
Abstract
Background
Fatty acids (FAs) with a chain length of more than 18 carbon atoms (> C18) are interesting for the production of specialty compounds derived from these FAs. These compounds include free FAs, like erucic acid (C22:1-Δ13), primary fatty alcohols (FOHs), like docosanol (C22:0-FOH), as well as jojoba-like wax esters (WEs) (C38-WE to C44-WE), which are esters of (very) long-chain FAs and (very) long-chain FOHs. In particular, FAs, FOHs and WEs are used in the production of chemicals, pharmaceuticals and cosmetic products. Jojoba seed oil is highly enriched in diunsaturated WEs with over 70 mol% being composed of C18:1–C24:1 monounsaturated FOH and monounsaturated FA moieties. In this study, we aim for the production of jojoba-like WEs in the yeast Saccharomyces cerevisiae by increasing the amount of very long-chain, monounsaturated FAs and simultaneously expressing enzymes required for WE synthesis.
Results
We show that the combined expression of a plant-derived fatty acid elongase (FAE/KCS) from Crambe abyssinica (CaKCS) together with the yeast intrinsic fatty acid desaturase (FAD) Ole1p leads to an increase in C20:1 and C22:1 FAs in S. cerevisiae. We also demonstrate that the best enzyme candidate for C24:1 FA production in S. cerevisiae is a FAE derived from Lunaria annua (LaKCS). The combined overexpression of CaKCS and Ole1p together with a fatty acyl reductase (FAR/FAldhR) from Marinobacter aquaeolei VT8 (MaFAldhR) and a wax synthase (WS) from Simmondsia chinensis (SciWS) in a S. cerevisiae strain, overexpressing a range of other enzymes involved in FA synthesis and elongation, leads to a yeast strain capable of producing high amounts of monounsaturated FOHs (up to C22:1-FOH) as well as diunsaturated WEs (up to C46:2-WE).
Conclusions
Changing the FA profile of the yeast S. cerevisiae towards very long-chain monounsaturated FAs is possible by combined overexpression of endogenous and heterologous enzymes derived from various sources (e.g. a marine copepod or plants). This strategy was used to produce jojoba-like WEs in S. cerevisiae and can potentially be extended towards other commercially interesting products derived from very long-chain FAs.
Similar content being viewed by others
Background
Fatty acids (FAs) play a major role during growth and maintenance of all living cells, since they are components of cell membrane lipids, organelle membrane lipids as well as storage lipids. Moreover, they can function as signaling compounds in the form of sphingolipids. The FAs naturally found in the yeast Saccharomyces cerevisiae comprise mostly C16 and C18 species. The most abundant one is palmitoleic acid (C16:1-Δ9), followed by oleic acid (C18:1-Δ9), palmitic acid (C16:0) and stearic acid (C18:0) [1]. The composition of FAs in yeast is strictly regulated and dependent, among other factors, on the cultivation temperature [2] as well as the media composition. Culture medium that contains a certain type of FA can change the distribution of FAs inside the yeast cell [3].
The total distribution of FAs is also reflected by the composition of membrane- as well as storage lipids in yeast. In case of phospholipids (PLs), which make up around 70% of the cell membrane, the main FA residues are C14:1-Δ9, C16:1-Δ9 and C18:1-Δ9, which shows the importance of those FA species for the integrity of cell membranes in S. cerevisiae. In case of triacylglycerols (TAGs), which function as storage lipids, the most abundant FA residues are C16:0, C16:1-Δ9, C18:0 and C18:1-Δ9 [4]. Besides having a storage function in yeast, TAGs also act as a buffer and sink for excess unsaturated FAs [5] and reflect the composition of added or synthesized FAs [4]. The addition of oleic acid to the growth medium of yeast cells has a strong effect on lipid metabolism and the composition of storage lipids. While cells grown on glucose show approximately a 50/50 accumulation of the storage lipids steryl esters (SEs) and TAGs, cells grown on oleate show a ratio of 1/99. Moreover, the composition of PLs and TAGs changes towards an increased incorporation of oleic acid in medium supplemented with oleate [4]. Yeast cells unable of producing storage lipids, by deletion of genes encoding SE and TAG formation enzymes (Lro1p, Dga1p, Are1p and Are2p), show a defective regulation of lipid synthesis, an extensive proliferation of intracellular membranes and finally cell death under excess of oleate [5].
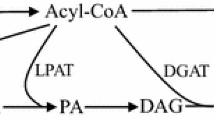
The de novo FA synthesis in S. cerevisiae is located in the cytosol and uses acetyl-coenzyme A (acetyl-CoA) as a building block. The first step of FA synthesis is the carboxylation of acetyl-CoA to malonyl-CoA by an acetyl-CoA carboxylase (Acc1p) [6]. Studies using a deregulated version of Acc1p, in which the serine 1157 was mutated to an alanine residue to lock the Acc1p in a non-phosphorylatable, highly active state, showed that the ratio of C18 to C16 FAs changed. In wildtype cells, the C18/C16 ratio was found to be around 0.5, while the deregulated Acc1p mutant strain showed a ratio of around 2 [7]. This mutation also led to an increase in very long-chain fatty acids (VLCFAs) (C20-C26). In the second step of FA synthesis, a C2 unit derived from malonyl-CoA is attached to acetyl–acyl carrier protein (acetyl-ACP) and in the further extensions to the acyl-ACP chain, leading to FAs up to C18. These reactions are in the cytosol catalyzed by the fatty acid synthase (FAS) consisting of Fas1p (β-subunit) and Fas2p (α-subunit), which form a hexameric α6β6 complex. When the FAs are released from the FAS enzyme complex, a coenzyme-A unit is attached to them [6]. Besides originating from de novo FA synthesis, external free FAs or free FAs derived from lipid or lipoprotein turnover can be activated to fatty acyl-CoAs (FACoAs) by FACoA synthetases [8,9,10,11]. In S. cerevisiae, FAs up to C18 can be further elongated up to C26 at the endoplasmic reticulum (ER). These reactions are catalyzed by the β-ketoacyl-CoA synthases (KCSs), also known as fatty acid elongases (FAEs), the β-ketoacyl-CoA reductase (KCR), the β-hydroxyacyl-CoA dehydratase (HCD) and the enoyl-CoA reductase (ECR) which are all integrated in the ER membrane and face the cytosol with their active site [6] (Fig. 1). S. cerevisiae possesses three KCS enzymes, Elo1p, Elo2p and Elo3p which all have a discrete substrate range. Elo1p elongates C12–C16 FACoAs to C16–C18 FACoAs. Elo2p is responsible for the biosynthesis of C20–C24 FACoAs and Elo3p specifically elongates C24 FACoAs to C26 FACoAs [12, 13]. In contrast to the KCS enzymes, which are substrate specific, the other elongation enzymes (KCR, HCD and ECR) show a broad specificity, accepting a wide range of substrates [6]. The elongation enzymes at the ER together fulfill the same function as the FAS complex in the cytosol, with the exception that the ER enzymes are distinct proteins in contrast to the FAS complex which contains two subunits with several functional domains.

De novo synthesis and elongation of fatty acids (FAs), fatty alcohols (FOHs) and wax esters (WEs) in S. cerevisiae. The first step shown in the reaction scheme is the carboxylation of acetyl-CoA to malonyl-CoA catalyzed by acetyl-CoA carboxylase (Acc1p) in the cytosol. In our study, a mutant version of Acc1p, containing two amino acid substitutions (S659A; S1157A), was used (Acc1p**). The next step in the synthesis of FAs in the cytosol is a series of reactions catalyzed by the fatty acid synthases 1 (Fas1p) and 2 (Fas2p) which leads to fatty acyl CoAs (FACoAs) with a chain length of C12-C18. The acyl-CoA binding protein (Acb1p) transports newly synthesized acyl-CoA esters from Fas1p–Fas2p to acyl-CoA-consuming processes. The elongation and desaturation of C12-C18 FACoAs is performed at the endoplasmic reticulum (ER). The desaturation of FAs in S. cerevisiae is catalyzed by Ole1p, a fatty acid desaturase (FAD) acting on C12-C19 FAs. This scheme also shows a heterologous FAD (green) which is able to insert a double bond into very long-chain FAs (C20-C26). The elongation of FAs in S. cerevisiae is catalyzed by a β-ketoacyl-CoA synthase (KCS), a β-ketoacyl-CoA reductase (KCR), a β-hydroxyacyl-CoA dehydratase (HCD) and an enoyl-CoA reductase (ECR) and leads to the production of C16-C26 FACoAs in yeast. FACoAs can be reduced by a heterologous fatty acyl-CoA reductase (FAR) to a fatty aldehyde and further to a FOH. A wax synthase (WS) catalyzes the esterification of a FOH with another FACoA molecule to form a WE. All yeast intrinsic enzymes are indicated in grey, whereas heterologous ones are indicated in green
Besides synthesis and elongation of FAs, also the desaturation of FAs plays an important role in the yeast cell metabolism. S. cerevisiae possesses only one fatty acid desaturase (FAD), Ole1p, which can integrate a double bond at the Δ9-position of FACoAs ranging from a carbon chain length of C12–C19 [14] (Fig. 1). Therefore, in contrast to other fungi, S. cerevisiae only contains monounsaturated fatty acids (MUFAs) with ~ 70–80% of its total glycerolipid acyl chains consisting of C14:1-Δ9, C16:1-Δ9 and C18:1-Δ9 [15].
