
Abstract
Poly (ADP-ribose) polymerase (PARP) inhibitors (PARPi) exploit the concept of synthetic lethality and offer great promise in the treatment of tumors with deficiencies in homologous recombination (HR) repair. PARPi exert antitumor activity by blocking Poly(ADP-ribosyl)ation (PARylation) and trapping PARP1 on damaged DNA. To date, the U.S. Food and Drug Administration (FDA) has approved four PARPi for the treatment of several cancer types including ovarian, breast, pancreatic and prostate cancer. Although patients with HR-deficient tumors benefit from PARPi, majority of tumors ultimately develop acquired resistance to PARPi. Furthermore, even though BRCA1/2 mutations are commonly used as markers of PARPi sensitivity in current clinical practice, not all patients with BRCA1/2 mutations have PARPi-sensitive disease. Thus, there is an urgent need to elucidate the molecular mechanisms of PARPi resistance to support the development of rational effective treatment strategies aimed at overcoming resistance to PARPi, as well as reliable biomarkers to accurately identify patients who will most likely benefit from treatment with PARPi, either as monotherapy or in combination with other agents, so called marker-guided effective therapy (Mget). In this review, we summarize the molecular mechanisms driving the efficacy of and resistance to PARPi as well as emerging therapeutic strategies to overcome PARPi resistance. We also highlight the identification of potential markers to predict PARPi resistance and guide promising PARPi-based combination strategies.
Similar content being viewed by others

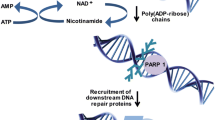

Background
The Poly (ADP-ribose) polymerase (PARP) family is essential for regulation of many critical cellular processes, including DNA damage response, transcription, chromatin remodeling, metabolism and inflammation [1,2,3]. To date, 17 members have been identified in the PARP family based on their homology to PARP1, the most well-characterized PARP protein which is responsible for more than 80% of Poly(ADP-ribosyl) (PAR) activity in the cell [4]. The well-known function of PARP1 is to initiate DNA repair by inducing PARylation, one of the post-translational modifications, in other proteins and itself [5]. PARP1 contains three functional domains including the DNA-binding domain, automodification domain and catalytic domain. The first two Zinc fingers in DNA-binding domain are critical for the binding of PARP1 on DNA damage sites and the third Zinc finger plays a key role in alteration of DNA-dependent PARP1 enzyme activity [6,7,8]. The glutamate and lysine residues in the central automodification domain are the acceptor sites for PARP1 to PARylate itself [9, 10]. Importantly, the catalytic domain in C-terminal of PARP1 is responsible for the transfer of ADP-ribose subunits from nicotinamide adenine dinucleotide (NAD+) to protein substrates and building up the negatively charged PAR chains [11]. The PARP1-mediated PARylation serve as a platform for recruiting the downstream repair proteins for repair DNA breaks [12]. Additionally, auto-PARylation of PARP1 is a critical step for successfully completing DNA repair and preventing the replication fork collapse caused by PARP1 trapped on damaged DNA [12]. In 2000s, the scientific focus on PARP1 transitioned from validating its molecular functions to identifying its physiological and pathological role in human cancer [13]. In addition, significantly increased expression of PARP1 and PARylation have been detected in malignant tumors of various cancer types [14, 15]. Based on these findings, PARP1 became the attractive therapeutic target for the treatment of cancer. Of note, remarkable studies in 2005 demonstrated that PARP inhibition selectively kills the BRCA1/2 mutant tumor cells [16, 17], leading to the rapid clinical development of PARP inhibitors (PARPi) for patients with homologous recombination (HR)-deficient cancer. PARPi are the nicotinamide analogs which compete with NAD+ for the catalytic binding sites on PARP molecules to inhibit the PARylatioin and induce PARP trapping activity [18]. Currently, there are four PARPi approved by the U.S. Food and Drug Administration (FDA) for the treatment of different types of cancer. Although these PARPi have promising clinical activity through prolonging the survival of a board population of cancer patients, resistance to PARPi remains a significant clinical challenge. Therefore, a better understanding of mechanisms of resistance to PARPi and identification of reliable biomarkers to predict PARPi resistance are necessary for the development of marker-guided effective therapy (Mget) to overcome PARPi resistance.
BRCA1/2 mutation and defective homologous recombination repair in cancer
Defects in many of genes encoding DNA repair proteins are commonly identified in human cancer [19]. Compromised DNA damage repair pathways in cancer cells result in genomic instability and leads to cancer development [20]. Of note, two tumor suppressor genes with critical roles in double-strand break (DSB) repair, BRCA1 and BRCA2, are frequently mutated or deleted in several cancer types [21]. In 1990, a geneticist, Mary-Claire King discovered the BRCA1 gene locus and its linkage to hereditary breast and ovarian cancer [22]. In 1994, the BRCA1 gene was cloned [23], and in the same year BRCA2 was also identified [24]. Till now, numerous studies have demonstrated that mutations of BRCA1/2 increase the lifetime risk of breast or ovarian cancer development. Specifically, a healthy woman who harbor germline mutations of BRCA1/2 have a 60–70% increased risk of breast cancer development and a 15–40% increased risk to develop ovarian cancer [25, 26]. Mutations of BRCA1/2 genes have also been found in many sporadic tumors including pancreatic [27, 28] and prostate cancer [29, 30]. In addition to BRCA1/2 aberrations, other genes involved in HR repair such as RAD51C, RAD51D, PALB2 and BRIP1, are known to be mutated in many cancer types [31]. HR repair is a critical process for repairing the most cytotoxic DNA lesions, DSBs. When DSBs occur, ATM kinase is activated by the MRN complex (MRE11, RAD50 and NBN) and then phosphorylates the down-stream effectors including BRCA1 to promote the HR activity [32,33,34,35]. BRCA1 is a key regulator required for generating single-strand DNA (ssDNA) and recruiting the PALB2-BRCA2 complex. With the help of the PALB2-BRCA2 complex at DNA repair sites, replication protein A (RPA) is displaced and then replaced by the RAD51 recombinase [36]. The assembly of RAD51 filaments promotes homology sequence searching and base pairing to accurate repair [37]. Dysfunction of HR repair resulting from these HR gene mutations leads to less effective, error-prone non-homologous end joining (NHEJ) repair and gives rise to severe chromosomal instability that is associated with tumor development. Moreover, alterations of essential HR repair factors can result in phenotypic features similar to those caused by BRCA1/2 mutations, giving rise to the term “BRCAness” [38]. Although the defects of DSB repair machinery due to BRCAness phenotype is associated with a higher risk of developing breast and ovarian cancer, patients with these tumors benefit from therapeutic strategies aimed at targeting the compromised DNA repair pathways to kill tumor cells through the accumulation of unrepaired DNA damage.
