
Abstract
Background
Poly (ADP-ribose) polymerase (PARP) inhibitors are approved for the treatment of breast cancer susceptibility genes 1 and 2 (BRCA1/2) mutant ovarian and breast cancers, and are now being evaluated in metastatic castration-resistant prostate cancer (mCRPC). Reversion mutations that restore BRCA1/2 function have been shown to be responsible for resistance to platinum-based chemotherapy and PARP inhibitors, however there is no information on the sequential use of these agents in prostate cancer.
Case presentation
A patient with mCRPC associated with a germline BRCA2 mutation was sequentially treated with carboplatin and the PARP inhibitor rucaparib. Genomic profiling of the available baseline tumor and progression blood samples using next-generation sequencing panel tests identified polyclonal BRCA2 reversion mutations post carboplatin treatment but prior to rucaparib treatment. A total of 12 somatic reversion mutations were detected and ranged from small indels to larger deletions of up to 387 amino acids. These alterations are all predicted to restore the BRCA2 open reading frame and potentially protein function. The patient received limited benefit while on rucaparib, likely due to these reversion mutations observed prior to treatment.
Conclusions
Here we report a case of a patient with prostate cancer who received a platinum agent and PARP inhibitor sequentially and in whom polyclonal BRCA2 reversion mutations were identified as the likely mechanism of acquired resistance to carboplatin and primary resistance to PARP inhibition. These findings suggest caution is warranted in sequencing these agents.
Similar content being viewed by others

Background
Results from the phase 2 TOPARP study (NCT01682772) suggest that the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib has activity in men with metastatic castration-resistant prostate cancer (mCRPC) who have a deleterious alteration in a DNA damage repair gene, such as BRCA2 [1]. Recently, preliminary results of the TRITON2 study (NCT02952534) showed that 52 and 44% of evaluable mCRPC patients with a deleterious BRCA1/2 mutation had a prostate-specific antigen (PSA) response and Response Evaluation Criteria In Solid Tumors response, respectively, when treated with the PARP inhibitor rucaparib [2]. Based on these encouraging results, the U.S. Food and Drug Administration granted Breakthrough Therapy designation to both olaparib and rucaparib in mCRPC, and there are many ongoing studies evaluating these and other PARP inhibitors in patients with prostate cancer.
PARP inhibitors have been approved for the treatment of BRCA1/2 mutant ovarian and breast cancers. A key mechanism of resistance to PARP inhibitors and platinum-based chemotherapy in these cancers is the acquisition of reversion mutations in BRCA1/2 that restore protein function [3, 4]. Reversion mutations in BRCA2 have also been observed in a small number of mCRPC patients treated with PARP inhibitors or carboplatin [5,6,7,8]. Acquired reversion mutations in BRCA1/2 resulting from exposure to platinum chemotherapy are likely to render tumors less sensitive to PARP inhibitor treatment. In a recent study of patients with ovarian cancer treated with rucaparib following platinum, patients without BRCA1/2 reversion mutations had a significantly longer median progression-free survival than patients with reversion mutations (9.0 vs. 1.8 months; hazard ratio, 0.12; P < 0.0001) [3]. However, there are limited data on the combination or sequential use of platinum and PARP inhibitors in prostate cancer.
In this manuscript, we describe a patient with mCRPC and a germline BRCA2 mutation who was sequentially treated with carboplatin and the PARP inhibitor rucaparib. We profiled the available baseline tumor and progression blood samples using next-generation sequencing panel tests and identified polyclonal BRCA2 reversion mutations post carboplatin treatment but prior to rucaparib treatment. The patient received limited benefit while on rucaparib, likely due to these reversion mutations observed prior to treatment.
Case presentation
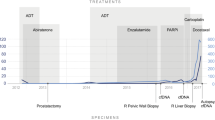
In May 2016, a 58-year-old patient presented with hematuria and rectal tenesmus. Baseline staging showed prostate cancer invading the mesorectum, pelvic lymphadenopathies, and high-volume bone metastases (T4N1M1); his serum PSA was 136 ng/mL, and his alkaline phosphatase (ALP) was 1106 IU/L (Fig. 1). A prostatic biopsy revealed a Gleason’s 5 + 5 prostate adenocarcinoma. His comorbidities included moderate aortic stenosis, left ventricular hypertrophy, left atrial dilatation, diabetes, hypercholesterolemia, and vitiligo. His Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) was 1.

