis of CLL patients [132].
On the tumor cells of CLL patients, CD32b is always produced at a considerable locus density, but this is not the case in non-B hematopoietic cells. CD32b CAR-T cells showed intense activity in both primary CLL cells and NSG mice transplanted with patient samples [133].
In a preclinical study, FcμR-specific CAR-T cells have successfully eliminated Mec-1 leukemic cells without affecting healthy B cells [134].
Receptor tyrosine kinase like orphan receptor (ROR) 1 is stably expressed in CLL patients and not on normal, healthy differentiated tissue [135]. ROR1 CAR-T cells are particular and could reduce side effects associated with treatment, such as B cell depletion and hypogammaglobulinemia. Therefore, it is an attractive target for CAR-T cell therapy (Fig. 2).
CAR-T cells targeting Siglec-6 and CD23 separately have been developed, and their effects will be confirmed in future experiments [136, 137].
Additionally, kappa and lambda chains are potential targets [138].
The application of bispecific CAR-T cells offers new hope for CLL treatment. In a phase 1 trial (NCT03019055), of 22 patients receiving CD19 and CD20-targeted CAR-T cells, 14 (64%) achieved CR, one (5%) developed grade 3–4 CRS, and three (14%) developed grade 3–4 neurotoxicity, suggesting that this therapeutic approach has good potential (CRS, American Society for Transplantation and Cellular Therapy criteria; other adverse events, CTCAE v5.0) [51].
Multiple myeloma (MM)
MM is a cancer of the plasma cells, second only to leukemia among hematologic malignancies. Despite substantial advancements over the last two decades, the prognosis for people with MM remains bleak. CAR-T cells have been demonstrated to have potential as a treatment option for patients with recurrent or refractory multiple myeloma (r/r MM).
BCMA is the most effective target for CAR-T cell therapy in MM among the numerous possible targets (Fig. 2). In normal cells, BCMA is primarily expressed by plasma cells and a small percentage of mature B cells, while it is absent from most B cells and other organs. BCMA is a highly desirable target for immunotherapy since it is extensively expressed in MM malignant cells [139]. In 2021, the FDA authorized idecabtagene vicleucel (Abecma) for use in patients with r/r MM who have failed fourth-line therapy. Idecabtagene vicleucel is the first FDA-approved CAR-T cell therapy to manage MM. In a phase 2 study including 128 patients with r/r MM (NCT03361748), patients had an ORR of 73%, a CRR of 33%, and a PFS of 8.8 months, and almost all had grade 3 or 4 toxicities (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.03) [140]. The FDA also approved a second BCMA-targeted CAR-T cell product, ciltacabtagene autoleucel (Carvykti), for the treatment of MM in 2022 [141]. The targeting domain of this CAR-T cell product is based on single-domain antibodies [142]. In a phase 1b/2 trial (CARTITUDE-1, NCT03548207), 67% of the 97 patients who received infusion ciltacabtagene autoleucel achieved CR. The rate at 12 months is 89%. Grade 3 and above hematological adverse events were common in patients, 21% of patients had neurotoxicity, and most patients who experienced CRS remitted, demonstrating the good-excellent efficacy and safety of the product (CRS, American Society for Transplantation and Cellular Therapy criteria; other adverse events, CTCAE v5.0) [142]. In the clinical trial of LCAR-B38M (NCT03090659), 100 participants were enrolled. A nanobody-based BCMA-redirected CAR-T cell treatment (LCAR-B38M) that targets two separate BCMAepitopes showed a 68% CRR, 15 months of PFS, and 65% rate of grade 3 and above adverse events in the patient population, suggesting its good performance (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.03) [143]. An autologous second-generation BCMA-redirected CAR-T constructed on humanized alpaca-derived anti-BCMA nanoantibodies demonstrated safety and efficacy in a trial of 16 patients with r/r MM (NCT03661554). Three patients with extramedullary lesions achieved PR within 1 month, and the overall response rate was 84.6% in the 13 patients without the extramedullary disease. Only two patients had CRS of grade 3 or above; the rest had mild CRS (grade 0 to 2) [144]. A separate report of the results of this clinical trial showed that as of 1 February 2021, 34 patients with MM had received this CAR-T cell with an overall response rate of 88.2% and an mPFS of more than 1 year, with haemocytopenia being the most common adverse effect and all greater than grade 3 (CTCAE v5.0). Twenty nine patients experienced CRS (any grade) (Lee 2014 criteria) [145]. This further confirms the efficacy and safety of nanobody-based BCMA retargeted CAR-T cell therapy for r/r MM patients. In 2021, a meta-analysis counted 22 studies using BCMA CAR-T cells for MM, with mean ORR and CRR of 85.2 and 47.0%, respectively [146]. Recent research found that suppressing elevated anti-apoptotic proteins in MM cells via bone marrow mesenchymal stromal cells could boost the efficiency of BCMA CAR-T cells (ChiCTR1800017051, ChiCTR2000033925) [147]. However, BCMA CAR-T cell therapy is associated with a high prevalence of toxic side effects and recurrence. Combining γ-secretase (GS) inhibitor (GSI) with CAR-T cells targeting BCMA is a possible solution. The GSI inhibited the decrease in antigen concentration caused by GS cleavage of BCMA on the tumor cell surface and the release of soluble BCMA fragments, which could hinder the function of CAR-T cells [148, 149]. In MM tumour-bearing NSG mice treated with GSI, BCMA expression on malignant cells was upregulated, soluble BCMA fragments in peripheral blood were reduced, and the efficacy of BCMA-targeted CAR-T cells was significantly enhanced [150]. Clinical trials combining GSI with CAR-T cells targeting BCMA are already underway (NCT03502577).
Exploring CAR-T cells that target new targets may also be an excellent way to address the problem of relapse. Two out of 10 patients showed significantly longer PFS after HSCT and CD19 CAR-T cell therapy compared with HSCT alone. No patient experienced severe CRS, which demonstrated the potential benefit of CD19 CAR-T cells for r/r MM patients (NCT02135406) [151] (Fig. 2). In a phase 1 trial, seven MM patients received κ light chain CAR-T therapy, four of whom had stable disease for two to seventeen months (NCT00881920). No toxicity attributed to CAR-T cells has been observed (CTCAE v4.03) [44].
Many potential targets are currently being explored (Fig. 2). For instance, CAR-T cells targeting signaling lymphocytic activation molecule F7 (SLAMF7) and signaling lymphocytic activation molecule F3 (SLAMF3) for untreated and chemo-resistant MM patients have shown efficient killing in both in vitro and in vivo experiments. Compared with BCMA, SLAMF7 is a surface glycoprotein and is more evenly expressed on myeloma cells and less on B cells [152]. Patients who relapsed after receiving BCMA CAR-T cells may benefit from treatment with SLAMF7 CAR-T cells. SLAMF7 CAR-T cells demonstrated its anti-myeloma-killing effect in mouse models [153]. Clinical trials targeting SLAMF7 are ongoing (NCT03958656, NCT04499339). SLAMF3 CAR-T cells showed strong cytotoxicity in patients’ primary tumor cells and MM cell lines U-266 and RPMI-8226. In a xenograft mouse model, CAR-T cells also demonstrated strong anti-tumor effects and significantly prolonged the survival of mice [154].
.In 52% of MM patients, the LewisY antigen is present [155]. LewisY CAR-T cells have been shown to have the potential to persist and exert anti-tumor effects after infusion into patients (Fig. 2). Their specific efficacy is yet to be verified in further experiments [156].
According to several reports, G protein-coupled receptor class-C group-5 member-D (GPRC5D) could be an essential target (Fig. 2). Hair follicles seem to be the only normal tissue in which GPRC5D expression has been discovered outside of cancerous bone marrow plasma cells [157]. Researchers developed a humanized GPRC5D CAR-T cell and found that it could eradicate tumor cells in a mouse MM model of BCMA antigen escape without causing significant toxic side effects [157].
CD44v6 is considered one of the tumor stem cell markers. Preclinical studies have shown that CD44v6 CAR-T cells exhibit potent antitumor activity against MM but lead to a reduction in beneficial monocytes in mouse models [158] (Fig. 2).
Besides, New York esophageal squamous cell carcinoma-1 (NY-ESO-1) is an intracellular protein whose peptide could be presented on the cell surface by MHC molecules when it is ubiquitinated and degraded in the cell [159]. NY-ESO-1 is expressed in about 60% of MM patients, with higher levels in individuals with relapses, suggesting that NY-ESO-1 is intimately linked to MM disease progression [160]. In the context of HLA-A*02:01, CARs that recognize the NY-ESO-1 immunodominant peptide 157–165 were made to redirect autologous CD8(+) T cells to NY-ESO-1(+) MM cells. Preclinical trials confirmed the targeting effect, cytokine secretion, and ability to induce immune memory in NY-ESO-1 CAR-T cells [161].
Given that NKG2DL is expressed in MM malignant cells but not in healthy tissues, it has huge prospects for clinical application (Fig. 2). However, existing NKG2DL CAR-T cells have limited amplification and persistence in MM patients. For improved clinical efficacy, more research is needed to improve NKG2DL CAR-T cell expansion [162].
Interestingly, it has been shown that CD126 CAR-T cells infiltrate, expand, and kill tumor cells in a MM xenograft model without producing toxic effects, suggesting its great potential [163].
Researchers have created nanobodies against CD38 and constructed CD38 CAR-T cells from them (Fig. 2). The cells showed strong toxic effects against CD38+ MM cell lines (LP-1, RPMI 8226, OPM2, MOLP8, and primary MM cells from patients) and inhibited tumor growth in mice inoculated with RPMI 8226 cells [164]. However, it should be borne in mind that CD38 is also expressed at moderate levels in hematopoietic progenitor cell subpopulations and some normal hematopoietic cells [165].
CD138 is a primary diagnostic marker for MM and is a desirable target for the treatment of MM [166] (Fig. 2). Nonetheless, CD138 CAR-T cells may also attack normal skin and mucosal tissues [167].
CD56 is a possible immunotherapeutic target strongly expressed by malignant plasma cells in 70% of MM patients [168] (Fig. 2). The expression of CD56 on the central and peripheral nervous system has raised neurotoxicity concerns.
Given that MM is phenotypically heterogeneous, a single CAR-T treatment targeting only one antigen is challenging to attain long-term CR. Bispecific CAR-T cells targeting BCMA and CD38 were found to lead to clinical responses and minimal residual disease negative in 87% of MM patients in a phase 1 experiment (ChiCTR1800018143, 23 participants). Grade 3–4 hematological toxicity is more common in patients, rarely reaching grade 3 CRS, without neurological symptoms (CRS, Lee 2014 criteria; other adverse events, CTCAE v5.0) [169]. In another phase 2 trial (ChiCTR1800017051, 22 participants), the treatment resulted in CR in 55% of patients, and 27.3% of patients experienced an adverse event more significant than or equal to grade 3 (CRS, American Society for Transplantation and Cellular Therapy criteria; other adverse events, CTCAE v4.03) [170]. CD19/BCMA CAR-T cell therapy showed promising results in a phase 2 trial (ChiCTR-OIC-17011272, 62 participants), with CR or better outcomes observed in 60% of patients, CRS in 95% of patients, of which 10% were grade 3 or higher, and neurotoxic events in 11% of patients, of which 3% were grade 3 or higher (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.03) [171]. Research on multi-specific CAR-T cells may lead to a breakthrough in the treatment of MM.
CAR-T cell therapy offers great promise in treating patients with hematological malignancies. Although there is still much room for development, it is currently showing exciting trends in B-ALL, MM, and B-NHL, especially in B-ALL and B-NHL. CD19 CAR-T cells have achieved excellent results in many cases of blood cancer. However, CAR-T cell therapy needs further exploration to treat patients with T-cell malignancies. The drug resistance of cancerous tissues to CAR-T cells and the possible side effects of treatment, such as severe inflammatory toxicity, are issues that need further research. Multi-target-specific CAR-T cells are currently the most commonly used treatment against drug resistance. HSCT and immunoglobulin transplantation could partially reduce the side effects of treatment, but they may have other adverse effects and may not be ideal solutions.
The most frequent CAR-T cell therapy targets in hematological malignancies are listed (Fig. 2).
Solid tumor
Breast cancer
Current evidence suggests that breast cancer (BC) accounted for 11.7% of all cancer types in 2020, surpassing lung cancer as the most prevalent cancer [172]. Recent advances in therapeutic approaches have improved BC patient survival and quality of life, but mortality rates remain high due to drug resistance limiting efficacy. Some members of the receptor tyrosine kinase (RTK) family and cell surface proteins are the primary targets for CAR-T cells to treat BC. Other targets include immune checkpoint, Ephrin type-A receptor (EphA) 10, stress ligands, disialoganglioside, and serum tumor markers [173].
Five RTKs, including human epidermal growth factor receptor 2 (HER2), epidermal growth factor receptor (EGFR), c-mesenchymal-epithelial transition factor (c-MET), ROR1, and AXL, are currently known to be targeted by CAR-T cells in BC to elicit therapeutic potential (Fig. 3). CAR-T cells have functioned in various preclinical BC models using these antigens (HER2, EGFR, c-MET, ROR1, AXL) as targets [174,175,176,177,178]. Four RTK targets have started clinical studies, including HER2, EGFR, c-MET, and ROR1. Currently, mRNA electroporation is considered the safest gene transduction method in T cells. The mRNA encoding the target gene is introduced into the cytoplasm by electroporation. It is also modified to increase stability and long-term expression. Although mRNA technology is efficient and easy to design in terms of transducing CARs compared to other transduction techniques, it also has a short lifespan [179]. In a phase 1 clinical trial, 3 × 107 or 3 × 108 c-MET CAR-T cells constructed via mRNA were administered to six patients with metastatic BC and showed well-tolerated results with inflammatory responses. None of the patients experienced more than grade 1 study drug-related adverse reactions (CTCAE v4.0). Only three patients developed grade 1 erythema (CTCAE v4.0). This trial used an intratumoral injection route, c-MET CAR-T cells, and an anti-tumor response could be detected at the injection site (NCT01837602) [176]. The median follow-up was ten months (range 3–28 months), with two patients progressing, three patients dying from the disease, and one with stable disease. The latest clinical trials of CAR-T cell therapies targeting HER2 (NCT05007379), EGFR (NCT05341492), and ROR1 (NCT05274451) for BC have begun between 2021 and 2022. But all clinical trials of CAR-T cells targeting HER2, EGFR, and ROR1 for BC have not reported results.

Common CAR-T cell therapy targets in solid tumors. EGFR: epidermal growth factor receptor; GD2: ganglioside2; MUC1: mucin 1; NKG2DL: natural killer group 2 member D ligand; HER2: human epidermal growth factor receptor 2; HNSCC: head and neck squamous cell carcinoma; TC: thyroid cancer; ICAM-1: intercellular adhesion molecule-1; LCa: lung cancer; PD-L1: programmed death-ligand 1; MSLN: mesothelin; ROR: receptor tyrosine kinase like orphan receptor; CEA: carcinoembryonic antigen; NY-ESO-1: New York esophageal squamous cell carcinoma-1; DLL3: delta-like ligand 3; BC: breast cancer; OS: osteosarcoma; EWS: Ewing’s sarcoma; MEL: melanoma; VEGFR-2: vascular endothelial growth factor receptor 2; RCC: renal cell carcinoma; CAIX: carbonic anhydrase IX; GPC3: glypican-3; OC: ovarian cancer; CC: cervical cancer; ALPPL2: alkaline phosphatase placental-like 2; PCa: prostate cancer; PSMA: prostate-specific membrane antigen; PSCA: prostate stem cell antigen; CRC: colorectal cancer; PC: pancreatic cancer; CLDN: claudin; GC: gastric cancer; GUCY2C: Guanylyl cyclase C; HCC: hepatocellular carcinoma; EC: esophageal cancer
Overexpressed proteins on the surface of BC cells suggest that they may be good candidates for CAR-targeted therapeutic interventions. At present, they mainly include mucin 1 (MUC1), mesothelin (MSLN), CD70, CD133, CD44v6, epithelial cell adhesion molecule (EpCAM), chondroitin sulfate proteoglycan 4 (CSPG4), intercellular adhesion molecule-1 (ICAM-1), Tumor endothelial marker 8 (TEM8), a trophoblast cell surface antigen 2 (TROP2), and folate receptor alpha (FRα) [173, 176, 180,181,182,183,184,185,186,187,188,189] (Fig. 3). Only six surface proteins have entered clinical studies, including MUC1 (NCT02580747), MSLN (NCT02414269), CD70 (NCT02830724), CD133 (NCT02541370), CD44v6 (NCT04430595), and EpCAM (NCT02915445). The above NCT numbers refer to each target’s most recently started or updated clinical trials. The safety and efficacy of CAR-T cell treatments targeting MSLN and EpCAM are evaluated in phase 1 clinical trials. The remaining CARs are in phase 1 and phase 2 clinical trials. In addition, the three CAR-T cells targeting MUC1 yielded heterogeneous effects in different clinical trials (NCT04020575, NCT04025216, and NCT02587689, respectively), targeting different structural domains of the cleaved form of MUC1, aberrant glycated MUC1, and entire MUC1 [173]. No preclinical evidence for CD44v6 CAR-T cell therapy for BC could be found. Still, it has also entered clinical trials due to its proven anti-tumor capacity in other preclinical cancer models [190]. In contrast, several preclinical studies on the remaining ten cell surface protein targets (CD133, MUC1, MSLN, CD70, EpCAM, CSPG4, ICAM-1, TEM8, TROP2, FRα) have shown potential as CAR-T targets for the treatment of BC [180,181,182,183,184,185,186,187,188,189].
Target selection is one of the determinants of CAR-T cell efficacy. Therefore, researchers have been working hard to identify new targets. Recent preclinical studies reported CAR therapy targeting EphA10 as a promising strategy for treating triple-negative BC [191].
Li et al. designed a novel PD-L1-targeted shark VNAR single-domain CAR-T cell. Shark VNAR is small that could bind epitopes difficult to conventional antibodies. They found that this type of CAR-T cell could lyse cancer cells in breast and liver cancer models by targeting immunosuppressive microenvironment antigen (PD-L1) [192].
Preclinical trials of ganglioside 2 (GD2), protein tyrosine kinase 7 (PTK7), and NKG2DL as CAR-T cell therapy targets also showed anti-BC activity [177, 193,194,195] (Fig. 3). Clinical trials on the safety and efficacy of GD2 CAR-T cells are underway (NCT04430595, NCT03635632).
No preclinical study of CAR-T cells for BC has targeted the serum tumor marker carcinoembryonic antigen (CEA), while a clinical trial of CAR-T cell therapy targeting CEA to remedy BC subjects is underway (NCT04348643) (Fig. 3).
In conclusion, although many institutions have registered many BC clinical trials of CAR-T cells over recent years, few results have been published. Accordingly, more researches are indispensable to validate and compare the effectiveness of different targets.
Lung cancer (LCa)
LCa is one of the most prevalent tumors globally, with a high degree of malignancy and poor prognosis [172]. Based on histological features, LCa could be divided into non-small cell lung cancer (NSCLC), which accounts for 85% of diagnosed LCa cases, and small cell lung cancer (SCLC) [196]. Although the prognosis of LCa has improved significantly in recent years with targeted and immune drugs, it is still unsatisfactory, and the mortality rate is high [196]. Mounting evidence suggests CAR-T cell therapy is effective in treating NSCLC [197].
According to the literature, CAR-T cells’ most common targeted antigens in NSCLC are EGFR, MSLN, CEA, PD-L1, ROR1, B7H3, MUC1, HER2, and Delta-like ligand 3 (DLL3) [177, 198,199,200,201,202,203,204] (Fig. 3). These targets have been proven to have tumor-suppressive effects in preclinical models and applied in clinical trials. Clinical studies of CAR-T therapy for LCa have published outcomes from a phase 1 trial in which EGFR CAR-T cells generated by the piggyBac transposon system were well tolerated by all patients with advanced relapsed/refractory EGFR(+) NSCLC (n = 9), with no reports of grade 4 adverse events or severe CRS (NCT03182816, CTCAE v5.0) [205]. The piggyBac transposon system was chosen to construct CAR-T cells in NCT03182816 because it is more straightforward and cost-effective than viruses. One patient had a sustained response of more than 13 months, while six and two patients had stable disease and progressive disease, respectively. The median progression-free survival (mPFS) was 7.13 months, with an mOS of 15.63 months. The above results indicate that this therapeutic approach is safe and effective [205]. Other than this, no remaining clinical trial results were reported. MSLN has the eleven LCa clinical trials registered on Clinicaltrials.gov, but no experimental results are currently available. CAR-T cells targeting MSLN killed NSCLC cells and exhibited greater anti-tumoural capacity than unmodified T cells in mouse models. Still, persistence is an issue that needs to be addressed [206].
DLL3 is considered a novel target for SCLC treatment; increased expression of DLL3 was found in SCLC and other neuroendocrine tumors, with lower expression levels in most normal tissues [207] (Fig. 3). However, clinical trials of DLL3 CAR-T cells for treating relapsed/refractory small cell LCa have been suspended due to the absence of active subjects in the trial (NCT03392064).
The NCT numbers of the latest initiated or updated clinical trials for the remaining targets are listed here for reference: CEA (NCT04348643), PD-L1 (NCT03330834), ROR1 (NCT05274451), EGFR/B7H3 (NCT05341492), MUC1 (NCT05239143), and HER2 (NCT04660929).
MAGE-A1 antigen, glypican-3 (GPC3), FRβ, CD44v6, CD133, c-MET, Olfactory receptor 2H1 (OR2H1), CD47, GD2, CD147, prostate stem cell antigen (PSCA), Fibroblast activating protein (FAP), EphA2 and PTK7 are also expected to be targets of CAR in the context of LCa, with preclinical studies completed [190, 195, 203, 204, 208,209,210,211,212,213,214,215,216,217,218] (Fig. 3). However, the relevant clinical trials are still not registered to be conducted.
Likewise, CLEC14A is an overexpressed tumor endothelial marker with relatively negligible physiological expression in normal endothelial cells. CLEC14A-redirected CAR-Ts sufficiently released IFN-γ and enforced anti-tumor effects in vitro. The intelligence behind the targeting of CLEC14A is that it is a glycoprotein with elevated expression in various solid tumors [219]. The treatment of CLEC14A-redirected CAR-Ts significantly inhibited tumor growth in Lewis lung carcinoma, Rip-Tag2, and mPDAC mouse models without signs of toxicity [220]. No registered clinical trials are using CLEC14A CAR-T cells to treat LCa.
Although CAR-T cell immunotherapy has demonstrated potential in various preclinical models of LCa, the pool of targeting antigens still needs to be expanded. More novel approaches need to be applied to find them. For example, antigens with a significantly higher expression on the surface of tumor cells could be used as targets. CXCR4 is highly expressed in LCa and is expected to be a novel target for NSCLC [221].
Anti-NY-ESO-1 TCR-transduced T cells have been shown to kill LCa cells (A549-A2-ESO) and depress the growth of tumors in xenograft mice models, but CAR-T cell clinical trials targeting this antigen have not been conducted [222].
