
Abstract
To date, two chimeric antigen receptors (CAR)-T cell products from autologous T cells have been approved by The United States Food and Drug Administration (FDA). The case-by-case autologous T cell generation setting is largely considered as a pivotal restraining cause for its large-scale clinical use because of the costly and prolonged manufacturing procedure. Further, activated CAR-T cells mainly express immune checkpoint molecules, including CTLA4, PD1, LAG3, abrogating CAR-T anti-tumor activity. In addition, CAR-T cell therapy potently results in some toxicity, such as cytokine releases syndrome (CRS). Therefore, the development of the universal allogeneic T cells with higher anti-tumor effects is of paramount importance. Thus, genome-editing technologies, in particular, clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 are currently being used to establish “off-the-shelf” CAR-T cells with robust resistance to immune cell-suppressive molecules. In fact, that simultaneous ablation of PD-1, T cell receptor alpha constant (TRAC or TCR), and also β-2 microglobulin (B2M) by CRISPR-Cas9 technique can support the manufacture of universal CAR-T cells with robust resistance to PD-L1. . Indeed, the ablation of β2M or TARC can severely hinder swift elimination of allogeneic T cells those express foreign HLA-I molecules, and thereby enables the generation of CAR-T cells from allogeneic healthy donors T cells with higher persistence in vivo. Herein, we will deliver a brief overview of the CAR-T cell application in the context of tumor immunotherapy. More importantly, we will discuss recent finding concerning the application of genome editing technologies for preparing universal CAR-T cells or cells that can effectively counter tumor escape, with a special focus on CRISPR-Cas9 technology.
Similar content being viewed by others
Introduction
Concerning the engineered or bacterial nucleases, the progress of genome editing machinery has provided the possibility of direct and specific recognition and modification of genomic sequences in practically all eukaryotic cells [1, 2]. Genome editing has resulted in the advancement of our knowledge respecting the finding of innovative therapeutic options for treating a wide spectrum of human disorders, ranging from infection to cancer. Current development in evolving programmable nucleases, including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), as well as clustered regularly interspaced short palindromic repeat (CRISPR)-CRISPR-associated protein 9 (Cas9), has critically accelerated the development of gene editing from notion to clinical practice [3]. As CRISPR-Cas9 has been suggested as an encouraging tool for generating gene knockouts, its competence to offer capable gene editing in primary T cells presents a pronounced study tool to support a paradigm shift in T cell-based immunotherapies, more importantly, next-generation chimeric antigen receptor (CAR)-T cells [4].
CAR-T cell therapy includes the genetic modification of patients’ autologous T cells or allograft cells to efficiently express a CAR involving a fusion protein of a selected single-chain fragment variable (ScFV) from a specific monoclonal antibody and one or more T cell receptor intracellular signaling domains. This chimer receptor can selectively and efficiently recognize the related tumor-associated antigen (TAA) expressed by tumor cells [5]. Nonetheless, severe and life-threatening toxicities, such as cytokine releases syndrome (CRS), graft-versus-host disease (GVHD), on-target/off-tumor toxicity, neurotoxicity, and tumor lysis syndrome, commonly constrain its clinical utility [6]. Correspondingly, it seems that further progress in the next-generation CAR-T cells with more optimized construction, promoted efficacy, and moderated toxicities is of paramount importance. Meanwhile, the production of the universal “off-the-shelf” CAR-T cells from healthy donors can circumvent the restraints and possibly be a milestone in the future. For overcoming the GVHD occurrence and potent rejection upon CAR-T cell, CRISPR/Cas9-mediated ablation of the endogenous αβ T cell receptor (TCR) has resulted in a pronounced success in preclinical studies [7]. The endogenous αβ TCR on adoptively transferred donor lymphocytes can identify alloantigens in human leukocyte antigen (HLA) mismatched recipients, and thereby leads to the GVHD; on the other hand, detection of foreign HLA molecules on donor T cells can cause rejection [7]. Further, ablation of beta-2-microglobulin (β2M), a pivotal subunit of HLA-I proteins, can potently avert swift eradication of allogeneic T cells those express foreign HLA-I molecules.
Also, it has been suggested that dual blockade of programmed cell death protein 1 (PD1), lymphocyte activation gene 3 (LAG-3), or cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) using genome editing technologies can sustain the improved T cell effector activities, facilitating an abrogation in tumor growth [8]. Moreover, knockout of diacylglycerol kinase (DGK), which metabolizes diacylglycerol to phosphatidic acid, using CRISPR/Cas9 supported CAR-T cell anti-tumor functions against U87MGvIII glioblastoma cell in vitro and xenografts [9].
Herein, we deliver a brief overview concerning the CAR-T cell-based therapy to treat human cancer, ranging from hematological malignancies to solid tumors. Also, we discuss recent findings respecting the application of genome editing platforms, in particular CRISP-Cas9, for potentiating the safety and efficacy of CAR-T cells in the context of tumor immunotherapy.
CRISPR/Cas9 therapeutic application
Early in 1987, CRISPRs were firstly discovered in E. coli and after that in a large number of other bacteria species [10]. Various investigations in 2005 displayed their likenesses to phage DNA, and succeeding studies indicated that these sequences contribute to bacterial and archaea adaptive immune responses toward offending foreign DNA by stimulating the RNA-guided DNA cleavage [11]. Today, the CRISPR-Cas systems are largely categorized into two main classes according to the structural dissimilarity of the Cas genes and their construction shape [12]. Meanwhile, a class 1 CRISPR-Cas system involves multiple effector complexes, while a class 2 system includes only a single effector protein. To date, six CRISPR-Cas types and approximately 29 subtypes have been discovered [13, 14]. The most commonly employed subtype of CRISPR systems is the type II CRISPR/Cas9 system, enabling targeting specific DNA sequences by a single Cas protein from Streptococcus pyogenes (SpCas9) [15]. The CRISPR/Cas9 system consists of two main parts, including a single-stranded guide RNA (sgRNA) as a particular 17–23 base-pair (bp) sequence intended for specific identification of target DNA region in a sequence-specific style, and also a Cas9 endonuclease [15]. The sgRNA sequence is required to be trailed by a short DNA sequence upstream to facilitate efficient compatibilization with the Cas9 protein [16]. Correspondingly, the sgRNA causes a connection with a target sequence by Watson-Crick base pairing and Cas9 exactly cuts the DNA for establishing a DNA double-strand break (DSB) [16]. Upon the DSB, DNA-DSB repair tools start genome repair. The DSBs can be repaired by one of the two main appliances that largely rein almost all cell types and organisms, including homology-directed repair (HDR) and nonhomologous end-joining (NHEJ), leading to the targeted integration or gene disruptions, respectively [17].
The further description concerning detailed mechanism of the CRISPR-Cas9 function and parameters implicated in the determining its efficacy is beyond the scopes of this article, and thereby audiences are referred to the some excellent review in this context [18,19,20].
