
Abstract
Background
Oxidized glutathione (GSSG) is the preferred form for industrial mass production of glutathione due to its high stability compared with reduced glutathione (GSH). In our previous study, over-expression of the mitochondrial thiol oxidase ERV1 gene was the most effective for high GSSG production in Saccharomyces cerevisiae cells among three types of different thiol oxidase genes.
Results
We improved Erv1 enzyme activity for oxidation of GSH and revealed that S32 and N34 residues are critical for the oxidation. Five engineered Erv1 variant proteins containing S32 and/or N34 replacements exhibited 1.7- to 2.4-fold higher in vitro GSH oxidation activity than that of parental Erv1, whereas the oxidation activities of these variants for γ-glutamylcysteine were comparable. According to three-dimensional structures of Erv1 and protein stability assays, S32 and N34 residues interact with nearby residues through hydrogen bonding and greatly contribute to protein stability. These results suggest that increased flexibility by amino acid replacements around the active center decrease inhibitory effects on GSH oxidation. Over-expressions of mutant genes coding these Erv1 variants also increased GSSG and consequently total glutathione production in S. cerevisiae cells. Over-expression of the ERV1 S32A gene was the most effective for GSSG production in S. cerevisiae cells among the parent and other mutant genes, and it increased GSSG production about 1.5-fold compared to that of the parental ERV1 gene.
Conclusions
This is the first study demonstrating the pivotal effects of S32 and N34 residues to high GSH oxidation activity of Erv1. Furthermore, in vivo validity of Erv1 variants containing these S32 and N34 replacements were also demonstrated. This study indicates potentials of Erv1 for high GSSG production.
Similar content being viewed by others


Background
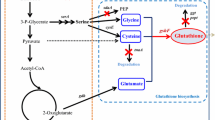
Glutathione is the most abundant thiol-containing tripeptide in all living organisms [1]. Glutathione is widely used in the medical, food, and cosmetic industries [2, 3] due to its various physiological functions such as acting as an antioxidant, a detoxifier of xenobiotics, and an immune booster [4–9]. Thus the demand for glutathione has increased in recent years. Glutathione is industrially produced mainly by fermentation using Saccharomyces cerevisiae, which contains a high concentration of glutathione and is accepted as a food-producing microorganism.
Glutathione is biologically synthesized by γ-glutamylcysteine (γ-GC) synthetase (GCS, EC 6.3.2.2) encoded by GSH1 and by glutathione synthetase (GS, EC 6.3.2.3) encoded by GSH2 from three precursor amino acids. GCS catalyzes the reaction to form γ-GC from l-glutamic acid and l-cysteine. GS catalyzes the reaction to form glutathione from γ-GC and glycine. Other glutathione-related enzymes include thiol oxidase (TO, EC 1.8.3.2) encoded by ERV1, ERV2, and ERO1, and glutathione-disulfide reductase (GR, EC 1.8.1.7) encoded by GLR1. TOs catalyze the reaction to form oxidized glutathione (GSSG) from reduced glutathione (GSH) by oxidizing the thiols, and GR catalyzes the reaction to form GSH from GSSG by reducing a disulfide bond.
In many cases, reduced glutathione (GSH) primarily exists to respond to oxidative stress in living organisms [10], and thus many glutathione-related studies have focused on GSH [11–13]. However, GSSG is of interest in glutathione production. In industrial mass production of glutathione, GSSG is preferable due to its higher stability. Furthermore, enhancements of GSSG production help to avoid a negative feedback regulation by GSH1, and consequently increase total glutathione production by fermentation with S. cerevisiae [14, 15]. GSSG also has advantages in utilization; it has the same extent of antioxidant activity in the intestines after dietary intake as GSH [16], and promotes plant growth more potently than GSH [17, 18].
In our previous study, deletion of the GLR1 gene and over-expression of the mitochondrial thiol oxidase ERV1 gene was the most effective for generating high GSSG production among three types of different thiol oxidase genes in S. cerevisiae cells [15]. Therefore, in this study, we improved the enzymatic activity of Erv1 for GSH oxidation by amino acid replacements, and consequently enhanced GSSG production in S. cerevisiae by using mutant ERV1 genes.