Clinical treatment course and PSA and ALP responses. Treatment and duration of treatment are denoted as arrows or colored areas, and time of sampling as diamonds. ALP, alkaline phosphatase; LHRH, luteinizing hormone-releasing hormone; PSA, prostate-specific antigen; RT, palliative radiotherapy
In June 2016, he commenced on luteinizing hormone-releasing hormone agonists with bicalutamide cover (PSA, 20 ng/mL; ALP, 1567 IU/L) and received his first cycle of docetaxel chemotherapy. In October 2016, docetaxel was discontinued after four cycles due to clinical and biochemical progression. Serum PSA was 41 ng/mL and ALP was 292 IU/L. In November 2016, the patient started on enzalutamide and shortly after received palliative radiotherapy to the lumbosacral spine and started zoledronic acid for prevention of skeletal-related events. He had a marked response to enzalutamide in terms of pain control and PSA and ALP decline (Fig. 1) until August 2017, when due to bone-related pain and PSA and ALP rise, treatment was stopped.
From August to November 2017, the patient received six cycles of second-line cabazitaxel chemotherapy, which were discontinued due to clinical and radiological progression. His ECOG Performance Status for the first time since his diagnosis declined to 2. Based on family history and the aggressive clinical behavior of the disease, in January 2018 he commenced third-line carboplatin chemotherapy (area under the concentration-time curve 5). His initial PSA and ALP levels were 24 ng/mL and 113 IU/L and reached a nadir of 10 ng/mL and 85 IU/L, respectively. Chemotherapy allowed better pain control and improved general condition. He received a total of six cycles of carboplatin, the last given in April 2018. Chemotherapy was discontinued for symptomatic progression and PSA progression, despite a stable ALP level (91 IU/L).
In May 2018, molecular testing was performed on the prostatic sample taken in June 2016 to determine if the patient was eligible for clinical trials. FoundationONE CDx (version T7) testing [9] identified a deleterious BRCA2 c.5727_5728insG (N1910fs*2) mutation in the original tumor biopsy (Fig. 2). This alteration was later confirmed to be a germline pathogenic variant in BRCA2 by Hereditary Cancer Solution testing. Based on published data suggesting that PARP inhibitors are active in patients with BRCA1/2-mutant mCRPC [1], in June 2018 the patient commenced on rucaparib 600 mg twice daily (BID) under a compassionate use program due to the lack of an approved standard of care or access to a clinical trial at that time. A baseline plasma sample for circulating tumor DNA (ctDNA) analysis was collected prior to the patient starting rucaparib and profiled using the FoundationACT assay [10]. In addition to the germline BRCA2 alteration, 12 other BRCA2 alterations were also observed. Six of the alterations were in close proximity (within ≈10 amino acids) of the original alteration (Fig. 2a, Table 1). All six alterations reestablished the BRCA2 open reading frame (ORF) by substitutions or short in-frame deletions. Five additional alterations were longer in-frame deletions ranging from 46 to 386 amino acids, four of which resulted in partial or complete loss of the BRC repeat sequences BRC5, BRC6, BRC7, and/or BRC8 (Fig. 2b). These alterations are also predicted to restore the BRCA2 ORF. The final BRCA2 alteration extended across the exon/intron border: nucleotides 5333–6841 of the coding region and the first 197 nucleotides of the intron (5333_6841 + 197del1706; Table 1). The 1509 base pair deletion within the coding region (6841–5333 + 1) would potentially remove the original mutation as part of an in-frame 503 amino acid deletion.

BRCA2 reversion mutations. Schematic of small indel (a) and large deletion mutations (b) detected. BRC repeats, interacting regions, and sequences are represented as yellow, blue, and orange boxes, respectively. Substitutions and deletions are represented as red text and black lines, respectively
The FoundationACT assay reported the variant allele fraction (VAF) for the detected alterations (Fig. 3, Table 1). The VAF for the baseline BRCA2 N1910fs*2 mutation was 83.7%, consistent with the confirmed germline alteration. The VAF of the reversion mutations ranged from 0.11–4.8%, with a total of 14.9%. A CDKN2A P114L alteration was observed at a VAF of 10.0%.

Graph of variant allele fractions for BRCA2 and CDKN2A mutations. BRCA2 reversion mutations are represented in legend
The patient received rucaparib 600 mg BID for a total of 47 days from June to July 2018. His general condition gradually deteriorated. Following hospital admission with sepsis and uncontrolled back pain, imaging confirmed disease progression (new nodal, pulmonary, and hepatic lesions), and rucaparib was discontinued permanently.
Discussion and conclusions
We report a case of a patient with mCRPC and a germline truncating mutation in BRCA2 who developed 12 different somatic reversion mutations that restored the protein ORF and would be expected to render the tumor insensitive to platinum-based chemotherapy or PARP inhibitor treatment. Consistent with this hypothesis, the patient had a limited response to subsequent treatment with rucaparib.