Compared to ZFN or TALEN tools, CRISPR-Cas9 is more suitable because of its flexibility and the capacity for multiple gene editing [21]. Indeed, endonuclease-based ZFN or TALEN technologies request the reengineering of a unique enzyme, which should be manufactured distinctly regarding each target sequence [21], but, as the nuclease protein Cas9 is the same in all cases, can be appropriately engineered to detect novel regions by varying the guide RNA sequences (sgRNA) [22]. Moreover, compared to CRISPR-Cas9, ZFNs and TALENs request much more labor and are more expensive. On the other hand, the unique competence of CRISPR/Cas9 to edit multiple loci concurrently signifies that this toll is easier, more efficient, and more scalable in comparison to the ZFNs and TALENs [23]. Thus, in the context of CAR-T cell-based targeted therapy, it is currently applicable to concurrently affect several genes and accomplish loss of function (LOF) of potentially any genetic or epigenetic target utilizing CRISPR-Cas9 [24].
CAR construction
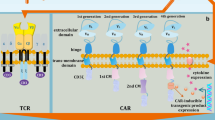
Concisely, CAR is an engineered modified fusion protein structurally similar to the TCR and involves an extracellular antigen detecting domain linked to one or more intracellular signaling domains [5]. The CAR extracellular domain is structurally an antibody single-chain variable fragment (scFv) and identifies the target antigen virtually overexpressed on the tumor cells in the HLA-independent manner [25]. The CAR intracellular domains typically involve CD28, 4-1BB, or OX40 to support effector cell activation, and also include CD3ζ for the exertion of the cytotoxicity against transformed cells. The first generation of CARs involves only an intracellular signal domain CD3ζ, while the second generation of CARs includes a costimulatory molecule in addition to CD3ζ, and also the third generation of CARs contains another costimulatory domain. The recently advanced fourth generation of CAR-T cells could potently stimulate the downstream transcription factor to trigger cytokine release following the detection of the tumor-associated antigen (TAA) with CAR. Importantly, the fifth generation of CARs which has been constructed respecting the second generation utilizes gene editing to inhibit the expression of the TCR (TRAC) gene, facilitating the ablation of TCR alpha and beta chains (Fig. 1) [26]. As described, CRISPR system is widely used during the recent years to establish novel generation of CAR-T cells. T cells are engineered to generate transgenic cytokines, such as interleukin (IL-12) within the targeted tumor and therefore attract higher quantities of anti-tumor immune cells (e.g., natural killer (NK) cells and macrophages) to provide next-generation CAR-T cells for better toxicity management [27]. Moreover, CAR-T cells are equipped with chemokine receptors to circumvent their poor homing to tumor sites. These strategies like knocking in cytokines or chemokine receptors eventually augment CAR-T cell cytotoxic functions against tumor cells. As well, approaches like knocking out immune checkpoint molecules, and also ablation of TRAC or B2M can ameliorate CAR cell persistent in vivo and also enables CAR-T cell generation form allogeneic donors [28]. As well, knocking out the endogenous TGF-β receptor II (TGFBR2) in CAR-T cells using CRISPR/Cas9 method largely attenuates the elicited Treg conversion and thus hinders the exhaustion of CAR-T cells [29].

The basic structure of CAR-T cell generations. The first generation of CARs contains only a CD3ζ as a well-documented intracellular signal domain. The second and third generations of CARs involve one or two costimulatory molecules in addition to CD3ζ, respectively. As well, the fourth generation of CAR-T cells strongly motivates the downstream transcription factor to prompt cytokine generation following interrelation between CAR and target antigen. Prominently, the genome edition technologies, such as CRISPR-Cas9, have been widely used to construct TRAC (TCR)-deficient CAR-T cells, establishing fifth generation of CAR-T cells
The CAR-bearing modified T cells can recognize CAR-targeted antigen and thus elicit T cell proliferation, cytokine manufacture, and critical and targeted cytotoxicity versus tumor cells [30]. Therefore, CAR-T cell treatment has supported appreciated attainment to treat hematological malignancies, including lymphoma, chronic lymphocytic leukemia (CLL), and acute lymphoblastic leukemia (ALL) [31, 32]. CARs deliver a wider array of functional impacts than transduced TCRs; however, CARs and TCRs have their advantages and disadvantages [33]. Although the flexibility and dynamic range of CARs are striking, existing CARs are restricted to identify cell surface antigens [33] while TCRs identify both cell surface and intracellular proteins. Nonetheless, antigen processing and presentation by HLA are not required for CARs, making them more applicable than TCRs to HLA-diverse patient populations [34].
The CAR’s engineering into T cells demands that T cells be cultivated to permit for transduction and succeeding expansion. Although the transduction can exploit diverse methods, steady gene transfer is essential to facilitate continued CAR expression in clonally expanding and persisting T cells.
CAR-T cells generation from autologous and allogeneic T cells
The genetic alteration of autologous or allogeneic peripheral blood T lymphocytes to create tumor-targeted T cells has become an inspiring therapeutic option. The great and pronounced competencies of TCR and CAR therapies are best exemplified through the stimulating clinical results achieved with NY-ESO-1 TCR [35] and CD19 CAR-T cells [36, 37]. CAR-T cell construction processes combine T cell activation and transduction stages for providing genetically targeted T cell products. Indeed, engineered T cells to express particular CARs can be generated from Ficoll-purified PBMCs followed by their activation with anti-CD3 monoclonal antibody (mAb) in the existence of irradiated allogeneic feeder cells, and finally efficient transduction with a vector encoding the CAR [38]. The encouraging clinical outcomes of CAR-T cell therapy may be more enlarged by establishing the potent and histocompatible T cells. Autologous methods have a confirmed track record, but personalized products can be challenging in some cases, for instance in patients with chemotherapy or HIV-mediated immune deficiency [39]. Accordingly, though T cells can be simply achieved from donors, their application is potently hindered by the high alloreactive capability. Indeed, TCRs have the natural competence to respond toward non-autologous tissues, identifying both allogeneic HLA molecules and other minor antigens [40]. This tendency inspires the incidence of graft rejection in transplant recipients and also the occurrence of GVHD in recipients of donor-isolated T cells [41]. Given these problems, inhibition of the alloreactive potential of allogeneic T cells to obtain an acceptable risk-benefit ratio is of paramount importance. To date, two main tactics have been designed to defeat the risk of graft-versus-host reaction (GVHR) concerning the selection of virus-specific TCRs devoid of GVHR or the ablation of TCR expression [39]. As described, three main technologies, containing ZFNs, TALEN, and CRISPR/Cas9, facilitate gene disruption in the human cell. Remarkably, the ablation of endogenous TCR expression largely obtained through utilizing genome-editing technologies abrogate the continuous districts of TRAC genes, and thereby offer the chance for manufacturing universal CAR-T cells [7, 42].