Methods
Strains, media, and primers
Saccharomyces cerevisiae GCIΔGLR1, GSH1/GSH2 cocktail δ-integrated and GLR1 deleted YPH499 (ABC1193/NBRC 10505) strain was previously constructed [19] and used for glutathione production in this study. Saccharomyces cerevisiae GCIΔGLR1 derivative strains were aerobically grown at 30 °C in yeast extract-peptonedextrose (YPD) medium (10 g l−1 yeast extract, 20 g l−1 bacto-peptone, and 20 g l−1 glucose) supplemented with 0.5 mg l−1 aureobasidin A (Aba). Escherichia coli NovaBlue (Novagen, Darmstadt, Germany) strain was used for DNA manipulation. E. coli Rosetta™(DE3)pLysS (Novagen) strain was used to produce recombinant proteins. E. coli strains were aerobically grown at 37 °C in Luria–Bertani (LB) medium (10 g l−1 tryptone, 5 g l−1 yeast extract, and 5 g l−1 sodium chloride). Ampicillin (Amp; 50 mg l−1) and chloramphenicol (Cm; 50 mg l−1) were added as necessary. The primer sequences used in this study are summarized in Additional file 1: Table S1.
Construction of plasmids
The ERV1 gene was amplified by polymerase chain reaction (PCR) from complementary DNA (cDNA) of S. cerevisiae YPH499 using primers ERV1F1 and ERV1R1. The cDNA was prepared by reverse transcription PCR using a PrimeScrip RT-PCR Kit (Takara Bio, Otsu, Japan) from total RNA extracted from S. cerevisiae YPH499 cells using NucleoSpin RNA (Takara Bio). The PCR product was cloned between SphI and BamHI sites of pUC19 (Takara Bio). After the sequence was checked, the ERV1 gene was subcloned between NdeI and XhoI sites of pET-22b (Novagen) to give pET-ERV1. pET-ERV1 was used for Erv1 protein preparation. The plasmids for preparation of Erv1 variant proteins were constructed by inverse PCR using corresponding primer pairs and templates (Additional file 1: Table S1). For expression of ERV1 and its mutant genes in S. cerevisiae cells, the ERV1 gene was amplified by PCR from pET-ERV1 using primers ERV1F2 and ERV1R2. The PCR product was cloned between NheI and BamHI sites of pGK406 designed for target gene expression in S. cerevisiae [20]. The mutant genes coding Erv1 variants were also amplified by the same primer pairs and cloned into pGK406.
Plasmid introduction into S. cerevisiae
Derivatives of pGK406 were introduced into S. cerevisiae cells using the lithium acetate method [21, 22]. Transformants were selected by uracil auxotrophy. Target gene insertion into the genomic DNA of each transformant was confirmed by PCR using the appropriate primer sets.
Preparation of recombinant proteins
The E. coli Rosetta™(DE3)pLysS strain harboring pET-ERV1 was aerobically grown in 5 ml of liquid LB medium supplemented with Amp and Cm at 37 °C for 18 h. The 1 ml of grown cells was inoculated into 100 ml of liquid LB medium supplemented with Amp, Cm, and 1% lactose, and aerobically grown at 20 °C for 48 h. The grown cells were then pelleted by centrifugation (16,000×g, 10 min) and resuspended in 20 mM potassium phosphate buffer (pH 7.0) containing 500 mM NaCl. The cell suspension was sonicated and centrifuged (16,000×g, 10 min) to remove cell debris. The His-tagged Erv1 protein in the supernatant was purified by TALON® Metal Affinity Resin (Takara Bio). The Erv1 variants were also prepared by the same method.
Enzyme assay
The activities of recombinant Erv1 and its variants were determined by measuring the initial velocity of product formation. The assay mixture containing 50 mM potassium phosphate buffer (pH 7.0), 10 mM GSH or γ-GC, and the purified recombinant protein was incubated at 30 °C for 10 min. The reaction was then stopped by adding an equal volume of 20% (w/v) trichloroacetic acid. The formed products were measured using high performance liquid chromatography (HPLC) as mentioned below. The protein concentrations were assayed by the Bradford method. All assays were separately performed three times.
Glutathione production
The S. cerevisiae GCIΔGLR1/ERV1 strain was streaked and grown on YPD solid media with Aba at 30 °C for 72 h. A single grown colony was inoculated into 5 ml of YPD liquid medium with Aba and aerobically grown at 30 °C for 18 h. The grown cells were inoculated into 20 ml of the same medium. The initial cell density (OD600) was 0.03, and cells were grown in a 200 ml baffled Erlenmeyer flask at 30 °C with agitation at 150 rpm for up to 48 h. The other mutant strains were grown by the same method. Intracellular GSH and GSSG were analyzed by HPLC as mentioned below. All fermentations for glutathione production were separately performed three times.