Although no definitive conclusions can be made due to the limited sampling, it is likely that the reversion mutations resulted from the 4-month course of carboplatin, as the reversion mutations were not detected in the tumor tissue sample obtained at primary diagnosis. We are also not aware of any reports describing de novo BRCA2 reversion mutations prior to platinum-based chemotherapy or PARP inhibitor treatment. The emergence of reversion mutations in BRCA1/2 has been associated with platinum drugs based on their mechanism of action of forming DNA-platinum adducts that leads to DNA lesions [11], whereas it has not been reported in patients treated with taxanes. Although anecdotal, it is remarkable that our patient developed reversion mutations after such a limited exposure to platinum, which suggests a different genomic or biological context in inducing secondary mutations among patients with prostate cancer compared to those with ovarian cancer.
All of the reversion mutations would result in unique, non-wild-type proteins that would restore the C-terminal end of BRCA2, including the DNA binding domains, the tower domain, oligonucleotide/oligosaccharide-binding folds, and nuclear localization sequence. However, several of the mutations resulted in large BRCA2 deletions (up to 387 amino acids), encompassing one or more of the BRC repeats BRC5–8. This region is known to stabilize the RAD51 filament and promote homologous recombination repair upon DNA damage [12]. A previous report suggests that BRC5–8 deletion may confer partial resistance to the DNA damaging agent mitomycin C using BRCA2-mutant cell lines [13]. Although the functional consequences of each of the reversion mutations cannot be determined without additional investigation, it is likely that many or all of them restore BRCA2 function.
The VAF for the BRCA2 reversion mutations ranged from 0.11 to 4.8%, totaling 14.9% overall. Although the limited activity observed with rucaparib cannot definitively be attributed to these low allele frequency reversion mutations, the identification of polyclonal reversion mutations in prostate cancer patients is consistent with that in other reports [5,6,7,8] and highlights the strong selective pressure to restore BRCA2 function. It is not possible to determine if these alterations are clonal or multiple reversion alterations in a single tumor cell, because shedding may not be similar from each tumor deposit. Interestingly, the patient’s PSA levels remained stable (ranging between 27 and 31 ng/L) throughout rucaparib treatment, indicating that perhaps not all tumor clones contained a reversion mutation and some were responding to treatment.
Another acquired alteration detected in ctDNA following carboplatin treatment was a CDKN2A P114L variant with a VAF of 10%, suggesting that it may have been a somatic tumor-specific variant. The CDKN2A gene encodes the p16(INK4A) and p14(ARF) proteins, which both function as tumor suppressors [14]. The P114L (c.341C > T) loss-of-function mutation would prevent p16 from inhibiting CDK4 and inducing cell cycle arrest [15]. CDKN2A is commonly altered in patients with metastatic melanoma and cutaneous squamous cell carcinoma [16, 17]. Although CDKN2A mutations are rarely observed in prostate cancer, a recent case study reported a CDKN2A P81L mutation (which would render the protein functionally defective) as the proposed mechanism underlying acquired resistance to enzalutamide in a patient with CRPC [18]. The emergence of the CDKN2A P114L alternation in our patient could have occurred during enzalutamide treatment. However, because the ctDNA sample was obtained after multiple therapies, including carboplatin and cabazitaxel, we cannot rule out the possibility that other agents may have caused the emergence of the CDKN2A mutation. Upregulation of cell-cycle pathway observed in patients resistant to enzalutamide along with genomic aberrations in the cell-cycle pathway observed in patients with prostate cancer (such as RB1 loss and CCDN1 amplification) suggest the potential importance of cell-cycle kinases in the development of prostate carcinoma and resistance to enzalutamide [19].
We acknowledge several limitations of our study regarding correlation to patient response. First, the patient’s ECOG PS at the start of rucaparib treatment was 2, and there are limited data on the effectiveness of PARP inhibitors in patients with an ECOG PS > 1. In addition, the patient was exposed to rucaparib for only 6 weeks. Although there is limited information on the time required for mCRPC patients to demonstrate a tumour response to a PARP inhibitor, it has previously been reported that 76% (19/25) of patients with a BRCA1/2 alteration treated with rucaparib in the TRITON2 trial had a radiographic response within 8 weeks of starting rucaparib [2].
The PARP inhibitor rucaparib is currently being evaluated in patients with mCRPC, where it has shown encouraging antitumor activity. An important question is how to use PARP inhibitors, as well as platinum-based chemotherapies, to maximize the clinical benefit in patients with mCRPC. This case study suggests that caution may be warranted in sequencing these agents.
Availability of data and materials
All available data generated or analyzed during this study are included in this published article.
