
Abstract
Due to the DNA repair defect, BRCA1/2 deficient tumor cells are more sensitive to PARP inhibitors (PARPi) through the mechanism of synthetic lethality. At present, several PAPRi targeting poly (ADP-ribose) polymerase (PARP) have been approved for ovarian cancer and breast cancer indications. However, PARPi resistance is ubiquitous in clinic. More than 40% BRCA1/2-deficient patients fail to respond to PARPi. In addition, lots of patients acquire PARPi resistance with prolonged oral administration of PARPi. Homologous recombination repair deficient (HRD), as an essential prerequisite of synthetic lethality, plays a vital role in killing tumor cells. Therefore, Homologous recombination repair restoration (HRR) becomes the predominant reason of PARPi resistance. Recently, it was reported that DNA replication fork protection also contributed to PARPi resistance in BRCA1/2-deficient cells and patients. Moreover, various factors, such as reversion mutations, epigenetic modification, restoration of ADP-ribosylation (PARylation) and pharmacological alteration lead to PARPi resistance as well. In this review, we reviewed the underlying mechanisms of PARP inhibitor resistance in detail and summarized the potential strategies to overcome PARPi resistance and increase PARPi sensitivity.
Similar content being viewed by others


Introduction
DNA damage response (DDR) is vital to maintaining genome stability [1]. When cells suffer from DNA damage, DDR is instigated and it can remove the damage by specified DNA repair pathways, including homologous recombination repair (HR), non-homologous end joining repair (NHEJ), single stranded break repair (SSBR) [2]. To cope with DNA single-strand breaks (SSB), base excision repair (BER) is activated in mammalian cells. Poly (ADP-ribose) polymerases (PARPs), especially PARP1, PARP2 and PARP3 are key to BER [3, 4]. As DNA damage sensors and signal transducers, they can bind damaged DNA at single strand DNA breaks sites, which result in the recruitment of DNA repair effectors to the sites of DNA breaks [4, 5]. NHEJ and HR are two mainly pathways to resolve the DNA double- strand breaks (DSB). NHEJ is an error prone pathway. In this mechanism, DSB sites are repaired by blunt end ligation with low fidelity [6]. While the use of NHEJ leads to accumulation of genetic aberrations, chromosomal instability, cell cycle arrest and apoptosis [7]. However, HR is a process of accurate restoration of the DSB with high fidelity [8]. BRCA1/2 proteins are crucial for the error-free repair of HR [9]. In the S/G2 phase, BRCA1 is recruited to the DSB sites, which counteracts 53BP1 and initiates ubiquitination of C-terminal binding protein interacting protein (CtIP) [10]. With the assistance of CtIP, the 5′ to 3′ resection occurs and generates 3′ overhangs. Afterwards, BRCA2 and PALB2 participate in the formation of the nucleoprotein filament and D-loop [11, 12] (Fig. 1). Given that DDR has the ability to overcome the cytotoxicity induced by chemo- and radiotherapy treatment, it’s important to uncover the underlying mechanisms of DNA repair pathway and exploit new drugs.

Schematic describing the function the principle of synthetic lethality interaction between PARPs and BRCA1/2. When cells suffer from DNA response, single-strand breaks emerge. PARPs, especially PARP1, bind to the DNA break sites, which result in the PARylation of target proteins and recrement of the DNA damage repair effectors. Then the auto-PARylation on PARPs leads to the dissociation of PARPs from DNA. Treating HR-deficient tumor cells with PARPi, NHEJ is the only pathway to use to repair double-strand break, which lead to accumulation of genome instability and cell death for the low fidelity
Germline mutations in BRCA1/2 (gBRCAm) predispose to ovarian cancer and breast cancer. Besides, somatic mutations of BRCA1/2 (sBRCAm) have also been suggested in various cancer types. Especially, nearly 20% of patients (16% gBRCAm and 4% sBRCAm) occur in ovarian cancer [13]. More importantly, up to 50% high-grade serious ovarian cancer (HGSOC) patients present as HRD [14]. Therefore, inhibition of PARPs may cause both SSBR deficient and HRD in BRCA1/2 deficient patients, leading to cell death [15, 16]. This is the so-called “synthetic lethality”, which is a concept proposed a century ago to describe the condition whereby a defect of either one of two genes have no/little effect but the combination of both genes (BRCA and PARPs) lead to cell death [17].
PARPi are the first agents designed to exploit synthetic lethality and permitted to use in clinic. They have the ability to bind and trap PARPs on DNA, preventing the release of PARPs from DNA break sites and removing PARPs from their normal catalytic cycle [5]. Due to more benefits and less adverse effects, olaparib (lynparza), niraparib (ZEJULA) and rucaparib (RUBRACA) are indicated for the maintenance treatment of recurrent ovarian cancer patients, who are in a complete or partial response to platinum-based chemotherapy in United states [18,19,20,21]. Olaparib is also approved to treating gBRCAm advanced ovarian cancer as four lines of chemotherapy [18]. It can also be used to treat gBRCAm, HER2-negative metastatic breast cancer patients, who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting [22, 23]. Recently, it’s suggested that carriers with HRD but not gBRCAm or sBRCAm, which is termed as “BRCAness”, are also sensitive to PARPi [24]. However, BRCA1/2 mutations remain the strongest genetic predictor of sensitivity of PARPi [25].
Similarity with other chemotherapy agents, PARPi also faced the drug resistance. More than 40% of BRCAm ovarian cancer patients failed to benefit from PARPi [26, 27]. Considering the important roles of HR repair pathway and protection of stalled replication forks in the effect of PARPi, we described the effects of DNA repair response and protection of stalled replication forks on PARPi resistance in detail. Besides, we reviewed the association between PARPi resistance and other factors, such as reversion mutations, epigenetic modification, restoration of PARPylation and pharmacological alteration. Finally, we summarized the feasible strategies to overcome PARPi resistance and enhance PARPi sensitivity in clinic.
Restoration of HR repair in PARPi resistance
Due to HRD is the main premise of anticancer effects of PARPi, it is crucial to understand the HR repair pathway. When DSB happen in mammalian cells, the DDR is activated. Coordinately, cells employ two typical mechanisms to repair DSB: HR and NHEJ. Normally, NHEJ is the mainly repair mechanism by ligating the broken DNA ends in a nonhomologous end-joining way and occurs throughout the cell cycle, especially in G0/G1 phase. However, HR predominates the S/G2 phase, due to the high DNA replication and available sister template [28]. In the process of HR, the DSB ends are firstly resected by Mre11-Rad50-Nbs1(MRN) complex together with CtIP and nucleases (EXO1, DNA2 and MUS8), leading to the formation of the single-strand DNA (ssDNA) and committing the cells to HR [29]. Afterwards, the resected DNA ends are coated by hyperphosphorylated single-strand DNA binding protein A (RPA) [30]. The variant H2AX (named γH2AX) is activated and phosphorylated by apical kinases, such as ataxia-telangiectasia mutated (ATM) and ATM- and Rad3-related (ATR). The spreading of γH2AX along the chromosome assists the recruitment and accumulation of additional DDR proteins, including p53-binding protein (53BP1) and BRCA1 to the DDR foci [31]. With the favor of PALB2, BRCA2 binds with BRCA1 and promotes the loading of recombinase RAD51 on the ssDNA [11]. The RAD51 mediates the invasion of the homologous sequence and formation of the nucleoprotein filament and D-loop by eliminating secondary structure formation and protecting DNA ends from degradation [32] (Fig. 2). Therefore, the restoration of HR pathway by inducing the process of DNA end resection and formation of nucleoprotein filament and D-loop may lead to PARPi resistance.

Homologous recombination repair in S/G2 phase. The double-strand break ends are resected by MRE11-RAD50-NBS1(MRN) complex together with CtIP. ATM is recruited to DSBs through MRN and phosphorylates targets such as 53BP1 and MDC1. MDC1 phosphorylation recruits the E3 ubiquitin ligase RNF8, which, through recruitment of a second E3 ubiquitin ligase (RNF168), leads to histone H2A ubiquitylation. This modification, together with H4K20 methylation, allows for 53BP1 recruitment. 53BP1 phosphorylation allows its interaction with RIF1 and PTIP, which can be blocked by WIP1. 53BP1 blocks DNA resection by recruiting shieldin and presents cells to NHEJ. While, BRCA1 counteracts the protection function of 53BP1, leading to the resection of DNA ends. Afterwards, the resected DNA ends are coated by PRA. With the favor of PALB2, BRCA2 binds with BRCA1 and promotes the loading of RAD51. The RAD51 mediates the invasion of the homologous sequence and formation of the nucleoprotein filament and D-loop by eliminating secondary structure formation. EMI and DDB2 mediate the degradation of RAD51. TOPBP1 phosphorylates RAD51. BRD4 and HORMAD1 are key regulators of RAD51 accumulation on chromatin. P, phosphorylation; Ub, ubiquitylation; Me, methylation, SUMO, SUMOylation, red arrows, resection
DNA end resection in PARPi resistance
Considering that DNA end resection is the key of different DNA repair pathways choices, it’s likely that DNA end resection dictates the different repair outcome and PARPi sensitivity. Recently, multiple reports have suggested that DNA end resection participated in the PARPi resistance (Fig. 2).