Analytical methods
To determine the cell concentration, the OD600 of the culture sample was measured using an UVmini-1240 Spectrometer (Shimadzu, Kyoto, Japan). Samples were prepared according to a previous report [19]. GSH and GSSG concentrations were determined by HPLC (Shimadzu) equipped with the YMC-Pack ODS-A column (YMC, Kyoto, Japan). The operating condition was 30 °C, with 50 mM potassium dihydrogen phosphate buffer (pH 2.8) and 10 mM sodium 1-heptanesulfonate as the mobile phase at a flow rate of 1.0 ml min−1, and detection was performed with a UV detector SPD-20A (Shimadzu) at 210 nm. Intracellular (reduced, oxidized, and total) glutathione content was calculated using the volumetric glutathione concentration (g l−1) divided by cell concentration (OD600 × 0.3) (g l−1), represented as % values (w/w).
Protein modeling
Protein structures and intramolecular interactions were simulated using PyMol software (https://www.pymol.org/). The structural data of the Erv1 variant protein of S. cerevisiae (PDB ID: 4E0I) was retrieved from RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do) and used for constructions of Erv1 and its variants structures.
Results
Catalytic activity of Erv1 and its variants
ERV1 and its mutants were successfully expressed in E. coli cells. The gene products were subsequently purified to homogeneity by metal affinity column chromatography. To evaluate their catalytic profiles, we first prepared and assayed Erv1 and its variants containing a replaced residue next to the catalytic cysteines (C30 and C33). Erv1S32A, Erv1S32T, and Erv1N34A oxidized GSH more efficiently than Erv1 (relative activity, 169, 178, and 201%, respectively) (Fig. 1a), whereas they oxidized γ-GC at comparable rates (relative activity, 119, 114, and 98%, respectively) (Fig. 1b). The other variants oxidized GSH and γ-GC at comparable or decreased rates (Fig. 1a, b). From the Erv1 protein structure, the side chain of N34 forms two hydrogen bonds to the main chain of D24, and the side chain of D24 also forms a hydrogen bond to the side chain of W132 located between C130 and C133, which are other catalytic cysteine residues (Fig. 2a, b) [23]. This fact suggests that these hydrogen bonds and the surrounding residues C130 and C133 play pivotal roles in the enzymatic oxidation of GSH. To investigate these implications, we additionally prepared and assayed Erv1D24A, Erv1P129A, Erv1N131A, and Erv1W132A, and these variants oxidized GSH and γ-GC at much lower rates (Fig. 1a, b) than did Erv1. Therefore, S32A, S32T, and N34A mutations are valuable for increasing Erv1 activity for GSH.

Relative activities of Erv1 and its variants. a GSH was used as a substrate; b γ-GC was used as a substrate. Erv1 and variants were assayed for 10 min at 30 °C and pH 7.0 (potassium phosphate buffer) using 10 mM substrates. The 100% relative activities of Erv1 using GSH and γ-GC as substrates were 1.62 and 1.59 mU mg−1, respectively. All assays were separately performed three times. Data are presented as the mean ± standard deviation (n = 3)

Structural models of the area surrounding catalytic cysteine residues. a Catalytic cysteine and its surrounding residues; b lateral view of the catalytic site; c, d surface around the active site. The structures were constructed from Erv1 variant protein of S. cerevisiae (PDB ID: 4E0I) by PyMOL software. Each subunit was separately rendered in green and pink. Oxygen, nitrogen, and sulfur atoms are rendered in red, blue, and yellow, respectively. Hydrogen bonds are shown as black dashed lines
To obtain effective Erv1 variants, we further prepared Erv1S32A/N34A and Erv1S32T/N34A, and assayed their specific activities. Erv1S32A/N34A showed the highest relative activity for GSH (240%) among all variants, and showed comparable activity for γ-GC (96%). On the other hand, Erv1S32T/N34A showed almost the same activity for GSH (192%) compared to Erv1S32A, Erv1S32T, and Erv1N34A, and high activity for γ-GC (161%).