To CAR-T cells hold potential as a safe and rapidly evolving therapeutic strategy for treating human malignancies, the development of methods to pharmacologically control them in vivo is required. Owing to this fact, some strategies, in particular, suicide mechanisms are developing [43, 44]. For example, Amatya and her colleagues designed a construction including CD28-containing anti-signaling lymphocytic activation molecule F7 (SLAMF7) CAR and a suicide gene [45]. SLAMF7 is a capable target for CAR-T cell treatment of multiple myeloma (MM) because of their robust expression on the surface of MM but not normal nonhematopoietic cells. The suicide gene encoded a dimerization domain bonded to a caspase-9 domain [45]. They showed that T cells expressing the SLAMF7-specific CAR accompanied with suicide-gene construct specifically identified and eradicated SLAMF7-positive cells in vitro and tumor cell-bearing mice. Interestingly, engineered T cells were eradicated on demand through injection of the dimerizing agent AP1903 [45]. However, as suicide strategies mainly result in the complete elimination of the CAT-T cells, they will possibly lead to the premature end of the intervention. Consequently, carrying out non-lethal control of CAR-T cells is required to expand the CAR-T cell both efficacy and safety [46]. In this regard, small molecule-based plans as described by Lim et al. can offer a possibility to turn the CAR-T cells “on” or “off” [47]. Further, synthetic splitting receptor [46], combinatorial target-antigen recognition [48], synthetic Notch receptors [49], and bispecific T cell engager [50] along with inhibitory chimeric antigen receptor (iCAR) [51] are other suggested strategies for improving the safety of engineered T cell.
CAR-T cell in clinical trails
Valuing the hopeful results achieved from a myriad of preclinical studies, numerous clinical trials have been conducted or are ongoing to address the safety, feasibility, and efficacy of CAR-T cells in patients suffering from hematological malignancies or solid tumors (Fig. 2) (Table 1).

Clinical trials concerning the CRISPR-Cas9 application in the context of CAR-T cell-based tumor immunotherapy registered in ClinicalTrials.gov (June 2021). The schematic presents conducted or ongoing clinical trials based on the CRISPR-Cas9-mediated genome edition in CAR-T cell depending on the study location (A), study phase (B), study status (C), and target antigen (D) registered in ClinicalTrials.gov (June 2021)
Hematological malignancies
Anti-CD19 CAR-T cell therapy has presented notable activity in patients with refractory or relapsed acute lymphocytic leukemia (ALL). Several anti-CD19 CAR-T cell constructs have been investigated and responses differ extensively among various studies [52]. In 2017, the Food and Drug Administration (FDA) granted regular approval to axicabtagene ciloleucel or Yescarta as a therapeutic option for large B cell lymphoma (BCL). Yescarta is a CD19-specific CAR-T cell mainly exploited for the treatment of adult patients with relapsed or refractory large BCL following two or more lines of systemic treatment. However, a trial in 101 patients with BCL who received a single injection of axicabtagene ciloleucel followed by lymphodepleting chemotherapy using cyclophosphamide and fludarabine indicated that intervention led to severe unwanted events in 52% of participants. Also, recurrence of the CRS and neurologic toxicities in 94% and 87% of participants, respectively, signified the importance of the operation of a risk assessment and mitigation strategy [53]. Nonetheless, infusion of the axicabtagene ciloleucel to 111 participants with diffuse large B cell lymphoma (DLBCL) at the dosage of 2 × 106 CD19-CAR-T cells/kg displayed significant efficacy. While the complete response rate was 54%, a significant number of patients experienced neutropenia, anemia accompanied by thrombocytopenia. Also, 13% and 28% of the patients experienced robust CRS and neurological effects, respectively [54]. Furthermore, brexucabtagene autoleucel (KTE-X19), another CD3ζ/CD28-based CD19-specific CAR-T cell, is specified for mantle cell lymphoma (MCL) therapy. A phase 2 trial in 74 participants with relapsed or refractory MCL revealed that brexucabtagene autoleucel could elicit durable remissions in a majority of patients who received 2 × 106 CD19-CAR-T cells/kg. However, similar to the previous reports, the intervention exerted severe and life-threatening toxic influences [55]. As well, KTE-C19 as an autologous CD3ζ/CD28-based CD19-specific CAR-T cell product at a target dose of 2 × 106 CAR-T cells/kg showed an acceptable safety profile along with an overall response rate of about 71%, and a complete response rate of about 57% in a participant with refractory DLBCL [56]. On the other hand, anti-B cell maturation antigen (BCMA) CAR-T cell therapy has been revealed to have desired activities in patients with relapsed or refractory multiple myeloma (MM) [57]. As well, a small subgroup of MM cells typically express CD19, and thereby CD19-CAR-T cell therapy has displayed a positive anti-tumor effect in some of these patients [57]. Evaluation of the safety and efficacy of combined treatment with anti-CD19 and anti-BCMA CAR-T cells in participants with relapsed or refractory MM have indicated that administration of humanized CD19-CAR-T cells accompanied by murine BCMA CAR-T cells at the similar dosage of 1 × 106 cells/kg following lymphocyte depletion may result in significant preliminary activity. But, the intervention led to the higher unwanted events, containing neutropenia, anemia, and thrombocytopenia in 86%, 62%, and 62% of enrolled participants, respectively, concomitant with one intervention-related death possibly due to the thrombocytopenia [57]. Besides, tisagenlecleucel, an autologous T cell with a lentiviral vector encoding a CD19-specific CAR, presented a significant efficacy along with a manageable safety profile in a subgroup of Japanese patients with relapsed/refractory (r/r) B-ALL [58] and DLBCL [59], making them a rational treatment strategy in patients with B-ALL and DLBCL.
In addition to the cited trails, a myriad of trials based on the targeting BCMA in MM ([60,61,62,63,64,65], CD19 in ALL [32, 66,67,68,69,70,71,72,73,74] and non-Hodgkin’s lymphoma (NHL) [69, 75,76,77,78,79], CD20 in BCL [70, 80,81,82], and CD22 in ALL [83,84,85,86] have shown the significant efficacy in the clinic.
Solid tumors
CAR-T cell therapy is more restricted in solid tumors than in hematological malignancies as CAR-T cells are circulated to the bloodstream and lymphatic system, and thereby have more interaction with blood tumor cells. Nevertheless, in solid tumors, these redirected effector cells may not be able to penetrate tumor tissue by the vascular endothelium [87]. Overall, studies have recognized various roadblocks for administered CAR-T cells, comprising a restricted spectrum of targetable antigens and heterogeneous antigen expression, restricted T cell survival before reaching tumor region, incapability of T cells to proficiently recruit to tumor region and penetrate physical barriers, and finally an immunosuppressive TME [88]. Nonetheless, various tumor-associated antigens (TAA) have been targeted by redirected effector immune cells to elicit an anti-tumor response in vitro and in vivo. For instance, anti-prostate-specific membrane antigen (PSMA) CAR-T cells could selectively target PSMA-positive cells in vitro and eradicate tumor cells in vivo [89]. A trial in 6 patients with prostate cancer revealed that infusion of the PSMA-specific autologous CAR-T cell led to no anti-PSMA toxicities and reactivities. Moreover, the use of PSMA-specific CAR-T cell plus IL-2 resulted in more prominent anti-tumor responses than monotherapy and thereby suggested that pharmacodynamics of “drug-drug” interactions could improve the efficacy of their co-application [90]. Further, it has been found that the potent activity of anti-PSMA CAR-T cells could be improved through the co-expression of a dominant-negative TGF-βRII (dnTGF-βRII). Meanwhile, expression of the dominant-negative TGF-βRII in CAR-T cells could support improved lymphocyte proliferation, augmented cytokine secretion, resistance to exhaustion, prolonged in vivo persistence, and also the stimulation of tumor elimination in vivo. As well, this strategy could be effective for the treatment of patients suffering from relapsed and refractory metastatic prostate cancer [91]. Interestingly, combine treatment with GD2 specific CAR-T cell with CD3ζ, CD28, and OX40 signaling domains and pembrolizumab (anti-PD-1 mAb) may augment the anti-tumor activity of the effector T cells by improving their persistence and expansion in patients with GD2-positive tumors, such as melanoma [92]. On the other hand, constructing and injecting anti-EGFRvIII CAR-T cells is feasible and safe, without indication of off-tumor toxicity or CRS [93, 94]. However, systemic injection of a single dose of EGFRvIII-specific CAR-T cells into 10 patients with glioblastoma mediated antigen loss and stimulated adaptive resistance in patients with recurrent glioblastoma [93]. These findings have shown that while systemic infusion could support on-target effect in the brain, defeating the adaptive variations in the local TME concurrently addressing the antigen heterogeneity are required to improve EGFRvIII-directed approaches in glioblastoma [93]. Moreover, a phase I/II clinical study in 19 patients with recurrent/refractory human epidermal growth factor receptor 2 (HER2)-positive sarcoma showed that injections were well tolerated in the lack of no dose-limiting toxicity [95]. This study was the first trial of the safety and efficacy of HER2-CAR-T cells in patients with tumors showing that the administrated cells persisted for 6 weeks without obvious toxicities [95]. Similarly, the safety and feasibility of HER2-CAR-T cell therapy were shown in patients with advanced biliary tract cancers (BTCs) and pancreatic cancers [96]. Besides, transplantation of the carboxy-anhydrase-IX (CAIX)-specific CAR-T cell into 12 patients with CAIX-expressing metastatic renal cell carcinoma (RCC) delivered in-patient proof that intervention could lead to positive anti-tumor responses [97].