Abbreviations
- ALP:
-
Alkaline phosphatase
- BID:
-
Twice daily
- BRCA1 :
-
Breast cancer susceptibility gene 1
- BRCA2 :
-
Breast cancer susceptibility gene 2
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- ctDNA:
-
Circulating-tumor DNA
- ECOG:
-
Eastern Cooperative Oncology Group
- LHRH:
-
Luteinizing hormone-releasing hormone
- mCRPC:
-
Metastatic castration-resistant prostate cancer
- ORF:
-
Open reading frame
- PARP:
-
Poly (ADP-ribose) polymerase
- PSA:
-
Prostate-specific antigen
- RT:
-
Palliative radiotherapy
- VAF:
-
Variant allele fraction
References
Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–708.
Abida W, Campbell D, Patnaik A, et al. Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)-deficient metastatic castration-resistant prostate cancer (mCRPC): Updated analyses. Ann Oncol. 2019;30(suppl 5):abst 846PD.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–9.
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W, et al. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol. 2011;29(22):3008–15.
Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006–17.
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7(9):999–1005.
Carneiro BA, Collier KA, Nagy RJ, Pamarthy S, Sagar V, Fairclough S, et al. Acquired resistance to poly (ADP-ribose) polymerase inhibitor olaparib in BRCA2-associated prostate cancer resulting from biallelic BRCA2 reversion mutations restores both germline and somatic loss-of-function mutations. JCO Precis Oncol. 2018;2:1–8.
Cheng HH, Salipante SJ, Nelson PS, Montgomery B, Pritchard CC. Polyclonal BRCA2 reversion mutations detected in circulating tumor DNA after platinum chemotherapy in a patient with metastatic prostate cancer. JCO Precis Oncol. 2018;2:1–5.
Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31(11):1023–31.
Clark TA, Chung JH, Kennedy M, Hughes JD, Chennagiri N, Lieber DS, et al. Analytical validation of a hybrid capture-based next-generation sequencing clinical assay for genomic profiling of cell-free circulating tumor DNA. J Mol Diagn. 2018;20(5):686–702.
Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–21.
Carreira A, Kowalczykowski SC. Two classes of BRC repeats in BRCA2 promote RAD51 nucleoprotein filament function by distinct mechanisms. Proc Natl Acad Sci U S A. 2011;108(26):10448–53.
Chatterjee G, Jimenez-Sainz J, Presti T, Nguyen T, Jensen RB. Distinct binding of BRCA2 BRC repeats to RAD51 generates differential DNA damage sensitivity. Nucleic Acids Res. 2016;44(11):5256–70.
Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of genetic and epigenetic alterations of CDKN2A (p16(INK4a)) in cancer. EBioMedicine. 2016;8:30–9.
Koh J, Enders GH, Dynlacht BD, Harlow E. Tumour-derived p16 alleles encoding proteins defective in cell-cycle inhibition. Nature. 1995;375(6531):506–10.
Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J Invest Dermatol. 2004;122(5):1284–92.
Helgadottir H, Ghiorzo P, van Doorn R, Puig S, Levin M, Kefford R, et al. Efficacy of novel immunotherapy regimens in patients with metastatic melanoma with germline CDKN2A mutations. J Med Genet. 2018. [Epub ahead of print]. https://doi.org/10.1136/jmedgenet-2018-105610.
Han GC, Hwang J, Wankowicz SAM, Zhang Z, Liu D, Cibulskis C, et al. Genomic resistance patterns to second-generation androgen blockade in paired tumor biopsies of metastatic castration-resistant prostate cancer. JCO Precis Oncol. 2017;1:1–11.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162(2):454.
Acknowledgments
We thank Linda Kelly and Catherine Smith for patient care, as well as Simon Watkins for his assistance in providing rucaparib for this patient. We appreciate Cheryl Chun of Clovis Oncology for her help in reviewing the manuscript. We thank the patient’s family for giving their consent to publish his case. Copy editing assistance funded by Clovis Oncology was provided by Shannon Davis of Ashfield Healthcare Communications (Middletown, CT, USA).
Funding
We would like to thank Clovis Oncology for funding the analysis of the ctDNA samples and Oxford University Hospitals NHS Foundation Trust for funding the germline analysis of the tumor DNA.
Author information
Authors and Affiliations
Contributions
EP was the treating physician, conceived of the case, and collected and assembled the data. ADS, MN, and EP analyzed and interpretated the data, drafted and revised the manuscript, and read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Patient written informed consent has been obtained. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
ADS and MN are employees of Clovis Oncology. EP reports a consulting or advisory role for Clovis Oncology; honoraria from Clovis Oncology, Astellas, Bristol-Myers Squibb, and Janssen; and travel support from Clovis Oncology, Astellas, Ipsen, and Janssen.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Simmons, A.D., Nguyen, M. & Pintus, E. Polyclonal BRCA2 mutations following carboplatin treatment confer resistance to the PARP inhibitor rucaparib in a patient with mCRPC: a case report. BMC Cancer 20, 215 (2020). https://doi.org/10.1186/s12885-020-6657-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-6657-2