Thermal and kinetic profiles of Erv1 and its variants
To investigate the effect of mutations on the Erv1 structure, thermal profiles of Erv1 and its variants that showed higher activities for GSH than parent Erv1 were assessed. Erv1, Erv1S32A, and Erv1S32T exhibited maximum activities at 60 °C (15.3, 22.9, and 27.8 mU mg−1, respectively) (Fig. 3a; Table 1). Erv1S32A and Erv1S32T were more activated by high temperature than parental Erv1. On the other hand, Erv1N34A, Erv1S32A/N34A, and Erv1S32T/N34A exhibited similar profiles to that of parental Erv1 with slightly decreased maximum temperatures at 55 °C (13.3, 16.5, and 15.8 mU mg−1, respectively) (Fig. 3b; Table 1). Thermostabilities of Erv1 and its variants were also assessed by incubating enzyme solutions for 1 h at each temperature before assaying. All tested variants showed obvious decreases in thermostability, whereas parental Erv1 was much more robust (Table 1). The variants containing N34A replacement showed lower thermostabilities than those of the variants containing S32 replacements (Table 1). These results suggest that both S32 and N34 residues greatly contribute not only to oxidations of GSH but also to thermostability of the enzyme.

Temperature effect on enzyme activity. a Specific activities of Erv1, Erv1S32A and Erv1S32T; b specific activities of Erv1N34A, Erv1S32A/N34A and Erv1S32T/N34A. Data are presented as the mean ± standard deviation (n = 3)
The kinetic profiles of Erv1 and its variants were also analyzed. All variants exhibited higher K m , V max , and K cat values than the parental enzyme (Table 1). The Erv1S32A and Erv1S32T containing only S32 replacement showed about 2.7- to 3.0- and 2.1- to 2.6-fold higher K m and V max values than those of the parental Erv1, respectively (Table 1). Furthermore, Erv1N34A, Erv1S32A/N34A and Erv1S32T/N34A containing N34A replacement showed about 7.6- to 9.4- and 4.3- to 7.6-fold higher K m and V max values than those of the parental enzyme, respectively (Table 1). These differences in thermal and kinetic profiles between variants containing S32 and/or N34 replacements suggest that effect of the N34A replacement to the Erv1 profile was greater than the S32 replacements.
Glutathione production by recombinant S. cerevisiae strains
To investigate the effects of mutant ERV1 genes for glutathione production by the S. cerevisiae GCIΔGLR1 strain, derivative strains expressing ERV1 and its mutant genes were constructed and grown. When the strains were grown for 24 h, GCIΔGLR1/ERV1 S32A produced much more intracellular GSSG (3.79%) and total glutathione (5.82%) than those of the other strains (Fig. 4b, c), whereas other derivative strains, especially GCIΔGLR1/ERV1, produced comparable GSH, GSSG and total glutathione to those of the vector control strain (Fig. 4). On the other hand, most of all strains expressing mutant ERV1 genes produced more intracellular GSSG and total glutathione than those of GCIΔGLR1/ERV1 at 48 h (Fig. 4b, c). Unlike the results of in vitro enzyme assays, GCIΔGLR1/ERV1 S32A produced the highest intracellular GSSG (5.30%) and total glutathione (6.57%) at 48 h (Fig. 4b, c). GCIΔGLR1/ERV1 S32T, GCIΔGLR1/ERV1 N34A, and GCIΔGLR1/ERV1 S32T/N34A produced only modestly high intracellular GSSG (4.13, 4.41, and 4.48%, respectively) and total glutathione (5.78, 6.02, and 6.20%, respectively) compared to GCIΔGLR1/ERV1 S32A (Fig. 4b, c). GCIΔGLR1/ERV1 S32A/N34A produced lower GSSG (3.32%) and total glutathione (5.26%) compared to GCIΔGLR1/ERV1, whereas the in vitro activity of its gene product for GSH was the highest among parental and other variant proteins (Figs. 1a, 4b, c). These variant strains also showed high volumetric GSSG and total glutathione production, though they showed slightly poor growth compared to the parental strain (Table 2). GCIΔGLR1/ERV1 S32T/N34A produced comparable volumetric total glutathione (105.7 mg l−1) to that of GCIΔGLR1/ERV1 S32A (105.5 mg l−1) due to their higher cell growth (1.71 g-cell l−1) than that of GCIΔGLR1/ERV1 S32A (1.61 g-cell l−1) (Table 2). However, GCIΔGLR1/ERV1 S32A also showed the highest volumetric GSSG (85.2 mg l−1) production among all tested strains (Table 2).