Cell cycle controls the choice of DSB repair pathways [33]. In the G1 phase, 53BP1 and RIF1 proteins localize to DSB sites, leading to the inhibition of BRCA1 recruitment, blocking DNA resection and promoting NHEJ repair pathway. Otherwise, DNA end resection is stimulated in the phase of S/G2 phase and promotes HR repair [34]. It is worth mentioning that DNA end resection is depended on cyclin-dependent kinases (CDKs) activity, which mediate phosphorylation of MRN complex and CtIP [35, 36]. It was reported that CDK5-silenced Hela cells were more sensitive to PARPi [36]. Besides, CDK12 was identified as a determinant of olaparib in the models of HGSOC by genome-wide synthetic lethal screen [37]. Loss-of functions (LOF) mutations in CDK12 disrupted HR repair and sensitized ovarian cancer cells to veliparib [38]. In triple-negative breast cancer (TNBC), deletion of CDK12 reversed both primary PARPi resistance and secondary PARPi resistance, no matter in BRCA wild-type and mutated models [39]. Besides, CDK18 facilitates ATR activation by interacting with ATR and regulating ATR-Rad9/ATR-ETAA1 interactions, promoting HR and PARPi resistance in glioblastoma stem-like cells [40]. Recently, a case report results indicated that PARPi combined with CDK4/6 inhibitor (palbociclib) revealed more excellent therapeutic effects than PARPi alone in the treating with BRCA-mutated, ER-positive breast cancer [41]. All these evidences suggested that CDKs blocked DNA end resection and lead to PARPi resistance and its inhibitors might overcome the PARPi resistance. Prospectively, the combination therapy of PARPi and CDKs inhibitors is applied in clinic.
In addition to Cell cycle and CDKs, accessory factors including 53BP1, REV7 and RIF1, contribute a lot to DNA end resection and PARPi resistance [42,43,44]. 53BP1, which is a chromatin-binding protein, blocks DNA resection by preventing the accession of CtIP to the DSB sites [45]. It has been suggested that the loss of 53BP1 induced DNA end resection and HR restoration, leading to PARPi resistance in various cancers, such as breast cancer [42], glioblastoma [46] and ovarian cancer [47]. Mainly, 53BP1 protects DNA ends from resection in two ways. One way is to strengthen the nucleosomal barrier to end-resection nucleases by recognizing and binding to the nucleosomes containing H4K20m2 and H2AK15ub [48]. The other way is to recruit effector complex proteins with end-protection activity49. Recently, it was demonstrated that shieldin, an effector complex composed by SHLD1, SHLD2, SHLD3 and REV7, were recruited by 53BP1 to the DSB sites in a 53BP1 and RIF1 depend manner [49]. Numerous evidences revealed that shieldin, as the key regulator of NHEJ repair and HR repair, was also associated with PARPi resistance [49,50,51]. REV7, as the component of shieldin, was also suggested to counteract DNA end resection and sensitize cells to PARPi [43]. Likewise, catalysed the inactivating conformational change of REV7 and dissociated REV7-Shieldin by TRIP13 ATPase promoted HR, leading to PARPi resistance [52]. The protection function of 53BP1 requires the interactions of PTIP and RIF1, which is depends on ATM [44, 53]. Hence, the interaction between 53BP1 and RIF1 plays pivotal roles in DNA end resection and PARPi resistance. As is known to us, only when 53BP1 is phosphorylated by ATM can it recruit RIF1 and PTIP [54]. It was demonstrated that ATM-deficient cancer cells was more sensitive to PARPi than ATM-proficient cells and the combination use of ATM inhibitors enhanced PARPi efficacy [55, 56]. Besides, multiple clinical trials results indicated that patients with low ATM proteins had a greater benefit from PARPi and more favorable prognosis [57,58,59]. Recently, it was disclosed that WIP1 dephosphorylated 53BP1 at Threonine 543 and attenuated its interaction with RIF1, leading to decreased sensitivity of cancer cells to PARPi [60], which confirmed the importance of the interaction between 5BP1 and RIF1 once more. Obviously, nucleases (i.e., MRE11 [61,62,63], DNA2 [64] and EXO1 [65, 66]), functioning as “DNA end clipping” in the process of DNA end resection, affected the sensitivity and resistance of PARPi.
Formation of RAD51-ssDNA filament and D-loop in PARPi resistance
The RAD51-ssDNA filament performs the central functions in homology search, DNA stand exchange and HR repair (Fig. 2). Especially, RAD51 foci is suggested to serve as a functional biomarker of HR repair and PAPRi resistance beyond BRCA mutation [67,68,69]. In the issue, the balance between RAD51 filament formation and disruption seem particularly important. By using a genetic screen, EMI1 was identified to constitutively target RAD51 for degradation and function as a modulator of PARPi sensitivity. Downregulation of EMI1 enhanced the RAD51 accumulation, leading to restoring HR and developing PARPi resistance in BRCA1-deficient TNBC cells [70]. Similarly, DNA damage binding protein 2 (DDB2), a DNA damage recognition factor, was reported to participate in the regulation of RAD51 degradation by physical interaction in TNBC cells. The inhibition of DDB2 induced RAD51 polyubiquitination and proteasomal degradation, leading to defective HR and sensitivity to PARPi [71]. Topoisomerase IIβ-binding protein 1 (TOPBP1) was essential for RAD51 phosphorylation at serine 14, which was necessary for RAD51 recruitment on chromatin and formation of RAD51 foci. Absent of TOPBP1 abrogated the HR and increased sensitivity of ovarian cancer cells to olaparib [72]. Bromodomain protein 4 (BRD4) is a kind of key chromosomal regulator of genome stability. The inhibition of BRD4 recruited RAD51 accumulation without activation of ATM/ATR-dependent DNA damage response [73]. It was mentioned that BRD4 was amplified in various cancer [74]. Growing evidence suggested that BRD4 inhibitors (JQ1, INCB054329) sensitized to PARPi and expanded the utility of PARPi in clinic [74,75,76,77]. In human lung adenocarcinoma (LUAD) tumors, patients expressing high HORMAD1 exhibited elevated mutational burden and poor survival. HORMAD1 were enriched for genes essential HR and promoted RAD51 filament formation. Accordingly, high expression of HORMAD1 contributed to PARPi resistance [78]. APRIN and PALB2 preferentially bind to D-loop structures and directly interact with RAD51 to stimulate strand invasion and promote HR. It has been shown that deletion of APRIN and PALB2 induced “BRCAness” and sensitized cells to PARPi [79, 80]. Moreover, Pol δ played vital roles in D-loop extension and inhibition of Pol δ also enhanced the sensitivity of HR-proficient cancer cells to PARPi [81].
Reversion mutations in PARPi resistance
In 2008, the influence of reversion mutations on PARPi resistance was independently discovered by two groups. Ashworth et al derived PARPi-resistant clones by deleting the BRCA2 c.6174delT frameshift mutation of human CAPAN1 pancreatic cancer cell line, a BRCA2-deficient cell line. Consequently, the reconstituted BRCA2-deficient cells acquired PARPi resistance [82]. Meanwhile, Sakai et al demonstrated that secondary mutations restored the wild-type BRCA2 reading frame was a major clinical mediator of acquired resistance to platinum and PARPi [83]. By using liquid biopsy or circulating cell-free DNA (cfDNA), lots of BRCA reversion mutations have been discovered to restore the open reading frame (ORF) of BRCA1/2 and confer resistance to PARPi-based therapy in various cancers [84,85,86,87,88,89,90] (Table 1).