Prostate cancer (PCa)
PCa is the most common tumor of the male genitourinary system, with more than 1.4 million cases and over 375,000 deaths worldwide [172]. Fatal metastatic debulking-resistant PCa is a late-stage sequela with only a median survival of 10 months to 21.7 months, a 30% five-year survival rate, and a poor prognosis. Although radiation, chemotherapy, and hormonal therapies have significantly progressed in treating PCa, limited treatment is available for patients with advanced diseases [223].
It is widely thought that prostate-specific membrane antigen (PSMA) is an attractive target that could be used to treat PCa (Fig. 3). PSMA is predominantly expressed in the healthy prostate and, to a lesser extent, in other tissues, including the intestine, brain, kidney, lacrimal gland, and salivary gland [224]. Notably, PSMA is expressed in almost all primary and metastatic PCas [225]. However, PSMA-directed CAR-T cells are less effective in lysis therapy. Indeed, CAR-T cells must overcome the immune-cold TME and efficiently transport and penetrate the site of tumor metastasis [226, 227]. Christopher C Kloss et al. improved the efficacy and safety by blocking transforming growth factor-β (TGFβ) signaling in T cells, allowing CAR-T cells to work better in PCa models [228]. They conducted a concurrent clinical trial with four therapeutic dose levels of TGFβ-insensitive armored CAR-T cells administered to 13 subjects (NCT03089203). Five patients were observed to develop grade ≥ 2 CRS, including one with prostate-specific antigen (PSA) reduction > 98%, and one died after experiencing grade 4 CRS complicated by sepsis (American Society for Transplantation and Cellular Therapy criteria). After adoptive cellular transfer, three other patients were found to have ≥30% reduction in PSA and CAR-T cell failure with simultaneous upregulation of multiple TME local suppressor molecules. The median of 15.9 months was good [228]. In conclusion, the clinical use of combining TGFβ blockade and PSMA CAR-T cells is promising and generally secure, and therapeutic approaches in combination with targeting inhibitory factors should be feasible. In addition, Claudia Arndt et al. built a modular platform called UniCAR. Here, they constructed a UniCAR epitope in combination with PSMA-11 to generate a compound that redirects UniCAR T cells to tumor cells. The advantage of UniCAR T cells is that bispecific bridging molecules, called target modules, could mediate them and do not interact directly with tumor cells like conventional CAR-T cells [225]. This finding provides a good tool and direction for developing diagnostic imaging and targeted therapy for PCa.
PSCA has gained significant attention as an important marker for bladder, prostate, and pancreatic cancers [227] (Fig. 3). Currently designed PSCA CAR-T cells have shown substantial antitumor effects in disease models of prostate and pancreatic cancers [229, 230].
Deng et al. demonstrated that in EpCAM CAR-T (Fig. 3), human peripheral blood lymphocytes have antitumor activity against PCa [231].
IL-7 was found to have an enhanced effect on NKG2DL CAR-T cell immunotherapy, which provides a therapeutic approach [232] (Fig. 3).
The clinical trials targeting PSCA (NCT03873805, NCT02744287), EpCAM (NCT03013712), and NKG2DL (NCT04107142) have not reported their results.
In addition, CAR-T cells targeting CEA, B7H3, MUC1, and CD126, respectively, have been found to play different roles, although all have antitumor activity [163, 233,234,235]. In addition to the therapies mentioned above, CAR-T cells with an inducible “ON” safety switch have recently been designed and shown to improve outcomes [236]. NCT04249947 is an ongoing phase 1 study targeting PSMA CAR-T cells using rimiducid as the “ON” or safety switch activator, which controls initiation and activation and could reduce toxic responses in a controlled manner.
Colorectal cancer (CRC)
CRC is the second leading cause of cancer-related death [172]. Disease control or cure could be achieved through early detection by screening and good results with conventional therapies for localized tumors. However, metastatic CRC remains a tricky problem [237], and patients with metastatic CRC have been the focus of CAR-T cell therapy. The first trial of CAR-T cells for solid tumors was conducted in the 1990s. Patients with metastatic CRC were treated with CAR-T cells targeting TAG72 in two phase 1 trials, one by intravenous infusion and the other by hepatic artery infusion (Fig. 3). Difficulties in T-cell transport to metastatic sites were found, but their relative safety was also demonstrated [238].
Many antigens targeted by CAR-T cell therapies for CRC have been tested and validated in preclinical studies and clinical trials in recent years. CEA is the most promising target for disseminated CEA CRC (Fig. 3). Current evidence suggests that CEA is overexpressed as a serum marker in 98.8% of CRC tissues [239]. Therefore, CEA is considered an attractive target for CAR-T therapy in CRC. Several clinical trials have been conducted for CAR-T therapies targeting CEA. In a phase 1 clinical trial (NCT02349724), CAR-T cells targeting CEA were applied systemically in 10 patients with metastatic CRC. The treatment was effective and well-tolerated even at high dose levels. Seven patients who experienced the progressive disease in previous treatments were stabilized, two of whom were stable for more than 30 weeks, and two others experienced tumor shrinkage. No serious adverse events associated with CAR-T cell therapy have been observed [240]. The efficacy could also be enhanced by lymphodepletion with cyclophosphamide/fludarabine chemotherapy [240]. This trial suggested that the safety profile of CEA CAR-T cell therapy is good, with only mild and manageable adverse effects associated with CRS, which was demonstrated by another trial [241]. Even during long-term observation, the decrease in serum CEA levels was evident in most patients. However, CEA CAR-T cells have been reported to induce transient colitis because CEA is expressed on normal gut epithelial cells [242]. To address this issue, Mark et al. designed CEA Tmod cells using a CAR activated by CEA and an LIR-1-based inhibitory receptor triggered by HLA-A*02 [243]. These cells could harness the loss of HLA heterozygous genes in tumors to safely and effectively kill tumor cells. However, unlike CEA CAR-T cells, Tmod cells still could specifically target tumor cells in the presence of cells expressing HLA-A*02 [243]. However, there are no clinical trials of Tmod cells.
CD133 is highly expressed on many solid tumors, and CD133 is a marker for cancer stem cells (CSCs) and endothelial progenitor cells [244]. Clinical trials for CAR-T cells targeting CD133 have also been published (Fig. 3). An antitumor response was observed in phase 1 clinical trial, NCT02541370, that recruited 23 patients suffering from HCC (n = 14), PC (n = 7), and CRC (n = 2), treated with CD133 CAR-T cells [245]. Three achieved PR, 14 patients were SD, and the mPFS was 5 months. The 2 CRC patients had SD. More extended periods of disease stabilization could be observed after repeated infusions of cells and are more effective in patients who have achieved some efficacy after the first cell infusion [245]. The primary toxicity is hemoglobin/thrombocytopenia (≤ grade 3), which recovers spontaneously within 1 week (CTCAE v4.0).
The NCT numbers of the latest initiated or updated clinical trials for the remaining targets are listed here for reference: MUC1 (NCT05239143), MSLN (NCT05089266), EpCAM (NCT05028933), HER2 (NCT04660929), NKG2DL (NCT04550663) and GUCY2C (NCT05287165). But there are no reports on the results of these experiments.
The combination of regorafenib and EpCAM CAR-NK cells performs more effectively in human CRC models than monotherapy with CAR-NK cells or regorafenib [246].
CAR-T cell therapy targeting HER2 showed potent results in animal models of CRC [247] (Fig. 3). Still, it resulted in acute respiratory failure syndrome in a case report, highlighting the need for further improvements [248].
In addition, the antitumor efficacy of NKG2DL RNA CAR-T cells was confirmed in a mouse model of peritoneal metastasis of colon cancer [249] (Fig. 3).
Guanylyl cyclase C (GUCY2C) CAR-T cells designed by Magee et al. identified and killed CRC cells that endogenously express GUCY2C [250].
MSLN, MUC1, placental alkaline phosphatase (PLAP), c-MET, and Cadherin-17 (CDH17) are also promising targets in CAR-T cell therapies for CRC, validated in several preclinical trials [251,252,253,254,255] (Fig. 3).
Gastric cancer (GC)
GC is a common malignancy globally, with gastric adenocarcinoma accounting for more than 90% of cases [256]. Despite the continuous improvement and innovation of therapeutic approaches for GC, treatment options for GC remain limited. CAR-T cell therapies are currently considered a promising therapeutic approach, with multiple target antigens that may be effective targets.
Claudin (CLDN) 18.2 was present in 70% of primary gastric adenocarcinomas and their metastases [257] (Fig. 3). In an ongoing, open-label, single-arm, phase 1 clinical trial, three different doses of CAR-T cells which aim at CLDN18.2 were employed for the treatment of CLDN18.2+ gastrointestinal cancers; 37 patients were treated, with 94.6% of patients experiencing grade 1 or grade 2 CRS but no serious adverse effects (NCT03874897, American Society for Transplantation and Cellular Therapy criteria); the ORR was 48.6%, and disease control rate (DCR) was 73.0%; the ORR and DCR of GC patients reached 57.1 and 75%, and the 6-month OS rate of GC patients reached 81.2% [258]. This finding corroborates the safeness and potency of CLDN18.2 CAR-T cells in CLDN18.2+ gastrointestinal cancers, especially in GC patients.
Many clinical trials of CAR-T cells targeting these targets (HER2, CEA, EpCAM, CLDN18.2, MSLN, MUC1, NKG2DL, EGFR, B7H3) have been registered and conducted [259] (Fig. 3). Clinical trials using CAR-T cells targeting ROR2 and CD44v6 for GC have also been reported to validate their feasibility and safety, but preclinical studies are scarce (NCT03960060, NCT04427449). However, Other than the CLDN18.2 results mentioned above, no other GC clinical trial results have been published.
Significantly, EpCAM is overexpressed in more than 90% of GC and has aroused interest due to its homogeneous expression [260]. In preclinical studies, CAR-T cell therapies targeting EpCAM have demonstrated antitumor effects [261].
HER2 is overexpressed in 10–20% of GCs and could affect CSCs (Fig. 3). Preclinical studies demonstrated that CAR-T cells targeting HER2 could recognize and lyse GC cells (N87, 7901, AGS, HGC27, MGC803, BGC823, MKN45, primary GC cells) with high affinity and significantly inhibited the in vivo tumorigenic capacity of CSCs [262].
CEA is also a potential target of CAR-T cells for treating GC since the high expression on the tumor cells and combining CEA CAR-T cells with recombinant human IL-12 significantly inhibited tumor growth [263] (Fig. 3).
In addition, the potential of CAR-T cells targeting MUC1, MSLN, NKG2DL, EGFR, and B7H3 has been validated in preclinical studies [264,265,266,267,268].
The NCT numbers of the latest initiated or updated clinical trials for these targets are listed here for reference: HER2 (NCT04660929), CEA (NCT05396300), EpCAM (NCT05028933), MUC1 (NCT05239143), MSLN (NCT03941626), EGFR (NCT03740256), B7H3 (NCT04864821), and NKG2DL (NCT04550663).
Indeed, CAR-T cell therapy still faces many problems, and finding new targets is the key to improving the therapeutic efficacy. Researchers substantiated the effectiveness of CAR-T cells targeting PSCA, FRα, PD-L1, c-MET, CD133, CDH17, ICAM-1, and urokinase plasminogen activator surface receptor (uPAR) in GC models in the last 2 years [255, 269,270,271,272,273,274,275].
In addition, antigens such as B7H6, ARP2/3, NRP-1, DSC2, AE1/2, TAG72, and CA19–9 have been suggested as possible targets for GC treatment with CAR-T cells [259, 276] (Fig. 3).
To further improve the efficacy, Zhao et al. designed bispecific Trop2/PD-L1 CAR-T cells with a significantly enhanced ability to inhibit tumor growth by intratumoral injection [277]. Because Trop2 and PD-L1 are highly expressed in various solid tumors, the bispecific cells could target two antigens (Trop2/PD-L1) with high specificity and be capable of blocking the PD-1/PD-L1 signaling pathway.
Liver cancer
Liver cancer currently ranks sixth in incidence among common malignancies worldwide and is the third leading cause of cancer-related deaths [172]. 85–90% of primary liver cancers are hepatocellular carcinoma (HCC), and surgery is often not indicated since most patients are diagnosed with HCC at an advanced stage [278]. Nowadays, targeted therapy and immunotherapy have achieved good results compared to the previous ones, but the prognosis of liver cancer is still poor.
Glypican-3 (GPC3) enhances HCC cell proliferation through the Wnt/β-catenin pathway and is the most commonly used target site of CAR-T cell therapies for HCC (Fig. 3). GPC3 has been documented in 72% of HCC patients, and 53% had significantly high serum GPC3 levels [279]. The high specificity and sensitivity of GPC3 have made it a target for diagnosing and treating HCC. Jiang et al. showed that CAR-T cells targeting GPC3 could inhibit tumor growth significantly in an in vivo model [280]. Many clinical studies of CAR-T cells targeting GPC3 for liver cancer are underway. In published phase 1 trial results, GPC3 CAR-T cells that could secrete IL-7 and CCL19 were injected intratumorally in a patient with advanced HCC. The tumor was eliminated within 30 days (NCT03198546). The patient developed severe fever, and no other serious side effects were observed [281].
CEA is also a target that has been studied in-depth (Fig. 3). In a phase 1b HITM-SIR clinical trial, Steven C. Katz et al. used CEA CAR-T cells to treat six patients with CEA+ liver metastases. CEA CAR-T cells infused via the hepatic artery were well tolerated. No grade 4 or 5 toxicities, severe CRS, or neurotoxicity were observed (CTCAE v4.03). And biological responses were demonstrated following conventional therapy (NCT02416466) failure with mOS for 8 months [282]. This clinical trial illustrates that CEA CAR-T cells infused by this delivery method could effectively treat liver cancer.
In addition to improving the CAR-T architecture, targeting CSCs is a strategy since CSCs play an essential role in promoting tumors. CD133 is considered a marker of CSCs (Fig. 3). In a single-arm, open-label phase 2 clinical trial, 21 advanced HCC patients were infused with CD133 CAR-T cells (NCT02541370). One was in PR, 14 individuals had SD for 2 to 16.3 months, and 6 had PD [283]. Four patients developed grade 3 hyperbilirubinemia, two had grade 3 anemia, and no other serious adverse events occurred (CTCAE v4.0). These findings indicate that CD133 CAR-T cells have antitumor efficacy and low toxicity in patients with advanced HCC [283]. NCT02541370 is a phase 1/2 clinical trial with phase 1 and phase 2 results published separately. The early results mentioned in the CRC section of the text are from the phase 1 trial, while the subsequent phase 2 trial report only mentions long-term clinical outcomes in HCC patients [245, 283].
In addition, CAR-T cells against DR5 (NCT03941626), MG7 (NCT02862704), HER2 (NCT04842812), and TGFβ (NCT03198546) are also being evaluated in clinical trials for the treatment of liver cancer, and the results are expected to be announced soon.
As molecular technology advances, more antigens are considered potential targets for CAR-T cells to treat liver cancer (Fig. 3). For example, CAR-T cells targeting AFP, EGFRvIII, B7H3, EpCAM, MUC1, NKG2DL, PD-L1, and CD147 were demonstrated in preclinical studies [189, 284,285,286,287,288,289]. Many clinical trials for these targets have also been registered and conducted. But there are still no reports about results. We list here the NCT numbers of the most recently initiated or updated clinical trials for these targets: AFP (NCT03253289), EGFRvIII (NCT03941626), B7H3 (NCT05323201), EpCAM (NCT03013712), MUC1 (NCT04842812), NKG2DL (NCT04550663), PD-L1 (NCT03672305), and CD147 (NCT03993743).
CD44 is a transmembrane glycoprotein that critically mediates cell adhesion, interaction, and migration [290]. scFv-based CD44-redirected CAR-Ts were potentially cytotoxic towards the HCC cell lines (Hep3B2, MHCC97H, SMMC-7721, HepG2, PLC8024) and secreted elevated levels of IL-2, IFN-γ, and TNF-α. Moreover, CD44-redirected CAR-Ts showed no signs of toxicities toward healthy tissues and significantly inhibited tumor growth in CD44-positive HCC xenograft mice [291]. There are no relevant clinical trials available.
To improve the therapeutic effect of CAR for HCC, researchers have developed novel strategies: combining immune checkpoint PD-L1 with CAR-T therapy and designing CAR-T cells targeting c-MET and PD-L1 simultaneously (Fig. 3). These dual-targeted T cells showed more vigorous growth inhibitory activity than single-targeted cells but also enhanced the ability of activated T cells to proliferate and produce INF-γ [292]. Another novel inducible CAR-T cell could control CAR expression. For example, the third-generation gene expression system, Tet-On 3G, could reversibly turn gene expression on or off, achieved by doxycycline (Dox) [289]. Experiments have proved that (Dox+) Tet-CD147 CAR-T cells generated by Tet-On 3G exhibited more potent cytotoxic effects and cytokine secretion than (Dox-) Tet-CD147 CAR-T cells [289].
Esophageal cancer (EC)
EC, which could be classified as esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC), is cancer with limited treatment options and a poor prognosis. EC ranks seventh in terms of incidence (604,000 new cases) and sixth in mortality overall (544,000 deaths) [172]. CAR-T cell therapy represents a potential therapeutic approach for EC. However, there are still no reports of clinical trial results on EC treatment with CAR-T cells.
B7H3 is strongly and uniformly expressed in ESCC and EAC malignant cells but rarely in healthy tissue. B7H3 CAR-T cells effectively kill ESCC tumor cells in human ESCC cell lines (EC109, KYSE150, TE-1, KYSE450, KYSE510, TE-7) and xenograft mouse models. CAR-T cells that induce tumor regression in a xenograft model prolong the survival of mice [293]. Tandem CAR-T cells targeting CD70 and B7H3 also exhibited anti-EC function [189].
HER2 was highly expressed in ESCC but at limited levels in normal esophageal tissues (Fig. 3). HER2 CAR-T cells demonstrated good therapeutic efficacy in HER2+ ESCC tumor cell lines (ECA109, TE-1) and xenograft mouse models [294].
In addition, EpCAM overexpression is associated with low survival in patients with ESCC [295] (Fig. 3). Research on CAR-T cells targeting HER2 and EpCAM-positive cancer is underway (NCT03740256, NCT03013712).
NY-ESO-1 TCR-engineered T cells have also been used in a clinical trial of EC (NCT03941626) (Fig. 3). However, no detailed information about this trial has been reported yet.
To enhance the activation and proliferation of CAR-T cells in solid tumors, Zhang et al. designed JAK-STAT domain-enhanced MUC1 CAR-T cells, which were found to induce the elimination of EC [296] (Fig. 3). This kind of CAR-T cell activated cytokine signaling pathways simultaneously while targeting MUC1. This is because the CAR structure of these cells integrated the IL2 receptor and the YXXQ motif of binding to STAT3, providing cytokine signals. A clinical trial using MUC1 CAR-T for EC is underway (NCT03706326).
Pancreatic cancer (PC)
PC is a malignant digestive system tumor with a distinct immunosuppressive environment. Due to the poor prognosis, the number of deaths from PC (466,000) is almost the same as the number of cases (496,000), which is the seventh most significant cause of cancer death in men and women [172].
The in-depth studied CAR targets in PC are MSLN, EGFR, HER2, CEA, and CD133, and the results of published clinical trials for these targets are summarized below (Fig. 3). A phase 1 study evaluated the toxicity and activity of CAR-T cells against MSLN in patients with malignant pleural mesothelioma (n = 5), ovarian cancer (n = 5), and pancreatic ductal adenocarcinoma (n = 5) (NCT02159716) [297]. In another clinical trial (NCT01897415), two of the six patients with chemotherapy-refractory metastatic pancreatic ductal adenocarcinoma were stable after treatment, and no patient developed dose-limiting toxicity, CRS, or neurological symptoms. The trial found that RNA CAR-T cells did not persist and lacked targeting toxicity [298]. Therefore, this clinical trial (NCT02159716) used MSLN CAR-T cells transduced with a lentiviral vector to increase CAR-T cell levels in peripheral blood. The results showed that MSLN CAR-T cells transduced with a lentiviral vector could expand in the blood, and pretreatment with cyclophosphamide promoted cell expansion. All patients well tolerated the therapy, but no additional clinical reactions were observed except for stable disease (11/15). A total of 20 grade 3 or higher adverse events were observed (CTCAE v4.0) [297]. This finding may be because only 3 of the 15 patients expressed MSLN on > 75% of the tumor cells, suggesting that a certain percentage of tumor cells expressing the target may be required to achieve significant clinical activity. This finding is consistent with observations from NCT01897415, where the lack of MSLN expression on tumor cells surface was associated with the limited efficacy of MSLN CAR-T cell therapy [298]. Therefore, patients could be screened prospectively for surface antigen expression to improve the efficiency of subsequent clinical trials. In another trial, Pang et al. designed a CAR-T cell (MSLN-7 × 19 CAR-T) targeting MSLN capable of secreting IL-7 and CCL19 in a patient with advanced PC. The tumor almost completely disappeared 240 days after intravenous infusion of MSLN-7 × 19 CAR-T cells (NCT03198546). No grade 2–4 adverse events or significant complications were observed [281].
EGFR is also a well-studied target in PC, and Yang et al. conducted a phase 1 clinical trial administering EGFR CAR-T cells to patients with metastatic PC and showed that the cells were safe and effective (NCT01869166) (Fig. 3). Of the 14 patients, 4 experienced a partial response for 2–4 months, and eight were stable for 2–4 months. Reduced EGFR expression on tumor cells was observed in patients with stable disease. Grade ≥ 3 adverse events included fever, nausea, etc., and were reversible (CTCAE v4.0) [299].
In addition, a phase 1 clinical trial targeting HER2 demonstrated the clinical activity of HER2 CAR-T cells in eleven enrolled patients with advanced biliary tract cancers and PC (NCT01935843) (Fig. 3). The mPFS was 4.8 months (range, 1.5–8.3 months); most adverse events were mild or moderate [300].
A patient with liver metastases secondary to stage IV pancreatic adenocarcinoma received locally infused CEA CAR-T cells (NCT02850536) (Fig. 3). The biological activity was demonstrated at 23.2 months, comparing favorably to the median survival time of 5 months for most stage IV patients. No serious adverse events above grade 3 were observed, indicating the treatment is safe [301].
In a phase 1 trial mentioned earlier (NCT02541370), two of the nine patients (7 of PC, 2 of CRC) treated with CD133 CAR-T cells achieved an OR, two achieved a PR, and five were stable, all with grade 2–4 lymphopenia (CRS, Lee 2014 criteria; other adverse events, CTCAE v4.0) [245].
These results provide data and experience into the future development of CAR-T strategies for treating PC.