Synthetic lethality between HR-deficiency and PARP inhibition
HR-deficient tumors have been found to be highly sensitive to DNA damage drugs such as platinum-based chemotherapy, which is frequently used as a part of the standard of care for patients with ovarian cancer. Two landmark studies published in 2005 [16, 17], first described the synthetic lethal interaction between BRCA1/2-deficiency and inhibition of PARP, offering a promising new approach over conventional chemotherapy for patients with HR-deficient tumors. PARP1 is a nuclear protein regulating base excision repair (BER) through PARylation [1]. Following DNA damage and sense of ssDNA breaks, PARP1 binds to the DNA damage sites and induces PARylation events to recruit multiple downstream DNA repair factors [12]. During this recruiting process, PARP1 auto-PARylates itself for releasing DNA-bounded PARP1 and allowing the DNA repair proteins to access and complete DNA repair [12]. Thus, inhibition of PARP1 results in the accumulation of unrepaired ssDNA break and replication fork collapse, which subsequently induce DSBs during DNA replication [17]. The persistence of DSBs is normally repaired by HR repair in the S phase of cell cycle [39]. BRCA1 and BRCA2 are the essential factors in regulating the HR repair pathway which is the largely error-free repair of DSBs [40]. For tumor cells with BRCA1/2- or HR- deficiency, PARP1 activity is important for preventing the spontaneous ssDNA breaks that results in accumulation of DSBs. Therefore, pharmacological inhibition of PARP selectively kills HR-deficient tumor cells by inducing the genomic instability and cell cycle arrest, ultimately leads to the synthetic lethality between PARP inhibition and HR deficiency. Together, these findings provide mechanistic insight and rationale for targeting compensatory DNA repair pathways as therapeutic strategies in cancer.
FDA approval of PARP inhibitors
Currently, four small-molecular PARPi have been approved by FDA for tumors with BRCA1/2 mutation or HR deficiency, including olaparib, rucaparib, talazoparib and niraparib (Table 1). Olaparib was the first PARP inhibitor approved for patients with BRCA1/2 mutant, advanced-stage ovarian cancers in 2014. Of note, retrospective analysis of results from a clinical trial demonstrated that olaparib also improve progression-free survival (PFS) in the patients with BRCA1/2-wild type ovarian cancer [41]. These data suggest that the expanded biomarkers are needed for identifying patients with BRCA1/2-wild type tumors who might benefit from PARPi maintenance therapy. PARPi maintenance therapy is the ongoing treatment of tumors with PARP inhibitor after tumor has responded to the first-line treatments of chemotherapies. In 2017 to 2018, niraparib [42], olaparib [43] and rucaparib [44] have been approved for maintenance therapy in patients with platinum-sensitive ovarian cancer due to the observations that PARPi maintenance therapy significantly improves the PFS of patients with ovarian cancer regardless of BRCA1/2 status. On the basis of above findings, some of tumors with wild type-BRCA1/2 might contain deficiency of other genes involved in the HR repair pathway. Indeed, niraparib was approved for patients with HR deficient-ovarian cancer in 2019 [45]. In addition to ovarian cancer, the use of PARPi was also extended to other cancer types including breast, pancreatic and prostate cancer. In 2018, olaparib became the first PARP inhibitor to be approved for patients with HER2-negative, germline BRCA1/2-mutated, metastatic breast cancer [46]. Following the approval of olaparib in breast cancer, the FDA also approved another PARP inhibitor, talazoparib for patients with germline BRCA-mutated, HER2‑negative locally advanced or metastatic breast cancer in the same year [47]. Most recently, olaparib was further approved for adjuvant treatment of patients with BRCA1/2-mutated, high-risk HER2-negative early breast cancer [48]. In 2019, olaparib was also received FDA approval for the maintenance therapy in patients with BRCA1/2 mutated pancreatic cancer [49]. Subsequently, olaparib and rucaparib were approved for patients with metastatic castration-resistant prostate cancer that is deficient in HR repair in 2020 [50, 51].