Since the composition of FAs is very important for the integrity of membrane lipid structures, it can be expected that the yeast FA pool can only be changed to a certain extent. Nevertheless, a change of the FA pool can be interesting if certain compounds with a specific chain length (> C18) shall be produced. Those products include free fatty acids (FFAs), fatty alcohols (FOHs), alkanes/alkenes, lipids as well as speciality FAs like eicosapentaenoic acid and have been produced by metabolic engineering of S. cerevisiae and other yeasts [16].
Several commercially interesting products are also derived from very long-chain fatty acids (VLCFAs) and comprise very long-chain fatty alcohols (VLCFOHs) like docosanol (C22:0-FOH) [17] as well as jojoba-like wax esters (WEs), which are mainly composed of C18–C24 monounsaturated fatty alcohol (MUFOH) and monounsaturated fatty acid (MUFA) residues [18] (Fig. 1). The application of jojoba oil ranges from cosmetic and personal care products over medical use to lubricants. The current production of jojoba oil is around 4000 tons/year, while the estimated demand is up to 200,000 tons/year [19]. Because of the increased need for jojoba oil, extraction from its natural source will not be enough to meet the demand in the future, even if growing regions for the jojoba plant are expanded extensively. Since S. cerevisiae has already been modified for the production of a wide range of chemicals [20], jojoba oil production in modified yeast represents a very promising approach.
In a previous study we could show that jojoba-like WEs can be synthesized in metabolically engineered S. cerevisiae by heterologous expression of a FAR from Marinobacter aquaeolei VT8 and a WS from Simmondsia chinensis, but the products were restricted to saturated very long-chain WEs (C40:0-WE and C42:0-WE) [21]. This study made clear that the pool of very long-chain monounsaturated fatty acids (VLCMUFAs) needs to increase in S. cerevisiae to enable the synthesis of jojoba-like diunsaturated wax esters (DUWEs).
To achieve this goal, we here expressed several endogenous and heterologous KCSs and FADs in S. cerevisiae and demonstrate that this can lead to a high increase in C20:1, C22:1 and C24:1 MUFAs. Moreover, it is shown that the combined expression of a plant KCS, a bacterial FAR, a plant WS and several yeast intrinsic enzymes leads to a final MUFOH and DUWE producing strain.
Results
Increasing the amount of MUFAs by expression of a heterologous KCS together with Ole1p of S. cerevisiae
Previously, we demonstrated that jojoba-like WEs can be synthesized in metabolically engineered S. cerevisiae strains by combined overexpression of the yeast intrinsic Elo2p together with a FAR from M. aquaeolei and a WS from S. chinensis, but the products were restricted to saturated very long-chain WEs (C40:0-WE and C42:0-WE) [21]. Based on this observation we concluded that the pool of VLCMUFAs is not high enough to enable the synthesis of jojoba-like DUWEs in S. cerevisiae.
Therefore, we investigated the effect of overexpressing the endogenous Ole1p either together with the yeast intrinsic Elo2p or together with a heterologous plant-derived FAE/KCS enzyme in S. cerevisiae. The heterologous FAE/KCS enzymes tested include those from Arabidopsis thaliana (AtFAE), Brassica napus (BnKCS), Crambe abyssinica (CaKCS), Lunaria annua (LaKCS), S. chinensis (SciFAE) and Tropaeolum majus (TmKCS). Combinations of a KCS and Ole1p were cloned into the episomal plasmid pYX212 via modular pathway engineering [22] (Additional file 1: Figure S1). The plasmids were transferred into the background strain S. cerevisiae CEN.PK 113-5D elo3Δ X-2::pMPC3::ACC1** X-3::IFA38::PHS1::TSC13::ACB1. This strain contains a constitutively active version of Acc1p to increase the supply of malonyl-CoA, a deletion of the ELO3 gene to prevent formation of C26 FAs and an additional copy of the genes IFA38, PHS1 and TSC13, encoding the yeast intrinsic elongation enzymes KCR, HCD and ECR, respectively. Moreover, it harbors an additional copy of the ACB1 gene encoding the acyl-CoA binding protein Acb1p to increase the acyl-CoA pool, which has been shown in a previous study to result in an increased fatty acid ethyl ester production [23].
The analysis of the FA profile of the resulting strains showed that the combined overexpression of a KCS together with the yeast intrinsic Ole1p leads to an increase in the pool of VLCMUFAs in case of the KCS enzymes Elo2p, CaKCS and LaKCS. The KCS enzymes from A. thaliana, B. napus and T. majus were not able to increase the pool of VLCMUFAs (Fig. 2).

Effect of expression of (heterologous) elongases (FAEs/KCSs) together with the yeast intrinsic Ole1p desaturase on the concentration of fatty acids (FAs) (mg/g CDW) in the background strain CEN.PK 113-5D elo3Δ X-2::pMPC3::ACC1** X-3::IFA38::PHS1::TSC13::ACB1 (LW03). The values represent the mean ± standard deviation (SD) of three biological replicates of strains LW04 (pYX212), LW05 (pYX212::ELO2::OLE1), LW06 (pYX212::AtFAE::OLE1), LW07 (pYX212::BnKCS::OLE1), LW08 (pYX212::CaKCS::OLE1), LW09 (pYX212::LaKCS::OLE1), LW10 (pYX212::SciFAE::OLE1) and LW11 (pYX212::TmKCS::OLE1), respectively. The strains were grown for 48 h in minimal medium containing 20 g/L glucose
The highest increase in C20:1 and C22:1 FAs in our study was obtained by combined expression of the KCS from C. abyssinica (CaKCS) together with the intrinsic Ole1p from S. cerevisiae. The level of C20:1 FAs and C22:1 FAs increased 20.6- and 95.7-fold, respectively, compared to the wildtype. In contrast to that, the highest increase in C24:1 FAs (25.6-fold compared to Elo2p overexpression strain) was observed by expression of the KCS from L. annua (LaKCS) together with Ole1p (Fig. 2).
Increasing the amount of MUFAs by expression of a heterologous KCS and a heterologous FAD
After having identified CaKCS and LaKCS as promising enzyme candidates to increase the C20:1, C22:1 and C24:1 FA concentrations in S. cerevisiae, we wanted to study the influence of heterologous FADs on the synthesis of VLCMUFAs. The heterologous FAD enzymes tested include those from C. hyperboreus (ChDes9-1) and S. chinensis (SciFAD-SP) as well as two acyl- CoA desaturase-like (ADS) proteins from A. thaliana (AtADS1.2 and AtADS1.4). Combinations of CaKCS or LaKCS and a heterologous FAD were cloned into the episomal plasmid pYX212 via modular pathway engineering [22] (Additional file 1: Figure S1). The plasmids were transferred into the background strain S. cerevisiae CEN.PK 113-5D elo3Δ X-2:pMPC3::ACC1** X-3::IFA38::PHS1::TSC13::ACB1.
The highest amount of C20:1 and C22:1 FAs (1.36 ± 0.20 mg FAs/g CDW and 2.91 ± 0.44 mg FAs/g CDW, respectively), accompanied by a decrease in C18:1 FAs of 23.3% (compared to strain LW04), was observed in a strain expressing CaKCS together with ChDes9-1. This is slightly less compared with the amount of VLCMUFAs produced in the strain expressing CaKCS together with Ole1p. The production of C24:1 FAs was highest in a strain expressing LaKCS together with an ADS protein from A. thaliana (AtADS1.2) (Fig. 3).