Moreover, several other potential targets have been studied (Fig. 3), including B7H3, PSCA, MUC1, roundabout homolog 1 (ROBO1), EpCAM, CLDN18.2, CD318, TSPAN 8, and FAP. Some studies have shown that CAR therapies against these targets could inhibit tumor growth in mouse models of PC [302,303,304,305,306,307,308]. Only B7H3, PSCA, MUC1, ROBO1, EpCAM, and CLDN18.2 have entered PC clinical studies, but there are still no reports. We list here the NCT numbers of the most recently initiated or updated clinical trials for these targets: B7H3 (NCT05143151), PSCA (NCT03267173), MUC1 (NCT05239143), ROBO1 (NCT03941457), EpCAM (NCT05028933), and CLDN18.2 (NCT04404595). Especially, NCT03941457 evaluates the safety and efficacy of ROBO1 CAR-NK cell immunotherapy for PC. In particular, enforced expression of CXC chemokine receptor type 6 (whose ligands are highly expressed in human and murine PC cells and tumor-infiltrating immune cells) in T cells could enhance the recognition and lysis of PC cells because we know chemokines and their receptors are essential for the migration and homing of lymphocytes [309].
EGFRvIII, GUCY2C, ROR2, and CD70 are promising targets (Fig. 3), and related clinical trials have been conducted (NCT03267173, NCT05287165, NCT03960060, NCT02830724). Indeed, further studies on these targets could provide a foothold for optimizing CAR-T cell therapies.
Podocalyxin (PODXL), also called TRA-1-60, is a type I membrane-bound glycoprotein. A murine PODXL-specific CasMab was successfully developed with exclusive reaction with the PODXL-overexpressing GBM cell line (LN229) and PC cell line (MIA PaCa-2). Then, a core fucose-deficient mAb, 60-mG2a-f, was developed by conferring augmented antibody-dependent cellular cytotoxicity (ADCC) to CasMab. 60-mG2a-f exhibited remarkable anti-tumor capacity in the MIA PaCa-2 xenograft mouse model of PC, suggesting a promising targeted immunotherapy approach [310]. There are no relevant clinical trials available.
Melanoma (MEL)
MEL is the most malignant skin cancer, formed mainly by the malignant proliferation of melanogenic cells (called melanocytes) in the skin. Several studies of different CAR-T cells have provided a theoretical basis for related clinical studies of MEL, including CD16, CD126, CD70, B7H3, HER2, Vascular endothelial growth factor receptor 2 (VEGFR-2), gp100/HLA-A2 complex, NY-ESO-1, CD20, mitochondrial-associated cysteine-rich protein, PD-L1, CSPG4, GD2 and GD3 have been investigated as promising targets of CAR-T cells [311,312,313,314,315,316,317,318,319,320] (Fig. 3).
For example, CD126 CAR-T cells showed potent tumor-suppressive activity in a metastatic MEL xenograft mouse model.
CSPG4 is overexpressed in most MEL cancer cell lines. One study designed a CSPG4 CAR-NK cell that released fewer cytokines than CAR-T cells but was capable of killing MEL cells and improving cytotoxicity [321].
αvβ3 integrins are reportedly overexpressed in many cancers, including MEL, breast, prostate, and PCs, and it plays a role in cancer survival and metastasis. Results of preclinical studies suggest that targeting αvβ3 integrins hold promise for treating MEL patients [322].
Many clinical trials have been registered and conducted for specific targets of MEL treatment with CAR-T cells, including VEGFR-2, GD2, c-MET, CD70, gp100, CD20, interleukin-13 receptor subunit alpha-2 (IL13Rα2), B7H3, and bispecific B7H3/CD19 (Fig. 3). Although clinical trials of CAR-T cells targeting IL13Rα2, c-MET, and CD20 for MEL are in progress to verify its feasibility and safety, few preclinical studies have been conducted. Most clinical trials have not been completed, or results have not been published. The following are all the published clinical trial results of CAR-T cell therapy for MEL. In phase 1 of the CARPETS trial (ACTRN12613000198729), GD2-positive metastatic MEL patients receiving CAR-T cell therapy showed upregulated LAG-3 and PD-1 expression in stimulated CAR-T cells [316]. Therefore, combined immune checkpoint blockade therapy may enhance the effect of CAR-T cell therapy. In a phase 1/2 study of 24 metastatic MEL patients (NCT01218867), no therapeutic effect was observed in patients treated with a combination of IL-2 and CAR-T cells targeting VEGFR-2, and 23 patients experienced adverse events, 5 of which were severe. In summary, the published clinical study results are not ideal.
CAR-T cell therapy could inhibit tumor growth through anti-angiogenic effects and direct cell killing while targeting multiple antigens simultaneously could also enhance efficacy. And these strategies have been applied to studies related to MEL. A study reported the simultaneous infusion of T cells specific for tumor antigen (gp100, TRP1, or TRP2) and CAR-T cells against the mesenchymal vascular system (VEGFR-2), which yielded a synergistic effect on a mouse model of MEL eradication and improved tumor-free survival versus treatment with either cell type alone or with T cells that co-express the two target molecules [314] (Fig. 3). To counter the immune escape mechanism of tumor cells, a specific combination of TCR against gp100 and CAR against CSPG4 was used [323]. Then CD8+ T cells expressing these two additional receptors were generated using electroporation receptor-encoding mRNA technology, and a strong effect was shown [323]. In addition, a Tandem CAR-T cell targeting both CD70/B7H3 yielded a significant cytolytic result in MEL model [189].
Overall, more studies are needed to assess the safety and efficacy of CAR-T cell therapeutic targets for MEL.
Ovarian cancer (OC)
The prognosis for OC is the poorest, and its fatality rate is the greatest of all gynecologic cancers. It is among the most prevalent forms of cancer found in women around the entire globe [324].
MSLN is the most intensively studied target and has the most significant number of relevant clinical trials. Studies have identified that MSLN is overexpressed in more than 75% of high-grade serous ovarian carcinoma tumors [325]. Preclinical studies focused on using MSLN CAR-T cells in subcutaneous or in situ mouse OC transplantation models and found them to inhibit tumor growth [326]. In a phase 1/2 clinical study (NCT03615313), a relapsed patient with epithelial OC was treated with αPD-1-meso CAR-T cells which contain MSLN CAR and antibody for PD-1 in combination with apatinib (a drug that inhibits angiogenesis), the patient experienced PR and survived for more than 17 months, grade 1 hypertension and fatigue are the only side effects (CTCAE v5.0) [327].
The 2022 American Association for Cancer Research (AACR) Annual Meeting recently announced advances in CAR-T therapies for solid tumors (Fig. 3). The first phase 1/2 clinical trial results to enhance CAR-T cell therapies’ activity via a CLDN6-encoding mRNA vaccine (CARVac) were presented at the annual meeting. CLDN6 is a tumor-specific antigen that is considered an ideal target. The vaccine provides a stimulus to the adoptively transferred CAR-Ts by making dendritic cells express the target antigen of the CAR [328]. In the 6th week of this trial, 14 patients with relapsed or refractory advanced could participate in the efficacy evaluation. Six patients (4 patients with testicular cancer and two with OC) had PR, and the ORR was nearly 43% [329]. About 40% of patients experienced a CRS and have no neurotoxicity.
Intravenous or intraperitoneal injections of MUC16 CAR-T cells could inhibit OC or eliminate tumors in mouse models [330]. 80% of epithelial OC cells express MUC16, a tumor marker [331]. Therefore, MUC16 may be an ideal antigenic target in CAR-T cell therapies for OC.
L1-CAM is highly overexpressed in OC, while studies have demonstrated the antitumor activity of L1-CAM CAR- T cells in OC xenograft models [332].
In addition, in OC in vivo or in vitro studies, CAR therapies targeting FRα, HER2, uPAR, 5 T4, ALPPL2, B7H3, PTK7, TAG72, CD47, OR2H1, and CDH6 have been suggested as promising therapeutic approaches [195, 213, 306, 333,334,335,336,337,338] (Fig. 3). FRα, HER2, ALPPL2, B7H3, and TAG72 have entered OC clinical studies, but there are still no reports. We list here the NCT numbers of the most recently initiated or updated clinical trials for these targets: FRα (NCT03585764), HER2 (NCT04660929), ALPPL2 (NCT04627740), B7H3 (NCT04670068), TAG72 (NCT05225363). Clinical trials targeting MUC1 and CD70 have also been carried out (NCT05239143, NCT02830724), but there is little related preclinical evidence for OC treatment.
Several strategies are available to improve the antitumor effects of CAR therapies for OC. For example, CXCR1 is a G protein-coupled receptor with a high affinity for binding IL-8. IL-8 production is increased in a wide range of solid tumor malignancies. It has been shown that CXCR1 expressed on CAR-NK cells enhances migration and infiltration of CAR-T cells by matching chemokines secreted by tumors and, more importantly, enhances anti-tumor responses in vivo [339]. In an OC mouse model, SynNotch CAR T cells showed better control of tumor load [334]. In addition, Song et al. investigated the FRα-specific site scFv (MOv19) binding to CD137 (4-1BB) co-stimulatory pattern (MOv19-BBζ). MOv19-BBζ CAR-T cells were used in animal models of FRα + OC in intraperitoneal, subcutaneous, and metastatic pulmonary models with positive therapeutic effects [340]. But CD137 signaling does not increase the anti-tumor effect in vivo despite improved T cell persistence.
Targeting tumor neovascularization and extracellular matrix is also a strategy, and recent studies have shown that Annexin A2 (ANXA2) has been detected in OC. Overexpression of ANXA2 mediates extracellular matrix degradation and neointima formation through fibrinolytic enzyme production and is associated with invasion and metastasis [341]. Elimination of CSCs is considered a promising strategy. Markers present on the surface of CSCs, such as CD133, CD44, and CD47, may be targets for CAR. The antitumor effects of CAR targeting CD133, CD44, and CD47 have been validated in OC models [342]. Combining CAR intervention with other therapy has been tried by several studies. For example, studies have shown synergistic effects of paclitaxel with ErbB CAR -T cells in vivo [343].
Glioma
As the most common primary tumors in the CNS, gliomas could be classified as low-grade gliomas or glioblastomas (GBM) based on histological and molecular features [344]. As one of the most malignant and recurrent solid tumors, GBM has a global incidence rate of 10/100000. Individuals with GBM have a five-year survival rate of fewer than 10% [345]. It has been established that patients with GBM have a poor prognosis after conventional therapy. As a promising therapeutic approach, CAR-T cell therapy has identified multiple specific targets that may address the challenge of treating glioma.
Up to now, well-studied CAR-T cell therapy targets in glioma include IL13Rα2, EGFRvIII, HER2, and GD2 (Fig. 3). IL13Rα2 is rarely expressed in normal brain cells but is highly expressed in GBM. This specificity makes them ideal targets for CAR-T cell therapy in GBM. The expression of IL13Rα2 in gliomas could also be used to assess prognosis [346]. In two phase 1 trial reported by Brown et al. in 2015 and 2016, several patients treated with IL13Rα2 CAR-T cells showed good tolerability (NCT02208362, NCT00730613) [347, 348]. Clinical trial (NCT02208362) reports clinical experience with one patient. After IL13Rα2 CAR T-cell treatment, regression of all intracranial and spinal tumors was observed. They observed therapeutic effects against GBM, with elevated immune cells and factor levels in the cerebrospinal fluid. This clinical response continued for 7.5 months after the initiation of CAR T-cell therapy [347]. In another clinical study (NCT00730613), three patients with recurrent GBM were treated with IL13Rα2-redirected CAR CD8+ T cells. Three patients with recurrent disease were well tolerated with controlled ephemeral brain inflammation. Transient evidence of antitumor response was observed in 2 of these patients [348]. Unfortunately, in this trial, one patient experienced shorter remission, which may be related to the loss of IL13Rα2 antigen on the relapsed tumor, leading to the poor response of IL13Rα2 CAR-T cells against GBM. In addition, the intraventricular infusion exhibited a better ability to eliminate distant tumor growth than intracranial tumor infusion. Recently, IL13Rα2 CAR-T has been shown to activate immune cells in vivo via IFN-γ-mediated pathways [349].
Tumor-specific antigens are relatively rare. EGFRvIII has received attention as a mutant of the EGFR receptor; it is expressed only on the surface of tumor cells but not in normal tissues (Fig. 3). In a phase 1 study, ten patients with recurrent GBM expressing EGFRvIII were treated with EGFRvIII CAR-T cells (NCT02209376). CAR-T cells exerted antitumor effects and mediated antigen deficiency and resistance in GBM. In these ten subjects, the median OS was 251 days (approximately 8 months), and no patients had severe adverse events [350].
HER2 has tyrosine kinase activity and belongs to the ErbB family; it could reportedly promote cell proliferation and further development of tumors [351] (Fig. 3). In a recent phase 1 clinical trial (NCT03500991), repeated topical administration of HER2 CAR-T cells to children and young people with recurrent or refractory CNS tumors, including diffuse midline gliomas, yielded no dose-limiting toxicity. High CXCL10 and CCL2 levels were detected in the cerebrospinal fluid [352]. This finding suggests CAR-T cell products with chemokine receptor expression may converge on CXCL10 and CCL2 expression sites to promote CAR-T cell binding to targets.
Another target that has been studied in depth is GD2 (Fig. 3). This disialoganglioside is usually expressed on peripheral neurons and parts of the CNS and acts as a promoter of intercellular adhesion [353]. It is highly and almost universally expressed in neuroblastoma tissue [354]. H3K27M+ diffuse intrinsic pontine glioma (DIPG) and spinal diffuse midline glioma (DMG) are highly aggressive, universally fatal tumors with few treatment options. A paper published in Nature reported the clinical experience of four patients with H3K27M mutated DIPG or spinal DMG who received intravenous infusion therapy with GD2 CAR-T cells [355]. Three patients demonstrated clinical and radiological improvement, and increased concentrations of inflammatory factors were found in cerebrospinal fluid and blood. Patients did not experience on-target, off-tumor toxicity. Observed improvements in neurological function highlight the potential of tumor cell-specific therapies in functional recovery [355].
Despite the advances in CAR-T cell therapy, patients that undergo single-targeted treatment are prone to relapse and subsequent resistance due to the molecular heterogeneity and evolution of tumors. Therefore, targeting multiple antigens or immunosuppressive cytokine antagonism is recommended [356]. In short, such combination therapies still require expanding the targeted antigen pool or developing new CAR-T cell types.
EphA2 is also overexpressed in GBM, enhancing tumorigenesis and migration. Lin et al. performed a phase 1 trial of EphA2 CAR-T cells in three EphA2-positive recurrent GBM patients with transient clinical efficacy and initial tolerability at the tested dose level (NCT03423992) (Fig. 3). One was SD (transit diminishment), and 2 were PD. No adverse events up to level 3 were reported [357].
B7H3, an immune checkpoint involved in tumor migration and invasion, has recently become a target of CAR-T cell therapy [358] (Fig. 3). Tang et al. reported using B7H3 CAR-T cells to treat a patient with recurrent GBM and showed that despite mediating a short-term antitumor response in situ, resistance quickly developed [359].
Moreover, NKG2DL is highly expressed in glioblastoma. One study substantiated synergy between NKG2DL CAR-T cells and radiation therapy in treating a mouse glioma model and the ability to lyse GBM tumor cell lines and CSCs (U-251 MG, T98G, U-87 MG, HTB185, GSC3) effectively [360, 361]. NKG2DL and B7H3 have also entered clinical studies to treat glioma (Fig. 3).
MUC1, CD147, and MMP2 have also been considered auspicious targeting sites, and related clinical trials are underway (Fig. 3). Here are the NCT numbers: MUC1 (NCT02617134, NCT02839954), CD147 (NCT04045847), and MMP2 (NCT04214392).
CSPG4 is also widely expressed in various malignancies [362]. CSPG4 CAR-T cells resisted brain tumor growth in cultured neurosphere and glioma xenograft mice models without signs of tumor escape [363].
A recent novel CAR-T cell, using the scorpion toxin peptide chlorotoxin (CLTX) as the targeting structural domain, has been reported to inhibit tumors in a xenograft GBM model with no observed off-target effects [364]. The pioneering design of the CLTX CAR-T suggests that designing CAR-T cells using tumor-binding peptides is promising and could potentially reduce tumor escape, further expanding the tumor-targeting antigen pool.
Besides, Podoplanin (PDPN) is a mucin-like glycoprotein whose overexpression has been associated with mesothelioma, EC, LCa, and mesenchymal GBM [365]. The third-generation PDPN-redirected CAR-Ts were reported to mediate efficient anti-tumor responses against PDPN-positive GBM cell lines (LN319, U87MG) in vitro and inhibit r growth in a glioma mouse xenograft model [366]. However, PDPN-redirected CAR-Ts were found with frequent toxicity in preclinical models by targeting normal tissue expressed PDPN. To solve this issue, a cancer-specific monoclonal antibody CasMab (LpMab-2), which only reacts with the aberrant tumor tissue expressed glycosylated PDPN, was developed [367].
Targeting the tumor stroma and vascular system embodies a novel strategy for tumor suppression. It is used to treat glioma.
It has been shown that p21-activated kinase 4 (PAK4) inhibition normalizes the tumor vascular microenvironment and makes GBM more sensitive to CAR-T cell therapy [368].
Moreover, P32 CAR-T cells have antitumor and anti-angiogenic effects in gliomas [369].
FAP has been reported in a recent study as an ideal immunotherapeutic antigen for targeting tumor cells and stroma [370].
Studies in animal models have indicated that the proteins CD133, AXL, c-mesenchymal-epithelialmesemesenchymal-epithelialnchymal-epithelial transition factor (c-MET), as well as factor-inducible 14 (Fn14) are specific targets for CAR-T cell treatment in GBM [371,372,373].
In conclusion, numerous TAA have significant potential therapeutic effects in GBM, and their applications need further development.
Targeted combinations of multiple tumor antigens mitigate tumor escape and enhance T-cell effects. For example, for the treatment of glioma, a tandem CAR-T cell functions more robustly and persistently and reduces antigen escape by crosslinking HER2 and IL13Rα2 receptors than a single targeted CAR-T cell [374]. In addition, a trivalent CAR-T cell combines three CAR molecules that could target HER2, IL13Rα2, and EphA2, respectively, and kill tumor cells expressing single or multiple antigens, expanding the therapeutic range [375]. The synNotch-CAR-T cells, which have become popular in recent years, could induce the expression of CARs by relying on a particular antigen (e.g., the GBM neoantigen EGFRvIII) to initiate the effect and kill based on a highly homogeneous antigen or set of antigens (e.g., EphA2 and IL13Rα2) [376]. This design allows CAR-T cells to display enhanced antitumor activity and persistence without causing extra-tumor killing. In a study, Lp2 CAR-T cells were designed to target PDPN-expressing glioma cells to exclude normal PDPN-expressing cells. Concurrent use of Lp2 CAR-T and third-generation recombinant herpes simplex virus-1 lytic virus G47Δ further inhibited tumor growth and improved survival [377].
Several different approaches are now proven to improve the safety and efficacy of CAR-T immunotherapy, including combination therapy between radiotherapy and CAR-T therapy and intracerebroventricular injection of CAR-T. These results provide new ideas for the future design of CAR-T in the treatment of solid tumors.
Head and neck squamous cell carcinoma (HNSCC)
Approximately 890,000 new HNSCCs are diagnosed each year [378]. Conventional treatment for HNSCC is primary radiotherapy, but resistance may develop due to tumor heterogeneity [379]. CAR-T cell treatments have advanced considerably in HNSCC in current history. The following are the possible targets of CAR-T therapies for HNSCC observed so far.
CSPG4 interacts with α4β1 integrin to directly regulate cell adhesion. CSPG4 CAR-T cells have been shown to inhibit the growth of various solid tumors in preclinical studies, including BC, HNSCC, and mesothelioma [185].
MUC1, PD-L1, CD70, and CD44v6 have been identified as promising targets for CAR-T cells in the treatment of HNSCC and have been confirmed in preclinical studies [380,381,382] (Fig. 3).
Several optimization strategies have been developed to address the problems faced by CAR-T cell therapies in HNSCC (Fig. 3). Rosewell et al. established a construct encoding a PD-L1-blocking antibody and IL-12p70 binary lysing adenovirus (CAd), and local treatment combined with CAd12_PD-L1 and systemic HER2 CAR-T cell infusion in an HNSCC xenograft model improved survival to > 100 days [383]. This suggests that CAd12_PD-L1 enhances the antitumor effects of HER2 CAR-T cells. Another study designed a novel CD98- or EGFR- redirected UniCAR-T cell to lyse radioresistant HNSCC cells effectively, thus potentially improving the prognosis of radioresistant cancer patients [384]. To improve the affinity, cetuximab-constructed CAR-T cells were highly responsive to EGFR-positive HNSCC cells [385].
CAR-T cell clinical studies targeting HER2 (NCT03740256) and MUC1 (NCT05239143) for the treatment of HNSCC have been performed (NCT03356795).
Other solid tumors
Most patients with thyroid cancer (TC) have a good prognosis, but metastatic and advanced TC have limited treatment options and a poor prognosis.
ICAM-1 plays a vital role in cell adhesion, cell signaling, and transendothelial migration of leukocytes to sites of inflammation [386] (Fig. 3). Irene et al. demonstrated that papillary TC and undifferentiated TC were associated with increased ICAM-1 [387]. ICAM-1 CAR-T cell showed substantial anti-tumor effects in TC cell lines (8505C, BCPAP, FRO, KHM-5 M) and xenograft mice models. They were also the first to report that ICAM-1 CAR-T cells could kill undifferentiated and hypofractionated TC cells in vitro and in vivo [387]. Interestingly, ICAM-1 CAR-T cells could induce increased ICAM-1 expression [388].
In addition, GFRα4 and TSHR are promising targets for treating TC and have been validated in preclinical models [389, 390] (Fig. 3).
Renal cell carcinoma (RCC) is currently one of the most malignant urological cancers [391]. CAR-T cell therapy has emerged as a new strategy for RCC treatment, but the current results are not satisfactory.
Cor H J Lamers et al. investigated the efficacy and safety of the first-generation CAIX CAR-T cells in 12 patients with carbonic anhydrase IX (CAIX)-positive metastatic renal cell carcinoma (mRCC) in a phase 1/2 trial (Fig. 3). Ultimately, no clinical responses were observed, and severe toxicity reactions occurred [392]. Nevertheless, the trial provided valuable lessons. A recent study has improved the design and composition of CAIX CAR-T cells. Their results suggest that CAIX BBζ CAR4/8 T cells are highly effective immunotherapy for RCC and promising for clinical application [393].
In addition, several preclinical studies have found that CAR cell therapies targeting L1-CAM, c-MET, PD-L1, EGFR, HER2, CD70, and B7H3 could suppress RCC [189, 320, 394,395,396,397]. CAR-T cells targeting EGFR, CAIX, CD70, B7H3, VEGFR-2, ROR2, MUC1, and GPC3 are currently in clinical trials to treat RCC (Fig. 3).
Cervical cancer (CC) is the fourth most prevalent malignancy and the fourth most significant cause of cancer mortality in women globally, according to a 2018 report [398]. Treatment with CAR-T cells for CC is in its infancy.
Preclinical research indicates that NKG2DL, MSLN, and PD-L1 are all excellent targets for CAR-T cells in the therapy of CC [399,400,401]. Furthermore, CAR-T cell clinical studies have been performed targeting MSLN, PD-L1, GD2, PSMA, and MUC1 (Fig. 3).
Osteosarcoma (OS) and Ewing’s sarcoma (EWS) have a poor prognosis once metastasis or recurrence occurs.