The efficacy of PARPi is associated with their binding activity to the NAD+ binding site of PARP1 and ability to induce trapping of PARP1 on DNA that impair BER activity and induce the stalled replication fork, respectively [18]. The mechanism of action of PARPi was originally attributed largely to catalytic inhibition of PARP1 activity, reducing PARylation and blocking the recruitment of repair proteins, such as XRCC1 and DNA ligase III, which eventually impair the single-strand break repair (Fig. 1a). In recent years, more and more studies demonstrate that PARP trapping is critical for the anti-tumor activities of PARPi. The key role of PARP trapping in PARP inhibitor-mediated anti-tumor activity is consistent with the observation that treatment with PARPi results in greater cytotoxicity compared with PARP1 depletion alone [52]. One of the working mechanisms of PARPi mediated PARP trapping is that PARPi competitively bind to NAD+ binding pocket on PARP molecules to inhibit the auto-PARylation of PARP and prevent the dissociation of PARP from DNA [53]. The trapped PARP on DNA damage sites results in replication fork collapse and subsequently leads to the formation of DSBs (Fig. 1a). In addition, a recent study further discovered the molecules which are important for cytotoxicity caused by PARPi-mediated PARP trapping by utilizing the CRISPR-based screening approach [54]. Mechanistically, the genome-embedded ribonucleotides serve as a source of DNA lesions for PARP trapping, which are removed by RNaseH2 through the ribonucleotide excision repair pathway [54]. Since RNaseH2 was found to remove the genome-embedded ribonucleotides, this study further determined the frequency of RNASEH2B deletions in cancer patients. Importantly, RNASEH2B deletions were present in 43% of chronic lymphocytic leukemia (CLL) and 34% of castration-resistant prostate cancers (CRPCs) samples, suggesting these tumors have higher frequency of genome-embedded ribonucleotides and hypersensitivity to PARP inhibitors [54]. Therefore, RNase H2 protects cells from such DNA lesions, while loss of RNaseH2 induces an alternative pathway mediated by the topoisomerase 1 that cleavages misincorporated nucleotides, thereby causing DNA lesions on which PARP is trapped after PARPi treatment [54].

The molecular mechanisms driving the efficacy of and resistance to PARPi. a PARPi compete with NAD+ to inhibit PARylation of PARP target proteins and induce PARP trapping on damaged DNA, which in turn results in replication fork collapse and accumulation of toxic DSBs in HR-deficient cells. b General PARPi-resistant mechanisms. (1) PARPi resistance caused by phosphorylation and binding partners of PARP1. (2) Restoration of HR capacity occurs via re-expressing functional HR repair proteins (upper panel) or acquiring DNA end resection (lower panel)
While these four FDA-approved PARP inhibitors have been widely used for treating tumor with HR deficiency, these first-generation PARP inhibitors are associated with hematologic toxicities due to inhibition of PARP2. Of note, a previous study demonstrated that loss of PARP2 but not PARP1, results in chronic anemia, highlighting the importance of developing selective PARP1 inhibitors [55] such as AZD5305 which has demonstrated a wide therapeutic index and limited toxicity in early clinical trials [56]. AZD5305 is known to exert anti-tumor efficacy by inhibition of PARylation, PARP trapping and growth inhibition. Importantly, AZD5305 selectively kills tumor cells with HR deficiency and exhibits limited cytotoxicity in normal cells [57]. Compared with first-generation FDA-approved PARPi, AZD5305 demonstrates better efficacy, greater target inhibition and improved tolerability [56].
Resistant mechanisms of PARPi
Although PARPi have been demonstrated to have the promising clinical activity in patients harboring HR-deficient tumors, resistance to PARPi remains a significant clinical challenge. PARPi resistance has been found to arise from inhibition of PARPi-PARP interaction and PARP1 trapping activity (Fig. 1b, left panel). Notably, a receptor tyrosine kinase (RTK), c-MET was demonstrated to directly interact with PARP1 and phosphorylate it at tyrosine 907 (Y907), which induces PARylation of PARP1 and decreases the binding activity of PARPi, thereby rendering tumors resistant to PARPi [58]. A study further provides the clinical evidence to link the PARPi resistance and cytotoxic trapped PARP [59]. The PARP1 p.R591C mutation that inhibits PARP1 trapping ability was identified in a patient with ovarian tumor resistant to olaparib [59]. Moreover, recent studies found that PARP1 associated proteins such as the ubiquitin-dependent ATPase, p97, [60] and HMGB3 [61] facilitate the removal of trapped PARP1 from chromatin. Inhibition of these two PARP1-binding partners prolongs PARP1 trapping and sensitized cancer cells to PARPi [60, 61]. A selective and orally bioavailable inhibitor of p97, CB-5083 led to marked increase of talazoparib sensitivity in a patient-derived tumor organoid model derived from a patient with BRCA1 mutated TNBC, suggesting the potential therapeutic effect of combined treatment of p97 and PARP inhibitors in cancer patients [60].
Restoration of HR activity in HR-deficient tumor cells is the most common mechanism of acquiring resistance to PARPi. Reactivation of HR through secondary mutations or epigenetic regulation of BRCA1/2 is frequently identified and has been found in patients with ovarian [62,63,64,65], breast [63, 64, 66], pancreatic [67] and prostate [68, 69] cancer with PARPi-resistant disease. The secondary mutations and epigenetic regulation of BRCA1/2 restore the functional BRCA proteins and contribute to PARPi resistance. Secondary mutations of BRCA2 have been shown to restore the open reading frame and expression of functional BRCA2 proteins [62]. Of note, recent studies further demonstrate that hypomorphic BRCA1 variants caused by genetic alterations are capable to regulate the HR activity [70], such as a BRCA1 alternative splicing isoform without exon 11 (BRCA1-Δ11q) can induce the foci formation of RAD51 in response to DNA damage and thereby lead to PARPi resistance [70]. Promoter demethylation is another mechanism by which the BRCA1 protein can be re-expressed through transcription of epigenetically silenced BRCA1 [71]. A preclinical study on PDX models with BRCA1-methylated ovarian cancer further showed that methylation status of all BRCA1 copies is associated with sensitivity of rucaparib [72], suggesting that complete methylation of BRCA1 promoter might be utilized to predict the PARPi response in the clinic. Notably, the reverse mutations and epigenetic alterations associated with PARPi resistance are not exclusively detected in BRCA1/2 but also observed in other genes involved in HR repair pathways, such as RAD51C, RAD51D and PALB2 [73,74,75], providing additional biomarkers to predict the response to PARPi (Fig. 1b, upper right panel).