Full length BRCA1 consists of N-terminal domains (BRCT), N-terminal RING domain and coiled-coil domain. BRCT is responsible for binding phosphorylated proteins such as CtIP. N-terminal RING domain can stabilize BRCA1 and ensure the E3 ligase activity [91]. Multiple evidence suggested that reversion mutations, which restored the functions of BRCT and N–terminal RING domain, played essentials roles in PAPRi resistance [92,93,94]. In addition, cancer cells lacking the exon 11 of BRCA1 promoted partial PARPi resistance [95]. BRCA2 contains a DNA-binding domain and eight BRC repeats that bind to RAD51 and mediates the recruitments of RAD51 and strand exchange in HR [91]. It was suggested that each BRC repeats was divided into two categories and only BRC 1–4 bound to RAD51 with high affinity and enhanced DNA strand exchange while BRC 5–8 bound to RAD51 with low affinity and did not affect DNA strand exchange [96]. However, an in vitro study indicated that BRCA2 mutations lacking BRC 6–8 also lead to PARPi resistance [82]. Recently, two reversion mutations (c.4434_5686delinsTT and c.4897_6807del) produced truncated BRCA2 protein were thought to be competent in conferring PARPi resistance [89]. In addition to reversion mutations in BRCA1/2, Secondary somatic mutations restoring Rad51C and Rad51D were also demonstrated to be associated with acquired resistance to the PARPi [84]. With the development of gene editing, CRISPR-Cas9 screens were recently used to identify point mutations in PARP1 conferring PARPi resistance. Several mutations in PARP1 including p.R591C and p.848delY, were identified to cause PARPi resistance. More importantly, the CRISPR-Cas9 “tad-mutate-enrich” mutagenesis screens approach could be employed in the analysis of other gene mutations [97].
Taken together with the growing body of data identifying reversion mutations in PARPi resistance, it seems to be the most well-validated mechanism of PARPi resistance in BRCAm cancer patients. However, we must notice that whether the reversion mutations are induced by PARPi itself or other anticancer drugs or even spontaneous is unclear. After all, cancer cells harboring BRCA mutations prefer to NHEJ repair, which lead to accumulation of genetic aberrations and increased risk of reversion mutations. Moreover, before or even during treating with PARPi-based therapy, other anticancer drugs, such as platinum, were also administered to patients, which invisibly make the study more difficult to investigate the influence of PARPi-based therapy on secondary mutation in clinic.
Furthermore, the frequency of reversion mutations occurred among patient population is still known. Recently, the prevalence of BRCA reversion mutations in metastatic castration-resistant prostate cancer (mCRPC) was estimated. By using a large genomic database, 24 gBRCAm carriers were selected from 1534 patients with mCRPC underwent ctDNA testing. At the time of the blood draw, 5 of these 24 patients were given either a PARP inhibitor or platinum-based chemotherapy. Two patients, one receiving olaparib and one carboplatin, had BRCA2 reversion mutations. Therefore, in this germline mutation–positive, platinum- or PARP-exposed cohort, the frequency of BRCA2 reversion mutations was 40% [98]. However, another clinical trial result showed that 8 of 97 HGSOC patients with gBRCAm or sBRCAm (8.2%) were identified to have BRCA reversion mutations before treating with rucaparib. After treating with rucaparib, only 8 of 78 postprogression patients had BRCA reversion mutations and the occurrence rate of reversion mutations was only 10.3% [99]. All these results reflected that the BRCA reversion mutations might be different in various cancers. Due to the small sample size, additional studies with more patients and various cancers are needed to carry out.
Protection of DNA replication fork in PARPi resistance
In addition to DNA repair. PARP1 and BRCA1/2 participate in DNA replication. PARP1 has a key role in mediating the accumulation of regressed forks and avoiding an untimely restart of reversed forks, leading to DSB formation [100]. Both BRCA1 and BRCA2 protect nascent DNA at stalled replication forks from MRE11/DNA2-dependent degradation [101, 102]. When PARP inhibitors trap PARP on DNA to block DNA replication, cells will rely on BRCA1/2 to stabilize their stalled replication forks and prevent them from being extensively degraded by nucleases (i.e., MRE11, DNA2, MUS81). As BRCA1/2 is defective, the absence of DNA replication forks protection leads to genome instability and cell death [103] (Fig. 3). Recently, more and more evidence suggested that DNA replication fork protection but not HRR caused PARPi resistance in BRCAm cells and patients, which challenged the HR dominance in synthetic lethality (Fig. 3). Rondinelli et al. showed that low EZH2 levels reduced H3K27 methylation, prevented MUS81 recruitment at stalled forks and caused fork stabilization, which promoted PARPi resistance in BRCA2-deficient cells but not in BRCA1-deficient cells [104]. Besides, Ray et al. demonstrated that PTIP, MELL3/4 and CHD4 deficiency did not restore HR activity at DSB. Instead, their absence inhibited the recruitment of the MRE11 nuclease to stalled replication forks and protected nascent DNA strands from extensive degradation, which in turn lead to acquisition of PARPi resistance in BRCA2-deficient cells [105]. FANCD2 suppresses MRE11-mediated fork degradation in a manner dependent on nucleoprotein filaments and plays an important role in the stabilization of stalled replication forks [106]. It’s reported that FANCD2 was highly expressed in BRCA1/2-mutated breast cancer, ovarian cancers and uterine cancers. FANCD2 overexpression conferred resistance to PARPi in BRCA1/2-mutated breast cancer cell lines [107, 108]. Due to the DNA translocase activity, SMARCAL1, a member of SNF2 family, could reverse the nascent DNA degradation induced by FANCD2 deficiency in BRCA1/2-mutated breast cancer cells. It promoted the formation of ssDNA gaps at replication forks and reversed forks catalyzed by SMARCAL1 was prone to be degraded by MRE11. More importantly, its deletion promoted PARPi and cisplatin resistance [109]. In addition to SMARCAL1, the SNF2-famlily DNA translocases ZRANB3 and HLTF exhibited fork-remodeling activities similar to SMARCAL1, indicating that they might be associated with PARPi resistance as well [110]. RADX deletion restored fork protection but not HR by regulating RAD51 at replication forks and conferred PARPi resistance in BRCA2-mutated cancer cell lines [111]. These collective results refocus our PARPi resistance spotlight onto fork protection, which might make significant contributions to PARPi resistance [112]. Consequently, it might provide us a novel strategy in considering the future cancer therapy.

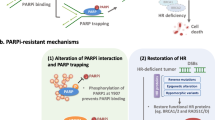
Schematic describing the function of PARP1 and BRCA1/2 in protection of DNA replication fork and mechanisms of protection of DNA replication fork leading to PARPi resistance. a PARPi trap PARP1 on DNA and cause fork stalling. After fork stalling, PRA is phosphorylated and ssDNA is coated by PRA rapidly. Then, RAD51 replaces RPA and mediates replication fork reversal. The revered fork can be degraded by MRE11 and MUS81. BRCA1/2 is relied on to protect nascent DNA replication forks from degradation. EZH2 induces H3K27 methylation and MUS81 recruitment at stalled forks. MELL3/4 induces H3K4 methylation increases the accumulation of PTIP, which leads to the recruitment of MRE11. FANCD2 suppresses MRE11-mediated fork degradation, which can be reversed by SMARCAL1. RADX blocks the recruitment of RAD51 at replication forks. b In BRCA1/2-deficient cells, low expression of PARP1, RADX, SMARCAL1, EZH2, PTIP, MELL3/4 and high expression of FANCD2 confer resistance of PARPi
Stalled replication forks are a major source of genome instability in proliferating cells, which need to be stabilized or restarted to promote cell survival. Through decades’ efforts, multitude of mechanisms were found to protect stalled replication forks to preserve genome stability under replication stress. Except for the pathways mentioned above, RecQ helicases and pathways involved in ATR/CHK1-dependent checkpoint activation also play essential roles in replication fork protection and genome stability maintenance [103]. Therefore, they might function as part of mechanisms of PARPi resistance. However, there is no relevant preclinical and clinical studies up to now, which are expected to be taken into consideration in the future.