In a phase 1/2 clinical study, 19 HER2 positive subjects (16 OS, 1 EWS, one primitive neuroectodermal tumor, and one protofibroblastic small round cell tumor) were treated with HER2 CAR-T cells (Fig. 3). Four of 17 evaluable patients were stable at 12 weeks to 14 months, three had their tumors removed, and one had ≥90% tumor necrosis (NCT00902044). No patient experienced adverse T-cell infusion-related events except one patient who developed a fever on the highest dose [402].
CAR-T cells targeting IL-11Rα, GD2, EphA2, ROR1, type I insulin-like growth factor receptor (IGF1R), B7H3, CD166, VEGFR-2, and NKG2D have also been shown to have a therapeutic effect in OS and EWS models [403,404,405,406,407,408,409,410,411] (Fig. 3). These new findings give hope for the future of CAR-T immunotherapy for solid tumors.
Although CAR-T cells have more alternative targets in treating solid tumors and their applications are up-and-coming, they have shown limited antitumor activity in clinical trials. Accordingly, the use of CAR-T cell therapies among solid tumors still needs to be further explored. This failure is due to multiple factors, such as lack of specific targets, inefficient homing and penetration in solid tumors, and the inhibitory effect of TME on CAR-T cells.
CAR-T cell therapy targets commonly used in solid tumors are summarized (Fig. 3).
Conclusions and outlooks
CAR-T cell therapy based on gene-editing technology has gained significant momentum in recent years, showing remarkable results in clinical applications and bringing a new dawn to immunotherapy for tumor patients.
Current evidence substantiates that CAR-T cell therapies are associated with excellent response rates in patients with hematologic malignancies [412]. Well-developed targets are available in B-ALL, B-NHL, and MM, and the FDA has approved four CAR-T products targeting CD19 and two targeting BCMA to treat these diseases. There are also targets in AML and HL that have good clinical outcomes, with CAR-T cell therapies targeting them. In T-ALL, CLL, and T-NHL, the therapeutic efficacy of CAR-T cell therapy has yet to be further improved, and suitable targets are still being explored [9]. The high heterogeneity of cancer and the tendency of malignant cells to recur through antigen-negative relapse mediated by antigen escape mechanisms under therapeutic pressure makes it difficult for CAR-T cell therapies targeting a single target to work well [9]. It should also be borne in mind that many targets are expressed in non-malignant tissues. This poor target specificity leads to severe consequences involving CAR-T cell attacks on normal hematopoietic tissues, such as T-cell self-mutilation caused by CAR-T cell therapies for T-cell tumors [5]. In addition, it has recently been found that in a mouse leukemia model, CAR-T cells could induce antigen transfer from cancer cells to T cells through a process called phagocytosis, resulting in antigen loss, reduced target density on tumor cells, and reduced effectiveness of therapy [413].
The solid tumor studied maturely mainly contains digestive system cancer, MEL, glioma, and so on. Treatment with CAR-T cells for CC and RCC is still in its infancy. Although research on CAR-T cell therapy has now shifted attention to solid tumors, studies on solid tumors continue to face difficulties due to multiple factors [414]. On the one hand, it is due to the characteristics of solid tumors. Solid tumor cells are heterogeneous, and few TSA has been found on the tumor cell’s surface. Therefore, targets are often selected for TAA that is highly expressed on the tumor surface and lowly expressed in normal tissues. Still, this approach may lead to off-target tumor toxicity, CRS, and antigen loss [415].
On the other hand, it is due to the complexity of the solid tumor microenvironment, which is highly abnormal with varying degrees of vascular collapse and dense rigid stromal structures [415]. These characteristics hinder the infiltration of CAR-T cells. In addition, there is a resistance of immunosuppressive cells and inhibitory factors in the tumor microenvironment [416]. The combination of the above factors resulted in difficult infiltration of CAR-T cells and aggravated the failure. To address the above problems, CAR-T modification and combination therapy are two good directions [417]. However, here we will still suggest possible directions for improvement based on the difficulties raised. First, we could enhance antigen-specific recognition of CAR-T cells and overcome the heterogeneity of tumor cell antigens. Second, targeting TME will be attractive, so improving TME infiltration of CAR-T cells and targeting immunosuppressive and metabolic tumor microenvironments would be helpful.
The manufacturing process of CAR-T cells may also be problematic. Circulating tumor cells are collected along with lymphocytes during collection. CAR could be transduced into these tumor cells, binding to targets expressed by the same cell, causing the tumor cells to escape [418]. The manufacture of CAR-T cells is also time-consuming and costly, and the use of allogeneic CAR-T cells from healthy donors, which are knocked out of HLA and endogenous TCR by CRISPR technology, seems to help us solve this problem [419].
In the current situation, the rational selection and application of CAR-T cells targeting different targets is the most important guarantee of achieving good clinical results. Many CAR-T targets are being discovered, and target selection is critical (Table 1). Regarding the number of clinical trials corresponding to the different targets for each tumor (Figs. 4 and 5), CD19, CD20, and CD22 are generally considered essential targets for lymphoma and leukemia. In contrast, BCMA is usually considered an important and necessary target for multiple myeloma. EGFRvIII and GPC3 are crucial targets in the solcriticalsection in glioma and HCC, respectively. Besides, tumor-associated glycoforms of conventional antigens, including SLAMF7, CLEC14A, PDPN, PODXL, and CD44, could also be ideal target antigens. These have been described in detail in the text. The perfect target should be highly expressed uniformly on the surface of tumors at different stages, not expressed in normal tissues, not subject to specific therapeutic pressures that may lead to downregulation or elimination, and participate in the pathophysiology of the disease [18]. If the target does not meet these requirements, side effects like tumor antigen-negative relapse and extratemporal tissue toxicity may lead to toxicity.

The proportions of clinical trials on CAR-T cell therapy targets in hematological malignancies. NHL: non-Hodgkin’s lymphoma; TRBC: T cell receptor β-chain constant domains; HL: Hodgkin’s lymphoma; B-ALL: B-acute lymphoblastic leukemia; BAFF-R: B-cell activating factor receptor; TSLPR: thymic stromal lymphopoietin receptor; T-ALL: T-acute lymphoblastic leukemia; AML: acute myeloid leukemia; WT1: wilms tumor 1; FLT3: FMS-like tyrosine kinase 3; NKG2DL: natural killer group 2 member D ligand; CLL1: C-type lectin like molecule 1; CLL: chronic lymphocytic leukemia; BCMA: B-cell maturation antigen; ROR: receptor tyrosine kinase like orphan receptor; MM: multiple myeloma; SLAMF7: signaling lymphocytic activation molecule F7; GPRC5D: G protein-coupled receptor class-C group-5 member-D

The proportions of clinical trials on CAR-T cell therapy targets in solid tumors. BC: breast cancer; MSLN: mesothelin; GD2: ganglioside2; ROR: receptor tyrosine kinase like orphan receptor; CEA: carcinoembryonic antigen; MUC1: mucin 1; EGFR: epidermal growth factor receptor; HER2: human epidermal growth factor receptor 2; LCa: lung cancer; NY-ESO-1: New York esophageal squamous cell carcinoma-1; DLL3: delta-like ligand 3; PD-L1: programmed death-ligand 1; PCa: prostate cancer; NKG2DL: natural killer group 2 member D ligand; PSCA: prostate stem cell antigen; PSMA: prostate-specific membrane antigen; CRC: colorectal cancer; GC: gastric cancer; CLDN: claudin; HCC: hepatocellular carcinoma; GPC3: glypican-3; EC: esophageal cancer; PC: pancreatic cancer; GUCY2C: Guanylyl cyclase C; MEL: melanoma; VEGFR-2: vascular endothelial growth factor receptor 2; OC: ovarian cancer; ALPPL2: alkaline phosphatase placental-like 2; HNSCC: head and neck squamous cell carcinoma; OS: osteosarcoma; EWS: Ewing’s sarcoma; TC: thyroid cancer; ICAM-1: intercellular adhesion molecule-1; RCC: renal cell carcinoma; CAIX: carbonic anhydrase IX; CC: cervical cancer
We should not stop the exploration of new targets. Proteomics, immunopeptidomics, and other techniques could search for new targets. The applicability of new targets to CAR-T cells should be evaluated mainly from stability, specificity, and pathophysiology. This is particularly important for the clinical application of CAR-T cells [18]. In preclinical experiments, some advanced technologies could also help us predict the effect of treatment, such as the tumor three-dimensional organoid model, which could well simulate the structural and functional heterogeneity of primary cells [420].
Further efforts to identify the optimal therapeutic target are essential to refine CAR-T cell therapy. Many other strategies have been used to deal with the lack of ideal antigens or loss of antigens (Fig. 6). First of all, the implementation of targeting multiple antigens could compensate for the lack of target coverage and stability (Tables 2 and 3). T cells transduced by tandem CARs or multiple cis-transgenic CARs are multispecific CAR-T cells, representing the most widely used strategy today [444]. Fcγ-CR based on chimeric receptors composed of FcγR and T-cell signaling molecules and a continuous infusion of CAR-T cells targeting different targets could also achieve this goal [444]. Compared to classical CAR-T cells, combining Fcγ-CR T cells with specific mAbs allows for diagnosing and eliminating cancer cells by ADCC. The advantage of Fcγ-CR T cells is that the same Fcγ-CR T cells could target a variety of different TAAs, and the withdrawal of mAbs could reduce the release of cytokines [445]. Besides, CAR-T cells could circumvent inhibitory immune checkpoint activity by gene-editing techniques, such as rendering cells unable to recognize PD-L1 by CRISPR/Cas9 [446]. In addition, CAR-T cells could be modified to acknowledge tumor chemokines to overcome the difficulty of CAR-T cell transport to malignant tissue in solid tumor therapy, prolong the duration of themselves in cancer tissue and enhance the antitumor effect [444]. The specificity of CAR-T cells could be improved by either raising the expression of the target antigen in tumor tissue or finding the ideal target with minimal expression in healthy cells [447]. CAR-T cells could be used to attack not only tumor cells but also the solid tumor’s TME stroma and blood vessels.

The strategies used to deal with the lack of ideal antigen or loss of antigen. iCAR: inhibitory CAR; DNR: dominant negative receptor; CSR: chimeric-switch receptor; FAP: fibroblast activation protein; VEGFR: vascular endothelial growth factor receptor; CAF: Cancer-associated fibroblast
Furthermore, incorporating switches could control CAR-T cell activity to reduce the toxic side effects. For example, certain types of CAR-T cells, including synNotch, and inhibitory CAR (iCAR), could regulate cell activity through endogenous switches when recognizing specific antigens, thereby reducing extra-tumor tissue toxicity [448]. iCAR also uses dual antigen targeting similar to Tandem CAR, but iCAR could inhibit the activation of active CARs by binding to a second inhibitory receptor [449]. On-Switch CAR could conversely regulate the intensity and duration of CAR-T cell activity by exogenous administration, which is more controllable [450].
In addition to the effects of target selection, we mention other side effects and corresponding remedies here. As many T cells are activated in a short period, the release of cytokines increases explosively over a short period, leading to serious adverse effects such as CRS and macrophage activation syndrome [451]. Neurotoxicity is another profound side effect of CAR-T treatment, which is usually treated clinically with tolimumab (IL-6 receptor inhibitor) or glucocorticoids [451]. Due to the low number of lymphocytes in cancer patients, autologous CAR-T cells take longer to produce and are difficult to expand. Still, allogeneic CAR-T is subject to graft versus host disease (GVHD) and rejection reactions [452]. By knocking down endogenous T-cell receptors (TCR) and leukocyte antigen class I molecules (HLA), universal CAR-T cells could both reduce immune rejection during allogeneic transplantation and avoid immune attack by allogeneic T cells on the host organ (GVHD) [419]. And methods such as lymphadenectomy could also reduce the risk of rejection. In addition, immunoglobulin injections could be used to maintain immune function, and allogeneic HSCT could be used to ensure HSC function.
Multiple combinatorial strategies are available to enhance therapeutic efficacy whileusingwhile using CAR-T therapies [453]. For example, mRNA technology could improve the expression of hidden antigens in tumor cells and encode target-specific CAR [321]. Oncolytic virus (OV) regulates TME and influences the ability of the host to mount an anti-tumor immune response [454]. Moreover, stimulating and enhancing endogenous DC activity will maximize T cell engagement and activation [455]. Nanotechnology has also been used to improve CAR-T cell therapy in recent years. Patients could be screened prospectively for surface antigen expression to enhance the efficiency of subsequent clinical trials.
CAR-T will eventually become a tumor buster by continuously exploring suitable targets and optimizing design solutions. CAR-T cell therapy is believed to bring a bright future to cancer patients.
Availability of data and materials
Not applicable.
Abbreviations
- AML:
-
acute myeloid leukemia
- ANXA2:
-
Annexin A2
- ADCC:
-
antibody-dependent cellular cytotoxicity
- B-ALL:
-
B-acute lymphoblastic leukemia
- BAFF-R:
-
B-cell activating factor receptor
- BCMA:
-
B-cell maturation antigen
- BL:
-
burkitt lymphoma
- B-NHL:
-
B-cell non-Hodgkin’s lymphoma
- BC:
-
breast cancer
- CAR:
-
chimeric antigen receptor
- CAR-T cell:
-
chimeric antigen receptor T cell
- CTCAE:
-
National Cancer Institute’s Common Terminology Criteria for Adverse Events
- CBEs:
-
cytosine base editors
- CNS:
-
central nervous system
- CRS:
-
cytokine release syndrome
- CRR:
-
complete remission rate
- CR:
-
complete remission
- CSPG4:
-
chondroitin sulfate proteoglycan 4
- CLL1:
-
C-type lectin like molecule 1
- CLL:
-
chronic lymphocytic leukemia
- CAIX:
-
carbonic anhydrase IX
- ccRCC:
-
clear cell renal cell carcinoma
- CLDN:
-
claudin
- CEA:
-
carcinoembryonic antigen
- CLTX:
-
chlorotoxin
- c-MET:
-
c-mesenchymal-epithelial transition factor
- CSCs:
-
cancer stem cells
- CXCR:
-
chemokine receptor
- CRC:
-
colorectal cancer
- CDH6:
-
cadherin 6
- CC:
-
cervical cancer
- CDH17:
-
cadherin 17
- DIPG:
-
diffuse intrinsic pontine glioma
- DLBCL:
-
diffuse large B-cell lymphoma
- DMG:
-
diffuse midline glioma
- Dox:
-
doxycycline
- DLL3:
-
delta-like ligand 3
- DSBs:
-
DNA double strand breaks
- EFS:
-
event-free survival
- EGFR:
-
epidermal growth factor receptor
- EphA:
-
Ephrin type-A receptor
- EpCAM:
-
epithelial cell adhesion molecule
- EC:
-
esophageal cancer
- ESCC:
-
esophageal squamous cell carcinoma
- EAC:
-
esophageal adenocarcinoma
- EWS:
-
Ewing’s sarcoma
- FDA:
-
U.S. Food and Drug Administration
- FRβ:
-
membrane-associated folate receptor β
- FLT3:
-
FMS-like tyrosine kinase 3
- FL:
-
follicular lymphoma
- FAP:
-
fibroblast activation protein
- Fn14:
-
factor-inducible 14
- FRα:
-
folate receptor α
- GVHD:
-
graft versus host disease
- GPRC5D:
-
G protein-coupled receptor class-C group-5 member-D
- GD2:
-
ganglioside2
- GBM:
-
glioblastoma
- GPC3:
-
glypican-3
- GUCY2C:
-
Guanylyl cyclase C
- GC:
-
gastric cancer
- GS:
-
γ-secretase
- GSI:
-
γ-secretase inhibitor
- HSC:
-
hematopoietic stem cells
- HL:
-
Hodgkin’s lymphoma
- HNSCC:
-
head and neck squamous cell carcinoma
- HER2:
-
human epidermal growth factor receptor 2
- HCC:
-
hepatocellular carcinoma
- HSCT:
-
hematopoietic stem cell transplantation
- ICAM-1:
-
intercellular adhesion molecule-1
- IGF1R:
-
type I insulin-like growth factor receptor
- iCAR:
-
inhibitory CAR
- LILRB4:
-
leukocyte immunoglobulin-like receptor-B4
- LCa:
-
lung cancer
- MHC:
-
major histocompatibility complex
- MSLN:
-
mesothelin
- MPL:
-
myeloproliferative leukemia protein
- mPFS:
-
median progression-free survival
- MM:
-
multiple myeloma
- MUC1:
-
mucin 1
- MEL:
-
melanoma
- NK cells:
-
natural killer cells
- NHL:
-
non-Hodgkin’s lymphoma
- NKG2D:
-
natural killer group 2 member D
- NY-ESO-1:
-
New York esophageal squamous cell carcinoma-1
- NSCLC:
-
non-small cell lung cancer
- NKG2DL:
-
natural killer group 2 member D ligand
- OC:
-
ovarian cancer
- OS:
-
osteosarcoma
- OR2H1:
-
olfactory receptor 2H1
- OR:
-
objective response
- ORR:
-
objective response rate
- PD-1:
-
programmed cell death 1
- PCNSL:
-
primary central nervous system lymphoma
- PODXL:
-
Podocalyxin
- PDPN:
-
Podoplanin
- PR:
-
partial remission
- PSMA:
-
prostate-specific membrane antigen
- PCa:
-
prostate cancer
- PSCA:
-
prostate stem cell antigen
- PAK4:
-
p21-activated kinase 4
- PD-L1:
-
programmed death-ligand 1
- PC:
-
pancreatic cancer
- PLAP:
-
placental alkaline phosphatase
- PTK7:
-
protein tyrosine kinase 7
- PFS:
-
progression-free survival
- r/r B-ALL:
-
relapsed or refractory B-acute lymphoblastic leukemia
- ROR:
-
receptor tyrosine kinase like orphan receptor
- r/r MM:
-
relapsed or refractory multiple myeloma
- RTK:
-
receptor tyrosine kinase
- RCC:
-
renal cell carcinoma
- scFv:
-
single-chain fragment variable
- SLAMF7:
-
signaling lymphocytic activation molecule F7
- SLAMF3:
-
signaling lymphocytic activation molecule F3
- SCLC:
-
small cell lung cancer
- TAA:
-
tumor-associated antigens
- TCR:
-
T cell receptor
- T-ALL:
-
T-acute lymphoblastic leukemia
- TSLPR:
-
thymic stromal lymphopoietin receptor
- TIM-3:
-
T cell immunoglobulin and mucin structural domain 3
- T-NHL:
-
T-cell non-Hodgkin’s lymphoma
- TRBC:
-
T cell receptor β-chain constant domains
- TME:
-
tumor microenvironment
- TGFβ:
-
transforming growth factor-β
- TROP2:
-
trophoblast cell surface antigen 2
- TC:
-
thyroid cancer
- VEGFR-2:
-
vascular endothelial growth factor receptor 2
- VHH:
-
single variable domain on a heavy chain
- WT1:
-
wilms tumor 1
References
Wang S, Sun J, Chen K, et al. Perspectives of tumor-infiltrating lymphocyte treatment in solid tumors. BMC Med. 2021;19(1):140.
Singh AK, McGuirk JP. CAR T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21(3):e168–78. https://doi.org/10.1016/S1470-2045(19)30823-X.
Newick K, O'Brien S, Moon E, et al. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–52. https://doi.org/10.1146/annurev-med-062315-120245.
Zhang C, Liu J, Zhong JF, et al. Engineering CAR-T cells. Biomark Res. 2017;5:22. https://doi.org/10.1186/s40364-017-0102-y.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3(4):388–98. https://doi.org/10.1158/2159-8290.CD-12-0548.
van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. https://doi.org/10.1038/nrd4597.
Marin V, Pizzitola I, Agostoni V, et al. Cytokine-induced killer cells for cell therapy of acute myeloid leukemia: improvement of their immune activity by expression of CD33-specific chimeric receptors. Haematologica. 2010;95(12):2144–52. https://doi.org/10.3324/haematol.2010.026310.
Roselli E, Boucher JC, Li G, et al. 4-1BB and optimized CD28 co-stimulation enhances function of human mono-specific and bi-specific third-generation CAR T cells. J Immunother Cancer. 2021;9(10). https://doi.org/10.1136/jitc-2021-003354.
Huang R, Li X, He Y, et al. Recent advances in CAR-T cell engineering. J Hematol Oncol. 2020;13(1):86. https://doi.org/10.1186/s13045-020-00910-5.
Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145–54. https://doi.org/10.1517/14712598.2015.1046430.
Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426–1438.e11. https://doi.org/10.1016/j.cell.2018.03.038.
Urbanska K, Lanitis E, Poussin M, et al. A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 2012;72(7):1844–52. https://doi.org/10.1158/0008-5472.CAN-11-3890.
Ma JS, Kim JY, Kazane SA, et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016;113(4):E450–8. https://doi.org/10.1073/pnas.1524193113.
Xie YJ, Dougan M, Jailkhani N, et al. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A. 2019;116(16):7624–31. https://doi.org/10.1073/pnas.1817147116.
Long AH, Haso WM, Shern JF, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90. https://doi.org/10.1038/nm.3838.
Weatherill EE, Cain KL, Heywood SP, et al. Towards a universal disulphide stabilised single chain Fv format: importance of interchain disulphide bond location and vL-vH orientation. Protein Eng Des Sel. 2012;25(7):321–9. https://doi.org/10.1093/protein/gzs021.
Zhylko A, Winiarska M, Graczyk-Jarzynka A. The great war of today: modifications of CAR-T cells to effectively combat malignancies. Cancers (Basel). 2020;12(8). https://doi.org/10.3390/cancers12082030.
Wei J, Han X, Bo J, et al. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12(1):62. https://doi.org/10.1186/s13045-019-0758-x.
Crump M, Neelapu SS, Farooq U, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017;130(16):1800–8. https://doi.org/10.1182/blood-2017-03-769620.
Mounier N, Canals C, Gisselbrecht C, et al. High-dose therapy and autologous stem cell transplantation in first relapse for diffuse large B cell lymphoma in the rituximab era: an analysis based on data from the European blood and marrow transplantation registry. Biol Blood Marrow Transplant. 2012;18(5):788–93. https://doi.org/10.1016/j.bbmt.2011.10.010.
Scheuermann RH, Racila E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma. 1995;18(5-6):385–97. https://doi.org/10.3109/10428199509059636.
Schuster SJ, Bishop MR, Tam CS, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56.
Abramson JS, Palomba ML, Gordon LI, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet (London, England). 2020;396(10254):839–52.
Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44.
Locke FL, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel as second-line therapy for large B-cell lymphoma. N Engl J Med. 2022;386(7):640–54. https://doi.org/10.1056/NEJMoa2116133.
Neelapu SS, Dickinson M, Munoz J, et al. Axicabtagene ciloleucel as first-line therapy in high-risk large B-cell lymphoma: the phase 2 ZUMA-12 trial. Nat Med. 2022;28(4):735–42. https://doi.org/10.1038/s41591-022-01731-4.
Ghione P, Palomba ML, Patel A, et al. Comparative effectiveness of ZUMA-5 (axi-cel) vs SCHOLAR-5 external control in relapsed/refractory follicular lymphoma. Blood. 2022. https://doi.org/10.1182/blood.2021014375.