In addition to the listed reports, CAR-T cell therapy based on the targeting tumor-associated glycoprotein (TAG)-72 in colorectal cancer [98], carcinoembryonic antigen (CEA) in lung cancer [99] and liver cancer [100], mesothelin [101], and EGFR [102] in pancreatic cancer, fibroblast activation protein (FAP) in mesothelioma [103], IL13Rα2 in glioblastoma [104], and mucin-1 (MUC1) in seminal vesicle cancer [105] have been conducted or are ongoing to address the safety and efficacy of redirected effector T cells in patients with tumors.
CRISPR/Cas9 potential to overcome potent challenges of CAR-T cell-based therapies
Currently, CRISPR/Cas9-mediated genome editing offers the potential of more effective immunotherapy, by manufacturing a universal “off-the-shelf” cellular product or modifying immune cells to defeat resistance in hematological or solid tumors (Table 2). Despite the existence of several challenges concerning the safety, efficiency, and scalability of this strategy, the CRISPR/Cas9 approach will undeniably reign in the context of CAR-T cell-based therapies for tumors [119].
Disruption of inhibitory molecules and signaling axis
It has been suggested that merging lentiviral delivery of CAR and electro-transfer of Cas9 mRNA and gRNAs targeting endogenous TCR, β-2 B2M, and PD-1 simultaneously cause preparing the universal “off-the-shelf” CAR-T cells. Meanwhile, TCR and HLA class I double-deficient T cells potentially show diminished alloreactivity and commonly cause no GVHD [109, 120]. Moreover, concurrent triple genome editing could support ameliorated in vivo anticancer functions of the gene-disrupted redirected effector T cells [109, 120]. Similarly, triple gene-disrupted CAR-T cells displayed raised activity in glioma mice models leading to the extended overall survival rate in mice bearing intracranial tumors following intracerebral, but not systemic administration [24]. Moreover, marked PD-1 gene disruption lonely can be an attractive plan to enhance the efficacy of CAR-T cell therapy in an immunosuppressive TME [110]. Hu et al. found that PD-1 gene disruption by CRISPR/Cas9 and using piggyBac transposon system for expressing CD133-specific CAR in one reaction resulted in the comparable rates of cytokine releases, while led to the promoted growth and cytotoxicity in vitro. Also, engineered CAR-T cells displayed robust resistance to inhibitory molecules in the glioma murine model compared to conventional CD133-CAR-T cells [110]. Likewise, PD-1-disrupted EGFRvIII-specific CAR-T cells exerted evident suppressive impacts in vitro on EGFRvIII positive glioblastoma cells (U-251MG and EGFRvIII-expressing DKMG) without any significant influence on the T cell phenotype and the expression of other checkpoint receptors [111]. Thereby, Nakazawa et al. suggested that the sgRNA/Cas9-mediated anti-tumor activities of EGFRvIII-specific CAR-T cells are intensely dependent on PD-1 disruption [111]. Besides, PD-1-deficient CD19-specific CAR-T cells showed elevated anti-tumor activity against and improved clearance of CD19+ PD-L1+ K562 myelogenous leukemia cells in NOD-SCID-IL-2Rγ−/− (NSG) mice compared to the conventional CD19-specific CAR-T cell [121]. Albeit, it was found that ectopic PD-L1 expression could not significantly modify intrinsic tumor proliferation in K562 cell-bearing mice since there was no alteration in growth kinetics of CD19+ and CD19+ PD-L1+ cells in the experimental model [121]. Too, PD-1 deficient mesothelin-specific CAR-T cell diminished PD-1+ population in triple-negative breast cancer (TNBC) [122]. Although observed attenuation had no significant impact on CAR-T cell proliferation, it stimulated CAR-T cell cytokine generation and cytotoxicity against PD-L1-expressing TNBC cells in vitro. More efficiently, PD-1 deficient mesothelin-specific CAR-T cells demonstrated a more prominent effect on tumor control and relapse prevention in the preclinical model than conventional CAR-T cells [122]. Besides, lymphocyte activation gene-3 (LAG-3) knockout CD19-specific CAR-T cells by CRISPR-Cas9 elicited strong antigen-specific anti-tumor effects in vitro and lymphoma Raji cell-bearing NOD-Prkdcscid Il2rgnull (NPG) mice. Nonetheless, LAG-3 knockout CAR-T cells showed no superiority in terms of the anti-tumor response and the reduction in tumor burden compared to the conventional CAR-T cells [112].
Reducing CRS and GVHD occurrence
As described, TCR and HLA class I double-deficient CAR-T cells robustly display attenuated alloreactivity and universally result in no GVHD occurrence. As well, these cells’ anti-tumor activity can be potently intensified by simultaneous ablation of PD-1 and CTLA-4 [123]. It has been documented that fratricide-resistant “off-the-shelf” CAR-T, known as UCART7, as a novel anti-CD7 CAR-T cell with a deficiency in TCR could exert robust cytotoxicity against CD7 expressing malignant cells in vitro and in vivo without GVHD development. Both UCART7 and anti-CD7 CAR-T cells could detect and eliminate CD7+ leukemic cell lines, MOLT3, CCRF-CEM, and HSB-2 in vitro with similar efficiencies, representative of no impairment in activity upon double deletion of CD7 and TCR [114]. Thereby, UCART7 as an allo-tolerant “off-the-shelf” CAR-T cell product signifies an efficient and applicable option for treating the relapsed and refractory T-ALL and non-Hodgkin’s T cell lymphoma [114].