Glutathione production by S. cerevisiae GCIΔGLR1 strains over-expressing ERV1 and its mutant genes. a Intracellular GSH content; b intracellular GSSG content; c intracellular GSH and GSSG content; d intracellular GSSG ratio to GSH and GSSG. All fermentation for glutathione production were separately performed three times. Data are presented as the mean ± standard deviation (n = 3). Asterisks indicate statistical significances determined by Student’s t test. One asterisk indicates a p value smaller than 0.05 (p < 0.05). Two asterisks indicate a p value smaller than 0.01 (p < 0.01)
Discussion
The ERV1 gene of S. cerevisiae has been studied mainly for its physiological roles due to its essentiality for respiration and growth [24–26]. In a series of studies, the active residues, cofactor binding residues, and three-dimensional structures of Erv1 have been revealed [23, 27, 28]. In our previous study, we first focused on the ERV1 gene for biomaterial production, and revealed that over-expression of the ERV1 gene enhanced GSSG and total glutathione production in S. cerevisiae cells [15]. In this study, we also first improved Erv1 for biomaterial production and revealed that S32 and N34 residues are critical for oxidation of GSH and protein stability. The side chain of N34 forms two hydrogen bonds to the main chain of D24 located in the loop covering the catalytic cysteines (Fig. 2a, b). The deletion of these hydrogen bonds by the N34A replacement led to an enhancement of Erv1 catalytic activity for GSH (Fig. 1a), whereas the catalytic activity for γ-GC remained unchanged. This result suggests that this covering loop inhibits oxidation of GSH. This idea is also supported by the comparable relative activities of Erv1N34Q for GSH (114%), because the side chain of Q34 in PyMol simulations also forms hydrogen bonds to the main chain of D24, similar to the hydrogen bonds between N34 and D24 in parental Erv1 (data not shown). The comparable activities of Erv1N34A and Erv1N34Q for γ-GC (98 and 85%, respectively) also support this idea. Inhibition by the covering loop for γ-GC may be lower than for GSH because γ-GC, a precursor of GSH, is a smaller molecule and has similar properties. On the other hand, Erv1S32A and Erv1S32T also showed high oxidation activities for GSH (Fig. 1a), though no interaction was found between S32 and other residues. In addition, the thermostabilities of these variants were obviously lower than that of parental Erv1 (Table 1). These results imply that the S32 residue interacts with other residues through solvent or metal ions. Therefore, we searched for and found an incomplete three-dimensional structure of S. cerevisiae Erv1 with solvents (Protein ID: 3W4Y). In this structure, a water molecule is located between corresponding S32 and P85 residues, and the main chain of P85 forms a hydrogen bond to the water molecule (Fig. 5d). This implies that the side chain of S32 and main chain of P85 form a hydrogen bond through a water molecule. However, Erv1S32T also showed similar activity and thermostability profiles to Erv1S32A, though the side chain of T32 in Erv1S32T also contains a hydroxyl group like the S32 residue in parental Erv1. In PyMol simulations, steric hindrance when the methyl moeity of the side chain of T32 was facing outward from the active center was small due to its bulkiness (Fig. 5c). This suggest that the hydrogen bond through a water molecule between S32 and P85 residues in Erv1 was deleted by the S32A and S32T replacements, and that these deletions resulted in increased flexibility around the active center and reduced steric hindrance for GSH (Fig. 2c, d).

Lateral view of the area surrounding catalytic cysteine residues of Erv1 and its variants. a Erv1; b Erv1S32A; c Erv1S32T; d different rendering of Erv1 with solvents. The structures were constructed from Erv1 mutant protein of S. cerevisiae (PDB ID: 4E0I) (a–c) and the Erv1 core of S. cerevisiae (PDB ID: 3W4Y) (d) by PyMOL software. Each subunit was separately rendered in green and pink. Oxygen, nitrogen, and sulfur atoms are rendered in red, blue, and yellow, respectively. The hydrogen bond and water molecule are shown as a black dashed line and cyan sphere, respectively
The kinetic profiles of Erv1 and its variants provided interesting insights into mechanisms of GSH oxidation by Erv1 variants (Table 1). Generally, increasing of K m value indicates decreasing of substrate binding affinities. However, V max values of Erv1 variants were also increased, and the over-expressions of mutant genes coding these Erv1 variants practically increased GSSG production in S. cerevisiae cells. These facts imply that Erv1 variants became hard to be binding to GSH compared with parental Erv1, due to instabilizations around the catalytic center by amino acid replacements. However, rates of GSH oxidations and/or GSSG dissociations from Erv1 variants became fast by decreasing of inhibitory effects as mentioned above.