Effect of expression of (heterologous) elongases (FAEs/KCSs) together with heterologous desaturases (FADs) on the concentration of fatty acids (FAs) (mg/g CDW) in the background strain CEN.PK 113-5D elo3Δ X-2::pMPC3::ACC1** X-3::IFA38::PHS1::TSC13::ACB1 (LW03). The values represent the mean ± standard deviation (SD) of three biological replicates of strains LW04 (pYX212), LW12 (pYX212::CaKCS::SciFAD-SP), LW13 (pYX212::CaKCS::ChDes9-1), LW14 (pYX212::CaKCS::AtADS1.2), LW15 (pYX212::CaKCS::AtADS1.4), LW16 (pYX212::LaKCS::SciFAD-SP), LW17 (pYX212::LaKCS::ChDes9-1), LW18 (pYX212::LaKCS::AtADS1.2) and LW19 (pYX212::LaKCS::AtADS1.4), respectively. The strains were grown for 48 h in minimal medium containing 20 g/L glucose
Therefore, after evaluation of the combined expression of CaKCS or LaKCS with various FADs, the combination of CaKCS and Ole1p was identified as best enzymes for high level production of C20:1 and C22:1 FAs and chosen to be expressed together with the WE biosynthesis enzymes MaFAldhR and SciWS to further boost WE production.
Integration of pathway genes to construct MUFOH and DUWE production strain
The overexpression cassettes for CaKCS, Elo1p, Faa1p and Ole1p were integrated into the genome of S. cerevisiae. The combined overexpression of CaKCS, Elo1p, Faa1p and Ole1p (strain LW22) led to a growth defect of the strain with a final OD600 of 2.62 ± 0.05 after 48 h, compared with the same strain lacking the overexpression of CaKCS and Ole1p (strain LW21) which showed a final OD600 of 10.9 ± 0.18 after 48 h. This growth defect was also observed in strain LW24, expressing CaKCS and Ole1p in combination with the WE synthesizing enzymes MaFAldhR and SciWS as well as Elo2p, but not in strain LW23, expressing MaFAldhR, SciWS and Elo2p, but lacking CaKCS and Ole1p (Additional file 1: Figure S3).
The integration-based expression of CaKCS, Elo1p, Faa1p and Ole1p together with the plasmid-based expression of MaFAldhR, Elo2p and SciWS led to appearance of two additional bands on the thin layer chromatography (TLC) plate in strain LW23 (expressing only Elo1p, Elo2p, Faa1p, MaFAldhR and SciWS) as well as in strain LW24 (expressing CaKCS, Elo1p, Elo2p, Faa1p, MaFAldhR, Ole1p and SciWS) (Additional file 1: Figures S4, S5).
The analysis via gas chromatography-flame ionization detector (GC-FID) and gas chromatography-mass spectrometry (GC/MS) showed that those additional bands on the TLC plate corresponded to FOHs and WEs, respectively (Additional file 1: Figures S6, S7). The control strains LW21 and LW22, which do not contain the plasmid pYX212::MaFAldhR::SciWS::ELO2, did not show peaks corresponding to FOHs or WEs. In contrast to that, strain LW23 produced C16:0-FOH, C18:0-FOH, C18:1-FOH, C20:0-FOH and C22:0-FOH (Additional file 1: Figure S6). These FOH species could also be detected in strain LW24, which additionally produced C16:1-FOH, C20:1-FOH and C22:1-FOH (Additional file 1: Figure S6). The analysis of WEs via GC/MS showed that both LW23 and LW24 strains produced WEs in the chain length range of C30-WE to C44-WE (Additional file 1: Figure S7), but only in case of strain LW24, DUWE synthesis could be detected. The synthesis of C38:2-WE, C40:2-WE, C42:2-WE and C44:2-WE was confirmed by analysis of the mass spectra of the corresponding GC peaks, which showed the appearance of m/z peaks corresponding to the molecular ions of these DUWEs (m/z = 561, 589, 617 and 645, respectively) (Additional file 1: Figure S8). Those DUWE specific m/z peaks could not be detected in the strain lacking the expression of CaKCS and Ole1p (strain LW23).
The quantification of FOHs and WEs showed that the expression of CaKCS and Ole1p in strain LW24 leads to the production of 1.27 ± 0.46 mg MUFOHs/g CDW, with a significant increase (p < 0.05) in the MUFOHs C18:1-FOH, C20:1-FOH and C22:1-FOH and a significant decrease (p < 0.05) in the saturated FOHs C16:0-FOH, C18:0-FOH and C20:0-FOH compared to strain LW23. In total, strain LW24 produced 2.46 ± 0.71 mg FOHs/g CDW. In contrast to that, the strain lacking the expression of CaKCS and Ole1p (strain LW23) produced 2.51 ± 0.29 mg FOHs/g CDW, of which 0.014 mg ± 0.005/g CDW were MUFOHs. This corresponds to an increase in MUFOHs by 89.5-fold in strain LW24 compared to strain LW23 (Fig. 4). The overall titer of FOHs was 6.92 ± 0.85 mg/L in strain LW23 and 2.10 ± 0.55 mg/L in strain LW24, which corresponds to a yield of 0.346 mg/g glucose and 0.105 mg/g glucose, respectively.

Concentration of fatty alcohols (FOHs) (mg/g CDW). The values represent the mean ± standard deviation (SD) of three biological replicates of strains LW23 (expressing only Elo1p, Elo2p, Faa1p, MaFAldhR and SciWS) and LW24 (expressing CaKCS, Elo1p, Elo2p, Faa1p, MaFAldhR, Ole1p and SciWS), respectively. The strains were grown for 48 h in minimal medium containing 20 g/L glucose. *p < 0.05 (Students t-test, two-tailed, unequal variance assumed)
This increase in MUFOHs was accompanied by the formation of DUWEs in strain LW24. The quantification of WEs in strains LW23 and LW24 showed that in total 13.13 ± 5.08 mg WEs/g CDW and 14.38 ± 1.76 mg WEs/g CDW, respectively, were produced. Strain LW24 showed a significant decrease (p < 0.05) in C34-WE species and a significant increase (p < 0.05) in C42-WE species (Fig. 5). The overall titer of WEs was 36.5 ± 16.71 mg/L in strain LW23 and 11.92 ± 1.47 mg/L in strain LW24, which corresponds to a yield of 1.83 mg/g glucose and 0.60 mg/g glucose, respectively. The C40-WE to C44-WE made up 36.6% in strain LW23, while in strain LW24, 76.4% of the WEs were C40-WE to C44-WE. The analysis of the molecular composition of WEs in strains LW23 and LW24 led to an overall identification of 73 WE species (Fig. 6). The most abundant WE species in strain LW23 were C18:0-C16:0 (25.30 ± 1.07 mol%), followed by C22:0–C18:1 (17.25 ± 1.24 mol%) and C18:0–C16:1 (10.92 ± 0.82 mol%). In contrast to that, strain LW24 produced WEs that contain longer and more unsaturated FOH and FA moieties, with the most abundant WE species being C22:0–C20:1 (12.52 ± 0.90 mol%), C22:1–C20:1 (10.67 ± 2.48 mol%) and C22:0–C18:1 (7.16 ± 1.38 mol%). Additionally, low amounts of C46-WE could be identified in strain LW24 (Additional file 1: Table S3). This represents the first study in which the production of DUWEs of a chain length of C38-WE to C46-WE was achieved in the yeast S. cerevisiae.

Concentration of wax esters (WEs) (mg/g CDW). The values represent the mean ± standard deviation (SD) of three biological replicates of strains LW23 (expressing only Elo1p, Elo2p, Faa1p, MaFAldhR and SciWS) and LW24 (expressing CaKCS, Elo1p, Elo2p, Faa1p, MaFAldhR, Ole1p and SciWS), respectively. The strains were grown for 48 h in minimal medium containing 20 g/L glucose. *p < 0.05 (Students t-test, two-tailed, unequal variance assumed)

Molecular composition of wax ester (WE) species (mol%). The values represent the mean ± standard deviation (SD) of three biological replicates of strains LW23 (expressing only Elo1p, Elo2p, Faa1p, MaFAldhR and SciWS) and LW24 (expressing CaKCS, Elo1p, Elo2p, Faa1p, MaFAldhR, Ole1p and SciWS), respectively. The strains were grown for 48 h in minimal medium containing 20 g/L glucose
Discussion
In this study we aimed for the increased production of VLCMUFAs in the yeast S. cerevisiae as precursors for the synthesis of jojoba-like DUWEs. For this purpose, we have screened a range of endogenous and heterologous FAEs/KCSs and FADs. It could be shown that the KCSs derived from the plants C. abyssinica (CaKCS) and L. annua (LaKCS) as well as the yeast intrinsic Elo2p are able to increase the pool of VLCMUFAs when expressed together with the yeast intrinsic FAD Ole1p. In contrast to that, the KCS enzymes from A. thaliana, B. napus, S. chinensis as well as T. majus failed to increase the concentration of FAs (Fig. 2). A. thaliana and B. napus enzymes have been expressed previously in S. cerevisiae and were shown to be able to increase the pool of VLCFAs [24, 25]. However, in those studies no codon optimized sequences were used, and the enzymes were expressed in a different S. cerevisiae background strain (INVSc1), which might explain the different results observed in our study.