Given the importance of the granulocyte-macrophage colony-stimulating factor (GM-CSF) in the simulation of CRS, some studies have focused on the attenuation of its effect on the CRS induction upon CAR-T cell therapy. GM-CSF is a colony-stimulating factor that adjusts the proliferation and differentiation of hematopoietic cells. This cytokine is abundantly generated by CAR-T cells following activation and exists in the TME at high levels [124]. In 2019, Sterner et al. investigated the use of CRISPR/Cas9 gene editing in CD19-specific CAR-T cells by transduction with a lentiviral construct including a guide RNA to GM-CSF and Cas9 [115]. They found that GM-CSF deficient anti-CD19 CAR-T cells efficiently released less GM-CSF, whereas maintained pivotal T cell function. Importantly, these redirected effector T cells exhibited a more prominent anti-tumor effect than wild-type CAR-T cells in vivo [115]. In another study, they found that GM-CSF neutralization with lenzilumab did not elicit any negative effect on anti-CD19 CAR-T cell activity in vitro and in vivo. Furthermore, anti-CD19 CAR-T cell proliferation was improved and durable control of ALL was ameliorated in patient-derived xenografts following GM-CSF neutralization with lenzilumab [116]. Finally, they found that GM-CSF deficient CAR-T cells upheld normal activity and had a superior anti-tumor function in vivo leading to an improved overall survival rate in comparison to the conventional anti-CD19 CAR-T cell [116].
Manufacturing allogeneic universal CAR-T cells
It is mainly difficult in newborn and elder patients to achieve sufficient and good quality T cells for manufacturing the patient-specific CAR-T cells. For providing more accessible CAR-T cells, it is greatly wanted to progress an allogeneic adoptive transfer plan, in which universal CAR-T cells are produced from healthy donor’s T cells to treat numerous patients [123, 125].
As cited, allogeneic universal CAR-T cells can potently be established by impairing TCR and B2M gene expression in CAR-T cells by genome editing strategies. Correspondingly, CAR+TCR_T cells seem to be a rational approach to introduce as the new generation CAR-T cell, providing an “off-the-shelf” therapy for the tentative treatment of B-lineage malignancies [114]. Genetically edition of anti-CD19 CAR-T cells to disrupt expression of the endogenous TCR for inhibition of GVHD progress could display the anticipated property of conventional CD19-specific CAR-T cells without responding to TCR stimulation [126]. Likewise, another report has implied that directing CD19-specific CAR to the TCR locus may sustain the uniform CAR expression in T cells and simultaneously improve T cell potency [117]. Remarkably, Eyquem et al. found that TCR-deficient CD19-specific CAR-T cells could trigger better anti-tumor response compared to conventional CAR-T cells in a mice model of ALL [117]. In addition, directing the CAR to the TCR locus prevents tonic CAR signaling and enables effective internalization and re-expression of the CAR upon the single or repeated exposure to antigen, which in turn leads to the delayed effector T cell differentiation and exhaustion. Indeed, targeting CARs to a TCR locus offers a safer therapeutic T cell by reducing the risk of insertional oncogenesis and TCR-stimulated autoimmunity and alloreactivity in addition to providing a more potent T cell, as documented by minimizing the constitutive signaling and abrogation of T cell depletion [117].
Resistance to the suppressive effects of TGF-β
Despite CAR-T cells’ remarkable activity against cancer, this therapeutic option still faces various challenges, in particular, immunosuppressive tumor microenvironment (TME) for eradicating solid tumors [29]. Although TGF-β exerts tumor-suppressive influences through inhibiting cell cycle development and inducing apoptosis in the early stages of tumors, TGF-β elicits tumor-promoting influences leading to the boosted tumor invasiveness as well as metastasis in late stages [127]. Besides, the TGF-β signaling axis creates interactions with other signaling axes in a synergistic or antagonistic mode and controls biological procedures. Taken together, given the critical role of TGF-β in tumor progress, this pathway is a rational target for tumor therapy. Various therapeutic strategies, comprising TGF-β antibodies, antisense oligonucleotides, and small molecules inhibitors of TGF-β receptor-1 (TGF-βR1), have exposed huge competence to negatively regulate TGF-β signaling [127].
It has been robustly evidenced that suppression of TGF-βR signaling improves the anti-tumor activities of receptor tyrosine kinase-like orphan receptor 1 (ROR1)-specific CAR-T cells toward TNBC. Meanwhile, blockade of the TGF-βR axis using the specific inhibitors could largely protect CD8+ and CD4+ ROR1-CAR-T cells from the suppressive impacts of TGF-β, facilitating their tumor-suppressive activity in the 3D tumor model [29]. Similarly, dominant-negative TGF-βR promotes PSMA-specific CAR-T cell proliferation and strongly increases prostate cancer elimination. These CAR-T cells demonstrate improved cytokine generation, resistance to exhaustion, and also prolonged persistence in vivo [91]. Moreover, the knocking out of the endogenous TGF-β receptor II (TGFBR2) in anti-mesothelin CAR-T cells using the CRISPR/Cas9 technique may decrease the activated Treg conversion and avoid CAR-T cells depletion [29]. Importantly, TGFBR2-edited CAR-T cells exhibited a more obvious capability to eliminate mesothelin-expressing CRL5826 and OVCAR-3 cells in tumor cell-bearing mice when injected locally or systemically [29]. As well, TGF-βRII-edited CAR-T cells are mainly resistant to TGF-β inhibition, and also elicit augmented cell killing compared to the conventional CAR-T cells in the existence of TGF-β against B cell maturation antigen (BCMA)-positive tumor cells [128]. Furthermore, CRISPR/Cas9-mediated knockout of the DGK, as a possible regulator of TGF-β, boosts the anti-tumor activity of the CAR-T versus U87MGvIII glioblastoma cell in vitro and murine models mainly by the triggering resistance to TGF-β and also PGE2 [9].
In addition to the CRISPR-Cas9 technology, other well-known genome-editing techniques have shown the pronounced capability to support the broader application of CAR-T cells (Table 3).
The off-target effects of CRISPR-Cas9 technology
Several classes of CRISPR-Cas systems have yet been advanced, while their comprehensive use can be hindered via off-target effects. Efforts are being accomplished to attenuate the off-target effects of CRISPR-Cas9 through establishing the multiple CRISPR/Cas systems with high fidelity and accuracy [137]. Thereby, a myriad of techniques have been utilized to identify off-target mutations, and restore the on-target effects and conversely reduce potent off-target effects. As the genomic frameworks of the targeted DNA concurrently the secondary structure of sgRNAs and their GC content are mainly contribute to determining cleavage efficiency, designing of the appropriate sgRNAs with high on-target activities using specific tools is severally suggested [137]. Recently, the amelioration of the specificity [138] of genome editing tools and the identification [139] of off-target effects are swiftly developing research areas. Such research incorporates designer nuclease development [140], discovery computational prediction programs and also databases [141] and also finding high-throughput sequencing [139] to diminish mutational occurrence. Overall, the amelioration of the off-target specificity in the CRISPR-Cas9 system undoubtedly will deliver solid genotype-phenotype associations, and therefore empower faithful interpretation of gene-editing statistics, facilitating the basic and clinical utility of this CRISPR-Cas9 technology [142].