Several studies have demonstrated that suppression of NHEJ activity in HR-deficient tumors can restore the HR activity and regulate the PARPi resistance [76]. HR and NHEJ are the two major repair pathways for DNA double strand break repair [77]. The DNA damage response factor, 53BP1 was shown to increase the activity of NHEJ and inhibit the HR repair [78]. Previous studies showed that BRCA1 is important for removing 53BP1 from DNA ends and facilitating the transition from NHEJ to HR when DSBs happened in the S phase [79, 80]. Loss of 53BP1 restored DNA end resection and rescued the HR defects, thereby rendering BRCA1-deficient mouse mammary tumors resistant to PARPi [81]. Additionally, 53BP1 deficiency has been reported in a patient with HR restored, BRCA1-deficient breast cancer after receiving therapy of a PARPi or platinum chemotherapy [82]. Thus, loss of BRCA1 promotes NHEJ activity and 53BP1-dependent formation of toxic chromosomal aberration in PARPi treated BRCA1-deficient cells, leading to hypersensitivity of PARPi [83] (Fig. 1b, lower right panel).
In addition to 53BP1 deficiency, EZH2-meditated epigenetic silencing of MAD2L2 (REV7), a critical factor involved in the 53BP1-dependent NHEJ repair pathway results in resistance to PARPi in ovarian cancer [84]. Similarly, a previous study showed that inhibiting PARylation of EZH2 promotes the EZH2-mediated epigenetic gene silencing and regulates tumor response to PARPi in BRCA-mutated breast cancer [85]. Moreover, loss of the end-resection antagonists, such as RIF1 [86,87,88] and the shieldin complex [89, 90] has been found to mediate resistance to PARP inhibitors in BRCA1-deficient tumors. PDX models with acquired resistance to PARPi were frequently associated with loss of shieldin components which comprised of SHLD1, SHLD2, SHLD3, and REV7 [90]. These end-resection antagonists were identified to block HR activity by locating at DSB sites and limiting DNA end resection [91]. Therefore, deficiency of these factors led to the recruitment of RAD51 and rescued the HR capacity in the absence of BRCA1 (Fig. 1b, lower right panel).
Emerging strategies to overcome PARP inhibitor resistance
Since several resistant mechanisms of PARPi have been identified, it is critical to discover the druggable targets for such mechanisms and develop the combinatorial strategies to overcome PARPi resistance. Based on the rationale of synthetic lethal interaction between PARPi and HR deficiency, therapeutic strategies that chemically induce the “BRCAness” phenotype were shown to (re)-sensitize HR-proficient or HR-restored tumors to PARPi in several cancer types. Recently, results of clinical trials evaluating PARPi in combination with inhibitors of DNA damage checkpoint proteins such as ATM, ATR, CHK1 or WEE1 demonstrated the significant efficacy of these combination by inducing synthetic lethality [92] (Fig. 2a). ATM and ATR play key roles in regulating cell cycle checkpoint signaling and induce cell cycle arrest in response to DNA damage. PARPi resistance caused by BRCA1-independent HR activity has been shown to rely on ATR-dependent RAD51 loading on DNA damage sites [93]. Of note, germline mutations of ATM compromise DSB repair and are associated with HR deficiency in patients with BRCA-wild type breast cancer [94]. Inhibition of another checkpoint kinase CHK1, a downstream target of ATM/ATR impairs foci formation of RAD51 and suppresses HR activity. Additionally, the G2/M checkpoint kinase, WEE1 is known to regulate G2-M cell cycle arrest to facilitate DNA repair prior to entering the mitotic phase. The combination of PARPi and WEE1 inhibitors has exhibited the significant synergistic effects in preclinical models [95], and currently, there are many ongoing clinical trials evaluating this combination in patients with different cancer types [92]. Furthermore, a recent study identified a novel druggable target, DNA polymerase theta (POLQ), which is highly expressed in HR-deficient ovarian and breast tumor [96]. POLQ has been shown to regulate DSB repair by the error-prone microhomology-mediated end-joining (MMEJ) pathway to compensate the impaired HR activity in HR-deficient tumors [96, 97]. Notably, preclinical studies demonstrated the synthetic lethal interaction between POLQ inhibitors and PARPi in HR-deficient tumors with acquired resistance to PARPi [98, 99] (Fig. 2b). The POLQ inhibitor, ART4215 recently entered the phase I/II clinical trial in combination with talazoparib for the treatment of patients with metastatic breast cancer. These findings suggest that POLQ inhibitors hold great potential to overcome the acquired resistance to PARPi in HR-deficient tumors. Although the pre-clinical and clinical studies combining DDR inhibitors with PARPi demonstrated significant anti-tumor effects [100], targeting multiple proteins in the DNA damage response pathways is frequently limited by overlapping toxicities to non-malignant cells [101].

Emerging strategies to overcome PARP inhibitor resistance. a Inhibition of DNA damage checkpoint proteins such as ATM, ATR, CHK1 or WEE1 induces synthetic lethality with PARP inhibitor. b Blocking POLQ mediated MMEJ repair sensitizes tumor to PARP inhibition. c PARPi enhances expression of PD-L1 on cell surface by inhibiting GS3Kβ and activating cGAS-STING pathway. Combination of PD-1/PD-L1 blockade and PARPi may be the potential approaches to increase the anti-tumor activity of PARPi
Due to the discovery of PARPi in regulating immune responses, combination of immune checkpoint inhibitors and PARPi may be the potential approaches to increase the anti-tumor activity of PARPi. In particular, PARPi is shown to enhance the PD-L1 expression and immunosuppressive effects via inhibition of GS3Kβ-mediated PD-L1 degradation [102]. Similarly, PARP inhibition induces the cytosolic accumulation of DNA fragments and activates the cGAS-STING signaling pathway to increase the surface expression of PD-L1 [103]. Furthermore, these studies also demonstrated that PARPi increases CD8+ T cell infiltration in tumors and promotes the anti-tumor effects of the PD-1/PD-L1 blockade in mouse models [102, 103] (Fig. 2c). Several clinical trials are currently underway investigating the anti-tumor effects of immune checkpoint inhibitors in combination with PARPi in several cancer types [104, 105]. Although results from a clinical trial demonstrated that the combination of niraparib and pembrolizumab is well tolerated and associated with promising signals of activity [104], further clinical studies are needed to validate these findings.