Jacobson CA, Chavez JC, Sehgal AR, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022;23(1):91–103. https://doi.org/10.1016/S1470-2045(21)00591-X.
Grommes C, DeAngelis LM. Primary CNS lymphoma. J Clin Oncol. 2017;35(21):2410–8. https://doi.org/10.1200/JCO.2017.72.7602.
Frigault MJ, Dietrich J, Gallagher K, et al. Safety and efficacy of tisagenlecleucel in primary CNS lymphoma: a phase 1/2 clinical trial. Blood. 2022;139(15):2306–15. https://doi.org/10.1182/blood.2021014738.
Wang M, Munoz J, Goy A, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–42. https://doi.org/10.1056/NEJMoa1914347.
Miles RR, Arnold S, Cairo MS. Risk factors and treatment of childhood and adolescent Burkitt lymphoma/leukaemia. Br J Haematol. 2012;156(6):730–43. https://doi.org/10.1111/j.1365-2141.2011.09024.x.
Russo-Carbolante EM, Picanço-Castro V, Alves DC, et al. Integration pattern of HIV-1 based lentiviral vector carrying recombinant coagulation factor VIII in Sk-Hep and 293T cells. Biotechnol Lett. 2011;33(1):23–31. https://doi.org/10.1007/s10529-010-0387-5.
Tao J, Zhou X, Jiang Z. cGAS-cGAMP-STING: the three musketeers of cytosolic DNA sensing and signaling. IUBMB Life. 2016;68(11):858–70. https://doi.org/10.1002/iub.1566.
Gándara C, Affleck V, Stoll EA. Manufacture of third-generation lentivirus for preclinical use, with process development considerations for translation to good manufacturing practice. Hum Gene Ther Methods. 2018;29(1):1–15. https://doi.org/10.1089/hgtb.2017.098.
Atianand MK, Fitzgerald KA. Molecular basis of DNA recognition in the immune system. J Immunol. 2013;190(5):1911–8. https://doi.org/10.4049/jimmunol.1203162.
Michieletto D, Lusic M, Marenduzzo D, et al. Physical principles of retroviral integration in the human genome. Nat Commun. 2019;10(1):575. https://doi.org/10.1038/s41467-019-08333-8.
Zhang J, Hu Y, Yang J, et al. Non-viral, specifically targeted CAR-T cells achieve high safety and efficacy in B-NHL. Nature. 2022. https://doi.org/10.1038/s41586-022-05140-y.
Doody GM, Justement LB, Delibrias CC, et al. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science. 1995;269(5221):242–4. https://doi.org/10.1126/science.7618087.
Baird JH, Frank MJ, Craig J, et al. CD22-directed CAR T-cell therapy induces complete remissions in CD19-directed CAR-refractory large B-cell lymphoma. Blood. 2021;137(17):2321–5. https://doi.org/10.1182/blood.2020009432.
Zhang WY, Wang Y, Guo YL, et al. Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal transduction and targeted. Therapy. 2016;1:16002.
Till BG, Jensen MC, Wang J, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. BLOOD. 2012;119(17):3940–50.
Du J, Zhang Y. Sequential anti-CD19, 22, and 20 autologous chimeric antigen receptor T-cell (CAR-T) treatments of a child with relapsed refractory Burkitt lymphoma: a case report and literature review. J Cancer Res Clin Oncol. 2020;146(6):1575–82. https://doi.org/10.1007/s00432-020-03198-7.
Ramos CA, Savoldo B, Torrano V, et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J Clin Invest. 2016;126(7):2588–96. https://doi.org/10.1172/JCI86000.
Ranganathan R, Shou P, Ahn S, et al. CAR T cells targeting human immunoglobulin light chains eradicate mature B-cell malignancies while sparing a subset of Normal B cells. Clin Cancer Res. 2021;27(21):5951–60. https://doi.org/10.1158/1078-0432.CCR-20-2754.
Bunse M, Pfeilschifter J, Bluhm J, et al. CXCR5 CAR-T cells simultaneously target B cell non-Hodgkin's lymphoma and tumor-supportive follicular T helper cells. Nat Commun. 2021;12(1):240. https://doi.org/10.1038/s41467-020-20488-3.
Spiegel JY, Patel S, Muffly L, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27(8):1419–31. https://doi.org/10.1038/s41591-021-01436-0.
Zhang Y, Li J, Lou X, et al. A prospective investigation of bispecific CD19/22 CAR T cell therapy in patients with relapsed or refractory B cell non-Hodgkin lymphoma. Front Oncol. 2021;11:664421. https://doi.org/10.3389/fonc.2021.664421.
Wei G, Zhang Y, Zhao H, et al. CD19/CD22 dual-targeted CAR T-cell therapy for relapsed/refractory aggressive B-cell lymphoma: a safety and efficacy study. Cancer Immunol Res. 2021;9(9):1061–70. https://doi.org/10.1158/2326-6066.CIR-20-0675.
Tong C, Zhang Y, Liu Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136(14):1632–44. https://doi.org/10.1182/blood.2020005278.
Shah NN, Johnson BD, Schneider D, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26(10):1569–75. https://doi.org/10.1038/s41591-020-1081-3.
Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR-T cell therapy in T-cell malignancies: is success a low-hanging fruit? Stem Cell Res Ther. 2021;12(1):527. https://doi.org/10.1186/s13287-021-02595-0.
Gomes-Silva D, Srinivasan M, Sharma S, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130(3):285–96. https://doi.org/10.1182/blood-2017-01-761320.
Berland R, Wortis HH. Origins and functions of B-1 cells with notes on the role of CD5. Annu Rev Immunol. 2002;20:253–300. https://doi.org/10.1146/annurev.immunol.20.100301.064833.
Sempowski GD, Lee DM, Kaufman RE, et al. Structure and function of the CD7 molecule. Crit Rev Immunol. 1999;19(4):331–48.
Leonard WJ. Cytokines and immunodeficiency diseases. Nat Rev Immunol. 2001;1(3):200–8. https://doi.org/10.1038/35105066.
Cooper ML, Choi J, Staser K, et al. An "off-the-shelf" fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32(9):1970–83. https://doi.org/10.1038/s41375-018-0065-5.
Mamonkin M, Rouce RH, Tashiro H, et al. A T-cell-directed chimeric antigen receptor for the selective treatment of T-cell malignancies. BLOOD. 2015;126(8):983–92.
Freitas C, Johnson DK, Weber KS. T cell calcium signaling regulation by the co-receptor CD5. Int J Mol Sci. 2018;19(5). https://doi.org/10.3390/ijms19051295.
Feng J, Xu H, Cinquina A, et al. Treatment of aggressive T cell lymphoblastic lymphoma/leukemia using anti-CD5 CAR T cells. Stem Cell Rev Rep. 2021;17(2):652–61. https://doi.org/10.1007/s12015-020-10092-9.
Lu X, Axtell RC, Collawn JF, et al. AP2 adaptor complex-dependent internalization of CD5: differential regulation in T and B cells. J Immunol. 2002;168(11):5612–20. https://doi.org/10.4049/jimmunol.168.11.5612.
Coppola C, Hopkins B, Huhn S, et al. Investigation of the impact from IL-2, IL-7, and IL-15 on the growth and signaling of activated CD4(+) T cells. Int J Mol Sci. 2020;21(21). https://doi.org/10.3390/ijms21217814.
Maciocia PM, Wawrzyniecka PA, Philip B, et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat Med. 2017;23(12):1416–23.
Guercio M, Orlando D, Di Cecca S, et al. CD28.OX40 co-stimulatory combination is associated with long in vivo persistence and high activity of CAR.CD30 T-cells. Haematologica. 2021;106(4):987–99. https://doi.org/10.3324/haematol.2019.231183.
Oluwasanjo A, Kartan S, Johnson W, et al. Peripheral T-cell lymphoma, not otherwise specified (PTCL-NOS). Cancer Treat Res. 2019;176:83–98. https://doi.org/10.1007/978-3-319-99716-2_4.
Dai Z, Mu W, Zhao Y, et al. T cells expressing CD5/CD7 bispecific chimeric antigen receptors with fully human heavy-chain-only domains mitigate tumor antigen escape. Signal Transduct Target Ther. 2022;7(1):85. https://doi.org/10.1038/s41392-022-00898-z.
Ansell SM. Hodgkin lymphoma: 2018 update on diagnosis, risk-stratification, and management. Am J Hematol. 2018;93(5):704–15. https://doi.org/10.1002/ajh.25071.
Ramos CA, Grover NS, Beaven AW, et al. Anti-CD30 CAR-T cell therapy in relapsed and refractory Hodgkin lymphoma. J Clin Oncol. 2020;38(32):3794–804. https://doi.org/10.1200/JCO.20.01342.
Voorhees TJ, Zhao B, Oldan J, et al. Pretherapy metabolic tumor volume is associated with response to CD30 CAR T cells in Hodgkin lymphoma. Blood Adv. 2022;6(4):1255–63. https://doi.org/10.1182/bloodadvances.2021005385.
Guo J, He S, Zhu Y, et al. Humanized CD30-targeted chimeric antigen receptor T cells exhibit potent preclinical activity against Hodgkin's lymphoma cells. Front Cell Dev Biol. 2021;9:775599. https://doi.org/10.3389/fcell.2021.775599.
Xue Y, Lai X, Li R, et al. CD19 and CD30 CAR T-cell immunotherapy for high-risk classical Hodgkin's lymphoma. Front Oncol. 2020;10:607362. https://doi.org/10.3389/fonc.2020.607362.
Beldjord K, Chevret S, Asnafi V, et al. Oncogenetics and minimal residual disease are independent outcome predictors in adult patients with acute lymphoblastic leukemia. Blood. 2014;123(24):3739–49. https://doi.org/10.1182/blood-2014-01-547695.
Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–48. https://doi.org/10.1056/NEJMoa1709866.
Maude SL, Pulsipher MA, Boyer MW, et al. Efficacy and safety of CTL019 in the first US phase II multicenter trial in pediatric relapsed/refractory acute lymphoblastic leukemia: results of an interim analysis. Blood. 2016;128(22):2801. https://doi.org/10.1182/blood.V128.22.2801.2801.
Laetsch TW, Myers GD, Baruchel A, et al. Patient-reported quality of life after tisagenlecleucel infusion in children and young adults with relapsed or refractory B-cell acute lymphoblastic leukaemia: a global, single-arm, phase 2 trial. Lancet Oncol. 2019;20(12):1710–8. https://doi.org/10.1016/S1470-2045(19)30493-0.
Shah BD, Ghobadi A, Oluwole OO, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398(10299):491–502. https://doi.org/10.1016/S0140-6736(21)01222-8.
Iwamoto S, Deguchi T, Ohta H, et al. Flow cytometric analysis of de novo acute lymphoblastic leukemia in childhood: report from the Japanese pediatric leukemia/lymphoma study group. Int J Hematol. 2011;94(2):185–92. https://doi.org/10.1007/s12185-011-0900-1.
Kantarjian H, Thomas D, Jorgensen J, et al. Results of inotuzumab ozogamicin, a CD22 monoclonal antibody, in refractory and relapsed acute lymphocytic leukemia. Cancer. 2013;119(15):2728–36. https://doi.org/10.1002/cncr.28136.
Pan J, Niu Q, Deng B, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33(12):2854–66. https://doi.org/10.1038/s41375-019-0488-7.
Guo Y, Feng K, Tong C, et al. Efficiency and side effects of anti-CD38 CAR T cells in an adult patient with relapsed B-ALL after failure of bi-specific CD19/CD22 CAR T cell treatment. Cell Mol Immunol. 2020;17(4):430–2. https://doi.org/10.1038/s41423-019-0355-5.
Testa U, Pelosi E, Frankel A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark Res. 2014;2(1):4. https://doi.org/10.1186/2050-7771-2-4.
Ruella M, Barrett DM, Kenderian SS, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126(10):3814–26. https://doi.org/10.1172/JCI87366.
Dong Z, Cheng WA, Smith DL, et al. Antitumor efficacy of BAFF-R targeting CAR T cells manufactured under clinic-ready conditions. Cancer Immunol Immunother. 2020;69(10):2139–45. https://doi.org/10.1007/s00262-020-02614-8.
Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–15. https://doi.org/10.1056/NEJMoa1403088.
Qin H, Cho M, Haso W, et al. Eradication of B-ALL using chimeric antigen receptor-expressing T cells targeting the TSLPR oncoprotein. Blood. 2015;126(5):629–39. https://doi.org/10.1182/blood-2014-11-612903.
Liu S, Deng B, Yin Z, et al. Combination of CD19 and CD22 CAR-T cell therapy in relapsed B-cell acute lymphoblastic leukemia after allogeneic transplantation. Am J Hematol. 2021;96(6):671–9. https://doi.org/10.1002/ajh.26160.
Dai H, Wu Z, Jia H, et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J Hematol Oncol. 2020;13(1):30. https://doi.org/10.1186/s13045-020-00856-8.
Yan N, Wang N, Wang G, et al. CAR19/22 T cell cocktail therapy for B-ALL relapsed after allogeneic hematopoietic stem cell transplantation. Cytotherapy. 2022;24(8):841–9.
Wang Y, Yang Y, Hong R, et al. A retrospective comparison of CD19 single and CD19/CD22 bispecific targeted chimeric antigen receptor T cell therapy in patients with relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2020;10(10):105. https://doi.org/10.1038/s41408-020-00371-6.
Fousek K, Watanabe J, Joseph SK, et al. CAR T-cells that target acute B-lineage leukemia irrespective of CD19 expression. Leukemia. 2021;35(1):75–89. https://doi.org/10.1038/s41375-020-0792-2.
Nix MA, Mandal K, Geng H, et al. Surface proteomics reveals CD72 as a target for in vitro-evolved Nanobody-based CAR-T cells in KMT2A/MLL1-rearranged B-ALL. Cancer Discov. 2021;11(8):2032–49. https://doi.org/10.1158/2159-8290.CD-20-0242.
Pan J, Tan Y, Wang G, et al. Donor-derived CD7 chimeric antigen receptor T cells for T-cell acute lymphoblastic leukemia: first-in-human. Phase I Trial J Clin Oncol. 2021;39(30):3340–51. https://doi.org/10.1200/JCO.21.00389.
Li S, Wang X, Yuan Z, et al. Eradication of T-ALL cells by CD7-targeted universal CAR-T cells and initial test of Ruxolitinib-based CRS management. Clin Cancer Res. 2021;27(5):1242–6.
Stadtmauer EA, Fraietta JA, Davis MM, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481). https://doi.org/10.1126/science.aba7365.
Yu Y, Leete TC, Born DA, et al. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat Commun. 2020;11(1):2052. https://doi.org/10.1038/s41467-020-15887-5.
Diorio C, Murray R, Naniong M, et al. Cytosine base editing enables quadruple-edited allogeneic CART cells for T-ALL. Blood. 2022;140(6):619–29. https://doi.org/10.1182/blood.2022015825.
Wada M, Zhang H, Fang L, et al. Characterization of an anti-CD5 directed CAR T-cell against T-cell malignancies. Stem Cell Rev Rep. 2020;16(2):369–84. https://doi.org/10.1007/s12015-019-09937-9.
Driouk L, Gicobi JK, Kamihara Y, et al. Chimeric antigen receptor T cells targeting NKG2D-ligands show robust efficacy against acute myeloid leukemia and T-cell acute lymphoblastic leukemia. Front Immunol. 2020;11:580328. https://doi.org/10.3389/fimmu.2020.580328.
Hassan G, Seno M. Blood and cancer: cancer stem cells as origin of hematopoietic cells in solid tumor microenvironments. Cells. 2020;9(5). https://doi.org/10.3390/cells9051293.
Cornelissen JJ, Blaise D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood. 2016;127(1):62–70. https://doi.org/10.1182/blood-2015-07-604546.
Mo JS, Park HW, Guan KL. The hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014;15(6):642–56. https://doi.org/10.15252/embr.201438638.
Ehninger A, Kramer M, Röllig C, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4(6):e218. https://doi.org/10.1038/bcj.2014.39.
Walter RB, Appelbaum FR, Estey EH, et al. Acute myeloid leukemia stem cells and CD33-targeted immunotherapy. Blood. 2012;119(26):6198–208. https://doi.org/10.1182/blood-2011-11-325050.
Willier S, Rothämel P, Hastreiter M, et al. CLEC12A and CD33 coexpression as a preferential target for pediatric AML combinatorial immunotherapy. Blood. 2021;137(8):1037–49. https://doi.org/10.1182/blood.2020006921.
Gill S, Tasian SK, Ruella M, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343–54. https://doi.org/10.1182/blood-2013-09-529537.
Petrov JC, Wada M, Pinz KG, et al. Compound CAR T-cells as a double-pronged approach for treating acute myeloid leukemia. Leukemia. 2018;32(6):1317–26. https://doi.org/10.1038/s41375-018-0075-3.
Wang QS, Wang Y, Lv HY, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23(1):184–91. https://doi.org/10.1038/mt.2014.164.
Baroni ML, Sanchez Martinez D, Gutierrez Aguera F, et al. 41BB-based and CD28-based CD123-redirected T-cells ablate human normal hematopoiesis in vivo. J Immunother Cancer. 2020;8(1). https://doi.org/10.1136/jitc-2020-000845.
Cui Q, Qian C, Xu N, et al. CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. 2021;14(1):82. https://doi.org/10.1186/s13045-021-01092-4.
Zhang H, Wang P, Li Z, et al. Anti-CLL1 chimeric antigen receptor T-cell therapy in children with relapsed/refractory acute myeloid leukemia. Clin Cancer Res. 2021;27(13):3549–55.
Ma YJ, Dai HP, Cui QY, et al. Successful application of PD-1 knockdown CLL-1 CAR-T therapy in two AML patients with post-transplant relapse and failure of anti-CD38 CAR-T cell treatment. Am J Cancer Res. 2022;12(2):615–21.
Ritchie DS, Neeson PJ, Khot A, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122–9. https://doi.org/10.1038/mt.2013.154.
Gomes-Silva D, Atilla E, Atilla PA, et al. CD7 CAR T cells for the therapy of acute myeloid leukemia. Mol Ther. 2019;27(1):272–80.
Zhang M, Chen D, Fu X, et al. Autologous Nanobody-derived fratricide-resistant CD7-CAR T-cell therapy for patients with relapsed and refractory T-cell acute lymphoblastic leukemia/lymphoma. Clin Cancer Res. 2022;28(13):2830–43. https://doi.org/10.1158/1078-0432.CCR-21-4097.
Sommer C, Cheng HY, Nguyen D, et al. Allogeneic FLT3 CAR T cells with an off-switch exhibit potent activity against AML and can be depleted to expedite bone marrow recovery. Mol Ther. 2020;28(10):2237–51. https://doi.org/10.1016/j.ymthe.2020.06.022.
Jetani H, Navarro-Bailón A, Maucher M, et al. Siglec-6 is a novel target for CAR T-cell therapy in acute myeloid leukemia. BLOOD. 2021;138(19):1830–42.
Myburgh R, Kiefer JD, Russkamp NF, et al. Anti-human CD117 CAR T-cells efficiently eliminate healthy and malignant CD117-expressing hematopoietic cells. Leukemia. 2020;34(10):2688–703.
Sauer T, Parikh K, Sharma S, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. 2021;138(4):318–30.
Zoine JT, Prince C, Story JY, et al. Thrombopoietin-based CAR-T cells demonstrate in vitro and in vivo cytotoxicity to MPL positive acute myelogenous leukemia and hematopoietic stem cells. Gene Ther. 2022;29(5):1–12.
Lee WS, Ye Z, Cheung A, et al. Effective killing of acute myeloid leukemia by TIM-3 targeted chimeric antigen receptor T cells. Mol Cancer Ther. 2021;20(9):1702–12. https://doi.org/10.1158/1535-7163.MCT-20-0155.
Casucci M, Nicolis di Robilant B, Falcone L, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122(20):3461–72. https://doi.org/10.1182/blood-2013-04-493361.
Lynn RC, Poussin M, Kalota A, et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood. 2015;125(22):3466–76. https://doi.org/10.1182/blood-2014-11-612721.
John S, Chen H, Deng M, et al. A novel anti-LILRB4 CAR-T cell for the treatment of Monocytic AML. Mol Ther. 2018;26(10):2487–95. https://doi.org/10.1016/j.ymthe.2018.08.001.
Rafiq S, Purdon TJ, Daniyan AF, et al. Optimized T-cell receptor-mimic chimeric antigen receptor T cells directed toward the intracellular Wilms tumor 1 antigen. Leukemia. 2017;31(8):1788–97. https://doi.org/10.1038/leu.2016.373.
Ma Q, Garber HR, Lu S, et al. A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. 2016;18(8):985–94. https://doi.org/10.1016/j.jcyt.2016.05.001.
Le Q, Castro S, Tang T, et al. Therapeutic targeting of Mesothelin with chimeric antigen receptor T cells in acute myeloid leukemia. Clin Cancer Res. 2021;27(20):5718–30.
Siddiqi T, Soumerai JD, Dorritie KA, et al. Phase 1 TRANSCEND CLL 004 study of lisocabtagene maraleucel in patients with relapsed/refractory CLL or SLL. Blood. 2022;139(12):1794–806. https://doi.org/10.1182/blood.2021011895.
Frey NV, Gill S, Hexner EO, et al. Long-term outcomes from a randomized dose optimization study of chimeric antigen receptor modified T cells in relapsed chronic lymphocytic leukemia. J Clin Oncol. 2020;38(25):2862–71. https://doi.org/10.1200/JCO.19.03237.
Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. https://doi.org/10.1126/scitranslmed.aac5415.
Turtle CJ, Hay KA, Hanafi LA, et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor-modified T cells after failure of Ibrutinib. J Clin Oncol. 2017;35(26):3010–20. https://doi.org/10.1200/JCO.2017.72.8519.
Bilich T, Nelde A, Bauer J, et al. Mass spectrometry-based identification of a B-cell maturation antigen-derived T-cell epitope for antigen-specific immunotherapy of multiple myeloma. Blood Cancer J. 2020;10(2):24. https://doi.org/10.1038/s41408-020-0288-3.
Udd KA, Bujarski S, Wirtschafter E, et al. Plasma B-cell maturation antigen levels are elevated and correlate with disease activity in patients with chronic lymphocytic leukemia. Target Oncol. 2019;14(5):551–61. https://doi.org/10.1007/s11523-019-00666-0.
Wang G, Sun X, Zuo S, et al. Homogeneously high expression of CD32b makes it a potential target for CAR-T therapy for chronic lymphocytic leukemia. J Hematol Oncol. 2021;14(1):149. https://doi.org/10.1186/s13045-021-01160-9.
Faitschuk E, Hombach AA, Frenzel LP, et al. Chimeric antigen receptor T cells targeting fc μ receptor selectively eliminate CLL cells while sparing healthy B cells. Blood. 2016;128(13):1711–22. https://doi.org/10.1182/blood-2016-01-692046.
Cui B, Ghia EM, Chen L, et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood. 2016;128(25):2931–40. https://doi.org/10.1182/blood-2016-04-712562.