Conclusion and prospect
The progress of genomic editing techniques enlarges the landscape of CAR-T cell-based therapies for adoptive cell therapy. Among the several technologies that can be exploited, CRISPR/Cas9 is comparatively easy to use, simple to design, and cost-effective concurrently remarkable multiplex genome engineering competencies [143]. Now, CRISPR/Cas9-based genome editing provides the capability of further streamlining immune cell-based therapies, more prominently, through the generation of a universal “off-the-shelf” cellular product or engineering these redirected effector cells to overcome resistance in human malignancies, ranging from hematological malignancies to solid tumors [144]. These findings have resulted in the execution of several clinical trials to evaluate the therapeutic safety and efficacy of CRISPR/Cas9-mediated genome editing in CAR-T cell therapy (Table 4). However, for further human trials, designing and expanding large-scale approaches for CRISPR/Cas9-mediated target ablation in mature T cells is of principal significance. These protocols must simplify the transference of sgRNA, and Cas9 concomitant with a gene encoding the CAR, maintain cell survival and support strong in vitro cultivation of modified T cells upon genetic manipulation [119]. These means may comprise transduction of CRISPR/Cas9 machinery and CAR transgenes employing the retroviruses or lentiviruses [145, 146] or using non-integrating viruses, including adenoviruses and adenovirus-associated viruses (AAV) [147, 148]. Further, the development of innovative strategies to attenuate off-target CRISPR/Cas9 editing, such as varying the Cas9 endonuclease using novel PAM specificities [149], applying the high-fidelity Cas9 variants, and also exploiting truncated sgRNAs can support more prominent consequences in vivo [119]. In sum, we guess that conduction of the more comprehensive studies based on the CRISPR-Cas9 application to improve CAR-T cell safety, efficacy, and accessibility could lead to the desired therapeutic outcomes in the clinic.
Availability of data and materials
Not applicable
Abbreviations
- CAR:
-
Chimeric antigen receptor
- GVHD:
-
Graft-versus-host disease
- CRS:
-
Cytokine release syndrome
- CRISPR:
-
Clustered regularly interspaced short palindromic repeat
- ZFN:
-
Zinc finger nuclease
- TALEN:
-
Transcription activator-like effector nuclease
- TGF-β:
-
Transforming growth factor beta
- TRAC:
-
T cell receptor alpha constant
- B2M:
-
Beta-2-microglobulin
- PD1:
-
Programmed cell death protein 1
- sgRNA:
-
Single-stranded guide RNA
References
Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun. 2018;9:1–13.
Bedell VM, Wang Y, Campbell JM, Poshusta TL, Starker CG, Krug RG II, et al. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–8.
Li H, Yang Y, Hong W, Huang M, Wu M, Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduc Targeted Ther. 2020;5:1.
Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J Exp Med. 2018;215:985–97.
Zhang C, Liu J, Zhong JF, Zhang X. Engineering car-t cells. Biomark Res. 2017;5:1–6.
Wang Z, Guo Y, Han W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell. 2017;8:896–925.
Li C, Mei H, Hu Y. Applications and explorations of CRISPR/Cas9 in CAR T-cell therapy. Brief Funct Genom. 2020;19:175–82.
Huang RY, Francois A, McGray AR, Miliotto A, Odunsi K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology. 2017;6:e1249561.
Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/Cas9-mediated knockout of DGK improves antitumor activities of human T cells. Cancer Res. 2018;78:4692–703.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–33.
Karginov FV, Hannon GJ. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell. 2010;37:7–19.
Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and evolution of type II CRISPR-Cas systems. Nucleic Acids Res. 2014;42:6091–105.
Liu Z, Dong H, Cui Y, Cong L, Zhang D. Application of different types of CRISPR/Cas-based systems in bacteria. Microb Cell Factories. 2020;19:172.
Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017;37:67–78.
Le Rhun A, Escalera-Maurer A, Bratovič M, Charpentier E. CRISPR-Cas in Streptococcus pyogenes. RNA Biol. 2019;16:380–9.
Xu H, Xiao T, Chen C-H, Li W, Meyer CA, Wu Q, et al. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–57.
O'Driscoll M, Jeggo PA. The role of double-strand break repair - insights from human genetics. Nat Rev Genet. 2006;7:45–54.
Jiang F, Doudna JA. CRISPR–Cas9 structures and mechanisms. Annu Rev Biophys. 2017;46:505–29.
Abadi S, Yan WX, Amar D, Mayrose I. A machine learning approach for predicting CRISPR-Cas9 cleavage efficiencies and patterns underlying its mechanism of action. PLoS Comput Biol. 2017;13:e1005807.
Zhu Y, Gao A, Zhan Q, Wang Y, Feng H, Liu S, et al. Diverse mechanisms of CRISPR-Cas9 inhibition by type IIC anti-CRISPR proteins. Mol Cell. 2019;74:296–309 e297.
Gaj T, Gersbach CA, Barbas CF 3rd. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405.
Ota S, Hisano Y, Ikawa Y, Kawahara A. Multiple genome modifications by the CRISPR/Cas9 system in zebrafish. Genes Cells. 2014;19:555–64.
Yang H, Wu J-J, Tang T, Liu K-D, Dai C. CRISPR/Cas9-mediated genome editing efficiently creates specific mutations at multiple loci using one sgRNA in Brassica napus. Sci Rep. 2017;7:7489.
Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019;7:304.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38:233–44.
Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med. 2012;14:405–15.
Andrea AE, Chiron A, Bessoles S, Hacein-Bey-Abina S. Engineering next-generation CAR-T cells for better toxicity management. Int J Mol Sci. 2020;21:8620.
Tian Y, Li Y, Shao Y, Zhang Y. Gene modification strategies for next-generation CAR T cells against solid cancers. J Hematol Oncol. 2020;13:54.
Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5.
Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR–T cells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med. 2012;18:377–84.
Hartmann J, Schüßler-Lenz M, Bondanza A, Buchholz CJ. Clinical development of CAR T cells—challenges and opportunities in translating innovative treatment concepts. EMBO Mol Med. 2017;9:1183–97.
Gardner RA, Finney O, Annesley C, Brakke H, Summers C, Leger K, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–31.
Sadelain M, Brentjens R, Rivière I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:388–98.
He Q, Jiang X, Zhou X, Weng J. Targeting cancers through TCR-peptide/MHC interactions. J Hematol Oncol. 2019;12:139.
Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29:917–24.
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra138.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra225.
Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32:689–702.
Themeli M, Rivière I, Sadelain M. New cell sources for T cell engineering and adoptive immunotherapy. Cell Stem Cell. 2015;16:357–66.
D'Orsogna LJ, Roelen DL, Doxiadis IIN, Claas FHJ. TCR cross-reactivity and allorecognition: new insights into the immunogenetics of allorecognition. Immunogenetics. 2012;64:77–85.
DeWolf S, Sykes M. Alloimmune T cells in transplantation. J Clin Invest. 2017;127:2473–81.
Alzubi J, Lock D, Rhiel M, Schmitz S, Wild S, Mussolino C, et al. Automated generation of gene-edited CAR T cells at clinical scale. Mol Ther Methods Clin Dev. 2021;20:379–88.