The over-expression of mutant ERV1 genes also increased GSSG and total glutathione production in S. cerevisiae cells as expected (Fig. 4b, c). These strains also showed higher volumetric GSSG and total glutathione production, whereas decreases in growth were observed in these recombinant strains (Table 2). In Table 2, the relationship between cell growth and GSH production is seemed to be proportional. This relationship is probably caused by reduced redox potentials of GSH in engineered strains. Generally, GSH works as a redox and antidotal agent in cells and therefore is an essential for various biological activities in all organisms [1]. Indeed, GSH1 or GSH2 deleted mutant S. cerevisiae strains lost their growth abilities in growth media without GSH [29], and genetically engineered S. cerevisiae strains that produce high concentration of GSH exhibited significant tolerance against diverse stresses such as high temperatures and presence of toxic agents [30, 31].
The in vivo GSSG production of the recombinant strains expressing ERV1 and its mutant genes was not necessarily reflected by in vitro enzyme activities (Figs. 1a, 4b). Generally, thiol oxidase oxidizes various substrates that are not only small molecules [27, 28], but also macromolecules such as mitochondrial intermembrane space (MIMS) proteins including Mia40, Cox19, and so on [24, 25]. Therefore, the Erv1 variants, especially Erv1S32A/N34A, may oxidize unexpected substrates and form disulfide bonds between GSH and thiols in other substrates in S. cerevisiae cells, due to a change in substrate specificity by the amino acid replacements. Indeed, the existence of unproductive oxidized MIMS proteins [25] and a wide variety of protein-glutathione adducts in yeast cells [32] have been reported in previous studies. Generally, Mia40 oxidizes thiols in proteins, and is consequently oxidized by Erv1 in the MIMS [26]. In this study, Erv1 protein was successfully improved for oxidation of GSH, and over-expression of its coding genes meaningfully increased GSSG production in S. cerevisiae cells. These results suggest that the Erv1 protein directly oxidized GSH in S. cerevisiae cells. The suppression of GSSG production by the simultaneous over-expression of ERV1 and MIA40 genes in our previous study [15] and the much lower oxidation activity of Mia40 for GSH [25] also support this idea.
GSSG and total glutathione production of S. cerevisiae were increased by over-expressing improved ERV1 genes. However, glutathione has been produced by industrial fermentation methods using high glutathione producing strains such as Candida utilis and S. cerevisiae, and their glutathione productions have been improved by screening from randomly mutated strains and metabolic engineering. These improved strains produce much higher amount of glutathione (e.g., S. cerevisiae K-2 strain produces 2700 mg l−1 glutathione for 24 h) than the host strain used in this study [3]. However, in many cases for improving glutathione production by metabolic engineering, genes involved in synthesis of GSH and its precursors, such as GSH1, PRO1, and CYS3 are frequently focused [19, 30, 33]. On contrast, ERV1 has never been applied for industrial glutathione production, and therefore Erv1 has even greater potential for industrial GSSG production when ERV1 and its mutant genes were overexpressed in the industrial glutathione producing S. cerevisiae host strain.
Conclusions
In this study, mitochondrial thiol oxidase Erv1 was applied for improvement of GSSG production by S. cerevisiae, and its enzyme activity for GSH oxidation was improved for the first time by site-directed mutations. The critical roles of S32 and N34 residues for GSH oxidation and protein stability were revealed with possible reasons. Five engineered Erv1 variants containing S32 and/or N34 replacements exhibited about 1.7- to 2.4-fold higher in vitro GSH oxidation activity than that of the parental Erv1. The over-expression of mutant ERV1 genes coding these variants also demonstrated in vivo validity, showing 1.5-fold higher GSSG production than that of the strain over-expressing ERV1 gene. This study indicates potential of Erv1 for high GSSG production by S. cerevisiae.