Tettamanti S, Rotiroti MC, Giordano Attianese G, et al. Lenalidomide enhances CD23.CAR T cell therapy in chronic lymphocytic leukemia. Leuk Lymphoma. 2022:1–14. https://doi.org/10.1080/10428194.2022.2043299.
Kovalovsky D, Yoon JH, Cyr MG, et al. Siglec-6 is a target for chimeric antigen receptor T-cell treatment of chronic lymphocytic leukemia. Leukemia. 2021;35(9):2581–91. https://doi.org/10.1038/s41375-021-01188-3.
Pricop L, Hatakeyama A, Isobe H, et al. Analysis of lambda repertoire in kappa-deficient mice. Clin Immunol Immunopathol. 1995;76(3 Pt 2):S179–87. https://doi.org/10.1016/s0090-1229(95)90162-0.
Carpenter RO, Evbuomwan MO, Pittaluga S, et al. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin Cancer Res. 2013;19(8):2048–60. https://doi.org/10.1158/1078-0432.CCR-12-2422.
Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene Vicleucel in relapsed and refractory multiple myeloma. N Engl J Med. 2021;384(8):705–16. https://doi.org/10.1056/NEJMoa2024850.
Mullard A. FDA approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discov. 2022;21(4):249. https://doi.org/10.1038/d41573-022-00048-8.
Berdeja JG, Madduri D, Usmani SZ, et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–24. https://doi.org/10.1016/S0140-6736(21)00933-8.
Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. https://doi.org/10.1186/s13045-018-0681-6.
Han L, Gao Q, Zhou K, et al. The phase I clinical study of CART targeting BCMA with humanized alpaca-derived single-domain antibody as antigen recognition domain. JCO. 2019;37(15_suppl):2535. https://doi.org/10.1200/JCO.2019.37.15_suppl.2535.
Han L, Gao Q, Zhou K, et al. The clinical study of anti-BCMA CAR-T with single-domain antibody as antigen binding domain. JCO. 2021;39(15_suppl):8025. https://doi.org/10.1200/JCO.2021.39.15_suppl.8025.
Zhang L, Shen X, Yu W, et al. Comprehensive meta-analysis of anti-BCMA chimeric antigen receptor T-cell therapy in relapsed or refractory multiple myeloma. Ann Med. 2021;53(1):1547–59. https://doi.org/10.1080/07853890.2021.1970218.
Deng H, Liu M, Yuan T, et al. Efficacy of humanized anti-BCMA CAR T cell therapy in relapsed/refractory multiple myeloma patients with and without extramedullary disease. Front Immunol. 2021;12:720571. https://doi.org/10.3389/fimmu.2021.720571.
Golde TE, Koo EH, Felsenstein KM, et al. γ-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828(12):2898–907. https://doi.org/10.1016/j.bbamem.2013.06.005.
Laurent SA, Hoffmann FS, Kuhn PH, et al. γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun. 2015;6:7333. https://doi.org/10.1038/ncomms8333.
Pont MJ, Hill T, Cole GO, et al. γ-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood. 2019;134(19):1585–97. https://doi.org/10.1182/blood.2019000050.
Garfall AL, Stadtmauer EA, Hwang WT, et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight. 2018;3(8). https://doi.org/10.1172/jci.insight.120505.
Hsi ED, Steinle R, Balasa B, et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin Cancer Res. 2008;14(9):2775–84. https://doi.org/10.1158/1078-0432.CCR-07-4246.
O'Neal J, Ritchey JK, Cooper ML, et al. CS1 CAR-T targeting the distal domain of CS1 (SLAMF7) shows efficacy in high tumor burden myeloma model despite fratricide of CD8+CS1 expressing CAR-T cells. Leukemia. 2022. https://doi.org/10.1038/s41375-022-01559-4.
Radhakrishnan SV, Luetkens T, Scherer SD, et al. CD229 CAR T cells eliminate multiple myeloma and tumor propagating cells without fratricide. Nat Commun. 2020;11(1):798. https://doi.org/10.1038/s41467-020-14619-z.
Peinert S, Prince HM, Guru PM, et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010;17(5):678–86. https://doi.org/10.1038/gt.2010.21.
Neeson P, Shin A, Tainton KM, et al. Ex vivo culture of chimeric antigen receptor T cells generates functional CD8+ T cells with effector and central memory-like phenotype. Gene Ther. 2010;17(9):1105–16. https://doi.org/10.1038/gt.2010.59.
Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019;11(485). https://doi.org/10.1126/scitranslmed.aau7746.
Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48. https://doi.org/10.1038/s41591-018-0036-4.
Gnjatic S, Nishikawa H, Jungbluth AA, et al. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30.
van Rhee F, Szmania SM, Zhan F, et al. NY-ESO-1 is highly expressed in poor-prognosis multiple myeloma and induces spontaneous humoral and cellular immune responses. Blood. 2005;105(10):3939–44. https://doi.org/10.1182/blood-2004-09-3707.
Schuberth PC, Jakka G, Jensen SM, et al. Effector memory and central memory NY-ESO-1-specific re-directed T cells for treatment of multiple myeloma. Gene Ther. 2013;20(4):386–95. https://doi.org/10.1038/gt.2012.48.
Baumeister SH, Murad J, Werner L, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100–12. https://doi.org/10.1158/2326-6066.CIR-18-0307.
Mishra AK, Kemler I, Dingli D. Preclinical development of CD126 CAR-T cells with broad antitumor activity. Blood Cancer J. 2021;11(1):3. https://doi.org/10.1038/s41408-020-00405-z.
An N, Hou YN, Zhang QX, et al. Anti-multiple myeloma activity of Nanobody-based anti-CD38 chimeric antigen receptor T cells. Mol Pharm. 2018;15(10):4577–88. https://doi.org/10.1021/acs.molpharmaceut.8b00584.
van de Donk N, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. 2018;9:2134. https://doi.org/10.3389/fimmu.2018.02134.
Nageshwari B, Merugu R. Effect of levamisole on expression of CD138 and interleukin-6 in human multiple myeloma cell lines. Indian J Cancer. 2017;54(3):566–71. https://doi.org/10.4103/ijc.IJC_349_17.
Sun C, Mahendravada A, Ballard B, et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget. 2019;10(24):2369–83. https://doi.org/10.18632/oncotarget.26792.
Skerget M, Skopec B, Zadnik V, et al. CD56 expression is an important prognostic factor in multiple myeloma even with Bortezomib induction. Acta Haematol. 2018;139(4):228–34. https://doi.org/10.1159/000489483.
Mei H, Li C, Jiang H, et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol. 2021;14(1):161. https://doi.org/10.1186/s13045-021-01170-7.
Zhang H, Liu M, Xiao X, et al. A combination of humanized anti-BCMA and murine anti-CD38 CAR-T cell therapy in patients with relapsed or refractory multiple myeloma. Leuk Lymphoma. 2022;63(6):1418–27. https://doi.org/10.1080/10428194.2022.2030476.
Wang Y, Cao J, Gu W, et al. Long-term follow-up of combination of B-cell maturation antigen and CD19 chimeric antigen receptor T cells in multiple myeloma. J Clin Oncol. 2022:JCO2101676. https://doi.org/10.1200/JCO.21.01676.
Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49. https://doi.org/10.3322/caac.21660.
Yang YH, Liu JW, Lu C, et al. CAR-T cell therapy for breast cancer: from basic research to clinical application. Int J Biol Sci. 2022;18(6):2609–26. https://doi.org/10.7150/ijbs.70120.
Szöőr Á, Tóth G, Zsebik B, et al. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020;484:1–8. https://doi.org/10.1016/j.canlet.2020.04.008.
Xia L, Zheng Z, Liu JY, et al. Targeting triple-negative breast cancer with combination therapy of EGFR CAR T cells and CDK7 inhibition. Cancer Immunol Res. 2021;9(6):707–22. https://doi.org/10.1158/2326-6066.CIR-20-0405.
Tchou J, Zhao Y, Levine BL, et al. Safety and efficacy of Intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res. 2017;5(12):1152–61. https://doi.org/10.1158/2326-6066.CIR-17-0189.
Wallstabe L, Göttlich C, Nelke LC, et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI. Insight. 2019;4(18). https://doi.org/10.1172/jci.insight.126345.
Wei J, Sun H, Zhang A, et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol. 2018;331:49–58. https://doi.org/10.1016/j.cellimm.2018.05.004.
Jayasooriya V, Ringwelski B, Dorsam G, et al. mRNA-based CAR T-cells manufactured by miniaturized two-step electroporation produce selective cytotoxicity toward target cancer cells. Lab Chip. 2021;21(19):3748–61. https://doi.org/10.1039/d1lc00219h.
Zhou R, Yazdanifar M, Roy LD, et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol. 2019;10:1149. https://doi.org/10.3389/fimmu.2019.01149.
Yang P, Cao X, Cai H, et al. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021;360:104262. https://doi.org/10.1016/j.cellimm.2020.104262.
Wang Z, Zhou G, Risu N, et al. Lenalidomide enhances CAR-T cell activity against solid tumor cells. Cell Transplant. 2020;29:963689720920825. https://doi.org/10.1177/0963689720920825.
Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61. https://doi.org/10.1007/s00262-012-1212-x.
Ye X, Deng X, Wen J, et al. Folate receptor-alpha targeted 7x19 CAR-γδT suppressed triple-negative breast cancer xenograft model in mice. J Oncol. 2022;2022:2112898. https://doi.org/10.1155/2022/2112898.
Geldres C, Savoldo B, Hoyos V, et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res. 2014;20(4):962–71. https://doi.org/10.1158/1078-0432.CCR-13-2218.
Wei H, Wang Z, Kuang Y, et al. Intercellular adhesion Molecule-1 as target for CAR-T-cell therapy of triple-negative breast cancer. Front Immunol. 2020;11:573823. https://doi.org/10.3389/fimmu.2020.573823.
Byrd TT, Fousek K, Pignata A, et al. TEM8/ANTXR1-specific CAR T cells as a targeted therapy for triple-negative breast cancer. Cancer Res. 2018;78(2):489–500. https://doi.org/10.1158/0008-5472.CAN-16-1911.
Chen H, Wei F, Yin M, et al. CD27 enhances the killing effect of CAR T cells targeting trophoblast cell surface antigen 2 in the treatment of solid tumors. Cancer Immunol Immunother. 2021;70(7):2059–71. https://doi.org/10.1007/s00262-020-02838-8.
Yang M, Tang X, Zhang Z, et al. Tandem CAR-T cells targeting CD70 and B7-H3 exhibit potent preclinical activity against multiple solid tumors. Theranostics. 2020;10(17):7622–34. https://doi.org/10.7150/thno.43991.
Porcellini S, Asperti C, Corna S, et al. CAR T cells redirected to CD44v6 control tumor growth in lung and ovary adenocarcinoma bearing mice. Front Immunol. 2020;11:99. https://doi.org/10.3389/fimmu.2020.00099.
Cha JH, Chan LC, Wang YN, et al. Ephrin receptor A10 monoclonal antibodies and the derived chimeric antigen receptor T cells exert an antitumor response in mouse models of triple-negative breast cancer. J Biol Chem. 2022;298(4):101817. https://doi.org/10.1016/j.jbc.2022.101817.
Li D, English H, Hong J, et al. A novel PD-L1-targeted shark V(NAR) single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer. Mol Ther Oncolytics. 2022;24:849–63. https://doi.org/10.1016/j.omto.2022.02.015.
Seitz CM, Schroeder S, Knopf P, et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology. 2020;9(1):1683345. https://doi.org/10.1080/2162402X.2019.1683345.
Zhang C, Röder J, Scherer A, et al. Bispecific antibody-mediated redirection of NKG2D-CAR natural killer cells facilitates dual targeting and enhances antitumor activity. J Immunother Cancer. 2021;9(10). https://doi.org/10.1136/jitc-2021-002980.
Jie Y, Liu G, Feng L, et al. PTK7-targeting CAR T-cells for the treatment of lung cancer and other malignancies. Front Immunol. 2021;12:665970. https://doi.org/10.3389/fimmu.2021.665970.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54. https://doi.org/10.1038/nature25183.
Doroshow DB, Sanmamed MF, Hastings K, et al. Immunotherapy in non-small cell lung cancer: facts and hopes. Clin Cancer Res. 2019;25(15):4592–602. https://doi.org/10.1158/1078-0432.CCR-18-1538.
Chen L, Chen F, Li J, et al. CAR-T cell therapy for lung cancer: potential and perspective. Thorac Cancer. 2022;13(7):889–99. https://doi.org/10.1111/1759-7714.14375.
Wang Y, Wang J, Yang X, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting Mesothelin in a non-small-cell lung carcinoma model. Front Immunol. 2021;12:628906. https://doi.org/10.3389/fimmu.2021.628906.
Qin L, Lai Y, Zhao R, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017;10(1):68. https://doi.org/10.1186/s13045-017-0437-8.
Li H, Huang Y, Jiang DQ, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018;9(2):177. https://doi.org/10.1038/s41419-017-0238-6.
Dai W, Wang L, Shi X. NK-92 cells modified with chimeric antigen receptor targeting PD-L1 inhibits the proliferation of A549 lung cancer cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2022;38(3):212–7.
Li H, Harrison EB, Li H, et al. Targeting brain lesions of non-small cell lung cancer by enhancing CCL2-mediated CAR-T cell migration. Nat Commun. 2022;13(1):2154. https://doi.org/10.1038/s41467-022-29647-0.
Wei X, Lai Y, Li J, et al. PSCA and MUC1 in non-small-cell lung cancer as targets of chimeric antigen receptor T cells. Oncoimmunology. 2017;6(3):e1284722. https://doi.org/10.1080/2162402X.2017.1284722.
Zhang Y, Zhang Z, Ding Y, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol. 2021;147(12):3725–34. https://doi.org/10.1007/s00432-021-03613-7.
Ye L, Lou Y, Lu L, et al. Mesothelin-targeted second generation CAR-T cells inhibit growth of mesothelin-expressing tumors in vivo. Exp Ther Med. 2019;17(1):739–47. https://doi.org/10.3892/etm.2018.7015.
Owen DH, Giffin MJ, Bailis JM, et al. DLL3: an emerging target in small cell lung cancer. J Hematol Oncol. 2019;12(1):61. https://doi.org/10.1186/s13045-019-0745-2.
Chu W, Zhou Y, Tang Q, et al. Bi-specific ligand-controlled chimeric antigen receptor T-cell therapy for non-small cell lung cancer. Biosci Trends. 2018;12(3):298–308. https://doi.org/10.5582/bst.2018.01048.
Mao Y, Fan W, Hu H, et al. MAGE-A1 in lung adenocarcinoma as a promising target of chimeric antigen receptor T cells. J Hematol Oncol. 2019;12(1):106. https://doi.org/10.1186/s13045-019-0793-7.
Shimizu Y, Suzuki T, Yoshikawa T, et al. Next-generation cancer immunotherapy targeting Glypican-3. Front Oncol. 2019;9:248. https://doi.org/10.3389/fonc.2019.00248.
Taromi S, Firat E, Simonis A, et al. Enhanced AC133-specific CAR T cell therapy induces durable remissions in mice with metastatic small cell lung cancer. Cancer Lett. 2022;538:215697. https://doi.org/10.1016/j.canlet.2022.215697.
Min J, Long C, Zhang L, et al. C-met specific CAR-T cells as a targeted therapy for non-small cell lung cancer cell A549. Bioengineered. 2022;13(4):9216–32. https://doi.org/10.1080/21655979.2022.2058149.
Martin AL, Anadon CM, Biswas S, et al. Olfactory receptor OR2H1 is an effective target for CAR T cells in human epithelial tumors. Mol Cancer Ther. 2022. https://doi.org/10.1158/1535-7163.MCT-21-0872.
Reppel L, Tsahouridis O, Akulian J, et al. Targeting disialoganglioside GD2 with chimeric antigen receptor-redirected T cells in lung cancer. J Immunother Cancer. 2022;10(1). https://doi.org/10.1136/jitc-2021-003897.
Chen XH, Chen R, Shi MY, et al. Chimeric antigen receptor T cells targeting CD147 for non-small cell lung cancer therapy. Transl Oncol. 2022;16:101309. https://doi.org/10.1016/j.tranon.2021.101309.
La HT, Tran D, Tran HM, et al. Third-generation anti-CD47-specific CAR-T cells effectively kill cancer cells and reduce the genes expression in lung cancer cell metastasis. J Immunol Res. 2021;2021:5575260. https://doi.org/10.1155/2021/5575260.
Kakarla S, Chow KK, Mata M, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. 2013;21(8):1611–20. https://doi.org/10.1038/mt.2013.110.
Li N, Liu S, Sun M, et al. Chimeric antigen receptor-modified T cells redirected to EphA2 for the immunotherapy of non-small cell lung cancer. Transl Oncol. 2018;11(1):11–7. https://doi.org/10.1016/j.tranon.2017.10.009.
Mura M, Swain RK, Zhuang X, et al. Identification and angiogenic role of the novel tumor endothelial marker CLEC14A. Oncogene. 2012;31(3):293–305. https://doi.org/10.1038/onc.2011.233.
Zhuang X, Maione F, Robinson J, et al. CAR T cells targeting tumor endothelial marker CLEC14A inhibit tumor growth. JCI Insight. 2020;5(19). https://doi.org/10.1172/jci.insight.138808.
Gangadhar T, Nandi S, Salgia R. The role of chemokine receptor CXCR4 in lung cancer. Cancer Biol Ther. 2010;9(6):409–16. https://doi.org/10.4161/cbt.9.6.11233.
Moon EK, Ranganathan R, Eruslanov E, et al. Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res. 2016;22(2):436–47. https://doi.org/10.1158/1078-0432.CCR-15-1070.
Powers E, Karachaliou GS, Kao C, et al. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J Hematol Oncol. 2020;13(1):144. https://doi.org/10.1186/s13045-020-00978-z.
Bouchelouche K, Choyke PL. Advances in prostate-specific membrane antigen PET of prostate cancer. Curr Opin Oncol. 2018;30(3):189–96. https://doi.org/10.1097/CCO.0000000000000439.
Arndt C, Feldmann A, Koristka S, et al. A theranostic PSMA ligand for PET imaging and retargeting of T cells expressing the universal chimeric antigen receptor UniCAR. Oncoimmunology. 2019;8(11):1659095. https://doi.org/10.1080/2162402X.2019.1659095.
Stultz J, Fong L. How to turn up the heat on the cold immune microenvironment of metastatic prostate cancer. Prostate Cancer Prostatic Dis. 2021;24(3):697–717. https://doi.org/10.1038/s41391-021-00340-5.
Berish RB, Ali AN, Telmer PG, et al. Translational models of prostate cancer bone metastasis. Nat Rev Urol. 2018;15(7):403–21. https://doi.org/10.1038/s41585-018-0020-2.
Narayan V, Barber-Rotenberg JS, Jung IY, et al. PSMA-targeting TGFβ-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. 2022;28(4):724–34. https://doi.org/10.1038/s41591-022-01726-1.
Arndt C, Bergmann R, Striese F, et al. Development and functional characterization of a versatile radio-/Immunotheranostic tool for prostate cancer management. Cancers (Basel). 2022;14(8). https://doi.org/10.3390/cancers14081996.
Abate-Daga D, Lagisetty KH, Tran E, et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum Gene Ther. 2014;25(12):1003–12. https://doi.org/10.1089/hum.2013.209.
Deng Z, Wu Y, Ma W, et al. Adoptive T-cell therapy of prostate cancer targeting the cancer stem cell antigen EpCAM. BMC Immunol. 2015;16(1):1. https://doi.org/10.1186/s12865-014-0064-x.
He C, Zhou Y, Li Z, et al. Co-expression of IL-7 improves NKG2D-based CAR T cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers (Basel). 2020;12(7). https://doi.org/10.3390/cancers12071969.
Zhang Y, He L, Sadagopan A, et al. Targeting radiation-resistant prostate cancer stem cells by B7-H3 CAR T cells. Mol Cancer Ther. 2021;20(3):577–88. https://doi.org/10.1158/1535-7163.MCT-20-0446.
Baek DS, Kim YJ, Vergara S, et al. A highly-specific fully-human antibody and CAR-T cells targeting CD66e/CEACAM5 are cytotoxic for CD66e-expressing cancer cells in vitro and in vivo. Cancer Lett. 2022;525:97–107. https://doi.org/10.1016/j.canlet.2021.10.041.
Sanchez C, Chan R, Bajgain P, et al. Combining T-cell immunotherapy and anti-androgen therapy for prostate cancer. Prostate Cancer Prostatic Dis. 2013;16(2):123–131, S1. https://doi.org/10.1038/pcan.2012.49.
Zheng Y, Nandakumar KS, Cheng K. Optimization of CAR-T cell-based therapies using small-molecule-based safety switches. J Med Chem. 2021;64(14):9577–91. https://doi.org/10.1021/acs.jmedchem.0c02054.
El Bali M, Bakkach J, Bennani MM. Colorectal cancer: from genetic landscape to targeted therapy. J Oncol. 2021;2021:9918116. https://doi.org/10.1155/2021/9918116.
Hege KM, Bergsland EK, Fisher GA, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22. https://doi.org/10.1186/s40425-017-0222-9.
Han ZW, Lyv ZW, Cui B, et al. Correction to: the old CEACAMs find their new role in tumor immunotherapy. Investig New Drugs. 2020;38(6):1899–900. https://doi.org/10.1007/s10637-020-00967-6.
Zhang C, Wang Z, Yang Z, et al. Phase I escalating-dose trial of CAR-T therapy targeting CEA(+) metastatic colorectal cancers. Mol Ther. 2017;25(5):1248–58. https://doi.org/10.1016/j.ymthe.2017.03.010.
Katz SC, Burga RA, McCormack E, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21(14):3149–59. https://doi.org/10.1158/1078-0432.CCR-14-1421.
Parkhurst MR, Yang JC, Langan RC, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–6. https://doi.org/10.1038/mt.2010.272.
Sandberg ML, Wang X, Martin AD, et al. A carcinoembryonic antigen-specific cell therapy selectively targets tumor cells with HLA loss of heterozygosity in vitro and in vivo. Sci Transl Med. 2022;14(634):eabm0306. https://doi.org/10.1126/scitranslmed.abm0306.
Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414(6859):105–11. https://doi.org/10.1038/35102167.
Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a phase I trial. Oncoimmunology. 2018;7(7):e1440169. https://doi.org/10.1080/2162402X.2018.1440169.
Zhang Q, Zhang H, Ding J, et al. Combination therapy with EpCAM-CAR-NK-92 cells and Regorafenib against human colorectal cancer models. J Immunol Res. 2018;2018:4263520. https://doi.org/10.1155/2018/4263520.
Xu J, Meng Q, Sun H, et al. HER2-specific chimeric antigen receptor-T cells for targeted therapy of metastatic colorectal cancer. Cell Death Dis. 2021;12(12):1109. https://doi.org/10.1038/s41419-021-04100-0.
Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51. https://doi.org/10.1038/mt.2010.24.