Marker-guided effective therapy (Mget) strategies to overcome resistance to PARPi
Although there are an increasing number of clinical trials evaluating PARPi in combination with other agents in several different cancer types [31], the lack of predictive biomarkers for guiding the combination therapy may limit their efficacy because responders and non-responders cannot be discriminated. Targeting oncogenic protein kinases have been shown to sensitize tumors to PARPi through regulating enzyme activity of PARP1 or indirectly inhibiting the HR machinery. Several studies reported that VEGFR [106], EGFR [107], or IGF1R [108] contribute to PARPi resistance through restoring the HR activity. Notably, recent studies further identified some of RTKs mediated phosphorylation of their downstream substrates could be utilized as biomarkers to predict the resistance to PARPi and guide rational combination of PARP and RTK inhibitors (Fig. 3). Specifically, c-MET is shown to directly interact with PARP1 and phosphorylate it at Y907 residue. The phosphorylation of Y907-PARP1 (p-Y907 PARP1) upregulates the enzymatic activity of PARP1 and prevents the binding of PARPi, thereby resulting in resistance to PARPi [58]. Importantly, expression of p-Y907 PARP1 is positively associated with expression of c-MET in the tumor tissues of breast cancer patients, and combination of c-MET and PARPi synergistically suppresses the growth of xenograft tumors which have high c-MET and p-Y907 PARP expression. In addition to breast cancer, c-MET/p-Y907 PARP axis meditated PARPi resistance has also been demonstrated in other cancer types, including ovarian [109] and pancreatic cancer [110]. Additionally, the abundant expression of p-Y907 PARP was also identified in the tumor tissues of patients with hepatocellular carcinoma (HCC) [97]. Interestingly, EGFR was found to interact with c-MET and phosphorylate PARP-Y907 in the HCC cells that have high EGFR and c-MET expression, and simultaneous inhibition of both EGFR and c-MET significantly increases the anti-tumor activity of PARPi in such HCC cells [111]. This finding has also been identified in the TNBC cells with acquired resistance to PARPi, suggesting that heterodimerization of EGFR and c-MET plays key role in PARPi resistance [112]. Most recently, another receptor tyrosine kinase, ALK was shown to promote HR activity and PARPi/platinum resistance through phosphorylating CDK9 at Y19 residue (p-Y19 CDK9) in ovarian and breast cancer [113]. Mechanistically, the phosphorylated ALK (p-ALK)/p-Y19 CDK9 kinase cascade stabilizes positive transcription elongation b complex (P-TEFb), and in turn, activates RNA Pol II-dependent transcription of genes involved in the HR pathway, resulting in PARPi resistance (Fig. 3a). Notably, combination of FDA-approved ALK and PARP inhibitors significantly suppressed tumor growth and prolonged animal survival in PARPi/platinum-resistant tumor xenograft models. Importantly, p-ALK expression is associated with resistance to PARPi and positively correlated with p-Y19-CDK9 expression in the human tumor tissues. This study provided the preclinical and clinical data in support of a marker-guided, PARPi-based combinatorial effective therapy which leverages synthetic lethality by targeting ALK [113]. Collectively, these findings suggest that expression of RTKs and their specific phosphorylated substrates (e.g. c-MET/p-Y907 PARP, EGFR/c-MET/p-Y907 PARP and p-ALK/p-Y19 CDK9) can be utilized to select patients whose tumors have a high likelihood of responding to combined inhibition of PARP and RTK (Fig. 3b). Furthermore, because these RTK inhibitors are currently used in the clinic, these promising combinatorial treatment strategies involving RTK and PARPi are expected to be rapidly translated into clinic.

Overcoming resistance to PARPi by Marker-guided effective therapy (Mget) strategies. a RTK-mediated PARPi resistance. (1) c-MET phosphorylates PARP1 at Y907 to upregulate PARylation and prevent the binding of PARPi, thereby resulting in PARPi resistance. (2) Heterodimerization of EGFR and c-MET contributes to PARPi resistance through phosphorylation of PARP1-Y907. (3) Phosphorylated ALK (p-ALK) promotes HR activity and PARPi resistance by directly phosphorylating CDK9 at Y19. p-Y19 CDK9 facilitates nuclear localization of CDK9 and interacts with cyclin T to form a p-TEFb (CDK9/cyclin T) complex, which turns on transcription of HR repair genes. b Marker-guided effective therapy (Mget) strategies to overcome resistance to PARPi. Expression of RTKs and their specific phosphorylated substrates detected in patient tumor tissues could be utilized to predict PARPi resistance and select the right patients for treatment with PARPi in combination with the specific RTK inhibitors
Conclusions and future perspective
In conclusion, the promising effects of PARPi in several cancer types have been highlighted by an increasing number of preclinical and clinical studies, showing their therapeutic benefits over conventional chemotherapy in a substantial population of patients. Moreover, knowledge of molecular mechanisms driving the efficacy of and resistance to PARPi has led to the development of multiple PARPi-based combination strategies. However, how to select the right patients for treatment with PARPi either as monotherapy or in combination with other agents remains an unmet need in the clinic. Therefore, further detailed mechanistic studies of PARPi resistance, along with pre- and post- treated patient samples from clinical trials will help us to maximize the use of PARPi in the clinic. Moreover, it is necessary to identify more reliable biomarkers for selecting appropriate patients, which may be identified by multi-omics strategies in patient samples with the corresponding clinical data.