Abbreviations
- Aba A:
-
aureobasidin A
- Amp:
-
ampicillin
- cDNA:
-
complementary deoxyribonucleic acid
- Cm:
-
chloramphenicol
- DNA:
-
deoxyribonucleic acid
- GCS:
-
γ-glutamylcysteine synthetase
- GR:
-
glutathione-disulfide reductase
- GS:
-
glutathione synthetase
- GSH:
-
reduced glutathione
- GSSG:
-
oxidized glutathione
- HPLC:
-
high performance liquid chromatography
- LB:
-
Luria–Bertani
- MIMS:
-
mitochondrial intermembrane space
- PCR:
-
polymerase chain reaction
- RNA:
-
ribonucleic acid
- TO:
-
thiol oxidase
- YPD:
-
yeast extract-peptonedextrose
- γ-GC:
-
γ-glutamylcysteine
References
Meister A, Andersen ME. Glutathione. Annu Rev Biochem. 1983;52:711–60.
Arjinpathana N, Asawanonda P. Glutathione as an oral whitening agent: a randomized, double-blind, placebo-controlled study. J Dermatolog Treat. 2012;23:97–102.
Li Y, Wei G, Chen J. Glutathione: a review on biotechnological production. Appl Microbiol Biotechnol. 2004;66:233–42.
DrögeW Breitkreutz R. Glutathione and immune function. Proc Nutr Soc. 2000;59:595–600.
Penninckx MJ. An overview on glutathione in Saccharomyces versus non-conventional yeasts. FEMS Yeast Res. 2002;2:295–305.
Vartanyan LS, Gurevich S, Kozachenko AI, Nagler LG, Lozovskaya EL, Burlakova EB. Changes in superoxide production rate and in superoxide dismutase and glutathione peroxidase activities in subcellular organelles in mouse liver under exposure to low doses of low-intensity radiation. Biochem (Mosc). 2000;65:442–6.
Ray S, Watkins DN, Misso NL, Thompson PJ. Oxidant stress induces gamma-glutamylcysteine synthetase and glutathione synthesis in human bronchial epithelial NCI-H292 cells. Clin Exp Allergy. 2002;32:571–7.
Rolseth V, Djurhuus R, Svardal AM. Additive toxicity of limonene and 50% oxygen and the role of glutathione in detoxification in human lung cells. Toxicology. 2002;170:75–88.
Singh RJ. Glutathione: a marker and antioxidant for aging. J Lab Clin Med. 2002;140:380–1.
Izawa S, Inoue Y, Kimura A. Oxidative stress response in yeast: effect of glutathione on adaptation to hydrogen peroxide stress in Saccharomyces cerevisiae. FEBS Lett. 1995;368:73–6.
Santos LO, Gonzales TA, Ubeda BT. Monte Alegre R. Influence of culture conditions on glutathione production by Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2007;77:763–9.
Wang M, Sun J, Xue F, Shang F, Wang Z, Tan T. The effect of intracellular amino acids on GSH production by high-cell-density cultivation of Saccharomyces cerevisiae. Appl Biochem Biotechnol. 2012;168:198–205.
Fei L, Wang Y, Chen S. Improved glutathione production by gene expression in Pichia pastoris. Bioprocess Biosyst Eng. 2009;32:729–35.
Kiriyama K, Hara KY, Kondo A. Oxidized glutathione fermentation using Saccharomyces cerevisiae engineered for glutathione metabolism. Appl Microbiol Biotechnol. 2013;97:7399–404.
Hara KY, Aoki N, Kobayashi J, Kiriyama K, Nishida K, Araki M, Kondo A. Improvement of oxidized glutathione fermentation by thiol redox metabolism engineering in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2015;99:9771–8.
Sugimura Y, Yamamoto K. Effect of orally administered reduced and oxidized-glutathione against acetaminophen-induced liver injury in rats. J Nutr Sci Vitaminol (Tokyo). 1998;44:613–24.
Roxas VP, Smith RK Jr, Allen ER, Allen RD. Overexpression of glutathione S-transferase/glutathione peroxidase enhances the growth of transgenic tobacco seedlings during stress. Nat Biotechnol. 1997;15:988–91.
Belmonte MF, Yeung EC. The effects of reduced and oxidized glutathione on white spruce somatic embryogenesis. In Vitro Cell Dev Biol Plant. 2004;40:61–6.
Hara KY, Kiriyama K, Inagaki A, Nakayama H, Kondo A. Improvement of glutathione production by metabolic engineering the sulfate assimilation pathway of Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2012;94:1313–9.