Li Z, Chi Z, Ang WX, et al. Experimental treatment of colorectal cancer in mice with human T cells electroporated with NKG2D RNA CAR. Immunotherapy. 2020;12(10):733–48. https://doi.org/10.2217/imt-2019-0137.
Magee MS, Abraham TS, Baybutt TR, et al. Human GUCY2C-targeted chimeric antigen receptor (CAR)-expressing T cells eliminate colorectal cancer metastases. Cancer Immunol Res. 2018;6(5):509–16. https://doi.org/10.1158/2326-6066.CIR-16-0362.
Zhang Q, Liu G, Liu J, et al. The antitumor capacity of mesothelin-CAR-T cells in targeting solid tumors in mice. Mol Ther Oncolytics. 2021;20:556–68. https://doi.org/10.1016/j.omto.2021.02.013.
Nabavinia MS, Gholoobi A, Charbgoo F, et al. Anti-MUC1 aptamer: a potential opportunity for cancer treatment. Med Res Rev. 2017;37(6):1518–39. https://doi.org/10.1002/med.21462.
Lee SJ, Lee J, Park SH, et al. C-MET overexpression in colorectal cancer: a poor prognostic factor for survival. Clin Colorectal Cancer. 2018;17(3):165–9. https://doi.org/10.1016/j.clcc.2018.02.013.
Li X, Berahovich R, Zhou H, et al. PLAP -CAR T cells mediate high specific cytotoxicity against colon cancer cells. Front Biosci (Landmark Ed). 2020;25(9):1765–86. https://doi.org/10.2741/4877.
Feng Z, He X, Zhang X, et al. Potent suppression of neuroendocrine tumors and gastrointestinal cancers by CDH17CAR T cells without toxicity to normal tissues. Nat Can. 2022. https://doi.org/10.1038/s43018-022-00344-7.
Grierson P, Lim KH, Amin M. Immunotherapy in gastrointestinal cancers. J Gastrointest Oncol. 2017;8(3):474–84. https://doi.org/10.21037/jgo.2017.05.01.
Lyons TG, Ku GY. Systemic therapy for esophagogastric cancer: targeted therapies. Chin. Clin Oncol. 2017;6(5):48. https://doi.org/10.21037/cco.2017.07.02.
Qi C, Gong J, Li J, et al. Claudin18.2-specific CAR T cells in gastrointestinal cancers: phase 1 trial interim results. Nat Med. 2022. https://doi.org/10.1038/s41591-022-01800-8.
Bębnowska D, Grywalska E, Niedźwiedzka-Rystwej P, et al. CAR-T cell therapy-An overview of targets in gastric cancer. J Clin Med. 2020;9(6). https://doi.org/10.3390/jcm9061894.
Dai M, Yuan F, Fu C, et al. Relationship between epithelial cell adhesion molecule (EpCAM) overexpression and gastric cancer patients: a systematic review and meta-analysis. PLoS One. 2017;12(4):e0175357. https://doi.org/10.1371/journal.pone.0175357.
Knödler M, Körfer J, Kunzmann V, et al. Randomised phase II trial to investigate catumaxomab (anti-EpCAM × anti-CD3) for treatment of peritoneal carcinomatosis in patients with gastric cancer. Br J Cancer. 2018;119(3):296–302. https://doi.org/10.1038/s41416-018-0150-6.
Song Y, Tong C, Wang Y, et al. Effective and persistent antitumor activity of HER2-directed CAR-T cells against gastric cancer cells in vitro and xenotransplanted tumors in vivo. Protein Cell. 2018;9(10):867–78. https://doi.org/10.1007/s13238-017-0384-8.
Chi X, Yang P, Zhang E, et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019;8(10):4753–65. https://doi.org/10.1002/cam4.2361.
Maher J, Wilkie S. CAR mechanics: driving T cells into the MUC of cancer. Cancer Res. 2009;69(11):4559–62. https://doi.org/10.1158/0008-5472.CAN-09-0564.
Lv J, Li P. Mesothelin as a biomarker for targeted therapy. Biomark Res. 2019;7:18. https://doi.org/10.1186/s40364-019-0169-8.
Tao K, He M, Tao F, et al. Development of NKG2D-based chimeric antigen receptor-T cells for gastric cancer treatment. Cancer Chemother Pharmacol. 2018;82(5):815–27. https://doi.org/10.1007/s00280-018-3670-0.
Zhou JT, Liu JH, Song TT, et al. EGLIF-CAR-T cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res. 2020;12:8893–902. https://doi.org/10.2147/CMAR.S260915.
Sun F, Yu X, Ju R, et al. Antitumor responses in gastric cancer by targeting B7H3 via chimeric antigen receptor T cells. Cancer Cell Int. 2022;22(1):50. https://doi.org/10.1186/s12935-022-02471-8.
Qin L, Zhao R, Chen D, et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark Res. 2020;8:19. https://doi.org/10.1186/s40364-020-00198-0.
Han Y, Sun B, Cai H, et al. Simultaneously target of normal and stem cells-like gastric cancer cells via cisplatin and anti-CD133 CAR-T combination therapy. Cancer Immunol Immunother. 2021;70(10):2795–803. https://doi.org/10.1007/s00262-021-02891-x.
Qin L, Wang L, Zhang J, et al. Therapeutic strategies targeting uPAR potentiate anti-PD-1 efficacy in diffuse-type gastric cancer. Sci Adv. 2022;8(21):eabn3774. https://doi.org/10.1126/sciadv.abn3774.
Kang CH, Kim Y, Lee DY, et al. C-met-specific chimeric antigen receptor T cells demonstrate anti-tumor effect in c-met positive gastric cancer. Cancers (Basel). 2021;13(22). https://doi.org/10.3390/cancers13225738.
Jung M, Yang Y, McCloskey JE, et al. Chimeric antigen receptor T cell therapy targeting ICAM-1 in gastric cancer. Mol Ther Oncolytics. 2020;18:587–601. https://doi.org/10.1016/j.omto.2020.08.009.
Kim M, Pyo S, Kang CH, et al. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 2018;13(6):e0198347. https://doi.org/10.1371/journal.pone.0198347.
Wu D, Lv J, Zhao R, et al. PSCA is a target of chimeric antigen receptor T cells in gastric cancer. Biomark Res. 2020;8:3. https://doi.org/10.1186/s40364-020-0183-x.
Zhang Q, Zhang Z, Peng M, et al. CAR-T cell therapy in gastrointestinal tumors and hepatic carcinoma: from bench to bedside. Oncoimmunology. 2016;5(12):e1251539. https://doi.org/10.1080/2162402X.2016.1251539.
Zhao W, Jia L, Zhang M, et al. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res. 2019;9(8):1846–56.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. https://doi.org/10.3322/caac.21590.
Capurro M, Wanless IR, Sherman M, et al. Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology. 2003;125(1):89–97. https://doi.org/10.1016/s0016-5085(03)00689-9.
Jiang Z, Jiang X, Chen S, et al. Anti-GPC3-CAR T cells suppress the growth of tumor cells in patient-derived xenografts of hepatocellular carcinoma. Front Immunol. 2016;7:690. https://doi.org/10.3389/fimmu.2016.00690.
Pang N, Shi J, Qin L, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol. 2021;14(1):118. https://doi.org/10.1186/s13045-021-01128-9.
Katz SC, Hardaway J, Prince E, et al. HITM-SIR: phase Ib trial of intraarterial chimeric antigen receptor T-cell therapy and selective internal radiation therapy for CEA(+) liver metastases. Cancer Gene Ther. 2020;27(5):341–55. https://doi.org/10.1038/s41417-019-0104-z.
Dai H, Tong C, Shi D, et al. Efficacy and biomarker analysis of CD133-directed CAR T cells in advanced hepatocellular carcinoma: a single-arm, open-label, phase II trial. Oncoimmunology. 2020;9(1):1846926. https://doi.org/10.1080/2162402X.2020.1846926.
Liu H, Xu Y, Xiang J, et al. Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res. 2017;23(2):478–88. https://doi.org/10.1158/1078-0432.CCR-16-1203.
Sun B, Yang D, Dai H, et al. Eradication of hepatocellular carcinoma by NKG2D-based CAR-T cells. Cancer Immunol Res. 2019;7(11):1813–23. https://doi.org/10.1158/2326-6066.CIR-19-0026.
Ma Y, Chen Y, Yan L, et al. EGFRvIII-specific CAR-T cells produced by piggyBac transposon exhibit efficient growth suppression against hepatocellular carcinoma. Int J Med Sci. 2020;17(10):1406–14. https://doi.org/10.7150/ijms.45603.
Haisma HJ, Pinedo HM, Rijswijk A, et al. Tumor-specific gene transfer via an adenoviral vector targeted to the pan-carcinoma antigen EpCAM. Gene Ther. 1999;6(8):1469–74. https://doi.org/10.1038/sj.gt.3300969.
Jiang Z, Cheng L, Wu Z, et al. Transforming primary human hepatocytes into hepatocellular carcinoma with genetically defined factors. EMBO Rep. 2022:e54275. https://doi.org/10.15252/embr.202154275.
Zhang RY, Wei D, Liu ZK, et al. Doxycycline inducible chimeric antigen receptor T cells targeting CD147 for hepatocellular carcinoma therapy. Front Cell Dev Biol. 2019;7:233. https://doi.org/10.3389/fcell.2019.00233.
Chen C, Zhao S, Karnad A, et al. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol. 2018;11(1):64. https://doi.org/10.1186/s13045-018-0605-5.
Wang H, Ye X, Ju Y, et al. Minicircle DNA-mediated CAR T cells targeting CD44 suppressed hepatocellular carcinoma both in vitro and in vivo. Onco Targets Ther. 2020;13:3703–16. https://doi.org/10.2147/OTT.S247836.
Xiong C, Mao Y, Wu T, et al. Optimized expression and characterization of a novel fully human bispecific single-chain Diabody targeting vascular endothelial growth Factor165 and programmed Death-1 in Pichia pastoris and evaluation of antitumor activity in vivo. Int J Mol Sci. 2018;19(10). https://doi.org/10.3390/ijms19102900.
Xuan Y, Sheng Y, Zhang D, et al. Targeting CD276 by CAR-T cells induces regression of esophagus squamous cell carcinoma in xenograft mouse models. Transl Oncol. 2021;14(8):101138. https://doi.org/10.1016/j.tranon.2021.101138.
Yu F, Wang X, Shi H, et al. Development of chimeric antigen receptor-modified T cells for the treatment of esophageal cancer. Tumori. 2021;107(4):341–52. https://doi.org/10.1177/0300891620960223.
Matsuda T, Takeuchi H, Matsuda S, et al. EpCAM, a potential therapeutic target for esophageal squamous cell carcinoma. Ann Surg Oncol. 2014;21 Suppl 3:S356–64. https://doi.org/10.1245/s10434-014-3579-8.
Zhang H, Zhao H, He X, et al. JAK-STAT domain enhanced MUC1-CAR-T cells induced esophageal cancer elimination. Cancer Manag Res. 2020;12:9813–24. https://doi.org/10.2147/CMAR.S264358.
Haas AR, Tanyi JL, O'Hara MH, et al. Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing Mesothelin in advanced solid cancers. Mol Ther. 2019;27(11):1919–29. https://doi.org/10.1016/j.ymthe.2019.07.015.
Beatty GL, O'Hara MH, Lacey SF, et al. Activity of Mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155(1):29–32. https://doi.org/10.1053/j.gastro.2018.03.029.
Liu Y, Guo Y, Wu Z, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy. 2020;22(10):573–80. https://doi.org/10.1016/j.jcyt.2020.04.088.
Feng K, Liu Y, Guo Y, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9(10):838–47. https://doi.org/10.1007/s13238-017-0440-4.
Katz SC, Moody AE, Guha P, et al. HITM-SURE: hepatic immunotherapy for metastases phase Ib anti-CEA CAR-T study utilizing pressure enabled drug delivery. J Immunother Cancer. 2020;8(2). https://doi.org/10.1136/jitc-2020-001097.
Xia N, Haopeng P, Gong JU, et al. Robo1-specific CAR-NK immunotherapy enhances efficacy of (125)I seed brachytherapy in an Orthotopic mouse model of human pancreatic carcinoma. Anticancer Res. 2019;39(11):5919–25. https://doi.org/10.21873/anticanres.13796.
Zhu G, Foletti D, Liu X, et al. Author correction: targeting CLDN18.2 by CD3 bispecific and ADC modalities for the treatments of gastric and pancreatic cancer. Sci Rep. 2019;9(1):16735. https://doi.org/10.1038/s41598-019-53130-4.
Teng KY, Mansour AG, Zhu Z, et al. Off-the-shelf prostate stem cell antigen-directed chimeric antigen receptor natural killer cell therapy to treat pancreatic cancer. Gastroenterology. 2022;162(4):1319–33. https://doi.org/10.1053/j.gastro.2021.12.281.
Posey AD Jr, Schwab RD, Boesteanu AC, et al. Engineered CAR T cells targeting the cancer-associated Tn-Glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity. 2016;44(6):1444–54. https://doi.org/10.1016/j.immuni.2016.05.014.
Du H, Hirabayashi K, Ahn S, et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 2019;35(2):221–237.e8. https://doi.org/10.1016/j.ccell.2019.01.002.
Lo A, Wang LS, Scholler J, et al. Tumor-promoting Desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75(14):2800–10. https://doi.org/10.1158/0008-5472.CAN-14-3041.
Schäfer D, Tomiuk S, Küster LN, et al. Identification of CD318, TSPAN8 and CD66c as target candidates for CAR T cell based immunotherapy of pancreatic adenocarcinoma. Nat Commun. 2021;12(1):1453. https://doi.org/10.1038/s41467-021-21774-4.
Lesch S, Blumenberg V, Stoiber S, et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat Biomed Eng. 2021;5(11):1246–60. https://doi.org/10.1038/s41551-021-00737-6.
Kaneko MK, Ohishi T, Kawada M, et al. A cancer-specific anti-podocalyxin monoclonal antibody (60-mG(2a)-f) exerts antitumor effects in mouse xenograft models of pancreatic carcinoma. Biochem Biophys Rep. 2020;24:100826. https://doi.org/10.1016/j.bbrep.2020.100826.
Rataj F, Jacobi SJ, Stoiber S, et al. High-affinity CD16-polymorphism and fc-engineered antibodies enable activity of CD16-chimeric antigen receptor-modified T cells for cancer therapy. Br J Cancer. 2019;120(1):79–87. https://doi.org/10.1038/s41416-018-0341-1.
Forsberg E, Lindberg MF, Jespersen H, et al. HER2 CAR-T cells eradicate uveal melanoma and T-cell therapy-resistant human melanoma in IL2 transgenic NOD/SCID IL2 receptor knockout mice. Cancer Res. 2019;79(5):899–904. https://doi.org/10.1158/0008-5472.CAN-18-3158.
Zhang G, Wang L, Cui H, et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep. 2014;4:3571. https://doi.org/10.1038/srep03571.
Chinnasamy D, Tran E, Yu Z, et al. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73(11):3371–80. https://doi.org/10.1158/0008-5472.CAN-12-3913.
Schmidt P, Kopecky C, Hombach A, et al. Eradication of melanomas by targeted elimination of a minor subset of tumor cells. Proc Natl Acad Sci U S A. 2011;108(6):2474–9. https://doi.org/10.1073/pnas.1009069108.
Gargett T, Yu W, Dotti G, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135–49. https://doi.org/10.1038/mt.2016.63.
Lo AS, Ma Q, Liu DL, et al. Anti-GD3 chimeric sFv-CD28/T-cell receptor zeta designer T cells for treatment of metastatic melanoma and other neuroectodermal tumors. Clin Cancer Res. 2010;16(10):2769–80. https://doi.org/10.1158/1078-0432.CCR-10-0043.
Zhang Z, Jiang C, Liu Z, et al. B7-H3-targeted CAR-T cells exhibit potent antitumor effects on hematologic and solid tumors. Mol Ther Oncolytics. 2020;17:180–9. https://doi.org/10.1016/j.omto.2020.03.019.
Liu X, Xu Y, Xiong W, et al. Development of a TCR-like antibody and chimeric antigen receptor against NY-ESO-1/HLA-A2 for cancer immunotherapy. J Immunother Cancer. 2022;10(3). https://doi.org/10.1136/jitc-2021-004035.
Parriott G, Deal K, Crean S, et al. T-cells expressing a chimeric-PD1-Dap10-CD3zeta receptor reduce tumour burden in multiple murine syngeneic models of solid cancer. Immunology. 2020;160(3):280–94. https://doi.org/10.1111/imm.13187.
Simon B, Wiesinger M, März J, et al. The generation of CAR-transfected natural killer T cells for the immunotherapy of melanoma. Int J Mol Sci. 2018;19(8). https://doi.org/10.3390/ijms19082365.
Wallstabe L, Mades A, Frenz S, et al. CAR T cells targeting α(v)β(3) integrin are effective against advanced cancer in preclinical models. Adv cell. Gene Ther. 2018;1(2). https://doi.org/10.1002/acg2.11.
Uslu U, Schuler G, Dörrie J, et al. Combining a chimeric antigen receptor and a conventional T-cell receptor to generate T cells expressing two additional receptors (TETARs) for a multi-hit immunotherapy of melanoma. Exp Dermatol. 2016;25(11):872–9. https://doi.org/10.1111/exd.13095.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. https://doi.org/10.3322/caac.21551.
Hassan R, Thomas A, Alewine C, et al. Mesothelin immunotherapy for cancer: ready for prime time? J Clin Oncol. 2016;34(34):4171–9. https://doi.org/10.1200/JCO.2016.68.3672.
Hung CF, Xu X, Li L, et al. Development of anti-human Mesothelin-targeted chimeric antigen receptor messenger RNA-transfected peripheral blood lymphocytes for ovarian cancer therapy. Hum Gene Ther. 2018;29(5):614–25. https://doi.org/10.1089/hum.2017.080.
Fang J, Ding N, Guo X, et al. αPD-1-mesoCAR-T cells partially inhibit the growth of advanced/refractory ovarian cancer in a patient along with daily apatinib. J Immunother Cancer. 2021;9(2). https://doi.org/10.1136/jitc-2020-001162.
Reinhard K, Rengstl B, Oehm P, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science. 2020;367(6476):446–53. https://doi.org/10.1126/science.aay5967.
AACR. New CAR T-cell Therapy for Solid Tumors Was Safe and Showed Early Efficacy.
Li T, Wang J. Therapeutic effect of dual CAR-T targeting PDL1 and MUC16 antigens on ovarian cancer cells in mice. BMC Cancer. 2020;20(1):678. https://doi.org/10.1186/s12885-020-07180-x.
Liu Q, Cheng Z, Luo L, et al. C-terminus of MUC16 activates Wnt signaling pathway through its interaction with β-catenin to promote tumorigenesis and metastasis. Oncotarget. 2016;7(24):36800–13. https://doi.org/10.18632/oncotarget.9191.
Hua T, Liu S, Xin X, et al. Prognostic significance of L1 cell adhesion molecule in cancer patients: a systematic review and meta-analysis. Oncotarget. 2016;7(51):85196–207. https://doi.org/10.18632/oncotarget.13236.
Liang Z, Dong J, Yang N, et al. Tandem CAR-T cells targeting FOLR1 and MSLN enhance the antitumor effects in ovarian cancer. Int J Biol Sci. 2021;17(15):4365–76. https://doi.org/10.7150/ijbs.63181.
Hyrenius-Wittsten A, Su Y, Park M, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med. 2021;13(591). https://doi.org/10.1126/scitranslmed.abd8836.
Owens GL, Sheard VE, Kalaitsidou M, et al. Preclinical assessment of CAR T-cell therapy targeting the tumor antigen 5T4 in ovarian cancer. J Immunother. 2018;41(3):130–40. https://doi.org/10.1097/CJI.0000000000000203.
Wang L, Yang R, Zhao L, et al. Basing on uPAR-binding fragment to design chimeric antigen receptors triggers antitumor efficacy against uPAR expressing ovarian cancer cells. Biomed Pharmacother. 2019;117:109173. https://doi.org/10.1016/j.biopha.2019.109173.
Pang L, Ren F, Xu X, et al. Construction and characterization of cadherin 6 (CDH6)-targeting chimeric antigen receptor (CAR) modified T cells. J Environ Pathol Toxicol Oncol. 2022;41(1):55–71. https://doi.org/10.1615/JEnvironPatholToxicolOncol.2021040339.
Shu R, Evtimov VJ, Hammett MV, et al. Engineered CAR-T cells targeting TAG-72 and CD47 in ovarian cancer. Mol Ther Oncolytics. 2021;20:325–41. https://doi.org/10.1016/j.omto.2021.01.002.
Ng YY, Tay J, Wang S. CXCR1 expression to improve anti-cancer efficacy of intravenously injected CAR-NK cells in mice with peritoneal xenografts. Mol Ther Oncolytics. 2020;16:75–85. https://doi.org/10.1016/j.omto.2019.12.006.
Song DG, Ye Q, Carpenito C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res. 2011;71(13):4617–27. https://doi.org/10.1158/0008-5472.CAN-11-0422.
Leong L, Tan HL, Cua S, et al. Preclinical activity of embryonic Annexin A2-specific chimeric antigen receptor T cells against ovarian cancer. Int J Mol Sci. 2020;21(2). https://doi.org/10.3390/ijms21020381.
Klapdor R, Wang S, Morgan M, et al. Characterization of a novel third-generation anti-CD24-CAR against ovarian cancer. Int J Mol Sci. 2019;20(3). https://doi.org/10.3390/ijms20030660.
Wahba J, Natoli M, Whilding LM, et al. Chemotherapy-induced apoptosis, autophagy and cell cycle arrest are key drivers of synergy in chemo-immunotherapy of epithelial ovarian cancer. Cancer Immunol Immunother. 2018;67(11):1753–65. https://doi.org/10.1007/s00262-018-2199-8.
Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842–50. https://doi.org/10.1001/jama.2013.280319.
Alexander BM, Cloughesy TF. Adult glioblastoma. J Clin Oncol. 2017;35(21):2402–9. https://doi.org/10.1200/JCO.2017.73.0119.
Brown CE, Warden CD, Starr R, et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. 2013;8(10):e77769. https://doi.org/10.1371/journal.pone.0077769.
Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–9. https://doi.org/10.1056/NEJMoa1610497.
Brown CE, Badie B, Barish ME, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–72. https://doi.org/10.1158/1078-0432.CCR-15-0428.
Alizadeh D, Wong RA, Gholamin S, et al. IFNγ is critical for CAR T cell-mediated myeloid activation and induction of endogenous immunity. Cancer Discov. 2021;11(9):2248–65. https://doi.org/10.1158/2159-8290.CD-20-1661.
O'Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399). https://doi.org/10.1126/scitranslmed.aaa0984.
Chi F, Wu R, Jin X, et al. HER2 induces cell proliferation and invasion of non-small-cell lung cancer by upregulating COX-2 expression via MEK/ERK signaling pathway. Onco Targets Ther. 2016;9:2709–16. https://doi.org/10.2147/OTT.S96197.
Vitanza NA, Johnson AJ, Wilson AL, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. 2021;27(9):1544–52. https://doi.org/10.1038/s41591-021-01404-8.