The first isolation and functional characterization of CaKCS has been performed by Mietkiewska et al. [26]. They heterologously expressed CaKCS in S. cerevisiae strain INVSc1 and showed that the strain produces the non-native to yeast FAs C20:1-Δ11, C20:1-Δ13, C22:1-Δ13, C22:1-∆15, C26:1-Δ17 and 26:1-∆19. Enzyme assays demonstrated that the preferred substrate of the enzyme is C20:1-CoA, and erucic acid (C22:1-Δ13) was synthesized as main product [26]. The heterologous expression of LaKCS in the S. cerevisiae strain INVSc1 led to the synthesis of C20:1-Δ11, C22:0, C22:1-Δ13, C24:0, C24:1-Δ15 and C26:1-Δ17 FAs [27].
A recent study in the oleaginous yeast Rhodosporidium toruloides, in which the influence of the expression of several plant derived KCS enzymes on the VLCFA pool was investigated, also showed that the highest amount of C22:1 FAs (24% of total fatty acids (TFAs)) was observed under expression of the KCS derived from C. abyssinica. This increase in C22:1 FAs was accompanied by a 50% decrease in C18:1 FAs. In our S. cerevisiae strain, the expression of CaKCS led to an erucic acid (C22:1-Δ13) content of 6% of TFAs and a 35.4% decrease in C18:1 FAs. In contrast to our study which identified the KCS from L. annua as best candidate for nervonic acid production, expression of LaKCS in R. toruloides did not lead to a high increase in nervonic acid, but a high increase in eicosenoic acid (21.4% of TFAs). In R. toruloides, the highest production of nervonic acid (10% of TFAs) was obtained by expression of the KCS from Cardamine graeca [28]. This comparison shows that the expression of plant derived KCS enzymes can lead to different results, depending on the yeast species.
The heterologous FADs we tested to increase the amount of MUFAs in S. cerevisiae included those from C. hyperboreus (ChDes9-1) and S. chinensis (SciFAD-SP) as well as two ADS proteins from A. thaliana (AtADS1.2 and AtADS1.4). Our results indicate that the combined expression of CaKCS and ChDes9-1 can increase the amount of C20:1 and C22:1 FAs, but not as strong as the combined expression of CaKCS and Ole1p. The production of C24:1 FAs was highest in a strain expressing LaKCS together with AtADS1.2 (Fig. 3).
ChDes9-1 was identified previously as a new type of Δ9-desaturase, which introduces a Δ9-double bond into VLCFAs ranging from C20:0 to C26:0 in its natural host, the marine copepod C. hyperboreus. When ChDes9-1 was expressed in the S. cerevisiae strain INVSc1, the yeast mainly produced two new FAs, C20:1-Δ9 and C26:1-Δ9. Upon supplementation of the medium with C18:0, also C22:1-Δ9, C22:1-Δ11, C24:1-Δ9, C24:1-Δ11 and C24:1-Δ13 could be detected, besides the production of C20:1-Δ9 and C26:1-Δ9. The highest conversion efficiency was observed for C20:0 [29].
AtADS1.2 was characterized recently in an attempt to determine the biological function of seven members of the ADS gene family in A. thaliana, which in total contains nine genes encoding FAD-like proteins. The enzymes were expressed in the yeast strain fat1Δ (BY4741 MATa his3D1 leu2D0 met15D0 ura3D0 YBR041w::kanMX4) from Invitrogen. Yeast cells expressing AtADS1.2 showed the synthesis of C22:1-Δ9, C24:1-Δ9 and C26:1-Δ9 FAs [30].
In contrast to the VLCMUFAs produced by ChDes9-1 and AtADS1.2, which mostly contain the double bond in the Δ9-position, jojoba oil consists of C20:1-Δ11, C22:1-Δ13 and C24:1-Δ15 FAs, which result from the elongation of C18:1-Δ9 [31, 32]. This means that the VLCMUFAs produced in a yeast strain heterologously expressing ChDes9-1 or AtADS1.2 probably do not represent the FA spectrum in jojoba oil. However, Ole1p introduces a double bond at the Δ9-position of FAs with a chain length up to C19 [14], indicating that a combined expression of a heterologous plant FAE/KCS together with the yeast intrinsic Ole1p can lead to the formation of jojoba-like VLCMUFAs in yeast by elongation of C18:1-Δ9 produced by Ole1p.
Therefore, the combination of CaKCS and Ole1p was identified as best enzymes for high level production of C20:1 and C22:1 FAs in S. cerevisiae and chosen to be expressed together with the WE biosynthesis enzymes MaFAldhR and SciWS to further boost WE production. The S. cerevisiae strain expressing CaKCS, Elo1p, Faa1p and Ole1p (strain LW22) showed a growth defect which was not detected in the same strain lacking the overexpression of CaKCS and Ole1p (strain LW21). This growth defect was also observed in strain LW24, expressing CaKCS and Ole1p in combination with the WE synthesizing enzymes MaFAldhR and SciWS as well as Elo2p, but not in strain LW23, expressing MaFAldhR, SciWS and Elo2p, but lacking CaKCS and Ole1p (Additional file 1: Figure S3). This observation indicates that the growth defect is due to an increased synthesis of VLCMUFAs in strains LW22 and LW24 which seem to be toxic to the cells as has been shown before for C16:1-Δ9 and C18:1-Δ9 FAs [5].
The synthesis of VLCMUFAs in a strain expressing a FAR from M. aquaeolei (MaFAldhR) and a WS from S. chinensis (SciWS) (strain LW24) triggered the synthesis of MUFOHs (C16:1-FOH, C18:1-FOH, C20:1-FOH and C22:1-FOH) and DUWEs (up to C44:2-WE) (Figs. 4, 5, Additional file 1: Figures S6, S7, S8). The analysis of the molecular composition of the WEs led to the identification of 73 WE species (Fig. 6). Whereas strain LW23 mostly showed the production of saturated C34-WE (25.30 ± 1.07 mol%), strain LW24 showed a clear trend towards longer and more unsaturated WEs with the most abundant one being C42:2-WE (18.26 ± 4.18 mol%), followed by C42:1-WE (17.43 ± 0.63 mol%), C40:1-WE (14.01 ± 1.55 mol%) and C40:2-WE (10.29 ± 0.41 mol%) (Additional file 1: Table S3). This composition of WEs is similar to the one of jojoba oil, which consists mostly of C42:2-WE (46.8 mol%), followed by C40:2-WE (20.7 mol%), C44:2-WE (6.0 mol%) and C38:2-WE (4.3 mol%), with the exception that jojoba oil only contains DUWEs [18]. These results clearly demonstrate that the production of jojoba-like WEs is possible in S. cerevisiae under supply of appropriate precursors and that MaFAldhR and SciWS are enzymes with a broad substrate specificity.
Conclusions
Our study shows that it is possible to drastically increase the amount of VLCMUFAs in S. cerevisiae by heterologous expression of a plant derived KCS together with a yeast intrinsic FAD. Moreover, we were able to show that the increase of VLCMUFAs can also trigger the synthesis of MUFOHs and DUWEs in a strain expressing a FAR and a WS. The general amount of jojoba-like WEs with a chain length of C40-WE to C44-WE was increased 2.3-fold by expression of a plant derived KCS together with a yeast intrinsic FAD compared to the same strain lacking the overexpression of those enzymes.
Methods
Strains and reagents
In this study, the strain S. cerevisiae CEN.PK 113-5D (MATa MAL2-8c SUC2 ura3-52), kindly provided by P. Kötter (University of Frankfurt, Germany), was used as the parental strain. Yeast strains constructed based on this strain are listed in Table 1. For construction and maintenance of plasmids, Escherichia coli DH5α was used.