Epigenetic modification, restoration of PARylation and pharmacological alteration in PARPi resistance
Epigenetic modification may affect PARPi sensitivity and lead to PARPi resistance. Multiple lines of treatment prior PARPi lead to loss of BRCA1 promoter methylation, which rescued the expression of BRCA1 and conferred resistance of PARPi [113]. MiR-622 and miR-493-5p induced PARPi resistance by suppressing NHEJ reapir and impacting multiple pathways pertinent to genome stability, respectively [114, 115]. Deubiquitination of BARD1 BRCT domain by USP15 assisted BRCA1 retention at DSBs and causes PARPi resistance [116]. Moreover, similar to deletion of 53BP1, acetylation of 53 bp1 inhibited NHEJ and promoted HR by negatively regulating 53 bp1 recruitment to DSBs, which made BRCA1-deficient cells acquire resistance to PARPi [117]. The role of N6-methyladenosine (m6A) modification in PARPi resistance was recently explored. Even though that there was no difference in total m6A-modified mRNA between parental and PARPi-resistant ovarian cancer PEO1 cells, the increased expression and N6-methylation modification of FZD10 were confirmed in resistant PEO1 cells. FZD10 contributed to PARPi resistance by upregulating the Wnt/β-catenin pathway [118].
As is known to us, PARPi kill tumor cells via PARPs activity inhibition and PARP trapping. PARPs activity increase and restoration of PARPylation are responsible to PARPi resistance. Phosphorylation of PARP1 at Tyr907, mediated by c-Met, increased PARP1 enzymatic activity and reduced its binding to PARPi, thereby rendering cancer cells resistant to PARPi [119]. By combing genetic screens with multi-omics analysis of matched PARP-sensitive and -resistance BRCA2-mutated mouse mammary tumors, PAR glycohydrolase (PARG) was found, the loss of which resulted in restoring PARylation formation and PARPi resistance [120]. Furthermore, the expression of PARP1 was significantly associated with PARPi toxicity. It has been revealed that both cells with low expression of PARP1 and cells harboring PARP1 LOF mutations were more resistant to PARPi [121, 122].
Pharmacological alteration also modulates PARPi inhibitor response. PARPi are substrates of multidrug resistance protein (MDR1, P-gp), encoded by ABCB1 gene [123]. Both in vivo and in vitro studies indicated the enhanced P-gp-mediated drug efflux contributed to the acquired resistance to PARPi [124, 125]. What’s more, the resistance could be reversed by coadministration of the P-gp inhibitors or genetic inactivation of P-gp [42, 123,124,125]. The overexpression of ABCB1 might be induced by long-term treating with PARPi but the mechanisms are still unclear. Compared to other factors, the weight of contribution in pharmacological changes to PARPi resistance in clinic is uncertain. More and more researches are needed to uncover the underlying mechanisms.
Clinical implications towards PARPi resistance
To enhance PARPi sensitivity and overcome PARPi resistance, several feasible strategies should be considered and implemented in the future (Table 2): 1) PARPi-oncolytic herpes simplex viruses (oHSVs) combination; oHSVs, approved by FDA for recurrent melanoma, are genetically engineered to selectively kill cancer cells, due to their characteristics of amplifying and spreading within the tumor but not normal tissue. They are actively involved in manipulating DDR [126]. Recently, MG18L, a newly identified activity of oHSV, was reported to proteasomally degrade RAD51 and sensitize glioblastoma stem cells (GSCs) to PARPi killing in synthetic lethal-like fashion in vivo and in vitro. The combination of olaparib with MG18L greatly increased survival in both PARPi-sensitive and -resistant GSC-derived tumors. The combination therapy not only overcomes PARPi resistance but also expands its use to tumors with HR-proficient. Most importantly, oHSVs only infect and kill tumor cells but not normal cells compared to conventional medicines, which means that they may have fewer side effects [127]. Due to its broad anti-tumor efficacy in most solid tumors, this novel combination therapy should be applicable to other cancer stem cells and tumors; 2) PARPi-ionizing radiation (IR) combination; Nuclear localization is required for BRCA1 to participate in HR-mediated DNA repair [128]. IR can initiate the export of BRCA1 from the nucleus to the cytoplasm, leading to increased sensitivity of PARPi in wild-type BRCA1 and HR-proficient tumor cells [129, 130] However, because of the synthetic lethality of the combination therapy is p53-depend, it can only be used in wild-type p53 patients [131]. Meanwhile, PARPi induce radiosensitization in vitro and in vivo models [132]. What’s even more refreshing is HR restoration by 53BP1 pathway inactivation further increased radiosensitivity in preclinical model systems. It was showed that BRCA1-mutated tumors, which acquired drug resistance due to BRCA1-independent HR restoration, could be sensitized to radiotherapy [133]. In addition to the preclinical results, clinical studies were also attempted to exploit the efficacy of PARPi-IR combination. A phase 1, open-label dose escalation study (NCT00649207) evaluating veliparib in combination with whole brain radiation therapy (WBRT) in patients with brain metastases were originated with Mehta and his colleagues [134]. The preliminary efficacy results were better than predicted outcome based on the graded prognostic factors in the published nomogram. Based on encouraging safety and preliminary efficacy results, a randomized, controlled phase 2b study is ongoing. Other two phase 1 trials (NCT01264432, NCT01589419) indicated that the PARPi-IR combination treatment was well-tolerated and show good responses as well [135, 136]. Undoubtedly, further evaluation of PARPi-IR combination treatments is currently underway in multiple phase 2 clinical trials in patients with NSCLC and breast cancer (NCT02412371, NCT01386385, NCT01618357). 3) PARPi-CDKs inhibitors; DNA end resection is depended on cyclin-dependent kinases (CDKs) activity. A number of studies indicated that CDKs played important roles in PARPi resistance [36,37,38,39,40,41]. CDK inhibitor dinaciclib resensitized TBNC cells, which had acquired resistance to niraparib. In addition to TBNC cells, synthetic lethal strategy combining dinaciclib with niraparib was also highly efficacious in ovarian, prostate, pancreatic, colon, and lung cancer cells [137]. Currently, CDK12 attracted more attentions in PARPi resistance, due to its inactivating somatic alterations were recurrently observed in various cancers. Numerous evidences proved that CDK12 mutation or deficiency lead to cancer cells sensitivity to PARPi [37]. Furthermore, CDK12 inhibitors reversed de novo and acquired PARPi resistance in BRCA1-mutant breast cancer cells [39]. 4) PARPi-immunotherapy; Jiao et al and her colleagues revealed that PARPi upregulated PD-L1 expression in breast cancer cell lines via inactivating GSK3β, which in return leading to attenuate anticancer immunity. Moreover, the combination of PARPi and anti-PD-L1 therapy showed better therapeutic efficacy than each alone [138]. PARPi-mediated modulation of the immune response contributes to their therapeutic effects independently of BRCA1/2 mutations. Recently results suggested that PARPi promoted accumulation of cytosolic DNA fragments because of unresolved DNA lesions, which in turn activated the DNA-sensing cGAS-STING pathway and stimulated production of type I interferons to induce antitumor immunity independent of BRCAness [139]. At present, several clinical trials (NCT02734004, NCT03824704 and NCT02849496) are ongoing. In this term, all these trails may be informative. 5) PARPi-epigenetic drugs; As previously mentioned, epigenetic modification was associated with PARPi sensitivity [113, 117, 118]. Acetylation and deacetylation of histones is one of the most important mechanisms of posttranslational regulation of gene expression [140]. So far, numerous studies have declared that treating with histone deacetylation inhibitors (HDACi) and PARPi exhibited synergy effects due to the induction of HDACi on HRD, which as a result sensitized cancer cells to PARPi [141,142,143,144]. Several mechanisms have been observed. Firstly, it was reported that HDACi decreased the expression of DNA repair genes such as RAD51, CHK1, BRCA1 and RAD21 mediated through transcription factor E2F1 [145]. Secondly, HDACi blocked the deacetylation and expression of HSP90, resulting in the degradation of its substrates BRCA1, Rad52, ATR and CHK1 [146]. Finally, recent studies showed that acetylation blocked DNA damage-induced chromatin PARylation and HDACi treatment significantly increased the trapping of PARP1 at DSB sites in chromatin [147, 148]. Additionally, low doses of DNA methyltransferase inhibitor (DNMTi) induced BRCAness phenotype through downregulating expression of key HR genes [149]. The combination DNMTi and PARPi enhanced the cytotoxic effect by increasing the PARP “trapping” on DSB sites independent on BRCA mutations [150, 151]. However, there is no clinical trial to evaluate its effect until now. 6) PARPi-other drugs; In addition to the above mentioned, PARPi was also suggested to combinate with HSP90 inhibitors, ATR/CHK1 inhibitors and WEE1 inhibitors [152, 153]. BRCA1 function is reliant on HSP90. HSP90 inhibitor, 17-AAG, could induce HRD and increase Olaparib sensitivity of HR-proficient ovarian cancer cell lines [154]. Treating PARPi-resistant cells with 7-dimethylaminoethylamino-17-demethoxygeldanamycin, a HSP90 inhibitor, reversed the resistance state by decreasing the quantity of BRCA1 protein [92]. ATR/CHK1 and WEE1 have emerged as putative BRCAness factors that function in both checkpoint activation and in replication fork stability. ATR/CHK1 inhibitors and WEE1 inhibitors treatment were recently shown to reverse PARPi resistance in cancer cells [152]. Currently, several trails to the safety and efficacy of these combination treatments in sporadic cancers are in progress (NCT03579316, NCT04197713, NCT02576444, NCT02511795, NCT04065269, NCT03787680, NCT03330847, NCT03878095, NCT03462342, NCT03428607, NCT03682289). In a word, the combination therapy to overcome PARPi resistance and enhance PARPi sensitivity is still in its infancy and has a long way to go. More and more studies are needed to investigate the feasibility in clinic.