Ishii J, Izawa K, Matsumura S, Wakamura K, Tanino T, Tanaka T, Ogino C, Fukuda H, Kondo A. A simple and immediate method for simultaneously evaluating expression level and plasmid maintenance in yeast. J Biochem. 2009;145:701–8.
Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153:163–8.
Chen DC, Yang BC, Kuo TT. One-step transformation of yeast in stationary phase. Curr Genet. 1992;21:83–4.
Guo PC, Ma JD, Jiang YL, Wang SJ, Bao ZZ, Yu XJ, Chen Y, Zhou CZ. Structure of yeast sulfhydryl oxidase Erv1 reveals electron transfer of the disulfide relay system in the mitochondrial intermembrane space. J Biol Chem. 2012;287:34961–9.
Allen S, Balabanidou V, Sideris DP, Lisowsky T, Tokatlidis K. Erv1 mediates the Mia40-dependent protein import pathway and provides a functional link to the respiratory chain by shuttling electrons to cytochrome c. J Mol Biol. 2005;353:937–44.
Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol Cell. 2010;37:516–28.
Böttinger L, Gornicka A, Czerwik T, Bragoszewski P, Loniewska-Lwowska A, Schulze-Specking A, Truscott KN, Guiard B, Milenkovic D, Chacinska A. In vivo evidence for cooperation of Mia40 and Erv1 in the oxidation of mitochondrial proteins. Mol Biol Cell. 2012;23:3957–69.
Hofhaus G, Lee JE, Tews I, Rosenberg B, Lisowsky T. The N-terminal cysteine pair of yeast sulfhydryl oxidase Erv1p is essential for in vivo activity and interacts with the primary redox centre. Eur J Biochem. 2003;270:1528–35.
Ang SK, Lu H. Deciphering structural and functional roles of individual disulfide bonds of the mitochondrial sulfhydryl oxidase Erv1p. J Biol Chem. 2009;284:28754–61.
Zechmann B, Liou LC, Koffler BE, Horvat L, Tomašić A, Fulgosi H, Zhang Z. Subcellular distribution of glutathione and its dynamic changes under oxidative stress in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2011;11:631–42.
Ask M, Mapelli V, Höck H, Olsson L, Bettiga M. Engineering glutathione biosynthesis of Saccharomyces cerevisiae increases robustness to inhibitors in pretreated lignocellulosic materials. Microb Cell Fact. 2013;12:87.
Qiu Z, Deng Z, Tan H, Zhou S, Cao L. Engineering the robustness of Saccharomyces cerevisiae by introducing bifunctional glutathione synthase gene. J Ind Microbiol Biotechnol. 2015;42:537–42.
Bulteau AL, Planamente S, Jornea L, Dur A, Lesuisse E, Camadro JM, Auchère F. Changes in mitochondrial glutathione levels and protein thiol oxidation in ∆yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim Biophys Acta. 2011;1822:212–25.
Tang L, Wang W, Zhou W, Cheng K, Yang Y, Liu M, Cheng K, Wang W. Three-pathway combination for glutathione biosynthesis in Saccharomyces cerevisiae. Microb Cell Fact. 2015;14:139.
Authors’ contributions
JK performed all experiments and wrote the manuscript. KYH planned the study. DS and KYH revised the manuscript. DS, KYH, TH and AK supervised the study. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Dr. J. Ishii (Organization of Technology and Innovation, Kobe University) for providing us with the pGK plasmid series.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The figures and tables supporting the conclusions of this study are included in this article. The primer sequences are listed in the Additional file 1.
Consent for publication
All authors consented to publication this paper.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Funding
This work was supported by a Special Coordination Fund for Promoting Science and Technology, Creation of Innovative Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction, Kobe) from the Ministry of Education, Culture, Sports, and Science and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Additional file
12934_2017_658_MOESM1_ESM.xlsx
Additional file 1: Table S1. Primer sequences, restriction sites, and DNA templates used for plasmid constructions in this study are summarized.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kobayashi, J., Sasaki, D., Hara, K.Y. et al. Enzymatic improvement of mitochondrial thiol oxidase Erv1 for oxidized glutathione fermentation by Saccharomyces cerevisiae . Microb Cell Fact 16, 44 (2017). https://doi.org/10.1186/s12934-017-0658-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-017-0658-0