Cultivation of strains
Saccharomyces cerevisiae CEN.PK 113-5D derived strains were grown in yeast extract-peptone-dextrose (YPD) medium containing 10 g/L yeast extract (Merck Millipore), 20 g/L peptone (Difco) and 20 g/L glucose (Merck Millipore) at 30 °C and 200 RPM. Yeast strains carrying a plasmid- or integration-based URA3 marker were selected on synthetic dextrose medium lacking uracil (SD-URA) plates, containing 6.9 g/L yeast nitrogen base (YNB) without amino acids (Formedium), 0.77 g/L complete supplement mixture (CSM) without uracil (Formedium), 20 g/L glucose and 20 g/L agar (Merck Millipore) with the pH adjusted to 5.5–6.0. To counter-select for the URA3 marker, strains were cultivated on plates with 5-fluoroorotic acid (5-FOA), containing 6.9 g/L YNB without amino acids, 0.77 g/L CSM without uracil, 0.05 g/L uracil, 20 g/L glucose, 20 g/L agar and 1.0 g/L 5-FOA (Sigma). Strains containing the kanMX marker were selected on YPD plates containing 200 mg/L G418 disulfate salt (Sigma Aldrich). To select for the natMX marker, yeast strains were grown on YPD plates containing 100 mg/L nourseothricin (Nordic Biosite).
To monitor FA, FOH and WE synthesis, growth experiments with S. cerevisiae strains were performed in modified minimal medium [33], containing 7.5 g/L (NH4)2SO4, 14.4 g/L KH2PO4, 20 g/L glucose and with the pH adjusted to 6.5 with 5 M NaOH. In case of strains not carrying a URA3 marker, 60 mg/L uracil (Alfa Aesar) were added to the medium. Single colonies were used to inoculate 5 mL of precultures, which were incubated for 2 days at 30 °C and 200 RPM before they were used to inoculate main cultures of 20–100 mL minimal medium in 100–500 mL shake flasks with an initial OD600 of 0.1. The growth rate of the strains was monitored by measuring the optical density of the cultures at 600 nm in a GENESYS™ 20 spectrophotometer (Thermo Fisher Scientific). The cells were harvested after 48 h of incubation by centrifugation at 1000×g for 5 min. After washing the cells with 5 mL phosphate buffer (10 mM KH2PO4, pH 7.5), the supernatant was removed, the pellet was frozen in liquid nitrogen and freeze-dried (Christ Alpha 2–4 LSC, Martin Christ Gefriertrocknungsanlagen GmbH) for 48 h.
Cultivations of E. coli were performed at 37 °C and 200 RPM in lysogeny broth (LB), containing 10 g/L tryptone (Merck Millipore), 5 g/L yeast extract (Merck Millipore), 10 g/L NaCl (Merck Millipore) or on plates containing additionally 20 g/L agar. E. coli strains carrying an AmpR marker were selected on LB plates containing 100 mg/L ampicillin (AppliChem).
Construction of plasmids and strains
In this study, we investigated genes coding for KCSs from A. thaliana (AtFAE) (NCBI accession no. AT4G34520), B. napus (BnKCS) (NCBI accession no. AF490459), C. abyssinica (CaKCS) (NCBI accession no. AY793549), L. annua (LaKCS) (NCBI accession no. EU871787), S. cerevisiae (ScElo2p) (NCBI accession no. YCR034 W), S. chinensis (SciFAE) (NCBI accession no. AAC49186) and T. majus (TmKCS) (NCBI accession no. AAC49186) as well as genes coding for FADs from Calanus hyperboreus (ChDes9-1) (NCBI accession no. AHL21604), S. cerevisiae (ScOle1p) (NCBI accession no. YGL055W) and S. chinensis (SciFAD-SP) (NCBI accession no. AAA33932). In case of SciFAD, a truncated version of the protein (SciFAD-SP), which did not include the 31 amino acids plastid localization signal at the N-terminus, was used. We also studied two acyl-coenzyme A (CoA) desaturase-like (ADS) proteins from A. thaliana (AtADS1.2/AtADS1.4) (NCBI accession no. AAC49186/AAC49186). Other genes investigated in this study include one coding for a FAR from M. aquaeolei VT8 (MaFAldhR = Maqu_2220) (NCBI accession no. YP_959486) as well as one coding for a WS from S. chinensis (SciWS) (NCBI accession no. AF149919) and the yeast intrinsic genes ACB1, ELO1, FAA1, IFA38, PHS1, TSC13.
The heterologous genes, which were codon optimized and synthesized by Genscript, are listed in Additional file 1: Table S1. The yeast intrinsic genes ACB1, ELO1, ELO2, FAA1, IFA38, OLE1, PHS1 and TSC13 (NCBI accession no. NP_011551.3/NP_012339.1/NP_009963.1/NP_014962.3/NP_009717.1/NP_011460.3/NP_012438.1/NP_010269.1) were amplified based on genomic DNA of S. cerevisiae CEN.PK 113-5D. Genes coding for a KCS/FAE as well as for Ole1p were integrated into the region 839226–840357 of chromosome XII (XII-5). Genes coding for Faa1p and Elo1p were integrated into the region 236336–237310 of chromosome X (X-4).
Oligonucleotides used for amplification of genes included specific overhangs to promoter and terminator sequences for fusion of promoter-gene-terminator parts, using PrimeStar DNA polymerase (TaKaRa Bio). They were custom synthesized by Eurofins and are listed in Additional file 1: Table S2. Episomal plasmids containing combinations of a KCS and a FAD were constructed via modular pathway engineering based on the background plasmid pYX212 [22] (Additional file 1: Figure S1). All episomal plasmids were first assembled in S. cerevisiae CEN.PK 113-5D and then extracted by using the Zymoprep Yeast Plasmid Miniprep II kit (Zymo Research Corp). The plasmids were re-transformed into E. coli DH5α competent cells based on the method by Inoue et al. [34]. After purification of the plasmids, they were verified by restriction analysis and sequencing. Finally, the plasmids were transformed into the desired yeast strain. Integrative plasmids were constructed based on the EasyClone(-Marker Free) vector toolkit [35,36,37] (Additional file 1: Figure S2). Yeast competent cells were prepared and transformed with 1 µg of plasmid DNA according to the lithium acetate/single-stranded carrier DNA/polyethylene glycol method [38]. Restriction enzymes, DNA gel purification and plasmid extraction kits were purchased from Thermo Fisher Scientific.
Construction of the background strain S. cerevisiae CEN.PK 113-5D elo3Δ X-2::pMPC3::ACC1** X-3::IFA38::PHS1::TSC13::ACB1 XI-5::Cas9
The background strain S. cerevisiae CEN.PK 113-5D elo3Δ was constructed in a previous study [21] as well as the plasmid containing the double mutated ACC1** gene under control of the S. cerevisiae MPC3 promoter and CYC1 terminator [39]. The plasmid contains a kanMX marker, flanked by loxP sites, for selection, and homologous sequences guiding the integration of the fragment into the region 194944–195980 of chromosome X (X-2) [37]. The genes ACB1, IFA38, PHS1 and TSC13 were integrated into the region 223616–224744 of chromosome X (X-3). The ACB1 gene is flanked by the PGK1 promoter and CYC1 terminator of S. cerevisiae, the IFA38 gene is flanked by the TEF1 promoter and FBA1 terminator of S. cerevisiae, the PHS1 gene is flanked by the truncated HXT7 promoter and TDH2 terminator of S. cerevisiae and the TSC13 gene is flanked by the TDH3 promoter and ADH1 terminator of S. cerevisiae. The plasmid contains a Kluyveromyces lactis URA3 marker, which is flanked by direct repeats [37]. All plasmids were linearized by NotI and the purified linear integration fragments were transformed into S. cerevisiae CEN.PK 113-5D elo3Δ by chemical transformation. To reuse the URA3 marker, the marker was looped out by cultivating the cells overnight in YPD medium and plating them twice on 5-FOA plates before looping out of the marker was confirmed by replica plating on YPD and SD-URA plates. The kanMX marker was removed by transforming the cells with a plasmid containing the cre-recombinase gene under control of the S. cerevisiae GAL1 promoter, cultivating the cells overnight in yeast extract-peptone-galactose (YPG) medium containing 10 g/L yeast extract (Merck Millipore), 20 g/L peptone (Difco) and 20 g/L galactose (Merck Millipore) and replica plating the cells on YPD and YPD + G418.