Minagawa K, Al-Obaidi M, Di Stasi A. Generation of suicide gene-modified chimeric antigen receptor-redirected T-cells for cancer immunotherapy. Methods Mol Biol. 2019;1895:57–73.
Minagawa K, Jamil MO, Al-Obaidi M, Pereboeva L, Salzman D, Erba HP, et al. In vitro pre-clinical validation of suicide gene modified anti-CD33 redirected chimeric antigen receptor T-cells for acute myeloid leukemia. PLoS One. 2016;11:e0166891.
Amatya C, Pegues MA, Lam N, Vanasse D, Geldres C, Choi S, et al. Development of CAR T cells expressing a suicide gene plus a chimeric antigen receptor targeting signaling lymphocytic-activation molecule F7. Mol Ther. 2021;29:702–17.
Juillerat A, Marechal A, Filhol J-M, Valton J, Duclert A, Poirot L, et al. Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep. 2016;6:18950.
Wu C-Y, Roybal KT, Puchner EM, Onuffer J, Lim WA. Remote control of therapeutic T cells through a small molecule–gated chimeric receptor. Science. 2015;350.
Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 2019;12:128.
Saffern M, Samstein R. Taking CAR T cells up a synthetic Notch. Nat Rev Immunol. 2021;21:135.
Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol. 2021;14:75.
Ren Y-B, Sun S-J, Han S-Y. Safety strategies of genetically engineered T cells in cancer immunotherapy. Curr Pharm Des. 2018;24:78–83.
Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol. 2020;7:e816–26.
Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin Cancer Res. 2019;25:1702–8.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382:1331–42.
Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 Results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25:285–95.
Yan Z, Cao J, Cheng H, Qiao J, Zhang H, Wang Y, et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: a single-arm, phase 2 trial. Lancet Haematol. 2019;6:e521–9.
Hiramatsu H, Adachi S, Umeda K, Kato I, Eldjerou L, Agostinho AC, et al. Efficacy and safety of tisagenlecleucel in Japanese pediatric and young adult patients with relapsed/refractory B cell acute lymphoblastic leukemia. Int J Hematol. 2020;111:303–10.
Goto H, Makita S, Kato K, Tokushige K, Fujita T, Akashi K, et al. Efficacy and safety of tisagenlecleucel in Japanese adult patients with relapsed/refractory diffuse large B-cell lymphoma. Int J Clin Oncol. 2020;25:1736–43.
Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380:1726–37.
Cohen AD, Garfall AL, Stadtmauer EA, Melenhorst JJ, Lacey SF, Lancaster E, et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J Clin Invest. 2019;129:2210–21.
Xu J, Chen LJ, Yang SS, Sun Y, Wu W, Liu YF, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci U S A. 2019;116:9543–51.
Ali SA, Shi V, Maric I, Wang M, Stroncek DF, Rose JJ, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016;128:1688–700.
Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11:141.
Da Vià MC, Dietrich O, Truger M, Arampatzi P, Duell J, Heidemeier A, et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat Med. 2021;27:616–9.
Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25:1408–14.
Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019;134:2361–8.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–63.
Itzhaki O, Jacoby E, Nissani A, Levi M, Nagler A, Kubi A, et al. Head-to-head comparison of in-house produced CD19 CAR-T cell in ALL and NHL patients. J Immunother Cancer. 2020;8.
Shah NN, Johnson BD, Schneider D, Zhu F, Szabo A, Keever-Taylor CA, et al. Bispecific anti-CD20, anti-CD19 CAR T cells for relapsed B cell malignancies: a phase 1 dose escalation and expansion trial. Nat Med. 2020;26:1569–75.
Zhang Q, Hu H, Chen SY, Liu CJ, Hu FF, Yu J, et al. Transcriptome and regulatory network analyses of CD19-CAR-T immunotherapy for B-ALL. Genom Proteom Bioinform. 2019;17:190–200.
Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26:270–80.
Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Invest. 2020;130:673–85.
Gardner R, Wu D, Cherian S, Fang M, Hanafi L-A, Finney O, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127:2406–10.
Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396:839–52.
Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–90.
Rossi J, Paczkowski P, Shen YW, Morse K, Flynn B, Kaiser A, et al. Preinfusion polyfunctional anti-CD19 chimeric antigen receptor T cells are associated with clinical outcomes in NHL. Blood. 2018;132:804–14.
Schubert ML, Schmitt A, Sellner L, Neuber B, Kunz J, Wuchter P, et al. Treatment of patients with relapsed or refractory CD19+ lymphoid disease with T lymphocytes transduced by RV-SFG.CD19.CD28.4-1BBzeta retroviral vector: a unicentre phase I/II clinical trial protocol. BMJ Open. 2019;9:e026644.
Sauter CS, Senechal B, Rivière I, Ni A, Bernal Y, Wang X, et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood. 2019;134:626–35.
Tong C, Zhang Y, Liu Y, Ji X, Zhang W, Guo Y, et al. Optimized tandem CD19/CD20 CAR-engineered T cells in refractory/relapsed B-cell lymphoma. Blood. 2020;136:1632–44.
Bhoj VG, Arhontoulis D, Wertheim G, Capobianchi J, Callahan CA, Ellebrecht CT, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood. 2016;128:360–70.
Michot JM, Annereau M, Danu A, Legoupil C, Bertin L, Chahine C, et al. High-dose cyclophosphamide for hard-to-treat patients with relapsed or refractory B-cell non-Hodgkin's lymphoma, a phase II result. Eur J Haematol. 2020;104:281–90.
Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20–8.
Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38:1938–50.
Pan J, Niu Q, Deng B, Liu S, Wu T, Gao Z, et al. CD22 CAR T-cell therapy in refractory or relapsed B acute lymphoblastic leukemia. Leukemia. 2019;33:2854–66.
Zhang Y, Chen H, Song Y, Tan X, Zhao Y, Liu X, et al. Chimeric antigens receptor T cell therapy as a bridge to haematopoietic stem cell transplantation for refractory/ relapsed B-cell acute lymphomalastic leukemia. Br J Haematol. 2020;189:146–52.
Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. 2021;12:81.
Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther. 2020;28:2320–39.
Hussein AA, Baban RS, Hussein AG. Prostate-specific antigen and (free prostate-specific antigen/prostate-specific antigen) ratio in patients with benign prostatic hyperplasia and prostate cancer. Baghdad J Biochem Appl Biol Sci. 2020;1:17–25.
Junghans RP, Ma Q, Rathore R, Gomes EM, Bais AJ, Lo AS, et al. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76:1257–70.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26:1855–66.
Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24:1135–49.
O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9.
Mousa NO, Gado M, Osman A. Multimodality of human epidermal growth factor receptor-2 antagonism restores the apoptotic capacity of liver cancer cells, Journal of Natural Science. Biol Med. 2020;11:118.
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) -specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33:1688–96.
Feng K, Liu Y, Guo Y, Qiu J, Wu Z, Dai H, et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein Cell. 2018;9:838–47.
Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21:904–12.
Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22.
Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66:1425–36.
Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21:3149–59.
Beatty GL, O'Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of mesothelin-specific chimeric antigen receptor T cells against pancreatic carcinoma metastases in a phase 1 trial. Gastroenterology. 2018;155:29–32.