Doronin II, Vishnyakova PA, Kholodenko IV, et al. Ganglioside GD2 in reception and transduction of cell death signal in tumor cells. BMC Cancer. 2014;14:295. https://doi.org/10.1186/1471-2407-14-295.
Mujoo K, Cheresh DA, Yang HM, et al. Disialoganglioside GD2 on human neuroblastoma cells: target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. 1987;47(4):1098–104.
Majzner RG, Ramakrishna S, Yeom KW, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–41. https://doi.org/10.1038/s41586-022-04489-4.
Yang K, Wu Z, Zhang H, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer. 2022;21(1):39. https://doi.org/10.1186/s12943-022-01513-z.
Lin Q, Ba T, Ho J, et al. First-in-human trial of EphA2-redirected CAR T-cells in patients with recurrent glioblastoma: a preliminary report of three cases at the starting dose. Front Oncol. 2021;11:694941. https://doi.org/10.3389/fonc.2021.694941.
Xie C, Liu D, Chen Q, et al. Soluble B7-H3 promotes the invasion and metastasis of pancreatic carcinoma cells through the TLR4/NF-κB pathway. Sci Rep. 2016;6:27528. https://doi.org/10.1038/srep27528.
Tang X, Wang Y, Huang J, et al. Administration of B7-H3 targeted chimeric antigen receptor-T cells induce regression of glioblastoma. Signal Transduct Target Ther. 2021;6(1):125. https://doi.org/10.1038/s41392-021-00505-7.
Weiss T, Weller M, Guckenberger M, et al. NKG2D-based CAR T cells and radiotherapy exert synergistic efficacy in glioblastoma. Cancer Res. 2018;78(4):1031–43. https://doi.org/10.1158/0008-5472.CAN-17-1788.
Yang D, Sun B, Dai H, et al. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J Immunother Cancer. 2019;7(1):171. https://doi.org/10.1186/s40425-019-0642-9.
Beard RE, Zheng Z, Lagisetty KH, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J Immunother Cancer. 2014;2:25. https://doi.org/10.1186/2051-1426-2-25.
CSPG4 shows promise for glioblastoma CAR T therapy. Cancer Discov. 2018;8(5):524–5. https://doi.org/10.1158/2159-8290.CD-NB2018-032.
Wang D, Starr R, Chang WC, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020;12(533). https://doi.org/10.1126/scitranslmed.aaw2672.
Waseda M, Kaneko S. Podoplanin as an attractive target of CAR T cell therapy. Cells. 2020;9(9). https://doi.org/10.3390/cells9091971.
Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–7. https://doi.org/10.1158/1078-0432.CCR-11-0116.
Kato Y, Kaneko MK. A cancer-specific monoclonal antibody recognizes the aberrantly glycosylated podoplanin. Sci Rep. 2014;4:5924. https://doi.org/10.1038/srep05924.
Ma W, Wang Y, Zhang R, et al. Targeting PAK4 to reprogram the vascular microenvironment and improve CAR-T immunotherapy for glioblastoma. Nat Can. 2021;2(1):83–97. https://doi.org/10.1038/s43018-020-00147-8.
Rousso-Noori L, Mastandrea I, Talmor S, et al. P32-specific CAR T cells with dual antitumor and antiangiogenic therapeutic potential in gliomas. Nat Commun. 2021;12(1):3615. https://doi.org/10.1038/s41467-021-23817-2.
Ebert LM, Yu W, Gargett T, et al. Endothelial, pericyte and tumor cell expression in glioblastoma identifies fibroblast activation protein (FAP) as an excellent target for immunotherapy. Clin Transl Immunol. 2020;9(10):e1191. https://doi.org/10.1002/cti2.1191.
Li G, Zhang Z, Cai L, et al. Fn14-targeted BiTE and CAR-T cells demonstrate potent preclinical activity against glioblastoma. Oncoimmunology. 2021;10(1):1983306. https://doi.org/10.1080/2162402X.2021.1983306.
Vora P, Venugopal C, Salim SK, et al. The rational development of CD133-targeting immunotherapies for glioblastoma. Cell Stem Cell. 2020;26(6):832–844.e6. https://doi.org/10.1016/j.stem.2020.04.008.
Kang CH, Kim Y, Lee SM, et al. Development of antigen-specific chimeric antigen receptor KHYG-1 cells for glioblastoma. Anticancer Res. 2021;41(4):1811–9. https://doi.org/10.21873/anticanres.14947.
Hegde M, Mukherjee M, Grada Z, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–52. https://doi.org/10.1172/JCI83416.
Bielamowicz K, Fousek K, Byrd TT, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology. 2018;20(4):506–18. https://doi.org/10.1093/neuonc/nox182.
Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591). https://doi.org/10.1126/scitranslmed.abe7378.
Chalise L, Kato A, Ohno M, et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Δ combination therapy against glioblastoma. Mol Ther Oncolytics. 2022;26:265–74. https://doi.org/10.1016/j.omto.2022.07.006.
Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–53. https://doi.org/10.1002/ijc.31937.
Linge A, Lohaus F, Löck S, et al. HPV status, cancer stem cell marker expression, hypoxia gene signatures and tumour volume identify good prognosis subgroups in patients with HNSCC after primary radiochemotherapy: a multicentre retrospective study of the German cancer consortium radiation oncology group (DKTK-ROG). Radiother Oncol. 2016;121(3):364–73. https://doi.org/10.1016/j.radonc.2016.11.008.
Mei Z, Zhang K, Lam AK, et al. MUC1 as a target for CAR-T therapy in head and neck squamous cell carinoma. Cancer Med. 2020;9(2):640–52. https://doi.org/10.1002/cam4.2733.
Haist C, Schulte E, Bartels N, et al. CD44v6-targeted CAR T-cells specifically eliminate CD44 isoform 6 expressing head/neck squamous cell carcinoma cells. Oral Oncol. 2021;116:105259. https://doi.org/10.1016/j.oraloncology.2021.105259.
Park YP, Jin L, Bennett KB, et al. CD70 as a target for chimeric antigen receptor T cells in head and neck squamous cell carcinoma. Oral Oncol. 2018;78:145–50. https://doi.org/10.1016/j.oraloncology.2018.01.024.
Rosewell Shaw A, Porter CE, Watanabe N, et al. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol Ther. 2017;25(11):2440–51. https://doi.org/10.1016/j.ymthe.2017.09.010.
Arndt C, Loureiro LR, Feldmann A, et al. UniCAR T cell immunotherapy enables efficient elimination of radioresistant cancer cells. Oncoimmunology. 2020;9(1):1743036. https://doi.org/10.1080/2162402X.2020.1743036.
Haist C, Poschinski Z, Bister A, et al. Engineering a single-chain variable fragment of cetuximab for CAR T-cell therapy against head and neck squamous cell carcinomas. Oral Oncol. 2022;129:105867. https://doi.org/10.1016/j.oraloncology.2022.105867.
Yang L, Froio RM, Sciuto TE, et al. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood. 2005;106(2):584–92. https://doi.org/10.1182/blood-2004-12-4942.
Min IM, Shevlin E, Vedvyas Y, et al. CAR T therapy targeting ICAM-1 eliminates advanced human thyroid tumors. Clin Cancer Res. 2017;23(24):7569–83. https://doi.org/10.1158/1078-0432.CCR-17-2008.
Gray KD, McCloskey JE, Vedvyas Y, et al. PD1 blockade enhances ICAM1-directed CAR T therapeutic efficacy in advanced thyroid cancer. Clin Cancer Res. 2020;26(22):6003–16. https://doi.org/10.1158/1078-0432.CCR-20-1523.
Li H, Zhou X, Wang G, et al. CAR-T cells targeting TSHR demonstrate safety and potent preclinical activity against differentiated thyroid cancer. J Clin Endocrinol Metab. 2022;107(4):1110–26. https://doi.org/10.1210/clinem/dgab819.
Bhoj VG, Li L, Parvathaneni K, et al. Adoptive T cell immunotherapy for medullary thyroid carcinoma targeting GDNF family receptor alpha 4. Mol Ther Oncolytics. 2021;20:387–98. https://doi.org/10.1016/j.omto.2021.01.012.
Kuusk T, Grivas N, de Bruijn R, et al. The current management of renal cell carcinoma. Minerva Med. 2017;108(4):357–69. https://doi.org/10.23736/S0026-4806.17.05058-3.
Lamers CH, Klaver Y, Gratama JW, et al. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochem Soc Trans. 2016;44(3):951–9. https://doi.org/10.1042/BST20160037.
Wang Y, Buck A, Grimaud M, et al. Anti-CAIX BBζ CAR4/8 T cells exhibit superior efficacy in a ccRCC mouse model. Mol Ther Oncolytics. 2022;24:385–99. https://doi.org/10.1016/j.omto.2021.12.019.
Hong H, Stastny M, Brown C, et al. Diverse solid tumors expressing a restricted epitope of L1-CAM can be targeted by chimeric antigen receptor redirected T lymphocytes. J Immunother. 2014;37(2):93–104. https://doi.org/10.1097/CJI.0000000000000018.
Mori JI, Adachi K, Sakoda Y, et al. Anti-tumor efficacy of human anti-c-met CAR-T cells against papillary renal cell carcinoma in an orthotopic model. Cancer Sci. 2021;112(4):1417–28. https://doi.org/10.1111/cas.14835.
Zhang Q, Tian K, Xu J, et al. Synergistic effects of Cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J Immunol Res. 2017;2017:6915912. https://doi.org/10.1155/2017/6915912.
Schönfeld K, Sahm C, Zhang C, et al. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol Ther. 2015;23(2):330–8. https://doi.org/10.1038/mt.2014.219.
Arbyn M, Weiderpass E, Bruni L, et al. Estimates of incidence and mortality of cervical cancer in 2018: a worldwide analysis. Lancet Glob Health. 2020;8(2):e191–203. https://doi.org/10.1016/S2214-109X(19)30482-6.
He Y, Li XM, Yin CH, et al. Killing cervical cancer cells by specific chimeric antigen receptor-modified T cells. J Reprod Immunol. 2020;139:103115. https://doi.org/10.1016/j.jri.2020.103115.
Zhang Y, Li X, Zhang J, et al. Novel cellular immunotherapy using NKG2D CAR-T for the treatment of cervical cancer. Biomed Pharmacother. 2020;131:110562. https://doi.org/10.1016/j.biopha.2020.110562.
Zheng J, Huang J, Ma W, et al. The antitumor activity of CAR-T-PD1 cells enhanced by HPV16mE7-pulsed and SOCS1-silenced DCs in cervical cancer models. Cancer Manag Res. 2021;13:6045–53. https://doi.org/10.2147/CMAR.S321402.
Ahmed N, Brawley VS, Hegde M, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–96. https://doi.org/10.1200/JCO.2014.58.0225.
Huang G, Yu L, Cooper LJ, et al. Genetically modified T cells targeting interleukin-11 receptor α-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012;72(1):271–81. https://doi.org/10.1158/0008-5472.CAN-11-2778.
Huang X, Park H, Greene J, et al. IGF1R- and ROR1-specific CAR T cells as a potential therapy for high risk sarcomas. PLoS One. 2015;10(7):e0133152. https://doi.org/10.1371/journal.pone.0133152.
Hsu K, Middlemiss S, Saletta F, et al. Chimeric antigen receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2021;28(3-4):321–34. https://doi.org/10.1038/s41417-020-00221-4.
Garcia-Monclús S, López-Alemany R, Almacellas-Rabaiget O, et al. EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int J Cancer. 2018;143(5):1188–201. https://doi.org/10.1002/ijc.31405.
Long AH, Highfill SL, Cui Y, et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol Res. 2016;4(10):869–80. https://doi.org/10.1158/2326-6066.CIR-15-0230.
Majzner RG, Theruvath JL, Nellan A, et al. CAR T cells targeting B7-H3, a Pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–74. https://doi.org/10.1158/1078-0432.CCR-18-0432.
Fernández L, Metais JY, Escudero A, et al. Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin Cancer Res. 2017;23(19):5824–35. https://doi.org/10.1158/1078-0432.CCR-17-0075.
Wang Y, Yu W, Zhu J, et al. Anti-CD166/4-1BB chimeric antigen receptor T cell therapy for the treatment of osteosarcoma. J Exp Clin Cancer Res. 2019;38(1):168. https://doi.org/10.1186/s13046-019-1147-6.
Englisch A, Altvater B, Kailayangiri S, et al. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr Blood Cancer. 2020;67(10):e28313. https://doi.org/10.1002/pbc.28313.
Roselli E, Faramand R, Davila ML. Insight into next-generation CAR therapeutics: designing CAR T cells to improve clinical outcomes. J Clin Invest. 2021;131(2). https://doi.org/10.1172/JCI142030.
Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–6. https://doi.org/10.1038/s41586-019-1054-1.
Slaney CY, von Scheidt B, Davenport AJ, et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res. 2017;23(10):2478–90. https://doi.org/10.1158/1078-0432.CCR-16-1860.
Ma S, Li X, Wang X, et al. Current Progress in CAR-T cell therapy for solid tumors. Int J Biol Sci. 2019;15(12):2548–60. https://doi.org/10.7150/ijbs.34213.
Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol. 2019;10:128. https://doi.org/10.3389/fimmu.2019.00128.
Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. Addressing the obstacles of CAR T cell migration in solid tumors: wishing a heavy traffic. Crit Rev Biotechnol. 2021:1–20. https://doi.org/10.1080/07388551.2021.1988509.
Ruella M, Xu J, Barrett DM, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24(10):1499–503. https://doi.org/10.1038/s41591-018-0201-9.
Ren J, Zhao Y. Advancing chimeric antigen receptor T cell therapy with CRISPR/Cas9. Protein Cell. 2017;8(9):634–43. https://doi.org/10.1007/s13238-017-0410-x.
Wang Z, McWilliams-Koeppen HP, Reza H, et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell. 2022;29(4):515–527.e8. https://doi.org/10.1016/j.stem.2022.02.009.
Schneider D, Xiong Y, Wu D, et al. Trispecific CD19-CD20-CD22-targeting duoCAR-T cells eliminate antigen-heterogeneous B cell tumors in preclinical models. Sci Transl Med. 2021;13(586). https://doi.org/10.1126/scitranslmed.abc6401.
Tu S, Zhou X, Guo Z, et al. CD19 and CD70 dual-target chimeric antigen receptor T-cell therapy for the treatment of relapsed and refractory primary central nervous system diffuse large B-cell lymphoma. Front Oncol. 2019;9:1350. https://doi.org/10.3389/fonc.2019.01350.
Golubovskaya V, Zhou H, Li F, et al. Novel CD37, humanized CD37 and bi-specific humanized CD37-CD19 CAR-T cells specifically target lymphoma. Cancers (Basel). 2021;13(5). https://doi.org/10.3390/cancers13050981.
Cordoba S, Onuoha S, Thomas S, et al. CAR T cells with dual targeting of CD19 and CD22 in pediatric and young adult patients with relapsed or refractory B cell acute lymphoblastic leukemia: a phase 1 trial. Nat Med. 2021;27(10):1797–805. https://doi.org/10.1038/s41591-021-01497-1.
Liang Z, Cui J, Chang AH, et al. Successful treatment of relapsed acute B-cell lymphoblastic leukemia with CD20/CD22 bispecific chimeric antigen receptor T-cell therapy. Regen Ther. 2020;15:281–4. https://doi.org/10.1016/j.reth.2020.11.001.
Wang X, Dong Z, Awuah D, et al. CD19/BAFF-R dual-targeted CAR T cells for the treatment of mixed antigen-negative variants of acute lymphoblastic leukemia. Leukemia. 2022;36(4):1015–24. https://doi.org/10.1038/s41375-021-01477-x.
Krawczyk E, Zolov SN, Huang K, et al. T-cell activity against AML improved by dual-targeted T cells stimulated through T-cell and IL7 receptors. Cancer Immunol Res. 2019;7(4):683–92. https://doi.org/10.1158/2326-6066.CIR-18-0748.
Ghamari A, Pakzad P, Majd A, et al. Design and production An effective bispecific tandem chimeric antigen receptor on T cells against CD123 and folate receptor ß towards B-acute myeloid Leukaemia blasts. Cell J. 2021;23(6):650–7. https://doi.org/10.22074/cellj.2021.7314.
Li KX, Wu HY, Pan WY, et al. A novel approach for relapsed/refractory FLT3(Mut+) acute myeloid leukaemia: synergistic effect of the combination of bispecific FLT3scFv/NKG2D-CAR T cells and gilteritinib. Mol Cancer. 2022;21(1):66. https://doi.org/10.1186/s12943-022-01541-9.
He X, Feng Z, Ma J, et al. Bispecific and split CAR T cells targeting CD13 and TIM3 eradicate acute myeloid leukemia. Blood. 2020;135(10):713–23. https://doi.org/10.1182/blood.2019002779.
Qin H, Edwards JP, Zaritskaya L, et al. Chimeric antigen receptors incorporating D domains targeting CD123 direct potent mono- and bi-specific antitumor activity of T cells. Mol Ther. 2019;27(7):1262–74. https://doi.org/10.1016/j.ymthe.2019.04.010.
Jiang W, Li T, Guo J, et al. Bispecific c-met/PD-L1 CAR-T cells have enhanced therapeutic effects on hepatocellular carcinoma. Front Oncol. 2021;11:546586. https://doi.org/10.3389/fonc.2021.546586.
Chen C, Li K, Jiang H, et al. Development of T cells carrying two complementary chimeric antigen receptors against glypican-3 and asialoglycoprotein receptor 1 for the treatment of hepatocellular carcinoma. Cancer Immunol Immunother. 2017;66(4):475–89. https://doi.org/10.1007/s00262-016-1949-8.
Tseng HC, Xiong W, Badeti S, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11(1):4810. https://doi.org/10.1038/s41467-020-18444-2.
Sabahi M, Jabbari P, Alizadeh Haghighi M, et al. Proposing a tandem AND-gate CAR T cell targeting glioblastoma multiforme. Med Hypotheses. 2020;137:109559. https://doi.org/10.1016/j.mehy.2020.109559.
Muhammad N, Wang R, Li W, et al. A novel TanCAR targeting IL13Rα2 and EphA2 for enhanced glioblastoma therapy. Mol Ther Oncolytics. 2022;24:729–41. https://doi.org/10.1016/j.omto.2022.02.012.
Wang G, Zhou X, Fucà G, et al. Fully human antibody V(H) domains to generate mono and bispecific CAR to target solid tumors. J Immunother Cancer. 2021;9(4). https://doi.org/10.1136/jitc-2020-002173.
Zhang E, Yang P, Gu J, et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11(1):102. https://doi.org/10.1186/s13045-018-0646-9.
Hombach AA, Rappl G, Abken H. Blocking CD30 on T cells by a dual specific CAR for CD30 and colon cancer antigens improves the CAR T cell response against CD30(-) tumors. Mol Ther. 2019;27(10):1825–35. https://doi.org/10.1016/j.ymthe.2019.06.007.
Cao YJ, Wang X, Wang Z, et al. Switchable CAR-T cells outperformed traditional antibody-redirected therapeutics targeting breast cancers. ACS Synth Biol. 2021;10(5):1176–83. https://doi.org/10.1021/acssynbio.1c00007.
Wilkie S, van Schalkwyk MC, Hobbs S, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–70. https://doi.org/10.1007/s10875-012-9689-9.
Srivastava S, Salter AI, Liggitt D, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to Normal tissues and enables selective tumor targeting. Cancer Cell. 2019;35(3):489–503.e8. https://doi.org/10.1016/j.ccell.2019.02.003.
Simon B, Harrer DC, Schuler-Thurner B, et al. Arming T cells with a gp100-specific TCR and a CSPG4-specific CAR using combined DNA- and RNA-based receptor transfer. Cancers (Basel). 2019;11(5). https://doi.org/10.3390/cancers11050696.
Wang Z, Wu Z, Liu Y, et al. New development in CAR-T cell therapy. J Hematol Oncol. 2017;10(1):53. https://doi.org/10.1186/s13045-017-0423-1.
Marei HE, Althani A, Caceci T, et al. Recent perspective on CAR and Fcγ-CR T cell immunotherapy for cancers: preclinical evidence versus clinical outcomes. Biochem Pharmacol. 2019;166:335–46. https://doi.org/10.1016/j.bcp.2019.06.002.
Razeghian E, Nasution M, Rahman HS, et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther. 2021;12(1):428. https://doi.org/10.1186/s13287-021-02510-7.
Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nat Biomed Eng. 2018;2(6):377–91. https://doi.org/10.1038/s41551-018-0235-9.
Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: mechanisms, manifestations and management. Blood Rev. 2019;34:45–55. https://doi.org/10.1016/j.blre.2018.11.002.
Fei F, Rong L, Jiang N, et al. Targeting HLA-DR loss in hematologic malignancies with an inhibitory chimeric antigen receptor. Mol Ther. 2022;30(3):1215–26. https://doi.org/10.1016/j.ymthe.2021.11.013.
Zajc CU, Dobersberger M, Schaffner I, et al. A conformation-specific ON-switch for controlling CAR T cells with an orally available drug. Proc Natl Acad Sci U S A. 2020;117(26):14926–35. https://doi.org/10.1073/pnas.1911154117.
Schubert ML, Schmitt M, Wang L, et al. Side-effect management of chimeric antigen receptor (CAR) T-cell therapy. Ann Oncol. 2021;32(1):34–48. https://doi.org/10.1016/j.annonc.2020.10.478.
Zhao J, Lin Q, Song Y, et al. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11(1):132. https://doi.org/10.1186/s13045-018-0677-2.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. https://doi.org/10.1038/s41408-021-00459-7.
Evgin L, Huff AL, Wongthida P, et al. Oncolytic virus-derived type I interferon restricts CAR T cell therapy. Nat Commun. 2020;11(1):3187. https://doi.org/10.1038/s41467-020-17011-z.
Ramachandran M, Dimberg A, Essand M. The cancer-immunity cycle as rational design for synthetic cancer drugs: novel DC vaccines and CAR T-cells. Semin Cancer Biol. 2017;45:23–35. https://doi.org/10.1016/j.semcancer.2017.02.010.
Acknowledgments
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (NO.82073893, 82172685, 81873635, 81703622). Hunan Provincial Natural Science Foundation of China (NO. 2022JJ20095, 2018SK2101, 2018JJ3838), China Postdoctoral Science Foundation (NO. 2018 M633002), Hunan Provincial Health Committee Foundation of China (NO. 202204044869). Xiangya Hospital Central South University postdoctoral foundation.
Author information
Authors and Affiliations
Contributions
C.Q. and H.Z. collected relevant literature, drafted manuscripts, and prepared figures and tables. C.Q., H.Z., H.C., L.T., H.M., F.L., L.Z., Z.Y., L.L., L.Y., Z.W., N.Z., P.L., J.Z., Z.L., and W.Y. reviewed and made significant revisions to the manuscript. Q.C. and Z.L. guided the preparation of this manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qu, C., Zhang, H., Cao, H. et al. Tumor buster - where will the CAR-T cell therapy ‘missile’ go?. Mol Cancer 21, 201 (2022). https://doi.org/10.1186/s12943-022-01669-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01669-8