The Cas9 gene was integrated as linear fragment under control of the TEF1 promoter and CYC1 terminator of S. cerevisiae into the region 117779–118957 of chromosome XI (XI-5). The fragment contains a K. lactis URA3 marker, which is flanked by direct repeats. The URA3 marker was removed in the final strains as described above.
TLC
For preparative TLC, 20–50 mg of freeze dried cells were weighed and mixed with 10 µg lauryl laurate as well as 10 µg heptadecanol as internal standards. The extraction of lipids was performed as described previously [40] with the exception that the sample was redissolved in 50 µL chloroform/methanol (2:1, v/v). The sample was loaded onto a TLC silica gel 60 F254 plate (Merck Millipore). The mobile phase used was composed of heptane, 2-propanol and acetic acid with a ratio of 95:5:1 (v/v/v). A standard composed of 100 µg each of cholesterol, oleic acid, triolein, methyl oleate and cholesteryl oleate as well as a separate WE standard composed of 50 µg lauryl laurate were loaded onto the plate for identification of sterols, FFAs, TAGs, fatty acid methyl esters (FAMEs), SEs and WEs. The analytical standards were purchased from Nu-Check Prep, Inc with an exact weight amount. After drying of the plate, the visualization of spots was performed by spraying the plate with 0.05% 2,7-dichlorofluorescein in ethanol and exposing it to ultraviolet radiation. The spots of the TLC plate corresponding to FOHs and WEs, respectively, were scraped off with a razor blade and extracted with a mix of 3 mL hexane, 2 mL methanol and 2 mL milliQ water. The tube was vigorously vortexed, and after centrifugation at 3000 RPM for 5 min, the upper layer was transferred to a clean tube. This solution, containing the FOHs and WEs, respectively, was dried by vacuum evaporation using a miVac concentrator (Genevac) before the samples were finally dissolved in 200 µL of hexane in case of WEs and 200 µL of ethyl acetate in case of FOHs.
Gas chromatography-mass spectrometry (GC/MS) analysis of FAMEs and WEs/
GC-flame ionization detector (GC-FID) analysis of FOHs
After separation of WEs from other lipids via TLC, the WE containing fraction was isolated, purified and analyzed by GC/MS (Focus GC ISQ™ single quadrupole GC; Thermo Fisher Scientific), using a ZB-50 column (L = 30 m, ID = 0.32 mm, df = 0.5 μm) (Phenomenex). The inlet temperature was set to 375 °C, the helium (carrier) gas flow to 1 mL/min splitless. The initial oven temperature was set to 150 °C and held for 10 min. Then the temperature was ramped to 350 °C by 7.5 °C/min and held for 10 min. The mass transfer line temperature was set to 250 °C, the ion source temperature was set to 250 °C and a full scan for m/z of 50 to 650 was performed. WEs were identified by comparison to analytical WE standards which were purchased from Nu-Check Prep, Inc. with an exact weight amount. They were dissolved in hexane and analyzed using the same protocol and column as for the samples. WEs are designated according to the nomenclature CX:Y-WE, with X indicating the total number of carbon atoms and Y indicating the total number of double bonds in the WE, e.g. C30:1, palmityl myristoleate. FOHs which were isolated and purified from TLC were analyzed via GC-FID as described previously [17]. FOH standards were purchased from Nu-Check Prep, Inc. with an exact weight amount. They were dissolved in ethyl acetate and analyzed using the same protocol and column as for the samples. FOHs are designated according to the nomenclature CX:Y-FOH, with X indicating the number of carbons and Y indicating the number of double bonds, e.g. C16:0, palmityl alcohol (hexadecanol); C16:1, palmitoleyl alcohol (hexadecenol). FAMEs were prepared and analyzed as described previously [41]. FAME standards were purchased from Sigma Aldrich. FAs are designated according to the nomenclature CX:Y, with X indicating the number of carbons and Y indicating the number of double bonds (e.g. C16:0, palmitic acid (hexadecanoic acid); C16:1-Δ9, palmitoleic acid (hexadecenoic acid)). If the position of the double bond is indicated, it is counted from the carboxyl group (Δ) and unless otherwise noted, the Z (cis) species (e.g. C16:1-Δ9, cis-Δ9 hexadecenoic acid).
Determination of molecular composition of WEs
Yeast cell lysis and lipid extraction were carried out at 4 °C as previously described [42]. In short, yeast cells (~ 10 ODu) were resuspended in 1 mL 155 mM ammonium formate and lysed with 400 μL of acid-washed glass beads using a cell disruptor.
Lipids were extracted from 0.4 ODu of yeast lysate in a total of 200 μL 155 mM ammonium formate by adding 990 μL chloroform/methanol (2:1 v/v) and mixing (1400 RPM, 2 h, 4 °C) using a ThermoMixer (Eppendorf). The lower organic phase was collected after centrifugation (1000×g, 2 min, 4 °C). Lipid extracts were dried and kept at − 20 °C until analyzed. Lipid extracts were analyzed by positive ion mode MSALL analysis, as previously described [43], using an Orbitrap Fusion Tribrid (Thermo Fisher Scientific) equipped with a robotic nanoflow ion source, TriVersa NanoMate (Advion Biosciences). In short, lipid extracts were dissolved in 100 μL chloroform/methanol (1:2 v/v). For MSALL analysis, 10 μL lipid extract was mixed with 12.9 μL 13.3 mM ammonium formate acetate in 2-propanol and infused using a back pressure of 1.25 psi and ionization voltage of + 0.96 kV. Survey Fourier Transform MS (FTMS) data were recorded using a target resolution setting of 500,000, a max injection time of 100 ms, automated gain control of 1e5, and three microscans. Sequential FTMS2 data were recorded in 1.0008 amu steps across the precursor m/z range 320.3–920.8 using a quadrupole ion isolation with a width of 1.0 amu, HCD fragmentation with collision energy at 22%, max injection time of 100 ms, automated gain control of 5e4, three microscans, and a target resolution of 30,000. All FTMS data were acquired in profile mode and using an ion transfer tube temperature of 275 °C. WE molecules detected by MSALL analysis were identified and quantified using ALEX123 software and SAS 9.3 [43,44,45]. Searches of FTMS and FTMS2 data were done with mass tolerances of 0.0025 and 0.0050 amu, respectively, and using an in silico-generated database of ammoniated, even-chained WE molecules having a total of 28 to 56 C-atoms and 0 to 2 double bonds in their FOH and FA chains. WE molecular species detected by FTMS and FTMS2 analysis in all samples were annotated by molecular composition nomenclature denoting the number of C-atoms and double bonds in both the FOH and the FA chain, following the general pattern CX1:Y1-CX2:Y2, with the first part of the abbreviation referring to the FOH moiety and the second part referring to the FA moiety of the WE (e.g. C16:1-C14:0, palmityl myristoleate). The “X” in the formula indicates the number of carbon atoms in the alcohol and acid moiety, respectively, and the “Y” indicates the number of double bonds in each moiety. Data are presented as mean ± standard deviation of three biological replicates, expressed as mol% per all monitored WE molecules (calculated by normalizing the intensity of individual WE molecules to the total intensity of all identified WE molecules).
Abbreviations
- At :
-
Arabidopsis thaliana
- Bn :
-
Brassica napus
- Ch :
-
Calanus hyperboreus
- CDW:
-
cell dry weight
- Ca :
-
Crambe abyssinica
- DUWE:
-
diunsaturated wax ester
- ER:
-
endoplasmic reticulum
- ECR:
-
enoyl-CoA reductase
- FAD:
-
fatty acid desaturase
- FAE (KCS):
-
fatty acid elongase
- FAME:
-
fatty acid methyl ester
- FAS:
-
fatty acid synthase
- FA:
-
fatty acid
- FAR/FAldhR:
-
fatty acyl reductase
- FACoA:
-
fatty acyl-CoA
- FOH:
-
fatty alcohol
- FID:
-
flame ionization detector
- FT:
-
fourier transform
- FFA:
-
free fatty acid
- GC:
-
gas chromatography
- La :
-
Lunaria annua
- Ma :
-
Marinobacter aquaeolei VT8
- MS:
-
mass spectrometry
- MUFA:
-
monounsaturated fatty acid
- MUFOH:
-
monounsaturated fatty alcohol
- ODu:
-
optical density (units)
- Sc :
-
Saccharomyces cerevisiae
- Sci :
-
Simmondsia chinensis
- SD:
-
standard deviation
- SE:
-
steryl ester
- TLC:
-
thin layer chromatography
- TFAs:
-
total fatty acids
- TAG:
-
triacylglycerol
- Tm :
-
Tropaeolum majus
- (V)LCFA:
-
(very) long-chain fatty acid
- (V)LCFOH:
-
(very) long-chain fatty alcohol
- (V)LCMUFA:
-
(very) long-chain monounsaturated fatty acid
- WE:
-
wax ester
- WS:
-
wax synthase
- HCD:
-
β-hydroxyacyl-CoA dehydratase
- KCR:
-
β-ketoacyl-CoA reductase
- KCS (FAE):
-
β-ketoacyl-CoA synthase (fatty acid elongase)
References
Welch JW, Burlingame AL. Very long-chain fatty acids in yeast. J Bacteriol. 1973;115:464–6.