Availability of data and materials
Not applicable.
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- BER:
-
Base excision repair
- c-MET:
-
Mesenchymal-epithelial transition factor
- DSBs:
-
Double-strand breaks
- EGFR:
-
Epidermal growth factor receptor
- HCC:
-
Hepatocellular carcinoma
- HR:
-
Homologous recombination
- NHEJ:
-
Non-homologous end joining
- PARP:
-
Poly (ADP-ribose) polymerase (PARP)
- PARylation:
-
Poly(ADP-ribosyl)ation
- P-TEFb:
-
Positive transcription elongation b complex
- PD-L1:
-
Programmed death-ligand 1
- POLQ:
-
DNA polymerase theta
- RTK:
-
Receptor tyrosine kinase
- TNBC:
-
Triple negative breast cancer
- Tyr or Y:
-
Tyrosine
References
Van Beek L, McClay É, Patel S, Schimpl M, Spagnolo L, Maia de Oliveira T. PARP power: A structural perspective on PARP1, PARP2, and PARP3 in DNA damage repair and nucleosome remodeling. Int J Mol Sci. 2021;22(10):5112. https://doi.org/10.3390/ijms22105112.
Kotova EY, Hsieh FK, Chang HW, Maluchenko NV, Langelier MF, Pascal JM, Luse DS, Feofanov AV, Studitsky VM. Human PARP1 facilitates transcription through a nucleosome and histone displacement by pol II In Vitro. Int J Mol Sci. 2022;23(13):7107. https://doi.org/10.3390/ijms23137107.
Kraus WL. Transcriptional control by PARP-1: chromatin modulation, enhancer-binding, coregulation, and insulation. Curr Opin Cell Biol. 2008;20(3):294–302.
Richard IA, Burgess JT, O’Byrne KJ, Bolderson E. Beyond PARP1: the potential of other members of the poly (ADP-Ribose) polymerase family in DNA repair and cancer therapeutics. Front Cell Dev Biol. 2021;9: 801200.
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301.
Gradwohl G, Ménissier de Murcia JM, Molinete M, Simonin F, Koken M, Hoeijmakers JH, de Murcia G. The second zinc-finger domain of poly(ADP-ribose) polymerase determines specificity for single-stranded breaks in DNA. Proc Natl Acad Sci U S A. 1990;87(8):2990–94. https://doi.org/10.1073/pnas.87.8.2990.
Langelier MF, Servent KM, Rogers EE, Pascal JM. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J Biol Chem. 2008;283(7):4105–14.
Tao Z, Gao P, Hoffman DW, Liu HW. Domain C of human poly(ADP-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry. 2008;47(21):5804–13.
Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37(11):3723–38.
Tao Z, Gao P, Liu HW. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J Am Chem Soc. 2009;131(40):14258–60.
Kameshita I, Matsuda Z, Taniguchi T, Shizuta Y. Poly (ADP-Ribose) synthetase. Separation and identification of three proteolytic fragments as the substrate-binding domain, the DNA-binding domain, and the automodification domain. J Biol Chem. 1984;259(8):4770–6.
Pascal JM, Ellenberger T. The rise and fall of poly(ADP-ribose): an enzymatic perspective. DNA Repair (Amst). 2015;32:10–6.
Kim DS, Camacho CV, Kraus WL. Alternate therapeutic pathways for PARP inhibitors and potential mechanisms of resistance. Exp Mol Med. 2021;53(1):42–51.
Ossovskaya V, Koo IC, Kaldjian EP, Alvares C, Sherman BM. Upregulation of poly (ADP-Ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer. 2010;1(8):812–21.
Zaremba T, Ketzer P, Cole M, Coulthard S, Plummer ER, Curtin NJ. Poly(ADP-ribose) polymerase-1 polymorphisms, expression and activity in selected human tumour cell lines. Br J Cancer. 2009;101(2):256–62.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Min A, Im SA. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers (Basel). 2020;12(2):394. https://doi.org/10.3390/cancers12020394.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644–56.
O’Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis. 2010;31(6):961–7.
Hurst JH. Pioneering geneticist Mary-Claire King receives the 2014 Lasker~Koshland Special Achievement Award in Medical Science. J Clin Invest. 2014;124(10):4148–51.
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71.
Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–92.
Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips KA, Mooij TM, Roos-Blom MJ, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–16.
Moschetta M, George A, Kaye SB, Banerjee S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016;27(8):1449–55.
Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, et al. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study. Genet Med. 2015;17(7):569–77.
Ferrone CR, Levine DA, Tang LH, Allen PJ, Jarnagin W, Brennan MF, et al. BRCA germline mutations in Jewish patients with pancreatic adenocarcinoma. J Clin Oncol. 2009;27(3):433–8.
Taylor RA, Fraser M, Rebello RJ, Boutros PC, Murphy DG, Bristow RG, et al. The influence of BRCA2 mutation on localized prostate cancer. Nat Rev Urol. 2019;16(5):281–90.
Castro E, Eeles R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J Androl. 2012;14(3):409–14.
Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16(2):81–104.
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316(5828):1160–6.
Syed A, Tainer JA. The MRE11-RAD50-NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem. 2018;87:263–94.
Foo TK, Vincelli G, Huselid E, Her J, Zheng H, Simhadri S, et al. ATR/ATM-mediated phosphorylation of BRCA1 T1394 promotes homologous recombinational repair and G2-M checkpoint maintenance. Cancer Res. 2021;81(18):4676–84.
Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–6.
Wu S, Zhou J, Zhang K, Chen H, Luo M, Lu Y, et al. Molecular mechanisms of PALB2 function and its role in breast cancer management. Front Oncol. 2020;10:301.