Conclusions and perspectives
In the past few decades, PARPi was successfully developed in treating BRCA mutation patients, which provided proof-of concept that synthetic lethal interactions could be translated into cancer therapy. However, the preclinical and clinical investigation of PRARi is far from complete. In terms of PARPi resistance, multiple potential resistance mechanisms, such as HR restoration and protection of DNA replication fork have been identified. Nonetheless, the contribution weight of them to PARPi resistance is incomprehensible. Recently, the PRIMA trial results suggested that among patients with newly diagnosed advanced ovarian cancer who had a response to platinum-based chemotherapy, those who received niraparib had significantly longer progression-free survival (PFS) than those who received placebo, regardless of the presence or absence of HRD [155]. Based on it, we assumed that PARPi might kill cancer cells in ways other than DNA repair. The association between PARPi resistance and protection of DNA replication fork confirmed this conjecture. Therefore, we should comprehensively understand how PARPi functions, especially, how do the roles of PARPi in processes unrelated to DNA repair influence the anti-cancer activity of PARPi, which would be conductive to understand the development of resistance. Also, to overcome PARPi resistance and increase PARPi sensitivity, the optimal combination of PARPi and other treatment regimens are urgently needed to identify.
In addition to PARPi resistance, a serious of unanswered questions that could guide the optimal use of PARPi in the future, are not addressed. For example, what other proteins beyond BRCA1 and BRCA2 contribute to the efficacy of PARPi? Currently, PTEN has received a lot of attention as a promising biomarker to predicting the sensitivity of PARPi. PTEN is one of the tumor suppressor genes most frequently inactivated in human cancers [156]. It is reported that loss of PTEN lead to HRD, increased genomic instability and replication fork collapse [157,158,159]. At present, there is a growing body of preclinical evidence that tumors with loss of PTEN function are defective in HR and may, therefore, be hypersensitive to PARPi [159,160,161]. Likewise, there are lots of conflicting results that PTEN deficiency has no effect on PARPi sensitivity [162,163,164]. In a word, vulnerabilities of PTEN-deficient sporadic cancers to PARP inhibition remain controversial.
Besides, due to additional biological process beyond HR related to sensitivity of PARPi, we need to redefine the concert of concept of “BRCAness” and exploit new techniques of companion diagnostics to predict the response of patients to PARPi [24, 152]. Current BRCAnalysis assay could not effectively identify BRCAness. For example, genomic scars of BRCAness, as they are currently measured, probably reflect the alteration of the genome in the absence of HR over the entire lifetime of a tumor, they might not provide an accurate estimation of whether HR is still defective in tumor cells at the time that treatment is delivered. Other proposed approaches such as the use of mRNA expression signatures and the individual analysis of genetic alterations in HR-related genes are both lack of specificity. RAD51 accumulation and the formation of RAD51-ssDNA play key roles in both HR and protection of stalled DNA replication fork, therefore, RAD51 assay may be feasible in identifying PARPi-sensitive cancer patients and broadening the population who may be response to PRAPi-based therapy.
In conclusion, if all these issues can be figured out, we firmly believe that a substantial subset of cancer patients could benefit from PARPi.
Availability of data and materials
All the data obtained and/or analyzed during the current study were available from the corresponding authors on reasonable request.
Abbreviations
- PARP:
-
Poly (ADP-ribose) polymerase
- PARPi:
-
PARP inhibitor
- HR:
-
Homologous recombination
- HRD:
-
Homologous recombination repair deficient
- PARylation:
-
ADP-ribosylation
- DDR:
-
DNA damage response
- NHEJ:
-
Non-homologous end joining repair
- SSBR:
-
Single stranded break repair
- SSB:
-
Single-strand breaks
- BER:
-
Base excision repair
- gBRCAm:
-
Germline mutations in BRCA1/2
- sBRCAm:
-
Somatic mutations of BRCA1/2
- HGSOC:
-
High-grade serious ovarian cancer
- BRCAm:
-
Mutation of BRCA1/2
- MRN:
-
Mre11-Rad50-Nbs1
- ssDNA:
-
Single-strand DNA
- CDKs:
-
Cyclin-dependent kinases
- LOF:
-
Loss-of functions
- TNBC:
-
Triple-negative breast cancer
- LUAD:
-
Lung adenocarcinoma
- cfDNA:
-
Circulating cell-free DNA
- ORF:
-
Open reading frame
- BRCT:
-
N-terminal domains of BRCA1
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- m6A:
-
N6-methyladenosine
- PARG:
-
PAR glycohydrolase
- MDR1:
-
Multidrug resistance protein
- oHSVs:
-
Oncolytic herpes simplex viruses
- GSCs:
-
Glioblastoma stem cells
- IR:
-
Ionizing radiation
- WBRT:
-
Whole brain radiation therapy
- HDACi:
-
Histone deacetylation inhibitors
- DNMTi:
-
DNA methyltransferase inhibitor
- PFS:
-
Progression-free survival
References
Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16:35–42.
Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen. 2017;58:235–63.
Langelier M-F, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–75.
Eustermann S, Wu W-F, Langelier M-F, Yang J-C, Easton LE, Riccio AA, Pascal JM, Neuhaus D. Structural basis of detection and signaling of DNA single-Strand breaks by human PARP-1. Mol Cell. 2015;60:742–54.
Langelier M-F, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl) ation by human PARP-1. Science (New York, NY). 2012;336:728–32.
Bétermier M, Bertrand P, Lopez BS. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014;10:e1004086.
Jiang X, Li X, Li W, Bai H, Zhang Z. PARP inhibitors in ovarian cancer: sensitivity prediction and resistance mechanisms. J Cell Mol Med. 2019;23:2303–13.
Heyer W-D, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–39.
Gudmundsdottir K, Ashworth A. The roles of BRCA1 and BRCA2 and associated proteins in the maintenance of genomic stability. Oncogene. 2006;25:5864–74.
Isono M, Niimi A, Oike T, Hagiwara Y, Sato H, Sekine R, Yoshida Y, Isobe S-Y, Obuse C, Nishi R, et al. BRCA1 directs the repair pathway to homologous recombination by promoting 53BP1 Dephosphorylation. Cell Rep. 2017;18:520–32.
Sy SMH, Huen MSY, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci U S A. 2009;106:7155–60.
Hanenberg H, Andreassen PR. PALB2 (partner and localizer of BRCA2). Atlas Genet Cytogenet Oncol Haematol. 2018;22:484–90.
Nielsen FC, van Overeem HT, Sørensen CS. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer. 2016;16:599–612.
Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7.
Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–8.
Friedlander M, Gebski V, Gibbs E, Davies L, Bloomfield R, Hilpert F, Wenzel LB, Eek D, Rodrigues M, Clamp A, et al. Health-related quality of life and patient-centred outcomes with olaparib maintenance after chemotherapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov-21): a placebo-controlled, phase 3 randomised trial. Lancet Oncol. 2018;19:1126–34.
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, Scott C, Meier W, Shapira-Frommer R, Safra T, et al. Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol. 2016;17:1579–89.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, Colombo N, Weberpals JI, Clamp A, Scambia G, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, Fabbro M, Ledermann JA, Lorusso D, Vergote I, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian Cancer. N Engl J Med. 2016;375:2154–64.
Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al. Olaparib for metastatic breast Cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33.
Robson ME, Tung N, Conte P, Im SA, Senkus E, Xu B, Masuda N, Delaloge S, Li W, Armstrong A, et al. OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician's choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30:558–66.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16:110–20.
Bouwman P, Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540–7.
Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, De Greve J, Lubinski J, Shanley S, Messiou C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9.
Audeh MW, Carmichael J, Penson RT, Friedlander M, Powell B, Bell-McGuinn KM, Scott C, Weitzel JN, Oaknin A, Loman N, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51.
Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell. 2012;47:320–9.
Cejka P. DNA end resection: nucleases team up with the right partners to initiate homologous recombination. J Biol Chem. 2015;290:22931–8.
Liu VF, Weaver DT. The ionizing radiation-induced replication protein a phosphorylation response differs between ataxia telangiectasia and normal human cells. Mol Cell Biol. 1993;13:7222–31.
Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–33.
Godin SK, Sullivan MR, Bernstein KA. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem Cell Biol. 2016;94:407–18.
Hustedt N, Durocher D. The control of DNA repair by the cell cycle. Nat Cell Biol. 2016;19:1–9.
Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu Rev Genet. 2011;45:247–71.
Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature. 2009;459:460–3.
Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, Burma S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun. 2014;5:3561.
Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 2014;74:287–97.
Joshi PM, Sutor SL, Huntoon CJ, Karnitz LM. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J Biol Chem. 2014;289:9247–53.
Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. CDK12 inhibition reverses De novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast Cancer. Cell Rep. 2016;17:2367–81.
Ning JF, Stanciu M, Humphrey MR, Gorham J, Wakimoto H, Nishihara R, Lees J, Zou L, Martuza RL, Wakimoto H, Rabkin SD. Myc targeted CDK18 promotes ATR and homologous recombination to mediate PARP inhibitor resistance in glioblastoma. Nat Commun. 2019;10:2910.
Militello AM, Zielli T, Boggiani D, Michiara M, Naldi N, Bortesi B, Zanelli P, Uliana V, Giuliotti S, Musolino A. Mechanism of action and clinical efficacy of CDK4/6 inhibitors in BRCA-mutated, estrogen receptor-positive breast cancers: case report and literature review. Front Oncol. 2019;9:759.
Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81.
Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M, et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 2015;521:541–4.
Chapman JR, Barral P, Vannier J-B, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–71.
Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JTF, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49:872–83.
Wang Y-T, Yuan B, Chen H-D, Xu L, Tian Y-N, Zhang A, He J-X, Miao Z-H. Acquired resistance of phosphatase and tensin homolog-deficient cells to poly(ADP-ribose) polymerase inhibitor and Ara-C mediated by 53BP1 loss and SAMHD1 overexpression. Cancer Sci. 2018;109:821–31.
Hurley RM, Wahner Hendrickson AE, Visscher DW, Ansell P, Harrell MI, Wagner JM, Negron V, Goergen KM, Maurer MJ, Oberg AL, et al. 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol Oncol. 2019;153:127–34.
Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry M-C, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–4.
Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, Olivieri M, Álvarez-Quilón A, Moatti N, Zimmermann M, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–21.
Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, Typas D, Lammers M, Mailand N, Nussenzweig A, et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell. 2018;173:972–88 e923.
Dev H, Chiang T-WW, Lescale C, de Krijger I, Martin AG, Pilger D, Coates J, Sczaniecka-Clift M, Wei W, Ostermaier M, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–65.
Clairmont CS, Sarangi P, Ponnienselvan K, Galli LD, Csete I, Moreau L, Adelmant G, Chowdhury D, Marto JA, D'Andrea AD. TRIP13 regulates DNA repair pathway choice through REV7 conformational change. Nat Cell Biol. 2020;22:87–96.
Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–4.
Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–5.
Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJS, Smith G, Powell JE, Rudzki Z, Kearns P, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87.
Schmitt A, Knittel G, Welcker D, Yang T-P, George J, Nowak M, Leeser U, Büttner R, Perner S, Peifer M, Reinhardt HC. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. 2017;77:3040–56.
Bang Y-J, Im S-A, Lee K-W, Cho JY, Song E-K, Lee KH, Kim YH, Park JO, Chun HG, Zang DY, et al. Randomized, double-blind phase II trial with prospective classification by ATM protein level to evaluate the efficacy and tolerability of Olaparib plus paclitaxel in patients with recurrent or metastatic gastric Cancer. J Clin Oncol. 2015;33:3858–65.
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N, et al. DNA-repair defects and Olaparib in metastatic prostate Cancer. N Engl J Med. 2015;373:1697–708.
Bang Y-J, Xu R-H, Chin K, Lee K-W, Park SH, Rha SY, Shen L, Qin S, Xu N, Im S-A, et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1637–51.
Burdova K, Storchova R, Palek M, Macurek L. WIP1 promotes homologous recombination and modulates sensitivity to PARP inhibitors. Cells. 2019;8:1258.
Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G, Gruber SB. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011;71:2632–42.
He YJ, Meghani K, Caron M-C, Yang C, Ronato DA, Bian J, Sharma A, Moore J, Niraj J, Detappe A, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563:522–6.
Li Z, Li J, Kong Y, Yan S, Ahmad N, Liu X. Plk1 phosphorylation of Mre11 antagonizes the DNA damage response. Cancer Res. 2017;77:3169–80.
Liu W, Zhou M, Li Z, Li H, Polaczek P, Dai H, Wu Q, Liu C, Karanja KK, Popuri V, et al. A selective small molecule DNA2 inhibitor for sensitization of human Cancer cells to chemotherapy. EBioMedicine. 2016;6:73–86.
Misenko SM, Patel DS, Her J, Bunting SF. DNA repair and cell cycle checkpoint defects in a mouse model of 'BRCAness' are partially rescued by 53BP1 deletion. Cell Cycle. 2018;17:881–91.
Tkáč J, Xu G, Adhikary H, Young JTF, Gallo D, Escribano-Díaz C, Krietsch J, Orthwein A, Munro M, Sol W, et al. HELB is a feedback inhibitor of DNA end resection. Mol Cell. 2016;61:405–18.
Liu Y, Burness ML, Martin-Trevino R, Guy J, Bai S, Harouaka R, Brooks MD, Shang L, Fox A, Luther TK, et al. RAD51 mediates resistance of Cancer stem cells to PARP inhibition in triple-negative breast Cancer. Clin Cancer Res. 2017;23:514–22.
Castroviejo-Bermejo M, Cruz C, Llop-Guevara A, Gutiérrez-Enríquez S, Ducy M, Ibrahim YH, Gris-Oliver A, Pellegrino B, Bruna A, Guzmán M, et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol Med. 2018;10:e9172.
Cruz C, Castroviejo-Bermejo M, Gutiérrez-Enríquez S, Llop-Guevara A, Ibrahim YH, Gris-Oliver A, Bonache S, Morancho B, Bruna A, Rueda OM, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol. 2018;29:1203–10.
Marzio A, Puccini J, Kwon Y, Maverakis NK, Arbini A, Sung P, Bar-Sagi D, Pagano M. The F-Box Domain-Dependent Activity of EMI1 Regulates PARPi Sensitivity in Triple-Negative Breast Cancers. Mol Cell. 2019;73:224–37 e226.
Zhao L, Si CS, Yu Y, Lu JW, Zhuang Y. Depletion of DNA damage binding protein 2 sensitizes triple-negative breast cancer cells to poly ADP-ribose polymerase inhibition by destabilizing Rad51. Cancer Sci. 2019;110:3543–52.
Moudry P, Watanabe K, Wolanin KM, Bartkova J, Wassing IE, Watanabe S, Strauss R, Troelsgaard Pedersen R, Oestergaard VH, Lisby M, et al. TOPBP1 regulates RAD51 phosphorylation and chromatin loading and determines PARP inhibitor sensitivity. J Cell Biol. 2016;212:281–8.
Bowry A, Piberger AL, Rojas P, Saponaro M, Petermann E. BET Inhibition Induces HEXIM1- and RAD51-Dependent Conflicts between Transcription and Replication. Cell Rep. 2018;25:2061–9 e2064.
Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, Wang Y, Fan L, Tang Z, Li C, et al. Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci Transl Med. 2017;9:eaal1645.
Miller AL, Fehling SC, Garcia PL, Gamblin TL, Council LN, van Waardenburg R, Yang ES, Bradner JE, Yoon KJ. The BET inhibitor JQ1 attenuates double-strand break repair and sensitizes models of pancreatic ductal adenocarcinoma to PARP inhibitors. EBioMedicine. 2019;44:419–30.
Wilson AJ, Stubbs M, Liu P, Ruggeri B, Khabele D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol Oncol. 2018;149:575–84.
Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell. 2018;33:401–16 e408.
Nichols BA, Oswald NW, McMillan EA, McGlynn K, Yan J, Kim MS, Saha J, Mallipeddi PL, LaDuke SA, Villalobos PA, et al. HORMAD1 is a negative prognostic Indicator in lung adenocarcinoma and specifies resistance to oxidative and genotoxic stress. Cancer Res. 2018;78:6196–208.
Couturier AM, Fleury H, Patenaude AM, Bentley VL, Rodrigue A, Coulombe Y, Niraj J, Pauty J, Berman JN, Dellaire G, et al. Roles for APRIN (PDS5B) in homologous recombination and in ovarian cancer prediction. Nucleic Acids Res. 2016;44:10879–97.
Buisson R, Dion-Côté AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17:1247–54.
Mishra B, Zhang S, Zhao H, Darzynkiewicz Z, Lee EYC, Lee M, Zhang Z. Discovery of a novel DNA polymerase inhibitor and characterization of its antiproliferative properties. Cancer Biol Ther. 2019;20:474–86.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7:984–98.
Simmons AD, Nguyen M, Pintus E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: a case report. BMC Cancer. 2020;20:215.
Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, Brody JR. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br J Cancer. 2017;116:1021–6.
Gornstein EL, Sandefur S, Chung JH, Gay LM, Holmes O, Erlich RL, Soman S, Martin LK, Rose AV, Stephens PJ, et al. BRCA2 reversion mutation associated with acquired resistance to Olaparib in estrogen receptor-positive breast Cancer detected by genomic profiling of tissue and liquid biopsy. Clin Breast Cancer. 2018;18:184–8.
Mayor P, Gay LM, Lele S, Elvin JA. BRCA1 reversion mutation acquired after treatment identified by liquid biopsy. Gynecol Oncol Rep. 2017;21:57–60.
Tao H, Liu S, Huang D, Han X, Wu X, Shao YW, Hu Y. Acquired multiple secondary BRCA2 mutations upon PARPi resistance in a metastatic pancreatic cancer patient harboring a BRCA2 germline mutation. Am J Transl Res. 2020;12:612–7.
Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, Assiotis I, Rodrigues DN, Reis Filho JS, Moreno V, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229:422–9.
Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12:68–78.
Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110:17041–6.
Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, Kim HH, George E, Swisher EM, Simpkins F, Johnson N. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest. 2016;126:3145–57.
Wang Y, Bernhardy AJ, Nacson J, Krais JJ, Tan YF, Nicolas E, Radke MR, Handorf E, Llop-Guevara A, Balmaña J, et al. BRCA1 intronic Alu elements drive gene rearrangements and PARP inhibitor resistance. Nat Commun. 2019;10:5661.
Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, van der Gulden H, van der Heijden I, O'Brien SW, et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76:2778–90.
Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci U S A. 2011;108:10448–53.
Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, Harrell MI, Menon M, Brough R, Campbell J, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9:1849.
Carneiro BA, Collier KA, Nagy RJ, Pamarthy S, Sagar V, Fairclough S, Odegaard J, Lanman RB, Costa R, Taxter T, et al. Acquired resistance to poly (ADP-ribose) polymerase inhibitor Olaparib in BRCA2-associated prostate Cancer resulting from Biallelic BRCA2 reversion mutations restores both germline and somatic loss-of-function mutations. JCO Precis Oncol. 2018;10:e9172.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, Helman E, Radke MR, Say C, Vo LT, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor Rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9:210–9.
Berti M, Ray Chaudhuri A, Thangavel S, Gomathinayagam S, Kenig S, Vujanovic M, Odreman F, Glatter T, Graziano S, Mendoza-Maldonado R, et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat Struct Mol Biol. 2013;20:347–54.
Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell. 2011;145:529–42.
Schlacher K, Wu H, Jasin M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell. 2012;22:106–16.
Liao H, Ji F, Helleday T, Ying S. Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018;19:e46263.
Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S, D'Andrea AD. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–8.
Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–7.
Kim TM, Son MY, Dodds S, Hu L, Luo G, Hasty P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015;43:893–903.
Kais Z, Rondinelli B, Holmes A, O'Leary C, Kozono D, D'Andrea AD, Ceccaldi R. FANCD2 maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA repair. Cell Rep. 2016;15:2488–99.
Michl J, Zimmer J, Buffa FM, McDermott U, Tarsounas M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat Struct Mol Biol. 2016;23:755–7.
Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell. 2017;68:414–30 e418.
Bétous R, Couch FB, Mason AC, Eichman BF, Manosas M, Cortez D. Substrate-selective repair and restart of replication forks by DNA translocases. Cell Rep. 2013;3:1958–69.
Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, Wessel SR, Sathe AA, Zhao R, Cortez D. RADX Promotes Genome Stability and Modulates Chemosensitivity by Regulating RAD51 at Replication Forks. Mol Cell. 2017;67:374–86 e375.
Schlacher K. PARPi focus the spotlight on replication fork protection in cancer. Nat Cell Biol. 2017;19:1309–10.
Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9:3970.
Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA, Elias KM, Drapkin R, Weinstock DM, Dao F, et al. Platinum and PARP inhibitor resistance due to overexpression of MicroRNA-622 in BRCA1-mutant ovarian Cancer. Cell Rep. 2016;14:429–39.
Meghani K, Fuchs W, Detappe A, Drané P, Gogola E, Rottenberg S, Jonkers J, Matulonis U, Swisher EM, Konstantinopoulos PA, Chowdhury D. Multifaceted impact of MicroRNA 493-5p on genome-stabilizing pathways induces platinum and PARP inhibitor resistance in BRCA2-mutated carcinomas. Cell Rep. 2018;23:100–11.
Peng Y, Liao Q, Tan W, Peng C, Hu Z, Chen Y, Li Z, Li J, Zhen B, Zhu W, et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat Commun. 2019;10:1224.
Guo X, Bai Y, Zhao M, Zhou M, Shen Q, Yun CH, Zhang H, Zhu WG, Wang J. Acetylation of 53BP1 dictates the DNA double strand break repair pathway. Nucleic Acids Res. 2018;46:689–703.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, Liu P, Kossenkov AV, Showe LC, Jean S, et al. N(6)-methylation of adenosine of FZD10 mRNA contributes to PARP inhibitor resistance. Cancer Res. 2019;79:2812–20.
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee GR, et al. Blocking c-met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22:194–201.
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S, et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018;33:1078–93 e1012.
Pettitt SJ, Rehman FL, Bajrami I, Brough R, Wallberg F, Kozarewa I, Fenwick K, Assiotis I, Chen L, Campbell J, et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS One. 2013;8:e61520.
Liu X, Han EK, Anderson M, Shi Y, Semizarov D, Wang G, McGonigal T, Roberts L, Lasko L, Palma J, et al. Acquired resistance to combination treatment with temozolomide and ABT-888 is mediated by both base excision repair and homologous recombination DNA repair pathways. Mol Cancer Res. 2009;7:1686–92.
Christie EL, Pattnaik S, Beach J, Copeland A, Rashoo N, Fereday S, Hendley J, Alsop K, Brady SL, Lamb G, et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat Commun. 2019;10:1295.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, Derksen PW, de Bruin M, Zevenhoven J, Lau A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84.
Oplustilova L, Wolanin K, Mistrik M, Korinkova G, Simkova D, Bouchal J, Lenobel R, Bartkova J, Lau A, O'Connor MJ, et al. Evaluation of candidate biomarkers to predict cancer cell sensitivity or resistance to PARP-1 inhibitor treatment. Cell Cycle. 2012;11:3837–50.
Kohlhapp FJ, Kaufman HL. Molecular pathways: mechanism of action for Talimogene Laherparepvec, a new oncolytic virus immunotherapy. Clin Cancer Res. 2016;22:1048–54.
Ning J, Wakimoto H, Peters C, Martuza RL, Rabkin SD. Rad51 degradation: role in oncolytic virus-poly(ADP-ribose) polymerase inhibitor combination therapy in glioblastoma. J Natl Cancer Inst. 2017;109:1–13.
Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA damage-induced cytotoxicity is dissociated from BRCA1's DNA repair function but is dependent on its cytosolic accumulation. Cancer Res. 2010;70:6258–67.
Jiang J, Yang ES, Jiang G, Nowsheen S, Wang H, Wang T, Wang Y, Billheimer D, Chakravarthy AB, Brown M, et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res. 2011;71:5546–57.