Okuyama H, Saito M. Regulation by temperature of the chain length of fatty acids in yeast. J Biol Chem. 1979;254:12281–4.
Suomalainen H, Keränen AJA. The fatty acid composition of baker’s and brewer’s yeast. Chem Phys Lipids. 1968;2:296–315.
Grillitsch K, Connerth M, Köfeler H, Arrey TN, Rietschel B, Wagner B, et al. Lipid particles/droplets of the yeast Saccharomyces cerevisiae revisited: lipidome meets proteome. Biochim Biophys Acta. 2011;1811:1165–75.
Petschnigg J, Wolinski H, Kolb D, Zelling G, Kurat CF, Natter K, et al. Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J Biol Chem. 2009;284:30981–93.
Tehlivets O, Scheuringer K, Kohlwein SD. Fatty acid synthesis and elongation in yeast. Biochim Biophys Acta. 2007;1771:255–70.
Hofbauer HF, Schopf FH, Schleifer H, Knittelfelder OL, Pieber B, Rechberger GN, et al. Regulation of gene expression through a transcriptional repressor that senses acyl-chain length in membrane phospholipids. Dev Cell. 2014;29:729–39.
Duronio RJ, Knoll LJ, Gordon JI. Isolation of a Saccharomyces cerevisiae long chain fatty acyl:CoA synthetase gene (FAA1) and assessment of its role in protein N-myristoylation. J Cell Biol. 1992;117:515–29.
Johnson DR, Knoll LJ, Rowley N, Gordon JI. Genetic analysis of the role of Saccharomyces cerevisiae acyl-CoA synthetase genes in regulating protein N-myristoylation. J Biol Chem. 1994;269:18037–46.
Johnson DR, Knoll LJ, Levin DE, Gordon JI. Saccharomyces cerevisiae contains four fatty acid activation (FAA) genes: an assessment of their role in regulating protein N-myristoylation and cellular lipid metabolism. J Cell Biol. 1994;127:751–62.
Faergeman NJ, DiRusso CC, Elberger A, Knudsen J, Black PN. Disruption of the Saccharomyces cerevisiae homologue to the murine fatty acid transport protein impairs uptake and growth on long-chain fatty acids. J Biol Chem. 1997;272:8531–8.
Toke D, Martin CE. Isolation and characterization of a gene affecting fatty acid elongation in Saccharomyces cerevisiae. J Biol Chem. 1996;271:18413–22.
Oh CS, Toke DA, Mandala S, Martin CE. ELO2 and ELO3, homologues of the Saccharomyces cerevisiae ELO1 gene, function in fatty acid elongation and are required for sphingolipid formation. J Biol Chem. 1997;272:17376–84.
Martin CE, Oh C-S, Kandasamy P, Chellapa R, Vemula M. Yeast desaturases. Biochem Soc Trans. 2002;30:1080–2.
Martin CE, Oh C-SS, Jiang Y. Regulation of long chain unsaturated fatty acid synthesis in yeast. Biochim Biophys Acta. 2007;1771:271–85.
Marella ER, Holkenbrink C, Siewers V, Borodina I. Engineering microbial fatty acid metabolism for biofuels and biochemicals. Curr Opin Biotechnol. 2018;50:39–46.
Yu T, Zhou YJ, Wenning L, Liu Q, Krivoruchko A, Siewers V, et al. Metabolic engineering of Saccharomyces cerevisiae for production of very long chain fatty acid-derived chemicals. Nat Commun. 2017;8:15587.
Iven T, Herrfurth C, Hornung E, Heilmann M, Hofvander P, Stymne S, et al. Wax ester profiling of seed oil by nano-electrospray ionization tandem mass spectrometry. Plant Methods. 2013;9:24.
Sánchez M, Avhad MR, Marchetti JM, Martínez M, Aracil J. Jojoba oil: a state of the art review and future prospects. Energy Convers Manag. 2016;129:293–304.
Nielsen J. Synthetic biology for engineering acetyl coenzyme A metabolism in yeast. MBio. 2014;5:e02153–214.
Wenning L, Yu T, David F, Nielsen J, Siewers V. Establishing very long-chain fatty alcohol and wax ester biosynthesis in Saccharomyces cerevisiae. Biotechnol Bioeng. 2017;114:1025–35.
Zhou YJ, Gao W, Rong Q, Jin G, Chu H, Liu W, et al. Modular pathway engineering of diterpenoid synthases and the mevalonic acid pathway for miltiradiene production. J Am Chem Soc. 2012;134:3234–41.
de Jong BW, Shi S, Valle-Rodríguez JO, Siewers V, Nielsen J. Metabolic pathway engineering for fatty acid ethyl ester production in Saccharomyces cerevisiae using stable chromosomal integration. J Ind Microbiol Biotechnol. 2015;42:477–86.
Katavic V, Mietkiewska E, Barton DL, Giblin EM, Reed DW, Taylor DC. Restoring enzyme activity in nonfunctional low erucic acid Brassica napus fatty acid elongase 1 by a single amino acid substitution. Eur J Biochem. 2002;269:5625–31.
Trenkamp S, Martin W, Tietjen K. Specific and differential inhibition of very-long-chain fatty acid elongases from Arabidopsis thaliana by different herbicides. Proc Natl Acad Sci USA. 2004;101:11903–8.
Mietkiewska E, Brost JM, Giblin EM, Barton DL, Taylor DC. Cloning and functional characterization of the fatty acid elongase 1 (FAE1) gene from high erucic Crambe abyssinica cv. Prophet. Plant Biotechnol J. 2007;5:636–45.
Guo Y, Mietkiewska E, Francis T, Katavic V, Brost JM, Giblin M, et al. Increase in nervonic acid content in transformed yeast and transgenic plants by introduction of a Lunaria annua L. 3-ketoacyl-CoA synthase (KCS) gene. Plant Mol Biol. 2009;69:565–75.
Fillet S, Ronchel C, Callejo C, Fajardo MJ, Moralejo H, Adrio JL. Engineering Rhodosporidium toruloides for the production of very long-chain monounsaturated fatty acid-rich oils. Appl Microbiol Biotechnol. 2017;101:7271–80.
Meesapyodsuk D, Qiu X. Structure determinants for the substrate specificity of acyl-CoA Δ9 desaturases from a marine copepod. ACS Chem Biol. 2014;9:922–34.
Smith MA, Dauk M, Ramadan H, Yang H, Seamons LE, Haslam RP, et al. Involvement of Arabidopsis acyl-coenzyme A desaturase-like2 (At2g31360) in the biosynthesis of the very-long-chain monounsaturated fatty acid components of membrane lipids. Plant Physiol. 2013;161:81–96.
Miwa TK. Jojoba oil wax esters and derived fatty acids and alcohols: gas chromatographic analyses. J Am Oil Chem Soc. 1971;48:259–64.
Miwa TK. Structural determination and uses of jojoba oil. J Am Oil Chem Soc. 1984;61:407–10.
Verduyn C, Postma E, Scheffers WA, van Dijken JP. Effect of benzoic acid on metabolic fluxes in yeasts: a continuous-culture study on the regulation of respiration and alcoholic fermentation. Yeast. 1992;8:501–17.
Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–8.
Jessop-Fabre MM, Jakočiūnas T, Stovicek V, Dai Z, Jensen MK, Keasling JD, et al. EasyClone-MARKERFree: a vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol J. 2016;11:1110–7.
Jensen NB, Strucko T, Kildegaard KR, David F, Maury J, Mortensen UH, et al. EasyClone: method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 2014;14:238–48.
Mikkelsen MD, Buron LD, Salomonsen B, Olsen CE, Hansen BG, Mortensen UH, et al. Microbial production of indolylglucosinolate through engineering of a multi-gene pathway in a versatile yeast expression platform. Metab Eng. 2012;14:104–11.
Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96.