Liu Y, Guo Y, Wu Z, Feng K, Tong C, Wang Y, et al. Anti-EGFR chimeric antigen receptor-modified T cells in metastatic pancreatic carcinoma: a phase I clinical trial. Cytotherapy. 2020;22:573–80.
Hiltbrunner S, Britschgi C, Schuberth P, Bankel L, Nguyen-Kim TDL, Gulati P, et al. Local delivery of CAR T cells targeting fibroblast activation protein is safe in patients with pleural mesothelioma: first report of FAPME, a phase I clinical trial. Ann Oncol. 2021;32:120–1.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21:4062–72.
You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao X, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated by modified Anti-MUC1 chimeric antigen receptor transduced T cells, Science China. Life Sci. 2016;59:386–97.
Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7:12320.
Schmidts A, Marsh LC, Srivastava AA, Bouffard AA, Boroughs AC, Scarfò I, et al. Cell-based artificial APC resistant to lentiviral transduction for efficient generation of CAR-T cells from various cell sources. J Immunother Cancer. 2020;8.
Kailayangiri S, Altvater B, Lesch S, Balbach S, Göttlich C, Kühnemundt J, et al. EZH2 inhibition in Ewing sarcoma upregulates G(D2) expression for targeting with gene-modified T cells. Mol Ther. 2019;27:933–46.
Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget. 2017;8:17002–11.
Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with plasmid DNA for CRISPR/Cas9-mediated inactivation of programmed cell death protein 1 in CD133-specific CAR T cells. Hum Gene Ther. 2019;30:446–58.
Nakazawa T, Natsume A, Nishimura F, Morimoto T, Matsuda R, Nakamura M, et al. Effect of CRISPR/Cas9-mediated PD-1-disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. Cells. 2020;9:998.
Zhang Y, Zhang X, Cheng C, Mu W, Liu X, Li N, et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front Med. 2017;11:554–62.
Dai X, Park JJ, Du Y, Kim HR, Wang G, Errami Y, et al. One-step generation of modular CAR-T cells with AAV-Cpf1. Nat Methods. 2019;16:247–54.
Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–83.
Sterner RM, Cox MJ, Sakemura R, Kenderian SS. Using CRISPR/Cas9 to knock out GM-CSF in CAR-T cells. JoVE. 2019.
Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133:697–709.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–7.
Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood. 2020;136:1407–18.
Salas-Mckee J, Kong W, Gladney WL, Jadlowsky JK, Plesa G, Davis MM, et al. CRISPR/Cas9-based genome editing in the era of CAR T cell immunotherapy. Hum Vaccin Immunother. 2019;15:1126–32.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66.
Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep. 2017;7:737.
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68:365–77.
Liu X, Zhao Y. CRISPR/Cas9 genome editing: fueling the revolution in cancer immunotherapy. Curr Res Translat Med. 2018;66:39–42.
Khadka RH, Sakemura R, Kenderian SS, Johnson AJ. Management of cytokine release syndrome: an update on emerging antigen-specific T cell engaging immunotherapies. Immunotherapy. 2019;11:851–7.
Georgiadis C, Preece R, Nickolay L, Etuk A, Petrova A, Ladon D, et al. Long terminal repeat CRISPR-CAR-coupled "universal" T cells mediate potent anti-leukemic effects. Mol Ther. 2018;26:1215–27.
Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–705.
Syed V. TGF-β Signaling in Cancer. J Cell Biochem. 2016;117:1279–87.
Welstead GG, Vong Q, Nye C, Hause R, Clouser C, Jones J, et al. Improving efficacy of CAR T cells through CRISPR/Cas9 mediated knockout of TGFBR2. In: Molecular Therapy. Cambridge: Cell Press; 2018. p. 61.
Sommer C, Boldajipour B, Kuo TC, Bentley T, Sutton J, Chen A, et al. Preclinical evaluation of allogeneic CAR T cells targeting BCMA for the treatment of multiple myeloma. Mol Ther. 2019;27:1126–38.
Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9.
Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex genome-edited T-cell manufacturing platform for "off-the-shelf" adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–64.
Sachdeva M, Busser BW, Temburni S, Jahangiri B, Gautron AS, Maréchal A, et al. Repurposing endogenous immune pathways to tailor and control chimeric antigen receptor T cell functionality. Nat Commun. 2019;10:5100.
Rasaiyaah J, Georgiadis C, Preece R, Mock U, Qasim W. TCRαβ/CD3 disruption enables CD3-specific antileukemic T cell immunotherapy. JCI Insight. 2018;3.
Gautron AS, Juillerat A, Guyot V, Filhol JM, Dessez E, Duclert A, et al. Fine and predictable tuning of TALEN gene editing targeting for improved T cell adoptive immunotherapy. Mol Ther Nucleic Acids. 2017;9:312–21.
Sachdeva M, Duchateau P, Depil S, Poirot L, Valton J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J Biol Chem. 2019;294:5430–7.
Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018;24:1499–503.
Liu X, Homma A, Sayadi J, Yang S, Ohashi J, Takumi T. Sequence features associated with the cleavage efficiency of CRISPR/Cas9 system. Sci Rep. 2016;6:1–9.
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32:279–84.
Tsai SQ, Nguyen NT, Malagon-Lopez J, Topkar VV, Aryee MJ, Joung JK. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat Methods. 2017;14:607–14.
Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32:569–76.
Singh R, Kuscu C, Quinlan A, Qi Y, Adli M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015;43:e118.
Zhang X-H, Tee LY, Wang X-G, Huang Q-S, Yang S-H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015;4:e264.
Savić N, Schwank G. Advances in therapeutic CRISPR/Cas9 genome editing. Transl Res. 2016;168:15–21.
Zhao J, Lin Q, Song Y, Liu D. Universal CARs, universal T cells, and universal CAR T cells. J Hematol Oncol. 2018;11:132.
Williams MR, Fricano-Kugler CJ, Getz SA, Skelton PD, Lee J, Rizzuto CP, et al. A retroviral CRISPR-Cas9 system for cellular autism-associated phenotype discovery in developing neurons. Sci Rep. 2016;6:25611.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–7.
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–91.
Swiech L, Heidenreich M, Banerjee A, Habib N, Li Y, Trombetta J, et al. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol. 2015;33:102–6.
Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, et al. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015;523:481–5.
Acknowledgements
Not applicable
Funding
No funders.
Author information
Authors and Affiliations
Contributions
All authors contributed to the conception and the main idea of the work. E.R, M.K.N, H.S.R, Z.R.G, W.K.A, S.A, D.O.B, W.S, P.N, SI.SH, F.M, M.Y, SO.SH, and R.M drafted the main text, figures, and tables. F.M.KH supervised the work and provided the comments and additional scientific information. F.M and F.M.KH also reviewed and revised the text. All authors read and approved the final version of the work to be published.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
There is no conflict of interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Razeghian, E., Nasution, M.K.M., Rahman, H.S. et al. A deep insight into CRISPR/Cas9 application in CAR-T cell-based tumor immunotherapies. Stem Cell Res Ther 12, 428 (2021). https://doi.org/10.1186/s13287-021-02510-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13287-021-02510-7