Baumann P, West SC. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem Sci. 1998;23(7):247–51.
Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20.
Saleh-Gohari N, Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004;32(12):3683–8.
Rodgers K, McVey M. Error-prone repair of DNA double-strand breaks. J Cell Physiol. 2016;231(1):15–24.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–84.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–61.
Moore KN, Secord AA, Geller MA, Miller DS, Cloven N, Fleming GF, et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019;20(5):636–48.
Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast ancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–33.
Litton JK, Rugo HS, Ettl J, Hurvitz SA, Goncalves A, Lee KH, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–63.
Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, et al. Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N Engl J Med. 2021;384(25):2394–405.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381(4):317–27.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. 2020;382(22):2091–102.
Abida W, Patnaik A, Campbell D, Shapiro J, Bryce AH, McDermott R, et al. Rucaparib in men with metastatic castration-resistant prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin Oncol. 2020;38(32):3763–72.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–99.
Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–15.
Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Z, et al. CRISPR screens identify genomic ribonucleotides as a source of PARP-trapping lesions. Nature. 2018;559(7713):285–9.
Farres J, Llacuna L, Martin-Caballero J, Martinez C, Lozano JJ, Ampurdanes C, et al. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015;22(7):1144–57.
AZD5305 More Tolerable than Earlier PARP Agents. Cancer Discov. 2022;12(7):1602. https://doi.org/10.1158/2159-8290.CD-NB2022-0039.
Illuzzi G, Staniszewska AD, Gill SJ, Pike A, McWilliams L, Critchlow SE, et al. Preclinical characterization of AZD5305, a next generation, highly selective PARP1 inhibitor and trapper. Clin Cancer Res. 2022. https://doi.org/10.1158/1078-0432.CCR-22-0301.
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22(2):194–201.
Pettitt SJ, Krastev DB, Brandsma I, Drean A, Song F, Aleksandrov R, Harrell MI, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9(1):1849.
Krastev DB, Li S, Sun Y, Wicks AJ, Hoslett G, Weekes D, et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nat Cell Biol. 2022;24(1):62–73.
Ma H, Qi G, Han F, Lu W, Peng J, Li R, Yan S, Yuan C, Kong B. HMGB3 promotes PARP inhibitor resistance through interacting with PARP1 in ovarian cancer. Cell Death Dis. 2022;13(3):263.
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5.
Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K, et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J Pathol. 2013;229(3):422–9.
Weigelt B, Comino-Mendez I, de Bruijn I, Tian L, Meisel JL, Garcia-Murillas I, et al. Diverse BRCA1 and BRCA2 reversion mutations in circulating cell-free DNA of therapy-resistant breast or ovarian cancer. Clin Cancer Res. 2017;23(21):6708–20.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–9.
Afghahi A, Timms KM, Vinayak S, Jensen KC, Kurian AW, Carlson RW, et al. Tumor BRCA1 reversion mutation arising during neoadjuvant platinum-based chemotherapy in triple-negative breast cancer is associated with therapy resistance. Clin Cancer Res. 2017;23(13):3365–70.
Pishvaian MJ, Biankin AV, Bailey P, Chang DK, Laheru D, Wolfgang CL, et al. BRCA2 secondary mutation-mediated resistance to platinum and PARP inhibitor-based therapy in pancreatic cancer. Br J Cancer. 2017;116(8):1021–6.
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov. 2017;7(9):999–1005.
Vidula N, Rich TA, Sartor O, Yen J, Hardin A, Nance T, et al. Routine plasma-based genotyping to comprehensively detect germline, somatic, and reversion BRCA mutations among patients with advanced solid tumors. Clin Cancer Res. 2020;26(11):2546–55.
Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, Peri S, et al. The BRCA1-delta11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76(9):2778–90.
Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J Natl Cancer Inst. 2016. https://doi.org/10.1093/jnci/djw148.
Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9(1):3970.
Nesic K, Kondrashova O, Hurley RM, McGehee CD, Vandenberg CJ, Ho GY, et al. Acquired RAD51C promoter methylation loss causes PARP inhibitor resistance in high-grade serous ovarian carcinoma. Cancer Res. 2021;81(18):4709–22.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2017;7(9):984–98.
Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating cell-free DNA to guide prostate cancer treatment with PARP inhibition. Cancer Discov. 2017;7(9):1006–17.
Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A. 2011;108(8):3406–11.
Brandsma I, Gent DC. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr. 2012;3(1):9.
Gupta A, Hunt CR, Chakraborty S, Pandita RK, Yordy J, Ramnarain DB, et al. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat Res. 2014;181(1):1–8.
Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54.
Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35(4):534–41.
Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3(1):68–81.
Waks AG, Cohen O, Kochupurakkal B, Kim D, Dunn CE, Buendia Buendia J, et al. Reversion and non-reversion mechanisms of resistance to PARP inhibitor or platinum chemotherapy in BRCA1/2-mutant metastatic breast cancer. Ann Oncol. 2020;31(5):590–8.
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17(6):688–95.
Karakashev S, Fukumoto T, Zhao B, Lin J, Wu S, Fatkhutdinov N, et al. EZH2 inhibition sensitizes CARM1-high, homologous recombination proficient ovarian cancers to PARP inhibition. Cancer Cell. 2020;37(2):157-167 e156.
Yamaguchi H, Du Y, Nakai K, Ding M, Chang SS, Hsu JL, et al. EZH2 contributes to the response to PARP inhibitors through its PARP-mediated poly-ADP ribosylation in breast cancer. Oncogene. 2018;37(2):208–17.
Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339(6120):700–4.
Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J Biol Chem. 2013;288(16):11135–43.
Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell. 2013;49(5):872–83.
Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, et al. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560(7716):122–7.
Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20(8):954–65.