Bergenholm D, Gossing M, Wei Y, Siewers V, Nielsen J. Modulation of saturation and chain length of fatty acids in Saccharomyces cerevisiae for production of cocoa butter-like lipids. Biotechnol Bioeng. 2018;115:932–42.
Khoomrung S, Chumnanpuen P, Jansa-Ard S, Ståhlman M, Nookaew I, Borén J, et al. Rapid quantification of yeast lipid using microwave-assisted total lipid extraction and HPLC-CAD. Anal Chem. 2013;85:4912–9.
Khoomrung S, Chumnanpuen P, Jansa-Ard S, Nookaew I, Nielsen J. Fast and accurate preparation fatty acid methyl esters by microwave-assisted derivatization in the yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2012;94:1637–46.
Ejsing CS, Sampaio JL, Surendranath V, Duchoslav E, Ekroos K, Klemm RW, et al. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc Natl Acad Sci USA. 2009;106:2136–41.
Almeida R, Pauling JK, Sokol E, Hannibal-Bach HK, Ejsing CS. Comprehensive lipidome analysis by shotgun lipidomics on a hybrid quadrupole-orbitrap-linear ion trap mass spectrometer. J Am Soc Mass Spectrom. 2015;26:133–48.
Ellis SR, Paine MRL, Eijkel GB, Pauling JK, Husen P, Jervelund MW, et al. Automated, parallel mass spectrometry imaging and structural identification of lipids. Nat Methods. 2018;15:515–8.
Husen P, Tarasov K, Katafiasz M, Sokol E, Vogt J, Baumgart J, et al. Analysis of lipid experiments (ALEX): a software framework for analysis of high-resolution shotgun lipidomics data. PLoS ONE. 2013;8:e79736.
Authors’ contributions
LW designed the study, conducted the experiments (except for the molecular species profile of the WEs), analyzed the results and wrote the manuscript. FD, VS and JN contributed to designing the study and writing the manuscript. RRS and CSE performed the quantitative analysis of molecular WE species. All authors read and approved the final manuscript.
Acknowledgements
Help with the analytics by the Chalmers Mass Spectrometry Infrastructure (CMSI) is much acknowledged.
Competing interests
LW, FD, VS and JN have filed for patent protection of part of the work presented here.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This research project received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA Grant Agreement No 606795, project acronym: YEASTCELL, from the Strategic Research Council (SSF) in Sweden, and from the Novo Nordisk Foundation (grant no. NNF10CC1016517). In addition, this project has received funding from the European Union’s Horizon 2020 Framework Programme for Research and Innovation—Grant Agreement No. 720824. We also acknowledge funding from the Knut and Alice Wallenberg Foundation. C.S.E. is supported by the VILLUM Foundation (VKR023439) and the VILLUM Center for Bioanalytical Sciences (VKR023179).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Table S1.
Codon optimized sequences of genes used in this study. Table S2. Sequences of oligonucleotides used in this study. Table S3. Distribution of wax ester species (mol%) in strains LW23 and LW24. The values represent the mean ± SD of three biological replicates of strains LW23 and LW24, respectively. Figure S1. DNA pathway assembly constructs. Genes coding for a heterologous fatty acyl reductase (FAR), a wax synthase (WS), a fatty acid elongase (FAE) or a fatty acid desaturase (FAD) were synthesized with a codon optimization for S. cerevisiae. The ELO2 gene and the OLE1 gene were amplified based on g-DNA from S. cerevisiae CEN.PK 113-5D. The promoter pTPI1 and the terminator pYX212t are homologous to the respective promoter and terminator on the pYX212 plasmid. All plasmids were constructed using the modular pathway engineering strategy [22]. Figure S2. Integration constructs. The gene coding for CaKCS was synthesized with a codon optimization for S. cerevisiae. The IFA38, PHS1, TSC13, ACB1, FAA1, ELO1 and OLE1 genes were amplified based on g-DNA from S. cerevisiae CEN.PK 113-5D. The ACC1** linear fragment, carrying a kanMX marker under the Ashbya gossypii TEF1 promoter/terminator and flanked by loxP sites, was integrated at position X-2. The TSC13/PHS1/IFA38/ACB1 linear fragment, carrying a Kluyveromyces lactis URA3 marker flanked by direct repeats, was integrated at position X-3. The FAA1/ELO1 linear fragment was integrated at position X-4. The Cas9 linear fragment was integrated at position XI-5 and the CaKCS/OLE1 linear fragment at position XII-5 in the genome. Integrative plasmids were constructed based on the EasyClone(-Marker Free) vector toolkit [35,36,37]. Figure S3. Growth behavior of strains LW21, LW22, LW23 and LW24 in minimal medium containing 20 g/L glucose. Figure S4. Thin layer chromatography. 1, TLC standard = 100 µg cholesterol, 100 µg oleic acid, 100 µg triolein, 100 µg methyl oleate, 100 µg cholesteryl oleate; 2, wax ester standard = 50 µg lauryl laurate (C24:0); 3, strain LW21 clone1; 4, strain LW21 clone 2; 5, strain LW21 clone 3; 6, strain LW23 clone 1; 7, strain LW23 clone 2; 8, strain LW23 clone 3. The TLC was performed as described in Materials and Methods. Figure S5. Thin layer chromatography. 1, TLC standard = 100 µg cholesterol, 100 µg oleic acid, 100 µg triolein, 100 µg methyl oleate, 100 µg cholesteryl oleate; 2, wax ester standard = 50 µg lauryl laurate (C24:0); 3, strain LW22 clone 1; 4, strain LW22 clone 2; 5, strain LW22 clone 3; 6, strain LW24 clone 1; 7, strain LW24 clone 2; 8, strain LW24 clone 3. The TLC was performed as described in Materials and Methods. Figure S6. GC-FID chromatograms of fatty alcohols (FOHs) isolated from strains LW21 (A), LW22 (B), LW23 (C) and LW24 (D). The peak labeled with (I) corresponds to the internal standard used (C17:0-FOH). A mixture of standards (c = 200 µg/mL) containing the saturated FOHs (II), C16:0-FOH; (III), C18:0-FOH; (IV), C20:0-FOH; (V), C22:0-FOH and (VI), C24:0-FOH is shown in (E). A mixture of standards (c = 200 µg/mL) containing the monounsaturated FOHs (II), C16:1-FOH; (III), C18:1-FOH; (IV), C20:1-FOH; (V), C22:1-FOH and (VI), C24:1-FOH is shown in (F). The chromatogram in (G) shows a combination of the standards (E), (c = 50 µg/mL); (F), (c = 50 µg/mL) and C17:0-FOH (c = 250 µg/mL). The strains were grown for 48 h in minimal medium containing 20 g/L glucose. Figure S7. GC/MS chromatograms of wax esters (WEs) isolated from strains LW21 (A), LW22 (B), LW23 (C) and LW24 (D). The peak labeled with (I) corresponds to the internal standard used (C24:0-WE). A mixture of standards (c = 10 µg/mL) containing the saturated WEs (I), C24:0-WE; (II), C30:0-WE; (III), C32:0-WE; (IV), C34:0-WE; (V), C36:0-WE, (VI), C38:0-WE; (VII), C40:0-WE and (VIII), C42:0-WE is shown in (E). A mixture of standards (c = 10 µg/mL) containing the diunsaturated WEs (III), C32:2-WE; (IV), C34:2-WE; (V), C36:2-WE; (VI), C38:2-WE; (VII), C40:2-WE; (VIII), C42:2-WE and (IX), C44:2-WE is shown in (F). The strains were grown for 48 h in minimal medium containing 20 g/L glucose. Figure S8. Mass spectra of selected peaks of total ion chromatograms of lipids extracted from strain LW24. Mass spectrum of the C38:2-WE (MW = 561 g/mol), eluting after 27.95 min with the specific m/z peak of 561 (A). Mass spectrum of the C40:2-WE (MW = 589 g/mol), eluting after 29.23 min with the specific m/z peak of 589 (B). Mass spectrum of the C42:2-WE (MW = 617 g/mol), eluting after 30.73 min with the specific m/z peak of 617 (C). Mass spectrum of the C44:2-WE (MW = 645 g/mol), eluting after 32.53 min with the specific m/z peak of 645 (D).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wenning, L., Ejsing, C.S., David, F. et al. Increasing jojoba-like wax ester production in Saccharomyces cerevisiae by enhancing very long-chain, monounsaturated fatty acid synthesis. Microb Cell Fact 18, 49 (2019). https://doi.org/10.1186/s12934-019-1098-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-019-1098-9