Yang ES, Nowsheen S, Rahman MA, Cook RS, Xia F. Targeting BRCA1 localization to augment breast tumor sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2012;72:5547–55.
Sizemore ST, Mohammad R, Sizemore GM, Nowsheen S, Yu H, Ostrowski MC, Chakravarti A, Xia F. Synthetic lethality of PARP inhibition and ionizing radiation is p53-dependent. Mol Cancer Res. 2018;16:1092–102.
Chalmers A, Johnston P, Woodcock M, Joiner M, Marples B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int J Radiat Oncol Biol Phys. 2004;58:410–9.
Barazas M, Gasparini A, Huang Y, Küçükosmanoğlu A, Annunziato S, Bouwman P, Sol W, Kersbergen A, Proost N, de Korte-Grimmerink R, et al. Radiosensitivity is an acquired vulnerability of PARPi-resistant BRCA1-deficient tumors. Cancer Res. 2019;79:452–60.
Mehta MP, Wang D, Wang F, Kleinberg L, Brade A, Robins HI, Turaka A, Leahy T, Medina D, Xiong H, et al. Veliparib in combination with whole brain radiation therapy in patients with brain metastases: results of a phase 1 study. J Neuro-Oncol. 2015;122:409–17.
Reiss KA, Herman JM, Zahurak M, Brade A, Dawson LA, Scardina A, Joffe C, Petito E, Hacker-Prietz A, Kinders RJ, et al. A phase I study of veliparib (ABT-888) in combination with low-dose fractionated whole abdominal radiation therapy in patients with advanced solid malignancies and peritoneal carcinomatosis. Clin Cancer Res. 2015;21:68–76.
Czito BG, Deming DA, Jameson GS, Mulcahy MF, Vaghefi H, Dudley MW, Holen KD, DeLuca A, Mittapalli RK, Munasinghe W, et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: a phase 1b study. Lancet Gastroenterol Hepatol. 2017;2:418–26.
Carey JPW, Karakas C, Bui T, Chen X, Vijayaraghavan S, Zhao Y, Wang J, Mikule K, Litton JK, Hunt KK, Keyomarsi K. Synthetic lethality of PARP inhibitors in combination with MYC blockade is independent of BRCA status in triple-negative breast Cancer. Cancer Res. 2018;78:742–57.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. PARP inhibitor upregulates PD-L1 expression and enhances Cancer-associated immunosuppression. Clin Cancer Res. 2017;23:3711–20.
Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G. PARPi triggers the STING-dependent immune response and enhances the therapeutic efficacy of immune checkpoint blockade independent of BRCAness. Cancer Res. 2019;79:311–9.
Audia JE, Campbell RM. Histone modifications and Cancer. Cold Spring Harb Perspect Biol. 2016;8:a019521.
Jasek E, Gajda M, Lis GJ, Jasińska M, Litwin JA. Combinatorial effects of PARP inhibitor PJ34 and histone deacetylase inhibitor vorinostat on leukemia cell lines. Anticancer Res. 2014;34:1849–56.
Baldan F, Mio C, Allegri L, Puppin C, Russo D, Filetti S, Damante G. Synergy between HDAC and PARP inhibitors on proliferation of a human anaplastic thyroid Cancer-derived cell line. Int J Endocrinol. 2015;2015:978371.
Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, et al. Histone deacetylase inhibitor treatment induces 'BRCAness' and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5:5637–50.
Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, Kim TY, Han SW, Oh DY, Kim TY, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015;17:33.
Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M, Shabbeer S, Mendonca J, Deangelis J, Marchionni L, et al. Downregulation of homologous recombination DNA repair genes by HDAC inhibition in prostate cancer is mediated through the E2F1 transcription factor. PLoS One. 2010;5:e11208.
Kim Y, Kim A, Sharip A, Sharip A, Jiang J, Yang Q, Xie Y. Reverse the resistance to PARP inhibitors. Int J Biol Sci. 2017;13:198–208.
Liszczak G, Diehl KL, Dann GP, Muir TW. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat Chem Biol. 2018;14:837–40.
Robert C, Nagaria PK, Pawar N, Adewuyi A, Gojo I, Meyers DJ, Cole PA, Rassool FV. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk Res. 2016;45:14–23.
Abbotts R, Topper MJ, Biondi C, Fontaine D, Goswami R, Stojanovic L, Choi EY, McLaughlin L, Kogan AA, Xia L, et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc Natl Acad Sci U S A. 2019;116:22609–18.
Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, Lyons J, Baylin SB, Matei D, Nakshatri H, et al. An effective epigenetic-PARP inhibitor combination therapy for breast and ovarian cancers independent of BRCA mutations. Clin Cancer Res. 2018;24:3163–75.
Muvarak NE, Chowdhury K, Xia L, Robert C, Choi EY, Cai Y, Bellani M, Zou Y, Singh ZN, Duong VH, et al. Enhancing the cytotoxic effects of PARP inhibitors with DNA Demethylating agents - a potential therapy for Cancer. Cancer Cell. 2016;30:637–50.
Byrum AK, Vindigni A, Mosammaparast N. Defining and modulating 'BRCAness'. Trends Cell Biol. 2019;29:740–51.
Dréan A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85.
Choi YE, Battelli C, Watson J, Liu J, Curtis J, Morse AN, Matulonis UA, Chowdhury D, Konstantinopoulos PA. Sublethal concentrations of 17-AAG suppress homologous recombination DNA repair and enhance sensitivity to carboplatin and olaparib in HR proficient ovarian cancer cells. Oncotarget. 2014;5:2678–87.
González-Martín A, Pothuri B, Vergote I, DePont CR, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. Niraparib in patients with newly diagnosed advanced ovarian Cancer. N Engl J Med. 2019;381:2391–402.
Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7.
Wang G, Li Y, Wang P, Liang H, Cui M, Zhu M, Guo L, Su Q, Sun Y, McNutt MA, Yin Y. PTEN regulates RPA1 and protects DNA replication forks. Cell Res. 2015;25:1189–204.
Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128:157–70.
McEllin B, Camacho CV, Mukherjee B, Hahm B, Tomimatsu N, Bachoo RM, Burma S. PTEN loss compromises homologous recombination repair in astrocytes: implications for glioblastoma therapy with temozolomide or poly(ADP-ribose) polymerase inhibitors. Cancer Res. 2010;70:5457–64.
Dedes KJ, Wetterskog D, Mendes-Pereira AM, Natrajan R, Lambros MB, Geyer FC, Vatcheva R, Savage K, Mackay A, Lord CJ, et al. PTEN deficiency in endometrioid endometrial adenocarcinomas predicts sensitivity to PARP inhibitors. Sci Transl Med. 2010;2:53ra75.
Forster MD, Dedes KJ, Sandhu S, Frentzas S, Kristeleit R, Ashworth A, Poole CJ, Weigelt B, Kaye SB, Molife LR. Treatment with olaparib in a patient with PTEN-deficient endometrioid endometrial cancer. Nat Rev Clin Oncol. 2011;8:302–6.
Fraser M, Zhao H, Luoto KR, Lundin C, Coackley C, Chan N, Joshua AM, Bismar TA, Evans A, Helleday T, Bristow RG. PTEN deletion in prostate cancer cells does not associate with loss of RAD51 function: implications for radiotherapy and chemotherapy. Clin Cancer Res. 2012;18:1015–27.
Sandhu SK, Schelman WR, Wilding G, Moreno V, Baird RD, Miranda S, Hylands L, Riisnaes R, Forster M, Omlin A, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14:882–92.
Bian X, Gao J, Luo F, Rui C, Zheng T, Wang D, Wang Y, Roberts TM, Liu P, Zhao JJ, Cheng H. PTEN deficiency sensitizes endometrioid endometrial cancer to compound PARP-PI3K inhibition but not PARP inhibition as monotherapy. Oncogene. 2018;37:341–51.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural Science Foundation of China (NO.81972836) and National Key R&D Program (2016YFC1303703).
Author information
Authors and Affiliations
Contributions
He Li and Jing Wang conceived the structure of manuscript and revised the manuscript. Zhao-Yi Liu and He Li made the figures and table. All authors revised the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors give consent for the publication of manuscript in Molecular Cancer.
Competing interests
The authors declare that there is no potential competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, H., Liu, ZY., Wu, N. et al. PARP inhibitor resistance: the underlying mechanisms and clinical implications. Mol Cancer 19, 107 (2020). https://doi.org/10.1186/s12943-020-01227-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01227-0