Setiaputra D, Durocher D. Shieldin—the protector of DNA ends. EMBO Rep. 2019. https://doi.org/10.15252/embr.201847560.
Gupta N, Huang TT, Horibata S, Lee JM. Cell cycle checkpoints and beyond: exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor-resistant cancer. Pharmacol Res. 2022;178: 106162.
Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31(3):318–32.
Renwick A, Thompson D, Seal S, Kelly P, Chagtai T, Ahmed M, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–5.
Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, et al. Sequential therapy with PARP and WEE1 inhibitors minimizes toxicity while maintaining efficacy. Cancer Cell. 2019;35(6):851-867 e857.
Ceccaldi R, Liu JC, Amunugama R, Hajdu I, Primack B, Petalcorin MI, et al. Homologous-recombination-deficient tumours are dependent on Poltheta-mediated repair. Nature. 2015;518(7538):258–62.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64.
Zhou J, Gelot C, Pantelidou C, Li A, Yucel H, Davis RE, et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat Cancer. 2021;2(6):598–610.
Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. 2021;12(1):3636.
Gourley C, Balmana J, Ledermann JA, Serra V, Dent R, Loibl S, et al. Moving from poly (ADP-Ribose) polymerase inhibition to targeting DNA repair and DNA damage response in cancer therapy. J Clin Oncol. 2019;37(25):2257–69.
O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60(4):547–60.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen MK, Hsu JM, Hsu JL, Yu WH, Du Y, Lee HH, et al. PARP inhibitor upregulates PD-L1 expression and enhances cancer-associated immunosuppression. Clin Cancer Res. 2017;23(14):3711–20.
Sen T, Rodriguez BL, Chen L, Corte CMD, Morikawa N, Fujimoto J, et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019;9(5):646–61.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5(8):1141–9.
Domchek SM, Postel-Vinay S, Im SA, Park YH, Delord JP, Italiano A, et al. Olaparib and durvalumab in patients with germline BRCA-mutated metastatic breast cancer (MEDIOLA): an open-label, multicentre, phase 1/2, basket study. Lancet Oncol. 2020;21(9):1155–64.
Bizzaro F, Fuso Nerini I, Taylor MA, Anastasia A, Russo M, Damia G, et al. VEGF pathway inhibition potentiates PARP inhibitor efficacy in ovarian cancer independent of BRCA status. J Hematol Oncol. 2021;14(1):186.
Nowsheen S, Cooper T, Stanley JA, Yang ES. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS ONE. 2012;7(10): e46614.
Amin O, Beauchamp MC, Nader PA, Laskov I, Iqbal S, Philip CA, et al. Suppression of homologous recombination by insulin-like growth factor-1 inhibition sensitizes cancer cells to PARP inhibitors. BMC Cancer. 2015;15:817.
Han Y, Chen MK, Wang HL, Hsu JL, Li CW, Chu YY, et al. Synergism of PARP inhibitor fluzoparib (HS10160) and MET inhibitor HS10241 in breast and ovarian cancer cells. Am J Cancer Res. 2019;9(3):608–18.
Gao Y, Chen MK, Chu YY, Yang L, Yu D, Liu Y, et al. Nuclear translocation of the receptor tyrosine kinase c-MET reduces the treatment efficacies of olaparib and gemcitabine in pancreatic ductal adenocarcinoma cells. Am J Cancer Res. 2021;11(1):236–50.
Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK, et al. EGFR and c-MET cooperate to enhance resistance to PARP inhibitors in hepatocellular carcinoma. Cancer Res. 2019;79(4):819–29.
Chu YY, Yam C, Chen MK, Chan LC, Xiao M, Wei YK, et al. Blocking c-Met and EGFR reverses acquired resistance of PARP inhibitors in triple-negative breast cancer. Am J Cancer Res. 2020;10(2):648–61.
Chu Y-Y, Chen M-K, Wei Y, Lee H-H, Xia W, Wang Y-N, et al. Targeting the ALK–CDK9-Tyr19 kinase cascade sensitizes ovarian and breast tumors to PARP inhibition via destabilization of the P-TEFb complex. Nat Cancer. 2022. https://doi.org/10.1038/s43018-022-00438-2.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the following: Breast Cancer Research Foundation (BCRF-17-069 to M.-C.H.); The MD Anderson-China Medical University Sister Institution Fund; The MD Anderson Moonshot for Breast Cancer; Drug Development Center, China Medical University from Ministry of Education in Taiwan (to M.-C.H.); The Ministry of Science and Technology in Taiwan (MOST 110-2314-B-039-060 to H.Y.); The National Science and Technology Council (NSTC-111-2639-B-039-001-ASP to M.-C.H.); a Conquer Cancer Development Award supported by Fleur Fairman (to C.Y.); and New Partnership Program for the Connection to the Top Labs in the World (Dragon Gate Program; 107-2911-I-006-519 to Y.-Y.C.); China Medical University for Yingtsai Scholar Award (CMU109-YT-06 to H.Y.).
Author information
Authors and Affiliations
Contributions
Y.-Y.C. initiated, drafted, and revised the manuscript. C.Y. and H.Y. provided scientific input. M.-C.H supervised the organization, writing, and revision of the article. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Overcoming PARP inhibitor resistance through the combination of RTK inhibitors and PARPi, are covered in the provisional patent UTSC.P1450US.P1. The provisional patent (UTSC.P1450US.P1) titled “Combinational Therapy Targeting PARP1 and RTK” was invented by M.-C.H., M.-K.C., and Y.-Y.C. and filed by The University of Texas.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chu, YY., Yam, C., Yamaguchi, H. et al. Biomarkers beyond BRCA: promising combinatorial treatment strategies in overcoming resistance to PARP inhibitors. J Biomed Sci 29, 86 (2022). https://doi.org/10.1186/s12929-022-00870-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00870-7